
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAddressing a future pandemic: how can non-biological complex drugs prepare us for antimicrobial resistance threats?
Lewis D.
Blackman
 *a,
Tara D.
Sutherland
b,
Paul J.
De Barro
c,
Helmut
Thissen
a and
Katherine E. S.
Locock
*a
*a,
Tara D.
Sutherland
b,
Paul J.
De Barro
c,
Helmut
Thissen
a and
Katherine E. S.
Locock
*a
aCSIRO Manufacturing, Research Way, Clayton, VIC 3168, Australia. E-mail: Lewis.Blackman@csiro.au; Katherine.Locock@csiro.au
bCSIRO Health & Biosecurity, Clunies Ross Street, Black Mountain, ACT 2601, Australia
cCSIRO Health & Biosecurity, Boggo Road, Dutton Park, QLD 4102, Australia
First published on 15th June 2022
Abstract
Loss of effective antibiotics through antimicrobial resistance (AMR) is one of the greatest threats to human health. By 2050, the annual death rate resulting from AMR infections is predicted to have climbed from 1.27 million per annum in 2019, up to 10 million per annum. It is therefore imperative to preserve the effectiveness of both existing and future antibiotics, such that they continue to save lives. One way to conserve the use of existing antibiotics and build further contingency against resistant strains is to develop alternatives. Non-biological complex drugs (NBCDs) are an emerging class of therapeutics that show multi-mechanistic antimicrobial activity and hold great promise as next generation antimicrobial agents. We critically outline the focal advancements for each key material class, including antimicrobial polymer materials, carbon nanomaterials, and inorganic nanomaterials, and highlight the potential for the development of antimicrobial resistance against each class. Finally, we outline remaining challenges for their clinical translation, including the need for specific regulatory pathways to be established in order to allow for more efficient clinical approval and adoption of these new technologies.
 Lewis D. Blackman | Dr Lewis Blackman leads the Biomedical Scaffolds Team within CSIRO Biomedical Manufacturing. Lewis joined CSIRO in 2017, initially as a CERC Postdoctoral Fellow, after obtaining an MChem from the University of Southampton (2012) and a PhD in polymer chemistry from the University of Warwick (awarded in 2018). Lewis was promoted to Research Scientist (2021), and later to Team Leader (2022), where he leads a portfolio of commercial and academic projects. Lewis’ interests include advanced polymeric and hybrid materials, spanning antimicrobial materials and other bioactive materials, drug delivery systems, and medical devices for a broad range of biomedical applications. |
 Tara D. Sutherland | Dr Tara Sutherland is the Science Lead for CSIRO's AMR Mission In-Development. Tara is a biotechnologist with expertise in the areas of AMR, diagnostics, biological materials, and biological advanced materials development with extensive experience in early stage product development, multi- and trans-disciplinary projects, assessing end user needs, and commercial engagement. She led CSIRO's discovery program for bio-manufactured advanced material templates, which successfully produced silk proteins at commercial scale. Her current research includes developing point of care devices for improved antimicrobial stewardship and a One Health AMR surveillance system in Fiji. She has co-authored over 80 journal publications and holds 14 patents, with an h-index of 30. |
 Paul De Barro | Dr Paul De Barro joined CSIRO in 1994 where he is a Senior Principal Research Scientist. An ecologist by training, Paul is a world authority on the globally important pest, Bemisia tabaci and recipient of the CSIRO Chairman's Medal for his research. He has led innovative multidisciplinary research in biosecurity science and technology since 2010 and in recent years has focused on human biosecurity. As part of this he has helped drive the Australian National Research Agency's focus on the threat posed by AMR to both people and agriculture. |
 Helmut Thissen | Dr Helmut Thissen leads a team at CSIRO Manufacturing in Melbourne, Australia that is focused on new and improved biomedical devices, and here in particular the effective control of biointerfacial interactions. He joined the premier Australian government research organisation after completing his PhD at RWTH Aachen University in Germany, where he also started to translate biomedical research into the clinic. While he has published more than 175 journal articles and book chapters, his strong translational focus is reflected by his role as the Director of CSIRO's Biomedical Materials Translational Facility (BMTF) and his contributions to multiple successful biomedical products. |
 Katherine E. S. Locock | Dr Katherine Locock leads the Drug Discovery Chemistry Team in the Manufacturing Business Unit of the CSIRO in Melbourne, Australia. Her research focuses on the development of biologically active polymers, with a particular focus on antimicrobial therapeutics. Prior to the CSIRO, Katherine held a position as an Associate Lecturer in Pharmacology at the University of Sydney, where undertook rational drug design to develop analogues to target memory and mood. In recognition of her contributions to the field she was elected as a Fellow of the Royal Australian Chemical Institute (2021) and AIPS Victorian Young Tall Poppy of the Year (2016). |
1. Introduction
Antibiotics are rapidly losing their efficacy against the majority of our most deadly and infectious bacterial and fungal pathogens.1,2 Since the dawn of modern antibiotics, with the discovery of penicillin in the 1920s, antimicrobial resistance (AMR) has been observed towards every class of antibiotics to be discovered.1 Indeed, AMR is an ancient phenomenon and microbial communities are preconditioned to become resistant to most conventional antibiotics through millennia of evolution.3,4 Recent overuse and misuse of antibiotics has seen an unprecedented rise in both the frequency and diversity of drug-resistant, multidrug-resistant, extensively drug-resistant, and pandrug-resistant strains.5,6 Looking ahead, we are now on track to see AMR become our next evermore significant pandemic, defined as the spread of an infectious disease across multiple continents. Among other factors, the ready availability of antibiotics and poor antibiotic use regulation have been identified as key contributing factors for driving AMR.6 Our ability to manage AMR risks is limited by lack of knowledge and poor surveillance.6,7 Furthermore, AMR is not only limited to the health domain; it is also closely connected to the agricultural and environmental domains, calling for a One Health approach for tackling the spread of AMR.7,8 The World Health Organization anticipates that AMR infections alone will lead to over 10 million human deaths annually by 2050,2 likely overtaking the cumulative mortality rate of all cancers combined. Additionally, in contrast to more deadly non-infectious diseases such as ischaemic heart disease and stroke, the threat of untreatable infectious diseases is arguably far greater threat to our social fabric. As evidenced by the current global viral pandemic, the risk of communicable transmission of infectious agents associated with social contact and human mobility forces the restriction of social interactions, human migration, and trade, which have drastic socioeconomic impacts. Therefore, effectively managing AMR threats now and into the future will be critical for preserving not only human lives, but also livelihoods and wellbeing in our modern interconnected world.Materials scientists are at the forefront of this challenge and will have a significant role to play in the development of intervention and monitoring technologies for tackling AMR. Three key areas in which advances in materials science are already showing great promise are in new therapies for the elimination of resistant pathogens, new coating technologies that destroy and prevent the formation of microbial biofilms, and sensing technologies for the diagnosis, monitoring, and surveillance of AMR. For example, materials intervention technologies include incorporation of antibiotics into coatings and devices to achieve localized and sustained delivery, as well as textured or patterned surfaces that topologically resist biofilm formation.9–11
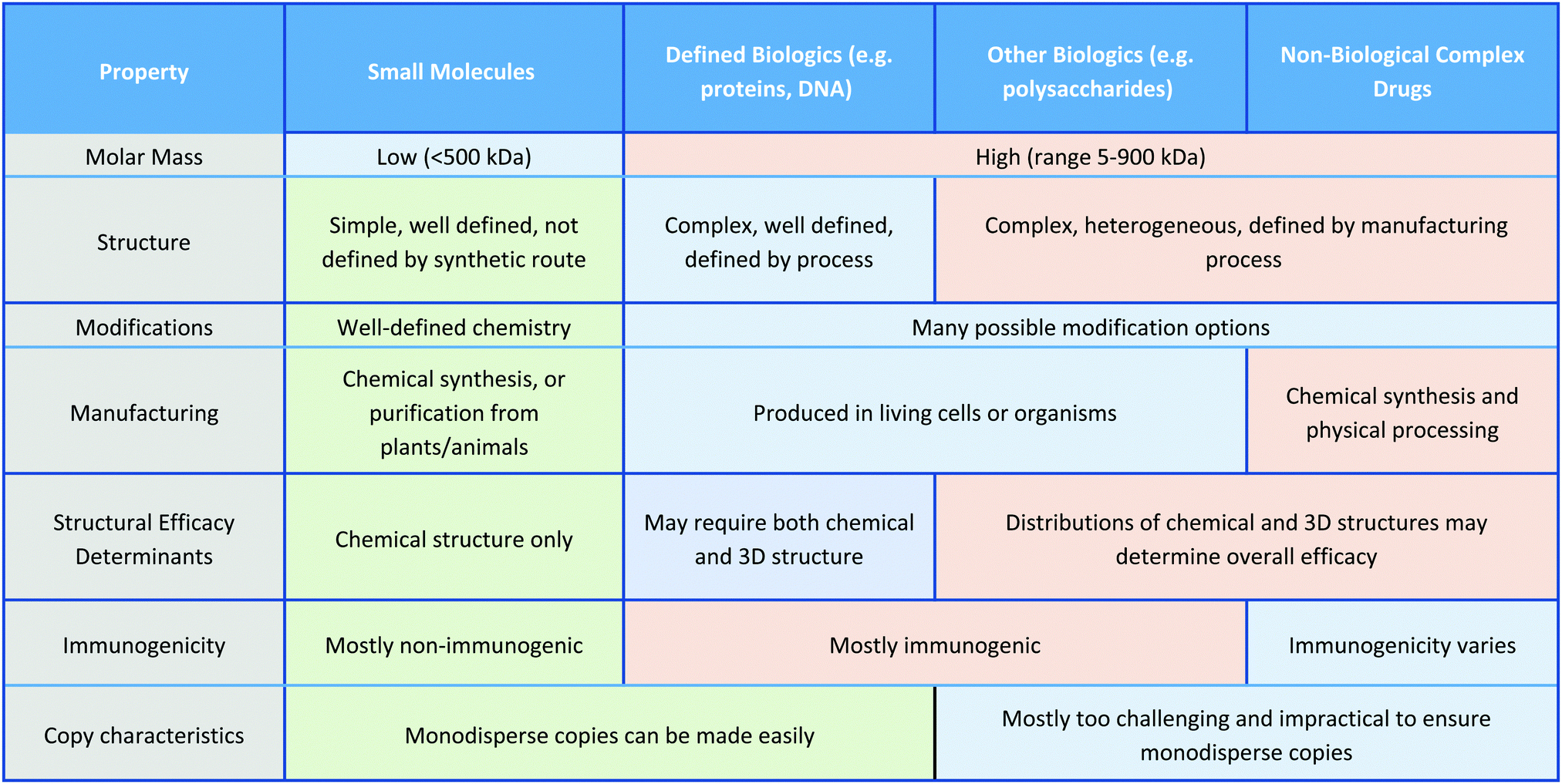
In this Review, we exclusively focus on solution-based therapeutic and disinfectant materials that are in development, which fall under the category of non-biological complex drugs (NBCDs). NBCDs are complex non-biologically derived drugs comprising high molar mass and/or nanoparticulate structures.12 Unlike small molecules, NBCDs are ill-defined and cannot be easily characterized or described by conventional physiochemical means. For example, these include polymers and polymeric nanoparticles, inorganic nanomaterials, and carbon-based nanomaterials, as well as hybrid materials comprized of two or more different NBCD classes. They may also contain small molecules/biologics through conjugation or loading within the structure. The broad range of antimicrobial NBCD materials discussed as part of this Review have far more complex structures than small molecule drugs. They are typically heterogeneous and difficult to fully characterize or precisely define by physiochemical means. Even simple homopolymer samples cannot be considered homo-molecular, as they contain a broad distribution of different polymer chain lengths and may contain different end group functionalities between chains, which can impact their solution behavior.13 Similar to biologic drugs that are derived from living organisms and are dependent on the host organism or expression system employed, the activity profile of these complex materials is highly dependent on the manufacturing process utilized. They contrast with biologics though, given they are derived from synthetic processes and typically do not have precise or sequence-defined structures. To reflect the unique profile of these complex materials (Table 1)14 the term NBCD was coined12 and appears to have gained traction. Note that whilst biologics can also include highly complex agents such as whole blood, cells, and tissues, for the purposes of this comparative discussion throughout this article, we only include isolated proteins, DNA/RNA, and polysaccharides, as biologic drugs that more closely resemble both NBCDs and small molecules.
|
While the novel properties of NBCDs underly their unique mechanisms of action, it is these same properties that can hinder their regulatory approval. Herein, we describe each NBCD material's attributes and challenges, including opportunities for development of AMR towards these agents, before discussing alternatives to existing regulatory pathways, which could occur to allow these materials to reach the market more effectively.
2. Resistance, AMR, tolerance, and persistence
Organisms can be intrinsically resistant to an antibiotic (natural antibiotic resistance), can become resistant to an antibiotic in certain environmental situations e.g. in biofilms (situational resistance), or organisms can acquire resistance through genetic modification (antimicrobial resistance, AMR). AMR arises if an organism obtains genes from other species (genetic transfer) or by accumulating mutations in their genome, see Fig. 1.16,17 There are a number of distinct mechanisms that confer AMR to organisms. The organism may contain modified antibiotic drug targets such that vital microbial processes targeted by the antibiotic can continue. Examples include modification of the penicillin-binding proteins responsible for peptidoglycan synthesis, which imparts resistance towards β-lactams; ribosomal mutation and/or methylation to impart resistance towards ribosome-targeting aminoglycosides, oxazolidinones, and streptogramins; and modifications in the active sites of enzymes responsible for metabolic processes, such as to impart resistance towards antibiotics that act as enzyme inhibitors.16,17 Organisms may upregulate enzymes that directly inactivate antibiotics, most notably exemplified by β-lactamase expression in pathogens resistant to the β-lactam class of antibiotics,18 encompassing penicillin derivatives, cephalosporins, monobactams, carbapenems, and carbacephems. The organism may produce drug efflux pumps16,19 to facilitate the removal of antibiotics from the microbial cell, thereby preventing them from reaching their intended drug target. Finally, the thickening and modification of barriers such as the cell wall, thereby limiting drug permeability, has also notably been observed in Staphyloccocus aureus resistance towards vancomycin.20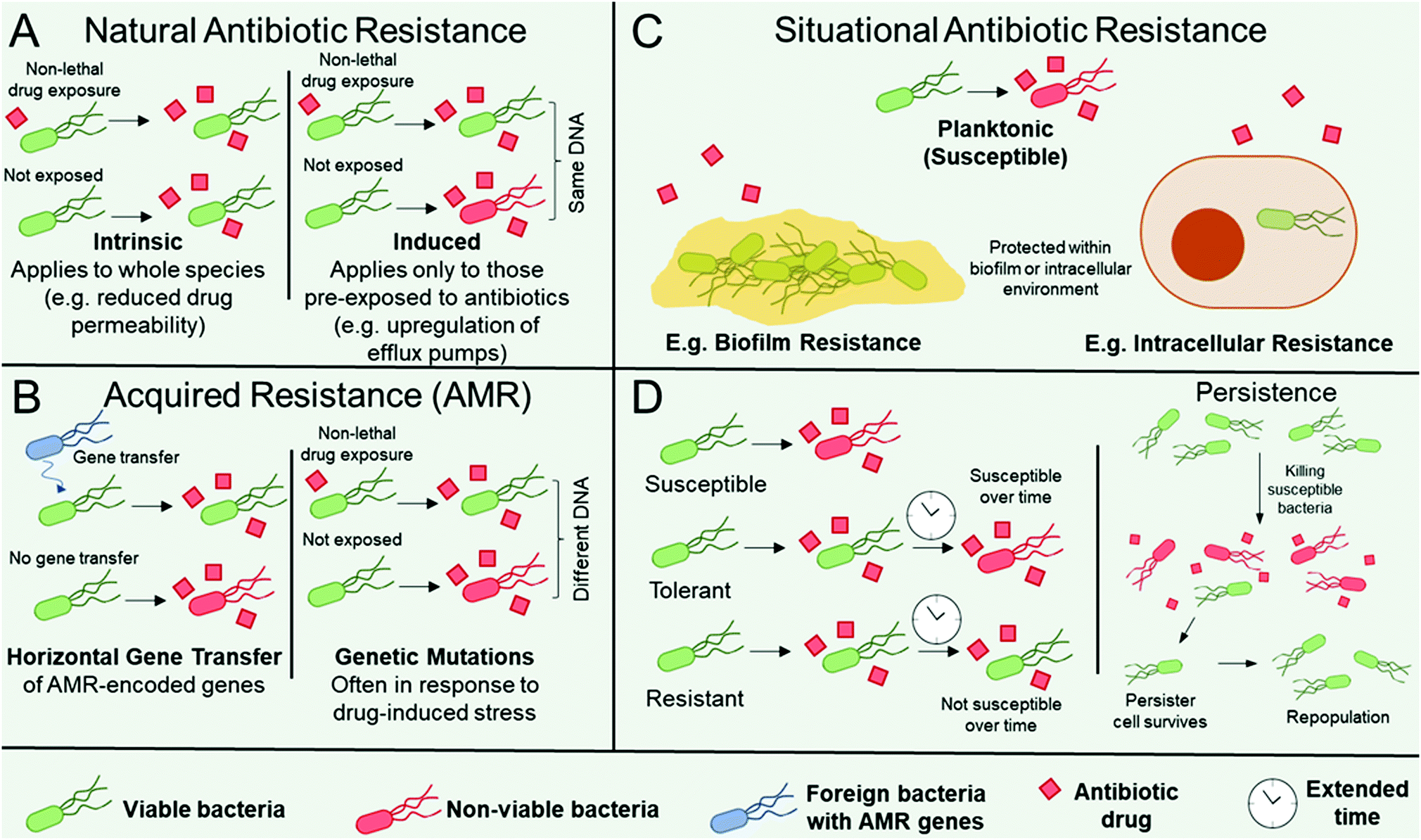 | ||
| Fig. 1 Illustrative outline of various ways that an organism can be resistant to an antibiotic (A–C) and the concept of tolerance and persistence. (A) Schematic showing different characteristics of natural resistance including intrinsic resistance and induced resistance. (B) Illustration showing genetic characteristics of AMR including horizontal gene transfer and genetic mutations. (C) Schematic showing examples of situational antibiotic resistance. (D) Schematic of the differences between resistance, tolerance, and persistence. Key: alive (green) and dead (red) bacteria are shown, as well as a foreign resistant (sub)species (blue), in the presence of an antibiotic (red squares). | ||
Often when AMR occurs as a result of genetic transfer from one microbe to another, the resistance genes accumulate on extracellular DNA (plasmids) that encode resistance to other classes of antibiotics, heavy metals, or biocides. In this instance, co-selection of AMR can occur through selection pressure from any of the other genes on the plasmid.21
Microbes can also become resistant to an antibiotic through changes in their immediate environment (i.e. situationally resistant, Fig. 1C). In this instance, the resistance is only observed when the microbe is within that environment, for example within intracellular environments, where the pathogen may be imparted temporary resistance by virtue of its infiltration within the host cell.22 A further example is when microbes are encapsulated within biofilms, which are surface-adhered communities each surrounded by a self-secreted matrix of extracellular polymeric substances (EPS).23,24 Microbes within biofilms are typically significantly more resistant to antibiotics than when in their planktonic form, owing to the additional protection imparted by the EPS and neighbouring microorganisms (Fig. 1C).24 Note that situational resistance is not AMR as no genetic changes have occurred and the resistance is only transient based on the environment of the microbe. The distinction between these three forms of resistance (natural, acquired, and situational) is important when discussing new therapeutics, as the mechanisms by which pathogens avoid destruction, as well as their ability to gain and pass on their resistance, must be considered in order to design effective antimicrobial materials. Although described in terms of resistance, whether the resistance is intrinsic, as a result of AMR, or through changes in the microbes’ environment, for all of the classes above a distinction can be made between the microbes’ tolerance, resistance, and persistence towards antimicrobial treatments. Higher resistance relates to a lower effectiveness of a given antibiotic (i.e. higher minimum inhibitory and biocidal concentrations), whereas higher tolerance relates to a longer required drug exposure time (Fig. 1D, LHS).25 Therefore, tolerant and non-tolerant strains can be equally susceptible to an antibiotic at the same minimum inhibitory concentration, but the tolerant strain will require a longer exposure for biocidal activity at a given concentration (Fig. 1D, LHS). This is an important consideration for antibiotics that rely on metabolic or growth processes, because species that transiently exhibit slow or interrupted growth, for example in their lag or stationary phase, will exhibit tolerance towards such antimicrobial mechanisms.25 Furthermore, biofilms display both antimicrobial resistance and tolerance.26 Interplay between resistance and tolerance is also of particular importance when designing combination therapy strategies.27
Persistence occurs when a small subset of a drug-susceptible microbial cohort survives treatment with high levels of antibiotics, despite being clonal and absent of any additional resistance mechanism (Fig. 1D, RHS).25,28 The surviving persister cells can subsequently repopulate the cohort to yield a clonal, drug-susceptible population. Although still debated,28 persister cells have been attributed to having dormant or low-level cellular processes, which aids their survival, however evidence is emerging that suggests that offspring of persister cells have greater susceptibility for genetic mutations and can contain greater subpopulations of antibiotic-resistant mutants than those from untreated controls.29
High priority species that have developed AMR are the ESKAPE pathogens, encompassing Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp.30 However, situational resistance mechanisms such as biofilm formation can be near-universally adopted by all microbial species. Therefore, whilst situational resistance mechanisms are of concern in healthcare settings, it is the rising occurrence of AMR observed in ESKAPE pathogens that is of the most concern. However, situational mechanisms provide a protective platform for greater survival of AMR pathogens and facilitate propagation of AMR genes through less effective antibiotic exposure. Therefore, we propose that a systematic approach for targeting both forms of resistance is imperative for curtailing the rise of AMR more generally.
Finally, although less studied, AMR amongst fungal pathogens is increasingly recognized as a key emerging threat for human health.31,32 Drawing parallels with bacterial AMR, fungal AMR is driven by factors such as nosocomial outbreak transmission, which aids the spread of fungal AMR, and the use of subtherapeutic concentrations or inadequate exposure of antifungal drugs, including large scale use of fungicides in agriculture.32 However, key differences include the fact that fungi have higher fidelity species boundaries, meaning there is currently limited evidence for horizontal gene transfer, and homologous recombination dominates as the primary mechanism. This means that AMR genes must generate individually for each species-drug combination, slowing the global rate of AMR development amongst different species, and there is currently no evidence supporting a pan-kingdom fungal “resistome”.32 Despite these differences, fungal AMR infections are estimated by the CDC to hospitalize 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 patients per year and are an increasing threat in the wake of the COVID-19 pandemic, where SARS-CoV-2 infection has been observed to be a comorbidity or risk factor for resistant fungal infections.33,34
000 patients per year and are an increasing threat in the wake of the COVID-19 pandemic, where SARS-CoV-2 infection has been observed to be a comorbidity or risk factor for resistant fungal infections.33,34
3. New antimicrobial NBCDs as therapeutics and biocides
Alternatives to conventional small molecule antibiotics are urgently required. New antimicrobial chemistry that exhibits similar mechanisms of action to previously designed antibiotics drives selection of resistance in pathogen populations through existing AMR mechanisms. Therefore, compounds or materials with a broad suite of antimicrobial mechanisms acting synergistically, or those that employ entirely new antimicrobial mechanisms, are critically required. The knowledge that pathogens can readily achieve resistance to small molecule antibiotics is driving strong healthcare policy to protect new antibiotics. The consequence of this is that return on investments are not sufficient to drive discovery and development of new antibiotics, which we will not discuss here as there have been several detailed texts and recent commentaries on this topic.35–39As an alternative to small molecule antibiotics, there is growing interest in the use of unconventional antimicrobial materials,40–51 including, for example, natural and synthetic peptides,52–54 antimicrobial enzymes,55 and bacteriophages,56–58 as well as lipid-based antibiotic delivery vehicles.59 In this paper we focus on specific subsets of these unconventional antimicrobial materials: polymeric compounds, carbon-based nanomaterials, and inorganic nanomaterials, each of which are discussed below.
3.1 Antimicrobial polymers and polymer nanoparticles
3.1.1.1 Polycation mechanisms and modes of action. Polycations primarily exhibit similar antimicrobial function to antimicrobial peptides, initially interacting with the bacteria via electrostatic interaction with anionic microbial membrane components, followed by facilitating cell lysis by disrupting the cell membranes (Fig. 2B). Although bacteriolysis has been understood to be the main mechanism for most materials in this class, some examples have been observed where no membrane lysis occurs66,73 indicating that other mechanisms are also possible. Furthermore, such polycations also show promise against fungal pathogens, for example cationic RAFT polymers were found to be effective against fungal biofilms,70 as well as cross-kingdom fungal/bacterial mixed biofilms,71 outperforming conventional antifungals in each case.
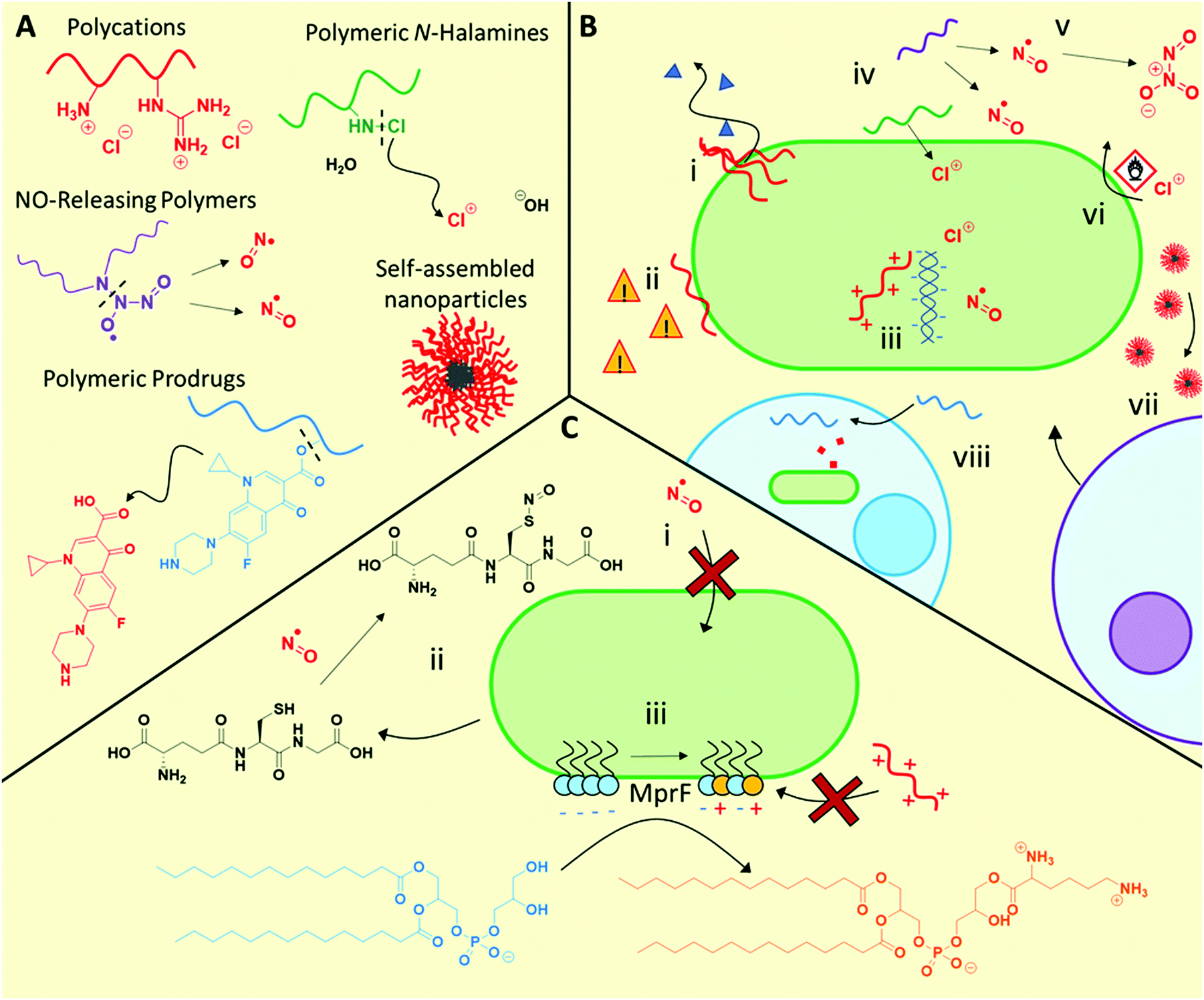 | ||
| Fig. 2 Structures and mechanisms of action of antimicrobial polymeric materials, as well as some described associated resistance mechanisms. (A) General structures and functions of polymeric antimicrobial materials. (B) Possible antimicrobial mechanisms of action including bacteriolysis through membrane disruption (i), leading to downstream stress signalling (ii), interaction of antimicrobial species with intracellular targets such as DNA and functional proteins (iii), release of antimicrobial compounds such a NO, oxidative halogen species, or conventional antibiotics (iv), some of which can often be converted into higher oxidation biocidal species (v), oxidative damage to membranes or essential biomolecules (vi), recruitment of host lymphocytes (vii) and mammalian cell entry to treat intracellular infections (viii). (C) Resistance mechanisms include modification/inactivation of trafficking systems to prevent biocide entry (i), inactivation of biocidal species using small molecule scavengers (ii), and modification of membrane lipid composition to reduce its interactions with polycations (iii). | ||
New elucidation of the complex and synergistical mechanisms for polycation antimicrobial activity has recently been examined.67,74 Employing super-resolution fluorescence microscopy, Locock and co-workers observed significant uptake of fluorescently tagged polycations containing either amine or guanidine side chain moieties, in contrast to neutral hydrophilic controls, into S. epidermidis.74 Using selective viability staining techniques, the authors deduced that in addition to disrupting bacterial membranes and causing leakage of ATP, the polymers also had possible intracellular targets such as nucleic acids, which led to their antimicrobial activity.74
These observations are broadly in agreement with those reported by Reynolds and Qiao et al. for E. coli treated with star-like cationic poly(peptide)s.67 The authors found that membrane association and disruption was a key antimicrobial mechanism, with some evidence of intracellular entry. The antimicrobial activity of these agents could be tapered by the addition of lipopolysaccharides, demonstrating the strong association between the star-like polymers and bacterial membrane components. In an in vivo model, the authors demonstrated that these synthetic polymers could also elicit neutrophil recruitment to the infection site of colistin-resistant and multidrug resistant (CMDR) A. baumannii infected mice, thereby providing evidence for both direct (bacteriolytic) and indirect (neutrophil recruitment) antimicrobial mechanisms.63
3.1.1.2 Development of AMR against polycations. To date, a number of studies have demonstrated that the acquisition of resistance towards such polycationic agents is negligible even after repeated exposure over many generations at sublethal concentrations.67,68,75 For example, the minimum bactericidal concentration (MBC) of the aforementioned star-like cationic polypeptides did not significantly rise even after the exposure of 600 bacterial generations to sublethal polymer concentrations.67 Similarly, Yan, Hedrick and Wu et al. found that sublethal exposure of A. baumannii to guanidinium-functional polycarbonates did not induce a change in minimum inhibitory concentration (MIC) after 30 passages.75 Furthermore, unlike for the imipenem β-lactam control, repeated exposure to these polymers did not induce a transcriptional response to known resistance pathways, and polymer-exposed bacteria showed significantly fewer differentially regulated genes than the antibiotic control.75
3.1.2.1 NO-Releasing materials mechanism and modes of action. NO exhibits a range of antimicrobial mechanisms. As a free radical compound, •NO reacts readily with proteins, nucleic acids, lipids, and other cell components, interfering with metabolic pathways.78 Furthermore, NO can react with endogenous species to form higher oxidation compounds such as S-nitrosothiols (RSNO, such as S-nitrosoglutathione), nitrogen dioxide (•NO2), and dinitrogen trioxide (N2O3), amongst others.84 The interplay between these numerous chemical species and the multiple drug targets with which they can interfere creates a complex and broad suite of antimicrobial mechanisms. However, whilst effective at high doses, low levels of NO are not biocidal, and can even give rise to increased bacterial protection against conventional antibiotics.85
3.1.2.2 Development of AMR against NO-releasing materials. NO is a gasotransmitter synthesized endogenously by many bacteria, and consequently microbial mechanisms exist to manage exposure to this molecule. Therefore, it is feasible that microbes could develop mechanisms to overcome higher, toxic levels of NO. Natural resistance mechanisms against NO include the production of scavenging thiols such as glutathione (and possibly other small molar mass thiols such as mycothiol and trypanothione), which sequester the reactive nitrogen species.84 For example, Salmonella typhimurium produces homocysteine. Mutants lacking the ability to produce homocysteine are hypersusceptible to treatment with S-nitrosothiols, suggesting that homocysteine production leads to increased resistance to NO.86 It is also plausible that blocking the entry of NO or the NO donor may further lead to resistance, as has been reported for S. typhimurium mutants with an inactivated specific peptide trafficking system, which showed higher resistance towards S-nitrosothiols.87 These examples highlight a potential means for development of AMR against NO. For a more detailed overview of these defence mechanisms against NO, the reader is referred to the following text.84 However, despite these mechanisms the multiple toxicity mechanisms of NO provide substantial hurdles for organisms to develop resistance to this drug. This notion was supported by Schoenfisch and co-workers, who found no significant increase in MIC for a range of different bacterial species exposed to one round of inhibitory, sublethal quantities of NO-releasing silica nanoparticles, as well as repeated exposure to sub-inhibitory concentrations for up to 20 passages.88 Given the low passage numbers in this study, further studies on resistance development for newly developed NO-releasing material systems are required to confirm whether resistance is possible at higher NO levels.
3.1.3.1 Polymeric N-halamine mechanism and modes of action. In spite of the moderately weak bond dissociation energies of N–X bonds such as N–Cl (200 kJ mol−1) and N–Br (243 kJ mol−1), N-halamines are stable in aqueous solution.90 Their antimicrobial properties are thought to arise from transfer of oxidative halogen ions (e.g. Cl+ or Br+) from the N atom of the N-halamine to the microbial cell wall upon direct contact, followed by oxidation, as opposed to aqueous dissociation of the N–X bond followed by diffusion of X+ through the media to reach the microorganism.96 Indeed, polymeric N-halamine nanoparticles were found to be effective after 24 h incubation in an organic-rich medium, as well as after several exposures of media inoculated with E. coli (effective after 6 repeated exposures over 9 days) or S. aureus (effective after 9 repeated exposures over 12 days). In contrast, NaOCl itself showed a minimum biocidal concentration of 0.36 M after it had been exposed to the non-inoculated organic-rich medium, 33 times that of the N-halamine nanoparticles.91 Using E. coli containing bioluminescence lux genes fused to stress response signals, Margel and Banin et al., demonstrated that sub-lethal exposure to N-halamine nanoparticles triggered sodA and micF genes associated with oxidative stress, implying oxidative stress as the biocidal mechanism. In contrast to the non-chlorinated nanoparticle control, the N-halamine particles formed specific interactions with the bacteria, as well as forming oxidative hydroxyl radicals during the antimicrobial process.91
3.1.3.2 Development of AMR towards polymeric N-halamines. These polymeric N-halamine species are active against multiple microbial targets,90,91 giving rise to synergistic antimicrobial activity. Therefore, the ability of a pathogen to develop resistance mechanisms is thought to be unlikely. To our knowledge no direct studies on resistance development towards N-halamine polymers have been performed, however studies on small molecule N-halamines have shown no increase in MIC after repeated exposure to subinhibitory concentrations.97 Greater study of AMR development against polymeric N-halamines would be highly valuable to the field.
3.2 Carbon nanomaterials
A range of materials based on carbon nanostructures are being developed as antimicrobial materials (Fig. 3).41,46,103,104 These are broadly categorized by their dimensionality, being either 0D (e.g. fullerenes, carbon dots), 1D (e.g. carbon nanotubes), 2D (e.g. graphene-based nanosheets), or 3D (e.g. sp3-hybridized nanodiamonds). These materials show efficacy against Gram-positive and Gram-negative bacteria, and fungi103,104 including (myco)bacterial strains with acquired multidrug-resistance.103,105 For example, both 1D and 2D carbon nanomaterials prevent M. tuberculosis infection by destruction of bacteria prior to infection, preventing intracellular entry of the bacteria, and inducing a host immune response.105 The specific antimicrobial activity of these materials is highly dependent on their physicochemical properties, including size, aspect ratio, conductivity, and presentation of surface functional groups.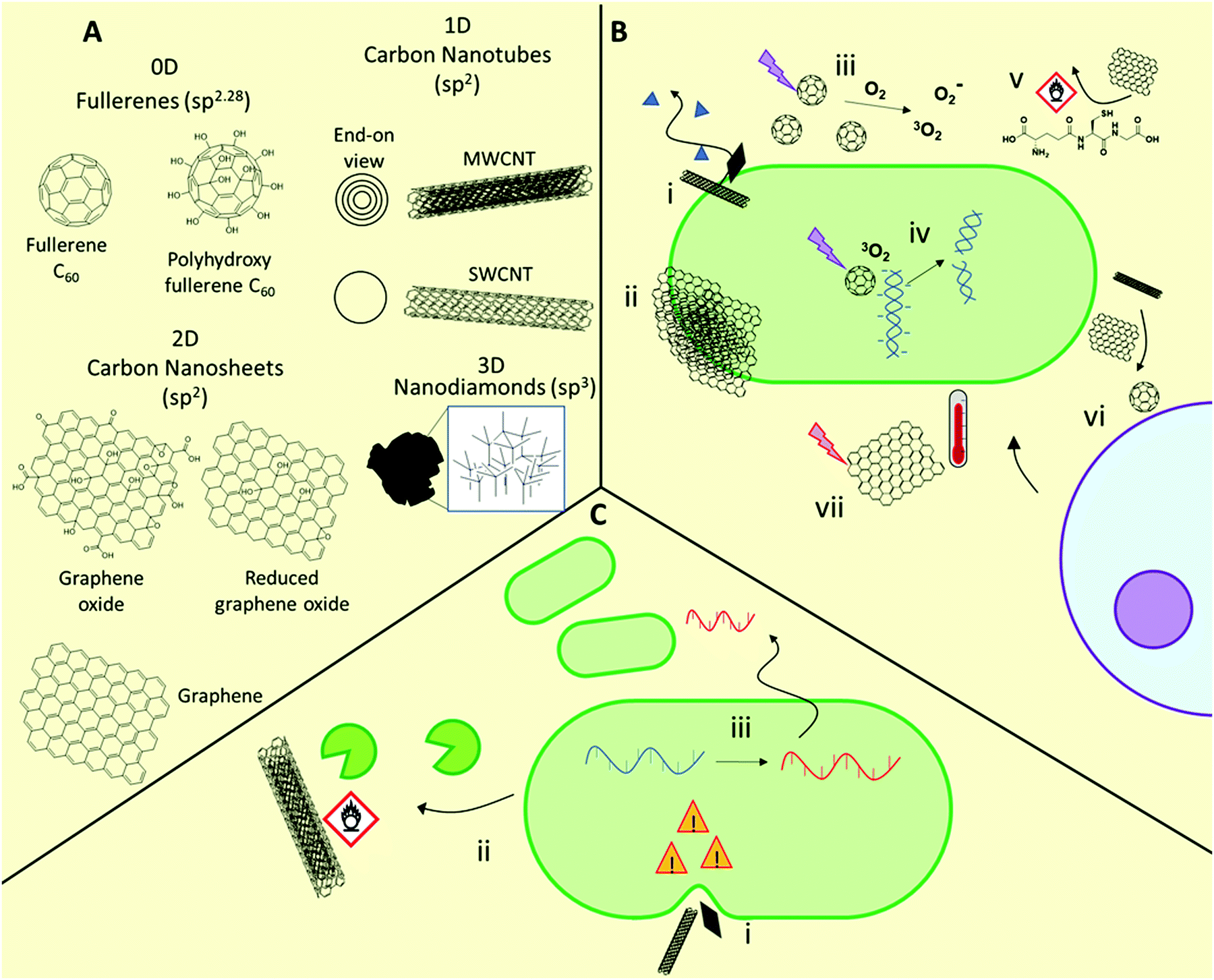 | ||
| Fig. 3 Structures and mechanisms of action of antimicrobial carbon nanomaterials, as well as some known associated resistance mechanisms. (A) General structures of common antimicrobial carbon nanomaterials. (B) Possible antimicrobial mechanisms of action including bacteriolysis through “nanoknife” membrane disruption (i), physical wrapping of microbes by 2D nanosheets (ii), photodynamic ROS production upon light irradiation (iii), damage of intracellular targets such as DNA and functional proteins (iv), oxidative damage to membranes or essential biomolecules (vi), recruitment of host lymphocytes (vi) and photothermal damage upon light irradiation (vii). (C) Resistance mechanisms include modification of membrane lipid composition to improve its mechanical resilience towards nanomaterial-induced rupture (i), enzymatic modification of nanomaterials through oxidation or other processes (ii), and adaptation through envelope stress responses (indicated by yellow danger signs) and subsequent horizontal gene transfer (iii). | ||
1D carbon nanomaterials include both single-walled and multi-walled carbon nanotubes (SWCNTs and MWCNTs, respectively). These elongated structures show potential as antimicrobial agents but there is growing consensus that the SWCNT morphology is vital for exhibiting strong antimicrobial behavior, with MWCNTs showing negligible or only minor antimicrobial activity.41,109 In carbon nanotubes, dimensional aspects such as the diameter and length play a major role in determining activity, in addition to surface functionality, with higher aspect ratio materials able to puncture bacterial membranes. This latter mechanism is sometimes referred to as the “nanoknife” mechanism.110 It has been proposed that nanotubes may also be less effective towards rod-shaped bacteria for this reason. As with fullerenes, production of ROS upon photoirradiation is also possible, with SWCNTs exhibiting greater ROS production than both MWCNTs and fullerenes.
2D carbon nanomaterials include graphene-based nanosheets, which are the thickness of a single carbon atom. As with 1D materials, the aspect ratio plays a major role in determining antimicrobial activity, with those with higher density of edges typically exhibiting higher activity through a nanoknife mechanism.111 Chen and co-workers compared the antimicrobial activities of micron-sized graphite (Gt) and graphite oxide (GtO) dispersions, with those of 2D graphene oxide (GO) and reduced graphene oxide (rGO) nanosheet dispersions, against E. coli.112 The authors demonstrated that while all formulations showed activity, GO and rGO nanosheets showed superior biocidal activity. They found that each nanosheet showed distinct interactions with bacteria, with GO typically forming thin sheets wrapping individual bacteria, whereas rGO formed large bacterial aggregates. Contrary to fullerenes, no materials investigated generated significant superoxide species, however each of the materials caused ROS-independent oxidative stress through glutathione oxidation. This was particularly true of rGO, which gave near quantitative in vitro glutathione oxidation after 2 h. The results indicated that both oxidative stress and stress on bacterial membranes caused by interaction with sharp nanometre-thin edges and corners were responsible for the high antimicrobial activity of GO and rGO dispersions.112 Additionally, certain 2D graphene-based materials, as well as 0D fullerenes and 1D carbon nanotubes, can exhibit photothermal activity, whereby they produce a highly localized heating effect when irradiated with near infrared light, which has been shown to be effective against a range of Gram-positive and Gram-negative bacteria, including methicillin-resistant S. aureus.113
3D carbon nanomaterials such as nanodiamonds have been less commonly studied as antimicrobial agents, yet their potential as therapeutics has recently become evident. The preparation and antimicrobial properties of these emerging materials are covered in detail in the following dedicated text.114
Very recently, Zhang et al. demonstrated that E. coli exposed to sublethal concentrations of graphene oxide over 200 passages resulted in significant alterations to both the transcriptome and the metabolic behavior through activation of an induced envelope stress response.118 These adaptations allowed improved survival in harsh oxidative and low pH environments and facilitated greater pathogenicity through increased biofilm formation and extracellular protease release, greater bacterial invasion and intracellular survival, and greater macrophage inflammatory response.117 This study highlights the unforeseen impact of low levels of such nanomaterials in the environment, with serious implications for human and animal health. With the rising interest in the use of carbon nanomaterials for environmental applications,119 such as pollution prevention, environmental sensing, high flux membranes and filtration, and as sorbents, as well as their use and disposal in electronic applications, the impact of this increased usage should be carefully considered in the context of the broader implications of such use.
3.3 Inorganic nanomaterials
A wide range of inorganic nanomaterials with antimicrobial activities have been extensively studied (Fig. 4), such as those based on Au, Ag, Cu, Co, Cr, Fe, Ga, Mg, Mo, Se, Ti, V, W, and Zn, amongst others, including oxides and/or sulfides thereof.42,120–122 Each elemental class has been reviewed in detail elsewhere and as such, herein we will discuss these generally, using specific examples to highlight their various modes of action before discussing the likelihood of antimicrobial resistance development towards such materials.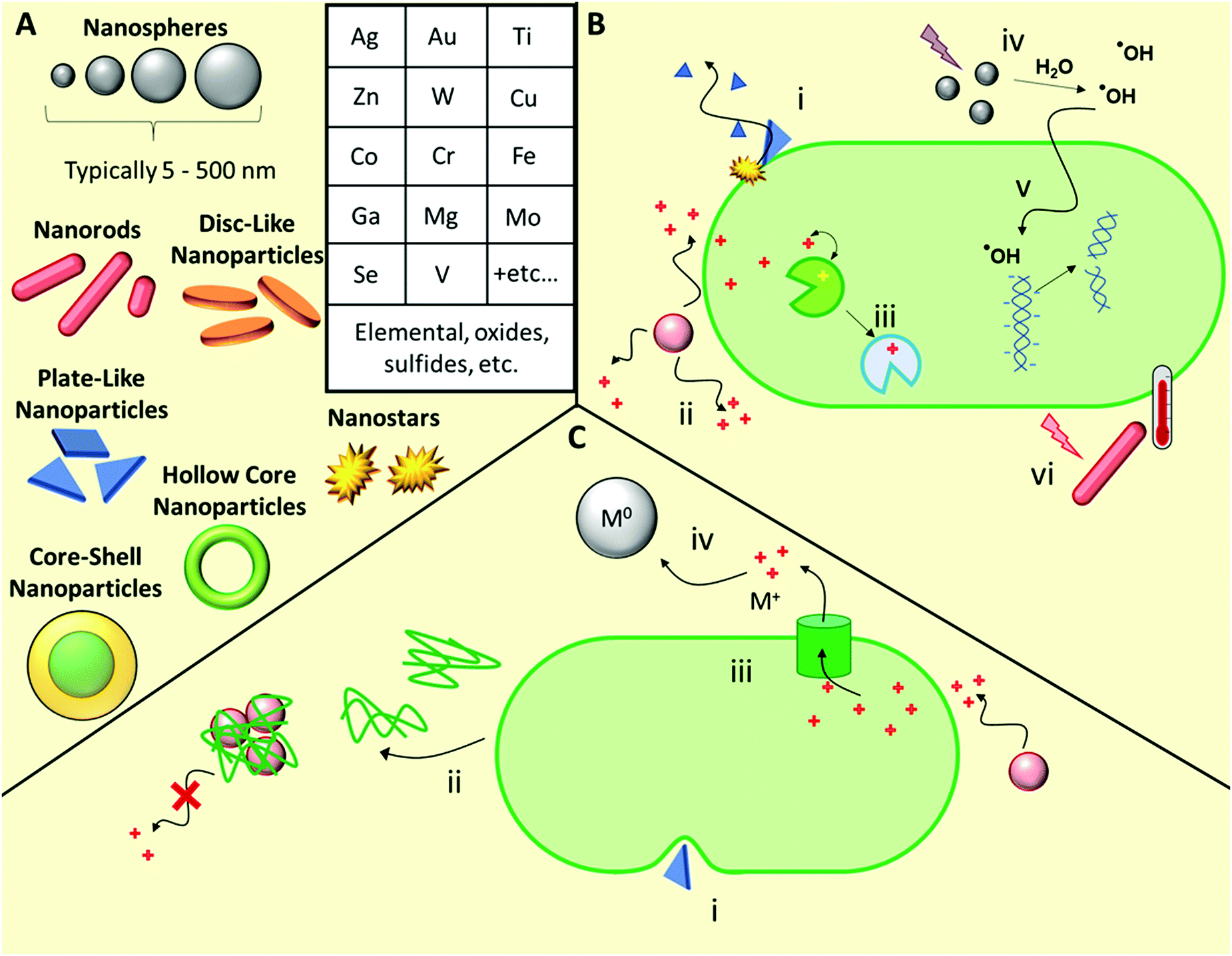 | ||
| Fig. 4 Structures and mechanisms of action of antimicrobial inorganic nanomaterials, as well as some known associated resistance mechanisms. (A) Examples of morphologies and elemental classes of common antimicrobial inorganic nanomaterials. (B) Possible antimicrobial mechanisms of action including bacteriolysis through membrane disruption (i), leaching of toxic cations (ii), ion mimicry through displacement of native ions in metalloenzymes, thereby leading to enzyme inactivation (iii), photodynamic production of ROS (iv), which can lead to damage of intracellular targets such as DNA and functional proteins (v), and photothermal damage upon light irradiation (vi). (C) Resistance mechanisms include modification of membrane lipid composition to improve its mechanical resilience towards nanomaterial-induced rupture (i), production of adhesive proteins that flocculate nanoparticles and reduce ion leaching (ii), adaptation of ion efflux pumps to remove intracellular ions (iii), and microbial processes that reduce toxic ions into relatively more inert neutral metal particles (iv). | ||
Ion release from metal and metal oxide/sulfide nanoparticles is a prominent antimicrobial mechanism for a range of elemental classes.122 Cationic metal ions are uptaken into microbial cells using both direct transport and co-transport (i.e. alongside other small molecules bound to the metal ion) mechanisms.122 Once uptaken, the ions act primarily through interfering with metabolic processes, for example through molecular exchange with cofactors naturally present in metalloenzymes in phenomenon known as molecular or ionic mimicry.123 Many ions are toxic to mammalian cell processes, hence identifying those specific to microbial processes is a key challenge when developing antimicrobial metallic nanomaterials.
Ag nanomaterials are the most broadly used with the greatest knowledge of efficacy and toxicology.124–127 A complete understanding of all the antimicrobial mechanisms of Ag-based nanomaterials is still yet to be achieved, however it is primarily the silver ions that are active. Ion mimicry is not a major mechanism of action for silver ions, however these silver ions interact with membrane constituent proteins and enzymes, and with DNA and RNA, while the nanoparticles themselves result in compromized bacterial membrane permeability and can also generate reactive oxygen species (ROS).128 Ag nanoparticles are effective against a wide range of drug-susceptible, drug-resistant and multi-drug resistant bacteria, both when used alone and in combination with conventional antibiotics.129 Though less reported, Ag nanomaterials are also effective against fungal pathogens, with silver-induced mitochondrial dysfunction and apoptotic features driven primarily by ROS and the production of hydroxyl radicals.130 Even though colloidal silver has been used extensively,131 resistance development towards Ag-based materials has also been widely reported, as will be discussed briefly later in the next subsection.124,126,132 While the antimicrobial properties of other ions are well known, a shift towards using other colloidal inorganic nanoparticles to replace silver, such as copper, has been brought into question, with some articles pointing to their limited direct antimicrobial activity.133 Here, the authors deduced that the antimicrobial properties of copper nanoparticles result solely from soluble ion leaching.133 However, further studies are needed to elucidate the relative importance of the various possible antimicrobial mechanisms in these materials and beyond.
Outside of Ag-based materials, typical ROS-producing metal nanoparticles exist in two main classes: catalysts that can facilitate Fenton-type autooxidation reactions, most notably Fe-, Cu-, Cr-, V-, and Co-based nanoparticles, and those that can undergo photodynamic ROS-production upon photoirradiation, most notably TiO2 and ZnO.42,45 For Fenton catalysts, antimicrobial properties arise from metal-mediated production of hydroxyl radical species, produced from endogenous hydrogen peroxide. Conversely, photodynamic species such as TiO2 and ZnO, upon activation by photoirradiation with UV or visible light sources, can either convert water to hydrogen and oxygen, subsequently producing ROS, or they can generate radicals at the material surface, subsequently producing hydroxyl radicals upon transfer to the surrounding water.42,45 ROS subsequently cause oxidative stress through radical reactions with metabolic enzymes, membrane components, and nucleic acids, thereby giving rise to antimicrobial activity.42 Similarly to Ag nanoparticles, other ROS-producing particles like ZnO show activity against fungal pathogens.134
Plasmonic nanoparticles such as Au, CuS, and MoS2, exhibit photothermal activity,113 whereby photoexcitation of electrons produces a highly localized heating effect through non-radiative relaxation. This localized heating often reaches several hundred degrees and causes nanosized bubbles of steam to form at the particle surface,135 which can physically rupture membranes, as well as imparting thermal stress on microbes. Photothermal antimicrobial therapy has been demonstrated to be effective against a wide range of drug-susceptible and drug-resistant microbial pathogens.113
Ga-based materials are a somewhat unique class of inorganic materials that target microbial siderophores, a specific microbial ion-transport mechanism for gaining intracellular entry.136 Siderophores are FeIII-chelators secreted by bacteria and fungi to facilitate uptake of Fe from their surroundings. Siderophores also bind and transport GaIII ions with high specificity. Once inside the cell, GaIII replaces FeIII in FeIII-binding sites, including in metalloenzymes, causing metabolic, respiratory, and DNA synthesis failures.136
Beyond simple elemental nanoparticles, mixed metallodrug compounds such as the class of polyhedral metal oxide clusters, polyoxometalates (POMs), also show promise as antibacterial materials.137 These compounds are similarly highly tunable in their size, shape, and composition. Their broad-ranging mechanisms of action have been outlined elsewhere.137 Only a few POMs, including polyoxotungstates, polyoxomolybdates, and polyoxovanadates, show direct antimicrobial activities, however a broader range can be used synergistically with conventional antibiotics.137
Development of resistance against both Ag nanoparticles and Ag+ ions has been observed in a number of pathogenic species.124,131 Resistance mechanisms include production of an adhesive protein (flagellin) that promotes aggregation of Ag nanoparticles thereby reducing rates of ion leaching,124 as well as mechanisms that reduce the anionic charge of the cell wall, and other envelope stress response mechanisms.126 Resistance to metal nanoparticles can also occur when microbes alter cell wall fatty acid composition, preventing the nanoparticles penetrating, or disrupting, the cell wall.140,141 For example, Heipieper and co-workers observed a significant rise in the trans/cis isomeric ratio of unsaturated fatty acids present in Pseudomonas putida upon exposure to increasing concentrations of both Ag nanoparticles and Ag+ ions, which was proposed to be an adaptive defence mechanism for overcoming silver-induced stress.141
3.4 Delivery vehicles for repurposing existing antibiotics and hybrid materials
Outside of the materials mentioned above, a smaller range of NBCD materials have been designed to deliver conventional small molecule antibiotics more effectively in order to subvert microbial resistance mechanisms or provide a pathway for improved or targeted local administration, thereby repurposing these existing drugs. These typically include polymeric prodrugs (sometimes referred to as “drugamers”),142,143 self-assembled polymeric nanoparticles and nanogels,43,143,144 hollow inorganic particles such as mesoporous silica nanoparticles,145,146 and drug conjugates of metallic nanomaterials.45 The advantage of these vehicles is that the mechanism, pharmacology and toxicity profile of the released active antimicrobial component is well-established, bearing in mind that these characteristics are still at risk of modification through the nature of the formulation itself, typically by design. These delivery vehicles provide targeted uptake or improved uptake through size or shape considerations, sustained release kinetics, and occasionally provide a platform for avoiding resistance mechanisms such as drug efflux or degradation. For example, the use of metallic nanoparticles has been shown to block efflux pumps, thereby synergistically enhancing and reviving the efficacy of conventional antibiotics.147 Reviews on the use of nanoparticles to overcome typical resistance mechanisms can be found in the following dedicated texts.147,148Targeted delivery can be used to improve the effectiveness of antibiotics against pathogens with situational resistance, such as those within biofilms or intracellular environments. For example, Stayton et al. designed a highly effective prodrug to treat intracellular lung infections where bacteria reside within host alveolar macrophages142 by conjugating mannose-functional polymers via both acid-labile and intracellular protease-cleavable linkers to ciprofloxacin, a potent antibiotic with poor pharmacokinetic properties. The mannose component greatly improved uptake into alveolar macrophages owing to the abundance of macrophage mannose receptors on macrophage cell surfaces; the intracellular specific protease-cleavable linkers ensured sustainable delivery of the antibiotic inside the local intracellular environment. The residence time of the released antibiotic inside the macrophages could be greatly increased relative to the free antibiotic, thereby prolonging the time during which the drug was above an effective biocidal concentration. When intratracheally administered into a lethal Francisella novicida intracellularly infected mouse model, these polymer prodrugs could increase the cohort survival from 0%, observed using the free antibiotic, to up to 100%, demonstrating the great influence of targeted and highly localized delivery for treating inherently resistant infections.142 Similarly, Wang and Hu et al. developed mannose-functional polymer nanogels with a selectively bacterial enzyme-degradable poly(phosphodiester) core loaded with vancomycin.149 These nanogels were stably taken up by macrophage mannose receptors and then degraded by intracellular phosphatase/phospholipase-secreting bacteria, to release the antibiotic intracellularly. This targeted delivery and on-demand bacteria-triggered release resulted in a 3-log reduction in intracellular MRSA viability, whereas vancomycin alone was ineffective at the same drug loading.
Nanoparticles designed to enable greater penetration and controlled release of antibiotics into established biofilms have also been developed. Strategies for achieving this include targeting of tissues within the infected site, and the introduction of stimuli-responsive functionality to allow specific drug release within the biofilm environment such as in response to changes in pH, enzymes, chemical concentration, or redox state.150,151 Furthermore, biofilms are susceptible to physical disruption and thermal ablation, which allows for the application of materials that can respond to external stimuli, such as magnetic fields, light, and ultrasound, which may play a synergistic role with antibiotic release.150 For example, superparamagnetic iron oxide nanoparticles (SPIONs) exhibit magnetic hyperthermia properties, whereby the particles convert the energy of a magnetic field into thermal energy.152 This property has been shown to improve their penetration into established biofilms and increase the lethality of such materials,153 as well as increasing the efficacy of conventional antibiotics when used in combination with SPIONs upon exposure to magnetic fields.154
Photothermal materials have also been used to facilitate antibiotic delivery. For example, Braeckmans and co-workers showed that laser-induced photothermal heating of Au nanoparticles resulted in formation of nanobubbles at the particle surface, which disrupted microbial biofilms and enhanced three-fold the efficacy of tobramycin against both Gram positive and Gram negative species.135 Similarly, Chen and Smeltzer et al. developed poly(dopamine)-coated Au nanocages loaded with daptomycin, which were conjugated to an antibody specific for S. aureus protein A.155 These bound to the cell surface of the bacterium and upon photoirradiation, enacted synergistic photothermal therapy and antibiotic release. Contrary to daptomycin alone, which was only effective after 24 h, treatment with these nanocages led to an immediate 6-log to 8-log reduction in methicillin-susceptible and methicillin-resistant S. aureus, which was sustained over a 24 h period post-irradiation. Non-loaded Au nanocages were effective after initial irradiation but did not prevent the regrowth of the susceptible bacteria 24 h post-irradiation, thus highlighting the synergy of these two mechanisms employed by the drug-loaded nanocages. Finally, these drug-loaded nanomaterials resulted in an immediate (0 h) and sustained (24 h) 7-log reduction in biofilm viability upon photoirradiation, in contrast to daptomycin alone, which showed negligible efficacy against such biofilms.155
Outside of delivering small molecule antibiotics, NBCDs and other drug delivery platforms can also be used to deliver biologics like enzymes,156–159 creating antimicrobial compounds locally on demand. Meier et al. prepared polymeric nanoreactors containing penicillin acylase and a porin (outer membrane protein F) to allow diffusion of small molecule substrates into the internal lumen.158 Using a prodrug approach, building block substrates could enter the lumen allowing in situ biosynthesis and local release of the antibiotic cephalexin.158 Blackman and Locock et al. prepared polymeric nanoreactors containing the antimicrobial enzyme glucose oxidase, which produced hydrogen peroxide in response to glucose.156 These nanoreactors showed a 5-log reduction in planktonic bacterial viability in both Gram-positive and Gram-negative bacteria (S. aureus, E. coli, K. pneumoniae, and S. epidermidis), as well as retaining the encapsulated enzyme's activity against established biofilms formed by methicillin resistant S. aureus.156 Building in complexity, Stevens et al. prepared dendrimersomes (vesicles formed by amphiphilic dendrimers) loaded with both glucose oxidase and myeloperoxidase.159 These enzymes act in a cascade, eventually resulting in the formation of highly biocidal hypochlorite ions (−OCl). The dual-loaded dendrimersome nanoreactors showed potency against multidrug resistant S. aureus and P. aeruginosa, which was not observed for dendrimersomes containing glucose oxidase alone.159 Encapsulation of biologics into semi-permeable nanocontainers in this way allows the activity and specificity of the biologic to be retained, while enhancing their stability in vivo and reducing immunogenicity.160 For an overview of vesicular nanocontainers used in antimicrobial applications, the reader is referred to this dedicated text by Sun et al.161
The above examples highlight some of the ways in which nanomaterials can better facilitate delivery of conventional antibiotics and antimicrobial biologics, thereby providing potential for overcoming situational and natural antibiotic resistance, as well as reducing the antibiotic dosage, reducing selection pressure that facilitates the spread of AMR. Aside from the antimicrobial properties of the nanomaterials themselves, they are able to act synergistically with conventional antibiotics, as well as with other non-conventional therapeutic components, such as hybrid materials comprized of metal-carbon, metal-polymer, and polymer-carbon combinations.98
4. Regulatory pathways and challenges for NBCDs
A lack of tailored regulatory pathways for NBCDs poses a major challenge to their translation. A summary of the potential approval pathways for NBCDs in a European and US context is provided in Table 2 and discussed in the following sections. Note that this is intended only as a review of some of the possible regulatory pathways and is not intended to serve as advice for translation of any therapeutic agent.| Body | Legislation | Type of medicines | Originator (full dossier) | Off-patent (abbreviated) |
|---|---|---|---|---|
| European Medicines Agency (EMA) centralized pathway | Annex I of directive 2001/83/EC | Small molecule, biologic and other | Stand-alone application | Generics application |
| Article 8(3) (also articles 10a, 10b & 10c) | Article 10(1) | |||
| Hybrid application | ||||
| Article 10(3) | ||||
| Biosimilar application | ||||
| Article 10(4) | ||||
| US Food & Drug Administration | Food, Drug & Cosmetics Act | Small molecule | New drug application (NDA) | Abbreviated new drug application (ANDA) |
| 505 (b) (1) (also 505(b) (2)) | 505 (j) | |||
| US Food & Drug Administration | Public Health Service Act | Biologic | Biologic license application (BLA) | Biologics price competition & innovation act (BPCI) |
| 351 (a) | 351 (k) |
4.1 European regulatory pathways for new medicines
Centralized European market approval of medicines is overseen by the European Medicine Agency (EMA). The registration of any new active substance for use in humans, including NBCDs, involves filing an ‘Originators Full Application’ as detailed in article 8(3) of Directive EMA/821278/2015.162,164,165 This includes a full dossier of pharmaceutical (biological and physicochemical) and non-clinical (pharmacological and toxicological) tests, clinical trial results and any supportive published literature. There are also related application pathways for new medicines summarized by Article 10a (well-established use), 10b (fixed combination) and 10c (informed consent) where existing data on products is taken into consideration.4.2 European regulatory pathways for off-patent medicines
Bodies that seek European market approval for off-patent medicines can undertake abridged applications that do not require the same breadth of primary evidence. There are three potential pathways (Directive 2001/83/EC article 10(1) Generics, 10(3) Hybrids, or 10(4) Biosimilars) for off-patent medicines, dictated by the medicine class.162,164,165 The first, 10(1) generics, includes medicines with the same active substance, in the same amount, with the same pharmaceutical form and bioequivalence. This pathway avoids the need for additional non-clinical tests or clinical trials. The Hybrid pathway (10(3)), is relevant for medicines that do not adhere to the definition of a generic, where bioequivalence cannot be demonstrated, or where there are changes to aspects such as therapeutic indication, route of administration etc. These applications can utilize some results from the initial reference medicine application in combination with additional non-clinical and/or clinical studies (where appropriate). Follow-on biological products that do not meet the criteria of a generic and are similar to an initial reference biological medicine are assessed via the 10(4) biosimilars pathway. This process recognizes that differences in starting materials or manufacturing processes can have an impact on the medicine's final biological profile. The application must be supported by appropriate non-clinical or clinical studies as well as a comparability exercise to demonstrate similarity to the reference product.None of these follow-on regulatory directives specifically cater for the NBCD class. A recent study, in fact, saw great variance in the selected pathway for approval for NBCD follow-ons, with 56% applying via Generic, 37% by Hybrid and 5% via the Biosimilar pathways. The authors did note, however, a recent trend towards the hybrid category.166 Such heterogeneity in the regulatory pathways taken could have significant impacts on the safety of these medicines.
4.3 US regulatory pathways for new and off-patent medicines
The US system for market approval, administered by the Food & Drug Administration (FDA) is structured differently to the EU, which causes further uncertainty. There are two different statutes governing this process, each encompassing an originator and a follow-on arm. The first, falls under the Food, Drug & Cosmetics Act and is for small molecule drugs. The second, falls under the Public Health Services Act, and is for biologic drugs. Neither has been specifically catered to the unique properties of NBCDs.162–164Given that NBCDs by nature are not derived from a biological source, they fall under the Food, Drug & Cosmetics Act. Originator medicines seeking market approval under this act are required to submit a full New Drug Application under 505 (b) (1) or 505 (b) (2).163 Both applications require a full report on the drug's animal and clinical trial results, potential drug abuse information, manufacturing, patient information and proposed labelling.167 The main difference between these pathways is that 505 (b) (2) can rely on previous study results, while 505 (b) (1) requires primary data.
Similarly, NBCD off-patent drugs are forced to adhere to the abbreviated new drug application pathway for approval, given they are not derived from biological sources. Approval at this stage requires evidence of a therapeutic equivalence to the originator163 through demonstration of pharmaceutical equivalence and bioequivalence.168 Pharmaceutical equivalence is evidenced by results from the administration of the same amount of an identical drug and dosage form, via the same route of administration. Bioequivalence is focussed on pharmacokinetic aspects, such that the same amount of drug is available at the target site when administered at the same molar dose and conditions as the originator drug. Give the properties of NBCDs, one or both of these aspects can be near impossible to demonstrate, greatly restricting the potential for follow-on NBCDs to find market approval.164
4.4 Outlook for antimicrobial NBCD market approval
The lack of regulatory pathways that specifically cater for the physical and biological profiles of NBCD medicines represents a significant concern. Alignment to a small molecule approval process can fail to adequately emphasize the relationship between the manufacture process and its influence on the final attributes of an NBCD. In the context of this article, this could mean that patients may simply not have access to new antimicrobial NBCDs currently in preclinical development, or to safe, effective, and affordable off-patent equivalents. For technological advancements more generally, this has often been referred to as “the pacing problem”, where technological innovation is increasingly observed to outpace the relatively incremental changes in applicable laws and regulations.169 Indeed, there may also be a negative feedback loop whereby the lack of fit-for-purpose regulatory pathway stunts further innovation in the complexity of AMR materials design. Certainly, it is expected that the pace in innovation required to overcome challenges associated with rapidly growing AMR will increasingly need to outstrip the pace of regulatory and legal amendments. Alongside economic barriers of antibiotic development, whereby the acute use profile of these drugs does not allow sufficient opportunity to recoup costs of research and development,37,39,170 these barriers will be further compounded by the lack of regulatory pathways that cater to NBCDs. If we are to see the full potential of antimicrobial NBCDs for AMR, we will first need to develop an evaluation of the regulatory pathways governing them.4.5 Materials design considerations for NBCD translation
As discussed, currently no specific NBCD translation pathway exists, whereby the dispersity in the materials’ size, shape, surface chemistry, and overall chemical composition is considered. However, one could envision a new or modified pathway that requires a full understanding of the complex relationships between physicochemical and pharmacological properties, catered to NBCDs. A specific pathway should require extensive characterization of NBCDs conducted using carefully considered complementary techniques. These techniques should include characterization of the materials at various hierarchical length scales, from the individual chemical components up to the 3D assembled nanostructure.13,171On the component level, for polymers, traditional chemical characterization techniques such as NMR, FTIR, and UV/Visible spectroscopy should be conducted to obtain average chemical composition. However, further analysis may be required such as understanding the monomer sequence for copolymers by obtaining reactivity ratios, as well as characterizing the end group functionality and fidelity. Molar mass distributions should also be obtained using size exclusion chromatography (SEC) fitted with refractive index, UV/Visible, or multiangle laser light scattering (MALLS) detectors. Newer paired in-line detection techniques such as SEC-NMR, SEC-FTIR, and SEC-fluorescence, among others can provide further information on the chemical distribution throughout the length distribution of a polymer. For carbon nanomaterials, chemical aspects such as the wrapping conformation of nanotubes should be obtained, as well as their electronic bandgap properties. Furthermore, the oxidation state and any residual functional groups remaining or introduced following a particular chemical process should be ascertained. Similarly, for inorganic NBCDs, trace elemental analysis should be conducted as well as understanding the distribution of oxidation states. If crystalline materials are used, the crystal structure should be obtained using X-ray diffraction techniques.
For nanoparticles, statistical assessment techniques should be employed that give structural information on the global distribution throughout the sample, such as one or more from dynamic and static light scattering, small angle X-ray and neutron scattering, and fluorescence correlated spectroscopy in the case of fluorescent materials. Additionally, zeta potential measurements should be conducted to reveal the average particle surface charge. Careful selection of these techniques should be conducted to ensure that the critical parameters that give optimal bioactivity and pharmacological properties can be accurately and reliably characterized. Many of these techniques are capable of analyzing over a billion particles in a single measurement to ensure statistical relevance. These should be coupled with microscopy techniques such as one or more from dry state, cryo-, or liquid-cell transmission electron microscopy, atomic force microscopy, and scanning electron microscopy, among others, and paired with elemental in-line techniques such as energy-dispersive X-ray spectroscopy (EDX) or electron energy loss spectroscopy (EELS) to determine composition with spatial resolution. Conversely, these techniques can only be practically used to analyze hundreds of particles per sample, however they reveal new information when used in conjunction with the aforementioned statistical assessment techniques. The gain of additional information on the materials’ properties using complementary techniques is well established to be greater than the sum of information gained from individual techniques.172
The physicochemical properties of NBCDs, and their associated size, shape, and chemical dispersities, can have dramatic effects on pharmacological outcomes such as biodistribution, efficacy, clearance, stability, toxicity, etc.13 Owing to the complexity of these parameters, simple NBCDs with minimally hierarchical designs, narrow dispersity (in size, shape, chemistry, etc.), and easily quantifiable key characteristics, show the greatest potential for translation. Unlike small molecules where multiple synthetic routes can afford compounds with identical physicochemical properties, the properties of NBCDs are strongly reliant on their manufacturing process. Validation of the process repeatability and reproducibility is also vital, therefore simple manufacturing processes with as few synthetic/fabrication steps as possible will again afford materials with the least variation and easiest route to clinical approval.
5. Remaining challenges and opportunities for antimicrobial NBCD materials
Even though a range of promising therapeutic materials have emerged from the academic literature, significant challenges limit their commercialization for clinical use. For many of these materials, considerable work is required to meet the standards of reproducibility of fabrication currently required for regulation. These regulations were designed for small chemical drugs and require clearly defined material composition and distribution, which is a significant challenge for economically priced NBCDs. For example, colloidal silver has been used extensively as additive materials for catheters, and for use in various other biomedical, medicinal, and consumer products.131 However, the commercialization of silver nanomaterials for clinical use remains limited, owing to historical irreproducibility and batch-to-batch variability preventing approval for clinical use from the Food and Drug Administration (FDA).131 Similar regulations have been imposed by the Therapeutic Goods Administration (TGA) in Australia, as well as the European Commission.131 Even if reproducible and non-variable materials could be fabricated, current regulation would require defining and quantifying each mechanism of action. The broad mechanisms of action that make these agents so advantageous also make it challenging to define precise mechanisms, off-target effects, residence half-life, and biodistribution required for regulatory approval. Hence, “the pacing problem” discussed in the previous section acts as a significant barrier for their clinical approval and adoption.For carbon nanomaterials and certain inorganic nanomaterials, safety concerns have been raised, both from the perspective of medical use but also their potential to damage and persist in the environment.173 In particular, materials that exhibit non-specific mechanisms of action involving physical “nanoknife” disruption of membranes and the production of ROS, issues around hemotoxicity and cytocompatibility may restrict clinical translation. Similarly, although some optimized examples of cationic antimicrobial polymers have been developed, the membrane-rupturing mechanism introduces risk for the development and translation of new materials in this class, particularly for in vivo systemic applications.173 Indeed, a detailed predictive toxicological paradigm has been proposed over a decade ago for polycationic, inorganic, and carbon nanomaterials,174 linking material properties and behavior in in vitro toxicological assays to in vivo scenarios using detailed compiled quantitative structure-activity relationship data. Establishing such predictive models for all new antimicrobial therapeutics, linking their structure and more easily accessible in vitro analysis data to both their in vivo behavior and environmental adverse toxicological risks, would allow more rapid and generalized in silico screening of new antimicrobial nanomaterials.175–177
The lack of degradability of many of the materials presented above poses a serious risk of extended environmental persistence. This is a particularly important risk when considering their possible widespread non-clinical use. Options for removing such antimicrobial materials from wastewater run-off after use are expensive and are not typically implemented in existing water treatment plants, and hence these long-lasting materials will enter and persist in the environment, likely having significant impacts on soil communities.178 Environmental persistence of compounds with antimicrobial activity will increase selection pressure and drive the evolution of AMR.
Finally, technical application-specific challenges also remain, particularly for stimuli-responsive materials. For example, whilst the photodynamic mechanism found for some carbon nanomaterials appears to be useful as an “on-demand” therapeutic approach for clinical applications, the path for effective translation of this technology into the clinic is unclear. Major unmet requirements for such approaches include the development of technologies to increase the depth of penetration of light (or given stimulus) into tissue, as well as methods to limit undesirable thermal damage/toxicity of the surrounding mammalian tissue. Important advances for these materials may include the design of colocalized energy transfer systems that give greater specificity over the spatiotemporal therapeutic control. Additionally, advances in light delivery systems may emerge from fields outside of materials chemistry, such as medical photonics.
As discussed in the previous section, globally there is a lack of unified regulations for NBCDs, often forcing an alignment to small molecule pathways, that do not adequately address how NBCD properties and manufacturing processes effect their activity profiles.162,167 There are also further issues governing the approval of NBCD off-patent drugs. Off-patent drugs, also termed ‘generic’ or ‘follow on’ drugs enter the market following the expiration of market exclusivity for an originator drug. Given the established nature of the originator drug, there is a much lower level of preclinical and clinical data to support their approval, sufficient only to prove therapeutic equivalence.162,163,166 This typically allows companies to provide these off-patent drugs at a much more competitive price than the originator. This is important for deriving broader patient access to this class of drug and reducing costs to the healthcare system. The current pathways for off-patent drugs can easily be rationalized for small molecules, where the chemical identity, properties, and pharmacokinetic profile is largely independent of the synthetic/processing route used to prepare such agents. However, these critical aspects are highly dependent on the manufacturing process for NBCDs such as nanomaterials or polymers, and as discussed, their properties are not easily determined or characterized such that appropriate comparisons to the originator material can be made. Given the lack of specific regulatory pathways for the approval of NBCD off-patent drugs, this can have potentially disastrous consequences if these mechanisms fail to recognize key NBCD properties.164 Take for instance a recent study that looked at patients receiving an iron sucrose off-patent product. Classed as a NBCD, this was a polymeric preparation of iron sucrose which stably delivered iron to deficient patients. When a comparison was made, it was shown that patients required on average 34% more iron for the off-patent product than the originator.179,180 While outside of the antimicrobial focus of this Review, this example does illustrate just how dramatic the treatment consequences can be if the unique properties of NBCDs are not addressed in the approval process.
In order to exploit the potential of NBCDs, further development is required in the following areas:
• Development of materials with short antimicrobial lifetimes, which rapidly revert to inert materials after their intended use; materials with activities that can be irreversibly switched off after use, in response to applied stimulus such as light, pH, or heat; or rapidly biodegradable materials with no active antimicrobial degradation products. Some examples are given in the following texts.75,181–184 The absence of selection pressure outside the immediate use window, and the lack of environmental persistence of these antimicrobials, would reduce the risk of microbes developing resistance to these materials.
• Understanding of the genetic mechanism behind development of resistance to any new technologies, to allow predictions of the rate of development of AMR to these materials.
• Development of materials that directly target established biofilms and persister cells,185 including those that exhibit non-lethal strategies such as anti-quorum sensing (quorum quenching)186,187 and other forms of non-lethal biofilm disruption, which place a smaller evolutionary strain on microbes subjected to such therapy. Even though a number of studies have investigated materials’ ability to inhibit biofilm formation when administered to planktonic bacteria at sublethal concentrations, the destruction or dispersal of established preformed biofilms is often more relevant for the development of clinical therapies.
• Investigation of cytocompatibility, hemotoxicity, understanding the formation and influence of the protein corona under different in vivo environments, immunogenicity, biodistribution, and degradation/clearance mechanisms, in addition to the material's synthetic reproducibility and quality assurance.
• Total impact studies including success of the intervention using clearly defined criteria, such as reduced prevalence of AMR genes in the local environment upon substitution of a conventional biocide with the new therapeutic material, or a specific reduction in antimicrobial resistant hospital-acquired infections in clinics employing the therapeutic in question.
• Less generalized regulatory pathways, which take into account the influence of material processing and dispersity on NBCD properties. This should also include a new evaluation of regulatory pathways for generic off-patent NBCDs prepared by new synthetic methods or processes.
Note that the final sections of this review have been focused on regulatory pathways towards clinical approval of NBCDs. However, such agents also show more immediate potential in non-clinical use settings, such as their use as disinfectants, in agriculture, in food technologies and packaging, and for personal care applications. With varying degrees, typically these use settings have a lower barrier for market entry than clinical settings and it may be expected that NBCDs enter these markets first. In doing so however, we must ensure that appropriate regulatory frameworks governing their use is established, thereby limiting their overuse and environmental exposure, in order to reduce AMR against these precious new antimicrobials.
6. Conclusions
NBCDs are a fascinating class of emerging antimicrobial materials with potential as therapeutics. Generally, these (nano)material classes exhibit antimicrobial mechanisms that are broad-spectrum, being indiscriminate between species or even across kingdoms, but perhaps more importantly are often unprecedented from a historical or evolutionary perspective. Their multimodal mechanisms of action make them ideally suited to overcoming antibiotic resistance pathways in key pathogens associated with AMR. Despite these properties, it is recognized that eventually AMR towards these technologies may still occur if these materials are used inappropriately.124,131,188,189 Therefore stewardship of these new technologies will be of paramount importance in preserving their long-term effectiveness.Though they have shown promise, many barriers for the clinical translation of NBCDs remain. In addition to existing issues facing any new antibiotics around inadequate return on research and development investment, NBCDs face considerable challenges in their regulatory approval pathways. In order to advance these alternatives to small molecules, it is becoming evident that specific approval pathways may be required that recognize the unique reliance on the materials’ manufacturing process, as well as their less well-defined physicochemical properties. Although non-clinical market entry is more easily achievable, care must be taken to ensure good stewardship, thereby limiting NBCD AMR, such that these technologies can eventually be used effectively in clinical applications. If these challenges can be addressed then it is envisioned that these next generation therapeutic agents will greatly enhance humanity's antimicrobial arsenal into the future, provided carefully considered policies and practices are established in order to limit their widespread use.
Author contributions
All authors contributed to the conceptualization, writing, and editing of this manuscript.Conflicts of interest
Please note that K. E. S. L. is listed as a co-inventor of a family of patents related to antibiofilm polymers (Worldwide Patent Application No. WO2016138558A1). No specific commercial activities in relation to these patents were underway at the time of writing. No funding from any external agencies, other than those listed in the acknowledgements section, has been obtained in relation to the preparation of this article.Acknowledgements
This work was supported by a CSIRO Early Research Career Postdoctoral Fellowship (L. D. B.) and a CSIRO Julius Career Award (K. E. S. L.).Notes and references
- C. L. Ventola, Phamacol. Ther., 2015, 40, 277–283 Search PubMed.
- Antimicrobial Resistance Fact Sheet, World Health Organization, 2021. https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance (accessed June 2022) Search PubMed.
- V. M. D’Costa, C. E. King, L. Kalan, M. Morar, W. W. L. Sung, C. Schwarz, D. Froese, G. Zazula, F. Calmels, R. Debruyne, G. B. Golding, H. N. Poinar and G. D. Wright, Nature, 2011, 477, 457–461 CrossRef PubMed.
- J. Perry, N. Waglechner and G. Wright, Cold Spring Harbor Perspect. Med., 2016, 6, a025197 CrossRef PubMed.
- A. P. Magiorakos, A. Srinivasan, R. B. Carey, Y. Carmeli, M. E. Falagas, C. G. Giske, S. Harbarth, J. F. Hindler, G. Kahlmeter, B. Olsson-Liljequist, D. L. Paterson, L. B. Rice, J. Stelling, M. J. Struelens, A. Vatopoulos, J. T. Weber and D. L. Monnet, Clin. Microbiol. Infect., 2012, 18, 268–281 CrossRef CAS PubMed.
- A. Chokshi, Z. Sifri, D. Cennimo and H. Horng, J. Global Infect. Dis., 2019, 11, 36–42 CrossRef PubMed.
- K. C. Caron, P. Richardson, M. B. Bodrossy, L. Voelcker, N. H. Thissen and H. T. D. Sutherland, Sensors, 2021, 21, 6625 CrossRef CAS PubMed.
- S. Hernando-Amado, T. M. Coque, F. Baquero and J. L. Martínez, Nat. Microbiol., 2019, 4, 1432–1442 CrossRef CAS PubMed.
- M. Salwiczek, Y. Qu, J. Gardiner, R. A. Strugnell, T. Lithgow, K. M. McLean and H. Thissen, Trends Biotechnol., 2014, 32, 82–90 CrossRef CAS PubMed.
- Z. K. Zander and M. L. Becker, ACS Macro Lett., 2018, 7, 16–25 CrossRef CAS PubMed.
- W.-D. Liu and B. Yang, Chin. Chem. Lett., 2017, 28, 675–690 CrossRef CAS.
- D. J. Crommelin, J. S. de Vlieger, V. Weinstein, S. Mühlebach, V. P. Shah and H. Schellekens, AAPS J., 2014, 16, 11–14 CrossRef CAS PubMed.
- K. E. B. Doncom, L. D. Blackman, D. B. Wright, M. I. Gibson and R. K. O’Reilly, Chem. Soc. Rev., 2017, 46, 4119–4134 RSC.
- B. Flühmann, I. Ntai, G. Borchard, S. Simoens and S. Mühlebach, Eur. J. Pharm. Sci., 2019, 128, 73–80 CrossRef PubMed.
- S. Mühlebach, Adv. Drug Delivery Rev., 2018, 131, 122–131 CrossRef PubMed.
- W. C. Reygaert, AIMS Microbiol., 2018, 4, 482–501 CAS.
- J. M. Munita and C. A. Arias, Microbiol. Spectrum, 2016, 4, 15 Search PubMed.
- K. Bush, Antimicrob. Agents Chemother., 2018, 62, e01076–e01018 CrossRef CAS PubMed.
- K. Poole, Ann. Med., 2007, 39, 162–176 CrossRef CAS PubMed.
- W. A. McGuinness, N. Malachowa and F. R. DeLeo, Yale J. Biol. Med., 2017, 90, 269–281 CAS.
- D. I. Andersson and D. Hughes, Nat. Rev. Microbiol., 2010, 8, 260–271 CrossRef CAS PubMed.
- N. F. Kamaruzzaman, S. Kendall and L. Good, Br. J. Pharmacol., 2017, 174, 2225–2236 CrossRef CAS PubMed.
- H.-C. Flemming and J. Wingender, Nat. Rev. Microbiol., 2010, 8, 623–633 CrossRef CAS PubMed.
- P. S. Stewart and J. William Costerton, Lancet, 2001, 358, 135–138 CrossRef CAS.
- A. Brauner, O. Fridman, O. Gefen and N. Q. Balaban, Nat. Rev. Microbiol., 2016, 14, 320–330 CrossRef CAS PubMed.
- H. Van Acker, P. Van Dijck and T. Coenye, Trends Microbiol., 2014, 22, 326–333 CrossRef CAS PubMed.
- J. Liu, O. Gefen, I. Ronin, M. Bar-Meir and N. Q. Balaban, Science, 2020, 367, 200 CrossRef CAS PubMed.
- T. K. Wood, S. J. Knabel and B. W. Kwan, Appl. Environ. Microbiol., 2013, 79, 7116–7121 CrossRef CAS PubMed.
- T. C. Barrett, W. W. K. Mok, A. M. Murawski and M. P. Brynildsen, Nat. Commun., 2019, 10, 1177 CrossRef PubMed.
- S. Santajit and N. Indrawattana, BioMed Res. Int., 2016, 2016, 2475067 Search PubMed.
- U. Gisi, Plant Pathol., 2022, 71, 131–149 CrossRef.
- M. C. Fisher, A. Alastruey-Izquierdo, J. Berman, T. Bicanic, E. M. Bignell, P. Bowyer, M. Bromley, R. Brüggemann, G. Garber, O. A. Cornely, S. J. Gurr, T. S. Harrison, E. Kuijper, J. Rhodes, D. C. Sheppard, A. Warris, P. L. White, J. Xu, B. Zwaan and P. E. Verweij, Nat. Rev. Microbiol., 2022 DOI:10.1038/s41579-022-00720-1.
- P. E. Verweij and F. L. van de Veerdonk, Lancet Respir. Med., 2022, 10, 127–128 CrossRef CAS PubMed.
- B. M. Hanson, A. Q. Dinh, T. T. Tran, S. Arenas, D. Pronty, H. B. Gershengorn, T. Ferreira, C. A. Arias and B. S. Shukla, Antimicrob. Agents Chemother., 2021, 65, e01146–e01121 CrossRef CAS PubMed.
- J. Conly and B. Johnston, Can. J. Infect. Dis. Med. Microbiol., 2005, 16, 159–160 CrossRef PubMed.
- M. McKenna, Nature, 2020, 584, 338–341 CrossRef CAS PubMed.
- M. Renwick and E. Mossialos, Expert Opin. Drug Discovery, 2018, 13, 889–892 CrossRef PubMed.
- L. S. J. Roope, R. D. Smith, K. B. Pouwels, J. Buchanan, L. Abel, P. Eibich, C. C. Butler, P. S. Tan, A. S. Walker, J. V. Robotham and S. Wordsworth, Science, 2019, 364, eaau4679 CrossRef CAS PubMed.
- B. Plackett, Nature, 2020, 586, S50–S52 CrossRef CAS.
- R. H. Stauber, S. Siemer, S. Becker, G.-B. Ding, S. Strieth and S. K. Knauer, ACS Nano, 2018, 12, 6351–6359 CrossRef CAS PubMed.
- S. Maleki Dizaj, A. Mennati, S. Jafari, K. Khezri and K. Adibkia, Adv. Pharm. Bull., 2015, 5, 19–23 Search PubMed.
- K. Gold, B. Slay, M. Knackstedt and A. K. Gaharwar, Adv. Ther., 2018, 1, 1700033 CrossRef.
- M. Álvarez-Paino, A. Muñoz-Bonilla and M. Fernández-García, Nanomaterials, 2017, 7, 48 CrossRef PubMed.
- A. Gupta, S. Mumtaz, C.-H. Li, I. Hussain and V. M. Rotello, Chem. Soc. Rev., 2019, 48, 415–427 RSC.
- K. P. Miller, L. Wang, B. C. Benicewicz and A. W. Decho, Chem. Soc. Rev., 2015, 44, 7787–7807 RSC.
- A. Al-Jumaili, S. Alancherry, K. Bazaka and M. V. Jacob, Materials, 2017, 10, 1066 CrossRef PubMed.
- E. F. Palermo, K. Lienkamp, E. R. Gillies and P. J. Ragogna, Angew. Chem., 2019, 131, 3728–3731 CrossRef.
- F. Siedenbiedel and J. C. Tiller, Polymers, 2012, 4, 46 CrossRef CAS.
- K. E. S. Locock, Aust. J. Chem., 2016, 69, 717–724 CrossRef CAS.
- C. Ergene, K. Yasuhara and E. F. Palermo, Polym. Chem., 2018, 9, 2407–2427 RSC.
- K.-S. Huang, C.-H. Yang, S.-L. Huang, C.-Y. Chen, Y.-Y. Lu and Y.-S. Lin, Int. J. Mol. Sci., 2016, 17, 1578 CrossRef PubMed.
- J. Lei, L. Sun, S. Huang, C. Zhu, P. Li, J. He, V. Mackey, D. H. Coy and Q. He, Am. J. Transl. Res., 2019, 11, 3919–3931 CAS.
- M. Mahlapuu, J. Håkansson, L. Ringstad and C. Björn, Front. Cell. Infect. Microbiol., 2016, 6, 194 Search PubMed.
- L.-j Zhang and R. L. Gallo, Curr. Biol., 2016, 26, R14–R19 CrossRef CAS PubMed.
- B. Thallinger, E. N. Prasetyo, G. S. Nyanhongo and G. M. Guebitz, Biotechnol. J., 2013, 8, 97–109 CrossRef CAS PubMed.
- B. K. Chan, S. T. Abedon and C. Loc-Carrillo, Future Microbiol., 2013, 8, 769–783 CrossRef CAS PubMed.
- F. L. Nobrega, A. R. Costa, L. D. Kluskens and J. Azeredo, Trends Microbiol., 2015, 23, 185–191 CrossRef CAS PubMed.
- D. Romero-Calle, R. Guimarães Benevides, A. Góes-Neto and C. Billington, Antibiotics, 2019, 8, 138 CrossRef CAS PubMed.
- D.-Y. Wang, H. C. van der Mei, Y. Ren, H. J. Busscher and L. Shi, Front. Chem., 2020, 7, 872 CrossRef PubMed.
- M. N. V. Ravi Kumar, React. Funct. Polym., 2000, 46, 1–27 CrossRef.
- Y. Yang, Z. Cai, Z. Huang, X. Tang and X. Zhang, Polym. J., 2018, 50, 33–44 CrossRef CAS.
- Y. Xue, H. Xiao and Y. Zhang, Int. J. Mol. Sci., 2015, 16, 3626–3655 CrossRef CAS PubMed.
- K. E. S. Locock, T. D. Michl, J. D. P. Valentin, K. Vasilev, J. D. Hayball, Y. Qu, A. Traven, H. J. Griesser, L. Meagher and M. Haeussler, Biomacromolecules, 2013, 14, 4021–4031 CrossRef CAS PubMed.
- E. F. Palermo and K. Kuroda, Appl. Microbiol. Biotechnol., 2010, 87, 1605–1615 CrossRef CAS PubMed.
- M. M. Konai, B. Bhattacharjee, S. Ghosh and J. Haldar, Biomacromolecules, 2018, 19, 1888–1917 CrossRef CAS PubMed.
- D. J. Phillips, J. Harrison, S.-J. Richards, D. E. Mitchell, E. Tichauer, A. T. M. Hubbard, C. Guy, I. Hands-Portman, E. Fullam and M. I. Gibson, Biomacromolecules, 2017, 18, 1592–1599 CrossRef CAS PubMed.
- S. J. Lam, N. M. O'Brien-Simpson, N. Pantarat, A. Sulistio, E. H. H. Wong, Y.-Y. Chen, J. C. Lenzo, J. A. Holden, A. Blencowe, E. C. Reynolds and G. G. Qiao, Nat. Microbiol., 2016, 1, 16162 CrossRef CAS PubMed.
- W. Lou, S. Venkataraman, G. Zhong, B. Ding, J. P. K. Tan, L. Xu, W. Fan and Y. Y. Yang, Acta Biomater., 2018, 78, 78–88 CrossRef CAS PubMed.
- Y. Qu, K. Locock, J. Verma-Gaur, I. D. Hay, L. Meagher and A. Traven, J. Antimicrob. Chemother., 2015, 71, 413–421 CrossRef PubMed.
- A. Kuroki, A. Kengmo Tchoupa, M. Hartlieb, R. Peltier, K. E. S. Locock, M. Unnikrishnan and S. Perrier, Biomaterials, 2019, 217, 119249 CrossRef CAS PubMed.
- G. J. Gabriel, A. E. Madkour, J. M. Dabkowski, C. F. Nelson, K. Nüsslein and G. N. Tew, Biomacromolecules, 2008, 9, 2980–2983 CrossRef CAS PubMed.
- X. Wu, S. Zhang, X. Xu, L. Shen, B. Xu, W. Qu, W. Zhuang, K. Locock, M. Deighton and Y. Qu, Front. Microbiol., 2019, 10, 2592 CrossRef PubMed.
- Y. Qu, K. Locock, J. Verma-Gaur, I. D. Hay, L. Meagher and A. Traven, J. Antimicrob. Chemother., 2015, 71, 413–421 CrossRef PubMed.
- T. D. Michl, B. Hibbs, L. Hyde, A. Postma, D. T. T. Tran, A. Zhalgasbaikyzy, K. Vasilev, L. Meagher, H. J. Griesser and K. E. S. Locock, Acta Biomater., 2020, 108, 168–177 CrossRef CAS PubMed.
- W. Chin, G. Zhong, Q. Pu, C. Yang, W. Lou, P. F. De Sessions, B. Periaswamy, A. Lee, Z. C. Liang, X. Ding, S. Gao, C. W. Chu, S. Bianco, C. Bao, Y. W. Tong, W. Fan, M. Wu, J. L. Hedrick and Y. Y. Yang, Nat. Commun., 2018, 9, 917 CrossRef PubMed.
- L. Yang, E. S. Feura, M. J. R. Ahonen and M. H. Schoenfisch, Adv. Healthcare Mater., 2018, 7, e1800155 CrossRef PubMed.
- Z. Sadrearhami, T.-K. Nguyen, R. Namivandi-Zangeneh, K. Jung, E. H. H. Wong and C. Boyer, J. Mater. Chem. B, 2018, 6, 2945–2959 RSC.
- F. T. Rong, Y. Wang, T. Feng, T. Song, J. Li and P. W. Huang, Antioxidants, 2019, 8, 556 CrossRef CAS PubMed.
- M. J. R. Ahonen, J. M. Dorrier and M. H. Schoenfisch, ACS Infect. Dis., 2019, 5, 1327–1335 CrossRef CAS PubMed.
- Z. Sadrearhami, J. Yeow, T.-K. Nguyen, K. K. K. Ho, N. Kumar and C. Boyer, Chem. Commun., 2017, 53, 12894–12897 RSC.
- Z. Shen, K. He, Z. Ding, M. Zhang, Y. Yu and J. Hu, Macromolecules, 2019, 52, 7668–7677 CrossRef CAS.
- S. P. Nichols, W. L. Storm, A. Koh and M. H. Schoenfisch, Adv. Drug Delivery Rev., 2012, 64, 1177–1188 CrossRef CAS PubMed.
- T. Yang, A. N. Zelikin and R. Chandrawati, Adv. Sci., 2018, 5, 1701043 CrossRef PubMed.
- F. C. Fang, J. Clin. Invest., 1997, 99, 2818–2825 CrossRef CAS PubMed.
- I. Gusarov, K. Shatalin, M. Starodubtseva and E. Nudler, Science, 2009, 325, 1380–1384 CrossRef CAS PubMed.
- M. A. De Groote, T. Testerman, Y. Xu, G. Stauffer and F. C. Fang, Science, 1996, 272, 414–417 CrossRef CAS PubMed.
- M. A. De Groote, D. Granger, Y. Xu, G. Campbell, R. Prince and F. C. Fang, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 6399–6403 CrossRef CAS PubMed.
- B. J. Privett, A. D. Broadnax, S. J. Bauman, D. A. Riccio and M. H. Schoenfisch, Nitric oxide, 2012, 26, 169–173 CrossRef CAS PubMed.
- F. Hui and C. Debiemme-Chouvy, Biomacromolecules, 2013, 14, 585–601 CrossRef CAS PubMed.
- A. Dong, Y.-J. Wang, Y. Gao, T. Gao and G. Gao, Chem. Rev., 2017, 117, 4806–4862 CrossRef CAS PubMed.
- M. Natan, O. Gutman, R. Lavi, S. Margel and E. Banin, ACS Nano, 2015, 9, 1175–1188 CrossRef CAS PubMed.
- C. Li, L. Xue, Q. Cai, S. Bao, T. Zhao, L. Xiao, G. Gao, C. Harnoode and A. Dong, RSC Adv., 2014, 4, 47853–47864 RSC.
- O. Gutman, M. Natan, E. Banin and S. Margel, Biomaterials, 2014, 35, 5079–5087 CrossRef CAS PubMed.
- Z. Cao and Y. Sun, ACS Appl. Mater. Interfaces, 2009, 1, 494–504 CrossRef CAS PubMed.
- J. Yao and Y. Sun, Ind. Eng. Chem. Res., 2008, 47, 5819–5824 CrossRef CAS.
- E.-R. Kenawy, S. D. Worley and R. Broughton, Biomacromolecules, 2007, 8, 1359–1384 CrossRef CAS PubMed.
- L. D'Lima, L. Friedman, L. Wang, P. Xu, M. Anderson and D. Debabov, Antimicrob. Agents Chemother., 2012, 56, 2753–2755 CrossRef PubMed.
- R. Yañez-Macías, A. Muñoz-Bonilla, M. A. De Jesús-Tellez, H. Maldonado-Textle, C. Guerrero-Sánchez, U. S. Schubert and R. Guerrero-Santos, Polymers, 2019, 11, 1789 CrossRef PubMed.
- Z. Hou, Y. V. Shankar, Y. Liu, F. Ding, J. L. Subramanion, V. Ravikumar, R. Zamudio-Vázquez, D. Keogh, H. Lim, M. Y. F. Tay, S. Bhattacharjya, S. A. Rice, J. Shi, H. Duan, X.-W. Liu, Y. Mu, N. S. Tan, K. C. Tam, K. Pethe and M. B. Chan-Park, ACS Appl. Mater. Interfaces, 2017, 9, 38288–38303 CrossRef CAS PubMed.
- Y. Su, L. Tian, M. Yu, Q. Gao, D. Wang, Y. Xi, P. Yang, B. Lei, P. X. Ma and P. Li, Polym. Chem., 2017, 8, 3788–3800 RSC.
- R. Namivandi-Zangeneh, Z. Sadrearhami, D. Dutta, M. Willcox, E. H. H. Wong and C. Boyer, ACS Infect. Dis., 2019, 5, 1357–1365 CrossRef CAS PubMed.
- Y. Wang, M. Yin, X. Lin, L. Li, Z. Li, X. Ren and Y. Sun, J. Colloid Interface Sci., 2019, 533, 604–611 CrossRef CAS PubMed.
- X. Dong, W. Liang, M. J. Meziani, Y.-P. Sun and L. Yang, Theranostics, 2020, 10, 671–686 CrossRef CAS PubMed.
- H. Ji, H. Sun and X. Qu, Adv. Drug Delivery Rev., 2016, 105, 176–189 CrossRef CAS PubMed.
- F. De Maio, V. Palmieri, M. De Spirito, G. Delogu and M. Papi, Expert Rev. Med. Devices, 2019, 16, 863–875 CrossRef CAS PubMed.
- Y. J. Tang, J. M. Ashcroft, D. Chen, G. Min, C.-H. Kim, B. Murkhejee, C. Larabell, J. D. Keasling and F. F. Chen, Nano Lett., 2007, 7, 754–760 CrossRef CAS PubMed.
- Y. Iwamoto and Y. Yamakoshi, Chem. Commun., 2006, 4805–4807 RSC.
- G. P. Tegos, T. N. Demidova, D. Arcila-Lopez, H. Lee, T. Wharton, H. Gali and M. R. Hamblin, Chem. Biol., 2005, 12, 1127–1135 CrossRef CAS PubMed.
- S. Kang, M. Herzberg, D. F. Rodrigues and M. Elimelech, Langmuir, 2008, 24, 6409–6413 CrossRef CAS PubMed.
- X. Zou, L. Zhang, Z. Wang and Y. Luo, J. Am. Chem. Soc., 2016, 138, 2064–2077 CrossRef CAS PubMed.
- V. T. H. Pham, V. K. Truong, M. D. J. Quinn, S. M. Notley, Y. Guo, V. A. Baulin, M. Al Kobaisi, R. J. Crawford and E. P. Ivanova, ACS Nano, 2015, 9, 8458–8467 CrossRef CAS PubMed.
- Q. Ye, F. Zhou and W. Liu, Chem. Soc. Rev., 2011, 40, 4244–4258 RSC.
- J.-W. Xu, K. Yao and Z.-K. Xu, Nanoscale, 2019, 11, 8680–8691 RSC.
- S. Szunerits, A. Barras and R. Boukherroub, Int. J. Environ. Res. Public Health, 2016, 13, 413 CrossRef PubMed.
- B. Zhu, X. Xia, N. Xia, S. Zhang and X. Guo, Environ. Sci. Technol., 2014, 48, 4086–4095 CrossRef CAS PubMed.
- J. Fang, D. Y. Lyon, M. R. Wiesner, J. Dong and P. J. J. Alvarez, Environ. Sci. Technol., 2007, 41, 2636–2642 CrossRef CAS PubMed.
- R. S. Chouhan, A. Qureshi, B. Yagci, M. A. Gülgün, V. Ozguz and J. H. Niazi, Chem. Eng. J., 2016, 298, 1–9 CrossRef CAS.
- Q. Zhang and C. Zhang, Environ. Sci. Technol., 2020, 54, 12412–12422 CrossRef CAS PubMed.
- M. S. Mauter and M. Elimelech, Environ. Sci. Technol., 2008, 42, 5843–5859 CrossRef CAS PubMed.
- P. V. Baptista, M. P. McCusker, A. Carvalho, D. A. Ferreira, N. M. Mohan, M. Martins and A. R. Fernandes, Front. Microbiol., 2018, 9, 1441 CrossRef PubMed.
- J. M. V. Makabenta, A. Nabawy, C.-H. Li, S. Schmidt-Malan, R. Patel and V. M. Rotello, Nat. Rev. Microbiol., 2021, 19, 23–36 CrossRef CAS PubMed.
- J. A. Lemire, J. J. Harrison and R. J. Turner, Nat. Rev. Microbiol., 2013, 11, 371–384 CrossRef CAS PubMed.
- T. W. Clarkson, Annu. Rev. Pharmacol. Toxicol., 1993, 33, 545–571 CrossRef CAS PubMed.
- A. Panáček, L. Kvítek, M. Smékalová, R. Večeřová, M. Kolář, M. Röderová, F. Dyčka, M. Šebela, R. Prucek, O. Tomanec and R. Zbořil, Nat. Nanotechnol., 2018, 13, 65–71 CrossRef PubMed.
- Z. Guo, G. Zeng, K. Cui and A. Chen, Environ. Chem. Lett., 2019, 17, 319–333 CrossRef CAS.
- M. Salas-Orozco, N. Niño-Martínez, G.-A. Martínez-Castañón, F. T. Méndez, M. E. C. Jasso and F. Ruiz, J. Nanomater., 2019, 2019, 7630316 Search PubMed.
- Z. Ferdous and A. Nemmar, Int. J. Mol. Sci., 2020, 21, 2375 CrossRef CAS PubMed.
- S. Prabhu and E. K. Poulose, Int. Nano Lett., 2012, 2, 32 CrossRef.
- T. C. Dakal, A. Kumar, R. S. Majumdar and V. Yadav, Front. Microbiol., 2016, 7, 1831 Search PubMed.
- I.-s Hwang, J. Lee, J. H. Hwang, K.-J. Kim and D. G. Lee, FEBS J., 2012, 279, 1327–1338 CrossRef CAS PubMed.
- W. B. Sim, R. T. Blaskovich and M. Z. M. Ziora, Antibiotics, 2018, 7, 93 CrossRef CAS PubMed.
- A. E.-D. M. Hosny, S. A. Rasmy, D. S. Aboul-Magd, M. T. Kashef and Z. E. El-Bazza, Infect. Drug Resist., 2019, 12, 1985–2001 CrossRef PubMed.
- C. A. P. Bastos, N. Faria, J. Wills, P. Malmberg, N. Scheers, P. Rees and J. J. Powell, NanoImpact, 2020, 17, 100192 CrossRef.
- R. K. Sharma and R. Ghose, Ceram. Int., 2015, 41, 967–975 CrossRef CAS.
- E. Teirlinck, R. Xiong, T. Brans, K. Forier, J. Fraire, H. Van Acker, N. Matthijs, R. De Rycke, S. C. De Smedt, T. Coenye and K. Braeckmans, Nat. Commun., 2018, 9, 4518 CrossRef PubMed.
- A. B. Kelson, M. Carnevali and V. Truong-Le, Curr. Opin. Pharmacol., 2013, 13, 707–716 CrossRef CAS PubMed.
- A. Bijelic, M. Aureliano and A. Rompel, Chem. Commun., 2018, 54, 1153–1169 RSC.
- D. H. Nies, Appl. Microbiol. Biotechnol., 1999, 51, 730–750 CrossRef CAS PubMed.
- H. Van Acker and T. Coenye, Trends Microbiol., 2017, 25, 456–466 CrossRef CAS PubMed.
- T. Pennanen, A. Frostegard, H. Fritze and E. Baath, Appl. Environ. Microbiol., 1996, 62, 420–428 CrossRef CAS PubMed.
- N. Hachicho, P. Hoffmann, K. Ahlert and H. J. Heipieper, FEMS Microbiol. Lett., 2014, 355, 71–77 CrossRef CAS PubMed.
- F.-Y. Su, S. Srinivasan, B. Lee, J. Chen, A. J. Convertine, T. E. West, D. M. Ratner, S. J. Skerrett and P. S. Stayton, J. Controlled Release, 2018, 287, 1–11 CrossRef CAS PubMed.
- M. R. E. Santos, A. C. Fonseca, P. V. Mendonça, R. Branco, A. C. Serra, P. V. Morais and J. F. J. Coelho, Materials, 2016, 9, 599 CrossRef PubMed.
- M.-H. Xiong, Y. Bao, X.-Z. Yang, Y.-H. Zhu and J. Wang, Adv. Drug Delivery Rev., 2014, 78, 63–76 CrossRef CAS PubMed.
- M. Martínez-Carmona, Y. K. Gun'ko and M. Vallet-Regí, Pharmaceutics, 2018, 10, 279 CrossRef PubMed.
- A. Bernardos, E. Piacenza, F. Sancenón, M. Hamidi, A. Maleki, R. J. Turner and R. Martínez-Máñez, Small, 2019, 15, 1900669 CrossRef PubMed.
- D. Gupta, A. Singh and A. U. Khan, Nanoscale Res. Lett., 2017, 12, 454 CrossRef PubMed.
- J. M. V. Makabenta, A. Nabawy, C.-H. Li, S. Schmidt-Malan, R. Patel and V. M. Rotello, Nat. Rev. Microbiol., 2021, 19, 23–36 CrossRef CAS PubMed.
- M.-H. Xiong, Y.-J. Li, Y. Bao, X.-Z. Yang, B. Hu and J. Wang, Adv. Mater., 2012, 24, 6175–6180 CrossRef CAS PubMed.
- Y.-C. Yeh, T.-H. Huang, S.-C. Yang, C.-C. Chen and J.-Y. Fang, Front. Chem., 2020, 8, 286 CrossRef CAS PubMed.
- M. A. Dos Santos Ramos, P. B. Da Silva, L. Spósito, L. G. De Toledo, B. V. Bonifácio, C. F. Rodero, K. C. Dos Santos, M. Chorilli and T. M. Bauab, Int. J. Nanomed., 2018, 13, 1179–1213 CrossRef CAS PubMed.
- A. B. Seabra, M. T. Pelegrino and P. S. Haddad, in Nanostructures for Antimicrobial Therapy, ed. A. Ficai and A. M. Grumezescu, Elsevier, 2017, ch. 24, pp. 531–550 Search PubMed.
- H. Park, H.-J. Park, J. A. Kim, S. H. Lee, J. H. Kim, J. Yoon and T. H. Park, J. Microbiol. Methods, 2011, 84, 41–45 CrossRef CAS PubMed.
- T.-K. Nguyen, H. T. T. Duong, R. Selvanayagam, C. Boyer and N. Barraud, Sci. Rep., 2015, 5, 18385 CrossRef CAS PubMed.
- D. G. Meeker, S. V. Jenkins, E. K. Miller, K. E. Beenken, A. J. Loughran, A. Powless, T. J. Muldoon, E. I. Galanzha, V. P. Zharov, M. S. Smeltzer and J. Chen, ACS Infect. Dis., 2016, 2, 241–250 CrossRef CAS PubMed.
- L. D. Blackman, Z. Y. Oo, Y. Qu, P. A. Gunatillake, P. Cass and K. E. S. Locock, ACS Appl. Mater. Interfaces, 2020, 12, 11353–11362 CrossRef CAS PubMed.
- T. Zhang, Y. Qu, P. A. Gunatillake, P. Cass, K. E. S. Locock and L. D. Blackman, Sci. Rep., 2020, 10, 15796 CrossRef CAS PubMed.
- K. Langowska, C. G. Palivan and W. Meier, Chem. Commun., 2013, 49, 128–130 RSC.
- M. Potter, A. Najer, A. Klöckner, S. Zhang, M. N. Holme, V. Nele, J. Che, L. Massi, J. Penders, C. Saunders, J. J. Doutch, A. M. Edwards, O. Ces and M. M. Stevens, ACS Nano, 2020, 14, 17333–17353 CrossRef CAS PubMed.
- L. D. Blackman, S. Varlas, M. C. Arno, Z. H. Houston, N. L. Fletcher, K. J. Thurecht, M. Hasan, M. I. Gibson and R. K. O’Reilly, ACS Cent. Sci., 2018, 4, 718–723 CrossRef CAS PubMed.
- H. Sun, Y. Wang and J. Song, Polymers, 2021, 13, 2903 CrossRef CAS PubMed.
- R. S. Gaspar, B. Silva-Lima, F. Magro, A. Alcobia, F. L. da Costa and J. Feio, Front. Med., 2020, 7, 590527 CrossRef PubMed.
- L. Hussaarts, S. Mühlebach, V. P. Shah, S. McNeil, G. Borchard, B. Flühmann, V. Weinstein, S. Neervannan, E. Griffiths, W. Jiang, E. Wolff-Holz, D. J. A. Crommelin and J. S. B. de Vlieger, Ann. N. Y. Acad. Sci., 2017, 1407, 39–49 CrossRef PubMed.
- S. Mühlebach, Adv. Drug Delivery Rev., 2018, 131, 122–131 CrossRef PubMed.
- B. Flühmann, I. Ntai, G. Borchard, S. Simoens and S. Mühlebach, Eur. J. Pharm. Sci., 2019, 128, 73–80 CrossRef PubMed.
- K. Klein, P. Stolk, M. L. De Bruin, H. G. M. Leufkens, D. J. A. Crommelin and J. S. B. De Vlieger, Eur. J. Pharm. Sci., 2019, 133, 228–235 CrossRef CAS PubMed.
- R. Foulkes, E. Man, J. Thind, S. Yeung, A. Joy and C. Hoskins, Biomater. Sci., 2020, 8, 4653–4664 RSC.
- S. P. Reddy, C. Ni and J. J. Wu, in Therapy for Severe Psoriasis, ed. J. J. Wu, S. R. Feldman and M. G. Lebwohl, Elsevier, 2016, ch. 14, pp. 163–170 Search PubMed.
- G. E. Marchant, B. R. Allenby and J. R. Herkert, The Growing Gap Between Emerging Technologies and Legal-Ethical Oversight, Springer, Dordrecht, 2011 Search PubMed.
- S. J. Projan, Curr. Opin. Microbiol., 2003, 6, 427–430 CrossRef PubMed.
- M. Faria, M. Björnmalm, K. J. Thurecht, S. J. Kent, R. G. Parton, M. Kavallaris, A. P. R. Johnston, J. J. Gooding, S. R. Corrie, B. J. Boyd, P. Thordarson, A. K. Whittaker, M. M. Stevens, C. A. Prestidge, C. J. H. Porter, W. J. Parak, T. P. Davis, E. J. Crampin and F. Caruso, Nat. Nanotechnol., 2018, 13, 777–785 CrossRef CAS PubMed.
- J. P. Patterson, M. P. Robin, C. Chassenieux, O. Colombani and R. K. O'Reilly, Chem. Soc. Rev., 2014, 43, 2412–2425 RSC.
- A. Ali, M. Ovais, X. Cui, Y. Rui and C. Chen, Chem. Res. Toxicol., 2020, 33, 1082–1109 Search PubMed.
- H. Meng, T. Xia, S. George and A. E. Nel, ACS Nano, 2009, 3, 1620–1627 CrossRef CAS PubMed.
- G. Chen, W. Peijnenburg, Y. Xiao and M. G. Vijver, Int. J. Mol. Sci., 2017, 18, 1504 CrossRef PubMed.
- E. Burello and A. Worth, Nat. Nanotechnol., 2011, 6, 138–139 CrossRef CAS PubMed.
- J.-S. Choi, M. K. Ha, T. X. Trinh, T. H. Yoon and H.-G. Byun, Sci. Rep., 2018, 8, 6110 CrossRef PubMed.
- M. S. McKee and J. Filser, Environ. Sci.: Nano, 2016, 3, 506–533 RSC.
- E. S. Lee, B. R. Park, J. S. Kim, G. Y. Choi, J. J. Lee and I. S. Lee, Curr. Med. Res. Opin., 2013, 29, 141–147 CrossRef CAS PubMed.
- J. Rottembourg, A. Kadri, E. Leonard, A. Dansaert and A. Lafuma, Nephrol., Dial., Transplant., 2011, 26, 3262–3267 CrossRef CAS PubMed.
- R. F. Landis, C.-H. Li, A. Gupta, Y.-W. Lee, M. Yazdani, N. Ngernyuang, I. Altinbasak, S. Mansoor, M. A. S. Khichi, A. Sanyal and V. M. Rotello, J. Am. Chem. Soc., 2018, 140, 6176–6182 CrossRef CAS PubMed.
- L. Chen, Y. Yang, P. Zhang, S. Wang, J.-F. Xu and X. Zhang, ACS Appl. Bio Mater., 2019, 2, 2920–2926 CrossRef CAS PubMed.
- C. A. Hae Cho, C. Liang, J. Perera, J. Liu, K. G. Varnava, V. Sarojini, R. P. Cooney, D. J. McGillivray, M. A. Brimble, S. Swift and J. Jin, Biomacromolecules, 2018, 19, 1389–1401 CrossRef CAS PubMed.
- M. Vatankhah-Varnosfaderani, X. Hu, Q. Li, H. Adelnia, M. Ina and S. S. Sheiko, ACS Appl. Mater. Interfaces, 2018, 10, 20869–20875 CrossRef CAS PubMed.
- L. D. Blackman, Y. Qu, P. Cass and K. E. S. Locock, Chem. Soc. Rev., 2021, 50, 1587–1616 RSC.
- D. Paul, Y. S. Kim, K. Ponnusamy and J. H. Kweon, Environ. Eng. Sci., 2009, 26, 1319–1324 CrossRef CAS.
- E. Paluch, J. Rewak-Soroczyńska, I. Jędrusik, E. Mazurkiewicz and K. Jermakow, Appl. Microbiol. Biotechnol., 2020, 104, 1871–1881 CrossRef CAS PubMed.
- N. Niño-Martínez, M. F. Salas Orozco, G.-A. Martínez-Castañón, F. Torres Méndez and F. Ruiz, Int. J. Mol. Sci., 2019, 20, 2808 CrossRef PubMed.
- C. Zhang, R. Sun and T. Xia, Nano Today, 2020, 34, 100909 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |