
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anodic generation of hydrogen peroxide in continuous flow†
Dhananjai
Pangotra
 ab,
Lénárd-István
Csepei
a,
Arne
Roth
a,
Volker
Sieber
ab and
Luciana
Vieira
*a
ab,
Lénárd-István
Csepei
a,
Arne
Roth
a,
Volker
Sieber
ab and
Luciana
Vieira
*a
aFraunhofer Institute of Interfacial Engineering and Biotechnology IGB, Bio-, Electro-, and Chemocatalysis BioCat, Straubing Branch, Schulgasse 11a, 94315, Straubing, Germany. E-mail: luciana.vieira@igb.fraunhofer.de
bChair of Chemistry for Biogenic Resources, Campus Straubing for Biotechnology and Sustainability, Technical University of Munich, Schulgasse 16, 94315, Straubing, Germany
First published on 18th August 2022
Abstract
The electrochemical production of hydrogen peroxide (H2O2) is an appealing green alternative to the classic anthraquinone process. Herein, we show the development of a process to produce H2O2 anodically in continuous flow at high current densities. The role of CO32− ion activity in enhancing the anodic H2O2 generation using a commercial boron-doped diamond (BDD) electrode is investigated in detail. The process development comprising the optimization of electrolyte flow type and flow rates enabled electrochemical operation at current densities up to 700 mA cm−2, with Faraday efficiencies up to 78%, and the highest-ever reported H2O2 production rate of 79 μmol min−1 cm−2. Continuous flow experiments are essential for technical applications, which is one of the first upscaling steps. A continuous and stable H2O2 productivity with constant production rates for up to 28 hours was achieved. Our experiments unfold the importance of electrochemical process development and the interplay of the electrode, electrolyte, and operating cell parameters to achieve a highly efficient, scalable, stable, and continuous system for the 2e− water oxidation to H2O2.
Introduction
Hydrogen peroxide (H2O2) is a widely used chemical with a market evaluated at $4 billion in 2020, forecasted to grow to $5.2 billion by 2026.1 Its strong oxidizing capability enables a broad application in the chemical industry. Moreover, because it releases only water (H2O) and oxygen (O2) as a side-product, H2O2 is extensively used as a green oxidant. Industrial applications include drinking water2,3 and wastewater treatment,4,5 bleaching,6 desulfurization of conventional energy carriers7 and bio-based feedstocks,8,9 sanitation,10 organic synthesis,11–13 and aerospace fuels.14 New niche of applications in catalyst synthesis,15 biofuel generation,16 and desulfurization of biogas17 are also expected to ramp up the demand for H2O2 in the future. Hence, in the context of these broad application spectra, H2O2 is among the 100 most essential chemicals globally.18,19The industrial production of H2O2 is mainly through the anthraquinone autoxidation (AO) process. The AO-process is considerably energy-demanding (aggregate consumption up to 17.6 kW h kg−1 H2O2),20 uses large amounts of organic solvents and involves remarkable risks associated with hydrogenation and oxidation reactions under high H2/O2 pressure.21 Furthermore, it requires expensive palladium-based catalysts and complex, large-scale equipment.22 The industrial AO process involves distillation of H2O2 to generate large volumes of concentrated solution (from 40 to 70 wt%),23 which is required for its transportation and very specific applications. Nevertheless, the primary use of H2O2 in bleaching and disinfectant requires H2O2 concentrations from 3 to 8 wt%.24,25 While transportation of H2O2 in high concentrations is risky, shipping in lower concentrations poses the disadvantage of moving mainly water, thus negatively affecting the carbon footprint and economic viability of the product. Therefore, a portable device for decentralized and on-demand H2O2 production only based on water, air, and (renewable) electric energy as input provides a desirable local and “green” solution for the supply of H2O2 where it is needed.
The electrochemical production of H2O2 is based on water, air, and renewable electric energy as primary feedstocks. Therefore, it is an elegant green solution for onsite and on-demand H2O2 production, also avoiding risks associated with large-scale storage and transportation of highly concentrated H2O2 solutions.22 There are two electrochemical routes for H2O2 production: the cathodic oxygen reduction reaction (ORR, eqn (1)) and the anodic water oxidation reaction (WOR, eqn (2)). Both pathways involve a two-electron transfer reaction.22 The WOR pathway at the anode is particularly attractive because it can be coupled with cathodic reactions running on large scales, such as the CO2 reduction reaction (CO2RR) and the water electrolysis for hydrogen evolution reaction (HER).26 Currently, both reactions are coupled with WOR to O2, which besides being harmless, is an economically insignificant half-cell reaction and utilizes expensive and critical electrode materials such as iridium.27,28 Combining anodic and cathodic processes that generate value-added products can enhance the overall cost efficiency of the whole production process.
| O2 + 2H+ + 2e− → H2O2 E° = 0.67 V vs. RHE | (1) |
| 2H2O ⇌ H2O2 + 2H+ + 2e− E° = 1.76 V vs. RHE | (2) |
The electrochemical WOR to H2O2 has gained massive interest in the last years. A strong focus on catalyst development has been crucial for reaction improvement leading to a significant increase in efficiency, selectivity, and stability. Catalysts based on metal oxides such as CaSnO3,29–31 BiVO4,32 and ZnO,33 as well as on metal porphyrin complexes of Ge34 and Al35 have been reported to be highly active catalysts for WOR. Another class of promising materials are boron-doped diamond (BDD) electrodes.36–38 BDDs are robust electrodes with remarkably high overpotential for oxygen evolution reaction (OER). By suppressing O2 evolution, high efficiency for H2O2 production is obtained on BDD.39 Such materials have been used as anode for waste-water treatment for a long time.40 Recent progress on the properties of BDD materials has significantly enhanced H2O2 production performance, leading to outstanding production rates of 74.6 μmol min−1 cm−2 H2O2 and peak faradaic efficiency (FE) of 87%.38
Nevertheless, all reports of WOR to H2O2 on BDD have so far been carried out in electrochemical H-cell setups.37,38 In this context, the process transfer to electrochemical flow cells would be the first step toward technical applications, continuous production and scale-up.41 Despite the remarkable benefits of electrochemical flow systems in terms of their operational conditions and scalability potential, only a few studies on WOR in flow cells have been reported up to date.36,42
Herein, we exploit the remarkable properties of a commercial BDD electrode for the anodic production of H2O2 in a scalable and continuous flow process. Our previous work reported a direct relationship between the ionic activity of CO32− and enhanced anodic H2O2 production on commercial carbon fiber paper (CFP) anodes.42 A cyclic mechanism of H2O2 production catalyzed by carbonate ions was proposed through the formation of peroxodicarbonate species (C2O62−) (eqn (3) and (4)).42
| 2CO32− → C2O62− + 2e− E° = 1.80 V vs. RHE | (3) |
| C2O62− + 2H2O → H2O2 + 2HCO3− | (4) |
This mechanism was based on the correlation between the calculated activity of carbonate ions (CO32−) in the electrolyte and the observed H2O2 formation rate, later confirmed by in situ infrared spectroscopy (ATR-FTIR) by Gill et al.43
In the present study, we extended the scope to show a continuous flow process using a commercial BDD electrode in combination with an optimized carbonate-based electrolyte system. We investigated and compared different flow setups (circular and single-pass) as well as the effect of flow rate on FE and production rate in continuous mode at current densities of up to 700 mA cm−2. Our experimental results show the crucial importance of the electrochemical process development beyond catalyst and electrolyte.
Experimental section
Materials
Potassium hydrogen carbonate (KHCO3, Sigma Aldrich, 99.5%), potassium carbonate (K2CO3, ReagentPlus®, 99%), titanium(IV) oxysulfate (TiOSO4, ≥29% Ti (as TiO2) basis, technical), and sodium metasilicate (Na2SiO3) were purchased from Sigma Aldrich. Potassium hydroxide (KOH, ≥85%) was obtained from Carl Roth. All materials and chemicals were used as received.Electrochemical experiments
Electrochemical measurements were performed using an Autolab PGSTAT128N potentiostat/galvanostat equipped with a 10 A booster. A two-compartment H-cell divided by Nafion 117 (Ion Power, Germany) cation exchange membrane (CEM) was used for preliminary studies. The anode was a 5 cm2 (geometric area) BDD electrode sputtered on a Si substrate and with boron doping between 2000 to 5000 ppm (DIACHEM®, Condias GmbH, Fig. S1, ESI†). The auxiliary cathode was an IrO2/Ti mesh, and the reference electrode a Ag/AgCl system in 3.5 mol L−1 KCl (eDAQ). The gap between cathode and anode was 6 cm. Each compartment of the H-cell was filled with 25 mL of electrolyte. Linear sweep voltammetry (LSV) was recorded at a scan rate of 100 mV s−1. Galvanostatic experiments were carried out at current densities ranging from 10 to 300 mA cm−2 using bicarbonate (HCO3−) and carbonate (CO32−) electrolytes. Current densities were calculated based on the geometric area of the electrode. Chronopotentiometry measurements were performed under a constant stirring rate of 1000 rpm with a Teflon coated magnetic stirrer. An ice bath was used to avoid thermal decomposition of generated H2O2. The potentials are reported against the reversible hydrogen electrode (RHE), calculated by the eqn (5).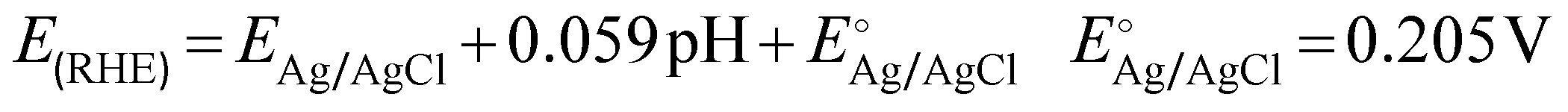 | (5) |
Experiments under flow were performed using a microflow cell (Electrocell, Denmark) with a unique design from Siemens Energy for the CO2EXIDE project (Horizon 2020, grant agreement no. 768789). The cell was equipped with 10 cm2 (3 × 3.5 cm) electrodes (cathode and anode) separated by a Nafion 117 membrane. A carbon electrode was used as a cathode, and DIACHEM® BDD/Ta (2000–5000 ppm B doping, Condias GmbH) was used as an anode, with an electrode distance of 8 mm. If not otherwise specified, the anolyte and catholyte volume were 200 mL for each half-cell. The electrolyte flow was controlled by a flow pump (Watson-Marlow) in a flow range varying from 2 to 120 mL min−1. The pH and conductivity of the electrolyte were monitored with a pH-meter (VWR pH 3210) and a conductometer (VWR pHenomenal® CO 3100 H), respectively. All flow experiments were carried out with 2 mol L−1 K2CO3. The electrolyte flow setup was varied as circular or single-pass. Circular flow experiments were performed at a constant flow rate of 100 mL min−1, with Na2SiO3 as a stabilizer in the electrolyte and regulated at a pH of 12.6. The single-pass flow rate was varied between 5 and 100 mL min−1. Moreover, in single-pass flow experiments, the electrolyte containing H2O2 was collected in a separate reservoir. Anolyte samples were collected periodically after having passed the cell and before collecting in the reservoir.
Quantitative product analysis
The quantification of the anodically generated H2O2 was performed using the TiOSO4 method as described in the literature.39,44,45 In short, 0.1 mol L−1 TiOSO4 in 2 mol L−1 H2SO4 solution was prepared prior to the H2O2 detection. 25 μL of anolyte sample was mixed with 975 μL of 0.1 mol L−1 TiOSO4 solution in a standard cuvette. In the presence of H2O2, a color change from colorless to yellow is observed due to the formation of pertitanic acid (eqn (6)). The absorbance of pertitanic acid was measured at 407 nm using a Shimadzu UV-1800 spectrophotometer. The molar extinction coefficient (ε407nm) is 6.89 × 102 L mol−1 cm−1.46| H2O2 + TiO2+ + H2O → H2TiO4 + 2H+ | (6) |
The faradaic efficiency (FE) was calculated with eqn (7):
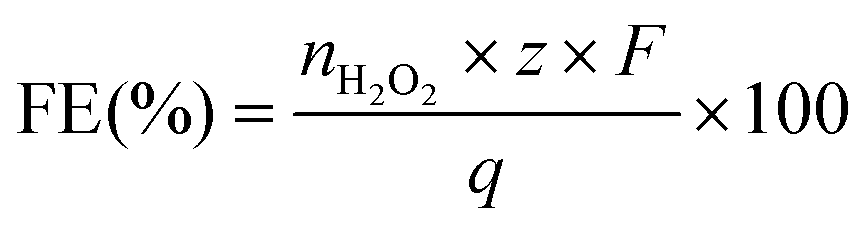 | (7) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), and q is the total charge passed (in Coulombs).
485 C mol−1), and q is the total charge passed (in Coulombs).
The H2O2 production rate is given by eqn (8):
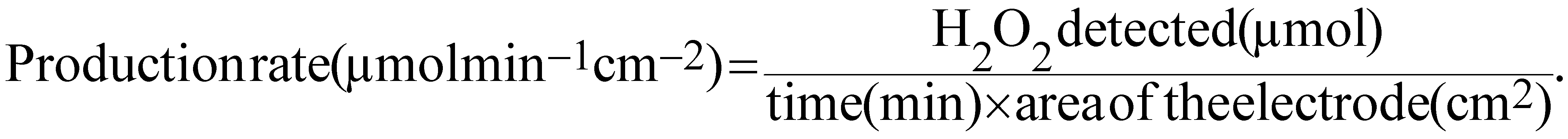 | (8) |
The energy consumption was calculated according to eqn (9).
 | (9) |
Calculation of ionic activities for HCO3− and CO32−
The ionic activity of the bicarbonate (a(HCO3−)) and carbonate (a(CO32−)) ions were calculated as previous reported (eqn (S1)–(S6), ESI†).42Results and discussion
With the objective of achieving unprecedented levels of performance for the anodic H2O2 generation under technically relevant and scalable operational conditions, this study is divided into two parts: (i) a preliminary evaluation of the electrolyte in an H-cell configuration and (ii) an electrochemical process optimization in a continuous flow reactor. In the first part, the effect of the electrolyte composition on WOR to H2O2 is qualitatively and quantitatively evaluated. In the second part, the process is transferred to a continuous flow reactor under optimized electrolyte conditions, where the effect of flow setup and flow rate is investigated and optimized towards higher anodic H2O2 productivity.The electrolyte effect
Bicarbonate solutions have been widely used for WOR to H2O2.37,43,47 The molar fraction of the bicarbonate (HCO3−)/carbonate (CO32−) species in solution is dependent on the pH of the electrolyte (eqn (10) and (11)).| H2CO3 ⇌ HCO3− + H+ (pKa,1 = 3.7) | (10) |
| HCO3− ⇌ CO32− + H+ (pKa,2 = 10.3) | (11) |
Basic pHs favor higher activity of carbonate ions, which promotes the WOR to H2O2 through the intermediate formation of peroxodicarbonate species (C2O62−, Scheme S1, ESI†).42,43 Although the benefits of high pH are significant, it can be detrimental due to the low stability of H2O2 under alkaline conditions. Therefore, a chemical stabilizer was added to the anolyte to decelerate the alkaline decomposition of H2O2.42
WOR in 2 mol L−1 solutions of bicarbonate (KHCO3, at pH 8.4) and carbonate (K2CO3, at pH 12.6) electrolytes were evaluated by linear sweep voltammetry (LSV) on BDD electrodes (Fig. 1a). The two electrolytes show a distinct current density profile. At +3.2 V vs. RHE, the current density is substantially higher for K2CO3 (155 mA cm−2) compared to KHCO3 (140 mA cm−2). The onset potential, defined as the potential where the current density starts to increase exponentially, is also considerably lower for carbonate (+2.45 V vs. RHE) than for bicarbonate solutions (+2.66 V vs. RHE). Both aspects, higher current density and low onset potential, indicate a higher electrochemical activity for WOR in K2CO3 electrolytes compared to KHCO3.
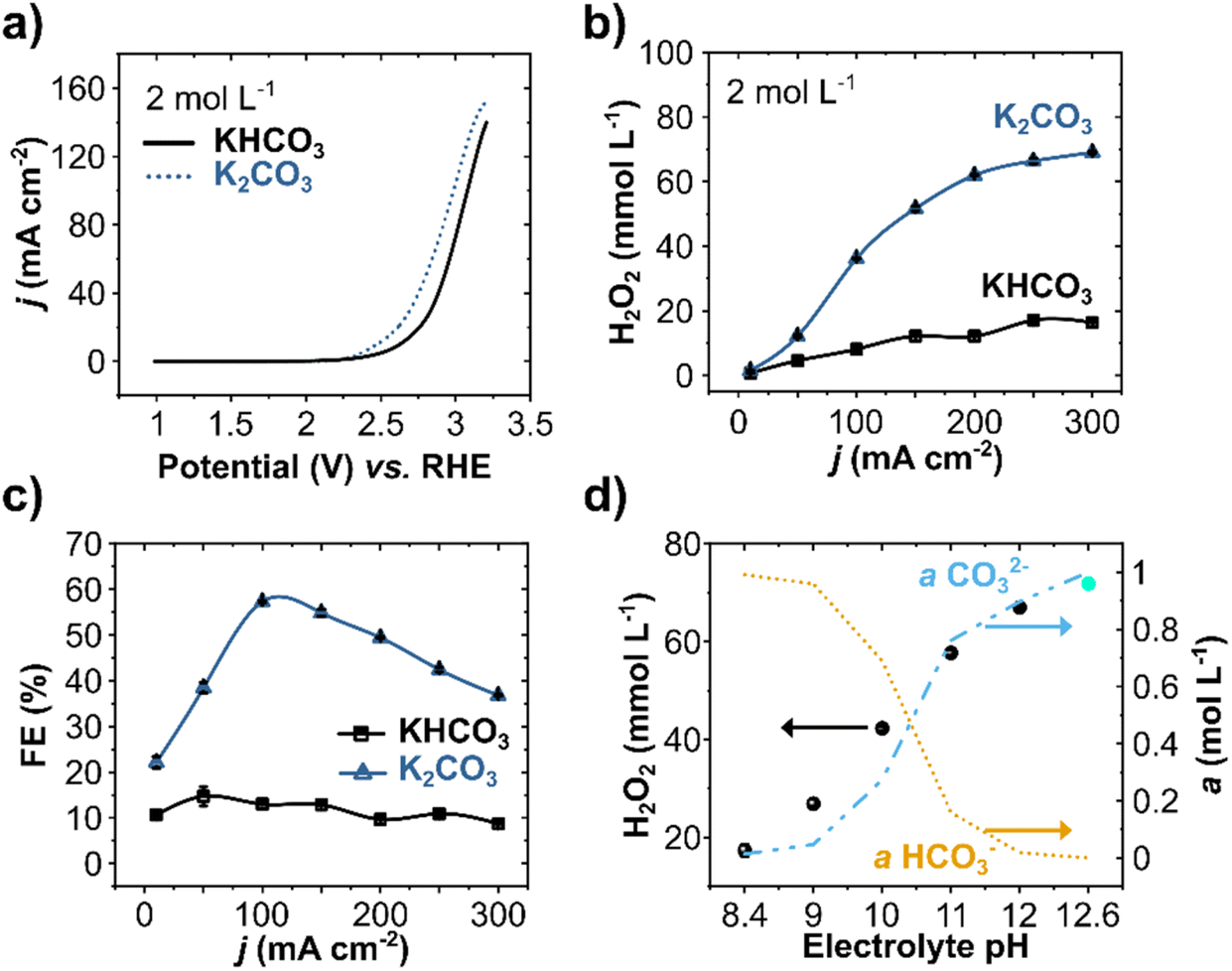 | ||
| Fig. 1 WOR to H2O2 in 2 mol L−1 KHCO3 and K2CO3 electrolytes. (a) Anodic LSV starting from +0.18 V vs. Ag/AgCl (calculated vs. RHE) with a scan rate of 100 mV s−1. (b) H2O2 concentration and (c) FE at the indicated applied current densities for 10 minutes at each step. (d) Galvanostatic polarization at 300 mA cm−2 for 10 minutes in 2 mol L−1 KHCO3 (at adjusted pH values from 8.4 to 12) and 2 mol L−1 K2CO3 (pH 12.6) and the corresponding calculated ionic activity for HCO3− and CO32− ions. The experiments were performed using a 5 cm2 BDD anode in a two-compartment H-cell cooled in an ice bath. The electrolyte was changed between the galvanostatic steps and kept under constant stirring of 1000 rpm. | ||
The effect of current density on H2O2 production for these two carbonate-based electrolytes (HCO3− and CO32−) was further evaluated by chronopotentiometry (CP) at pH 8.4 and pH 12.6 (Fig. 1b). Increasing the current density results in a higher H2O2 concentration for both electrolytes, yet carbonate-based electrolytes lead to higher overall H2O2 concentrations.38,42,48 A maximum of 70 mmol L−1 of H2O2 was obtained in carbonate at 300 mA cm−2, compared to only 16.4 mmol L−1 H2O2 in bicarbonate.
The FE at applied different current densities showed a peak of 57% in carbonate solutions at 100 mA cm−2, whereas the FEs in bicarbonate electrolytes lay between 10 and 15% for all current densities (Fig. 1c). Higher FE for carbonate solutions (at least four times higher than in bicarbonate) confirms that higher pH regimes enhance the electrochemical production of H2O2 on BDD anodes, consistent with our previous studies with carbon anodes.42 At current densities higher than 100 mA cm−2, the FE decreases, reaching 36% at 300 mA cm−2, where the highest H2O2 concentration was achieved. This lower FE, despite higher net H2O2 concentrations, indicates a combined effect of enhanced oxygen evolution and electrolytic H2O2 decomposition.37 Notwithstanding, anodic H2O2 formation reached a partial current density (jH2O2) of 110 mA cm−2 and a production rate of 35 μmol min−1 cm−2 at 300 mA cm−2 (Fig. S3, ESI†).
As different carbonate species (H2CO3, HCO3−, and CO32−) can be formed by varying the pH of the solution, the ionic activity for HCO3− (a(HCO3−)) and CO32− (a(CO32−)) species in the electrolyte was calculated as a function of pH for the range of pH 8.4 to 12.6 (Fig. 1d). In the same pH range, the anodic H2O2 production at 300 mA cm−2 was evaluated experimentally. The pH of the 2 mol L−1 KHCO3 was adjusted using KOH salt. The calculated activity profile for a(HCO3−) and a(CO32−) show a remarkable correlation with the experimentally measured anodic H2O2 production. Higher a(CO32−) at higher pH values lead to significantly higher H2O2 concentrations. A considerable visual difference in the anode was also observed at higher pH values, as the O2 evolution substantially decreased. Higher H2O2 production at higher pH is consistent with our proposed mechanism, which involves the oxidation of carbonate ions to peroxodicarbonate species as intermediates.42
WOR in a continuous flow reactor
Envisioning the scale-up and technical application of anodic water oxidation to H2O2, a flow cell setup was used for further investigations. The electrochemical flow mode/setup (circular or single-pass), the electrolyte flow rate, and the long-term stability of the system were investigated and optimized.Anodic H2O2 production in circular flow
Circular flow experiments allow the increase of product concentration in the electrolyte reservoir by recirculating it through the cell multiple times (Fig. 2a and Fig. S4, ESI†). Galvanostatic experiments on circular flow were carried out at current densities of 100, 200, and 300 mA cm−2 (Fig. 2b) with a constant flow rate of 100 mL min−1. The highest H2O2 concentration was obtained at 200 mA cm−2 (58 mmol L−1) after recirculating for 150 minutes. In all experiments, the FE reached its maximum peak at 15 minutes, and longer reaction times resulted in lower FE and formation rates (Fig. 2c and Fig. S5a, ESI†), indicating not only WOR to O2 as also the electrochemical decomposition of H2O2. Such production rate decrease with time was also observed in H-cell experiments (Fig. S6, ESI†), and it is consistent with the literature.37,42,49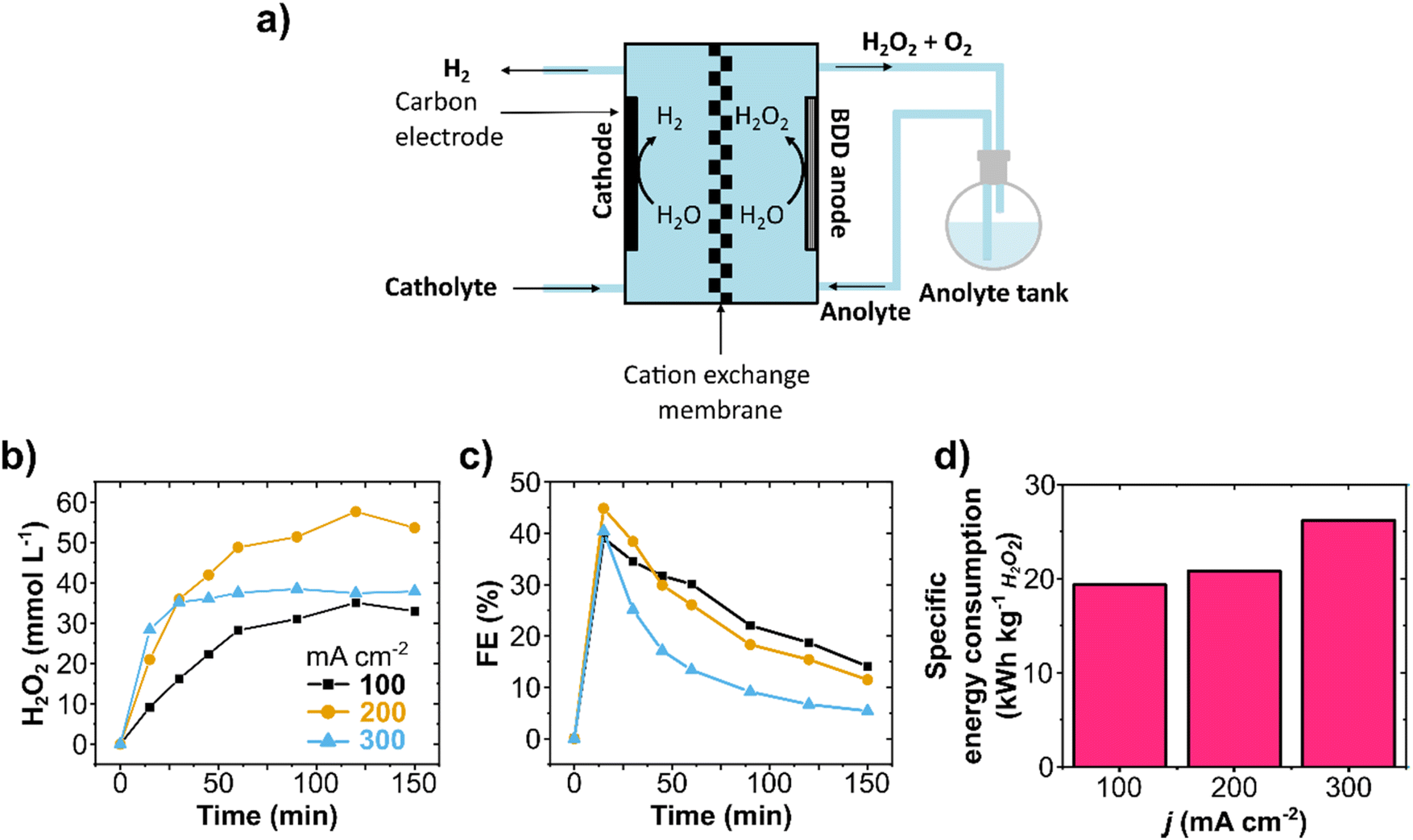 | ||
| Fig. 2 Anodic H2O2 production in a circular flow reactor. (a) Schematic representation of the flow cell with circular flow. (b) H2O2 concentration and (c) FE obtained at different applied current densities. (d) Energy consumption to produce 1 kg of H2O2 based on the peak FE (at 15 min) at each applied current density. The flow cell was equipped with a carbon cathode and a BDD/Ta anode (both with a 10 cm2 geometric area) separated by a Nafion 117 membrane. Each cell compartment contained a reservoir with 200 mL of 2 mol L−1 K2CO3 electrolyte circulating at 100 mL min−1 flow rate. 90 mmol L−1 Na2SiO3 was added to the anolyte as a chemical stabilizer to avoid H2O2 decomposition. The pH of the anolyte was kept constant at 12.6. | ||
The specific electric energy consumption for the anodic H2O2 generation was calculated for the first reaction stage of high formation rate (first 15 minutes, Fig. 2d) and for the whole reaction time (150 minutes, Fig. S5c and Table S1, ESI†). The specific energy consumption of a product is one of the most basic approaches to determining the unit energy consumption.50 The first reaction stage at 100 mA cm−2 had a specific energy consumption of 19.3 kW h kg−1, which raised considerably to 26 kW h kg−1 at 300 mA cm−2. After 150 minutes of reaction with recirculation (75 cycles), the specific energy consumption was as high as 196 kW h kg−1 at 300 mA cm−2 (Fig. S5c and Table S1, ESI†). At a flow rate of 100 mL min−1 and 200 mL anolyte volume, each recirculation cycle takes 2 minutes. The increase in energy consumption after 15 minutes is mainly due to the electrochemical H2O2 decomposition at the electrode surface, which can be particularly prominent at higher current densities (as observed at 300 mA cm−2). It is important to notice that the aggregate energy consumption for classical anthraquinone process is in about 17.6 kW h kg−1 H2O2.20 Hence, energy efficiency needs to be improved further for WOR-based H2O2 production. Therefore, a compromise in process parameters (current density, cell potential, cell design, membrane resistivity, etc.) should be found.
The effect of recirculating the electrolyte on the anodic generation and electrodecomposition of H2O2 was investigated stepwise by decreasing the electrolyte flow rate from 100 to 10 mL min−1 and limiting the recirculating steps to four (Fig. 3). Each cycle lasted for 20 minutes, thereafter the electrolyte was recirculated for a new pass cycle. An increase in H2O2 concentration was observed with each consecutive cycle at 100 and 300 mA cm−2, albeit FE and production rate decreased. The H2O2 concentration in the anolyte reached 46 and 110 mmol L−1 after four cycles at 100 and 300 mA cm−2, respectively (Fig. 3a). However, the FE decreased from 40% to 33% (1st to 4th cycle) at 100 mA cm−2 and from 45% (1st cycle) to 6% during the 4th cycle at 300 mA cm−2 (Fig. 3b). A drastic decrease in production rate (Fig. 3c) from 41 to 7 μmol min−1 cm−2 (70% decrease) at 300 mA cm−2 in comparison to only 12% decrease at 100 mA cm−2 was observed. The decrease in FE and formation rate with cycle is an effect of the decomposition of electrochemically produced H2O2 on the electrode surface after recirculation, particularly at high current densities, as also observed in continuous circular flow experiments. Additionally, the pH of the electrolyte also decreased with each cycle (Fig. 3d) due to the constant oxidation of OH− ions to O2. This pH decrease was more pronounced for higher current densities. Thus, the remarkable decline in production rate observed at 300 mA cm2 can also be due to the drop in pH, as it plays a direct role in the a(CO32−), consequently, in the peroxodicarbonate formation and H2O2.42 A considerable drop in the electrolyte conductivity was observed at a high current density (Fig. S7a, ESI†). The cell potential remained, nevertheless, essentially constant during all four cycles (Fig. S7b, ESI†). Increasing the number of cycles increases the overall product concentration, however it has the drawback of decreasing the production rate, and consequently increasing the specific energy consumption for H2O2 production (Fig. S7c, ESI†).
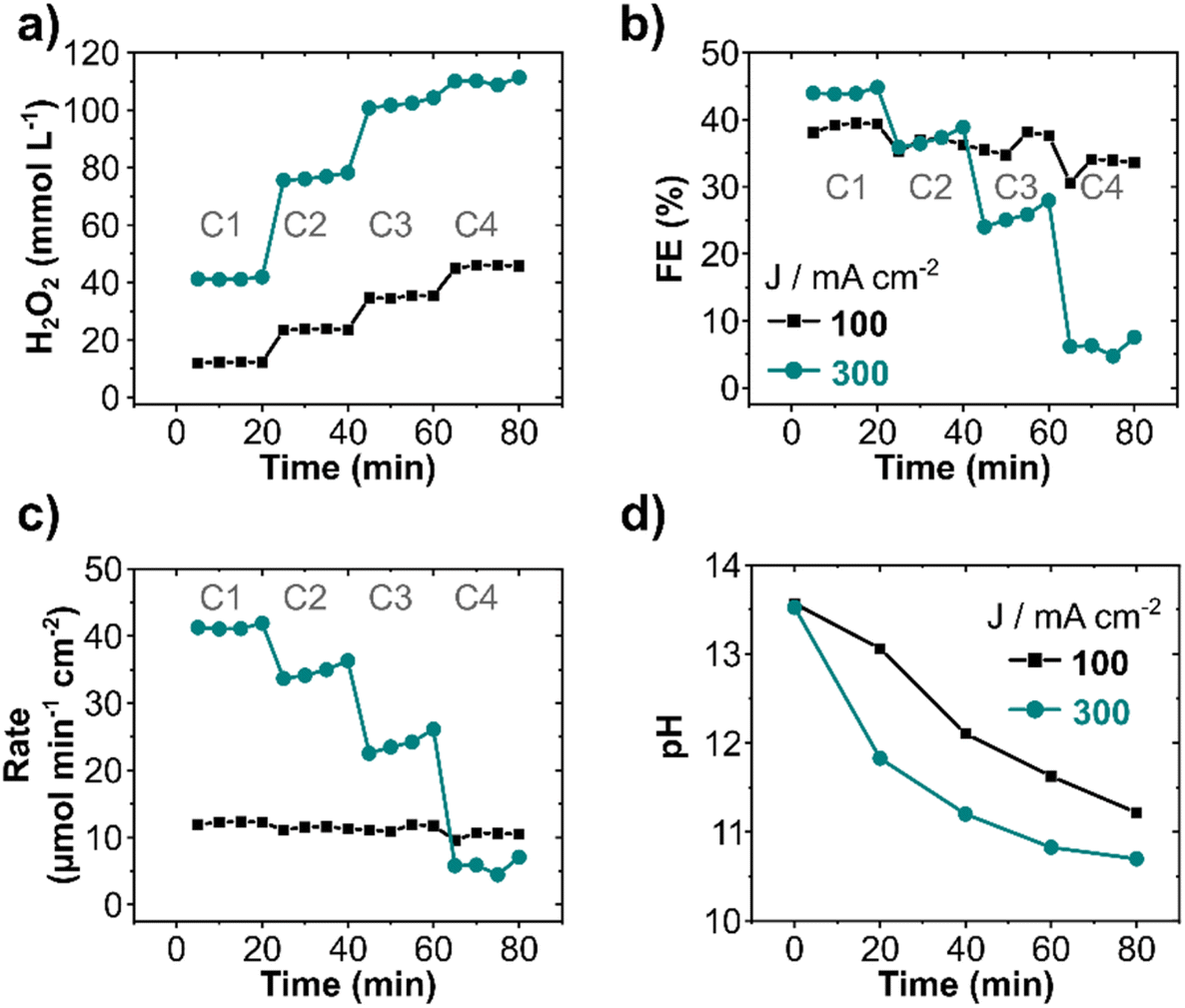 | ||
| Fig. 3 H2O2 generation under electrolyte flow with multiple recirculation cycles. (a) Anodic H2O2 concentration, (b) FE, (c) production rate, and (d) pH change. The experiments were carried out in a flow cell equipped with 10 cm2 BDD as anode and recirculated with 2 mol L−1 K2CO3 containing 90 mmol L−1 Na2SiO3 at 10 mL min−1 flow rate. The total volume of the electrolyte was 200 mL. Experiments were performed in 4 cycles of electrolyte flow (indicated as C1 to C4) of 20 minutes each. | ||
Chemical stabilizers can slightly decrease the decomposition of H2O2 in the electrolyte. Sodium silicate (Na2SiO3) is a particularly attractive stabilizer because it is not redox-active, thus it does not affect the electrode reactions.36,42 Circular flow experiments using anolyte with and without Na2SiO3 revealed that the silicate stabilizer considerably enhances H2O2 stability upon recirculation (Fig. S8, ESI†). Nevertheless, the challenge of anodic H2O2 decomposition can be tackled by regularly replacing the electrolyte in batches or optimizing the flow rate for maximum and continuous H2O2 productivity without recirculation (single-pass flow).
Single-pass flow
A single-pass flow mode was employed to minimize performance losses through electrochemical decomposition upon anolyte recirculation (Fig. 4a and Fig. S9, ESI†). In this setup, the anolyte containing the electrochemically generated H2O2 is not recirculated but optimized for maximum continuous production.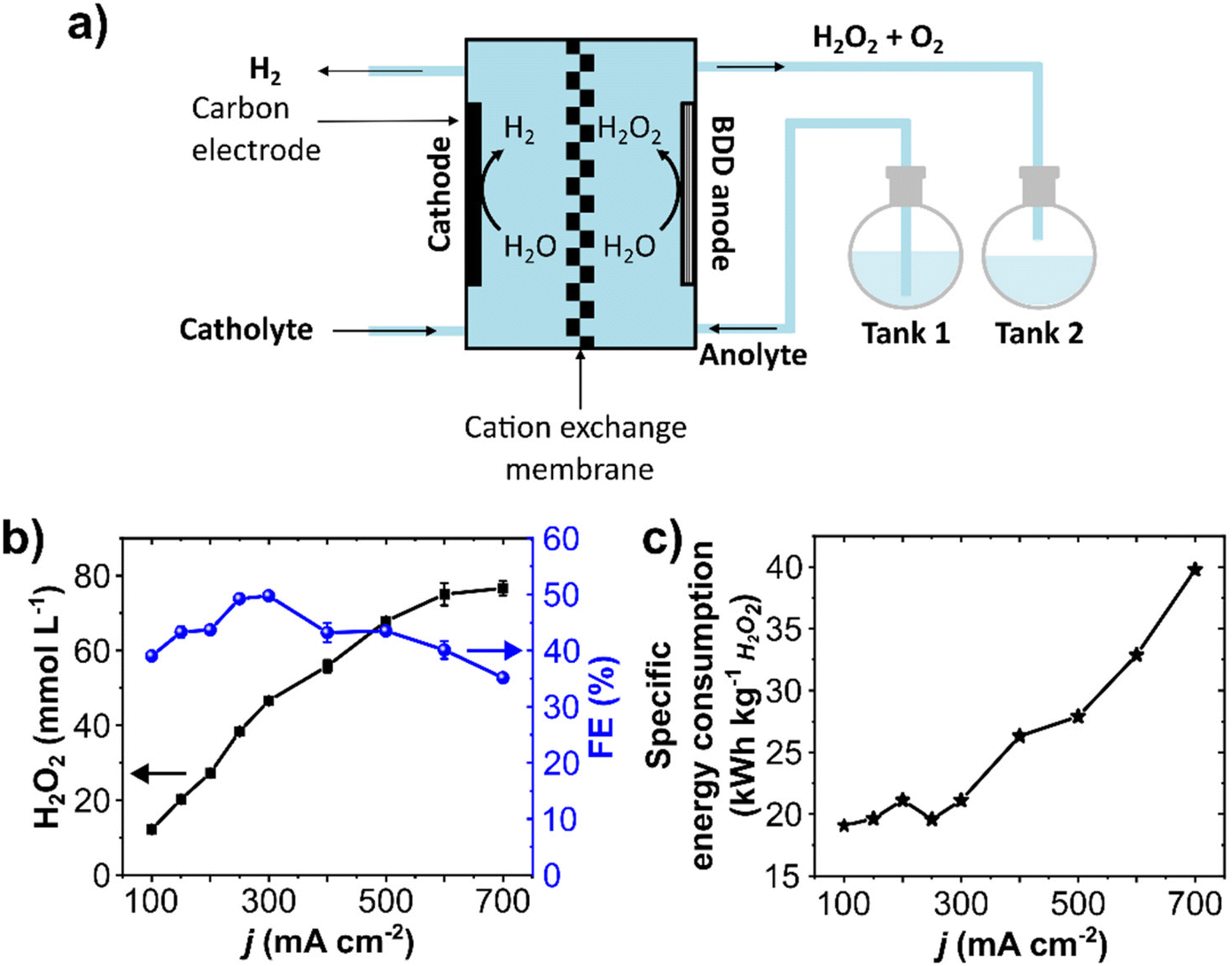 | ||
| Fig. 4 Anodic H2O2 production in a single-pass mode flow reactor. (a) Schematic representation of the flow cell with a single-pass flow setup. (b) Average H2O2 concentration, FE, and (c) specific energy consumption at different current densities (20 minutes steps). 200 mL of 2 mol L−1 K2CO3 + 90 mmol L−1 Na2SiO3 was used as an anolyte at a 10 mL min−1 flow rate without recirculation. The flow cell was equipped with a BDD/Ta working electrode (10 cm2 geometric area) and a carbon cathode. | ||
Electrolytic steps of 20 minutes at 10 mL min−1 electrolyte flow rate were carried out at current densities ranging from 100 to 700 mA cm2. The concentration of anodically generated H2O2 was directly proportional to the current density (Fig. 4b). Starting at 12 mmol L−1 at 100 mA cm−2, a maximum of 79 mmol L−1 H2O2 was obtained at 700 mA cm−2. The H2O2 production rate was stable and constant during the 20 minutes step, and a maximum of 79 μmol min−1 cm−2 was obtained at 700 mA cm2 (Fig. S10b and c, ESI†). To the best of our knowledge, this is the highest production rate for the anodic H2O2 generation so far reported. A detailed literature comparison of production rates is shown in Table S2, ESI.† The partial current density to produce H2O2 (jH2O2) reached 250 mA cm−2 (Fig. S10d, ESI†). It is important to remark that this single-pass setup allowed reaching considerably higher current densities, up to 700 mA cm−2. In this regard, techno-economic studies show that the economic (cost-related) performance of anodic H2O2 generation can be enhanced by increasing the current density.39,51
A maximum FE of 50% was achieved at 300 mA cm−2 (Fig. 4b), which dropped to 35% at 700 mA cm−1. This FE decreases upon increasing current density is likely due to an increase in O2 evolution. Although the cell potential increased at higher current densities (from 4.7 V at 100 mA cm−2 to 9 V at 700 mA cm−2, Fig. S11, ESI†), the total cell potential is still below the maximum recommended (11 V) to enable the technical feasibility of the setup in combination with HER.39
The specific energy consumption was around 20 kW h kg−1 for current densities up to 300 mA cm−2 (Fig. 4c). However, the energy consumption takes a steady steep at higher current densities, increasing to 40 kW h kg−1 at 700 mA cm−2, as a considerable share of energy consumption is used for O2 production.42 Since the highest FE and relatively low specific energy consumption were obtained at 300 mA cm−2, further optimizations with the single-pass flow were performed at this current density.
Effect of stabilizers at single-pass flow
A chemical stabilizer is essential to avoid chemical and electrochemical decomposition of H2O2, particularly in a circular flow setup, as observed in our experiments with and without Na2SiO3, where higher H2O2 concentrations were obtained in the presence of the silicate (Fig. S8, ESI†).42 However, in a single-pass flow mode, the electrochemical H2O2 decomposition can be minimized by optimizing the residence time of the electrolyte in contact with the electrode surface. Hence, a stabilizer may no longer be necessary.To validate this hypothesis and to elucidate the effect of chemically stabilizing agents in a single-pass flow system, experiments at 300 mA cm−2 were carried out with and without a stabilizer. In a single-pass mode, a similar H2O2 concentration of 52 mmol L−1 was obtained, independently of the presence of Na2SiO3. However, when the electrochemically produced H2O2 was stored for 3 hours without any applied current, the H2O2 concentration decreased by 42% (from 52 to 29 mmol L−1) in the absence of the stabilizer, compared to only 8% (53 to 49 mmol L−1) in its presence (Fig. 5). Thus, the use of Na2SiO3 as a stabilizer in single-pass mode does not necessarily enhance the electrochemical H2O2 formation, it rather inhibits the chemical decomposition of H2O2 during storage and thereby increases its bench life. Hence, immediate use of H2O2 in on-site applications can abstain from chemical stabilizers, which could consequently influence the total process costs. In cases requiring intermediate storage of the generated H2O2, however, stabilizers are recommended to avoid substantial chemical losses.
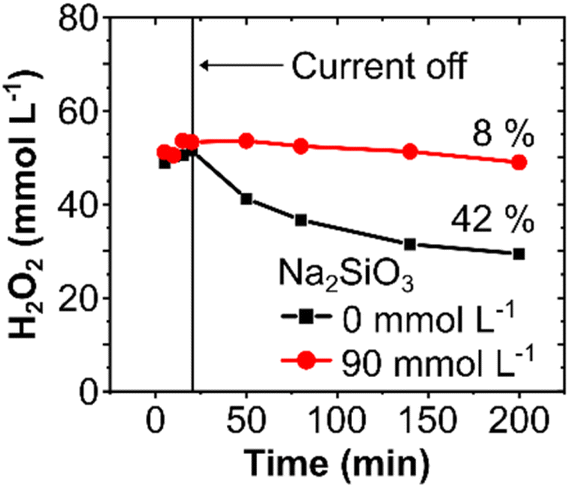 | ||
Fig. 5 Effect of stabilizer on the stability of electrochemically produced H2O2. Change in H2O2 concentration during electrolyte storage in the absence (■) and presence ( ) of 90 mmol L−1 Na2SiO3 in 2 mol L−1 K2CO3. The H2O2 was anodically generated for 20 minutes at 300 mA cm−2. Thereafter, electrolyte samples were taken at 30, 60, 120, and 180 minutes from the storage reservoir. ) of 90 mmol L−1 Na2SiO3 in 2 mol L−1 K2CO3. The H2O2 was anodically generated for 20 minutes at 300 mA cm−2. Thereafter, electrolyte samples were taken at 30, 60, 120, and 180 minutes from the storage reservoir. | ||
Effect of flow rate
The flow rate optimization maximizes the transient electrolyte time in the electrochemical cell and, consequently, the continuous anodic H2O2 production in single-pass flow. The experiments described to this point had a constant flow rate of 10 mL min−1. Variation of the flow-rate in the range of 5 to 100 mL min−1 in single-pass flow at a constant current density of 300 mA cm−2 shows contrasting shapes for H2O2 concentrations and FE at different flow rates (Fig. 6a). Slower flow rates (5 mL min−1) lead to higher H2O2 concentrations (75 mmol L−1), however, to lower FE (40%). Speeding the flow rate decreases the residence time of a specific electrolyte volume in the vicinity of the electrode, thus minimizing relevant side-reactions, such as O2 evolution and electrodecomposition of H2O2, and resulting in higher FE and production rate (Fig. 6b). Nevertheless, higher flow rates also decrease the H2O2 concentration and increase the required electrolyte volume per time, which can be costly. Therefore, a compromise between H2O2 concentration and FE must be considered depending on the application of anodic H2O2 production in continuous flow.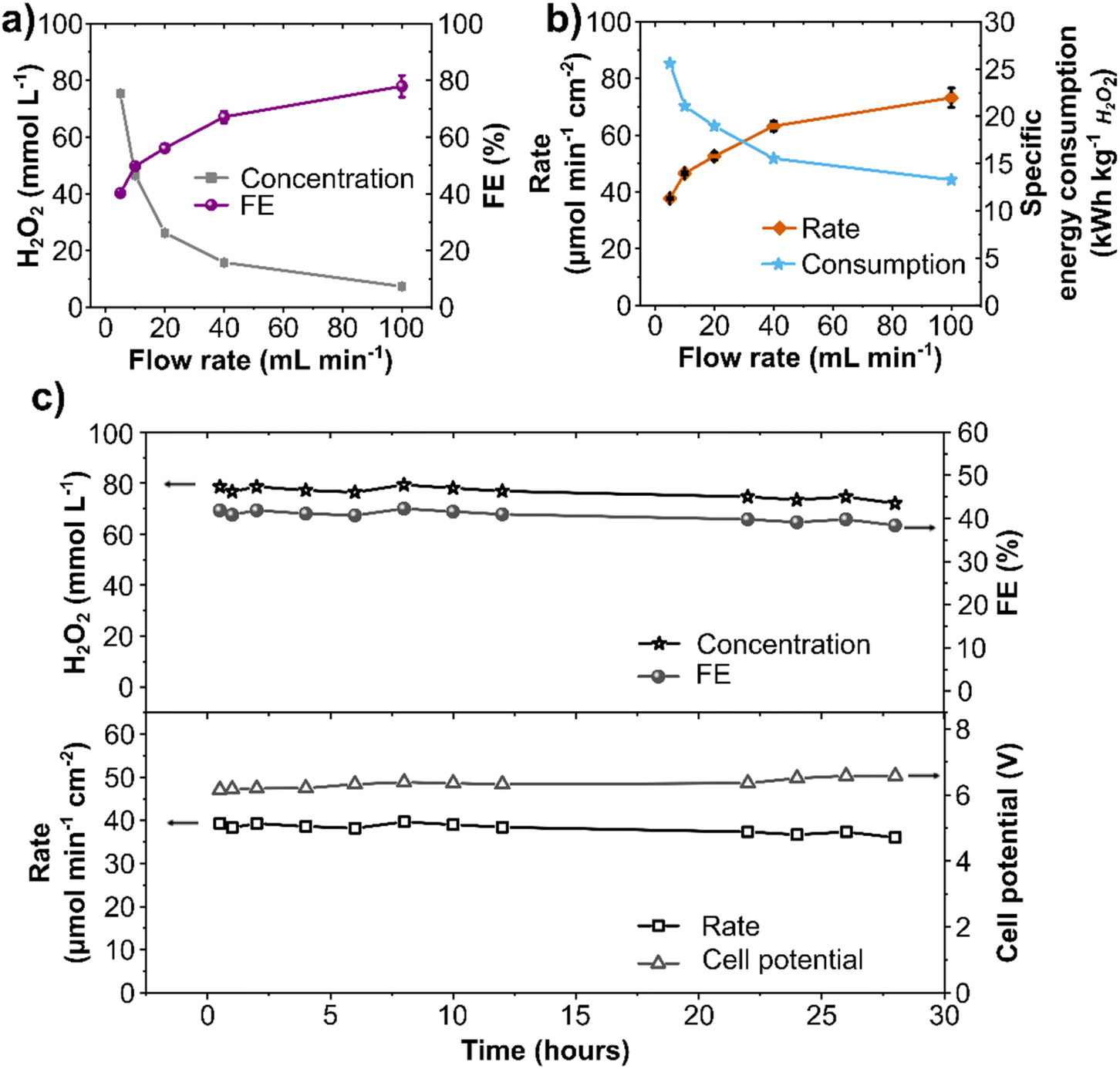 | ||
| Fig. 6 Flow rate effect on the continuous anodic H2O2 generation. (a) Anodic H2O2 concentration and FE, (b) production rate and specific energy consumption to produce 1 kg H2O2 at different electrolyte flow rates. Experimental conditions: single-pass flow of 200 mL anolyte (2 mol L−1 K2CO3) at a constant current density of 300 mA cm−2. The same electrolyte volume was used for each flow rate, resulting in different durations of the individual experiments. (c) Electrode stability test in a continuous flow setup: H2O2 concentration, FE, production rate, and cell potential at a 10 cm2 BDD at a constant current density of 300 mA cm−2 with a single-pass flow mode of 2 mol L−1 K2CO3 electrolyte at a flow rate of 5 mL min−1. | ||
Moreover, the corresponding specific energy consumption decreased by around 50% (from 25 kW h kg−1 to 13 kW h kg−1) upon increasing the flow rate from 5 to 100 mL min−1 (Fig. 6b). Considering the cost of renewable electricity of 3 cents($) per kW per h,36,52 the specific electricity-related cost of anodic H2O2 generation ($ per kg H2O2) at 300 mA cm−2 is estimated at $0.77 per kg at 5 mL min−1 and $0.4 per kg at 100 mL min−1 (Fig. S12, ESI†). Commercial H2O2 from the anthraquinone process is currently priced at $1.50 per kg without transportation costs.36 Nonetheless, it should be noted that our figures for calculated specific energy consumption only include the electric energy required for electrochemical cell operation. Hence, the estimated electricity cost reported in this study is specific to this lab-scale electrochemical cell. Certainly, other operating costs (e.g., material consumption, cell design, cathode material, membrane type, and downstream processing) could and will substantially contribute to the overall cost structure of H2O2 production via WOR. Yet, the cost of electricity to produce H2O2 at the anode can be further reduced by combining it with an optimized cathodic half-cell reaction such as HER, CO2RR, or ORR to replace the current WOR to O2.
Finally, a long-term stability study using the optimized single-pass flow setup was performed to produce H2O2 continuously. The stability of the electrochemical system is essential for its technical application, as it reduces the occurrence of component failures and the associated need to disassemble and reassemble the reactor.53 Galvanostatic experiment at 300 mA cm−2 was performed in 2 mol L−1 K2CO3 at a 5 mL min−1 flow rate for 28 hours (Fig. 6c). Anolyte samples were taken directly from the electrochemical cell outlet, showing a continuous generation of H2O2 for 28 hours with a constant concentration of about 80 mmol L−1 and a constant FE of 40%. An average production rate of 40 μmol min−1 cm−2 was observed with a stable cell potential of 6.3 V throughout the experiment. This stability experiment highlights the effectiveness of an optimized electrochemical setup. Previous reports on electrode stability for WOR to H2O2 using carbonaceous electrodes are restricted to batch mode (H-cells) with low electrolyte volume and at current densities from 100 to 200 mA cm−2 with a maximum time of 10 hours.31,36,38 Our last study using commercial carbon fiber paper electrodes reported stable operation for up to 17.5 hours at 100 mA cm−2.42 Herein, we expanded the potential of process development for WOR to H2O2 to reach considerably higher current densities and continuous stable production at a robust commercial BDD electrode. This is also the first comprehensive study of an electrochemical flow system for WOR to H2O2, which remains stable for 28 hours. Such investigations are essential for future technical electrochemical applications. Moreover, considering that BDD anodes are already used in several electrochemical industrial processes with a lifetime up to several months,54 it can be expected that H2O2 generation through WOR at BDD anodes can also be operated with high stability and performance for a considerably longer time. Electrode and process stability is an important step forward in terms of scale-up and utilization.
Conclusions
This research work demonstrates that beyond the catalyst development, the process parameters (including electrolyte type, pH, flow type, and flow rate) can dramatically affect the efficiency of anodic H2O2 production and, consequently, the technical process implementation. The continuous flow process has been developed with commercially available and scalable BDD anodes. Hence, it highlights a robust, stable, and promising foundation for piloting. Future work includes the combination of the anodic H2O2 production with value-added cathodic reactions, with the objective of utilizing the invested redox equivalents most efficiently by exploiting both half-cells. Considering the still high capital expenses for BDD electrodes, other low-cost carbon-based materials are a promising area for further exploration in searching for efficient and scalable WOR to H2O2. In this context, such research should be performed with a clear perspective of future industrial applications, focusing on relevant and scalable process configurations, as well as resource efficiency, investment, operational cost, and operational stability.The electrochemical H2O2 production is nonetheless not a replacement for the current industrial AO process. From the perspective of mobile electrochemical prototypes, this is rather an expansion of the H2O2 market to accommodate decentralized solutions, especially in remote areas where transportation of H2O2 is difficult. As an additional benefit, the anodic H2O2 production could be combined with hydrogen production, thereby also diversifying and adding value to the hydrogen industry.
Author contributions
Dhananjai Pangotra: conceptualization, investigation, formal analysis, validation, writing – original draft. Lénárd-István Csepei: conceptualization, writing – review & editing. Arne Roth: conceptualization, supervision, writing – review & editing. Volker Sieber: supervision, writing – review & editing. Luciana Vieira: conceptualization, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Prof. Cordt Zollfrank (TUM-CS) for allowing access to SEM. Dr Tobias Graßl from Condias GmbH is deeply acknowledged for sharing scientific know-how on BDD electrodes. We thank Dr Leonardo Castañeda Losada for reading this manuscript and providing constructive feedback. All authors express their gratitude to the European Commission for the financial support of this research within the European Framework Programme for Research and Innovation Horizon 2020 (Grant No. 768789).References
- Global Hydrogen Peroxide Market Outlook, https://www.expertmarketresearch.com/reports/hydrogen-peroxide-market, (accessed 20 August 2021, 2021).
- A. Rubio-Clemente, E. Chica and G. Peñuela, Environ. Sci. Pollut. Res., 2019, 26, 4462–4473 CrossRef CAS PubMed.
- Chlorine or Hydrogen Peroxide – Which is Better for Treating Water?, https://www.uswatersystems.com/blog/chlorine-or-hydrogen-peroxide, (accessed 17 May 2022, 2021).
- D. Horová, J. Nováková, L. Pelíšková, J. Kohout, J. Šafář, K. Hrachovcová and V. Tokarová, React. Kinet., Mech. Catal., 2020, 130, 1077–1092 CrossRef.
- A. D. Ortiz-Marin, E. R. Bandala, K. Ramírez, G. Moeller-Chávez, L. Pérez-Estrada, B. Ramírez-Pereda and L. E. Amabilis-Sosa, React. Kinet., Mech. Catal., 2022, 135, 639–654 CrossRef CAS.
- R. Hage and A. Lienke, Angew. Chem., Int. Ed., 2005, 45, 206–222 CrossRef PubMed.
- N. de la Fuente, L. Chen, J. A. Wang, J. González and J. Navarrete, React. Kinet., Mech. Catal., 2021, 132, 1119–1135 CrossRef CAS.
- H. Shuangyang, Hi Tech Chemical Co Ltd, A kind of deodorization of crude sulfate turpentine and the production method of sulfate turpentine, CN106281041B, 2016.
- Institute of Chemical Industry of Forest Products of CAF, Refining method for desulphurizing and deodorizing crude sulphate turpentine, CN101654597B, 2009.
- J. C. Rubio-Romero, M. D. C. Pardo-Ferreira, J. A. Torrecilla-Garcia and S. Calero-Castro, Saf. Sci., 2020, 129, 104830 CrossRef PubMed.
- G. Lewandowski, M. Kujbida and A. Wróblewska, React. Kinet., Mech. Catal., 2021, 132, 983–1001 CrossRef CAS.
- L. Pierri, A. Gemenetzi, A. Mavrogiorgou, J. Borges Regitano, Y. Deligiannakis and M. Louloudi, Mol. Catal., 2020, 489, 110946 CrossRef CAS.
- J. D. Tibbetts, W. B. Cunningham, M. Vezzoli, P. Plucinski and S. D. Bull, Green Chem., 2021, 23, 5449–5455 RSC.
- A. S. Gohardani, J. Stanojev, A. Demairé, K. Anflo, M. Persson, N. Wingborg and C. Nilsson, Prog. Aerosp. Sci., 2014, 71, 128–149 CrossRef.
- M. Hävecker, S. Wrabetz, J. Kröhnert, L.-I. Csepei, R. Naumann d'Alnoncourt, Y. V. Kolen'ko, F. Girgsdies, R. Schlögl and A. Trunschke, J. Catal., 2012, 285, 48–60 CrossRef.
- S. T. Renu Singh, M. Srivastava and A. Shukla, Agric. Eng. Int.: CIGR J., 2014, 16, 173–181 Search PubMed.
- J. Krischan, A. Makaruk and M. Harasek, J. Hazard. Mater., 2012, 215–216, 49–56 CrossRef CAS PubMed.
- R. L. Myers, The 100 Most Important Chemical Compounds: A Reference Guide, Greenwood Press, 2007 Search PubMed.
- L. Pesterfield, J. Chem. Educ., 2009, 86, 1182 CrossRef CAS.
- A. T. Murray, S. Voskian, M. Schreier, T. A. Hatton and Y. Surendranath, Joule, 2019, 3, 2942–2954 CrossRef CAS.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- S. C. Perry, D. Pangotra, L. Vieira, L.-I. Csepei, V. Sieber, L. Wang, C. Ponce de León and F. C. Walsh, Nat. Rev. Chem., 2019, 3, 442–458 CrossRef CAS.
- J. García-Serna, T. Moreno, P. Biasi, M. J. Cocero, J.-P. Mikkola and T. O. Salmi, Green Chem., 2014, 16, 2320–2343 RSC.
- X. Shi, S. Back, T. M. Gill, S. Siahrostami and X. Zheng, Chem, 2021, 7, 38–63 CAS.
- Evonik, Hydrogen Peroxide for Pulp and Paper Industry, https://active-oxygens.evonik.com/product/h2o2/downloads/applications-hydrogen-peroxide-for-the-pulp-and-paper-industry-en.pdf, (accessed 29 March 2022).
- K. Wenderich, B. A. M. Nieuweweme, G. Mul and B. T. Mei, ACS Sustainable Chem. Eng., 2021, 9, 7803–7812 CrossRef CAS.
- C. Minke, M. Suermann, B. Bensmann and R. Hanke-Rauschenbach, Int. J. Hydrogen Energy, 2021, 46, 23581–23590 CrossRef CAS.
- S. Kiemel, T. Smolinka, F. Lehner, J. Full, A. Sauer and R. Miehe, Int. J. Energy Res., 2021, 45, 9914–9935 CrossRef CAS.
- T. Kang, B. Li, Q. Hao, W. Gao, F. Bin, K. N. Hui, D. Fu and B. Dou, ACS Sustainable Chem. Eng., 2020, 8, 15005–15012 CrossRef CAS.
- S. Y. Park, H. Abroshan, X. Shi, H. S. Jung, S. Siahrostami and X. Zheng, ACS Energy Lett., 2019, 4, 352–357 CrossRef CAS.
- C. Zhang, R. Lu, C. Liu, L. Yuan, J. Wang, Y. Zhao and C. Yu, Adv. Funct. Mater., 2021, 31, 2100099 CrossRef CAS.
- X. Shi, S. Siahrostami, G. L. Li, Y. Zhang, P. Chakthranont, F. Studt, T. F. Jaramillo, X. Zheng and J. K. Norskov, Nat. Commun., 2017, 8, 701 CrossRef PubMed.
- S. R. Kelly, X. Shi, S. Back, L. Vallez, S. Y. Park, S. Siahrostami, X. Zheng and J. K. Nørskov, ACS Catal., 2019, 9, 4593–4599 CrossRef CAS.
- T. Shiragami, H. Nakamura, J. Matsumoto, M. Yasuda, Y. Suzuri, H. Tachibana and H. Inoue, J. Photochem. Photobiol., A, 2015, 313, 131–136 CrossRef CAS.
- F. Kuttassery, S. Mathew, S. Sagawa, S. N. Remello, A. Thomas, D. Yamamoto, S. Onuki, Y. Nabetani, H. Tachibana and H. Inoue, ChemSusChem, 2017, 10, 1909–1915 CrossRef CAS PubMed.
- C. Xia, S. Back, S. Ringe, K. Jiang, F. Chen, X. Sun, S. Siahrostami, K. Chan and H. Wang, Nat. Catal., 2020, 3, 125–134 CrossRef CAS.
- S. Mavrikis, M. Göltz, S. Rosiwal, L. Wang and C. Ponce de León, ACS Appl. Energy Mater., 2020, 3, 3169–3173 CrossRef CAS.
- S. Mavrikis, M. Göltz, S. C. Perry, F. Bogdan, P. K. Leung, S. Rosiwal, L. Wang and C. Ponce de León, ACS Energy Lett., 2021, 6, 2369–2377 CrossRef CAS.
- K. Wenderich, B. A. M. Nieuweweme, G. Mul and B. T. Mei, ACS Sustainable Chem. Eng., 2021, 9, 7803–7812 CrossRef CAS.
- C. P. Chardon, T. Matthée, R. Neuber, M. Fryda and C. Comninellis, ChemistrySelect, 2017, 2, 1037–1040 CrossRef CAS.
- S. Maljuric, W. Jud, C. O. Kappe and D. Cantillo, J. Flow Chem., 2020, 10, 181–190 CrossRef CAS.
- D. Pangotra, L.-I. Csepei, A. Roth, C. Ponce de León, V. Sieber and L. Vieira, Appl. Catal., B, 2022, 303, 120848 CrossRef CAS.
- T. M. Gill, L. Vallez and X. Zheng, ACS Energy Lett., 2021, 6, 2854–2862 CrossRef CAS.
- B. J. Deadman, K. Hellgardt and K. K. Hii, React. Chem. Eng., 2017, 2, 462–466 RSC.
- G. Eisenberg, Ind. Eng. Chem., Anal. Ed., 1943, 15, 327–328 CrossRef CAS.
- Z. Machala, B. Tarabova, K. Hensel, E. Spetlikova, L. Sikurova and P. Lukes, Plasma Processes Polym., 2013, 10, 649–659 CrossRef CAS.
- K. Fuku, Y. Miyase, Y. Miseki, T. Gunji and K. Sayama, ChemistrySelect, 2016, 1, 5721–5726 CrossRef CAS.
- L. Fan, X. Bai, C. Xia, X. Zhang, X. Zhao, Y. Xia, Z.-Y. Wu, Y. Lu, Y. Liu and H. Wang, Nat. Commun., 2022, 13, 2668 CrossRef CAS PubMed.
- P. A. Michaud, M. Panizza, L. Ouattara, T. Diaco, G. Foti and C. Comninellis, J. Appl. Electrochem., 2003, 33, 151–154 CrossRef CAS.
- S. Palamutcu, in Handbook of Life Cycle Assessment (LCA) of Textiles and Clothing, ed. S. S. Muthu, Woodhead Publishing, 2015, pp. 31–61 Search PubMed.
- K. Wenderich, W. Kwak, A. Grimm, G. J. Kramer, G. Mul and B. Mei, Sustainable Energy Fuels, 2020, 4, 3143–3156 RSC.
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef PubMed.
- S. C. Perry, S. Mavrikis, L. Wang and C. Ponce de León, Curr. Opin. Electrochem., 2021, 30, 100792 CrossRef CAS.
- J. V. Macpherson, Phys. Chem. Chem. Phys., 2015, 17, 2935–2949 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc02575b |
| This journal is © The Royal Society of Chemistry 2022 |