
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembly of colloidal nanoparticles into 2D arrays at water–oil interfaces: rational construction of stable SERS substrates with accessible enhancing surfaces and tailored plasmonic response
Ziwei
Ye
,
Chunchun
Li
,
Qinglu
Chen
,
Yikai
Xu
 * and
Steven E. J.
Bell
*
* and
Steven E. J.
Bell
*
School of Chemistry and Chemical Engineering, Queen's University Belfast, University Road, Belfast, BT7 1NN, UK. E-mail: yxu18@qub.ac.uk; S.Bell@qub.ac.uk
First published on 16th February 2021
Abstract
Self-assembly at water–oil interfaces has been shown to be a cheap, convenient and efficient route to obtain densely packed layers of plasmonic nanoparticles which have small interparticle distances. This creates highly plasmonically active materials that can be used to give strong SERS enhancement and whose structure means that they are well suited to creating the highly stable, reproducible and uniform substrates that are needed to allow routine and accurate quantitative SERS measurements. A variety of methods have been developed to induce nanoparticle self-assembly at water–oil interfaces, fine tune the surface chemistry and adjust the position of the nanoparticles at the interface but only some of these are compatible with eventual use in SERS, where it is important that target molecules can access the active surface unimpeded. Similarly, it is useful to transform liquid plasmonic arrays into easy-to-handle free-standing solid films but these can only be used as solid SERS substrates if the process leaves the surface nanoparticles exposed. Here, we review the progress made in these research areas and discuss how these developments may lead towards achieving rational construction of tailored SERS substrates for sensitive and quantitative SERS analysis.
 Ziwei Ye | Dr Ziwei Ye received his BS degree in applied chemistry at East China University of Science and Technology in China. He proceeded to study in chemistry at Queen’s University Belfast and obtained his PhD degree in 2019. He then continued to pursue his research in materials chemistry and currently holds a position as research assistant at Queen’s University Belfast. His current research focuses on fabrication of multidimensional nanomaterials and their applications in photonics and photocatalysis. |
 Chunchun Li | Chunchun Li obtained her BSc in 2017 from East China University of Science and Technology in Applied Chemistry. Currently, she is a PhD student in chemistry under the supervision of Prof Steven E. J. Bell at Queen’s University Belfast. Her research focuses on understanding molecular adsorption and related phenomenon on noble metal nanomaterial surfaces via SE(R)RS for the construction of nanomaterials with tailored surface properties. |
 Qinglu Chen | Qinglu Chen obtained his BSc in 2019 from East China University of Science and Technology in Applied Chemistry. Currently, he is a PhD student in chemistry under the supervision of Prof Steven E. J. Bell at Queen’s University Belfast. His research focuses on the construction of plasmonic nanomaterials for SE(R)RS applications. |
 Yikai Xu | Dr Yikai Xu is currently an independent Leverhulme Early Career Fellow at Queen’s University Belfast. He obtained his BSc degree in Applied Chemistry at East China University of Science and Technology and completed his PhD research at QUB. Dr Xu is the recipient of the 2019 Kathleen Lonsdale Royal Irish Academy Prize for the most outstanding PhD research in chemical science in Ireland. His research interests are in the preparation and applications of plasmonic nanomaterials, particularly the preparation of hybrid materials containing plasmonic nanoparticle assemblies. |
 Steven E. J. Bell | Prof Steven E. J. Bell received his PhD from Queen's University Belfast and worked at the Rutherford-Appleton Laboratory and the University of York before returning to QUB where he is a Professor of Physical Chemistry and Head of the School of Chemistry and Chemical Engineering. His research centres on nanomaterials and Raman spectroscopy. He has a particular interest in the application of Raman methods to real world problems including medical and security applications and was founder/director of a successful company manufacturing Raman spectrometers. |
1 Introduction
Surface-electron oscillations on noble metal (particularly Au and Ag) nanoparticles can be excited by light to generate an electromagnetic field, which is localized on the surface of the nanoparticles (often referred to as localized surface plasmon resonance, LSPR). The Raman scattering of molecules which are adsorbed on the surface of such nanoparticles are significantly enhanced by the LSPR. The electromagnetic enhancement provided by LSPR together with other less significant enhancing mechanisms, mainly chemical enhancement, gives rise to surface-enhanced Raman spectroscopy (SERS). More details of the mechanism of SERS have been summarized in many excellent past reviews.1–3 More importantly, with its unique combination of sensitivity and molecular specificity, surface-enhanced Raman spectroscopy (SERS) has grown into a powerful analytical technique which allows vibrational information on molecules to be obtained down to even single-molecule level.4 However, real-world applications of SERS are still very limited, mainly due to the difficulty associated with finding methods for preparing SERS substrates.5–7 that can not only give the required sensitivity and reproducibility but are widely available to the potential user community. There are no universally accepted standard enhancing materials because no suitable materials have been demonstrated to either be capable of being prepared successfully in any standard laboratory or available commercially at an affordable price. Current strategies for producing SERS enhancing substrates generally fall into two broad classes: top–down lithography, which typically involves carving nanostructures on bulk plasmonic metals, and bottom-up synthesis, which typically involves chemical synthesis of plasmonic metal nanoparticles and subsequent assembly into hierarchical nanostructures.8 While top-down lithographic strategies are intrinsically more suited for the production of highly uniform and reproducible solid SERS substrates, the sophisticated equipment and procedures required for producing them greatly increases the overall cost of this approach and therefore hinders their routine use in most real-world applications. In contrast, bottom-up synthesis offers a considerably cheaper method for producing highly plasmonically active substrate materials and has therefore been the predominant strategy for fabricating SERS substrates, particularly those intended for chemical analysis.Unfortunately, the electromagnetic enhancement obtained from an individual colloidal metal nanoparticle is usually very limited. For example, it has been shown that individual Au/Ag spheres only give enhancement of ca. 10×, while individual anisotropic plasmonic metal nanoparticles with sharp edges and tips, such as a nanostar/triangle, provide electromagnetic enhancement up to 105×.9 The largest electromagnetic enhancement is obtained when plasmonic nanoparticles are assembled to form densely packed structures to create nanojunctions, in which the electromagnetic fields of adjacent particles overlap. In these plasmonic “hot-spots” the electromagnetic enhancement can reach 1010×, which allows the vibrational signals of individual molecules to be detected.10
The simplest way to create nanoparticle assemblies is by adding inorganic salts to electrostatically stabilized Ag/Au colloids to prepare aggregates.† These aggregated colloids were used to produce the earliest single-molecule SERS data and remain a popular substrate for SERS due to their simple fabrication and strong signal enhancement.11,12 The main disadvantages of colloidal aggregates as enhancing substrates are the low uniformity of their structure and poor stability, which can cause major issues in acquiring SERS signals with the reproducible absolute intensities that are critical for quantitative SERS measurements if internal standards are to be avoided. This has meant that a significant amount of effort has been devoted by the SERS community to develop methods for fabricating stable colloidal assemblies which offer strong plasmonic enhancement along with good signal reproducibility.13 Typical examples of these include colloidal dimers/trimers,14,15 1-dimensional (1D) arrays of nanoparticles i.e. nanoparticle chains,16,17 2-dimensional (2D) arrays of nanoparticles18,19 and 3-dimensional (3D) arrays of nanoparticles,20,21 such as multilayer particle assemblies and core–shell systems with particle satellites. Depending on their specific structures each type of nanoparticle assembly possesses unique advantages and drawbacks. For example, colloidal dimers/trimers normally provide weaker plasmonic enhancement than other aggregates but are appropriately sized for cell uptake which makes them useful for in vivo SERS studies. Conversely, 3D arrays possess a high density of hot-spots which increases the enhancement they can provide, but the associated increase in sensitivity comes at the price of complicated fabrication procedures and reduced reproducibility. In this review we are most concerned with 2D arrays since they offer an appropriate balance between sensitivity, stability, reproducibility, and affordability, which make them arguably the most promising candidate for general SERS analysis in real-life applications.
To date, a huge variety of methods have been developed to fabricate highly ordered 2D arrays of plasmonic nanoparticles,22 amongst which self-assembly of colloidal nanoparticles at water–oil interfaces stands out as an excellent technique, particularly for the fabrication of substrates for routing real-life SERS analysis. This is mainly because a wide range of plasmonic nanoparticles can be induced to assemble at water–oil interfaces into 2D arrays with procedures which are mild, rapid, simple, and reproducible. Moreover, these interfacial arrays can be used for multiphase detection, or as the precursor to produce solid plasmonically active films. However, despite the number of excellent recent reviews which discuss the fabrication of SERS substrates using nanoparticle self-assembly,23–25 reviews which are dedicated specifically to the discussion of nanoparticle self-assembly at water–oil interfaces for SERS remain scarce. In this review, we will introduce the general strategies for assembling plasmonic nanoparticles into densely packed 2D arrays at water–oil interfaces, as well as the associated advantages/disadvantages of using these materials for SERS. This will be followed by discussions of the current applications of 2D interfacial arrays in SERS and their transformation into more robust solid 2D films which are more convenient for routine SERS measurements and applications in the field. Finally, the current state-of-the-art in controlling the structure of self-assembled interfacial 2D arrays will be discussed with particular emphasis on the potential impact this might have on future SERS applications.
2 Self-assembly of nanoparticles at water–oil interfaces for SERS
The field of interfacial self-assembly has witnessed tremendous development in the past few decades and has now become a reliable and versatile technique for creating highly ordered nanoparticle arrays from plasmonic nanoparticles of different sizes, shapes and compositions. Although the exact procedure used to achieve interfacial self-assembly often varies between research groups, they are all governed by the same basic principles which underlie the migration of nanoparticles to, and localisation at, the water–oil interface. In general, the driving force is the reduction of the Helmholtz free energy, ΔE, which is given by the following equation:ΔE = −πR2γ(1 ± cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ)2 θ)2 | (1) |
Simple inspection of eqn (1) shows that the reduction in the energy of the system reaches a maximum when the particles are adsorbed at the interface with a 90° contact angle, which is easy to understand as this is the situation where the particles reduce the contact between oil and water to the maximum extent. This has led to a substantial amount of research on modifying the surface of nanoparticles with amphiphilic capping ligands with the aim of driving interfacial self-assembly.28–30 However, closer inspection of eqn (1) shows that self-assembly of nanoparticles at water–oil interfaces is always energetically favoured (provided θ° ≠ 0 or 180°). Indeed, we along with other groups, have shown that nanoparticles are spontaneously driven to the water–oil interface regardless of their surface hydrophobicity.31–33 However, it is important to point out that the hydrophobicity of the nanoparticle does affect its stability at the interface since particles with extremely small/large contact angles are more subject to thermal fluctuations and in turn are more likely to desorb from the interface.33
Often, the nanoparticles used for self-assembly carry a surface charge, which is important because the resulting interparticle electrostatic repulsion prevents aggregation and therefore stabilizes the particles in solution. However, this same effect prevents charged nanoparticles from forming closely packed arrays at the interface, although very loosely packed arrays are permitted since electrostatic repulsion between nanoparticles becomes negligible at long distances.31,34 This is particularly relevant for SERS since many commonly used types of Ag/Au colloidal particles, such as citrate- and hydroxylamine-reduced colloids, are charged but they need to be packed tightly to form interparticle nanojunctions which are <2 nm wide to provide strong SERS enhancement. Therefore, a vital part of interfacial self-assembly to create SERS-active 2D arrays is to overcome interparticle electrostatic repulsion to form plasmonically-coupled films. Based on this line of thought, methods for inducing interfacial self-assembly of plasmonic nanoparticles at water–oil interfaces can be divided into two broad categories: charge-reduction methods and charge-screening methods, as illustrated in Fig. 1. The charge reduction method can be further divided into subcategories, where the charge from the surface-species is neutralized or the charged surface-species are replaced.
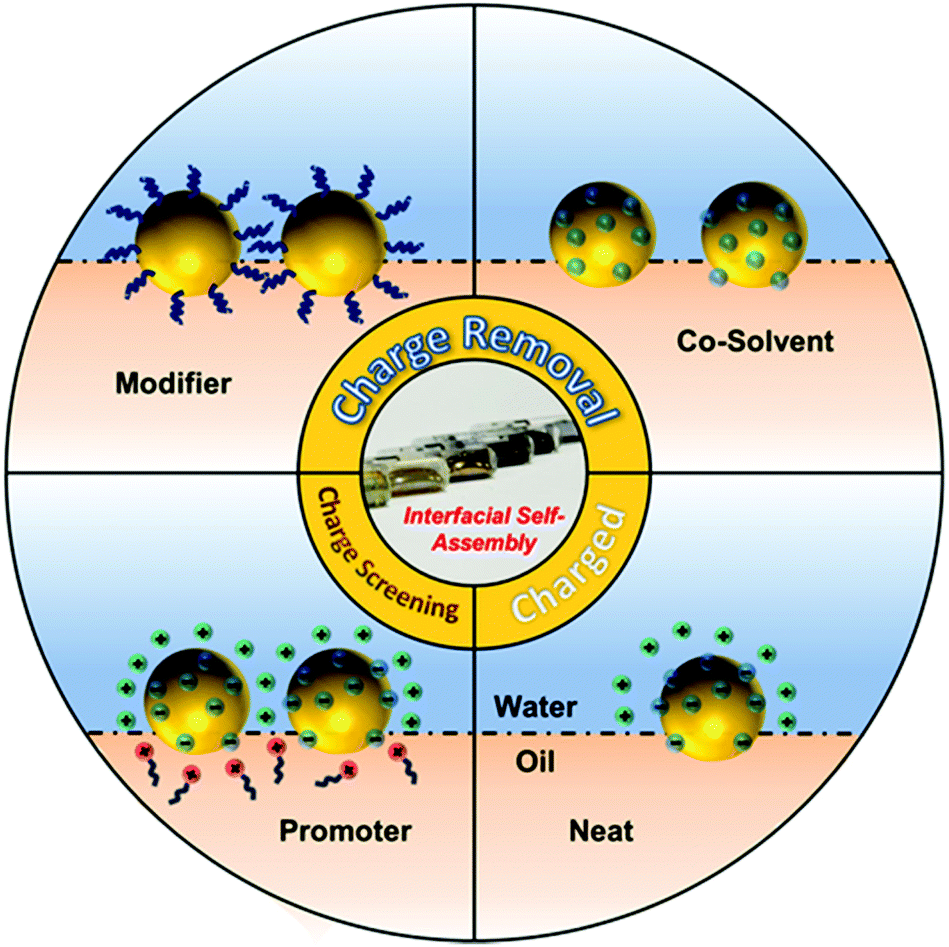 | ||
| Fig. 1 Schematic illustration of a charged nanoparticle sitting at the water–oil interface, contrasted with charged nanoparticles assembled at the water–oil interface with the aid of promoters and charge-neutral nanoparticles assembled with the aid of modifiers or co-solvents. | ||
2.1 Charge-reduction with chemical modifiers
The most direct way to overcome the electrostatic repulsion between plasmonic nanoparticles at water–oil interfaces is by chemically substituting the charged ligands on the surface of the particles with charge-neutral ligands. Following initial reports from Efrima et al. of interfacial nanoparticle arrays formed by generating Ag nanoparticles in situ at water–oil interfaces,35 in 1989 McGarvey et al. first reported the formation of metal–liquid like films (MeLLFs) with pre-formed citrate-reduced Ag nanoparticles at water–oil interfaces through the addition of a range of metal complexes, some of which were later identified as charge-neutral modifying molecules.32,36 Later, Wang et al. demonstrated the use of 2,2′-dithiobis[1-2-bromo-2-methyl-propionyloxyethane] as modifiers to induce the formation of densely packed citrate-reduced Au nanoparticle arrays at water-toluene interfaces (Fig. 2a).28 At the time it was believed that the self-assembly was a result of the contact angle of the particles at the interface being moved from 60° to 90° due to the adsorption of the thiol ligands rather than the removal of electrostatic repulsion. Nonetheless, this work pioneered the use of charge-neutral thiols as organic modifiers to produce noble metal nanoparticle arrays at water–oil interfaces, which remains a widely employed approach.37–39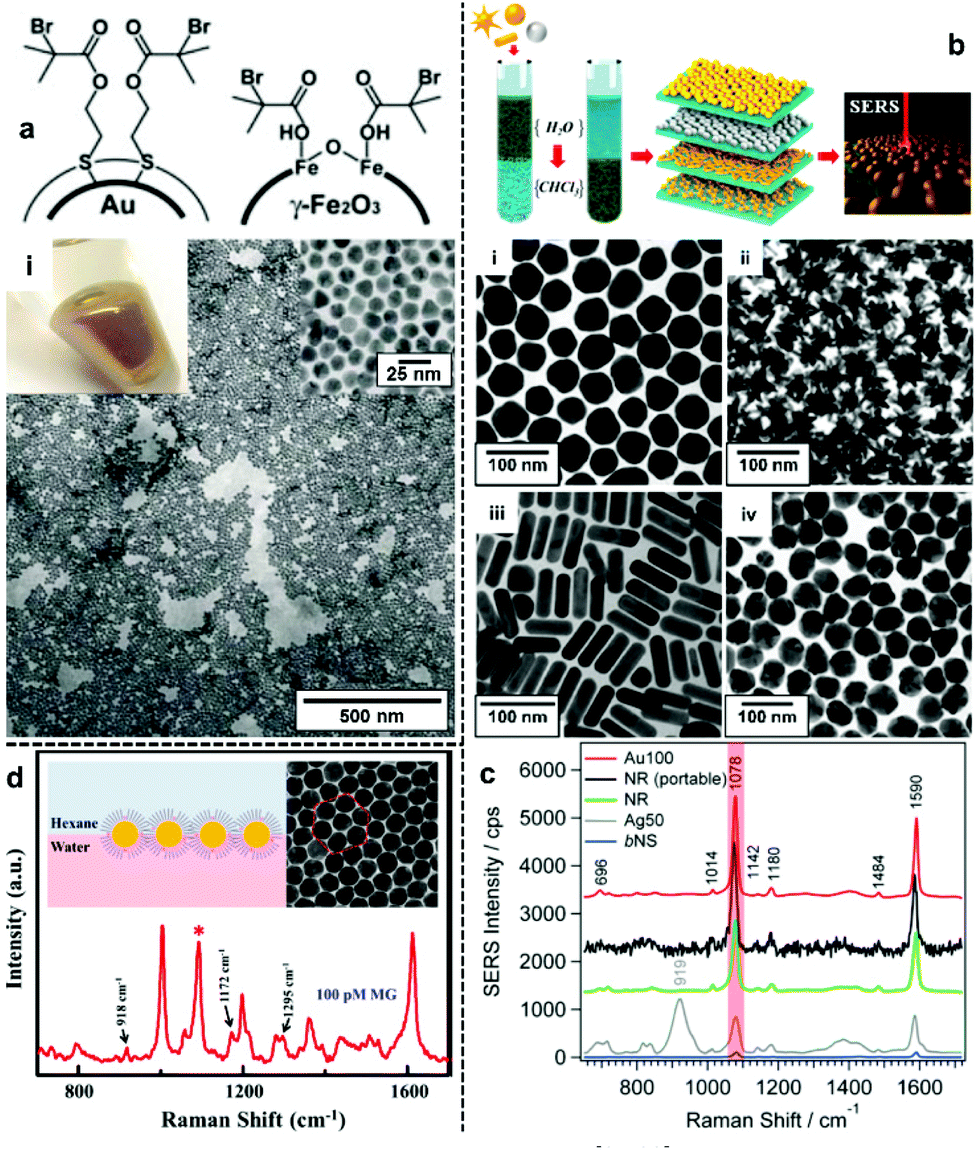 | ||
| Fig. 2 (a) Schematic illustrations of thiol modifiers adsorbed on the surface of Au and Fe2O3 nanoparticles to induce interfacial self-assembly. Optical image and electron micrograph of a 2D Au array formed using thiol modifiers. (b) Schematic illustrations of the phase-transfer and interfacial self-assembly process induced by a combination of PEG-SH and dodecane thiol modifiers. Transmission electron microscopy (TEM) images of 2D arrays formed with (i) Au nanospheres, (ii) Au nanostars, (iii) Au nanorods and (iv) Ag nanospheres. (c) SERS spectra of 4-mercaptobenzoic acid obtained using the different types of 2D nanoparticle arrays shown in (b). (d) Schematic illustrations and SEM images of 2D Au nanoparticle arrays formed at water–oil interfaces using modifiers. The spectrum shows the SERS signal obtained by using the nanoparticle array as enhancing substrate for malachite green. Panel (a) adapted with permission from ref. 28. Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. Panel (b and c) reproduced with permission from ref. 42. Copyright 2015 American Chemical Society. Panel (d) reproduced with permission from ref. 39. Copyright 2019 American Chemical Society. | ||
In principle, it should be possible to remove charged surface ligands to reduce electrostatic repulsion between Ag/Au nanoparticles with any type of charge-neutral thiol modifier. However, in practice it has been found that the molecular structure of the modifier is very important and that the use of small molecules, which are unable to provide sufficient interparticle steric repulsion as modifiers, typically leads to uncontrolled aggregation of the nanoparticles, resulting in the formation of arrays with numerous defects, which is undesirable for SERS measurements. An effective approach to overcome this problem is to use bulky polymeric molecules, such as polyvinylpyrrolidone (PVP), poly(N-isopropylacrylamide) (NIPAM), thiolated poly (ethylene glycol) (SH-PEG) and thiolated polystyrene (SH-PS), as modifiers to provide interparticle steric repulsion to prevent aggregation.40–43 For example, Liz-Marzán et al. reported a method for the assembly of a wide variety of Au and Ag nanoparticles of different sizes (up to 200 nm) and shapes (including spheres, rods and stars), as shown in Fig. 2b.42 In their work, a combination of PEG-SH and dodecane thiol were used as modifiers, since it was discovered that the use of dodecane thiols alone led to particle aggregation. As shown in Fig. 2c, the 2D arrays showed varying plasmonic properties depending on the type of nanoparticle building block used. Using 4-mercaptobenzoic acid as the test analyte, the best signal enhancement was obtained from 2D arrays formed from Au nanospheres 100 nm in diameter. Alternatively, thiol modifiers with bulky tail groups have also been shown to be able to effectively prevent uncontrolled aggregation of nanoparticles at interfaces to form uniform 2D arrays. For example, Tian and Fang et al. have shown that 2D arrays of Au nanoparticles can be generated at the water–hexane interface with a mixture of 4-mercaptopyridine and dodecanethiol modifiers (Fig. 2d).39 Within this system the bulky dodecanthiols acts as a spacer to prevent aggregation of the nanoparticles while the 4-mercaptopyridine acts as an internal standard for quantitative SERS measurements.
Although using modifiers to remove surface-charge is an efficient method to fabricate densely packed 2D arrays of plasmonic nanoparticles which exhibit strong plasmonic properties, one major drawback with this approach is that the modifier molecules will inevitably compete with analyte molecules for the enhancing surface, which is particularly undesirable for SERS studies of analytes at low concentrations and/or with weak affinity for the enhancing surface. As a result, the use of 2D nanoparticle arrays fabricated with modifiers in SERS has been mostly limited to studying model analytes with strong affinity for metal surfaces.
2.2 Charge-reduction with co-solvents
An alternative method of reducing the charge on nanoparticles so that they self-assemble into densely packed arrays at water–oil interfaces is to add a third co-solvent, typically ethanol, as shown in Fig. 3a.44–46 This method is arguably the most popular method for producing 2D nanoparticle arrays for SERS since it has been shown to be effective for different types of Ag/Au nanoparticles and does not involve the use of strongly adsorbing modifiers. Despite its widespread use, the detailed mechanism for this self-assembly process has remained elusive.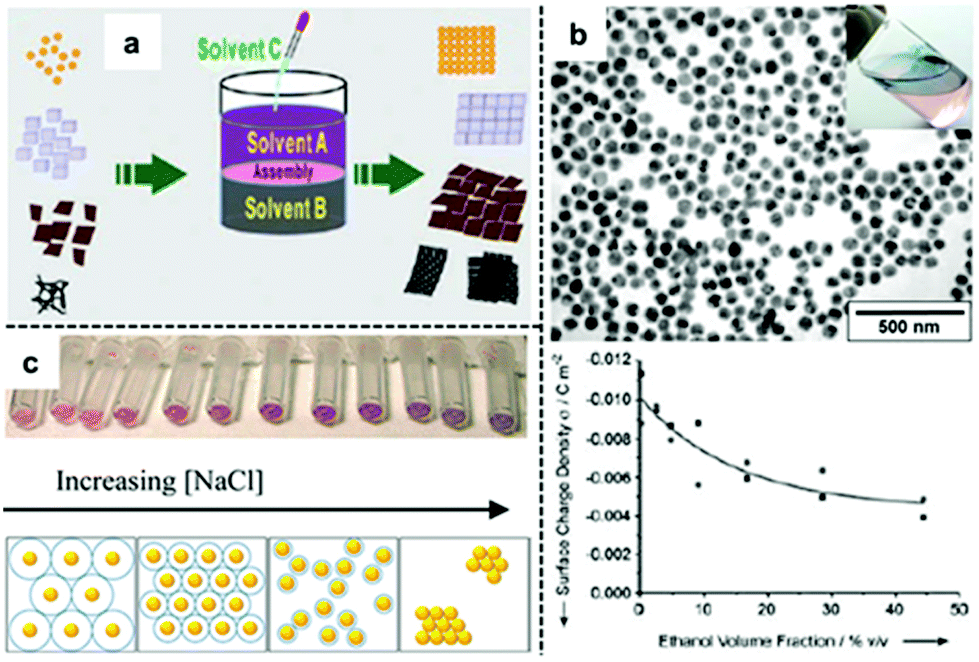 | ||
| Fig. 3 (a) Schematic illustration of co-solvent induced self-assembly of nanoparticles at water–oil interfaces. (b) Scanning electron microscopy (SEM) and optical image of 2D Au nanoparticle arrays formed at water–oil interfaces with ethanol. Plot showing the decrease of surface-charge density of Au nanoparticles on adding ethanol to the aqueous Au nanoparticle colloid. (c) Optical image and schematic illustrations showing Au nanoparticles packing tighter at the water–oil interface with thinner electric-double layers caused by increased ionic concentration in the solution. Panel (a) adapted with permission from ref. 49. Copyright 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. Panel (b) adapted with permission from ref. 47. Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. Panel (c) reproduced with permission from ref. 52. Copyright 2012 American Chemical Society. | ||
The use of ethanol to induce self-assembly of Au nanoparticles at water–oil interfaces was first reported by Vanmaekelbergh et al.47 In their pioneering work, it was suggested that the ethanol induced nanoparticle self-assembly by competitively adsorbing onto the surface of nanoparticles, reducing surface-charge in a manner similar as modifiers. This was supported by the observation of a gradual decrease in the surface-charge density of their citrate-capped Au nanoparticles when increased amounts of ethanol were added to the colloid, as shown in Fig. 3b. Although no 3D aggregates were observed within the product 2D arrays, the resulting 2D arrays lacked long-range uniformity due to the presence of cracks and voids (Fig. 3b). This method was later perfected by Sun et al. who were able to fabricate densely packed and highly-ordered 2D arrays from several types of nanoparticles (including Au) by fine-tuning the addition rate and total amount of ethanol used for self-assembly.48 In their experiments, it was observed that large amounts/high addition rate of ethanol resulted in the formation of defects and aggregates in the product film while the packing order of the particles within the interfacial arrays was highly dependent on the charge and type of capping ligand present on the surface of the particles. For example, it was observed that nanoparticle arrays formed from Au nanoparticles carrying charged citrate ligands contained congeries and defects while Pt nanoparticles carrying weakly-charged and bulky PVP polymer ligands had larger interparticle gap-distances and less defects. The relevance of capping ligands to particle packing order was also discussed by Yu et al. who in their work on assembly of nanoparticles capped with different surface ligands observed voids in the interfacial arrays assembled with citrate-capped Au nanoparticles while those formed from PVP-capped Ag nanoparticles were defect-free.49 Moreover, a range of co-solvents including ethanol, acetone, tetrahydrofuran, dimethylformamide and isopropyl alcohol were found to be effective in inducing nanoparticle self-assembly at water–oil interfaces with no observable difference in packing order of the final product arrays. The diversity in the chemical structure and the general lack of strongly adsorbing functional groups within the co-solvents both suggested that the co-solvents were not acting as adsorbing modifiers during the self-assembly process. In this context, Park et al. proposed that ethanol did not reduce the surface-charge of nanoparticles through surface adsorption but instead acted to decrease the dielectric constant of the water that it was mixed with, which in turn affected the dissociation constant of the charged ligands on the surface of the particles.50,51
Apart from the addition of a third co-solvent the surface-charge of electrostatically stabilized nanoparticles can also be reduced with the addition of appropriate amounts of aqueous salts. Similar to the addition of a co-solvent, the aqueous salt is believed to reduce interparticle electrostatic repulsion not by adsorbing directly onto the particles surface but by disrupting the electric-double layer which surrounds the electrostatically-stabilized nanoparticles.52,53 Indeed, Edel, Kornyshev et al., have shown that it is possible to alter the Debye length of the electric double layer surrounding the particles by varying the amount of NaCl added to Au colloids. As shown in Fig. 3c, the authors showed that the density of nanoparticles at the interface increased with increasing salt concentration.52 This was reflected by a red-shift in the optical reflectance maxima of the interfacial 2D array which showed an increased plasmonic coupling between the nanoparticles.53
In summary, in contrast to strongly adsorbing modifiers, the addition of co-solvents or aqueous salts, leaves the surface of the nanoparticles in the product arrays chemically exposed for further interactions with analytes, which is a significant advantage for SERS. However, the reduction of surface-charge inevitably leads to uncontrolled aggregation due to reduction of electrostatic repulsion between nanoparticles. This leads to arrays with defects and, in turn, non-uniform and irreproducible SERS measurements. Conversely, while many surface modifiers have been found to provide the appropriate balance of steric repulsion and intermolecular interactions to allow highly uniform and reproducible interfacial arrays to be formed, the addition of such modifiers and the surface blocking they create means that these approaches are still not ideal for achieving highly reproducible quantitative SERS.
2.3 Charge-screening with promoters
An alternative way to overcome electrostatic repulsion between particles located at the interface is through charge-screening provided by “promoters” which are ions carrying the opposite charge to the particles.54–56Fig. 4a illustrates the mechanism underlying this approach, which was first reported by our group in 2016.32 In this work we showed that since hydrophilic ions with opposite charge to the nanoparticles are inherently present in electrostatically-stabilized colloids, the key to successful self-assembly is the addition of micromolar concentrations of hydrophobic promoter ions (Fig. 4e), which are oppositely charged to the nanoparticles, such as tetrabutylammonium (TBA+) for negative particles and tetraphenylborate (TPB−) for positive particles. As shown by contrast SERS studies in Fig. 4d, these promoters screen the electrostatic repulsion between the parts of the particles which are immersed in the oil phase, allowing densely packed nanoparticle arrays to be formed without passivating the surface of the particles with strongly adsorbing chemical modifiers. Moreover, since the surface-charge of the particles is retained, uncontrolled aggregation of the electrostatically stabilized nanoparticles is prevented, which allows highly reproducible and defect-free 2D arrays to be obtained even with poly-disperse Ag nanoparticles. As a result, the SERS enhancement provided by the product interfacial films was found to be extremely uniform and stable.57 As shown in Fig. 4b, the SERS signals obtained from the product arrays remained unchanged for at least 20 hours. Moreover, the standard deviation of the absolute signal intensities recorded on different areas of the product array was only 1.1% (Fig. 4c), which was significantly lower than the typical point-to-point variation for state-of-the-art SERS substrates fabricated through other bottom-up approaches.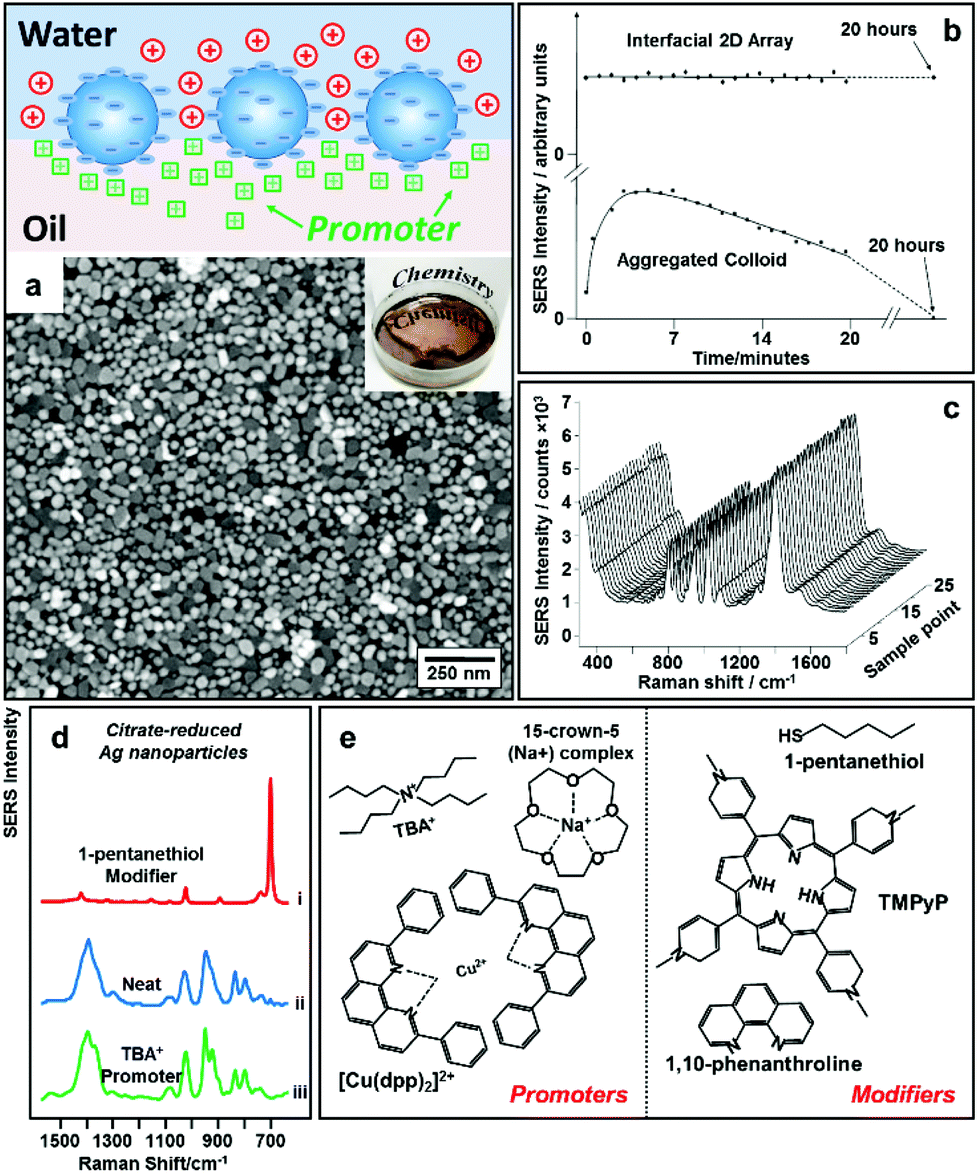 | ||
| Fig. 4 (a) Schematic illustrations of “promoter” induced self-assembly of charged colloidal nanoparticles at water–oil interfaces. SEM and optical image of 2D Au nanoparticle array formed at water–oil interfaces with promoters. (b) Plot comparing the SERS signal stability of 2D Ag nanoparticle arrays formed with promoters and aggregated colloid. (c) SERS signals obtained on 25 random sampling points on a 2D Ag nanoparticle array sample formed with promoters. (d) SERS spectra obtained from 2D arrays of citrate-reduced Ag nanoparticles fabricated using 1-pentanethiol modifiers (i), aggregated citrate-reduced Ag nanoparticles (ii), and 2D arrays of citrate-reduced Ag nanoparticles fabricated using TBA+ promoters (iii). (e) Molecular structures of typical modifiers and promoters. Panel (a), (d) and (e) adapted with permission from ref. 32. Copyright 2016 American Chemical Society. Panel (b and c) adapted with permission from ref. 57. Copyright 2012 American Chemical Society. | ||
Early reports by Fermín et al. also suggested that it was possible to overcome interparticle electrostatic repulsion electrochemically by applying a biasing potential between electrodes immersed in the aqueous and organic phase.58 However, the organic phase in these experiments contained organo-electrolytes similar to TBA+ promoters, which introduces some ambiguity since it is possible that the observed self-assembly behaviour of nanoparticles at the water–oil interface was triggered at least partly by the presence of promoters. Indeed, recent computational studies performed by Král, Lin, Schlossman et al. showed that ion condensation, particularly condensation of counterions present in the oil phase of electrochemical interfacial self-assembly systems play a critical role in lowering the electrostatic barrier that prevents the assembly of charged Au nanoparticles at water–oil interfaces.59
In contrast to charge-reduction methods, charge-screening provides a general route to assemble various types of plasmonic metal nanoparticles into stable and densely packed interfacial arrays with chemically exposed surfaces for further analyte adsorption, which allows stable and reproducible SERS measurements to be performed. As a result, this method has been adopted by various research groups for SERS analysis in both applied and fundamental research.
2.4 Combined techniques for self-assembly
The search for methods to generate stable and uniform 2D nanoparticle arrays at water–oil interfaces has also led to attempts to induce self-assembly using combinations of the different approaches to overcome interparticle electrostatic repulsion. As shown in Fig. 5a, the most widely reported examples of these combined approaches are those where co-solvents/aqueous salts were used to reduce surface-charge to induce self-assembly while modifiers were used to prevent aggregation of the nanoparticles.39,50–53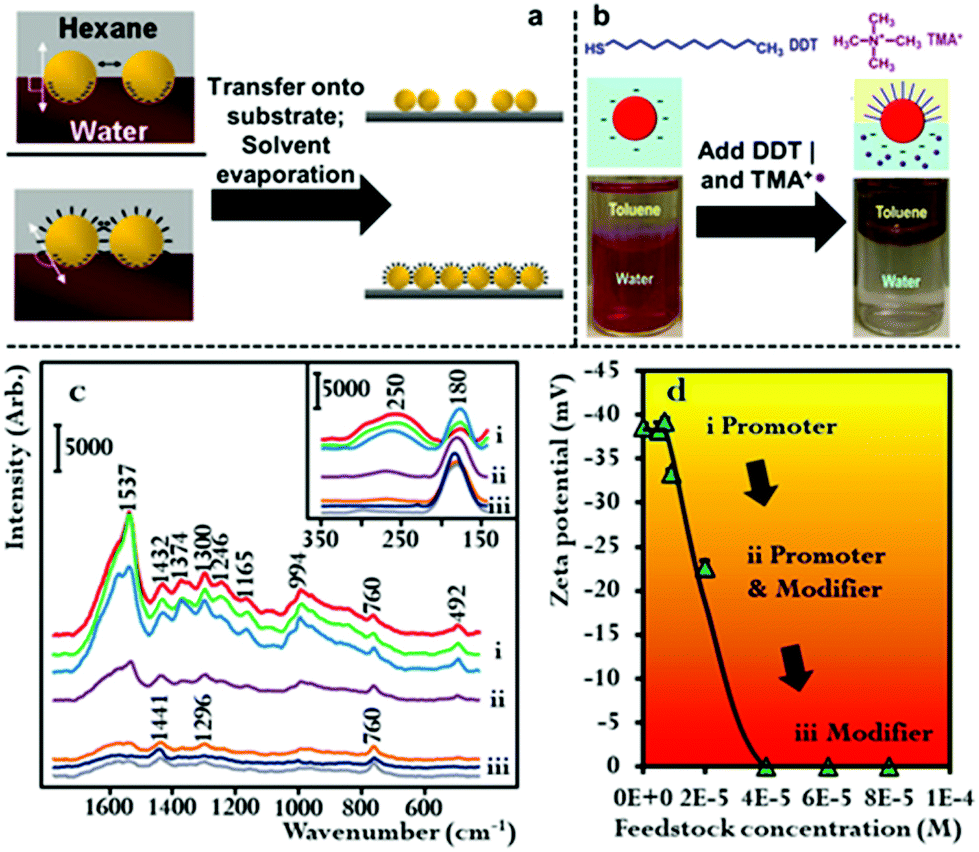 | ||
| Fig. 5 (a) Schematic illustrations comparing the difference between particle packing in 2D arrays formed at water–oil interfaces with just ethanol or with a combination of ethanol and thiol modifiers. (b) Schematic illustrations of a nanoparticle stabilized at water–oil interfaces with the aid of promoters and modifiers. Optical image shows 2D arrays of Au nanoparticles formed at the water–oil interface with a combination of promoters and modifiers. (c) SERS spectra showing the gradual change in the composition of the capping agents adsorbed on the surface of Au nanoparticles with increasing concentration of cetyltrimethylammonium bromide (CTAB). (d) Zeta potential data showing the decrease in surface potential of the Au nanoparticles with increasing amounts of CTAB added to the Au colloid. Panel (a) adapted with permission from ref. 51. Copyright 2008 American Chemical Society. Panel (b) adapted with permission from ref. 60. Copyright 2015 American Chemical Society. Panel (c and d) reproduced with permission from ref. 61. Copyright 2020 the authors, published by Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
The combined use of promoters and modifiers for producing plasmonic interfacial nanoparticle arrays for SERS has also been demonstrated.53,59,60 As shown in Fig. 5b, He et al. found that the use of tetramethylammonium (TMA+) promoters or 1-dodecane thiol (DDT) modifiers alone led to the formation of sparse and aggregated 2D arrays of Au nanoparticles, respectively, and that optimal results were achieved when the modifier and promoter were used in combination.60 It was determined in their work that the TMA+ promoter did not provide sufficient charge screening to generate densely packed 2D arrays. This could be due to the low solubility of TMA+ in the toluene oil phase which they used in their work.
In addition to the work using combinations of agents for nanoparticle self-assembly, we have recently shown that it is also possible for a single compound to assume dual roles in nanoparticle self-assembly. In this work we showed that hexadecyltrimethylammonium bromide (CTAB), which is an amphiphilic surfactant molecule commonly used as a growth directing agent and stabilizing agent in the synthesis of plasmonic nanoparticles can also induce self-assembly of Au nanoparticles at final concentrations between 3.5 × 10−7–5.6 × 10−6 M.61 Moreover, we showed that depending on its concentration CTAB acted as a modifier and/or promoter in the self-assembly system. This is possible because a proportion of the CTAB added to the oil–water system will dissolve into the oil phase as CTA+ anion and act as a promoter while the remainder of the CTAB will adsorb onto the surface of Au particles and act as charge-neutral CTAB modifiers. As shown in Fig. 5c and d, using SERS and zeta potential measurements it was found that at CTAB concentrations between 3.5 × 10−7–1.4 × 10−6 M the modifier effect was negligible, which meant that self-assembly was induced by charge-screening. However, as the concentration of CTAB was increased from 1.4 × 10−6–2.8 × 10−6 M, a gradual but clear reduction in the net surface-charge of the Au particles was observed, which showed the CTAB molecules were acting as both modifiers and promoters. At CTAB concentrations between 2.8 × 10−6–5.6 × 10−6 M, the surface-charge of the Au nanoparticles was completely removed, which showed that the CTAB molecules were acting as modifiers. Consistent with discussions above, it was found that highly uniform 2D arrays were formed when the concentration was sufficiently low that the CTAB acted predominantly as CTA+ promoter, while defected and aggregated 2D arrays were formed when the surface-charge of the particles was reduced due to the addition of higher concentrations of CTAB modifiers.
In general, up to now, interfacial plasmonic 2D nanoparticle arrays fabricated using combined techniques have not shown significant advantages in SERS over their traditional counterparts. However, these methods provide interesting fundamental insights into the self-assembly of plasmonic nanoparticles at water–oil interfaces and add to the versatility of this approach for fabricating stable, uniform and active SERS substrates.
2.5 SERS applications of 2D nanoparticle arrays at water–oil interfaces
To date, interfacial 2D nanoparticle arrays have found applications as SERS substrates for the detection of various important analytes.62–64 For example, Yang etc. showed indirect quantitative SERS detection of a variety of microRNAs using 2D Au nanoparticle arrays formed at the water–hexane interface (Fig. 6a).65 In their work, the target miRNAs were added to a carefully designed sampling mixture which contained free carboxyrhodamine (ROX) Raman reporters as well as DNA strands functionalized with methylene blue (MB) Raman reporters and magnetic particles. The interactions between the target miRNA and DNA led to the release of the MB reporter into solution along with the binding of ROX reporter onto the DNA, which led to the simultaneous increase in the SERS signal of MB and decrease in the SERS signals of ROX. By measuring the intensity ratio between the characteristic peaks of MB and ROX, the authors showed that miRNA 155 could be quantitatively detected down to 1.10 aM, even though miRNA 155 did not generate any detectable SERS signals alone. Using L-cysteine modifiers, Liu et al. showed SERS detection of trinitrotoluene (explosive) with 2D Au nanoparticle arrays formed at the interface between water and dimethyl carbonate as the enhancing substrate.66 In their work, it was shown that the trinitrotoluene molecules adsorbed onto the Au surface by forming Meisenheimer complexes with the L-cysteine modifiers while molecules of similar structures, such as dinitrotoluene and 3-nitrotoluene could not. This allowed highly selective and sensitive detection of TNT down to 50 fM. Edel, Kornyshev et al. demonstrated that a droplet of Au nanoparticle array formed with NaCl solution on a coverslip can serve as a versatile substrate for quantitative SERS detection of analytes including toxins, narcotics, explosives and other hazardous chemicals.67 Even with simple spherical Au nanoparticles, the limits of detection for the majority of the analytes were below nanomoles. Moreover, they showed that the interfacial arrays were also effective at detecting airborne analytes which could be captured by the liquid droplets and transferred onto the enhancing surface.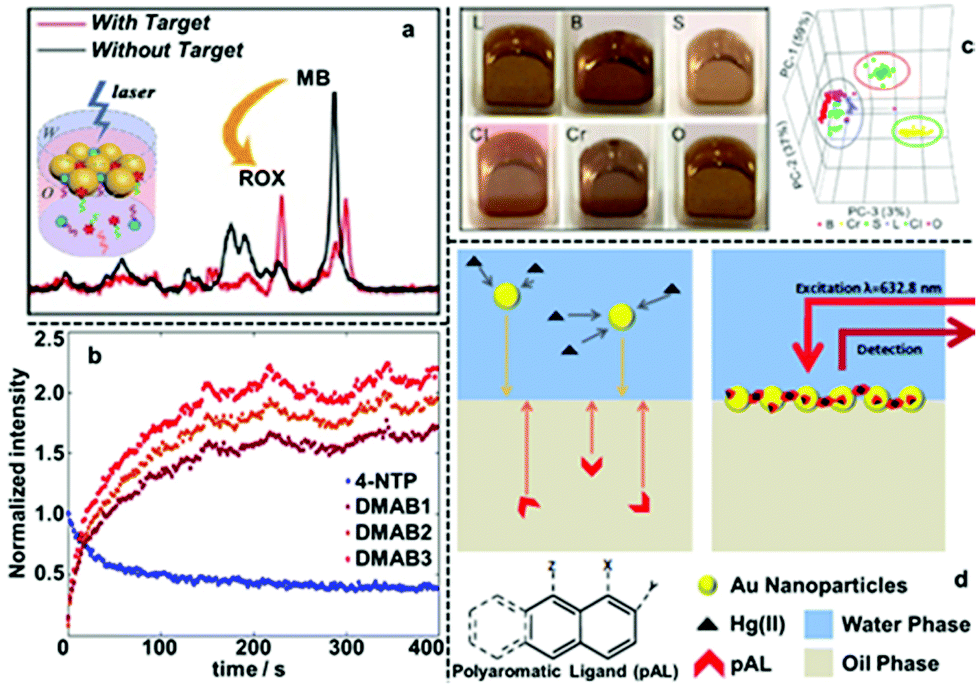 | ||
| Fig. 6 (a) SERS spectra of two Raman tags, carboxyrhodamine (also known as “ROX”) and methylene blue (MB) obtained using 2D Au arrays formed at water–oil interfaces for ratiometric quantitative detection of miRNA. (b) The SERS signal intensities of the characteristic peaks of 4-nitrophenol (4-NTP) and three different characteristic peaks of 4,4-dimercaptoazobenzene (DMAB) obtained using 2D Ag arrays formed at water–oil interfaces and plotted versus time. (c) Optical images of 2D Au arrays formed at water–oil interfaces containing different types of edible oils. 3D principle component analysis score plots obtained from the SERS signals of the 2D arrays containing different oils. (d) Schematic illustrations showing SERS detection of Hg(II) in aqueous solution using 2D Au arrays functionalized with polyaromatic ligands as the enhancing substrate. Panel (a) adapted with permission from ref. 65. Copyright 2020 American Chemical Society. Panel (b) adapted with permission from ref. 68. Copyright 2020 American Chemical Society. Panel (c) reproduced with permission from ref. 72. Copyright 2019 American Chemical Society. Panel (d) adapted with permission from ref. 76. Copyright 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
In addition to being highly plasmonically active, the high stability of 2D nanoparticle arrays compared to other liquid-based SERS substrates makes them excellent candidates for performing in situ kinetic SERS studies. For example, Haisch et al. showed that Ag nanoparticle arrays formed with TBA+ promoters can be used as a convenient and highly stable SERS substrate for mechanistic studies of surface-plasmon resonance driven reduction of 4-nitrothiophenol to 4-aminothiophenol.68 As shown in Fig. 6b, the authors were able to simultaneously monitor evolution of the SERS signals of the reactant (4-nitrophenol), intermediate (4,4-dimercaptoazobenzene) and product (4-aminophenol). Moreover, by modifying the surface of particles with PVP polymer, highly stable 2D arrays were produced which allowed the effect of different ionic concentrations to be probed without affecting the reproducibility of the SERS measurements.
Another significant advantage of 2D arrays formed at water–oil interfaces as SERS substrates is that the enhancing particles are open to the adsorption of molecules present in both the organic and aqueous side of the interface. Moreover, since the chemical composition of the oil is unimportant, as long as it provides a high energy interface with water for self-assembly, a variety of organic solvents can be used as the oil phase, which provides an extremely versatile SERS platform, especially for studying dipolar analyte molecules with low solubility in water.69–75 For example, Liu et al. showed that six edible oils can be distinguished using SERS and interfacial 2D Au nanoparticle arrays as enhancing substrates (Fig. 6c).72 Moreover, they showed that oxidation and adulteration of the edible oils could also be detected with the assistance of principle component analysis.66,75 In a separate study from the same research group, it was shown that four different polycyclic aromatic hydrocarbons (PAHs) could be detected with a detection limit of 10 ppb using 2D Au nanoparticle arrays assembled at water-chloroform interfaces as the SERS enhancing substrate.73 More impressively, they showed that multiple PAHs could be detected simultaneously from different types of edible oils, which provides a convenient and sensitive platform for food quality monitoring. It should be noted that even for analyte molecules which are soluble in water, the solubility difference of the analyte in water and oil can still be exploited to improve SERS analysis. For example, Tan and Liu et al. showed that convenient and highly quantitative SERS analysis of thiabendazole fungicide in apple juice samples can be achieved by using the oil phase (chloroform) as an extraction phase.74 This was achieved by simply shaking the apple juice containing thiabendazole with chloroform to facilitate phase transfer of thiabendazole molecules into chloroform, which was then used as the oil phase for self-assembly of plasmonic Au nanoparticles.
Apart from introducing analytes with low solubility in water, the oil phase of 2D plasmonic arrays has also been exploited as the reaction medium for modifying the surface of the enhancing particles with dipolar modifiers to facilitate detection of non-adsorbing analytes. For example, Edel, Wilton-Ely et al. exploited the oil phase of 2D Au nanoparticle arrays to functionalize the surface of the plasmonic nanoparticles with polyaromatic ligands (PAL), such as 1,8-diaminonaphthalene, which had low solubility in water.76 As shown in Fig. 6d, this allowed successful SERS detection of Hg(II) and Ag(I), which do not adsorb directly to Au nanoparticles, by forming metal-PAL complexes. The concentration of Hg(II) could be assessed through a systematic change of PAL SERS spectrum. Moreover, it was demonstrated that it was possible to detect Hg(II) present both in water and air using these PAL functionalized 2D Au nanoparticle arrays as the SERS substrate.
2.6 2D nanoparticle arrays at curved water–oil interfaces
Finally, it is useful to note that it is possible to prepare structures in which 2D nanoparticle arrays at water–oil interfaces are curved into a 3D spheres, the best known being Pickering emulsions, which are composed of particle-coated liquid droplets sitting within an immiscible liquid. In general, the forces driving the interfacial particle assembly to form Pickering emulsions are similar to those governing formation of the analogous planar 2D nanoparticle arrays. However, in addition to these forces it is also necessary to consider the properties which the nanoparticles need to possess in order to stabilise a curved liquid meniscus,77 which are related to the particle hydrophobicity and have been extensively investigated, forming a large research area in their own right. In practice, stable plasmonic Pickering emulsions can be formed readily from a variety of particle types and have been shown to be highly active substrates for a variety of SERS applications ranging from reaction monitoring to bioanalysis.78–80 Moreover, plasmonic Pickering emulsions can also serve as template materials to form multi-dimensional substrates for SERS. For example, evaporation of the oil core of plasmonically active emulsions produces spherical particle-aggregates which are stable and give very high SERS enhancements81,82 while pre-doping the oil with polymer before evaporation yields SERS-active particles fixed onto a solid polymer core.83,843 Transforming soft 2D plasmonic arrays into solid SERS films
Despite the many advantages provided by plasmonic arrays sitting at water–oil interfaces these materials also have limitations associated with both stability and ease of handling which are particularly significant for real-life applications. Specifically, the arrangement and stability of the particles at the interface and in turn their plasmonic response, is closely associated with the chemical and physical properties of the nanoparticles’ surface, the water–oil interface and the bulk liquid environment. Moreover, 2D nanoparticle arrays at liquid–liquid interfaces are inherently less convenient to store, handle and dispose of than solid substrates. As a result, considerable effort has been devoted towards transforming such arrays into solid films for routine SERS analysis.3.1 Transferring soft-arrays onto rigid-substrates
The most straightforward and widely applied strategy to transform nanoparticle arrays assembled at water–oil interfaces into solid films is by physically depositing pre-formed nanoparticle arrays onto solid substrates. For example, we and other research groups have demonstrated that the interfacial arrays can be transferred onto solid substrates, such as silicon wafers or quartz slides, by sliding the solid substrate material beneath the particles at a tilted angle and then carefully lifting the particle array out horizontally.85–90 Similarly, it has been shown that arrays can be deposited onto the substrate material by placing the substrate at the bottom of the reaction vessel and then draining away the liquid phases.40,91,92The biggest advantage of the physical deposition strategy lies in its versatility, which allows the nanoparticle arrays to be transferred onto almost any type of solid surface with ease, and the way in which the product material can combine the functionalities of both the 2D arrays and the solid substrates. For example, Millyard et al. demonstrated that by depositing Au nanoparticle arrays on polydimethylsiloxane (PMDS), the interparticle distance could be varied by applying external mechanical strain to the PDMS substrate, which enabled reversible modulation of the plasmonic response of the SERS substrate (Fig. 7a).93 Liu and Ji et al. demonstrated that when chromatographic paper was used as the solid substrate, the target component can be separated from the sample mixture based on polarity differences and concentrated at the tip of the paper substrate, which is coated with plasmonic 2D nanoparticle arrays, by lateral flow (Fig. 7b). As an example, they showed that this allowed the detection of rhodamine 6G down to attomolar level with just microliters of sample (Fig. 7c).94 We have recently shown that 2D arrays of plasmonic nanoparticles can be deposited onto the hydrophilic tips of copper needles with superhydrophobic side-walls, which allowed sub-micromolar SERS detection of analytes, such as crystal violet, trinitrotoluene and adenine deposited from sub-microliter volumes of analyte solutions.95
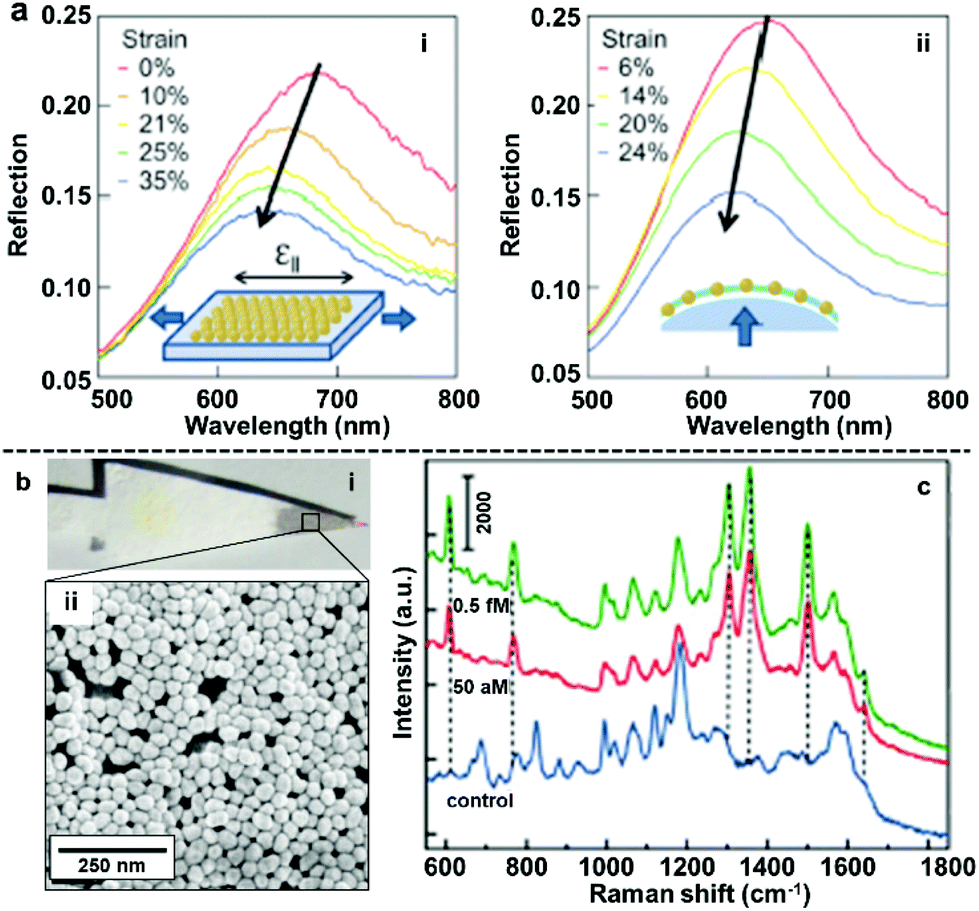 | ||
| Fig. 7 (a) Reflection spectra obtained by uniaxial stretching Au nanoparticle mats parallel to optical polarization (left) and biaxial stretching Au nanoparticle mats under unpolarised light. (b) Optical images showing filter paper after being dip-coated with 2D Au arrays (i) SEM image of the 2D Au layer on the surface of the filter paper substrate (ii). (c) SERS spectra of rhodamine 6G obtained at different concentrations using filter paper dip-coated with 2D Au arrays as enhancing substrate. Panel (a) reproduced with permission from ref. 93. Copyright 2012 American Institute of Physics. Panel (b and c) adapted with permission from ref. 94. Copyright 2015 American Chemical Society. | ||
3.2 Converting soft-arrays into flexible polymer-based films
Although the physical deposition method for converting interfacial 2D arrays into solid films is convenient and versatile it does have a major disadvantage, which is that the particles are only weakly attached to the surface of the substrate.96 As a result, it is extremely difficult to preserve the long-range uniformity of the 2D arrays during the deposition process, which leads to poor signal uniformity and batch-to-batch reproducibility in SERS measurements. Moreover, the weak interaction between the plasmonic particle layer and the substrate material also means that the product material is unable to sustain more challenging sampling environments and manipulation, such as sonication or continuous agitation, which are often required if the SERS analysis is more challenging than simple detection of pure analytes under ideal laboratory environments.A potential strategy to maintain the long-range uniformity of the 2D arrays during physical deposition is by locking the relative positions of the nanoparticles within the 2D array at the interface, either through ligand–ligand interactions between neighbouring particles or particle-particle sintering.97,98 For example, Fiałkowski et al. demonstrated that interfacial Au nanoparticle arrays can be deposited as a highly uniform and defect-free monolayer onto silicon wafers by chemically crosslinking the particles at the interface prior to physical deposition.99 As shown in Fig. 8a, this was achieved by functionalizing the surface of the nanoparticles with aminothiolate ligands which could be used along with a crosslinking agent to connect neighbouring particles and increase the stability of the 2D array during physical deposition. Similarly, Cheng et al. have shown that using thiolated-polystyrene as the capping agent yielded mechanically strengthened Au nanoparticle arrays at water–oil interfaces.43,100–103 The resulting films could then be transferred onto TEM grids or silicon wafers to form SERS substrates with excellent uniformity and reproducibility.103
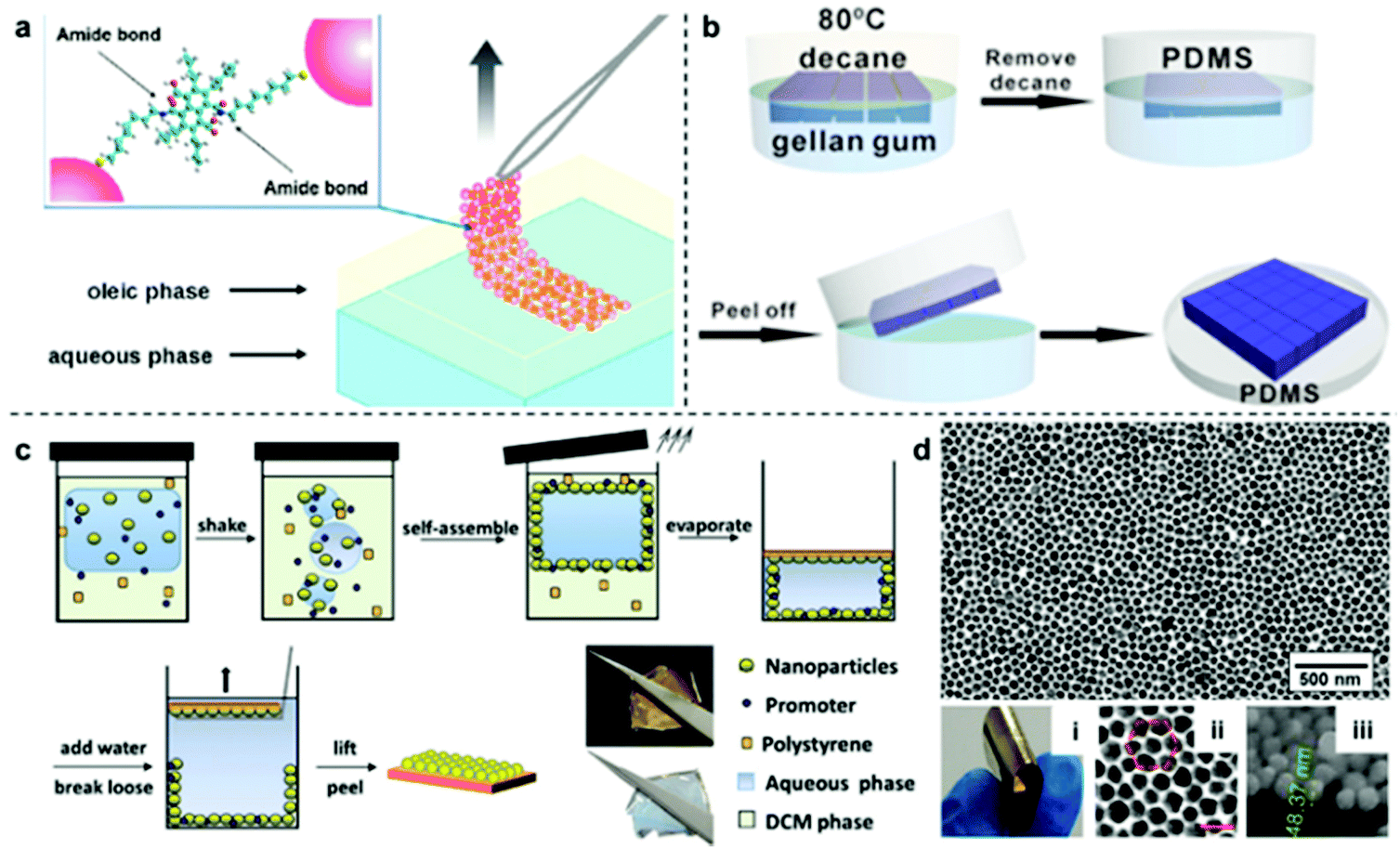 | ||
| Fig. 8 (a) Schematic illustrations of freestanding interfacial 2D Au arrays fabricated via interparticle crosslinking. (b) Schematic illustrations of the two-step fabrication of PDMS supported nanoparticle arrays via gel-trapping. (c) Schematic illustrations of the one-pot fabrication of polystyrene supported nanoparticle arrays via solvent evaporation induced polymer deposition. (d) Optical and SEM images of the product polystyrene supported Au nanoparticle array fabricated with the one-pot approach illustrated in (c). Panel (a) reproduced with permission from ref. 99. Copyright 2016 American Chemical Society. Panel (b) reproduced with permission from ref. 109. Copyright 2016 American Chemical Society. Panel (c) reproduced with permission from ref. 110. Copyright 2016 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. Panel (d) adapted with permission from ref. 111. Copyright 2018 Elsevier Ltd. | ||
However, since ligand–ligand interactions requires the surface of the plasmonic particles to be covered with strongly adsorbing modifiers, the enhanced uniformity and integrity comes at the price of surface accessibility, which is crucial for SERS. To mitigate the effect of modifiers on the SERS performance of the product substrates, various methods, such as oxygen-plasma and NaBH4 treatment have been proposed.104,105 However, these methods add to the complexity of the substrate fabrication process, which lowers their applicability. Therefore, the ideal method to transform interfacial 2D arrays into solid 2D substrates should allow both the initial structure and accessibility of the parent interfacial array to be preserved while adding the ease of use and robustness of solid substrates. This can be achieved by partially trapping the 2D arrays in gels/polymers, such as PDMS, polystyrene, poly(methyl methacrylate) and polyvinyl chloride, to create polymer films with arrays of exposed nanoparticles anchored on their surfaces.106–108 As shown in Fig. 8b, Ling et al. adopted a two-step method initially created to measure the three-phase contact angle of nanoparticles at fluid interfaces to anchor interfacial 2D arrays of Ag nanocubes onto PDMS films for SERS.109 In this method, the nanoparticles are first assembled at the water–oil interface with gellan gum dissolved in the aqueous phase. After formation of the 2D array, gelling of the aqueous phase was initiated by lowering the temperature of the solution, which resulted in the relative position of the particles at the interface being locked by the solidified gel. Finally, the oil phase was replaced with PDMS solution which cures slowly at room temperature to form a robust and flexible film that could be peeled off with the particle layer anchored on the surface. More recently, we have shown a one-pot method which takes advantage of the solubility of polystyrene in oils, such as dichloromethane and chloroform, to produce 2D Au/Ag plasmonic arrays anchored on polystyrene films.110 As shown in Fig. 8c, in this method the polystyrene was predissolved into the oil phase prior to self-assembly and could be triggered to deposit as a film by evaporating the oil at room-temperature. Since polystyrene is immiscible in water, this meant that the polystyrene film formed only on the oil side of the interface, which allowed the particle arrays to be anchored tightly into the product polystyrene film with the bulk of their surfaces exposed for interaction with SERS analytes. The strong attachment between nanoparticles and the polystyrene substrate was demonstrated with ASTM pencil tests, which showed that the particle array could not be scraped off the polymer surface without first breaking the polymer film. Moreover, this method allowed the long-range packing and in turn, the SERS uniformity of the parent interfacial arrays to be preserved with excellent batch-to-batch reproducibility, as shown by measuring SERS signals of thiophenol model analytes on two different batches of nanoparticle–polymer films.111,112
An emerging application of nanoparticle–polymer enhancing substrates in SERS that came with the unique combination of robustness, flexibility and transparency of polymer substrates is for the in situ detection of analyte molecules dispersed on uneven surfaces. This typically involves stamping the nanoparticle–polymer substrate onto the sample surface with the particle side contacting the analytes while the SERS signals are collected from the polymer side of the substrate.96,103,107,111 As shown in Fig. 9a, Zheng et al. reported that stamping flexible Au-PMMA films onto fish allowed detection of malachite green isothiocyanate down to 0.1 μM.107 By taking advantage of the flexibility of the polymer substrate and accessibility of the plasmonic nanoparticle array, we have recently shown that SERS of solid analytes can even be obtained by stamping the particle side of nanoparticle–polymer films directly onto completely dry solid analytes to press the analytes into plasmonic hots-spots, as shown in Fig. 9b.111 Since the analytes are physically pressed into hot-spots rather than being adsorbed, the method can be readily adopted for SERS detection of various weak/non-adsorbing analyte molecules, which have already included explosives, novel psychoactive substances (“legal highs”), biomolecules and pharmaceutical drugs. Moreover, the excellent signal uniformity of the nanoparticle–polymer films allowed not only highly reproducible quantitative SERS detection of the analyte molecules down to nano/picogram levels but also the spatial distribution of the analyte molecule on the sample surface to be mapped.
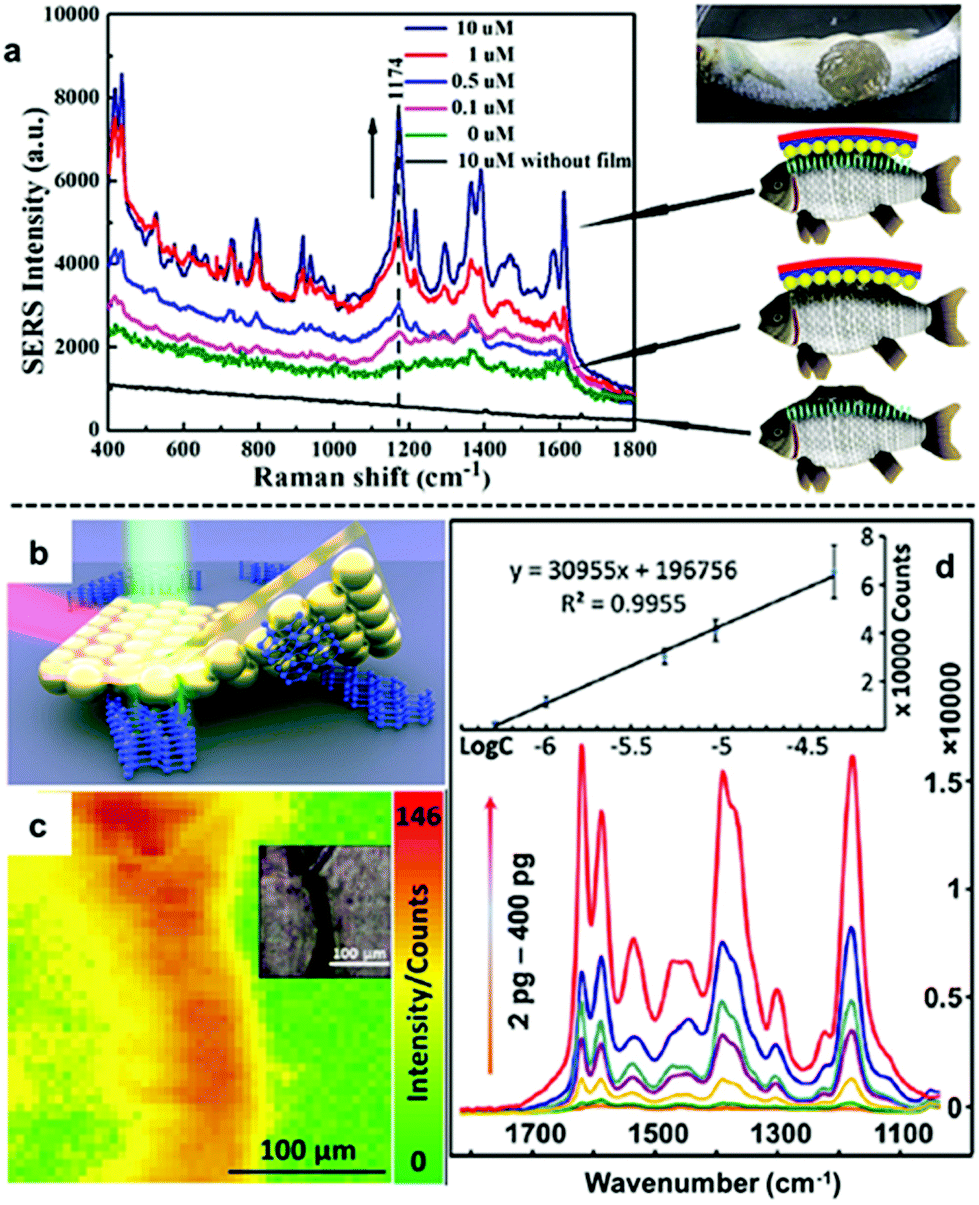 | ||
| Fig. 9 (a) In situ SERS detection of different concentrations of malachite green on fish. (b) Schematic illustration of solvent-free SERS achieved by physically pressing solid analyte crystals into plasmonic hot-spots. (c) SERS heat-signature map obtained by stamping a solid line of crystal deposits with polystyrene–Au nanoparticle films. Inset shows an optical image of the sampled area. (d) SERS spectra obtained by stamping different amounts of crystal violet crystals with polystyrene–Au nanoparticle films. Inset shows the calibration plot of SERS signals versus the logarithm of amount of crystal violet. Panel (a) reproduced with permission from ref. 107. Copyright 2014 American Chemical Society. Panel (b–d) reproduced with permission from ref. 111. Copyright 2018 Elsevier Ltd. | ||
It is worth noting that before this, stamping with nanoparticle–polymer films has always been performed on a wet sample surface to facilitate the adsorption of the analyte molecules into the plasmonic hot-spots. In fact, the majority of SERS in general has required the analyte to be in the liquid or vapour phases so that analyte molecules can diffuse into plasmonic hot-spots which are typically only a few nanometres wide. The ability to carry out solvent-free SERS with nanoparticle–polymer enhancing substrates opens new possibilities for SERS analysis of crystal structures, precious samples or non-soluble materials.
4 Future directions: towards fine-controlled nanoparticle behaviour at water–oil interfaces for tailored plasmonic properties
With the establishment of methods to produce stable, uniform and surface-accessible plasmonic 2D arrays and to further transform these soft-arrays into robust easy to handle hybrid materials, increasing effort is now being devoted towards developing methods to accurately manipulate the behaviour of nanoparticles at water–oil interfaces and in turn their plasmonic properties. In this context, the most widely researched area has been the fine tuning of the gap distance between adjacent nanoparticles.113–115 For example, by modifying the surface of the Au nanoparticles with acrylamine, Huang and Chen et al. demonstrated that the size of the interfacial films formed with these particles can shrink by more than 50% upon UV light irradiation due to the crosslinking of acrylamine on the surface of neighbouring particles (Fig. 10a).116 In their work, the authors showed that the crosslinking led to gradual decrease in gap distance between adjacent nanoparticles from ca. 5 nm to ca. 0.5 nm with increasing reaction time, which resulted in an increase in the SERS intensity of 4-aminothiophenol signal recorded on the assembled films. In addition, since the shrinkage of interfacial films effectively reduced the number of voids, the uniformity of SERS signals across the films also increased. Similarly, other groups have demonstrated that the gap distance between nanoparticles in interfacial arrays can be tuned by changing the length of ligands, such as alkyl thiols, alkyl amines and polymers, adsorbed on the surface of the plasmonic nanoparticles.113,115,117 While adjusting the length of ligands on the surface of Au nanoparticles provides a simple method to manipulate the interparticle distance of 2D nanoparticle interfacial arrays, the addition of strongly adsorbing ligands inevitability decreases the accessibility of the plasmonic surface, which is undesirable for SERS. In an effort to circumvent this issue, Kang et al. recently demonstrated a method based on Au@SiO2 nanoparticles which allowed the distance between the plasmonic cores in interfacial arrays to be controlled without the use of surface-passivating molecular modifiers.118 As shown in Fig. 10b, this was achieved with a multi-step method which involved assembling Au@SiO2 nanoparticles at water–oil interfaces, followed by transferring the soft-arrays onto a solid substrate and finally etching away the SiO2 shell with NaOH solution. By controlling the NaOH etching time the distance between the plasmonic cores could be controlled with ∼1 nm precision. However, it was observed that the gap distance between adjacent nanoparticles upon complete removal of the SiO2 shell did not resemble the thickness of the initial SiO2 layers between the plasmonic cores, but was set at ca. 2 nm. This was attributed to an increase in attractive van der Waals forces between the bare Au particles compared to Au@SiO2 particles, which led to the particle moving closer towards each other. Despite this unexpected result, the authors showed that the formation of small ∼2 nm gaps led to increased SERS enhancement which allowed two model test analytes to be detected down to nanomolar concentrations.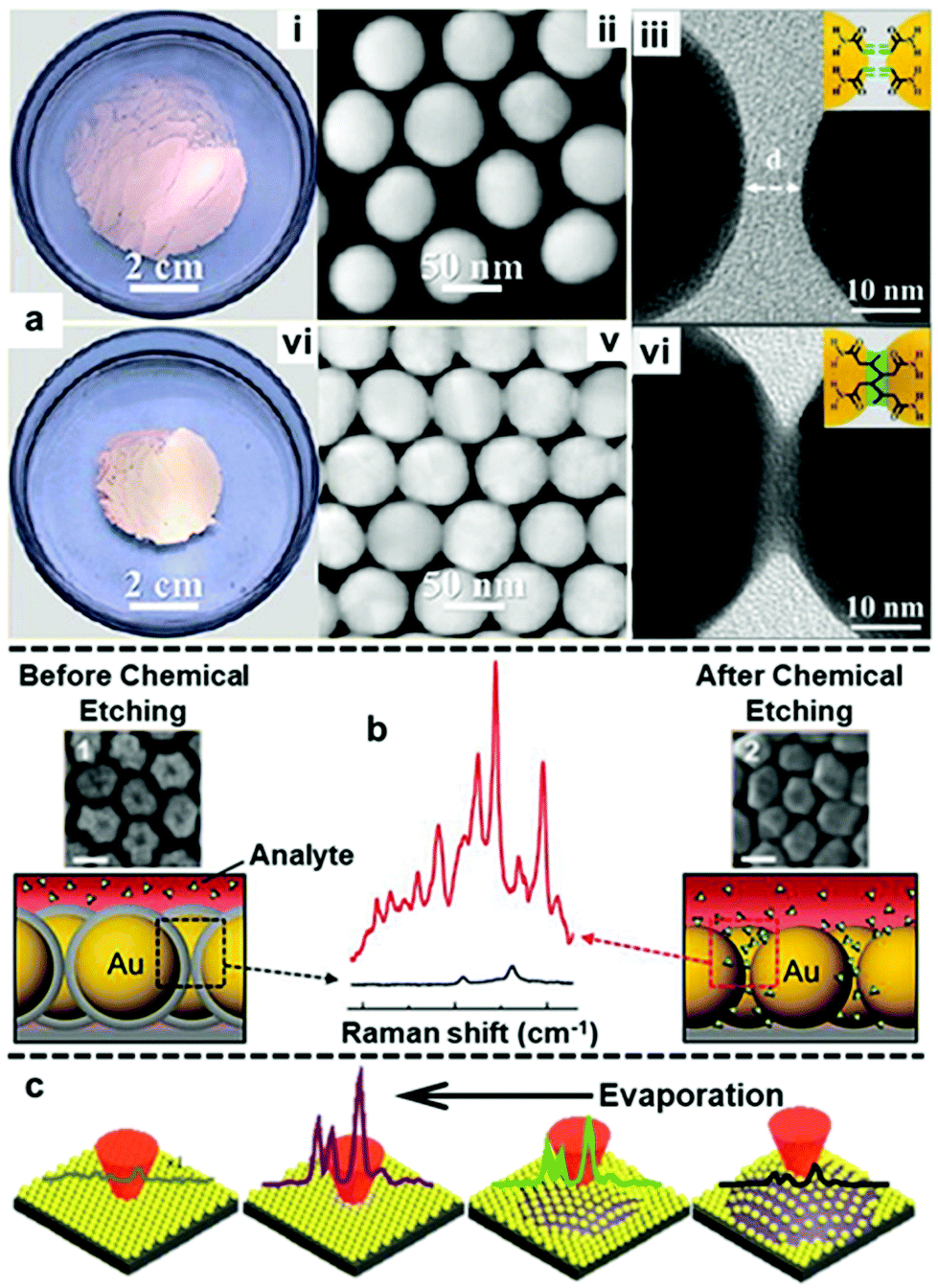 | ||
| Fig. 10 (a) Optical and TEM images of 2D Au arrays formed at water–oil interfaces before (i–iii) and after (iv–vi) photo-crosslinking. (b) Schematic illustrations and SEM images of Au@SiO2 nanoparticle arrays before and after chemical etching. The scale bars in the SEM images correspond to 50 nm. SERS spectra showing the difference in signal enhancement provided by the 2D arrays before and after chemical etching. (c) Schematic illustrations of the arrangement and corresponding SERS signals of nanoparticles on the surface of a droplet throughout droplet evaporation. Panel (a) reproduced with permission from ref. 116. Copyright 2018 American Chemical Society. Panel (b) adapted with permission from ref. 118. Copyright 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. Panel (c) adapted with permission from ref. 119. Copyright 2017 The Royal Society of Chemistry. | ||
With increased understanding of the forces controlling the gap distance between adjacent particles at the interface, Yuan, Yao et al. have been able to demonstrate that the plasmonic properties of interfacial 2D Au nanoparticle arrays can be dynamically controlled by altering the size of the liquid interface.119 As shown in Fig. 10c, it was observed that the signals of the Raman reporter, thiophenol, increased with continuous evaporation of the aqueous phase supporting the 2D plasmonic array. This was attributed to the shrinking of the interface which led to tighter packing of the Au particles at the interface and, in turn, increased SERS activity. While in this case the change in the observed SERS activity reflects only the average gap distance of the plasmonic array, it does give a clear indication of the significance of gap distance in SERS. Although methods to dynamically and precisely control interparticle gap distance in plasmonic interfacial arrays have been demonstrated, current limitations in the size of the Au/Ag nanoparticles that can be manipulated using these methods mean that they cannot be used for SERS applications. For example, Ding, Baumberg et al. showed that the interparticle distance of polymer functionalized Au nanoparticle arrays at water–oil interfaces could be dynamically controlled with temperature.41 More specifically, Au nanoparticles 15 nm in diameter were modified with poly-N-isopropylacrylamide (PNIPAM), which reversibly contracts and relaxes depending on the environment temperature and allowed the gap distance between nanoparticles at the interface to be manipulated. With this method, it was possible to reversibly adjust the peak of the elastic scattering of the interfacial 2D array from 640 nm to 690 nm by adjusting the temperature from 35 °C to 25 °C. Varying the electric potential of the interface with externally applied voltages has also been explored as an approach to control the gap distance of interfacial 2D arrays.58,59,120 For example, Kornyshev, Edel et al. recently showed an electrochemical nanoplasmonic platform which allowed reversible fine-tuning of the gap distances in 2D Au nanoparticle arrays at water–oil interfaces.121 With 16 nm diameter Au nanoparticles functionalized with 12-mercaptododecanoic acid, the authors demonstrated that 2D plasmonic arrays could be reversibly controlled from full assembly (2.8 nm gap distance) to complete disassembly, where the nanoparticles are returned to the bulk aqueous phase as a colloidal dispersion.
In addition to controlling gap distance, another widely researched method to obtain interfacial 2D arrays with improved plasmonic properties is the use of anisotropic nanoparticles as building blocks. Some examples of these anisotropic nanoparticles include rods, stars, octahedra, plates and boxes.42,122–124 In general, the basic forces which govern the assembly of anisotropic nanoparticles at water–oil interfaces is the same compared to isotropic nanoparticles. However, the added geometric complexity of anisotropic nanoparticles makes their orientation also a deciding factor in their plasmonic properties at the water–oil interface. For example, Zhang, Li et al. compared the SERS activity of Ag nanorods with different particle lengths assembled into horizontal or vertical 2D arrays at water–oil interfaces.122 As shown in Fig. 11a, it was found that the SERS enhancement of the horizontally aligned arrays was independent to the length of the nanorods while the SERS enhancement of the vertically aligned arrays increased near-linearly with the length of the nanorods. As a result, although the SERS enhancement observed for vertically and horizontally aligned arrays were similar for nanorods that were ca. 150 nm in length, with ca. 350 nm nanorods the vertically aligned arrays was found to give >2× the SERS enhancement of horizontally aligned nanorods.
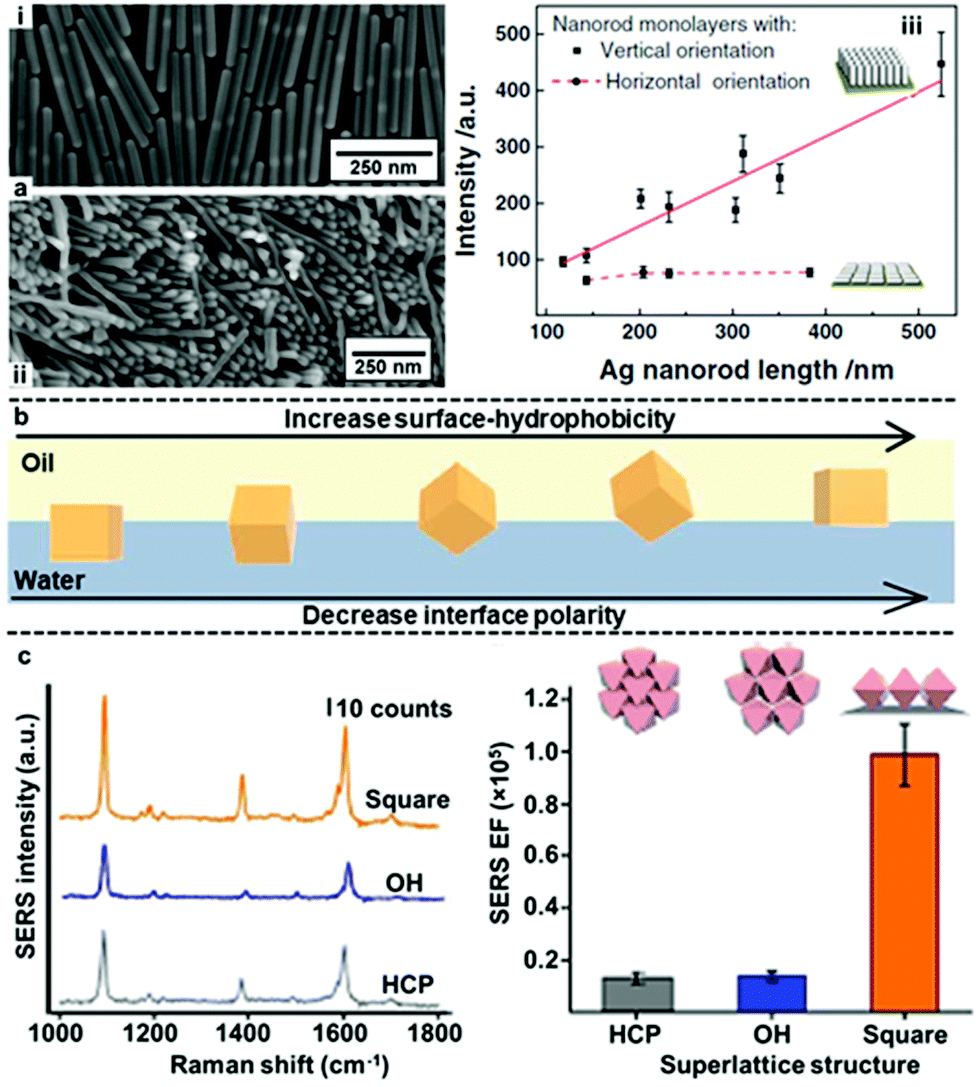 | ||
| Fig. 11 (a) SEM image of (i) horizontally and (ii) vertically assembled interfacial Ag nanorods arrays. (iii) Plot showing the trend in SERS signal enhancement versus the length of nanorods in vertically and horizontally aligned Ag nanorod arrays. (b) Schematic illustration of the orientation of Ag nanocubes at water–oil interfaces in relation to the surface-hydrophobicity of the nanocubes or the polarity of the interface. (c) Plots showing the SERS signals and signal enhancement obtained from arrays of Ag nanocubes packed as open hexagonal (OH), hexagonal close-packed (HCP) and square superlattices. Panel (a) adapted with permission from ref. 122. Copyright 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. Panel (b) adapted with permission from ref. 125. Copyright 2017 American Chemical Society. Panel (c) reproduced with permission from ref. 123. Copyright 2015 Macmillan Publishers Limited. | ||
Notably, Lee, Ling et al. have systematically investigated the orientation of highly plasmonically active Ag octahedrons at water–oil interfaces.109,123,125–127 They showed that several distinctive interfacial packing configurations could be obtained by tuning the surface hydrophobicity of the Ag octahedrons with different types/compositions of modifiers or by tuning the polarity of the oil phase used for self-assembly (Fig. 11b).109,123,125 Importantly, they demonstrated with molecular dynamic simulations that the different nanoparticle configurations observed corresponded to different thermodynamic equilibrium states rather than random transient states.126 Moreover, it was found that the different configurations of the nanoparticles at the interface had very different plasmonic properties. Surprisingly, as shown in Fig. 11c, it was found that 2D arrays formed with octahedrons in the standing orientation, which had low packing density, exhibited the highest SERS activity.109,123,127 This was attributed to large-area electromagnetic field delocalization effects and shows the significance of particle orientation in SERS.
5 Conclusions and perspectives
In conclusion, significant advances have been made in the assembly of nanoparticles at water–oil interfaces which can be used directly as uniform and sensitive SERS substrates or further fabricated into solid substrates which are even more stable and easy-to-use. Notably, the development of promoters for inducing assembly allows high quality arrays of particles with exposed surfaces to be formed. In addition, in situ gel/polymer trapping techniques allow the liquid-arrays to be converted into solid nanoparticle films, while preserving the plasmonic properties and surface-accessibility of the parent arrays, which paves the way for a range of fundamental and applied applications of SERS.To further enhance the plasmonic properties of interfacial 2D arrays and to build the next generation of affordable and smart SERS substrates will require development of methods to manipulate the position of individual nanoparticles at the interface. Some exciting progress has already been made in this area with preliminary results demonstrating that significant improvements in SERS performance could potentially be obtained by fine-tuning the gap-distance and orientation of particles in plasmonic assemblies. The next challenge is to build on these results by developing methods to achieve more precise control of the behaviour of nanoparticles at interfaces without passivating the surface of the particles and to transform these interfacial 2D arrays with tailored gap-distance into solid substrates for convenient application. To fully recognize the development in the above fields, it will be equally important to establish standard protocols to compare the SERS activity and uniformity of different plasmonic 2D arrays.
Author contributions
Z. Y. contributed to conceptualization, data curation and drafting of the manuscript. C. L. and Q. C. contributed to reviewing and editing. Y. X. contributed to conceptualization, data curation, drafting, supervision, reviewing, funding acquisition and editing of the manuscript. S. B. contributed to funding acquisition, conceptualization, supervision and reviewing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Z. Y. and C. L. acknowledges the University Special Research Scholarship (Q. U. B.) for financial support. Y. X. acknowledges the Leverhulme Trust (Early Career Fellowship) for funding support.Notes and references
- J. Langer, D. J. de Aberasturi, J. Aizpurua, R. A. Alvarez-Puebla, B. Auguié, J. J. Baumberg, G. C. Bazan, S. E. J. Bell, A. Boisen, A. G. Brolo, J. Choo, D. Cialla-May, V. Deckert, L. Fabris, K. Faulds, J. G. de Abajo, R. Goodacre, D. Graham, A. J. Haes, C. L. Haynes, C. Huck, T. Itoh, M. Kall, J. Kneipp, N. A. Kotov, H. Kuang, E. C. Le Ru, H. K. Lee, J.-F. Li, X. Y. Ling, S. A. Maier, T. Mayerhöfer, M. Moskovits, K. Murakoshi, J.-M. Nam, S. Nie, Y. Ozaki, I. Pastoriza-Santos, J. Perez-Juste, J. Popp, A. Pucci, S. Reich, B. Ren, G. C. Schatz, T. Shegai, S. Schlücker, L.-L. Tay, K. G. Thomas, Z.-Q. Tian, R. P. Van Duyne, T. Vo-Dinh, Y. Wang, K. A. Willets, C. Xu, H. Xu, Y. Xu, Y. S. Yamamoto, B. Zhao and L. M. Liz-Marzán, ACS Nano, 2020, 14, 28–117 CrossRef CAS.
- S. E. J. Bell, G. Charron, E. Cortés, J. Kneipp, M. L. de la Chapelle, J. Langer, M. Procházka, V. Tran and S. Schlücker, Angew. Chem., 2020, 59, 5454–5462 CrossRef CAS.
- X. Wang, S.-C. Huang, S. Hu, S. Yan and B. Ren, Nat. Rev. Phys., 2020, 2, 253–271 CrossRef.
- D. Radziuk and H. Moehwald, Phys. Chem. Chem. Phys., 2015, 17, 21072–21093 RSC.
- S. M. Restaino and I. M. White, Anal. Chim. Acta, 2019, 1060, 17–29 CrossRef CAS.
- J. Cailletauda, C. De Bleyea, E. Dumont, P.-Y. Sacré, L. Netchacovitch, Y. Gut, M. Boiret, Y.-M. Ginot, Ph. Hubert and E. Ziemons, J. Pharm. Biomed. Anal., 2018, 147, 458–472 CrossRef.
- M. Li, Y. Qiu, C. Fan, K. Cui, Y. Zhang and Z. Xiao, Acta Pharm. Sin. B, 2018, 8, 381–389 CrossRef.
- A. Shiohara, Y. Wang and L. M. Liz-Marzán, J. Photochem. Photobiol., C, 2014, 21, 2–25 CrossRef CAS.
- V. Tran, C. Thiel, J. T. Svejda, M. Jalali, B. Walkenfort, D. Erni and S. Schlücker, Nanoscale, 2018, 10, 21721–21731 RSC.
- Y. Fang, N.-H. Seong and D. D. Dlott, Science, 2008, 321, 388–392 CrossRef CAS.
- K. Kneipp, Y. Wang, H. Kneipp, L. T. Perelman, I. Itzkan, R. R. Dasari and M. S. Feld, Phys. Rev. Lett., 1997, 78, 1667–1670 CrossRef CAS.
- S. Nie and S. R. Emory, Science, 1997, 275, 1102–1106 CrossRef CAS.
- R. Tantra, R. J. C. Brown and M. J. T. Milton, J. Raman Spectrosc., 2007, 38, 1469–1479 CrossRef CAS.
- K. L. Wustholz, A.-I. Henry, J. M. McMahon, R. G. Freeman, N. Valley, M. E. Piotti, M. J. Natan, G. C. Schatz and R. P. Van Duyne, J. Am. Chem. Soc., 2010, 132, 10903–10910 CrossRef CAS.
- T. Chung, T. Koker and F. Pinaud, Small, 2016, 12, 5891–5901 CrossRef CAS.
- F. Zou, H. Zhou, T. V. Tan, J. Kim, K. Koh and J. Lee, ACS Appl. Mater. Interfaces, 2015, 7, 12168–12175 CrossRef CAS.
- R. W. Taylor, T.-C. Lee, O. A. Scherman, R. Esteban, J. Aizpurua, F. M. Huang, J. J. Baumberg and S. Mahajan, ACS Nano, 2011, 5, 3878–3887 CrossRef CAS.
- J. B. Edel, A. A. Kornyshev and M. Urbakh, ACS Nano, 2013, 7, 9526–9532 CrossRef CAS.
- L. Song, Y. Huang, Z. Nie and T. Chen, Nanoscale, 2020, 12, 7433–7460 RSC.
- W. Xie and S. Schlücker, Nat. Commun., 2015, 6, 7570 CrossRef.
- R. A. Alvarez-Pueblaa, A. Agarwal, P. Manna, B. P. Khanal, P. Aldeanueva-Potel, E. Carbó-Argibay, N. Pazos-Pérez, L. Vigderman, E. R. Zubarev, N. A. Kotov and L. M. Liz-Marzán, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8157–8161 CrossRef.
- N. Vogel, M. Retsch, C.-A. Fustin, A. del Campo and U. Jonas, Chem. Rev., 2015, 115, 6265–6311 CrossRef CAS.
- M. Fan, G. F. S. Andrade and A. G. Brolo, Anal. Chim. Acta, 2011, 693, 7–25 CrossRef CAS.
- R. Liu, S. Li and J.-F. Liu, Trends Anal. Chem., 2017, 97, 188–200 CrossRef CAS.
- J. Chen, L. Guo, B. Qiu, Z. Lin and T. Wang, Mater. Chem. Front., 2018, 2, 835–860 RSC.
- R. McGorty, J. Fung, D. Kaz and V. N. Manoharan, Mater. Today, 2010, 13, 34–42 CrossRef CAS.
- S. G. Booth and R. A. W. Dryfe, J. Phys. Chem. C, 2015, 119, 23295–23309 CrossRef CAS.
- H. Duan, D. Wang, D. G. Kurth and H. Möhwald, Angew. Chem., Int. Ed., 2004, 43, 5639–5642 CrossRef CAS.
- D. M. Andala, S. H. R. Shin, H.-Y. Lee and K. J. M. Bishop, ACS Nano, 2012, 6, 1044–1050 CrossRef CAS.
- H.-J. Kim, C.-R. Lee, D.-G. Park, J. W. Chang and H.-Y. Lee, J. Phys. Chem. C, 2020, 124, 16423–16430 CrossRef CAS.
- F. Reincke, W. K. Kegel, H. Zhang, M. Nolte, D. Wang, D. Vanmaekelbergh and H. Möhwald, Phys. Chem. Chem. Phys., 2006, 8, 3828–3835 RSC.
- Y. Xu, M. P. Konrad, W. W. Y. Lee, Z. Ye and S. E. J. Bell, Nano Lett., 2016, 16, 5255–5260 CrossRef CAS.
- B. P. Binks, Curr. Opin. Colloid Interface Sci., 2002, 7, 21–41 CrossRef CAS.
- T. S. Horozov, R. Aveyard, J. H. Clint and B. P. Binks, Langmuir, 2003, 19, 2822–2829 CrossRef CAS.
- D. Yogev and S. Efrima, J. Phys. Chem., 1988, 92, 5754–5760 CrossRef CAS.
- K. C. Gordon, J. J. McGarvey and K. P. Taylor, J. Phys. Chem., 1989, 93, 6814–6817 CrossRef CAS.
- M. Gadogbe, S. M. Ansar, I.-W. Chu, S. Zou and D. Zhang, Langmuir, 2014, 30, 11520–11527 CrossRef CAS.
- M. N. Martin, J. I. Basham, P. Chando and S.-K. Eah, Langmuir, 2010, 26, 7410–7417 CrossRef CAS.
- H. Tian, H. Li and Y. Fang, ACS Appl. Mater. Interfaces, 2019, 11, 16207–16213 CrossRef CAS.
- M. Mao, B. Zhou, X. Tang, C. Chen, M. Ge, P. Li, X. Huang, L. Yang and J. Liu, Chem. – Eur. J., 2018, 24, 4094–4102 CrossRef CAS.
- T. Ding, A. W. Rudrum, L. O. Herrmann, V. Turek and J. J. Baumberg, Nanoscale, 2016, 8, 15864–15869 RSC.
- A. B. Serrano-Montes, D. J. de Aberasturi, J. Langer, J. J. Giner-Casares, L. Scarabelli, A. Herrero and L. M. Liz-Marzán, Langmuir, 2015, 31, 9205–9213 CrossRef CAS.
- Y. Chen, J. Fu, K. C. Ng, Y. Tang and W. Cheng, Cryst. Growth Des., 2011, 11, 4742–4746 CrossRef CAS.
- E. Smirnov, P. Peljo, M. D. Scanlon, F. Gumy and H. H. Girault, Nanoscale, 2016, 8, 7723–7737 RSC.
- S. Si, W. Liang, Y. Sun, J. Huang, W. Ma, Z. Liang, Q. Bao and L. Jiang, Adv. Funct. Mater., 2016, 26, 8137–8145 CrossRef CAS.
- Y. Du, W. Wei, X. Zhang and Y. Li, J. Phys. Chem. C, 2018, 122, 7997–8002 CrossRef CAS.
- F. Reincke, S. G. Hickey, W. K. Kegel and D. Vanmaekelbergh, Angew. Chem., Int. Ed., 2004, 43, 458–462 CrossRef CAS.
- Y.-J. Li, W.-J. Huang and S.-G. Sun, Angew. Chem., Int. Ed., 2006, 45, 2537–2539 CrossRef CAS.
- J.-W. Liu, S.-Y. Zhang, H. Qi, W.-C. Wen and S.-H. Yu, Small, 2012, 8, 2412–2420 CrossRef CAS.
- Y.-K. Park, S.-H. Yoo and S. Park, Langmuir, 2007, 23, 10505–10510 CrossRef CAS.
- Y.-K. Park and S. Park, Chem. Mater., 2008, 20, 2388–2393 CrossRef CAS.
- V. A. Turek, M. P. Cecchini, J. Paget, A. R. Kucernak, A. A. Kornyshev and J. B. Edel, ACS Nano, 2012, 6, 7789–7799 CrossRef CAS.
- L. Velleman, D. Sikdar, V. A. Turek, A. R. Kucernak, S. J. Roser, A. A. Kornyshev and J. B. Edel, Nanoscale, 2016, 8, 19229–19241 RSC.
- R. Wang, X.-R. Shen, M. Zhang, R. Devasenathipathy, R. Pang, D.-Y. Wu, J. Zhang, J. Ulstrup and Z.-Q. Tian, J. Phys. Chem. C, 2019, 123, 23026–23036 CrossRef CAS.
- W.-L.-J. Hasi, S. Lin, X. Lin, X.-T. Lou, F. Yang, D.-Y. Lin and Z.-W. Lu, Anal. Methods, 2014, 6, 9547–9553 RSC.
- S. Lin, W.-L.-J. Hasi, X. Lin, S.-Q.-G.-W. Han, X.-T. Lou, F. Yang, D.-Y. Lin and Z.-W. Lu, Anal. Methods, 2015, 7, 5289–5294 RSC.
- M. P. Konrad, A. P. Doherty and S. E. J. Bell, Anal. Chem., 2013, 85, 6783–6789 CrossRef CAS.
- B. Su, J.-P. Abid, D. J. Fermín, H. H. Girault, H. Hoffmannová, P. Krtil and Z. Samec, J. Am. Chem. Soc., 2004, 126, 915–919 CrossRef CAS.
- M. K. Bera, H. Chan, D. F. Moyano, H. Yu, S. Tatur, D. Amoanu, W. Bu, V. M. Rotello, M. Meron, P. Král, B. Lin and M. L. Schlossman, Nano Lett., 2014, 14, 6816–6822 CrossRef CAS.
- M. Wang, Z. Zhang and J. He, Langmuir, 2015, 31, 12911–12919 CrossRef CAS.
- C. Li, Y. Xu, X. Li, Z. Ye, C Yao, Q. Chen, Y. Zhang and S. E. J. Bell, Adv. Mater. Interfaces, 2020, 7, 2000391 CrossRef CAS.
- L. Zhang, F. Liu, Y. Zou, X. Hu, S. Huang, Y. Xu, L. Zhang, Q. Dong, Z. Liu, L Chen, Z. Chen and W. Tan, Anal. Chem., 2018, 90, 11183–11187 CrossRef CAS.
- M. Su, X. Li, S. Zhang, F. Yu, L. Tian, Y. Jiang and H. Liu, Anal. Chem., 2019, 91, 2288–2295 CrossRef CAS.
- K. Kim, H. S. Han, I. Choi, C. Lee, S. Hong, S.-H. Suh, L. P. Lee and T. Kang, Nat. Commun., 2013, 4, 2182 CrossRef.
- W. Luo, C. Wu, S. Huang, X. Luo, R. Yuan and X. Yang, Anal. Chem., 2020, 92, 15573–15578 CrossRef CAS.
- K. Zhang, J. Ji, Y. Li and B. Liu, Anal. Chem., 2014, 86, 6660–6665 CrossRef CAS.
- M. P. Cecchini, V. A. Turek, J. Paget, A. A. Kornyshev and J. B. Edel, Nat. Mater., 2013, 12, 165–171 CrossRef CAS.
- L. Qiu, G. A. Pang, G. Zheng, D. Bauer, K. Wieland and C. Haisch, ACS Appl. Mater. Interfaces, 2020, 12, 21133–21142 CrossRef CAS.
- Y. Shin, K. Whang, J. Chang, J. Jang, S. Yoo, J. Lee, Y. Choi, I. Choi and T. Kang, Adv. Mater. Interfaces, 2018, 5, 1800981 CrossRef.
- Y. Ma, H. Liu, M. Mao, J. Meng, L. Yang and J. Liu, Anal. Chem., 2016, 88, 8145–8151 CrossRef CAS.
- F. Yu, M. Su, L. Tian, H. Wang and H. Liu, Anal. Chem., 2018, 90, 5232–5238 CrossRef CAS.
- S. Du, M. Su, Y. Jiang, F. Yu, Y. Xu, X. Lou, T. Yu and H. Liu, ACS Sens., 2019, 4, 1798–1805 CrossRef CAS.
- M. Su, C. Wang, T. Wang, Y. Jiang, Y. Xu and H. Liu, Anal. Chem., 2020, 92, 6941–6948 CrossRef CAS.
- L. Tian, M. Su, F. Yu, Y. Xu, X. Li, L. Li, H. Liu and W. Tan, Nat. Commun., 2018, 9, 3642 CrossRef.
- Y. Jiang, M. Su, T. Yu, S. Du, L. Liao, H. Wang, Y. Wu and H. Liu, Food Chem., 2021, 344, 128709 CrossRef CAS.
- M. P. Cecchini, V. A. Turek, A. Demetriadou, G. Britovsek, T. Welton, A. A. Kornyshev, J. D. E. T. Wilton-Ely and J. B. Edel, Adv. Opt. Mater., 2014, 2, 966–977 CrossRef CAS.
- N. D. Denkov, I. B. Ivanov and P. A. Kralchevsky, J. Colloid Interface Sci., 1992, 150, 589–593 CrossRef CAS.
- G. C. Phan-Quang, E. H. Z. Wee, F. Yang, H. K. Lee, I. Y. Phang, X. Feng, R. A. Alvarez-Puebla and X. Y. Ling, Angew. Chem., 2017, 129, 5657–5661 CrossRef.
- A. S. Sarycheva, N. A. Brazhe, A. A. Baizhumanov, E. I. Nikelshparg, A. A. Semenova, A. V. Garshev, A. E. Baranchikov, V. K. Ivanov, G. V. Maksimov, O. Sosnovtseva and E. A. Goodilin, J. Mater. Chem. B, 2016, 4, 539–546 RSC.
- L. Ouyang, D. Li, L. Zhu, W. Yang and H. Tang, J. Mater. Chem. C, 2016, 4, 736–744 RSC.
- G. C. Phan-Quang, H. K. Lee, H. W. Teng, C. S. L. Koh, B. Q. Yim, E. K. M. Tan, W. L. Tok, I. Y. Phang and X. Y. Ling, Angew. Chem., Int. Ed., 2018, 57, 5792–5796 CrossRef CAS.
- Z. Ye, C. Li, Q. Chen, Y. Xu and S. E. J. Bell, Angew. Chem., Int. Ed., 2019, 58, 19054–19059 CrossRef CAS.
- Z. Ye, C. Li, N. Skillen, Y. Xu, H. McCabe, J. Kelly, P. Robertson and S. E. J. Bell, Appl. Mater. Today, 2019, 15, 398–404 CrossRef.
- K. Wang, L. Feng, X. Li and W. Wang, J. Mater. Res., 2019, 34, 2137–2145 CrossRef CAS.
- W. Zhao, Y. Zhang, J. Yang, J. Li, Y. Feng, M. Quan, Z. Yang and S. Xiao, Nanoscale, 2020, 12, 18056–18066 RSC.
- X. Lin, S. Lin, Y. Liu, H. Zhao, B. Liu and L. Wang, J. Raman Spectrosc., 2019, 50, 916–925 CrossRef CAS.
- S. Lin, X. Lin, S. Han, Y. Liu, W. Hasi and L. Wang, Anal. Chim. Acta, 2020, 1108, 167–176 CrossRef CAS.
- J. Kelly, R. Patrick, S. Patrick and S. E. J. Bell, Angew. Chem., Int. Ed., 2018, 57, 15686–15690 CrossRef CAS.
- G. Yang, J. Nanda, B. Wang, G. Chen and D. T. Hallinan, Jr., ACS Appl. Mater. Interfaces, 2017, 9, 13457–13470 CrossRef CAS.
- L. Zhang, X. Li, W. Liu, R. Hao, H. Jia, Y. Dai, M. U. Amin, H. You, T. Li and J. Fang, J. Mater. Chem. C, 2019, 7, 15259–15268 RSC.
- G. Yang and D. T. H. Jr., Nanotechnology, 2016, 27, 22 Search PubMed.
- Y. Qu, C. Tan, Z. Zhang and L. He, Analyst, 2017, 142, 4075–4082 RSC.
- M. G. Millyard, F. M. Huang, R. White, E. Spigone, J. Kivioja and J. J. Baumberg, Appl. Phys. Lett., 2012, 100, 073101 CrossRef.
- K. Zhang, J. Zhao, H. Xu, Y. Li, J. Ji and B. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 16767–16774 CrossRef CAS.
- C. Li, L. Chai, Q. Chen, Z. Ye, Y. Xu and S. E. J. Bell, J. Raman Spectrosc., 2020, 52, 386–393 CrossRef.
- P. Wu, L.-B. Zhong, Q. Liu, X. Zhou and Y.-M. Zheng, Nanoscale, 2019, 11, 12829–12836 RSC.
- H. Xia and D. Wang, Adv. Mater., 2008, 20, 4253–4256 CrossRef CAS.
- A. Liu, M. V. de Ruiter, W. Zhu, S. J. Maassen, L. Yang and J. J. L. M. Cornelissen, Adv. Funct. Mater., 2018, 28, 1801574 CrossRef.
- T. Andryszewski, M. Iwan, M. Hołdynski and M. Fiałkowski, Chem. Mater., 2016, 28, 5304–5313 CrossRef CAS.
- S. Rao, K. J. Si, L. W. Yap, Y. Xiang and W. Cheng, ACS Nano, 2015, 9, 11218–11224 CrossRef CAS.
- K. Chaw Ng, I. B. Udagedara, I. D. Rukhlenko, Y. Chen, Y. Tang, M. Premaratne and W. Cheng, ACS Nano, 2012, 6, 925–934 CrossRef.
- K. J. Si, D. Sikdar, Y. Chen, F. Eftekhari, Z. Xu, Y. Tang, W. Xiong, P. Guo, S. Zhang, Y. Lu, Q. Bao, W. Zhu, M. Premaratne and W. Cheng, ACS Nano, 2014, 8, 11086–11093 CrossRef CAS.
- Y. Chen, K. J. Si, D. Sikdar, Y. Tang, M. Premaratne and W. Cheng, Adv. Opt. Mater., 2015, 3, 919–924 CrossRef CAS.
- J. Song, W. Nam and W. Zhou, Adv. Mater. Technol., 2019, 4, 1800689 CrossRef.
- H. J. Yin, Z. Y. Chen, Y. M. Zhao, M. Y. Lv, C. A. Shi, Z. L. Wu, X. Zhang, L. Liu, M. L. Wang and H. J. Xu, Sci. Rep., 2015, 5, 14502 CrossRef.
- X. Lin, W.-L.-J. Hasi, S.-Q.-G.-W. Han, X.-T. Lou, D.-Y. Lin and Z.-W. Lu, Phys. Chem. Chem. Phys., 2015, 17, 31324–31331 RSC.
- L.-B. Zhong, J. Yin, Y.-M. Zheng, Q. Liu, X.-X. Cheng and F.-H. Luo, Anal. Chem., 2014, 86, 6262–6267 CrossRef CAS.
- L. N. Arnaudov, O. J. Cayre, M. A. C. Stuart, S. D. Stoyanov and V. N. Paunov, Phys. Chem. Chem. Phys., 2010, 12, 328–331 RSC.
- Y. Yang, Y. H. Lee, I. Y. Phang, R. Jiang, H. Y. F. Sim, J. Wang and X. Y. Ling, Nano Lett., 2016, 16, 3872–3878 CrossRef CAS.
- Y. Xu, M. P. Konrad, J. L. Trotter, C. P. McCoy and S. E. J. Bell, Small, 2016, 13, 1602163 CrossRef.
- Y. Xu, Z. Ye, C. Li, H. McCabe, J. Kelly and S. E. J. Bell, Appl. Mater. Today, 2018, 13, 352–358 CrossRef.
- C. Li, Q. Chen, H. McCabe, J. Kelly, Z. Ye, Y. Xu and S. E. J. Bell, Data Brief, 2019, 23, 103746 CrossRef.
- I.-H. Kim, J. H. Kim, J.-Y. Choi, C. H. Shin, J.-H. Kim, G.-T. Bae and K. S. Shin, Chem. Phys. Lett., 2019, 715, 91–99 CrossRef CAS.
- V. Sashuk, K. Winkler, A. Żywociński, T. Wojciechowski, E. Górecka and M. Fiałkowski, ACS Nano, 2013, 7, 8833–8839 CrossRef CAS.
- F. Schulz, S. Tober and H. Lange, Langmuir, 2017, 33, 14437–14444 CrossRef CAS.
- X. Lu, Y. Huang, B. Liu, L. Zhang, L. Song, J. Zhang, A. Zhang and T. Chen, Chem. Mater., 2018, 30, 1989–1997 CrossRef CAS.
- G. Yang, L. Hu, T. D. Keiper, P. Xiong and D. T. Hallinan, Jr., Langmuir, 2016, 32, 4022–4033 CrossRef CAS.
- Y. Shin, J. Song, D. Kim and T. Kang, Adv. Mater., 2015, 27, 4344–4350 CrossRef CAS.
- C. Zhang, E. You, Q. Jin, Y. Yuan, M. Xu, S. Ding, J. Yao and Z. Tian, Chem. Commun., 2017, 53, 6788–6791 RSC.
- M. E. Flatté, A. A. Kornyshev and M. Urbakh, J. Phys. Chem. C, 2010, 114, 1735–1747 CrossRef.
- Y. Montelongo, D. Sikdar, Y. Ma, A. J. S. McIntosh, L. Velleman, A. R. Kucernak, J. B. Edel and A. A. Kornyshev, Nat. Mater., 2017, 16, 1127–1135 CrossRef CAS.
- X.-D. Tian, Y. Lin, J.-C. Dong, Y.-J. Zhang, S.-R. Wu, S.-Y. Liu, Y. Zhang, J.-F. Li and Z.-Q. Tian, Adv. Opt. Mater., 2017, 5, 1700581 CrossRef.
- Y. H. Lee, W. Shi, H. K. Lee, R. Jiang, I. Y. Phang, Y. Cui, L. Isa, Y. Yang, J. Wang, S. Li and X. Y. Ling, Nat. Commun., 2015, 6, 6990 CrossRef CAS.
- L. Scarabelli, M. Coronado-Puchau, J. J. Giner-Casares, J. Langer and L. M. Liz-Marzán, ACS Nano, 2014, 8, 5833–5842 CrossRef CAS.
- Y. Yang, Y. H. Lee, C. L. Lay and X. Y. Ling, Chem. Mater., 2017, 29, 6137–6144 CrossRef CAS.
- W. Shi, Y. H. Lee, X. Y. Ling and S. Li, Nanoscale, 2017, 9, 11239–11248 RSC.
- Y. H. Lee, H. K. Lee, J. Y. C. Ho, Y. Yang and X. Y. Ling, Analyst, 2016, 141, 5107–5112 RSC.
Footnote |
| † Here we use the term “aggregated” for consistency with the literature although “agglomerated” is a better term for collections of particles which retain their individual identity. In the broader context, aggregates would normally be expected to have joined or fused and therefore would not be expected to be highly SERS active. |
| This journal is © The Royal Society of Chemistry 2021 |