
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceShape retaining self-healing metal-coordinated hydrogels†
Alvaro
Charlet
 a,
Viviane
Lutz-Bueno
b,
Raffaele
Mezzenga
bc and
Esther
Amstad
*a
a,
Viviane
Lutz-Bueno
b,
Raffaele
Mezzenga
bc and
Esther
Amstad
*a
aSoft Materials Laboratory, Institute of Materials, EPFL Lausanne, Lausanne 1015, Switzerland. E-mail: esther.amstad@epfl.ch
bLaboratory of Food and Soft Materials Science, Department of Health Sciences and Technology, ETH Zurich, Zurich 8092, Switzerland
cDepartment of Materials, ETH Zurich, Zurich 8093, Switzerland
First published on 28th January 2021
Abstract
Metal-coordinated hydrogels are physical hydrogels entirely crosslinked by complexes between ligand decorated polymers and metal ions. The mechanical properties of these hydrogels strongly depend on the density and dynamics of metal-coordinated interactions. Most commonly, telechelic metal-coordinated hydrogels contain catechol or histidine ligands, although hydrogels containing a stronger complexation agent, nitrocatechol, have been reported. Here, we introduce a pyrogallol end-functionalized polymer that can be crosslinked with di- and trivalent ions, in contrast to previously reported metal-coordinated hydrogels. We can tune the mechanical properties of the hydrogels with the types of ions used and the density of crosslinking sites. Ions form nm-sized precipitates that bind to pyrogallols and impart distinct properties to the hydrogels: strong ion–pyrogallol interactions that form in the presence of Al3+, V3+, Mn2+, Fe3+, Co2+, Ni2+ and Cu2+ result in long relaxation times. The resulting hydrogels display solid-like yet reversible mechanical properties, such that they can be processed into macroscopic 3D structures that retain their shapes. Weak ion–pyrogallol interactions that form in the presence of Ca2+ or Zn2+ result in short relaxation times. The resulting hydrogels display a fast self-healing behavior, suited for underwater glues, for example. The flexibility of tuning the mechanical properties of hydrogels simply by selecting the adequate ion–pyrogallol pair broadens the mechanical properties of metal-coordinated hydrogels to suit a wide range of applications that require them to retain their shape for a given time to act as dampers.
Introduction
The use of hydrogels is consolidated in biomedicine, as wound healing agents,1 synthetic cartilages and tendons,2 biomechanical actuators,3 or tissue scaffolds.4,5 The key for a successful application is adequate mechanical properties, which depend on the type and density of crosslinks. Hydrogel networks are classified as chemical if they are crosslinked by permanent covalent bonds, such that they are often stiff but rather brittle, or physical if they are formed by reversible interactions, such that they are often tough but rather weak. Dissipative networks6,7 and supramolecular interactions8–11 are often included to increase the toughness of covalent hydrogels. Natural soft load-bearing materials, such as mussel holdfast threads, possess a combination of both crosslinking methods:12 domains that are covalently crosslinked, and others that are reversibly crosslinked through metal complexes.13–16 Metal complexes are composed of ligand molecules attracted to a central metallic ion. These complexes form reversible crosslinks with binding strengths that can approach those of covalent bonds. This high binding strength of metal coordination contrasts the relatively weak ionic interactions of physical hydrogels.17Inspired by nature, metal-coordinated telechelic hydrogels, composed of polymer chains end-functionalized by ligands and crosslinked by metal complexes, have potential to generate mechanically active, reversible networks.18–26 The dynamic properties of these hydrogels can be conveniently tuned with the choice of the cationic crosslinker, its relative concentration, and the solution pH.25,27,28 The reversible nature of their constituting crosslinks gives them self-healing properties, such that a damaged network can heal defects. To obtain this self-healing behavior, the ions must complex the ligand sufficiently strongly to result in an integral hydrogel. Unfortunately, the selection of ions that fulfil this criterion is very limited, narrowing the range of mechanical properties of metal-coordinated hydrogels.
Hydrogels with long relaxation times have solid-like behaviors and are capable of retaining their shape, yet are able to self-heal.29 The relaxation time of metal-ion coordination sites increases if ions are replaced by their corresponding nanoparticles,29 which must have adequate surface chemistry to interact with the surrounding ligands. Polysaccharides functionalized with a high density of ligands can also increase the relaxation time.30–32 These ligands improve the adhesion of the polysaccharides towards surfaces,33 which reinforce the mechanical strength,30,33 and impart self-healing properties to the hydrogels.34,35 However, these systems can often only be used in a very limited pH range mainly because the most commonly used ligands, catechols, and some of its derivatives have a high propensity to oxidize. The oxidation of catechols can lead to inter-catechol covalent bonds, which may result in hydrogels possessing a mixture of covalent and ionic bonds.20 The combination of transient ion–ligand interactions and the oxidation of the ligand complicates their characterization, limiting their applications to non-oxidizing environments.
Several natural organisms, some of which live in quite harsh environments, employ metal complexes for mechanical reinforcement, hard coatings or energy dissipation.13,16,36,37 A prominent example is a class of marine water-filtering invertebrates, ascidians, which employ pyrogallol groups complexed by vanadium ions to heal their cellulosic tunic.38–40 Pyrogallols are catechol derivatives possessing an additional alcohol group on the carbon ring. This electronegative group reduces their propensity to oxidize when compared to catechols. They are known to complex a broad range of ions of d-block elements.41 This feature has been exploited, for example, by using tannic acid, which contains several pyrogallols, to form coatings,42–45 metallogels,28,46,47 and capsules.42,48 Pyrogallols have also been employed in synthetic hydrogels, however in the absence of ionic crosslinks: they were oxidized to crosslink polysaccharides.30,31,49,50 The contribution of pyrogallols to the dynamic and reversible mechanical properties of polysaccharide-based hydrogels remains unclear.31 The formulation of ionically crosslinked hydrogels whose mechanical properties can be tuned over a much wider range than what is currently possible requires a better understanding of the influence of chelators, such as pyrogallols that can bind to a wide range of ions, on the mechanical properties of the resulting hydrogels.
In this work, we introduce shape retaining self-healing metal-coordinated telechelic hydrogels with well-defined, solid-like and tunable mechanical properties. To achieve this goal, we end-functionalize linear polyethylene glycol (PEG) with pyrogallols (2gPEG). We investigate the influence of the type and concentration of ions used as crosslinkers on the macroscopic properties and structure of the hydrogel. The mechanical properties of these metal-coordinated hydrogels, such as their storage modulus at low frequencies, vary over an order of magnitude if adequate ions are selected. Remarkably, these telechelic PEGs can be crosslinked with di- and trivalent ions, thereby broadening the selection of ions and the range of accessible mechanical properties. During the ion–2gPEG interaction, the ions preferentially form small particles that act as multivalent binding sites, thereby facilitating the formation of a percolating network and increasing the effective cross-link density. We demonstrate the formation of viscoelastic macroscopic freestanding 3D structures, which can be applied as underwater adhesives that can bear load even under tension. Additionally, these hydrogels can be functionalized with inorganic particles to respond to external stimuli, such as external magnetic fields.
Results and discussion
To ensure scalability, we end-functionalize commercially available hydroxy-terminated linear PEG, analogous to nitrocatechol-functionalized linear PEGs.51 We produce the hydrogels by dissolving 10 wt% 2gPEG in water and adding iron chloride (FeCl3), selected because of the high affinity between pyrogallol and iron(III) (Fe3+). For each pyrogallol end-group, we add twice the molar concentration of Fe3+ to achieve gelation, a ratio higher than the stoichiometric ratio of 3 ligands per ion, known for catechol hydrogels,20,23 resulting in a solution pH of 1.5. The complex formation is triggered by increasing the pH to deprotonate pyrogallols. When the pH is increased to 4.8 by adding sodium hydroxide (NaOH), the system instantaneously gels, as shown in Fig. 1a and b. Importantly, the system remains gelled even if the pH is increased to physiological values, such that these hydrogels can be used for biomedical applications, in stark contrast to catechol-functionalized counterparts. The hydrogel remains stable in deionized water for 24 h, before it starts to dissociate. Note that the crosslinking is reversible: if HCl is added to lower the pH, the hydrogel liquefies, as shown in Fig. S1 and 1b.† Unfortunately, this procedure cannot be repeated because further additions of bases or acids dilute the system below the gelation point.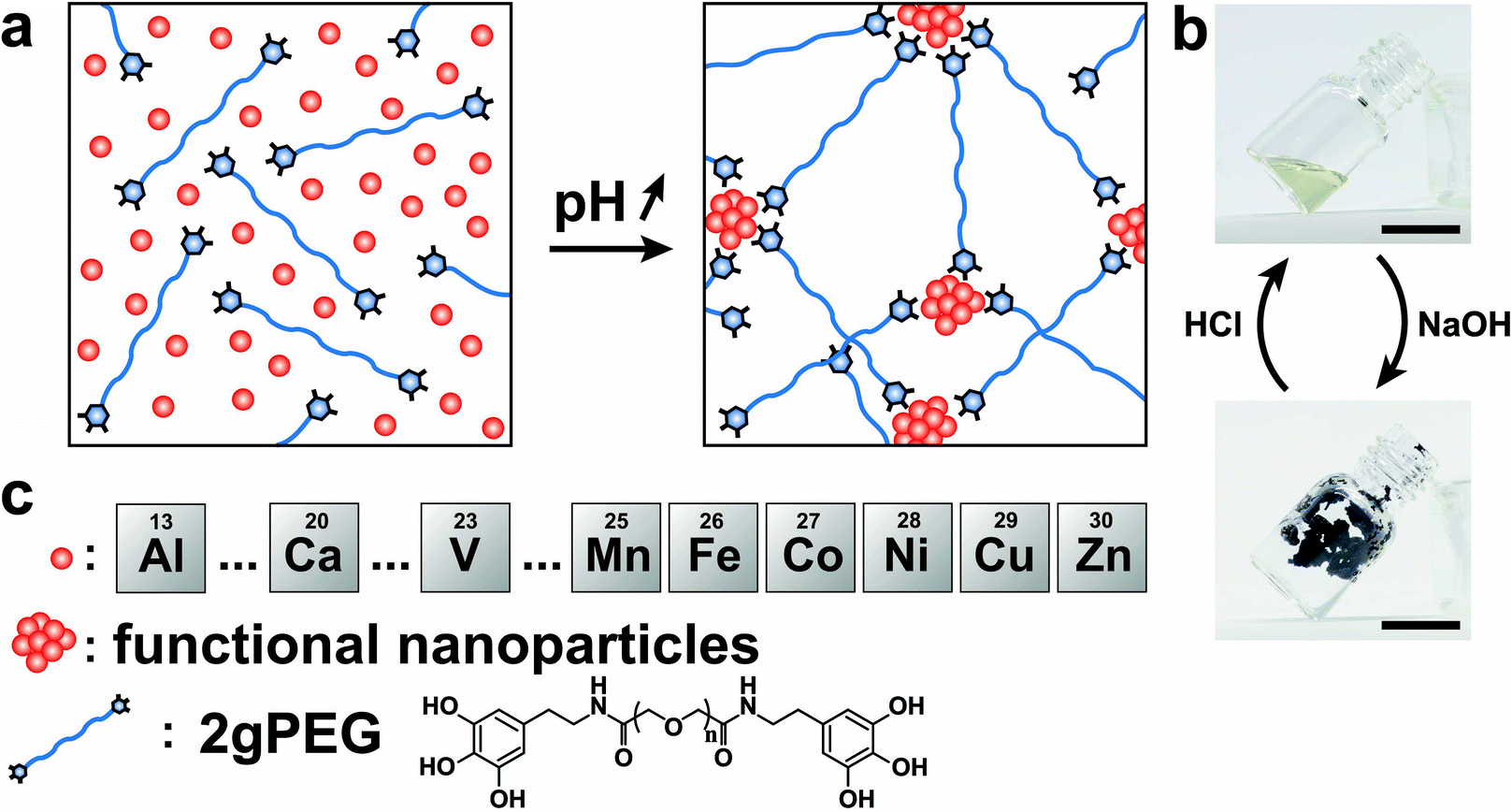 | ||
| Fig. 1 Gelation mechanism of 2gPEG hydrogels. (a) Schematic illustration of the hydrogel formation. The blue line terminated by blue hexagons represents the 2gPEG polymer, end-functionalized by the pyrogallol molecule. The ions are sketched as red circles. The increase in pH causes the precipitation of the ions into nanoparticles and the deprotonation of the pyrogallol groups that bind to the precipitates. The nanoparticles act as multivalent crosslinking sites of the hydrogel network. (b) Photographs of the solution of 2gPEG and iron ions before (top) and after gelation (bottom) induced through the addition of NaOH. The coordination bond between pyrogallols and iron ions has strong absorption in the visible range, which results in a dark purple hydrogel. After adding HCl, the hydrogel liquefies to a light-yellow solution, indicating that some pyrogallol groups oxidized. The scale bar is 1 cm. (c) Schematic illustration of the selection of bivalent and trivalent ions that can crosslink 2gPEG, the nanoparticles, and the 2gPEG molecule. | ||
Impact of ion valency
To expand the selection of ions that can be used to crosslink telechelic molecules, we crosslink 2gPEG with a divalent ion, calcium(II) (Ca2+). If the same stoichiometry is maintained as for Fe3+, no gelation is observed. However, if we increase the Ca2+ concentration 5-fold and increase the pH to 12, we observe the gelation of the hydrogel. A similar behavior occurs if 2gPEG is crosslinked with other divalent d-block elements such as manganese(II) (Mn2+), cobalt(II) (Co2+), nickel(II) (Ni2+), copper(II) (Cu2+), and zinc(II) (Zn2+). This is a remarkable property of these hydrogels, because metal-coordinated telechelic hydrogels have not been shown to crosslink with both trivalent and divalent ions. Typically, 4-armed catechol-functionalized PEG molecules are crosslinked only by trivalent ions such as Fe3+, vanadium(III) (V3+), and aluminum(III) (Al3+).20,23 In contrast, histidine-functionalized PEG molecules can only be crosslinked with divalent ions,24,25 but not with trivalent ones. The fact that 2gPEG gels in the presence of divalent and trivalent ions opens up new possibilities to tune the mechanical properties of such metal-coordinated hydrogels over a much wider range. We confirm that the gelling is caused by the ions and not the pH change by increasing the pH of an ion-free 2gPEG solution to the same value, as shown in Fig. S2.† These results suggest that the gelation is primarily caused by the ion–pyrogallol interactions even though we cannot exclude that a small fraction of pyrogallols oxidizes at this high pH.Our results suggest that the ion–pyrogallol interactions are key in the hydrogel formation. To investigate the implications of these ionic interactions on the mechanical properties of the hydrogels, we systematically measure their rheological behaviors. The dynamic mechanical properties of ionically crosslinked telechelic hydrogels depend on the average lifetime of the crosslinks, and hence, the ion–ligand affinity. We use frequency sweeps to quantify the lifetime of these crosslinks by determining the frequency where the storage modulus (G′) equals the loss modulus (G′′).20 2gPEG hydrogels crosslinked with Fe3+ display a solid-like behavior: their G′ is higher than G′′ over the entire measured frequency range, as shown in Fig. 2a. As a reference, we end-functionalize linear PEG molecules possessing the same molecular weight as the 2gPEGs with catechols (2cPEG). 2cPEG hydrogels crosslinked with Fe3+ that have been prepared with the same protocol as the 2gPEG counterparts, only display a solid-like behavior for frequencies higher than 10 rad per s (Fig. 2a). Hence, 2cPEG hydrogels are unable to retain their shape over time. This behavior reveals that 2gPEG hydrogels have much longer relaxation times than their 2cPEG analogues at a given pH. Furthermore, the observed G′ of 2gPEG is an order of magnitude above the one of 2cPEG, confirming that pyrogallol–Fe3+ crosslinks are significantly stronger or more numerous than their catechol–Fe3+ counterparts.
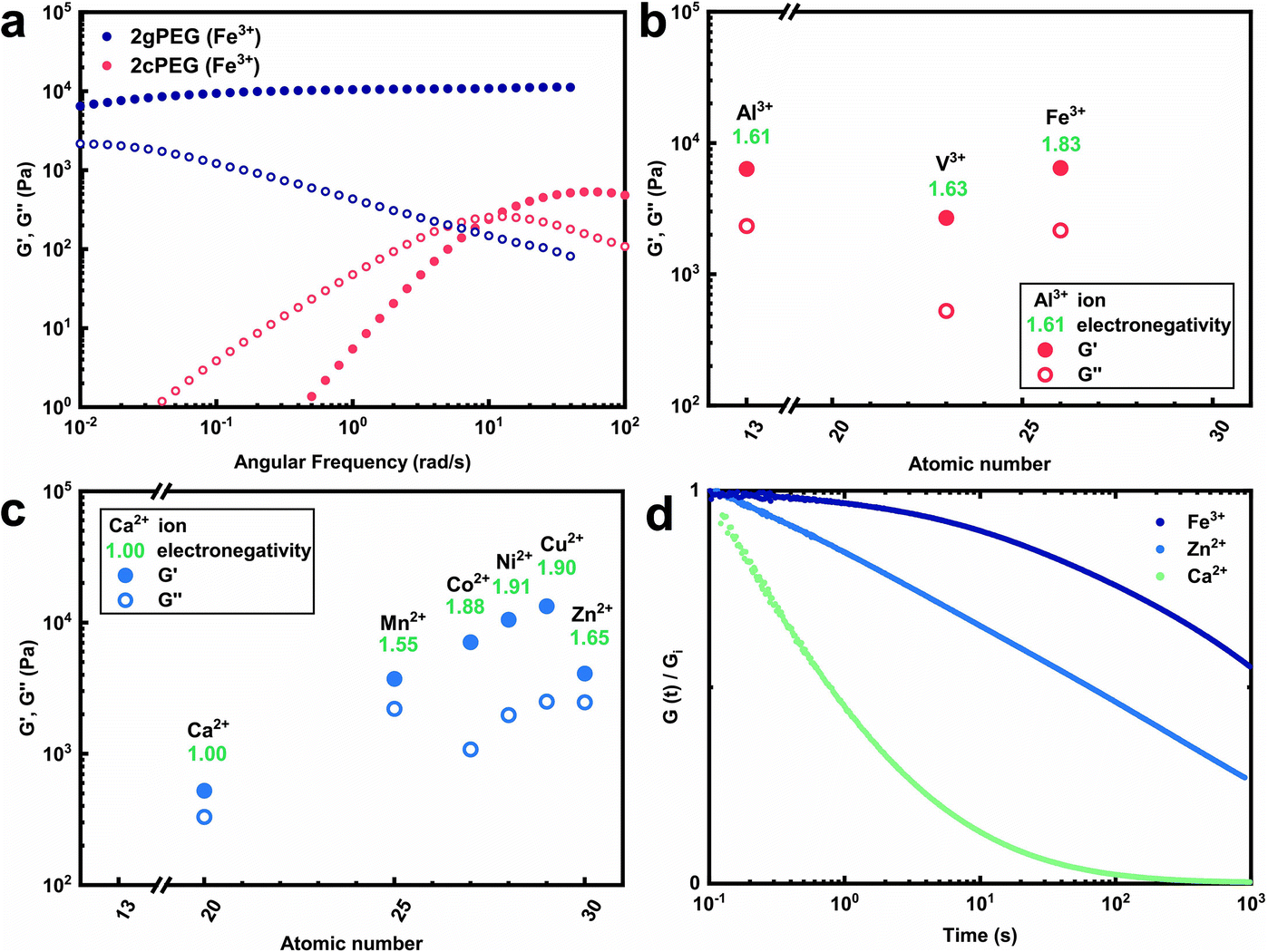 | ||
| Fig. 2 Rheology of 2gPEG hydrogels. (a) Frequency sweep of 2cPEG and 2gPEG crosslinked with Fe3+, where we keep the molar ratio of pyrogallols to Fe3+ constant at 2. Both hydrogels contain the same concentration of ligand-functionalized polymer, the same concentration of ions, and pH. The different mechanical properties are attributed to different ion–ligand binding affinities. (b and c) The storage and loss moduli at 10−2 rad s of 2gPEG crosslinked with different (b) trivalent and (c) divalent ions. (d) Stress relaxation measurements of 2gPEG hydrogels at 25 °C. The modulus is normalized to the initial modulus measured at 0.1 s. The different binding affinities lead to different relaxation behaviors. Filled symbols represent G′ and empty symbols represent G′′. | ||
We explore the range of accessible mechanical properties by crosslinking 2gPEG with other trivalent and divalent ions. Al3+-, V3+-, and Fe3+-functionalized hydrogels have similar mechanical properties, as shown in Fig. 2b. In contrast, the mechanical properties of 2gPEG hydrogels crosslinked with divalent ions vary over an order of magnitude: G′ increases with the electronegativity (χ) of the complexing ion from Ca2+ (χ = 1) to Cu2+ (χ = 1.9), as shown in Fig. 2c. Note that this trend is not observed for the trivalent ions.
The solid-like mechanical properties displayed by 2gPEG hydrogels, combined with a decaying G′ at low frequencies, suggest that ion–pyrogallol crosslinks have longer relaxation times than ion–catechols. Despite these longer lifetimes, the ion–2gPEG crosslinks remain dynamic, and the network can rearrange to dissipate stresses. The rate of relaxation can be described based on the relaxation time, which is longest for pyrogallols complexed with Fe3+ and shortest for those complexed with Ca2+, as measured by stress relaxation and summarized in Fig. 2d. We assign this decrease in relaxation times to the decreasing binding affinities of the cations to pyrogallols.
Why do we need so much more divalent ions than trivalent ones to gel the 2gPEG network? This significant difference in stoichiometry hints towards a particle based crosslinking mechanism. To explore this hint, we measure the energy barrier of bond dissociation and the spread in relaxation times; both these parameters are significantly larger if nanoparticles act as crosslinkers.29 We select two divalent ions, of a low (Ca2+) and high (Zn2+) atomic number, and Fe3+ to represent the trivalent ions. We perform temperature-dependent strain relaxation measurements with a step strain of 10%,29 as exemplified in Fig. 3a for Ca2+ and extrapolate the dissociation energy of the crosslink, as shown in Fig. 3b for Ca2+. The dissociation energy for 2gPEG–Fe3+ hydrogels is similar to that of 2cPEG–Fe3+ hydrogels (Fig. 3c), and in good agreement with the literature,29 confirming that our hydrogels are mainly ionically crosslinked. Note that the dissociation energy is related to the dynamics of the hydrogel and not its elastic behavior. The comparison molecule, 2cPEG, cannot be crosslinked by divalent ions such as Ca2+ and Zn2+, such that the dissociation energies are considered to be below kBT. For the crosslinked samples, we extrapolate the Kohlrausch exponent of our model that can be understood as the spreading of relaxation times,52 as shown in Fig. 3d and e and described in the methods. At Kohlrausch exponent values close to 1, the stress relaxation is fitted to a single relaxation time that is typical for ion–catechol tris complexes. At low Kohlrausch exponent values, the relaxation time spectrum broadens, as observed in the 2gPEG hydrogels. A broadening of the relaxation time spectrum hints at different relaxation mechanisms, which are present in these hydrogels. A single ion in a tris complex leads to a single relaxation time. By contrast, nanoparticles result in different binding affinities with ligands, and hence, different relaxation times, because their size distribution varies and their surfaces are rough. The observed lower values for the Kohlrausch exponents therefore suggest that divalent ions precipitate at the crosslinking sites to form nanoparticles. Following the same argument, we postulate that nanoparticles also form in the case of trivalent ions. Note that even though our hydrogels are primarily crosslinked by nanoparticles, we call them metal-coordinated hydrogels as the chelator–nanoparticle interactions are based on coordination chemistries. The smaller concentration of trivalent ions and the lower pH required to form the hydrogel are likely related to their stronger affinity to pyrogallols, their poorer solubility that results in the formation of precipitates at lower pH values, or a combination of the two aspects.
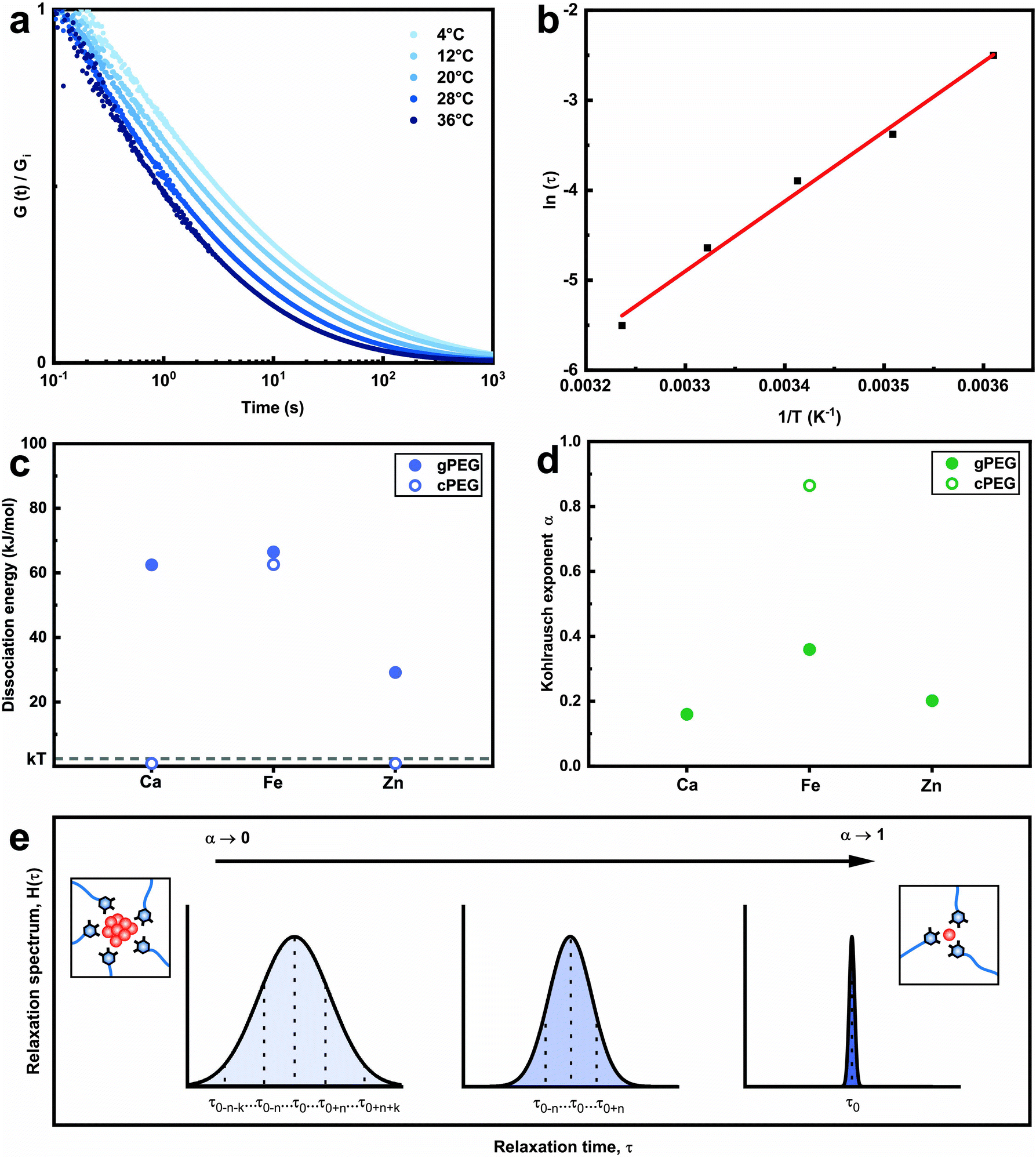 | ||
| Fig. 3 Ionic bond relaxation. (a) Temperature-dependent stress relaxation of 2gPEG crosslinked with Ca2+. Higher temperatures lead to faster stress relaxation. (b) The curves in (a) are fitted using a stretched exponential decay function, and the natural logarithm of the average relaxation time coefficient is plotted against the inverse temperature. The Arrhenius plot is fitted using a linear function to extract the dissociation energy of the network. (c) Calculated dissociation energies of 2gPEG crosslinked with Ca2+, Fe3+ and Zn2+ ions and 2cPEG crosslinked with Fe3+. 2cPEG does not gel with Ca2+ and Zn2+, indicating that their dissociation energy is below kBT at room temperature. (d) The calculated average Kohlrausch exponent of the stretched exponential decay. (e) Schematic illustration of the relaxation spectrum as a function of the Kohlrausch exponent α. Larger Kohlrausch values lead to a narrower distribution of relaxation times, corresponding to a narrower distribution of relaxation mechanisms in the hydrogel network. | ||
Structure of ion–2gPEG hydrogels
To assess the effect of crosslinking chemistry on the network structure, we perform small-angle X-ray scattering (SAXS) on selected samples. The scattering of the network of telechelic crosslinked polymers, such as 2gPEG, reveals structures within 1–33 nm. To determine the contribution of ions and polymers to the scattering signal, we measure a solution of ions and 2gPEG under acidic conditions. To determine the scattering signal of the ions in solution, we subtract the signal of pure 2gPEG in solution. There is no structuring for ions under acidic conditions even in the presence of 2gPEG, indicating that they remain in solution and do not form precipitates or structures in the size range of 1–33 nm that can be detected by SAXS (Fig. S4a†). With increasing pH, the ion-2gPEG systems start to gel, and clear differences appear in the plot of the scattering intensity (I) as a function of the scattering vector (q) (Fig. 4a). The strong increase in the scattering intensity measured at low q-values indicates that larger scattering objects are formed within the network's structure. At the length scale covered by SAXS, two characteristic dimensions are observed in the crosslinked hydrogel networks: (i) a short correlation length (ξ) describes rapid fluctuations of the position of the polymer chains in solution, represented by a single Lorentzian curve at high q-values; and (ii) a long correlation length (ψ) describes the static distance between the crosslinking points, and is estimated by a Gaussian that shows the formation of a network with stationary crosslinking points at low q-values.53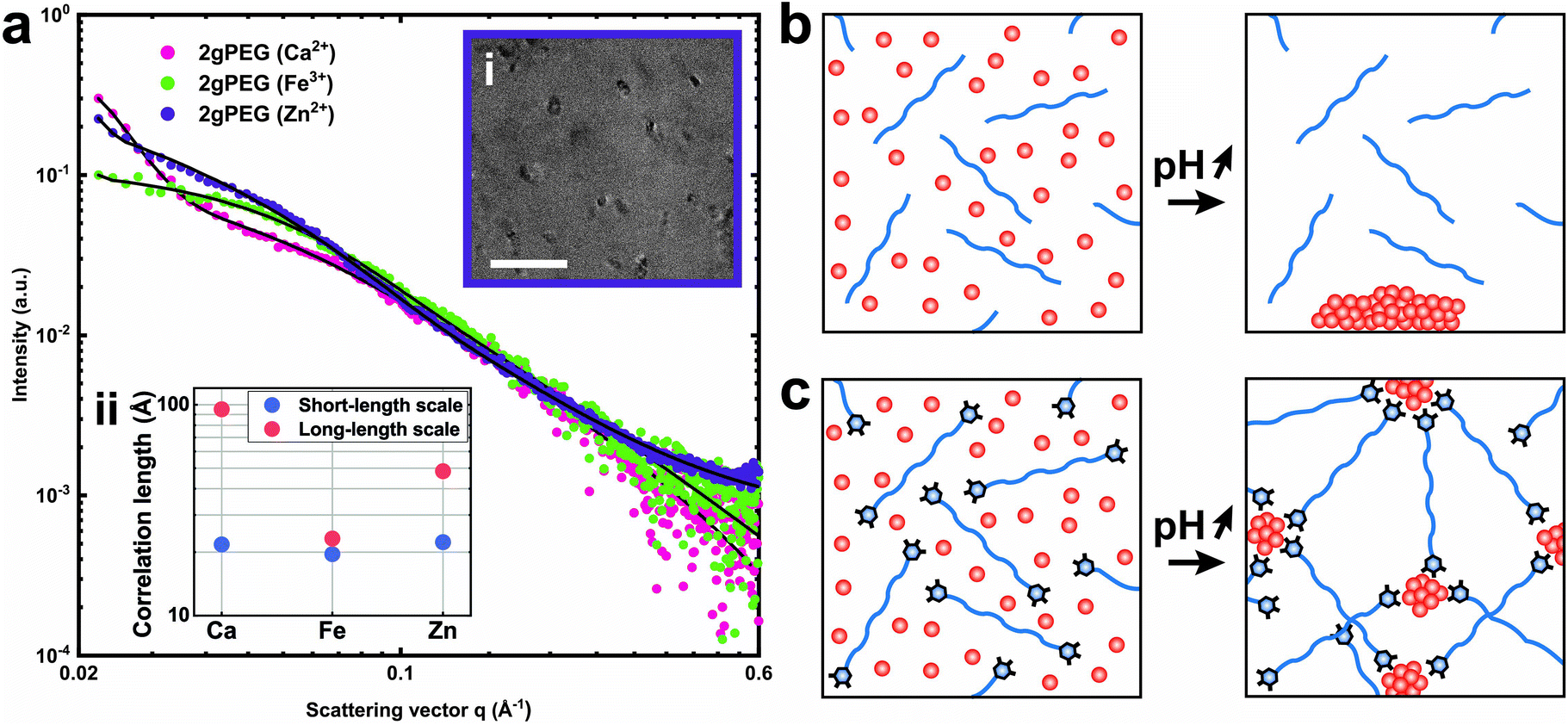 | ||
| Fig. 4 Characterization of the crosslinking mechanism in 2gPEG networks. (a) SAXS measurements of 2gPEG hydrogels crosslinked with Ca2+, Fe3+, and Zn2+. The spectra are fitted using a Lorentzian–Gaussian model (black line). TEM of freeze-dried 2gPEG hydrogels crosslinked with Zn2+ is shown in inset (i). Polydisperse nanoparticles are observed; the scale bar is 100 nm. The fitting parameters of the short-length scale and long-length scale scattering are shown in inset (ii). All samples have a similar short-length scale parameter ξ because they are composed of the same polymer. In contrast, the long-length scale parameter ψ, which is attributed to the average distance between crosslinks, differs significantly. (b) Schematic illustration of the formation of macroscopic inorganic precipitates upon pH increase in the absence of ligands on the polymer chains. The ions precipitate and agglomerate, without interacting with the polymer such that the solution remains liquid. (c) Schematic illustration of the hydrogel gelation when the polymer chains are functionalized with pyrogallol groups that act as nanoparticle stabilizers and metal coordinating sites. | ||
With increasing volume fraction of the polymer, ξ and ψ tend to decrease because the polymer network becomes denser such that the crosslinking density increases, thereby reducing the mobility of the polymer chains.53 The type of ions used to crosslink 2gPEG gels does not significantly influence the short correlation length ξ. This result indicates that the mobility of the individual polymer segments is independent of the ionic crosslinker, as can be expected for linear telechelic polymers. By contrast, the type of ions used to crosslink 2gPEG influences the long correlation length ψ attributed to the crosslinking sites: Fe3+ ions result in the smallest values of ψ, indicating that these ions result in the highest crosslinking density. This finding is in good agreement with our rheology results. It hints at the fact that Fe3+ crosslinked samples have the least dangling bonds, which could be a result of the long dissipation times of Fe3+-2gPEG hydrogels. This finding could also indicate that the valency of Fe3+-containing crosslinking sites is the highest, which could be the case if ions are transformed into nanoparticles at the crosslinking sites and the density of pyrogallols that bind to each nanoparticle is comparably high. The lack of structure and form factor peaks in these SAXS curves suggest that nanoparticles, if present, are polydisperse. We confirm the presence of polydisperse nanoparticles in the size range of 20 nm by Transmission Electron Microscopy (TEM), as shown in Fig. 4b the inset of Fig. 4a. To test if smaller or crystalline nanoparticles form at the crosslinking sites, we perform X-ray diffraction (XRD) on dried samples. We only obtain diffraction peaks from PEG and NaCl, both formed during the drying process of the XRD samples, as shown in Fig. S3.† The lack of additional peaks indicates that the nanoparticles formed at the cross-link sites do not possess crystalline domains or that they are not sufficiently large or numerous to be detected by XRD.
Crosslinking mechanism
To understand the mechanism of formation of these nanoparticles, we prepare a solution containing unfunctionalized PEG and ions and increase the pH, as we did to gel 2gPEG samples. If unfunctionalized PEG is used, we observe the precipitation of macroscopic particles (Fig. 4a and S5†). This result suggests that the pyrogallol groups strongly bind to the surfaces of the forming particles, thereby slowing down or even arresting their growth, as schematically shown in Fig. 4c. This observation is well in agreement with previous reports on pyrogallol-functionalized individually dispersed particles.49,54,55 We measure resonance Raman spectroscopy on Fe3+ crosslinked 2gPEG hydrogels to assess the ligand–ion interactions, as shown in Fig. 5. Three signature peaks are known for catechols if excited with a 785 nm laser source.13,20,56 They are assigned to the charge transfer (533 cm−1) and the bidentate chelation of Fe3+ by oxygens on the catechol ring (590, 633 cm−1). Due to the chemical similarity of pyrogallols and catechols, we observe similar resonance peaks in our 2gPEG hydrogels. The Fe3+ complexed 2gPEG hydrogel displays a strong charge transfer peak at 537 cm−1 and the bidentate chelation peaks at 588 and 620 cm−1 (Fig. 5). Remarkably, the two latter resonance peaks are broader than those of catechol-functionalized hydrogels, suggesting that pyrogallol–iron complexes possess a range of different binding affinities.20,29 This result is in good agreement with the observed small values of the Kohlrausch exponent. The Raman peaks are weaker and broader if 2gPEG is crosslinked with V3+, in stark contrast to catechol-functionalized hydrogels crosslinked with V3+ that result in stronger Raman signals.23 We cannot excite any phonon resonance in the 500–680 cm−1 region with a 785 nm laser if 2gPEG is crosslinked with Zn2+ and Ca2+, as shown in Fig. S6.† This result hints at clear differences in the metal–pyrogallol interactions that are likely responsible for the observed differences in mechanical properties.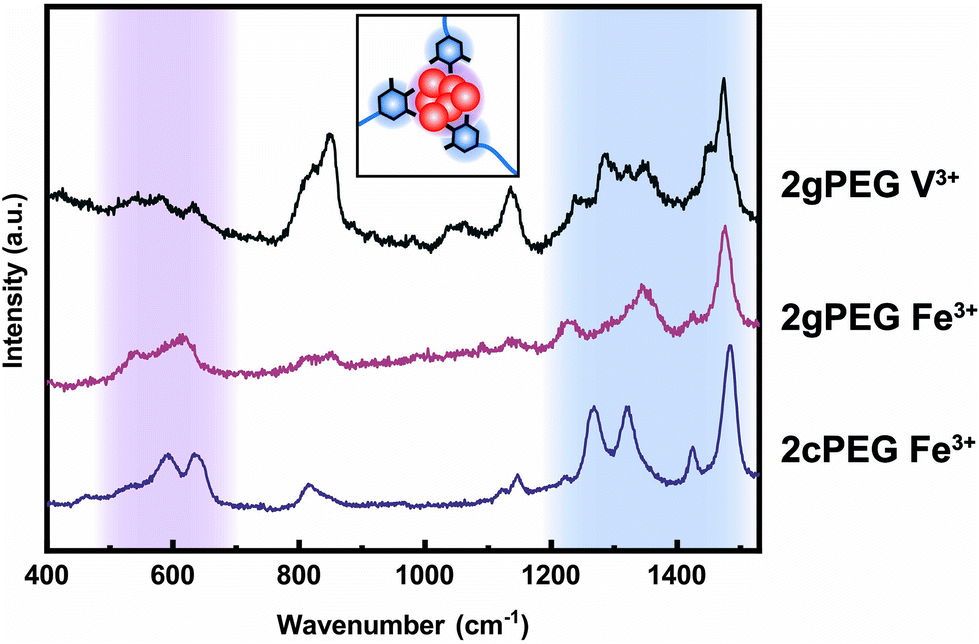 | ||
| Fig. 5 Raman characterization of 2gPEG hydrogels. Resonance Raman spectra of 2gPEG crosslinked with V3+ and Fe3+, and 2cPEG crosslinked with Fe3+. The peaks between 500 and 680 cm−1 are attributed to the catechol–Fe3+ coordination or pyrogallol–Fe3+ coordination, while the peaks between 1230 and 1500 cm−1 are due to the carbon ring vibration. | ||
Application as underwater adhesives
Ligands present in functionalized hydrogels can bind to the surface ions of bulk materials, making them interesting candidates for underwater adhesives. Catechols, commonly found in the marine mussel foot plaque, which holds onto rocks under harsh conditions, are the text book example of underwater adhesion. Synthetic catechol-functionalized telechelic PEGs have already been applied as medical sealing for the amniotic sac after fetal surgery.57 The adhesion of catechols and pyrogallols to wet surfaces is increasingly reversible if ions are added.31,58,59 Here we demonstrate that our 2gPEG hydrogels can be employed as self-healing underwater adhesives. We crosslink 2gPEG with a weak crosslinker, Ca2+, such that it displays a fast relaxation and can adapt its shape quickly to the roughness of the solid surface. We deposit the hydrogel onto an inox steel surface, immerse it in water, and place it in contact with the surface of a 500 g brass weight. After the two metal surfaces are hand-pressed against each other for 10 s, the weight can be lifted out of the water, as shown in Fig. 6a and Movie S1.† This result demonstrates that the thin hydrogel layer sustains a pressure of 50 kPa. Note that the tested hydrogel is one of the weaker ones we introduced such that it shows a fast self-healing behavior. Nevertheless, its mechanical properties are similar to recently reported underwater adhesives and superior to the state-of-the-art cyanoacrylates.60 This is only possible if the adhesion between the hydrogel and the solid surfaces is strong such that the load can efficiently be transferred, and the cohesion within this rather dynamic hydrogel is still sufficiently high. | ||
| Fig. 6 Applications of 2gPEG hydrogels. (a) Photographs of 2gPEG crosslinked with Ca2+, acting as an underwater glue. The hydrogel glue is able to lift a 500 g copper weight outside of the water, corresponding to a 50 kPa pressure. (b) Time-lapse photographs of the self-healing behavior of hydrogels possessing various relaxation times. The left image is taken 10 s after cutting through the hydrogels, and the right image 20 min thereafter. (c) Photograph of 2gPEG crosslinked by iron oxide precipitates formed by exposing the 2gPEG solution containing an excess of Fe2+ and Fe3+ ions to ammonia. The hydrogel forms macroscopic particles when mechanically mixed on Parafilm. The particles are casted in an Eppendorf tube using centrifugation (left) and merged to yield a single integral solid hydrogel piece. Upon removal from the mold, the hydrogel retains its shape (right). (d) The iron oxide precipitates impart magnetic properties to this hydrogel: if immersed in water (left), it can be displaced by an external magnetic field (right). | ||
Self-healing properties
A key feature of our ion–2gPEG hydrogels is the possibility to tune the relaxation times and hence the time scales over which they self-heal and start to flow over a much wider range than currently known. To demonstrate this feature, we cut the hydrogels that have been crosslinked with different ions, and monitor their self-healing as a function of time. Hydrogels that are crosslinked with the most commonly employed metal coordination motif, catechol–Fe3+, start to lose their shape within seconds. By contrast, 2gPEG that is crosslinked with the same ion, Fe3+, retains its shape for at least 20 min, which is the duration of our experiment, as shown in Fig. 6b. We attribute the much higher shape stability of 2gPEG–Fe3+ hydrogels to the longer dissipation times of pyrogallol–Fe3+ complexes compared to their catechol–Fe3+ counterparts. However, the good shape stability comes at the expense of the self-healing properties: these hydrogels only self-heal within an hour when placed in contact. If faster self-healing is required, 2gPEG can be crosslinked with weaker complexation agents, such as divalent ions. For example, if 2gPEG is crosslinked with Zn2+, the hydrogel self-heals within 15 minutes, whereas those crosslinked with Ca2+ self-heal within a minute, as shown in Fig. 6b and Movie S2.† Note that, despite the relatively weak interactions of pyrogallol and Ca2+, the time scale over which this sample loses its shape is much longer than that of the much more commonly used catechol–Fe3+ crosslinked counterpart. These results illustrate the potential of 2gPEG-based hydrogels to adjust their dynamic mechanical properties to the needs of the specific application.Functionalization with nanoparticles
The wide range of ions that can be used to crosslink 2gPEG hydrogels opens up new possibilities to functionalize them with nanoparticles. To demonstrate this feature, we crosslink 2gPEG hydrogels with iron oxide nanoparticles. To achieve this goal, we mix 2gPEG with an aqueous solution of Fe3+ and Fe2+ ions where we fix the molar ratio of Fe3+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe2+ to 2:1. Upon exposure to ammonia, the cations precipitate to form iron oxide nanoparticles. The resulting hydrogel is brittle and forms pieces when mechanically mixed. If densely packed using centrifugation, these pieces can be casted, resulting in an integral part within a minute that retains its shape upon demolding for more than 24 h. Furthermore, the resulting nanoparticles impart the hydrogels with magnetic properties, as illustrated by the movement of the hydrogel towards the externally applied magnetic field, shown in Fig. 6c, d and Movie S3.† This result demonstrates the possibility to introduce functionalities into these hydrogels without sacrificing their mechanical properties.
:Fe2+ to 2:1. Upon exposure to ammonia, the cations precipitate to form iron oxide nanoparticles. The resulting hydrogel is brittle and forms pieces when mechanically mixed. If densely packed using centrifugation, these pieces can be casted, resulting in an integral part within a minute that retains its shape upon demolding for more than 24 h. Furthermore, the resulting nanoparticles impart the hydrogels with magnetic properties, as illustrated by the movement of the hydrogel towards the externally applied magnetic field, shown in Fig. 6c, d and Movie S3.† This result demonstrates the possibility to introduce functionalities into these hydrogels without sacrificing their mechanical properties.
Conclusion
We introduce a metal-coordinated hydrogel whose mechanical properties can be tuned over a wide range by selecting appropriate ions. The broadening in the dynamic mechanical properties of the hydrogel is achieved using telechelic linear PEGs that are end-functionalized with pyrogallols. Pyrogallol–ion complexes have longer dissipation times than the more commonly used catechol–ion or histidine–ion counterparts, and consequently they can retain their shapes much longer. This asset opens up new possibilities to construct truly 3D self-healing materials from metal-coordinated hydrogels. We attribute the long dissipation times of the metal-coordination sites to the precipitation of nanoparticles at the crosslinking sites. The pyrogallols slow down or even prevent the growth of precipitates, resulting in multivalent crosslinking sites that impart excellent mechanical properties to the dynamic networks. Importantly, the mechanical properties of these networks can be tuned with the choice of divalent and trivalent ions used to create the crosslinking sites. The tunable mechanical properties promise to advance the design of multifunctional mechanically robust dynamic hydrogels and might find applications in biomedicine, for example if antibacterial nanoparticles are included, or in optics, if high refractive index nanoparticles are employed. Finally, the inherent adhesion of pyrogallols to wet surfaces allows the creation of medical seals with well-defined mesh structures, which offer a tight control over the diffusion of essential small molecules.Experimental section
Materials
All chemicals are obtained from Sigma-Aldrich and are used as received unless otherwise specified. Deionized water with a resistivity of 18.2 MOhm cm−1 was used.Synthesis of PEG-COOH
PEG-OH is functionalized with carboxylic groups, using protocols previously established.25 In short, we add 30 g of OH-terminated linear PEG (6 kDa) and 600 mL of H2O to a 1 L round-bottom flask and stir the solution. Once the PEG has dissolved, 200 mg TEMPO and 200 mg sodium bromide (NaBr) are added. After a homogeneous solution is formed, 110 mL of aqueous sodium hypochlorite (NaClO) (10–20% available chlorine) is added. The solution is stirred for 30 min while continuously monitoring the pH. The pH is maintained between 10 and 11 with 1 M NaOH. To halt the reaction, 50 mL of ethanol is added, and the pH is decreased to 2 by adding 12.1 M HCl. The aqueous mixture is extracted with four portions of 150–200 mL dichloromethane (DCM). The combined organic layers are extracted with 700 mL Milli-Q H2O and dried with MgSO4. After filtering, the DCM is removed by rotary evaporation. The crude PEG-COOH is purified by precipitating in −20 °C methanol and freeze-dried for 24 h. Any PEG-COOH not immediately used is stored at −20 °C.Synthesis of 2gPEG
PEG-COOH is converted to 2gPEG, using protocols previously established.25 In brief, 1.39 g PEG-COOH, 50 mL DMF, and 25 mL CH2Cl2 are added to a 250 mL round-bottom flask and mixed until the PEG-COOH is completely dissolved. 5-Hydroxydopamine chloride (1.05× mol eq. relative to –COOH) and PyBOP (1.05× mol eq. relative to –COOH) are sequentially added and mixed until completely dissolved. N,N-Diisopropylethylamine (DIPEA) (1.05× mol eq. relative to –COOH) is added to start the reaction, which proceeds at room temperature for 2 h. The reaction solution is reduced via rotary evaporation to remove the DCM. The crude 2gPEG (di-hydroxydopamine PEG) is purified by precipitation in acidified diethyl ether, and dried by rotary evaporation. The product is dissolved in Milli-Q water (pH = 5) in a Falcon tube (50 mL). The tube is centrifuged for 10 min at 4000 rpm, and filtered using filter paper. The clear solution is extracted in DCM and dried with MgSO4. The organic phase is dried by rotary evaporation. The crude 2gPEG is purified by precipitation in diethyl ether.Synthesis of 2cPEG
2cPEG is prepared similarly to 2gPEG, by replacing 5-hydroxydopamine chloride with dopamine hydrochloride.Spectroscopy of synthesis products
To investigate the coupling of 5-hydroxydopamine and PEG, we perform FTIR spectroscopy on dry PEG, PEG-di-COOH and 2gPEG using a 6700 Nicolet (Thermo Fischer Scientific) spectrometer. We confirm the successful 2-step synthesis by the observation of the vibrational stretch of the carboxylic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond at 1750 cm−1 in PEG-di-COOH and the observation of the amide I and II stretches at 1650 and 1550 cm−1, respectively, of 2gPEG, as shown in Fig. S7.† To further confirm the presence of catechol and pyrogallol coupled onto PEG, we dissolve 2cPEG and 2gPEG in D2O, and the 1H spectra are recorded on a Bruker Avance III 400 MHz spectrometer and processed using MestReNova software (http://www.mestrelab.com). Chemical shifts are reported in ppm relative to tetramethylsilane. The presence of catechol and pyrogallol are confirmed by the NMR spectra for 2cPEG and 2gPEG, respectively, as shown in Fig. S8.† Spectrum of 2cPEG: 1H NMR (400 MHz, deuterium oxide) δ 6.78 (d, J = 8.1 Hz, 1H), 6.72 (s, 1H), 6.64 (dd, J = 8.1, 2.1 Hz, 1H). Spectrum of 2gPEG: 1H NMR (400 MHz, deuterium oxide) δ 6.32 (s, 2H).
O bond at 1750 cm−1 in PEG-di-COOH and the observation of the amide I and II stretches at 1650 and 1550 cm−1, respectively, of 2gPEG, as shown in Fig. S7.† To further confirm the presence of catechol and pyrogallol coupled onto PEG, we dissolve 2cPEG and 2gPEG in D2O, and the 1H spectra are recorded on a Bruker Avance III 400 MHz spectrometer and processed using MestReNova software (http://www.mestrelab.com). Chemical shifts are reported in ppm relative to tetramethylsilane. The presence of catechol and pyrogallol are confirmed by the NMR spectra for 2cPEG and 2gPEG, respectively, as shown in Fig. S8.† Spectrum of 2cPEG: 1H NMR (400 MHz, deuterium oxide) δ 6.78 (d, J = 8.1 Hz, 1H), 6.72 (s, 1H), 6.64 (dd, J = 8.1, 2.1 Hz, 1H). Spectrum of 2gPEG: 1H NMR (400 MHz, deuterium oxide) δ 6.32 (s, 2H).
Preparation of 2gPEG hydrogels
For a 100 μL hydrogel sample, 10 mg of 2gPEG is dissolved in Milli-Q water, wherein Vwater = 100 μL − Vion − Vbase. A solution containing 1 M of the appropriate ion chloride solution is prepared separately and diluted in the water solution. The resulting solution is deposited on a piece of Parafilm. An adequate volume of 1 M NaOH is added, and manually mixed with the solution by folding the Parafilm onto itself until the hydrogel appears homogeneous.Rheological characterization
Rheometry is performed using a DHR-3 TA Instruments rheometer, with an 8 mm diameter plate–plate steel geometry. The gap is set to 1000 μm. Stress relaxation measurements are performed using an initial 10% step strain, followed by a continued monitoring of the modulus as a function of time over 1000 s. The sample is allowed to relax for 600 s at the set temperature before starting a measurement. Frequency sweeps are made at 25 °C, using a 1% strain; the sample is allowed to initially relax for 600 s.Stress relaxation model
The recorded modulus (G(t)) is fitted using the Kohlrausch stretched exponential relaxation model, G(t) = G0exp[−(t/τ)α], where 0 < α < 1 and τ(T) = τ0exp[Ea/kT]; here, G0 is the plateau modulus, τ the characteristic relaxation time, α the Kohlrausch exponent, and Ea the dissociation energy of the metal-ion complex. The characteristic relaxation time is temperature-dependent (Fig. 3b for Ca2+). We use this dependence to extract the dissociation energy by fitting the data to the model. The final Kohlrausch exponent is averaged over the measured temperature.
Rheometric data analysis
Data are exported using TA Instrument TRIOS software (http://www.tainstruments.com), and extracted using a python script into DAT files (http://www.python.org). The fitting is done with the Anaconda library (docs.anaconda.com), using the curve_fit function.SAXS method and data analysis
The particle size is estimated from transmission electron microscopy (TEM) images, shown in Fig. 4b. The correlation lengths are deducted from small-angle X-ray scattering (SAXS) of the hydrogels in capillaries. We use a Kratky system (Bruker). The low pH 2gPEG solution is used as the background for the solutions at low pH. Only the empty sample holder is used as the background correction for the gels. All curves are corrected by sample transmission and exposure time. All measurements are performed at room temperature. The intensity I is radially integrated as a function of the scattering vector q. The scattering intensity I(q) is fitted non-linearly by:53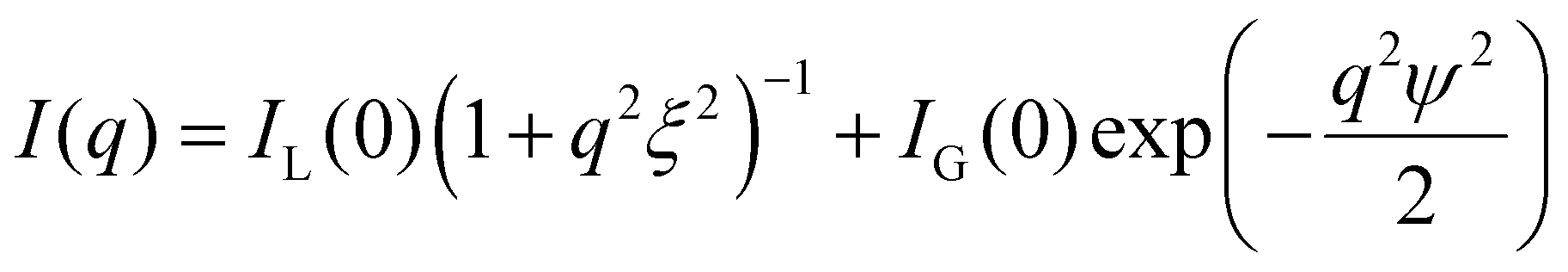 | (1) |
Resonance Raman characterization
A Renishaw Raman spectrometer, equipped with a confocal microscope, a 785 nm laser line, a 1600 l mm−1 grating and a Renishaw camera, is used. For each spectrum, an average of 60 exposures of 1 s each is taken. The peaks at 806 and 1140 cm−1 can be assigned to PEG.62 The peaks at 1251–1270, 1312–1322, 1416–1423 and 1468–1476 cm−1 can be assigned to the catechol ring vibration.13 The Raman spectrum of the synthesized dry 2gPEG is shown in Fig. S10.†XRD characterization
Measurements were performed on an Empyrean system (Theta-Theta, 240 mm) equipped with a PIXcel-1D detector, Bragg–Brentano beam optics (including a hybrid monochromator) and parallel beam optics. A reflection transmission spinner was used.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The authors thank Niels Andersen-Holten for the insightful discussions on the relaxation mechanics of hydrogels. We thank Matthew Harrington and Emmanouela Filippidi for the discussions on resonance Raman spectroscopy. We also thank Lucas Güniat for the TEM imaging. Finally, we would like to thank Matteo Hirsch, Mathias Steinacher, Michael Kessler and the rest of the Soft Materials Laboratory for their fruitful discussions. The work was financially supported by the Swiss National Science Foundation (200020_182662).References
- H. Yuk, et al., Dry double-sided tape for adhesion of wet tissues and devices, Nature, 2019, 575, 169–174 CrossRef CAS.
- J. Liu, et al., Fatigue-resistant adhesion of hydrogels, Nat. Commun., 2020, 11, 1–9 Search PubMed.
- X. Liu, J. Liu, S. Lin and X. Zhao, Hydrogel machines, Mater. Today, 2020, 36, 102–124 CrossRef CAS.
- K. Y. Lee and D. J. Mooney, Hydrogels for Tissue Engineering, Chem. Rev., 2001, 101, 1869–1880 CrossRef CAS.
- J. M. de Rutte, J. Koh and D. Di Carlo, Scalable High–Throughput Production of Modular Microgels for In Situ Assembly of Microporous Tissue Scaffolds, Adv. Funct. Mater., 2019, 29, 1900071 CrossRef.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Double-Network Hydrogels with Extremely High Mechanical Strength, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- J. P. Gong, Why are double network hydrogels so tough?, Soft Matter, 2010, 6, 2583–2590 RSC.
- J.-Y. Sun, et al., Highly stretchable and tough hydrogels, Nature, 2012, 489, 133–136 CrossRef CAS.
- R. Eelkema and A. Pich, Pros and Cons: Supramolecular or Macromolecular: What Is Best for Functional Hydrogels with Advanced Properties?, Adv. Mater., 2020, n/a, 1906012 CrossRef.
- X. Zhao, Multi-scale multi-mechanism design of tough hydrogels: building dissipation into stretchy networks, Soft Matter, 2014, 10, 672–687 RSC.
- C. Creton, 50th Anniversary Perspective: Networks and Gels: Soft but Dynamic and Tough, Macromolecules, 2017, 50, 8297–8316 CrossRef CAS.
- H. G. Silverman and F. F. Roberto, Understanding Marine Mussel Adhesion, Mar. Biotechnol., 2007, 9, 661–681 CrossRef CAS.
- M. J. Harrington, A. Masic and N. Holten-andersen, Iron-Clad Fibers : A Metal-Based, Science, 2010, 328, 216–220 CrossRef CAS.
- M. H. Wilhelm, E. Filippidi, J. H. Waite and M. T. Valentine, Influence of multi-cycle loading on the structure and mechanics of marine mussel plaques, Soft Matter, 2017, 13, 7381–7388 RSC.
- T. Priemel, E. Degtyar, M. N. Dean and M. J. Harrington, Rapid self-assembly of complex biomolecular architectures during mussel byssus biofabrication, Nat. Commun., 2017, 8, 14539–14539 CrossRef.
- M. J. Harrington, H. S. Gupta, P. Fratzl and J. H. Waite, Collagen insulated from tensile damage by domains that unfold reversibly: In situ X-ray investigation of mechanical yield and damage repair in the mussel byssus, J. Struct. Biol., 2009, 167, 47–54 CrossRef CAS.
- H. Lee, N. F. Scherer and P. B. Messersmith, Single-molecule mechanics of mussel adhesion, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12999–13003 CrossRef CAS.
- K. Huang, B. P. Lee, D. R. Ingram and P. B. Messersmith, Synthesis and Characterization of Self-Assembling Block Copolymers Containing Bioadhesive End Groups, Biomacromolecules, 2002, 3, 397–406 CrossRef CAS.
- B. P. Lee, J. L. Dalsin and P. B. Messersmith, Synthesis and Gelation of DOPA-Modified Poly(ethylene glycol) Hydrogels, Biomacromolecules, 2002, 3, 1038–1047 CrossRef CAS.
- N. Holten-Andersen, et al., pH-induced metal-ligand cross-links inspired by mussel yield self-healing polymer networks with near-covalent elastic moduli, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655 CrossRef CAS.
- Z. Shafiq, et al., Bioinspired underwater bonding and debonding on demand, Angew. Chem., Int. Ed., 2012, 51, 4332–4335 CrossRef CAS.
- M. S. Menyo, C. J. Hawker and J. H. Waite, Versatile tuning of supramolecular hydrogels through metal complexation of oxidation-resistant catechol-inspired ligands, Soft Matter, 2013, 9, 10314–10323 RSC.
- N. Holten-Andersen, et al., Metal-coordination: using one of nature's tricks to control soft material mechanics, J. Mater. Chem. B, 2014, 2, 2467–2467 RSC.
- S. Grindy, M. Lenz and N. Holten-Andersen, Engineering Elasticity and Relaxation Time in Metal-Coordinate Cross-Linked Hydrogels, Macromolecules, 2016, 49, 8306–8312 CrossRef CAS.
- S. Grindy, et al., Control of hierarchical polymer mechanics with bioinspired metal-coordination dynamics, Nat. Mater., 2015, 14, 1210–1216 CrossRef CAS.
- L. García–Fernández, et al., Antibacterial Strategies from the Sea: Polymer-Bound Cl-Catechols for Prevention of Biofilm Formation, Adv. Mater., 2013, 25, 529–533 CrossRef.
- M. Krogsgaard, M. A. Behrens, J. S. Pedersen and H. Birkedal, Self-Healing Mussel-Inspired Multi-pH-Responsive Hydrogels, Biomacromolecules, 2013, 14, 297–301 CrossRef CAS.
- M. Krogsgaard, V. Nue and H. Birkedal, Mussel-Inspired Materials: Self-Healing through Coordination Chemistry, Chem. – Eur. J., 2016, 22, 844–857 CrossRef CAS.
- Q. Li, D. G. Barrett, P. B. Messersmith and N. Holten-Andersen, Controlling Hydrogel Mechanics via Bio-Inspired Polymer–Nanoparticle Bond Dynamics, ACS Nano, 2016, 10, 1317–1324 CrossRef CAS.
- J. P. Park, et al., Vanadyl–Catecholamine Hydrogels Inspired by Ascidians and Mussels, Chem. Mater., 2015, 27, 105–111 CrossRef CAS.
- D. X. Oh, S. Kim, D. Lee and D. S. Hwang, Tunicate-mimetic nanofibrous hydrogel adhesive with improved wet adhesion, Acta Biomater., 2015, 20, 104–112 CrossRef CAS.
- Z. Guo, S. Mi and W. Sun, A Facile Strategy for Preparing Tough, Self-Healing Double-Network Hyaluronic Acid Hydrogels Inspired by Mussel Cuticles, Macromol. Mater. Eng., 2019, 304, 1800715 CrossRef.
- J. H. Ryu, S. Hong and H. Lee, Bio-inspired adhesive catechol-conjugated chitosan for biomedical applications: A mini review, Acta Biomater., 2015, 27, 101–115 CrossRef CAS.
- M. Krogsgaard, M. R. Hansen and H. Birkedal, Metals & polymers in the mix: fine-tuning the mechanical properties & color of self-healing mussel-inspired hydrogels, J. Mater. Chem. B, 2014, 2, 8292–8297 RSC.
- P. S. Yavvari and A. Srivastava, Robust, self-healing hydrogels synthesised from catechol rich polymers, J. Mater. Chem. B, 2015, 3, 899–910 RSC.
- N. Holten-Andersen, H. Zhao and J. H. Waite, Stiff Coatings on Compliant Biofibers: The Cuticle of Mytilus californianus Byssal Threads, Biochemistry, 2009, 48, 2752–2759 CrossRef CAS.
- M. J. Harrington and J. H. Waite, pH-Dependent Locking of Giant Mesogens in Fibers Drawn from Mussel Byssal Collagens, Biomacromolecules, 2008, 9, 1480–1486 CrossRef CAS.
- E. Kime-Hunt, K. Spartalian, S. Holmes, M. Mohan and C. J. Carrano, Vanadium metabolism in tunicates: The coordination chemistry of V(III), V(IV), and V(V) with models for the tunichromes, J. Inorg. Biochem., 1991, 41, 125–141 CrossRef CAS.
- S. W. Taylor, M. M. Ross and J. H. Waite, Novel 3,4-Di- and 3,4,5-Trihydroxyphenylalanine-Containing Polypeptides from the Blood Cells of the AscidiansAscidia ceratodesandMolgula manhattensis, Arch. Biochem. Biophys., 1995, 324, 228–240 CrossRef CAS.
- S. W. Taylor, B. Kammerer and E. Bayer, New Perspectives in the Chemistry and Biochemistry of the Tunichromes and Related Compounds, Chem. Rev., 1997, 97, 333–346 CrossRef CAS.
- A. Üçer, A. Uyanik and Ş.F Aygün, Adsorption of Cu(II), Cd(II), Zn(II), Mn(II) and Fe(III) ions by tannic acid immobilised activated carbon, Sep. Purif. Technol., 2006, 47, 113–118 CrossRef.
- H. Ejima, et al., One-Step Assembly of Coordination Complexes for Versatile Film and Particle Engineering, Science, 2013, 341, 154–157 CrossRef CAS.
- Q.-Z. Zhong, et al., Spray Assembly of Metal–Phenolic Networks: Formation, Growth, and Applications, ACS Appl. Mater. Interfaces, 2018, 10, 33721–33729 CrossRef CAS.
- H. J. Kim, et al., Polyphenol/FeIII Complex Coated Membranes Having Multifunctional Properties Prepared by a One-Step Fast Assembly, Adv. Mater. Interfaces, 2015, 2, 1500298 CrossRef.
- A.-K. Koopmann, et al., Tannin-Based Hybrid Materials and Their Applications: A Review, Molecules, 2020, 25, 4910 CrossRef CAS.
- M. A. Rahim, et al., Metal–Phenolic Supramolecular Gelation, Angew. Chem., Int. Ed., 2016, 55, 13803–13807 CrossRef CAS.
- M. Krogsgaard, A. Andersen and H. Birkedal, Gels and threads: mussel-inspired one-pot route to advanced responsive materials, Chem. Commun., 2014, 50, 13278–13281 RSC.
- B. J. Kim, S. Han, K.-B. Lee and I. S. Choi, Biphasic Supramolecular Self-Assembly of Ferric Ions and Tannic Acid across Interfaces for Nanofilm Formation, Adv. Mater., 2017, 29, 1700784 CrossRef.
- M. Shin, K. H. Song, J. C. Burrell, D. K. Cullen and J. A. Burdick, Injectable and Conductive Granular Hydrogels for 3D Printing and Electroactive Tissue Support, Adv. Sci., 2019, 6, 1901229 CrossRef CAS.
- J. S. Lee, et al., In Situ Self-Cross-Linkable, Long-Term Stable Hyaluronic Acid Filler by Gallol Autoxidation for Tissue Augmentation and Wrinkle Correction, Chem. Mater., 2019, 31, 9614–9624 CrossRef CAS.
- S. A. Cazzell and N. Holten-Andersen, Expanding the stoichiometric window for metal cross-linked gel assembly using competition, Proc. Natl. Acad. Sci. U. S. A., 2019, 201906349, DOI:10.1073/pnas.1906349116.
- S. Palato, N. Metatla and A. Soldera, Temperature behavior of the Kohlrausch exponent for a series of vinylic polymers modelled by an all-atomistic approach, Eur. Phys. J. E, 2011, 34, 90 CrossRef CAS.
- S. Mallam, F. Horkay, A. M. Hecht, A. R. Rennie and E. Geissler, Microscopic and macroscopic thermodynamic observations in swollen poly(dimethylsiloxane) networks, Macromolecules, 1991, 24, 543–548 CrossRef CAS.
- G. Milczarek, T. Rebis and J. Fabianska, One-step synthesis of lignosulfonate-stabilized silver nanoparticles, Colloids Surf., B, 2013, 105, 335–341 CrossRef CAS.
- D. Gan, et al., Plant-inspired adhesive and tough hydrogel based on Ag-Lignin nanoparticles-triggered dynamic redox catechol chemistry, Nat. Commun., 2019, 10, 1487 CrossRef.
- E. Filippidi, et al., Toughening elastomers using mussel-inspired iron-catechol complexes, Science, 2017, 358, 502–505 CrossRef CAS.
- D. W. R. Balkenende, S. M. Winkler and P. B. Messersmith, Marine-inspired polymers in medical adhesion, Eur. Polym. J., 2019, 116, 134–143 CrossRef CAS.
- S.-B. Lee, C. González-Cabezas, K.-M. Kim, K.-N. Kim and K. Kuroda, Catechol-Functionalized Synthetic Polymer as a Dental Adhesive to Contaminated Dentin Surface for a Composite Restoration, Biomacromolecules, 2015, 16, 2265–2275 CrossRef CAS.
- C. Shao, et al., Mussel-Inspired Cellulose Nanocomposite Tough Hydrogels with Synergistic Self-Healing, Adhesive, and Strain-Sensitive Properties, Chem. Mater., 2018, 30, 3110–3121 CrossRef CAS.
- D. Lee, et al., VATA: Poly(vinyl alcohol)- and Tannic Acid-based Nontoxic Underwater Adhesive, ACS Appl. Mater. Interfaces, 2020, 12, 20933–20941 CrossRef CAS.
- G. Evmenenko, E. Theunissen, K. Mortensen and H. Reynaers, SANS study of surfactant ordering in κ-carrageenan/cetylpyridinium chloride complexes, Polymer, 2001, 42, 2907–2913 CrossRef CAS.
- E. A. Sagitova, et al., Raman analysis of polyethylene glycols and polyethylene oxides, J. Phys. Conf. Ser., 2018, 999, 012002 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr08351h |
| This journal is © The Royal Society of Chemistry 2021 |