
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent trends in biocatalysis
Dong
Yi†
 ,
Thomas
Bayer†
,
Christoffel P. S.
Badenhorst†
,
Shuke
Wu†
,
Mark
Doerr†
,
Matthias
Höhne†
and
Uwe T.
Bornscheuer
*
,
Thomas
Bayer†
,
Christoffel P. S.
Badenhorst†
,
Shuke
Wu†
,
Mark
Doerr†
,
Matthias
Höhne†
and
Uwe T.
Bornscheuer
*
Department of Biotechnology & Enzyme Catalysis, Institute of Biochemistry, University Greifswald, Felix-Hausdorff-Str. 4, D-17487 Greifswald, Germany. E-mail: uwe.bornscheuer@uni-greifswald.de
First published on 18th June 2021
Abstract
Biocatalysis has undergone revolutionary progress in the past century. Benefited by the integration of multidisciplinary technologies, natural enzymatic reactions are constantly being explored. Protein engineering gives birth to robust biocatalysts that are widely used in industrial production. These research achievements have gradually constructed a network containing natural enzymatic synthesis pathways and artificially designed enzymatic cascades. Nowadays, the development of artificial intelligence, automation, and ultra-high-throughput technology provides infinite possibilities for the discovery of novel enzymes, enzymatic mechanisms and enzymatic cascades, and gradually complements the lack of remaining key steps in the pathway design of enzymatic total synthesis. Therefore, the research of biocatalysis is gradually moving towards the era of novel technology integration, intelligent manufacturing and enzymatic total synthesis.
 Dong Yi | Dong Yi studied chemistry supported by the Deutscher Akademischer Austauschdienst (DAAD) with a doctoral scholarship at the TU Darmstadt (Germany) and obtained his PhD (summa cum laude) under supervision of Prof. Wolf-Dieter Fessner in 2012. He worked as Postdoctoral Teaching Fellow at the East China University of Science and Technology (China) from 2013 and was involved in establishing a pharmaceutical company. In 2018, he joined the Bornscheuer group at the University of Greifswald (Germany) as a postdoc and worked within the EU funded project “SynBio4Flav”. He currently works as the Head of Systems Biosynthesis in the Shanghai Institute of Pharmaceutical Industry (China). |
 Thomas Bayer | Thomas Bayer graduated in the Molecular Biology programme from the University of Vienna and received his PhD in (Bio)organic Chemistry at the TU Wien (Austria) before joining the Bornscheuer group as a post-doctoral fellow. Thomas Bayer received the Erwin Schrödinger fellowship by The Austrian Science Fund (FWF), which included scientific stays at the University of Greifswald (Germany) and the Albert Einstein College of Medicine in New York City (USA) in 2019–2020. Since 2021, he works at the Institute of Molecular Biotechnology at the TU Graz (Austria). |
 Christoffel P. S. Badenhorst | Chris Badenhorst received a BSc in biochemistry and microbiology from the University of South Africa. He then joined Prof. Albie van Dijk's laboratory at the Potchefstroom Campus of the North West University, where he obtained his BSc Honours, MSc, and PhD degrees in biochemistry. He was subsequently awarded an Alexander von Humboldt Research fellowship to join the Bornscheuer group, where he has been working on the development of new ultrahigh-throughput screening methods. |
 Shuke Wu | Shuke Wu obtained his PhD from the National University of Singapore (2015) with Prof. Zhi Li (NUS) and Prof. Daniel I. C. Wang (MIT). In 2017, he moved to Switzerland to work with Prof. Thomas R. Ward at the University of Basel sponsored by a Swiss government excellence scholarship. Until 2020, he worked in the Bornscheuer group at the University of Greifswald as an Alexander von Humboldt fellow. Now he is a professor at the Huazhong Agricultural University in China. |
 Mark Doerr | Mark Doerr studied chemistry and biology at the Universities of Mainz and Jena. He received his PhD at the Institute of Inorganic Chemistry at the University in Jena, Germany (2004). He then joined research projects in the groups of Peter Nielsen in Copenhagen, Denmark (2006–2009) and Steen Rasmussen in Odense, Denmark (2009–2012) working on fundamental bio-/inorganic-chemical questions as a postdoctoral research associate. Since 2012 he is staff scientist in the Bornscheuer group working on high-throughput robotic screening, lab automation, molecular modelling and the combination of machine learning with protein engineering. |
 Matthias Höhne | Matthias Höhne studied biochemistry and obtained his PhD in biotechnology in 2010 at Greifswald University under the supervision of Prof. Uwe Bornscheuer. He then moved to Heidelberg for his postdoc to work with Prof. Andres Jäschke and then started his Junior Professorship focussing on enzyme discovery and application with a focus on chiral amine synthesis. Since 2018, he continues with an ERC starting grant as group leader at the Institute of Biochemistry in Greifswald and orients his research towards photobiocatalysis and the characterisation of binding and transport proteins involved in marine glycan degradation. |
 Uwe T. Bornscheuer | Uwe Bornscheuer studied chemistry and received his PhD in 1993 at Hannover University followed by a postdoc at Nagoya University (Japan). In 1998, he completed his Habilitation at Stuttgart University about the use of lipases and esterases in organic synthesis. He has been Professor at the Institute of Biochemistry at Greifswald University since 1999. In 2008, he received – beside other awards – the BioCat2008 Award. He was just recognised as ‘Chemistry Europe Fellow’. His current research interest focuses on the discovery and engineering of enzymes from various classes for applications in organic synthesis, lipid modification, degradation of plastics or complex marine polysaccharides. |
Introduction
More than a century ago, Rosenthaler synthesised (R)-mandelonitrile from benzaldehyde and hydrogen cyanide using a plant extract containing a hydroxynitrile lyase and opened the door to a completely new research field: biocatalysis.1 Over the next hundred years, this research field had developed rapidly and experienced three distinct waves,2 advancing from the use of crude extract to purified enzymes and then to recombinant enzyme systems. The screening of biocatalysts had evolved from natural source extraction to gene mining with bioinformatic methods. Finally, sequence—structure—function analysis benefited from the developments in structural biology. The engineering of biocatalysts had shifted from modifying reaction conditions to suit enzymatic properties to directed evolution of enzymes to adapt to reaction conditions and molecular structures of substrates.2 These technological revolutions all profited from the development and integration of multidisciplinary technologies and theories which cover most scientific fields including chemistry, biology, pharmaceutics, food, physics, mathematics, computer science, automation and engineering, and prospered the in-depth study of single-step enzymatic reactions, including enzymatic properties, reaction mechanisms, and protein engineering. As a result, a large number of enzymes used for biocatalysis have been identified from nature including almost all enzyme classes: oxidoreductases, transferases, hydrolases, lyases, isomerases, and ligases. The reactions catalysed by these enzymes cover common chemical reaction types, such as redox, substitution, addition, elimination, rearrangement and pericyclic reactions. Based on understanding enzymatic catalytic mechanisms well, these common enzymes and basic enzymatic reactions can be combined into a biocatalytic network, in which the upper edges and nodes are connected in series to form multi-step enzymatic cascades. These cascades made the biosynthesis of chemicals with complex structures feasible and thus have greatly enriched the achievements and application scope of biocatalysis. Therefore, more and more studies have focused on novel enzymatic reactions and advanced cascades for the biosynthesis of complex natural and non-natural products, which led to a remarkable research trend turning from enzymatic synthesis of compounds with simple structures to enzymatic total synthesis of challenging targets.However, since the natural synthetic pathways of many natural products have not been fully elucidated, coupled with the challenges of heterologous expression of enzymes, it is not easy to achieve the enzymatic total synthesis of natural products in vitro or in model host cells. Complex drug compounds are mostly artificially designed molecular structures, which require well designed enzymatic synthesis pathways and heavy protein engineering to reduce the synthetic steps and solve the problem of low enzyme activity towards non-natural substrates. Comfortingly, innovative results of novel enzymes, enzymatic reaction mechanisms, and enzymatic cascades make the de novo biocatalytic synthesis of natural products and their derivatives easier. Especially, at present, the fourth scientific and technological revolution characterised by informatization, networking, and intellectualization is in full swing. Following Moore's Law,3 the integration and computing power of electronic chips are being continuously doubled, which enable big data mining, network relational processes, and in silico protein design that require heavy computation to be realised. Machine learning (ML), which has recently entered the era of deep learning (DL), has opened up the intelligence of data analysis and network construction on big data processing and extended their application in genomics, proteomics and metabolomics.4–6 This brings us effective tools for the discovery of novel enzymes, biocatalytic reactions, and enzymatic synthetic pathways from massive gene, protein and chemical data analysis. In addition, ML also guides the rational design and protein engineering to create artificially evolved enzymes.7,8 Moreover, the rapid development of quantum computers, automation and ultrahigh-throughput (UHTP) screening technology has brought unlimited possibilities for biocatalysis research. Impressively, quantum computing has been oriented towards practical use for calculating quantum chemistry models at an incredibly highspeed.9 Automated screening robots have gained popularity in many laboratories. The next generation experiment robot has begun to serve chemical experiments with an efficiency hundreds of times higher than that of humans.10 Combined with fluorescence-activated cell sorting (FACS) and microfluidics, these advancements increase the screening throughput of enzyme mutagenesis libraries by several orders of magnitude.11–13 It is foreseeable that biocatalysis research will enter a new era characterised by the integration of novel technologies and intelligent manufacturing in the next decade. Therefore, enzymes and enzymatic pathway designs by artificial intelligence, as well as adaptive evolution assisted by UHTP screening, have simplified synthetic pathways of complex compounds and already have been adapted to model host cells. Together with the latest achievements in synthetic biology, several important natural products, their derivatives, and non-natural compounds with pharmaceutical value have been fully enzymatically synthesised, which has elevated the field of biocatalysis substantially. Eventually, historical borders between classical biocatalysis – using isolated and often immobilised enzymes – and biotransformation – using (engineered) whole-cell microorganisms – will disappear since nowadays all available tools can be integrated to create product-oriented designer pathways and only efficiency in terms of productivity in grams per litre and time will decide which type of reaction system is superior.
In this review, we briefly summarize the application of novel advanced technologies applied in biocatalysis, highlight the identification of novel enzymes and enzymatic reactions/cascades, and collect the innovative artificial enzymatic total synthesis of natural products and valuable drug compounds to elaborate the features of the new trends in biocatalysis: integration of novel technologies, intelligent manufacturing, and enzymatic total synthesis of complex molecules (Fig. 1).
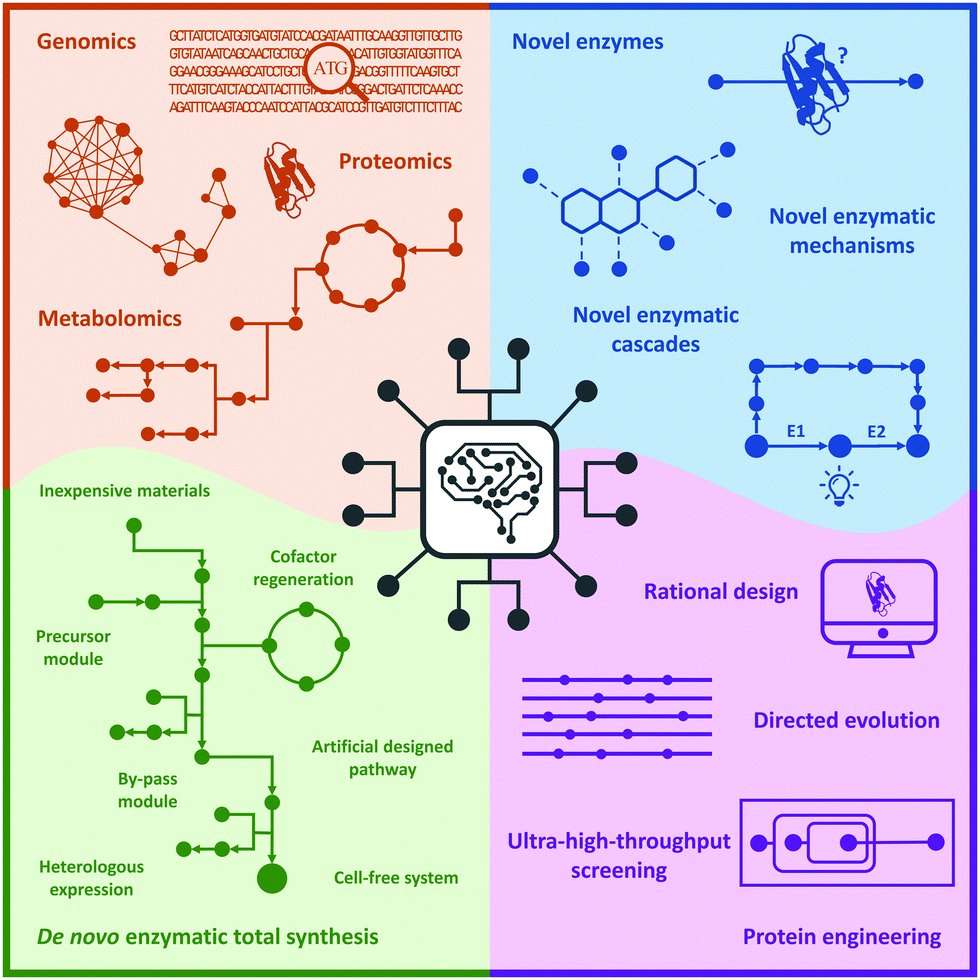 | ||
| Fig. 1 New trends in biocatalysis: integration of novel technologies, intelligent manufacturing, and enzymatic total synthesis. | ||
Advanced technologies for the discovery and design of novel enzymes and enzymatic cascades
Machine learning
The rapid development of technologies related to high-throughput (HTP) gene sequencing, structural biology, screening, and bioinformatics, has accumulated a series of huge databases for genomics, proteomics, and metabolomics. This treasure trove of data provides us a wealth of research materials, but effectively analysing and revealing the interrelationships between the data is a current challenge. Our human brain is very good at recognising simple two- or three-dimensional (2D or 3D) relationships. However, it is powerless to deal with big data processing on a higher-dimensional level. Computing machines, in contrast, can be very well instructed to evaluate and find much higher dimensional relations, incomprehensible to most human minds. Therefore, ML has become an effective tool for biological data processing. Here, we would like to highlight some complementary aspects that are underestimated by non-ML experts.Most ML algorithms assist in finding rules or patterns in very high dimensional data, e.g., millions of 3D molecular coordinates of atoms of amino acid residues, and reduce these rules to likely (low dimensional) predictions of the behaviour of an investigated system, e.g., a (single) enzyme activity. The downside of current computer systems is that they have no experience and understanding of our experimental work, the objects of investigation, the relation of parts of an experiment and the conditions under which a certain experiment was performed and data acquired. All this information about the details and the process of experiments that lead to a certain outcome is called “meta-data”. Meta-data is therefore all information required for an uninstructed entity (like a machine) to reproduce a scientific experiment: exactly what components (e.g., down to the lot of a chemical), what devices (e.g., exact type and firmware version), what experimental conditions (e.g., temperature, pH, pressure, solvents, metabolites, metal ions), the sequence of operations, and additional information about the experiment itself, such as who did the experiments where and when, is meta-information that helps a machine to relate the outcome of different experiments and judge possible sources of error. The predictive power of “learning” machines is highly related to the quality and reliability of the data – and especially the meta-data – it “learned” from.14,15 Therefore, the future success of the application of ML strategies, also in protein engineering and biocatalysis, will heavily rely on proper data generation in conjunction with association of as much meta-data as possible. Meta-data and data linkage are the keys to good predictions since they describe many aspects under which a certain data set was generated and puts data into a context that can be “understood” by a machine.
If not instructed, machines have no notion of meaning, the “semantics”, of these objects and relations. With a formal language derived from the theories of logics and reasoning, we can provide a tiny subset of descriptive relations to objects and operations of experiments that enable the machine to perform a limited amount of logical reasoning and “understanding” of an underlying experiment or data set. This subset is called “ontology”, which is derived from the ancient Greek terms “being” and “logical discourse” – so a logical discourse about the being, e.g., of an experiment or measurement. Creating more and more fine grained logical descriptive term networks (“ontologies” – Google.com also calls them “knowledge graphs”) and associating them with the performed, biochemical experiments will help machines to more and more “understand” the underlying experiments and perform logical reasoning and relating information from very different sources (e.g., international databases such as UniProt, PDB, BRENDA, KEGG, PubMed, and PubChem) to particular experiments. Therefore, the development of precise and consistent ontologies in the realm of protein engineering or biocatalysis and connecting these terms with ontological terms from related disciplines, such as chemistry, molecular biology, and biology, is paramount for a very powerful aggregation of data to further improve the predictive power of machine-based reasoning and ML approaches. This also requires not only a standardised terminology and (fitting) ontologies, but also standardised means of (meta-)data transfer and a standardised query language. Good sources for actively developed ontologies are the EMBL-EBI Ontology Lookup Service (https://www.ebi.ac.uk/ols/index), ontologies of the Pistoia Alliance (https://www.pistoiaalliance.org/), and Allotrope foundation (https://www.allotrope.org/ontologies). Furthermore, a very recent initiative of the German Research Foundation (DFG), NFDI4Cat, is currently assembling a workgroup to develop and refine ontologies for (bio-)catalytic processes, which stresses their importance. In the case of database query language development, the international W3C consortium gathered a standardization workgroup, which is developing the Graph Query Language (GQL, https://www.gqlstandards.org/), a very promising candidate to aggregate data from large, public chemistry and biochemistry databases.
A foreboding of the power of combining knowledge of different databases is given by the recently published EnzymeMiner webtool (https://loschmidt.chemi.muni.cz/enzymeminer/) of the Damborsky group.16 Based on ML predictions, EnzymeMiner proposes a selection of promising enzymes originating from a broad range of organisms that have a potentially similar activity as a provided template and are very likely solubly expressible.17 This can dramatically enhance the exploration of new enzyme types that were not in the focus of current research and knowledge.
Another webtool of this kind in the realm of synthetic biology pathway design is Galaxy SynbioCAD (https://galaxy-synbiocad.org/).18 SynbioCAD starts with a retrosynthetic pathway of a target compound. It then proposes the best biosynthetic pathways for the transformation, cloning strategies of the corresponding enzymes into target vectors and finally liquid handling protocols to execute the assembly of the complex vector constructs.
In current ML-based protein engineering approaches, ML experts need to work tightly with biochemists to first extract the relevant features from a protein engineering question and build a numeric feature set from that to train the ML algorithms. This requires a lot of experience and deep knowledge in modelling, the right questions from the ML side as well as some general understanding from the biochemist's side. With good ontological systems in place, many aspects of this model building processes might be possibly automated in the future, so that ML can become a standard tool for enzyme engineers.
Semantics, derived from ontologies and logical reasoning, is currently a very fast-growing part to improve the information retrieval for (bio-)catalytic information, but it is only one aspect. Another very related aspect that is also rapidly changing and which will influence how we engineer novel enzymes in the future is the development of lab automation standards and data file formats and data exchange protocols. It is very important that these standards are open, freely accessible and royalty free to achieve a global adoption overcoming the monopolism of proprietary island solutions of large instrument vendors. Such free and open lab automation and data standards are developed by the SiLA (https://sila-standard.com/) and AnIML (https://animl.org/) workgroups to support reproducible and reliable data transfer and automated documentation. The quality of the collected data also highly relies on a reasonable number of replicates, consistency checks, personal responsibility of researchers, a systematic way of reporting data (e.g., Standards for reporting Enzyme Data – STRENDA19). It should be also stressed that reports about experiments with negative results are of a very high value for the community – not only that they reduce the number of redundant trials, but also enable machines to learn what did not work. It should also be noted that results might get a completely different context, if our knowledge proceeds: unexplainable results might become very understandable in the future, when new knowledge is created. Results of our biocatalytic exploration should therefore be handled neutral, avoiding human bias.
If ML shall be successful in the future, the machines need standardised, consistent data with the semantics to interpret them. Therefore, a shift in paradigm in the way how we design experiments is required: a high level of automation – with validated, reproducible devices, to get good statistics about the intrinsic variation of a measurement (e.g., three technical replicates of an activity measurement are not enough). Variations related to the expression host, growth medium, composition, expression vector, etc., need to be much more explored.
A shift in thinking will also be necessary in modelling ML questions for protein engineering. Most aspects of current ML in protein engineering have been covered by very recent and exhaustive reviews from the groups of Frances Arnold,8 Jiri Damborsky,15 and Manfred Reetz.14 Briefly, ML has been successfully applied to improve enzyme activities,20 stereoselectivity by the AR algorithm21 which is related to the promising Adaptive Substituent Reordering Algorithm (ASRA),22 enzyme thermostabilities,17 and soluble expression using the SoluProt tool.16 Currently, most ML approaches use too crude assumptions about an enzymatic system despite experimental experience that in many cases, enzyme reactions rely on a very fine-tuned arrangement and dynamics of residues in the active site and very small levels of energy differences play crucial roles in their activities. Enzymes are complex multi-dimensional objects (at least three spatial and one temporal). The exchange of one single amino acid can sometimes completely alter the reactivity or stereo-/enantio-selectivity of an enzyme.23 Predicting these small effects on a quantum mechanical level seemed for a long time inaccessible. Very recent developments of ML derived/generated force fields with quantum chemical accuracy might very soon lead to much better predictions of enzymatic activities and lead to a deeper understanding of enzyme mechanisms.24
A very contrasting, holistic approach is followed by Alley et al.,25 who applied deep learning to approximately 24 million unlabelled natural amino-acid sequences to extract important features, like protein stability and – to some extent – activity.
These unified representations contain short and long term “memories” of a protein sequence, allowing forecasting of structural features with high accuracy even of completely de novo proteins unknown in nature. The model contains a hidden knowledge of protein architecture and could be by that a very powerful tool to annotate or even improve protein activity. Very powerful algorithms for predicting protein structures – one foundation of enzyme activity prediction – have been developed by DeepMind, a Google.com company: AlphaFold 2,26 a community version of the AlphaFold algorithms with open source code is maintained by Wendy Billings et al.18 AlphaFold and related algorithms learn and predict distances between pairs of amino acids and bond angles within amino acid residues from large protein structure databases, like the PDB, by feeding deep neural networks. AlphaFold 2 won the 2020 Critical Assessment of Protein Structure Prediction contest (CASP14) with a highest Global Distance Test (GDT) score of 92.4. One of the currently fastest implementations of folding algorithms, based on Recurrent Geometric Networks (RGN), was published by Mohammed AlQuraishi.27 His algorithm can predict protein structures in milliseconds and accuracies in similar orders as the AlphaFold algorithm.
One important aspect of recent ML-based protein engineering is the question of epistatic mutations28 and their pivotal role as enablers for the emergence of new enzymatic activities and means of leaving local activity optima towards higher enzymatic activities. More experimental data exploring folding and enzyme activity transitions via epistatic mutations should be extended to explore the full power of this concept.
Enzymes in a biological system, either a cell or an in vitro system, also have a very dynamically changing environment, such as different metal ions, protonation states, electron transfers, interactions with small molecules, oligomers and other proteins or (poly-)nucleic acids, which modulate their activities. Therefore, reducing enzymes to the bare one-dimensional sequence information is just a tribute to our present limitations in computational power – and the lack of knowledge of the structural dynamics, molecular interactions and electrodynamics of the associated components. Advances in high-performance computing and quantum computing might overcome these current restrictions in the future.
Microfluidics
Screening novel catalysts from rapidly growing gene and protein databases and large enzyme mutagenesis libraries requires HTP and UHTP screening platforms, which have thus become an important topic in protein engineering and biocatalyst discovery. The microtiter plate format (96- and 384-well) currently dominates screening assays and will likely continue to do so for some time. The reasons for this are that microtiter plate readers are commonly available, microtiter plates offer unmatched flexibility in sample manipulation, a vast collection of protocols have been published, and it is easy to increase throughput by robotic automation. In practice, microtiter plate assays are limited to screening several tens of thousands of clones. This can be sufficient if single site-saturation or small combinatorial libraries have to be screened.29,30 While these methods are still applicable to larger random and combinatorial libraries, they are not capable of screening a significant fraction of the library diversity.31 One solution to this problem is to make the wells smaller, going through 1536-well plates to microcapillary arrays. Chen et al. reported the use of microcapillary arrays for HTP screening. While the flexibility of microtiter plates is lost, this method potentially has much higher throughput than using plates.32,33 Continuing this miniaturisation down a third dimension turns the capillaries into droplets. Droplets of an aqueous phase in an oil phase can be created by simple vortexing (polydisperse) or using sophisticated pumps and microfluidic chips.34,35 Microfluidic droplets are highly monodisperse, commonly with less than 2% variation between droplets. This allows enzyme activity to be quantitatively measured in droplets that are only a few picolitres in volume.35,36 Based on activity measurements, droplets can then be sorted, at rates up to 30 kHz, into active and inactive pools by application of dielectrophoretic force using embedded electrodes (Fig. 2).36,37 Fluorescence is the most commonly used readout and it is even possible to simultaneously sort droplets into multiple populations.38 Equally important is the ability to sort droplets based on more than one fluorescence measurement, which facilitates for example the evolution of enantioselective enzymes (Fig. 2G and H).39,40 The combination of multiple fluorescence measurements with multiple sorting channels should enable very sophisticated screening, but this has not been applied to directed evolution.32,38,40–42 The major drawback of these advanced methods is that they require complex setups that are intimidating if not unavailable. Few laboratories have the necessary expertise in microfluidics, optics, electronics, and programming to set up fully functional sorting rigs. Fortunately, there is an alternative that is accessible to a much larger number of laboratories. The actual droplet generation step is relatively straight forward, and the necessary droplet-generation chips are commercially available. Instead of using electronic sorting chips, water-in-oil droplets can be subjected to a second droplet formation step, generating water-in-oil-in-water or “double emulsion” droplets (Fig. 2B). The advantage of these droplets is that they are stable enough to be sorted using standard flow cytometry equipment, just like cells.43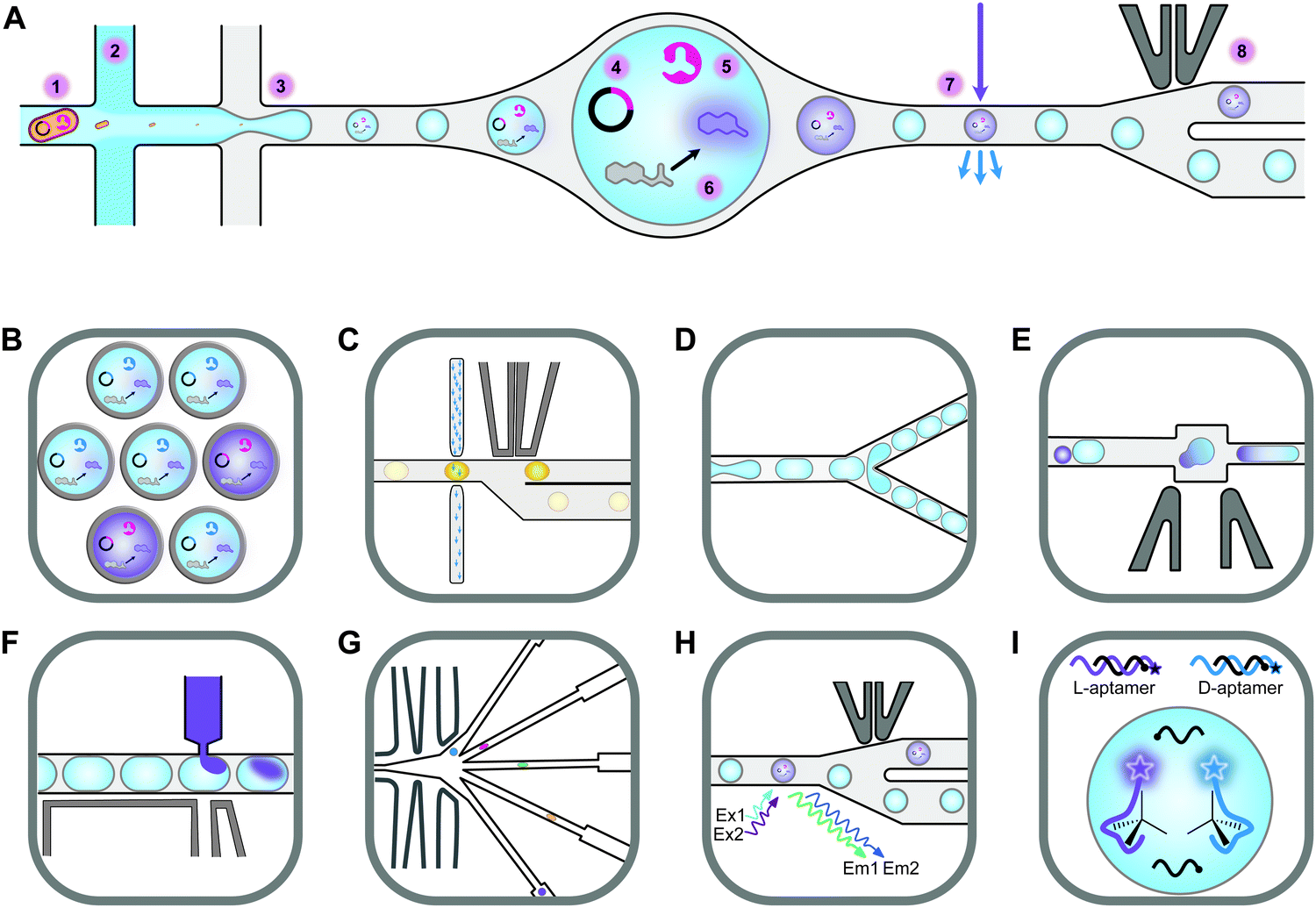 | ||
| Fig. 2 An overview of droplet microfluidic technologies relevant to ultrahigh-throughput screening. (A) A schematic representation of droplet formation using two aqueous components and subsequent fluorescence activated droplet sorting (FADS). (1) Single cells are injected simultaneously with (2) a mixture of assay components, usually containing a fluorogenic substrate and a lysis agent. (3) A fluorinated oil containing a surfactant is injected into the third inlet. This breaks up the aqueous mixture into monodisperse water-in-oil droplets. (4) The single-cell lysate contains both the genotype (plasmid) and (5) the phenotype (enzyme). (6) Active variants convert the substrate to a fluorescent product, which (7) can be detected using the appropriate lasers and photomultiplier tubes (not shown). (8) A fluorescence signal triggers an electric pulse, delivered through imbedded electrodes. The dielectrophoretic force pulls the droplet from the path of least resistance into the “sorted” channel. DNA is subsequently recovered from sorted droplets by plasmid isolation or PCR (not shown). (B) Droplets can be reinjected into a second chip to make double emulsions. In this setup, inlet 1 would contain the droplets, inlet 2 some oil to space the droplets, and inlet 3 the outer aqueous phase containing a surfactant like Tween.43 The resulting double emulsion droplets can be sorted using flow cytometers, similar to FACS. (C) As an alternative to fluorescence measurements, imbedded fibre optic cables have been used to measure the absorbance of droplets. An increased absorbance signal triggers an electric pulse that moves the droplet into the sorting channel.44,45 (D) Droplets can be split, evenly or unevenly, into smaller droplets. This is useful for example if a destructive detection method like mass spectrometry is used, so the first droplet is sacrificed, and the second droplet sorted.46,47 (E) Droplets can also be fused, which is useful, for example, for delivering larger volumes of substrate or lysis agents. The chip first aligns droplets and then uses an electric pulse to merge them.48 (F) Smaller volumes of liquid can also be added to droplets using a method called picoinjection. Droplets flow past an aqueous inlet and an electric field is used to combine the two aqueous phases.49 (G) Droplets can also be sorted into multiple channels38 based on (H) at least two fluorescence measurements.40 (I) An interesting and promising approach allows the label free detection of molecules in droplets using RNA or DNA aptamers.50,51 A unique feature of aptamers is that they allow different enantiomers to be detected using the L- and D-forms of the nucleic acid aptamers. In the example shown, fluorescently labelled DNA aptamers are hybridised with antisense oligonucleotides labelled with quenchers. Binding to the target molecule separates the fluorophores and quenchers, resulting in a fluorescence signal. This allows both the concentration and enantiopurity of the substance to be determined. Importantly, if an aptamer for one enantiomer is available, the other enantiomer is easily detectable by synthesising the opposite enantiomer of the aptamer.50 This figure was inspired by Kintses et al. and Neun et al.52,53 | ||
Despite the technical simplicity of double emulsion sorting by flow cytometry, droplet microfluidics has made a relatively minor impact on biocatalyst discovery and engineering. While droplets have been used to screen hydrolase, oxidoreductase, aldolase, transferase, and isomerase activities, the hydrolases dominate by far (lipase, esterase, phosphatase, phosphonate hydrolase, sulfatase, β-glucosidase, β-galactosidase, and more).42,53–56 The reason for this is simply that most droplet sorting systems require a fluorescent signal and that it is relatively simple to design and synthesise fluorogenic hydrolase substrates. Four years ago, the Hollfelder group broadened the applicability of droplet sorting by introducing absorbance-activated droplet sorting (AADS) (Fig. 2B). They used a chip with embedded optic fibres to measure the absorbance of and sort individual ∼80 μm droplets at a rate of about 300 Hz. This enabled them to evolve an NAD+-dependent amino acid dehydrogenase.44 AADS has attracted significant attention and the paper has been highly cited. Just recently, the Hollfelder group published the first follow up AADS papers, albeit for the same type of reaction and detection system.45,57 As far as we know, no other group has published the use of AADS for directed evolution. There might be several reasons for this. The droplets used are rather large (∼80 μm compared to ∼20 μm for fluorescence sorting, representing a >50-fold larger volume), resulting in a much lower final enzyme concentration and making the method unsuitable for the detection of very low activities. Furthermore, the detection limit was about 10 μM for a strongly absorbing formazan dye (extinction coefficient >37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1), meaning that much higher concentrations of dyes with lower extinction coefficients, like 4-nitrophenolate (18500 M−1 cm−1), would be needed. Unfortunately, this brings us to a much bigger problem, which affects not only absorbance assays but also assays based on fluorescence or any other type of detection system. Despite being predominantly charged at alkaline pH, 4-nitrophenolate is known to “leak” between droplets, meaning that it can transiently enter the oil phase and then move to neighbouring droplets.58 Some dyes leak within seconds (aminocoumarin), some in minutes (rhodamine 6G), some over hours (resorufin) and some over days (fluorescein).59,60 While adding charged groups to a dye is known to dramatically slow leaking (from seconds to days), the case of 4-nitrophenolate demonstrates that this is a complex and often counterintuitive phenomenon.58,60 Not only hydrophilicity but also size matters, demonstrated by the ability of water molecules to diffuse over the fluorinated oil barrier rather easily.43 For simple hydrophobic molecules like haloalkanes there is a correlation between hydrophobicity (logP) and a tendency to partition into the oil phase.61 However, for more complex structures like fluorescent dyes, leakage seems to be related to surfactant concentration, with lower surfactant concentrations slowing down the leaking process, probably due to lower rates of micellar transport.59 Unfortunately, surfactants are needed to facilitate droplet formation and to stabilise droplets during incubation. Sindy Tang's group introduced the use of amphiphilic silica nanoparticles to address this problem. The partially fluorophilic nanoparticles adsorb to the aqueous phase, forming very stable pickering emulsions.62,63 Unlike surfactants, the nanoparticles cannot escape from the droplet surface and can therefore not facilitate the transport of molecules between droplets. This exciting technology has been commercialised by Dolomite microfluidics as a ready-to-use mix in HFE-7500 called Fluoro-Phase. While this approach, combined with alternative oils like perfluoro(methyldecalin), has been demonstrated to significantly (but not completely) reduce leakage, we are not yet aware of a publication describing the use of Fluoro-Phase in high-throughput screening (HTS).62,63
000 M−1 cm−1), meaning that much higher concentrations of dyes with lower extinction coefficients, like 4-nitrophenolate (18500 M−1 cm−1), would be needed. Unfortunately, this brings us to a much bigger problem, which affects not only absorbance assays but also assays based on fluorescence or any other type of detection system. Despite being predominantly charged at alkaline pH, 4-nitrophenolate is known to “leak” between droplets, meaning that it can transiently enter the oil phase and then move to neighbouring droplets.58 Some dyes leak within seconds (aminocoumarin), some in minutes (rhodamine 6G), some over hours (resorufin) and some over days (fluorescein).59,60 While adding charged groups to a dye is known to dramatically slow leaking (from seconds to days), the case of 4-nitrophenolate demonstrates that this is a complex and often counterintuitive phenomenon.58,60 Not only hydrophilicity but also size matters, demonstrated by the ability of water molecules to diffuse over the fluorinated oil barrier rather easily.43 For simple hydrophobic molecules like haloalkanes there is a correlation between hydrophobicity (logP) and a tendency to partition into the oil phase.61 However, for more complex structures like fluorescent dyes, leakage seems to be related to surfactant concentration, with lower surfactant concentrations slowing down the leaking process, probably due to lower rates of micellar transport.59 Unfortunately, surfactants are needed to facilitate droplet formation and to stabilise droplets during incubation. Sindy Tang's group introduced the use of amphiphilic silica nanoparticles to address this problem. The partially fluorophilic nanoparticles adsorb to the aqueous phase, forming very stable pickering emulsions.62,63 Unlike surfactants, the nanoparticles cannot escape from the droplet surface and can therefore not facilitate the transport of molecules between droplets. This exciting technology has been commercialised by Dolomite microfluidics as a ready-to-use mix in HFE-7500 called Fluoro-Phase. While this approach, combined with alternative oils like perfluoro(methyldecalin), has been demonstrated to significantly (but not completely) reduce leakage, we are not yet aware of a publication describing the use of Fluoro-Phase in high-throughput screening (HTS).62,63
Beyond leakage, limited detection options are certainly one of the most serious challenges of microfluidic droplet sorting. As mentioned before, fluorescence and absorbance assays are not always applicable, and when they are, the use of chromogenic surrogate substrates tends to bias the screening outcome (“you get what you screen for”).64 Therefore, there is intense interest in developing alternative detection strategies. Surface-enhanced Raman spectroscopy, light scattering, image analysis, mass spectrometry, impedance measurements, electrochemical detection, and even NMR have been used to detect droplet contents. Wang et al. recently reported Raman-activated droplet sorting (RADS) at a frequency of about 1–2 Hz. They achieved sensitive detection of intracellular triacylglycerols by using an electric field to temporarily halt a moving cell, allowing enough time for an accurate Raman measurement. They claim to have detected intracellular TAG levels previously undetectable using fluorescent stains like Nile Red. However, because they do measurements on single cells before droplet encapsulation, it is not clear whether this method will find use for analysis of products that do not accumulate intracellularly.65 Mass spectrometry (MS) has been used to detect droplet contents at rates up to 30 Hz,66 and sorting was reported at rates of about 6 Hz, using electrospray ionization (ESI)-MS to analyse ∼15000 droplets in 6 h.46 While droplets containing an in vitro expressed transaminase could be sorted, the large droplet volume (25 nanolitre compared to 2 picolitre for fluorescence activated droplet sorting (FADS)) means that this detection strategy would not be applicable to biocatalysts derived from single DNA molecules or single cells.53 However, recent advances in in vitro DNA amplification, transcription, translation, and assay in microfluidic droplets could address this issue.67 Another limitation of MS detection is that it is destructive, so droplets have to be split before analysis. Droplets stored in a delay line are later sorted based on the outcome of the MS analysis.46 This means that the sequence of droplets is critically important and that the fusion of any two droplets would desynchronise the droplet trains, resulting in the loss of all hits. While this problem can be dealt with by co-injection of reference droplets, the system is far from straight-forward. An excellent summary by Neun et al. shows that despite all the progress, only fluorescence, absorbance, and electrochemical detection has been used for the sorting of actual directed evolution-derived or metagenome libraries.53 Furthermore, fluorescence detection is still unique in being the only format that uses droplets of only a few picolitres (high enzyme concentration) and is capable of detecting low nanomolar product concentrations and sorting at frequencies of several kHz. High-speed absorbance measurements on picolitre and femtolitre droplets is possible using differential detection photothermal interferometry (DDPI). This recent technology allows 100 picolitre droplets to be analysed at 1 kHz, with a detection limit of 1.4 μM for Erythrosin B (82500 M−1 cm−1).68 DDPI therefore has the potential to dramatically expand the scope of AADS. However, due to its complexity, the technique will likely remain limited to a few specialist laboratories.
Smart libraries, powered by advances in rational design and DNA synthesis, reduce but have yet to abolish the need for UHTP screening in directed evolution. Furthermore, HTP screening will remain important for the identification of novel biocatalysts from metagenome libraries.42,69 Until DNA synthesis becomes significantly cheaper than it currently is, functional metagenomics will remain essential for exploring the rich functional diversity of nature, which is more relevant to this review than UHTP screening of mutant libraries. Therefore, continued research and development is critical, and it is important that the basic techniques become more accessible to larger numbers of researchers. Commercialisation of key technologies would certainly facilitate this process. Affordable commercially available devices capable of generating and sorting droplets based on fluorescence measurements, combined with simple and user-friendly software, would significantly encourage more researchers to start working with droplet microfluidics.
Biosensors
Traditional HTP screening methods for enzyme activity mainly use absorption, ultraviolet, or fluorescence spectroscopy to determine the concentrations of substrates, products, or cofactors. However, many of these methods cannot meet the requirements of UHTP screening and it is also difficult to accurately evaluate the performance of each enzymatic step in a multi-enzyme cascade in real-time. Likewise, efficient manufacturing by engineered microbial cell factories regularly suffers from metabolic imbalances arising from the introduction and expression of (heterologous) pathways, impaired microbial growth due to the drain of cellular resources, the cytotoxicity of substrates and metabolites, as well as unintended contextual effects, ultimately lowering yields and product titres.70–72 Hence, fine-tuning of (heterologous) pathway expression, maintaining cell viability, and maximising metabolic fluxes towards the desired product are equally important.70,72 The performance of microbial cell factories is usually assessed by the qualitative and quantitative analysis of the metabolites produced through chromatographical methods. However, these can be time-consuming at only low to moderate sample throughput, therefore, impeding the development and optimisation of microbial cell factories.73–75This bottleneck has been addressed by the implementation of genetically encoded biosensors such as (allosteric) transcription factors (TFs) or riboswitches (Fig. 3). Although TFs have already long been used to construct inducible gene expression systems for different prokaryotic and eukaryotic hosts,76–79 their added value as biosensors for the detection of small molecules has only been recognised in the last decade.80–82 Since then, genetically encoded biosensors facilitated the directed evolution of enzymes83–86 and the engineering of (natural) metabolic pathways by the high-throughput detection of metabolites,87–89 as well as the dynamic regulation of genetic circuits to improve overall pathway performance, among other applications.71,80,90–94
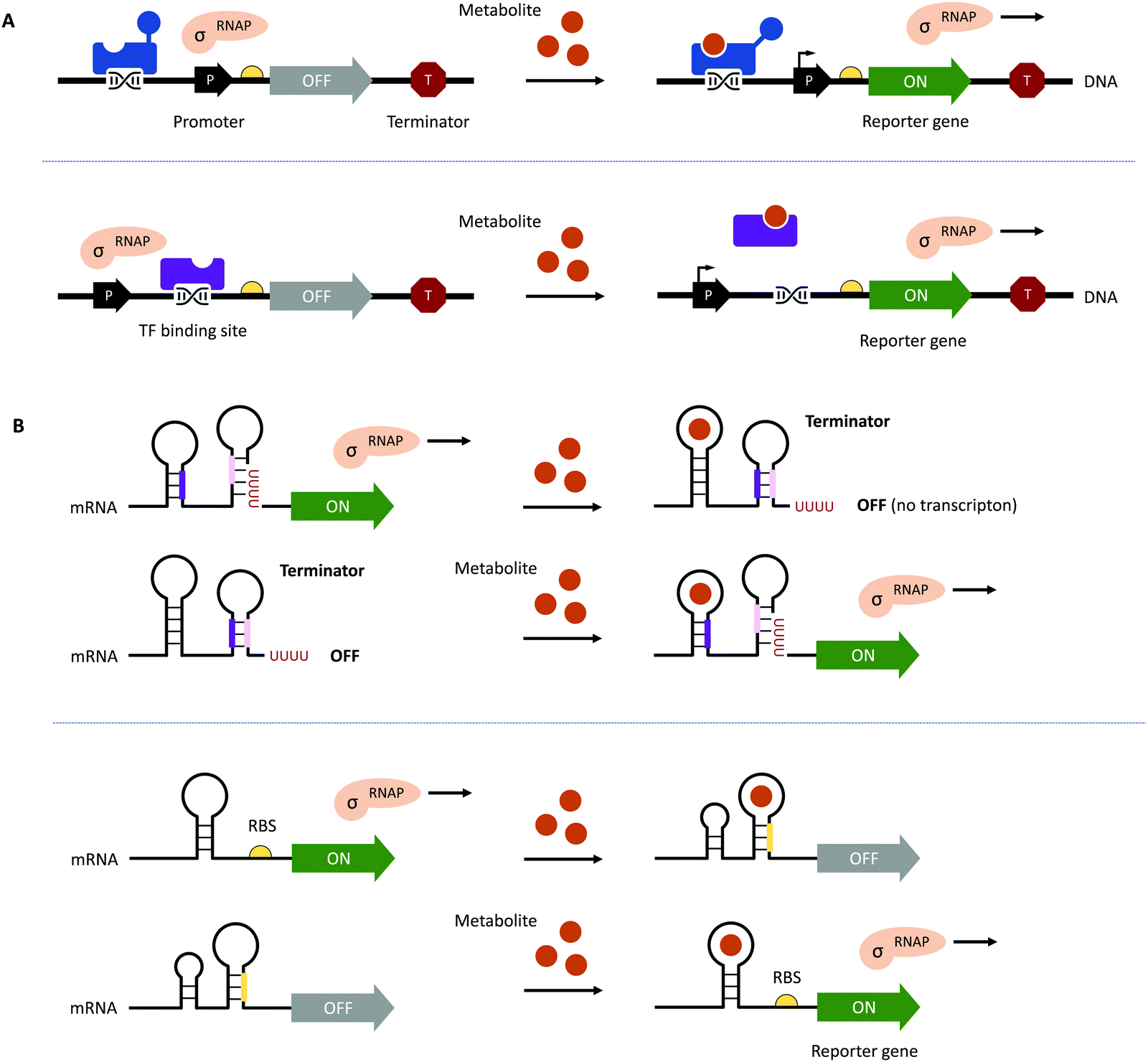 | ||
| Fig. 3 Genetically encoded biosensors. (A) (Allosteric) TFs which bind metabolites can act as transcriptional activators (shown in blue at the top) or repressors (shown in purple at the bottom). TFs can also recruit other activators or repressors to regulate the activity of RNA polymerase (RNAP; not shown).71 (B) Upon binding a ligand or metabolite, riboswitches can act on the levels of transcription and translation by the formation/resolution of a terminator hairpin (top) and the sequestration/release of the ribosome binding site (RBS, bottom, shown in yellow). | ||
To function as biosensors, TFs contain a ligand-binding domain (LBD) and a DNA-binding domain (DBD). The LBD detects the presence of a chemical compound in the environment or a metabolite inside the cell, whereas the DBD facilitates the association with the cognate nucleotide sequence or the dissociation of the TF upon binding of a ligand (Fig. 3A). TFs can act as transcriptional activators or repressors and have been successfully identified by gene expression and protein profiling in the presence of a desired small molecule through combined transcriptome and proteome analyses95–97 and the computer-assisted mining of databases.74,98,99 Furthermore, (microbial) TFs can be responsive to different but structurally related compounds, which has inspired both random mutagenesis and rational design of various LBDs and DBDs.100–105 For example, the TtgR regulatory protein from Pseudomonas putida was engineered by directed evolution and subjected to repeating rounds of FACS, yielding variants with enhanced response to resveratrol.102,103 The Keasling group employed a chemoinformatic approach inspired by small molecule drug discovery. By scouting catabolisable chemicals with molecular shapes similar to the metabolic engineering target and subsequent gene cluster analysis, the ChnR/Pb TF-promoter pair was identified as a suitable biosensor for lactams.106 Besides the identification of TF-based biosensors, improving their performance in terms of selectivity, sensitivity, and operational range can be challenging since additional regulatory elements in the 5′ and 3′ untranslated region (UTR) including (natural and synthetic) promoters, the context of RBS, and transcriptional terminators will influence the functionality of the biosensor.71,72,80 The operational range is defined as the concentration of the metabolite of interest (i.e., the input signal) required for the biosensor to provide a significant change in the output signal (e.g., fluorescence).89 It is not only affected by the attributes of TF including affinity for the ligand and the cognate DNA sequence and its concentration to saturate operator binding sites; expression levels of the reporter have to be carefully adjusted as well. Alternatively, the output signal can be amplified by the implementation of an enzymatic reporter.71,89 The dynamic range of the output signal can be expanded by TF, riboswitch, promoter, and RBS engineering.94,107–114 Curated databases for prokaryotic TFs, their associated regulatory elements and target genes may be helpful for initial designs but might not avoid the necessity of iterative rounds permutating different combinations of genetic parts.71,99,115,116 All these strategies are often time-consuming and results nonintuitive. Berepiki et al. addressed this issue and used a design of experiments (DoE) methodology to efficiently map gene expression levels and provide biosensors for protocatechuic acid and ferulic acid with maximised signal output, improved dynamic range, expanded sensing range and sensitivity.117
In contrast to the multicomponent design of TF-based biosensors, riboswitches comprise of an RNA aptamer – an oligonucleotide sequence with a length of 30–80 nucleobases located in the 5′- or 3′-UTR of mRNA – specifically binding a target molecule (Fig. 3B). Due to the physical proximity, binding of the ligand leads to a conformational change, which can directly affect the binding of ribosomes to RBS on the mRNA upstream of a reporter gene or facilitate the formation of a terminator.71,107 Although riboswitch-based biosensors seem to have a simple architecture and exclusively act at the post-transcriptional level, their rational design is still in its infancy due to the limited understanding of ligand-induced structural changes and the frequently encountered small operational window of riboswitches.118–120 Accessible sources for riboswitches are the RiboD,121 Rfam,122 and RiboGap123 databases, compiling information about prokaryotic riboswitches and their ligands, sequence alignments and conserved secondary structures, and intergenic regions harboring noncoding RNAs and Rho-independent terminators, respectively.
Recently, Calero et al. connected a synthetic fluoride-responsive riboswitch (FRS) to the induction of artificial metabolic pathways for the biosynthesis of fluoronucleotides and fluorosugars in engineered P. putida using inorganic fluoride as both the only fluorine source (i.e., substrate) and as the inducer of the genetic circuit.124 The FRS post-transcriptionally (Fig. 3B, bottom) binds fluoride ions, which triggers the translation of the orthogonal T7 RNA polymerase, subsequently enabling the T7 promoter-controlled production of fluorinases and a purine nucleotide phosphorylase.
Regarding the construction of artificial riboswitches, the systematic evolution of ligands by exponential enrichment (SELEX) has been successfully employed. During SELEX, a library of oligonucleotides specifically binding a target ligand or ligands are produced, selected, and enriched in vitro.73,125,126 Furthermore, natural riboswitches can be engineered like TFs and enzymes.127 Examples include the engineering of a set of riboswitch-based genetic devices to enable the control of gene expression according to changes in the environmental pH128 and the switching of a thiamine pyrophosphate-sensing riboswitch from a device for the repression of downstream genes to an activator.129 Lastly, the physicochemical stability of DNA can be used to detect natural products such as biotin, vitamin D, and folate at nanomolar levels by strand displacement reaction-based biosensors, which have been shown to exhibit increased sensitivity, low interference, and high controllability.130,131
The application of (small-molecule) biosensors and the development and engineering of new sensory devices is certainly of interest for different industries to meet performance criteria through the directed evolution of enzymes,132,133 for the optimisation of microbial cell factories,134–136 and the real-time monitoring of the production of target molecules137 including (aromatic) alcohols, aldehydes, and acids,112,113,138–141 precursors for the synthesis fatty acids and their derivatives,84,92,93,142–148 isoprene and terpenoids,149,150 steroids, as well as flavonoids. Biosensor systems for the last two will be highlighted in the following.
Steroids are polycyclic and highly functionalised compounds and their production is of high interest due to their broad significance as active pharmaceutical ingredients (APIs). However, the synthesis of steroids is demanding and often low-yielding and advanced bio-based procedures are desirable as addressed later in this review. The analysis of steroids usually involves time-consuming sample preparation and analysis by chromatographic methods that limits sample throughput and the efficient development of production strains. Consequently, the development of biosensors for steroids offers advantages to established methods. Mazumder and McMillen constructed a dual-mode promoter in yeast that comprises five steroid hormone responsive elements and one lac operator upstream and downstream of the TATA box, respectively, in a minimal cytochrome C promoter. This dual controller is activated by testosterone (see also Scheme 8) and repressed by IPTG.151 More recently, Chamas et al. created biosensors for the detection of estrogens, progestogens (see also Scheme 8), and androgens in Arxula adeninivorans yeast strains by coupling human hormone receptors and different fluorescent reporter proteins.152 A complementary approach was followed by Maser and Xiong and put into perspective of alternative steroid-sensing methods.153 Their Comamonas testosteroni steroid-sensor (COSS) system is based on the insertion of a green fluorescent protein (gfp) gene upstream of the regulatory region of the hsdA gene encoding a 3α-hydroxysteroid dehydrogenase/carbonyl reductase. Upon steroid exposure, GFP is produced. Disadvantages of the COSS assay were the high background of fluorescence observed in both cellular and cell-free assays. Lastly, the Galagan group identified a progesterone-sensing TF from Pimelobacter simplex by exposure of cultures to different steroids, subsequent RNA sequencing, and bioinformatic analysis. The allosteric TF was ultimately implemented into an optical biosensor consisting of quantum dots coated with the TF and oligonucleotides. The latter resemble the TF binding site and are conjugated to a fluorescence resonance energy transfer (FRET) acceptor. Upon ligand binding to the TF, the DNA probe is released and the FRET signal quenched, corresponding to the concentration of progesterone.154 Optical and other emerging strategies for the design of biosensors for the detection of natural products were recently reviewed by Piroozmand and co-workers.155
For similar reasons, the synthesis and sensing of flavonoids has gained attention in the last decades. Hence, studies aimed at the design of flavonoid-biosensors and the improvement of microbial production. Siedler and colleagues developed a FdeR-based biosensor for naringenin and a QdoR-based sensing device to detect quercetin and kaempferol in real-time.135 Recently, De Paepe et al. followed two strategies for the development of chimeric LysR-type biosensors with customised ligand specificities towards the flavonoids naringenin, apigenin, and luteolin. The first strategy involved the construction of chimeric promoter regions to tune TF binding; the second approach created chimeric TFs by engineering and customization of the LBDs.156 Although DBDs and linker sequences connecting them to the LBDs as well as chimeric TFs were constructed previously,101,105,157,158 the combination of both strategies certainly points towards the expansion of the repertoire of (chimeric) biosensors for the detection of flavonoids and other natural products.159
Thus, biosensors are a powerful tool not only for the engineering of enzymes and the set-up of HTS by monitoring the presence of metabolites in real-time; biosensor systems can time and precisely control the expression levels of pathway enzymes.71 Most of the selected examples of biosensor applications in living cells sensed target molecules in the cytosol. However, recent efforts have been made to sense natural products secreted into the extracellular environment as well.137,146,160–162 An elegant biosensor set-up was realised by Mukherjee et al. who coupled a medium-chain fatty acid (MCFA)-responsive G protein to a receptor on the cellular membrane, enabling the transduction of subsequent signals in the presence of extracellular MCFAs.147 Similarly, the group of Peralta-Yahya engineered a human serotonin G protein-coupled receptor to detect serotonin secreted by a serotonin producing yeast strain.163
Remaining challenges involve the contextualization of novel biosensor designs in terms of their operational range and the functional implementation in heterologous hosts, especially transferring prokaryotic TFs into eukaryotic hosts.164,165 The continuous advancements in bioinformatics and synthetic biology provide a solid foundation to discover TFs and riboswitches and their cognate natural genetic parts or rationally combine them with artificial regulatory elements. DoE methodologies have already shortened this process.117 Furthermore, the combination of TFs and riboswitches that complement each other's shortcomings have emerged as exemplified by Wang et al. who reported a hybrid controller consisting of a riboswitch-based detector and a protein regulator for compensating the low dynamic range of the riboswitch.111 Current and future strategies address feedback control and aim at synchronizing a cell population, reducing the metabolic burden, and balancing the expression of multiple pathway genes depending on the input signals.166–169 Ceroni et al. designed a dCas9-based feedback-regulation system in which the promoter automatically adjusts the downstream gene expression in response to burden167 and Liu et al. constructed quorum sensing-controlled CRISPRi systems, which can dynamically program bacterial consortia.166 To control multigene expression by one chemical signal, Cunningham-Bryant and co-workers reported a genetic controller that consists of catalytically inactive Cas9 and an RNA-binding protein fused to an inducible TF.168 These last examples not only highlight the versatility and variations of the CRISPR/Cas9 technology (which won the Nobel Prize in Chemistry in 2020) but showcase how far our understanding of biosensors as integral parts in regulatory networks and associated metabolic pathways has already advanced.
Novel enzymes and enzymatic reactions leading to total synthesis of complex natural products
Natural products have a wide range of pharmacological activities to humans. However, the extraction of natural products from original organisms often requires huge planting and breeding resources with limitations to season and region. Therefore, heterologous biosynthesis of natural products in vitro or in model microorganisms is a more promising way and has been a research hotspot for decades. At present, the use of heterologous pathways for natural products still mainly replicates the biosynthetic pathways present in the original hosts. However, due to the dependence of enzyme expression and coenzyme/energy cycles in the original host organism and the intracellular environment, the same synthetic pathway is hard to replicate in model microorganisms or in vitro. Therefore, some optimisation strategies from the viewpoint of biochemistry and organic chemistry are widely used, including but not limited to (i) discovery of novel enzymes and protein interactions in the original metabolic pathways with well-studied catalytic and interaction mechanisms to provide novel enzyme tools for synthetic pathway design, (ii) substitution of enzymes in natural synthetic pathways with isoenzymes derived from microorganisms, (iii) replacement of natural synthetic pathways with microbial degradation pathways and artificial enzymatic cascades, (iv) design of artificial coenzyme regeneration pathways, and (v) protein engineering of key enzymes by rational design and/or directed evolution to improve their activity, specificity, substrate scope as well as expression level and stability. Here, we only concentrate on some excellent examples to elaborate the key effects of novel enzymes, novel enzymatic mechanisms, and novel enzymatic cascades on the optimisation of synthetic pathways for the heterologous and in vitro biosynthesis of typical natural products.Flavonoids
Flavonoids are some of the main secondary metabolites of plants, which are a class of polyphenol compounds mainly containing a C6–C3–C6 backbone with more than 9000 structures found in nature.170 Besides their important roles in the physiology of plants, flavonoids also have a variety of pharmacological effects on humans and are one of the main active substance classes in herbal medicine.171 At present, the heterologous biosynthetic pathway of flavonoids mainly contains the precursor module for the synthesis of p-coumaric acid (p-CA) and malonate, a central and diversification module for the synthesis of dihydrochalcones, flavanones, flavanonols, flavones and flavanols, and a functionalization module for hydroxylation, methylation, prenylation, glycosylation, acetylation, sulfonation or glucuronidation (Scheme 1).172–176 However, the metabolic pathways of flavonoids are not limited to plants. Biosynthesis and biodegradation pathways of flavonoids have also been found in fungi and bacteria, bringing novel enzyme tools and de novo synthetic pathways to the heterologous biosynthesis of flavonoids.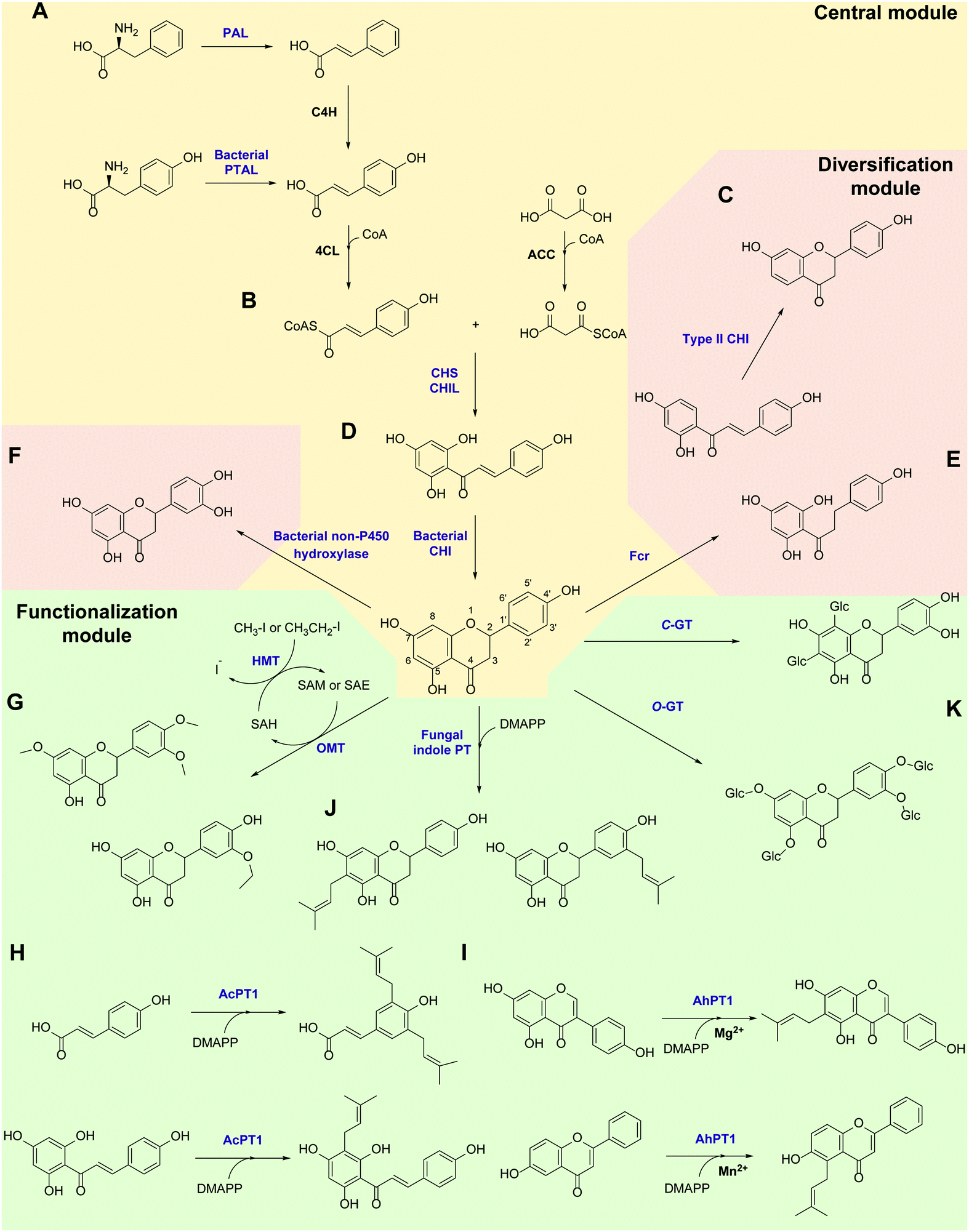 | ||
| Scheme 1 Novel enzymes, enzymatic mechanisms and cascades in flavonoid synthesis. PAL: phenylalanine ammonia lyase; C4H: cinnamate 4-hydroxylase; PTAL: bifunctional phenylalanine/tyrosine ammonia lyase; CHS: chalcone synthase; CHIL: non-catalytic chalcone isomerase-like protein; CHI: chalcone isomerase; Fcr: flavanone- and flavanonol-cleaving reductase; OMT: O-methyltransferase; HMT: halide methyltransferase; PT: aromatic prenyltransferase; GT: glycosyltransferase. | ||
In the precursor synthesis pathway, the biosynthesis of p-CA mainly relies on phenylalanine or tyrosine as starting materials (Scheme 1A). Aromatic ammonia lyases catalyse the deamination of phenylalanine or tyrosine to form cinnamic acid or p-CA, respectively. Cinnamic acid can be further hydroxylated by cinnamate 4-hydroxylase (C4H) to obtain p-CA. Phenylalanine ammonia lyase (PAL) and bifunctional phenylalanine/tyrosine ammonia lyase (PTAL) have been identified in dicotyledonous plants and some monocots, respectively.177,178 Both have quite high activity towards phenylalanine, and the latter can also accept tyrosine but with lower affinity.177 Plant-derived PALs and PTALs are good biocatalyst candidates for the synthesis of p-CA,178 but may require chaperones for better expression in prokaryotic hosts, which is a limiting factor for their synthetic application.179 On the other hand, PAL, PTAL and monofunctional tyrosine ammonia lyase (TAL) have been reported in microorganisms, especially fungi and bacteria that produce antibiotic phenylpropanoids or utilize phenylalanine and tyrosine as carbon and nitrogen sources.177,180 Among them, the PTAL from Rhodotorula glutinis (RgPTAL) shows impressive activity towards tyrosine181 and thus has been used for the biosynthesis of several phenylpropanoids.182–184 Furthermore, a mutant of RgPTAL (S9N/A11T/E518V), obtained through random mutagenesis, probably anchors the flexible loop region (Glu325–Arg336) to maintain the active-site pocket opening which ensures easy access by tyrosine and thus significantly improved its activity and the yield of p-CA.185 The regioselectivity of ammonia lyases to phenylalanine and tyrosine is also affected by the amino acid residue that binds to the para-hydroxyl group on the benzene ring. Substitution of this position by polar residues can obviously increase the specificity of PAL towards tyrosine.186 Recently, two novel TALs were identified from actinomycetes and achieved the productivity of p-CA up to 2.88 g L−1 h−1) using recombinant Escherichia coli as a whole-cell biocatalyst, which currently represents the highest efficiency for microbial production of p-CA.187 Therefore, these microbial-derived ammonia lyases and their mutants have become the preferred enzymes for the synthesis of p-CA in the precursor pathway with significantly higher expression level, catalytic activity and p-CA yield.
In the central synthesis pathway, chalcone synthase (CHS) catalyses the synthesis of a chalcone, 2′,4,4′,6′-tetrahydroxychalcone (THC), from p-coumaroyl-CoA and malonyl-CoA (Scheme 1B).188 CHS is a plant-specific promiscuous type III polyketide synthase (PKS) which also produces other polyketides such as the p-coumaroyltriacetic acid lactone (CTAL). Therefore, how to improve its product specificity is of great significance for optimising metabolic flux and increasing flavonoid production. Very recently, a conserved strategy was uncovered by which non-catalytic chalcone isomerase-like proteins (CHILs), which are ubiquitous in plants, are able to bind to CHS a rectifier and increased the kcat value (2–15 times higher) for the THC production potentially through binding to the tetraketide-CoA intermediate in an energetically favourable manner, and thus enhance THC production and decrease CTAL formation.189 Since CHILs perform macromolecular interaction with other enzymes in plant specialised metabolism, this result brings us a revelation that protein–protein interactions could be widespread in the biosynthesis of natural products by a broader effect on promoting the activity and specificity of enzymes and the regulation of metabolic flux, which can provide an important tool for optimising heterologous biosynthesis.
Chalcone isomerase (CHI) catalyses the intramolecular cyclisation of THC and generates the flavanone naringenin which is a key intermediate for the structural differentiation to other flavonoids. Plant CHIs are considered to have evolved from fatty acid binding proteins,190 which shows the key role of protein evolution in modifying the catalytic mechanism of enzymes and broadening the source of novel enzymes by mutagenesis.191 According to substrate selectivity and catalytic mechanism, plant CHIs can be divided into type I and II. Both can accept 6′-hydroxychalcones as substrates, while the latter also has high activity towards 6′-deoxychalcones (Scheme 1C).192 Recently, the reaction mechanisms of enantioselective oxa-Michael cyclisation performed by type I and II CHIs have been revealed by X-ray crystal structure and molecular dynamics simulations, wherein the guanidinium ion of a conserved arginine positions the nucleophilic phenoxide and activates the electrophilic enone for cyclisation through Brønsted and Lewis acid interactions.193 This mechanism presents a new enantioselective Michael-type reaction in natural product biosynthesis that efficiently constructs C–O bonds. The crystal structure of type II CHI also revealed two unique water molecules in the active pocket which form an ordered hydrogen bond network with the polar amino acids in the pocket. This extended hydrogen bond network supports the role of ordered water in the destruction of the intramolecular interaction between ketone oxygen and 2′-OH and further provides a ring flip of 6′-deoxychalcone. Therefore, the catalytic efficiency towards 6′-deoxychalcone has been greatly improved.193 These results provide a theoretical basis for screening novel CHIs and broadening the substrate tolerance of CHIs through mutagenesis.
Besides in plants, CHIs also occur in some anaerobic intestinal bacteria as key enzymes for the degradation of flavonoids. The first bacterial CHI was isolated and cloned from Eubacterium ramulus (ErCHI), which has activity towards THC, isoliquiritigenin, butein, eriodictyol chalcone, and hesperetin chalcone (Scheme 1D).194,195 However, this bacterial CHI has no homology to plant CHIs and is even rare in protein databases, which shows its unique evolutionary origin.191 The protein structure of ErCHI consists of two ferredoxin domains as catalytic domains and a solvent-exposed domain.196 Unlike plant CHIs, the intramolecular cyclisation of chalcones is catalysed by bacterial CHI via a reversible Michael addition catalysed by histidine.196 Therefore, ErCHI can also catalyse the isomerisation of the flavanonol taxifolin to the auronol alphitionin.197 The study of these novel enzymatic mechanisms shows the impressive diversity of isoenzymes from various sources related to flavonoid metabolic pathways. In-depth studies on bacterial enzymes in the degradation pathway of flavonoids may enable them to replace plant-derived enzymes and enable the design of novel biosynthetic pathways for flavanones. For example, a flavanone- and flavanonol-cleaving reductase (Fcr) was recently identified from E. ramulus, which is an iron-sulfur flavoprotein containing an intramolecular electron transfer chain. It performs a cofactor-mediated hydride transfer from nicotinamide adenine dinucleotide (NADH) onto C2 of the respective substrate via flavin adenine dinucleotide (FAD), a 4Fe–4S cluster, and flavin mononucleotide (FMN), and further directly attaches the C2 of flavanones and flavanonols and cleaves the heterocyclic C-ring, which provides a novel pathway to synthesise dihydrochalcones and dihydroflavonols from flavanones and flavanonols, respectively (Scheme 1E).198
Hydroxylation and methylation greatly extend the structural differentiation of flavonoids. The hydroxylation mainly occurs on C3 of the A-ring and para- and ortho-positions of the B-ring catalysed by plant-derived flavanone 3-hydroxylase, 3′-hydroxylase, and 3′,5′-hydroxylase, respectively. Because of the low expression of P450 enzymes and the lack of effective electron transport systems in prokaryotic host cells, bacterial hydroxylases, such as an endogenous non-P450 hydroxylase complex from E. coli (HpaBC), have shown their advantages in cell factory construction and have been reported to additionally hydroxylate the ortho-position of the B-ring to achieve conversion of naringenin and afzelechin to eriodictyol and catechin, respectively, with high yields (Scheme 1F).199 Moreover, O-methylation of hydroxyl groups is a common modification of flavonoids catalysed by O-methyltransferases (OMTs) using S-adenosyl-L-methionine (SAM) as cofactor for providing the methyl group, which mostly takes place on the 7,3′,4′,5′-hydroxyl groups of flavonoids and the 7,4′-hydroxyl groups of isoflavonoids. OMTs have been widely found in plants, showing diverse substrate specificity and regioselectivity.200 Meanwhile, some flavonoid OMTs were also discovered in microorganisms, such as Bacillus and Streptomyces.201–203 Many OMTs have been recombinantly produced and used for the biosynthesis of flavonoids due to their superior chemo-, regio- and stereo-selectivity.204,205 However, the bulk demand of the methyl group donating cofactor SAM has hindered the industrial applications of OMTs.204 Therefore, in situ regeneration of SAM is one of the key factors affecting methylation biosynthesis. A early attempt of SAM regeneration was a complex SAM recycling cascade involving five additional enzymes on the basis of the physiological cycle of the metabolites in cells.206,207S-adenosyl-L-homocysteine (SAH) produced after transferring the methyl group is hydrolysed to adenosine and homocysteine by a SAH hydrolase, after which adenosine is sequentially phosphorylated by adenosine kinase, polyphosphate kinases 2 I and II, producing adenosine triphosphate (ATP). After that, SAM is reproduced from ATP and L-methionine by a methionine adenosyltransferase. Although this is a feasible way to regenerate SAM, such a long and energy-consuming coenzyme regeneration pathway is not suitable for biocatalytic methylation, at least in vitro. A newly established and more efficient SAM recycling system consists of only one enzyme, halide methyltransferase (HMT), which produces SAM directly from SAH with methyl iodide as donor (Scheme 1G).208 This novel cascade shows that a simpler cofactor regeneration system can be designed and realised by introducing non-natural donors and off-path tool enzymes, which helps to partly depart from the original metabolic pathway and simplify the biosynthetic pathways of natural products. In addition to methylation, the structural and functional diversity of flavonoids can be dramatically expanded via hydroxyl group bioalkylation with SAM analogues and promiscuous MTs. SAM analogues containing different alkyl substituents can be produced via chemical method or by a chemoenzymatic methods using L-methionine analogues catalysed by methionine adenosyltransferases or halogenases.209–211 A more advanced way is to explore promiscuous or engineered HMTs for the production of SAM analogues and to achieve flavonoid bioalkylation on the basis of the MT-HMT cofactor regeneration system.30,212 This artificial cofactor regeneration pathway provides a novel inspiration and solution for solving the problem of low efficiency of SAM regeneration in biosynthesis.
Prenylation is another structural modification for the functionalization of flavonoids catalysed by aromatic prenyltransferases (PTs). Plant-derived PTs generally have high regiospecificity, transferring the prenyl moiety on the C6 and C8 of the final flavonoid skeleton, as well as the C3′ of chalcones and the C3 and C5 of p-CA in the intermediate biosynthetic step (Scheme 1H).174 Recently, a novel di-PT was isolated from Artemisia capillaris which can accept p-CA as its specific substrate and transfers two prenyl residues stepwise to yield artepillin C.213 This is the first plant PT involved in the biosynthesis of phenylpropanes and capable to introduce multiple prenyl residues to native substrates with different regiospecificity. The plant-derived PTs are transmembrane enzymes. Due to the lack of high-resolution protein crystal structures, the substrate binding pocket and catalytic mechanism of PTs are currently unclear, which limits the protein engineering studies of PTs, such as widening the donor-binding pocket to accept longer chain prenyl donors and thereby broadening the diversity of product structures.174 Plant PTs prefer magnesium ions (Mg2+) to stabilize the pyrophosphate group of the donor. However, a recent study reveals that metal ions can change the substrate specificity of a flavonoid PT from Artocarpus heterophyllus (AhPT1). AhPT1 could catalyse 6-C-prenylation of genistein when Mg2+ served as cofactor but without any activity towards 6-hydroxyflavone. However, 5-C-prenylation of 6-hydroxyflavone was identified by AhPT1 when Mn2+ was used (Scheme 1I).214 This new discovery shows that metal ions play a key role in the substrate specificity, prenylation sites and catalytic mechanism of PTs, rather than just stabilizing the donor. Besides, the prenylation products on the O-site have also been found in plants. However, O-specific PTs have not been discovered yet, which revealed that O-specific PTs might have no homology with the C-specific ones.174 Therefore, the intelligent analysis of genomic, proteomic and metabolomic data could most likely bring new opportunities for the discovery of PTs with O-specificity. In addition, soluble PTs from bacteria show their catalytic capability towards flavonoids and prenylation specificity. For example, indole PT 7-DMATS from the fungus Aspergillus fumigatus accepted chalcones, isoflavonoids, and flavanones, and mainly catalysed prenylation at C6, while another indole PT, AnaPT, prefers prenylations at C6 or C3′ of flavanones and isoflavones (Scheme 1J). These fungal PTs have replaced plant-derived PTs for the heterologous biosynthesis of prenylated flavonoids.215 In addition, dimethylallyl diphosphate (DMAPP) is the preferred donor for PTs and is synthesised through the mevalonate (MEV) pathway and the methylerythritol phosphate (MEP) pathway in vivo. Ensuring an adequate donor supply is one of the limiting conditions of prenylation. Besides the optimisation of the natural donor synthesis pathway, one step phosphorylation of dimethylallyl alcohol by acid phosphatase and isopentenylphosphate kinase with ATP as high-energy phosphate donor offers a simplified pathway to improve the efficiency of prenylation.216
Glycosylation is a major structural modification for flavonoids to increase their solubility, reduce toxicity, and improve bioavailability. Glycosylation takes place mainly on the multi-hydroxyl groups of the flavonoid structure (3-OH, 5-OH, 7-OH, 3′-OH, 4′-OH and 5′-OH) with glucose, mannose or galactose and their 6-deoxy derivatives, arabinose, apiose, and xylose (Scheme 1K). Some dideoxyhexosides, such as pyranoside and bovino pyranoside, have also been reported as sugar moieties.217 In addition, the carbon atoms of the benzene ring can also be glycosylated to form C-glycosides. Glycosylation is mainly catalysed by glycosyltransferases (GTs), which generally have high regio- and stereo-selectivity towards donors and acceptors. Therefore, mining novel GTs in whole genomes and the CAZy database (http://www.cazy.org) via bioinformatic methods is the main concept to find novel enzymes with specificity towards flavonoids.218 Meanwhile, protein engineering has been carried out on GTs to excise the transmembrane domain of GTs to improve soluble expression, optimize the substrate binding pocket to extend substrate scope and improve the efficiency of glycosyl transfer, and to reduce the flexibility of enzyme structures to improve the stability of GTs.218 The use of transglycosylation activity catalysed by glycosidases is another way to achieve the O-glycosylation of flavonoids. Recently, an amylosucrase obtained from Deinococcus geothermalis (DgASase) exhibited its unique transglycosylation activity towards various hydroxyflavones and hydroxyflavanones with high site specificity at the 6-OH and 4′-OH positions, leaving the 3-OH and 7-OH positions unchanged.219 This provides a reference for catalytic mechanism and glycosylation site specificity for predicting and screening more glycosidases. On the other hand, as more C-glycoside flavonoids have been discovered from plants and show great medicinal potential, C-glycosylation has become a hot topic in the study of flavonoid glycosylation. Some C-GTs derived from plants and fungi were cloned and confirmed to catalyse the C-glycosylation of flavonoids at positions C6 and C8 (Scheme 1K).220–225 Very recently, a promiscuous C-GT from Aloe barbadensis was identified to be capable of C-glycosylating scaffolds lacking an acyl group. With dihydrochalcones as substrates, di-C-glycosylation can occur at the C6 and C8 positions.226 Remarkably, a promiscuous C-glycosyltransferase from Trollius chinensis can accept multiple structures of flavones, flavonols, flavanones, flavanonols and dihydrochalcones, and introduce a glycosyl moiety at the C8 position. Meanwhile, it showed O-glycosylation activity on the C7 position when the C8 is already substituted by methoxyl or prenyl moieties.220 The study of the catalytic mechanism and site mutagenesis at two positions (I94E and G284K) switched its C- to O-glycosylation, which provides an important reference for the rational design and directed evolution of C- to O-GTs for synthetic purposes.220
As natural products with the most extensive pharmacological activity, the potential medical use of flavonoids has recently been expanded to treat infections by the coronavirus.227 The discovery of each enzymatic step in the natural synthesis of flavonoids and the replacement of designed de novo enzymatic reactions/cascades are completing the map of heterologous flavonoid synthesis. Under the guidance of the Design–Build–Test–Learn (DBTL) concept and the application of ML,227 the construction of in vitro biosynthesis and establishing new cell factories for flavonoid syntheses provides an efficient biosynthesis program for natural flavonoids and their novel structural derivatives.
Alkaloids
Alkaloids are a large class of nitrogen-containing natural products with the heterocycle nitrogen atom derived from an amino acid (e.g., tyrosine, tryptophan, lysine or ornithine).228–230 The majority of alkaloids originate higher plants with natural functions to interact with other organisms. Thus, many alkaloids are privileged compounds exhibiting biological and pharmacological activities (e.g., analgesic, anticancer, antibacterial, stimulant, etc.). Although the majority of important alkaloids are currently produced and extracted from the native or engineered plants or plant tissue culture, it is more promising to produce them in heterologous microbes (in vivo) or in vitro with biosynthetic enzymes because of (i) advanced technologies for engineering and optimising microbial production; (ii) much cleaner targeted products with less byproducts, and (iii) the potential to access analogues and derivatives.231–236 The recent advances of enzyme discovery/engineering and synthetic biology for several important groups of alkaloids are discussed below.Benzylisoquinoline alkaloids (BIAs) are one of the most important plant alkaloids derived from tyrosine or phenylalanine. Because of several important drugs, including morphine, codeine, berberine and noscapine, the biosynthesis pathways of BIAs have been intensively investigated and almost all the key steps were elucidated in opium poppy recently (Scheme 2A).237 In brief, dopamine and 4-hydroxyphenylacetaldehyde (both derived from tyrosine) undergo a Pictet–Spengler reaction with a Norcoclaurine synthase (NCS) to yield (S)-Norcoclaurine, the first committed intermediate in the BIAs biosynthesis. (S)-Norcoclaurine is subjected to a hydroxylation and three methylation steps to form (S)-reticuline, the pivotal intermediate of many BIAs. The further synthesis of key BIAs branches here. For the synthesis of berberine and noscapine, a Berberine bridge enzyme (BBE) catalyses the oxidative C–C bond formation to give (S)-scoulerine, which is further transformed into berberine or noscapine via enzymes from a 10-gene cluster in opium poppy.238 For the synthesis of morphine, (S)-reticuline is epimerised to (R)-reticuline by a unique (S)- to (R)-reticuline epimerase (STORR) with a fused cytochrome P450 monooxygenase (CYP) domain and a reductase domain, which was revealed by three research groups in 2015.239–241 (R)-Reticuline is transformed to salutaridine via a C–C phenol-coupling mediated by salutaridine synthase (SalSyn), and then further converted to thebaine and morphine via several tailoring enzymes.242 The almost full elucidation of enzymes in the biosynthetic pathway of several BIAs has significantly facilitated the engineering of fast-growing microbes for efficient heterogenous production.243
 | ||
| Scheme 2 Novel enzymes, enzymatic mechanisms and cascades for the synthesis of benzylisoquinoline alkaloids and related tetrahydroisoquinolines. NCS: norcoclaurine synthase; BBE: berberine bridge enzyme; STORR: (S)- to (R)-reticuline epimerase; SalSyn: salutaridine synthase; TfNCS: norcoclaurine synthase from Thalictrum flavum; IR45: imine reductase from Streptomyces aurantiacus; CNMT: coclaurine N-methyltransferase; IRED: imine reductase; MAO-N: monoamine oxidase from Aspergillus niger; CHAO: cyclohexylamine oxidase; DAAO: D-amino acid oxidase; DpkA: piperidine-2-carboxylate reductase from Pseudomonas putida. | ||
In 2015, the Smolke group reported the complete biosynthesis of opioids from glucose in yeast, a major milestone in heterogenous BIA production.240 In total, more than 20 enzymes from plants, mammals, bacteria, and yeast were over-expressed to access thebaine and hydrocodone. Although the final titres of thebaine and hydrocodone were only on the 0.3–6.4 μg L−1 scale, it represents a ground-breaking advance for the total biosynthesis of complex natural products. One year later, the total biosynthesis of opiates was also achieved via stepwise conversion using four engineered E. coli strains giving thebaine in 2.1 mg L−1.244 For the other branch of BIAs, the Smolke group reported the first total biosynthesis of the anticancer drug noscapine (2.2 mg L−1) in yeast via expression of >30 enzymes from various sources.245 In addition, by feeding 3-halogenated tyrosines, the yeast produced several 8-halogenated (S)-N-methylcoclaurines and (S)-reticulines. A much more practical synthesis of the BIA intermediate was reported very recently: 4.6 g L−1 of (S)-reticuline was successfully produced from sugar via extensive engineering of yeast and using more efficient key enzymes (e.g., NCS).246 Furthermore, by feeding dopamine and different L-amino acids (precursors for aldehydes) to a simplified version of yeast, an array of non-natural tetrahydroisoquinolines (THIQs) were produced, illustrating the broad substrate scope of NCS and methylation enzymes.
Besides engineering BIA pathways in heterogenous hosts, many enzymes (especially the C–C bond forming NCS and BBE) in the biosynthetic pathway could be engineered and evolved for the in vitro synthesis of novel THIQs.247–249 NCS is well-known for its broad scope for accepting different aldehydes to give THIQs. In 2017, Lichman et al. discovered that the TfNCS from Thalictrum flavum catalysed the Pictet–Spengler reaction between dopamine and ketones, leading to novel chiral 1,1′-disubstituted and spiro-THIQs (Scheme 2B).250 The 1,1′-disubstituted THIQ was featured with a chiral quaternary carbon centre, which is challenging to form in organic chemistry and unattainable through imine reductases (IREDs) or monoamine oxidases (MAOs).251 Several variants of TfNCS were explored to ensure high conversion and preparation of these unique THIQs. The Ward and Hailes groups continued to explore TfNCS and its variants for kinetic resolution of α-methyl aldehydes leading to (1S,1′R)-THIQs with two chiral centres in a single step (Scheme 2C).252 The broad scope (aldehyde and ketone), enantioselectivity and mechanism of NCS were investigated and explained in a recent quantum chemical study.253 Furthermore, NCS was combined with other enzymatic or chemical transformations as in vitro (chemo)-enzymatic cascades for the synthesis of THIQ analogues: (i) a carboligase-transaminase-NCS cascade to access chiral 1,3,4-trisubstituted THIQs;254 (ii) an NCS-catalysed Pictet–Spengler reaction and Na2CO3-mediated cyclisation to afford (S)-trolline;255 (iii) a network of tyrosinase, decarboxylase, transaminase and NCS for efficient synthesis of several natural and non-natural BIAs.256 Besides NCS, another important C–C bond forming enzyme, BBE, has been explored for synthetic purposes, such as preparation of (S)-scoulerine and its analogues via kinetic resolution257 or deracemization258 of the corresponding THIQs, and enantioselective dealkylation of N-ethyl THIQs.259
Given the importance of the chiral THIQ scaffolds, many other biocatalytic approaches (besides NCS and BBE in the BIA pathway) have also been developed. One facile approach is direct asymmetric reduction of chemically synthesised 3,4-dihydroisoquinolines (DHIQs) with natural IREDs260,261 or artificial transferhydrogenases.262,263 Many natural IREDs were able to enantioselectively reduce DHIQs with a 1-methyl- or simple 1-alkyl substituent.264 To produce chiral bulky 1-aryl-THIQs, the Qu group assayed a large number of diverse IREDs and found several (R)-selective IREDs and one unique (S)-selective IRED (IR45) converting chloro-, methyl-, and methoxyl-benzyl DHIQ into the corresponding (R)- or (S)-THIQ in high-to-excellent conversions and optical purities.265 To access plant-sourced alkaloids, they further engineered IR45 to improve its activity and combined it with coclaurine N-methyltransferase (CNMT) to achieve the one-pot synthesis of five N-methyl THIQ alkaloids (Scheme 2D).266 The THIQ analogue, 1-benzyl-1,2,3,4,5,6,7,8-octahydroisoquinoline (1-benzyl-OHIQ), is an important synthon for synthetic morphinan drugs (Scheme 2E). Recently, the Zhu group identified two IREDs with complementary enantioselectivity to produce (S)- or (R)-1-benzyl-OHIQs in high optical purity and yield from the corresponding imines.267
Another approach for accessing chiral THIQs is via enantioselective oxidation with MAO.268,269 Although the natural substrates for MAO are usually small primary amines, the Turner group had pioneered in engineering MAO-N from Aspergillus niger (A. niger) for bulky secondary and tertiary bulky amines, such as 1-phenyltetrahydroisoquinoline (1-phenyl-THIQ, Scheme 2F).270 By enantioselective oxidation with MAO-N D11 and simultaneous reduction with BH3-NH3, racemic 1-phenyl-THIQ was deracemised to (S)-1-phenyl-THIQ (a precursor for Solifenacin) in excellent optical purity and yield. Reetz et al. simultaneously engineered the entrance tunnel and active site of MAO-N for efficient deracemization of several 1-substituted THIQs.271 Recently, the Hilvert group applied a UHTP microfluidic assay for single-round remodelling of a cyclohexylamine oxidase (CHAO).54 A highly active CHAO variant was obtained for the synthesis of several (S)-1-substituted THIQs via deracemization. For the 1- and 3-carboxyl-THIQs, a D-amino acid oxidase (DAAO) was successfully employed for deracemization.272 Furthermore, by combining DAAO-catalysed oxidation and a reductase (DpkA)-mediated reduction, the Wu group developed a fully biocatalytic deracemization process to produce (S)-1-carboxyl-THIQs in excellent enantiomeric excess (e.e.) and yield (Scheme 2G).273 This is similar to a previously developed MAO-artificial transferhydrogenase system for deracemization of simple THIQs.274
Another very famous natural THIQ alkaloid, Colchicine, is a potent microtubule inhibitor used for the treatment of inflammatory disorders as well as a research tool for many years. Early feeding studies on Colchicum plants suggested its biosynthesis from tyrosine and phenylalanine,275 but most of the enzymes remained mysterious until very recently. The Sattely group applied metabolomics, transcriptomics and heterologous expression to fully elucidate the near-complete biosynthetic pathway of colchicine in Gloriosa superba (Scheme 3).276 In brief, dopamine (from Tyr) and 4-hydroxydihydrocinnamaldehyde (from Phe) were condensed to a 1-phenethylisoquinoline scaffold, which undergoes methylations and hydroxylations to (S)-autumnaline. Next, P450-catalysed phenol coupling created the bridged tetracyclic isoandrocymbine, which was further subjected to methylation and a unique P450-catalysed oxidative ring expansion to generate N-formyldemecolcine with the hallmark tropolone ring. Further N-modifications gave colchicine. In this ground-breaking study, they not only elucidated the enzymes, but also reconstituted the pathway to N-formyldemecolcine in heterologous Nicotiana benthamiana.
 | ||
| Scheme 3 Novel enzymes, enzymatic mechanisms and cascades for the synthesis of Colchicine. NCS: norcoclaurine synthase; P450: cytochrome P450 monooxygenase. | ||
Monoterpenoid indole alkaloids (MIAs) are another very important class of alkaloids, including the anti-cancer drugs vincristine, camptothecin, anti-arrhythmic ajmaline, and anti-malarial quinine. The key intermediate of MIAs, strictosidine, is constructed from tryptophan-derived tryptamine and monoterpenoid secologanin via a C–C bond-forming strictosidine synthase (STR).277 This key intermediate undergoes different transformations to several sub-classes of MIAs (Scheme 4A). In the pathway to vincristine and vinblastine in Catharanthus roseus, the conversion of strictosidine to tabersonine and catharanthine was still mysterious (it involves many unstable intermediates) until very recently, the final missing enzymes were fully elucidated by two groups.278–280 Tabersonine is further converted by seven enzymes to vindoline, which is coupled with catharanthine to form vinblastine and vincristine by a peroxidase. Although the main research focus of MIAs was still identification of enzymes and elucidation of pathways in native plants, several studies managed to reconstitute parts of the pathway in yeast. The O’Connor group introduced >20 different genes (including 14 from the MIA pathway) into yeast, and de novo produced the key intermediate strictosidine at ∼0.5 mg L−1.281 For the downstream part, the De Luca group discovered seven enzymes and reconstituted the pathway from tabersonine to vindoline (up to 2.7 mg L−1) in yeast.282 It is still very challenging to reconstitute the whole pathway of complex MIAs in yeast, yet several halogenated derivatives of MIAs have been cleverly accessed by introducing bacterial tryptophan halogenases into the hairy root culture of C. roseus.283
 | ||
| Scheme 4 Novel enzymes, enzymatic mechanisms and cascades for the synthesis of monoterpenoid indole alkaloids. STR: strictosidine synthase. | ||
The strictosidine synthase (STR) in the biosynthesis pathway of MIAs has recently been explored in the synthesis of 1-substituted tetrahydro-β-carbolines (THBCs) by the group of Kroutil. Different from the natural (S)-strictosidine produced from tryptamine and secologanin, replacement of secologanin with several simple aliphatic aldehydes produced (R)-1-alkyl-THBCs in medium to high optical purity by several STRs (Scheme 4B).284 The STR-reaction was combined with a chemical reduction to achieve a facile two-step synthesis of (R)-harmicine. The switch of enantioselectivity was explained as the inverted binding of short-chain aliphatic aldehydes in STR through a structural and computational study.285 They further employed a substrate-walking strategy to engineer the STR to accept benzaldehydes and produce (R)-1-aryl-THBCs.286 Besides STR, IREDs have been used to produce simple 1-methyl-THBCs from the corresponding imines287 and MAO-N mediated deracemization has been explored to access a variety of chiral 1-substituted-THBCs.288
Terpenoids
Terpenoids (isoprenoids) consisting of C5 isoprene units are the most diverse group of natural products, with more than 80000 known structures to date.289,290 They are prevalent in all kingdoms of living organisms, and many of them possess indispensable biological functions and activities (e.g., light-harvesting, electron transfer, membrane constituents) with broad potential applications as fragrances, nutraceuticals, pharmaceuticals, etc. Despite their astonishing structural diversity and widespread occurrence, the canonical biosynthetic route of terpenoids is highly modular (Scheme 5):291,292 two key C5 precursors, isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP), are synthesised from common primary metabolites via either the MEP (methylerythritol phosphate) pathway in bacteria and plant plastids or the MVA pathway in animals, fungi, and the cytosol of plants;293 IPP and DMAPP are assembled by prenyltransferases to give polyisoprenoid diphosphates, such as geranyl diphosphate (GPP, C10), farnesyl diphosphate (FPP, C15), or geranylgeranyl diphosphate (GGPP, C20); polyisoprenoid diphosphates are usually cyclised with terpene synthases to generate terpenoid skeletons;290 structures are further tailored with various enzymes, such as P450s.294 Due to the importance of many terpenoids and existence of native MEP/MVA pathways in microbes, terpenoids have been very popular targets in metabolic engineering and synthetic biology for more than 20 years.295–298 Several early studies focused on engineering E. coli to produce lycopene and carotenoids due to easy detection and lack of complex tailoring.299 The engineering of S. cerevisiae to produce artemisinic acid (precursor for the anti-malarial drug artemisinin) by the Keasling group and Amyris is a landmark in metabolic engineering/synthetic biology.300 More recent progress includes engineering S. cerevisiae for the production of farnesene (bulk chemical)301 and tocotrienols (vitamin E),302 as well as engineering E. coli for the production of taxadiene (precursor for Taxol)303 and viridiflorol (fine chemical).304 However, achieving a productive synthesis of terpenoids (e.g., >10 g L−1) in microbes is often very challenging and requires extensive engineering and optimisation efforts due to the toxicity of intermediates/products and the complex regulation of native pathways.
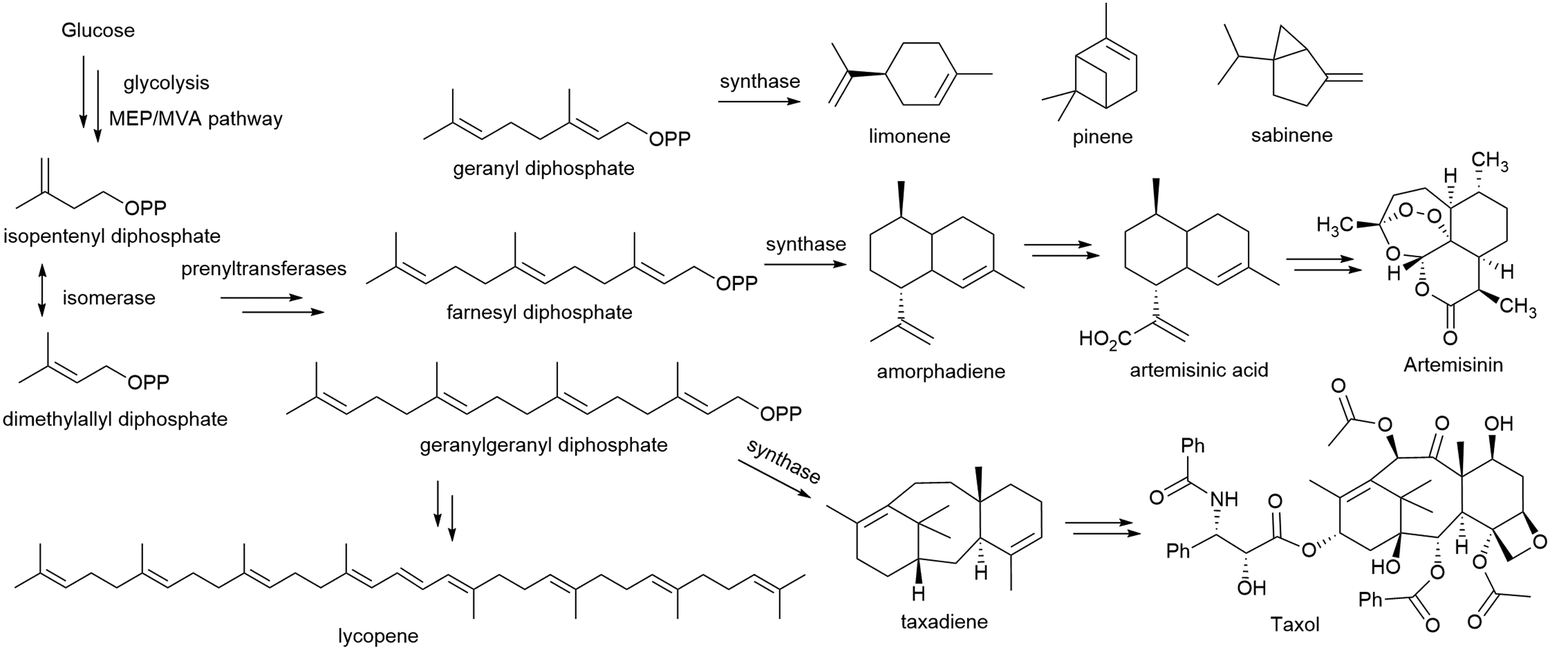 | ||
| Scheme 5 Novel enzymes, enzymatic mechanisms and cascades for the synthesis of terpenoids. MEP: methylerythritol phosphate; MVA: mevalonate. | ||
Complementary to the biosynthesis of terpenoids in microbes, cell-free in vitro multi-enzymatic synthesis avoids many problems in cells (e.g., toxicity, complex regulation), minimizes side reactions and genetic engineering efforts, and often offers higher yields of final products in a cleaner reaction system.305–308 These advantages were clearly demonstrated in the pioneering work on the cell-free one-pot production of monoterpenes from glucose by the Bowie group.309 Simply combining standard Embden–Meyerhof–Parnas glycolysis and the MVA pathway leads to imbalance of cofactors: the glycolysis generates ATP and NADH (excess), while the MVA pathway consumes ATP and NADPH. The authors cleverly tackled this issue by creating enzymatic purge valve nodes:310 an NAD+-utilizing glyceraldehyde-3-phosphate dehydrogenase (GAPDH), an NADP+-utilizing mutated GAPDH and an NADH oxidase. Mathematic modelling was used to identify the potential bottlenecks (hexokinase, pyruvate dehydrogenase, and phosphate), and these key parameters were experimentally optimised. With careful consideration and optimisation, the final in vitro system comprising 27 enzymes converted glucose (500 mM) to limonene (12.5 g L−1) with a theoretical yield of 88% over 7 days. By replacing the terpene synthase, pinene and sabinene were also produced at 14.9 and 15.9 g L−1 and almost quantitative yields, respectively. These product titres are at least 10 times higher than those from microbial production, far exceeding the toxicity limits of these compounds. In vitro multi-enzymatic synthesis has been reported for other terpenoids, such as the production of amorphadiene from mevalonic acid (6 steps with ATP recycling)311 and the production of geosmin and patchoulol from acetic acid (10 steps with cofactor regeneration).312 Cell-free in vitro multi-enzymatic synthesis has made great progress recently, but it is still difficult to work on some complex and difficult enzymes (e.g., membrane enzymes, P450s), and large-scale application is hampered by relatively high costs (e.g., enzyme purification, cofactors).
Currently, the biosynthetic pathways for the majority of natural terpenoids have not been fully elucidated. Thanks to the advances in genomic sequencing and bioinformatics, many putative terpenoid synthetic enzymes could be identified in silico. To verify and characterize these putative enzymes, heterologous expression in suitable (engineered) hosts could often enable efficient production of terpenoid products for characterisation. For rather simple and small bacterial terpenes, heterologous expression in E. coli is often sufficient for rapid and facile characterisation.313,314 While for more complex plant-origin terpenoids, heterologous expression of the enzymes in yeast or plants is necessary.315–317 For example, plant diterpene labdanes and clerodanes are often synthesised by a pair of distinct monofunctional class I and class II diterpene synthases (diTPSs). By mimicking the modular diterpene biosynthesis, the Hamberger group tested every combination of 9 class I and 11 class II diTPS from 10 plant species in N. benthamiana by A. tumefaciens-mediated transient expression.318 51 Diterpene skeletons were stereo-selectively biosynthesised, including 41 new-to-nature ones. By engineering S. cerevisiae, four useful diterpenes were produced at a scale relevant for industrial applications. To quickly access highly diverse oxygenated plant triterpenes (>20000 reported so far),319 the Osbourn group developed a translational synthetic biology platform based on transient expression in the whole plant of N. benthamiana (Scheme 6).320 Initially, a feedback-insensitive version of an HMG-CoA reductase (tHMGR) in the MVA pathway was found to significantly boost the production of β-amyrin when co-expressing with the oat β-amyrin synthase (SAD1) in N. benthamiana. To provide a gram-scale synthesis of triterpenes, the authors developed a vacuum agro-infiltration system for transient expression in the whole plant rather than individual leaves. Co-expression of tHMGR and SAD1 in about 460 N. benthamiana plants and cultivation for 5 days allowed successful isolation of 800 mg of β-amyrin with >98% purity. This platform (tHMGR and SAD1 in N. benthamiana) was combined with one or a pair of five different β-amyrin-oxidising P450 enzymes to offer 41 different oxygenated triterpenes (some new-to-nature). A handful of them were isolated on 10 mg scale and further evaluated for antiproliferative and anti-inflammatory activities. Besides the proper functional expression of plant enzymes, another advantage of transient expression in N. benthamiana is that multiple genes can be co-expressed by simply co-infiltrating multiple A. tumefaciens strains. This feature allows a quick test of combinations of enzymes either to generate new products (as shown in the two examples above) or to elucidate the biosynthetic pathway, which was demonstrated in a recent study of root triterpenes in A. thaliana.321
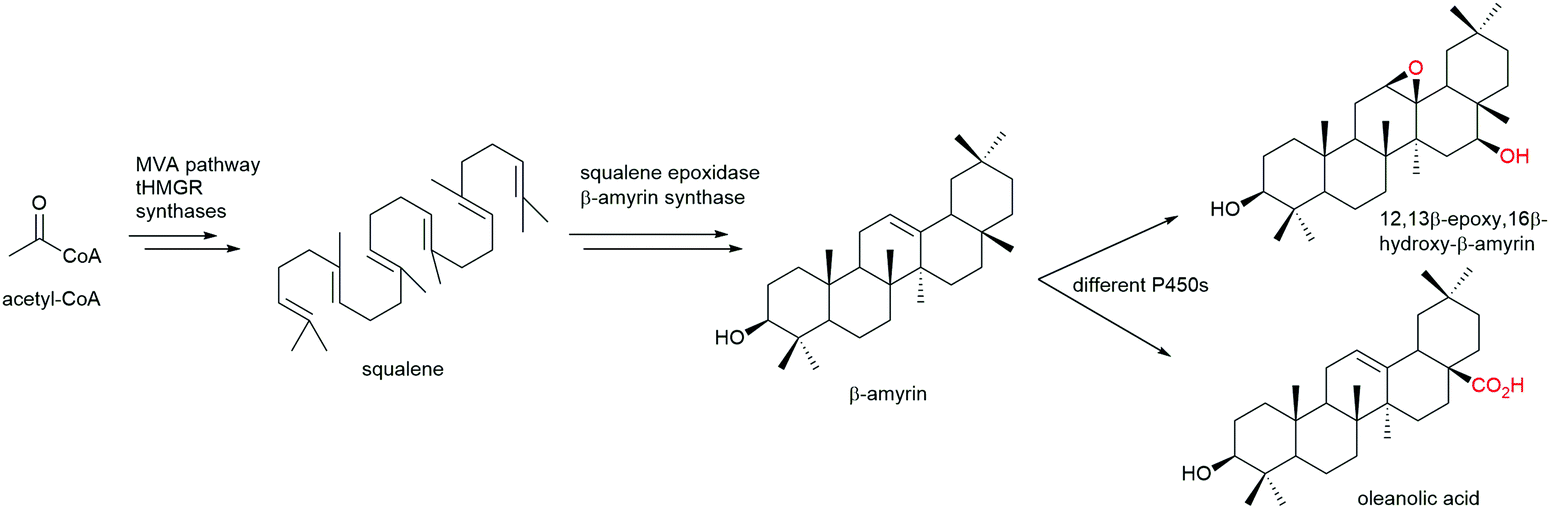 | ||
| Scheme 6 Novel enzymes, enzymatic mechanisms and cascades for the synthesis of oxygenated plant triterpenes. tHMGR: feedback-insensitive version of HMG-CoA reductase; P450: cytochrome P450 monooxygenase. | ||
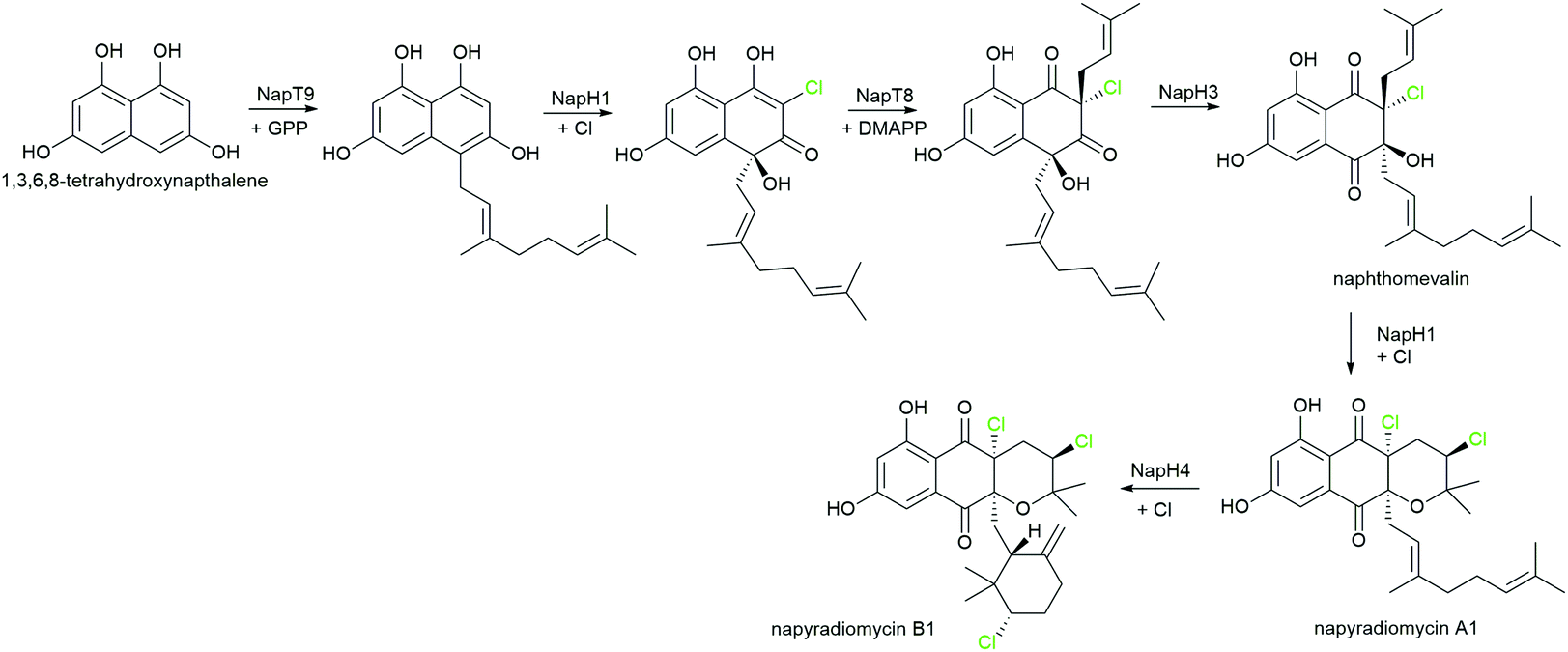 | ||
| Scheme 7 Novel enzymes, enzymatic mechanisms and cascades for the synthesis of Napyradiomycins. GPP: geranyl diphosphate; DMAPP: dimethylallyl diphosphate; NapH1, H3, and H9: vanadium-dependent haloperoxidases; NapT8 and T9: aromatic prenyltransferases. | ||
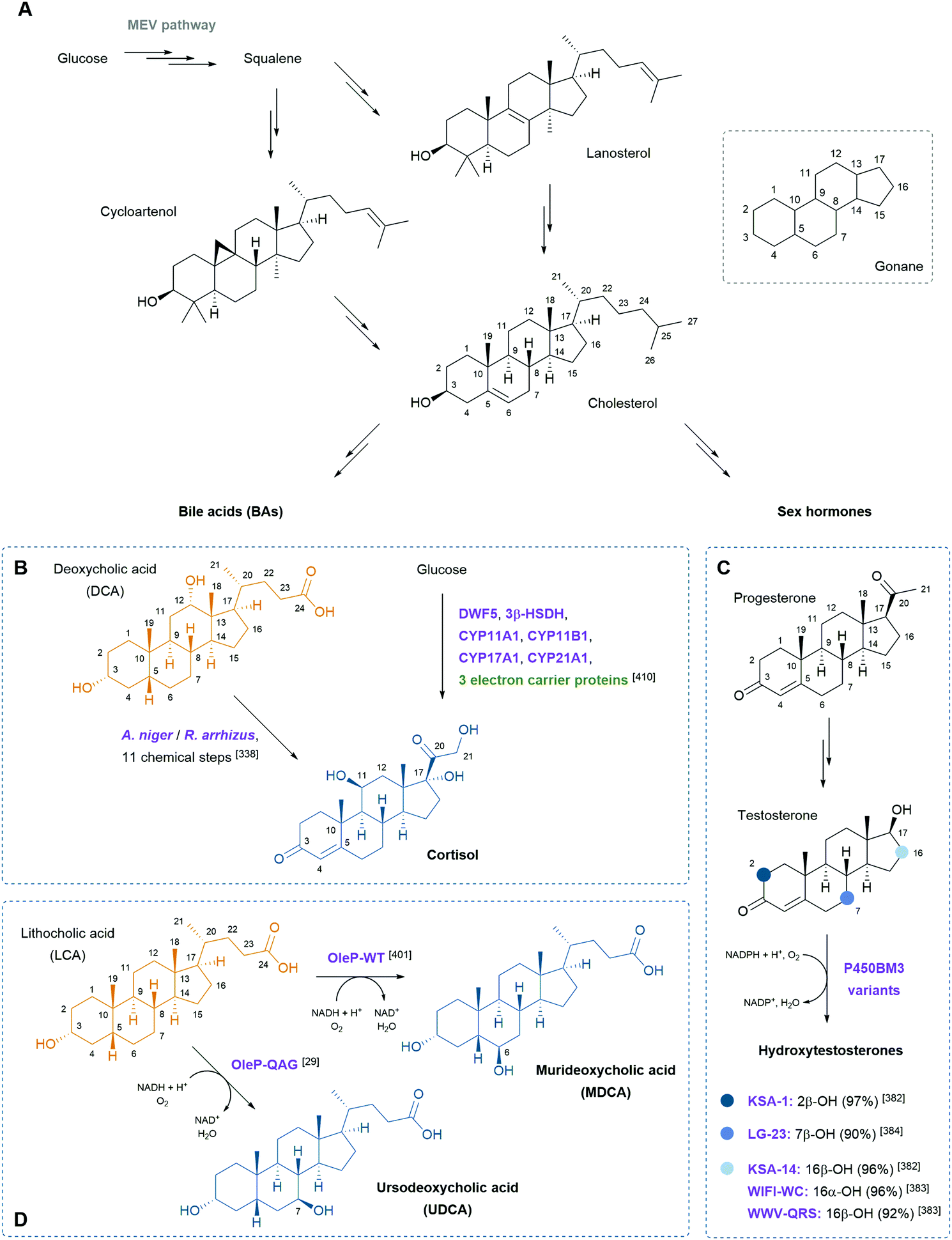 | ||
| Scheme 8 Steroidal precursors and APIs. (A) The biosynthesis of steroids through the MEV pathway leads to precursors like lanosterol, cycloartenol, and cholesterol, an important intermediate towards BAs and sex hormones. (B) Cortisol production from DCA involving fermentations with A. niger, R. arrhizus, and 11 chemical steps in total or from glucose through a heterologous pathway mimicking human steroid biosynthesis. DWF5: 7-dehydrocholesterol reductase. (C) Progesterone and testosterone are important APIs; the latter can be hydroxylated by P450BM3 variants in the positions 2, 7, and 16 (shades of blue), for example. Regiospecificity given in parentheses for KSA-1, KSA-14, and LG-23; stereospecificity for WIFI-WC and WWV-QRS. (D) OleP wild-type (OleP-WT) exclusively yields MDCA from LCA by 6β-hydroxylation. The triple mutant OleP-QAG yields the 7β-product UDCA, a reaction new to the biocatalytic toolbox of single enzymes. Important steroid substrates are numbered and modifications in target products indicated. Structures of BA precursors are highlighted in orange, products in blue, key enzymes in purple and accessory proteins in green. | ||
Merochlorin A and B are common C4-prenylated meroterpenes, while certain vanadium-dependent haloperoxidases mediate an α-hydroxyketone rearrangement, leading to naphthomevalin with a unique C3-prenylation pattern (see also Section Meroterpenoids, Scheme 14).322 These enzymes were recently employed for total enzymatic syntheses of antimicrobial and cytotoxic meroterpenoids, napyradiomycin A1 and napyradiomycin B1 from 1,3,6,8-tetrahydroxynapthalene, GPP and DMAPP (Scheme 7).323 By applying two aromatic prenyltransferases (NapT8 and T9) and two vanadium-dependent haloperoxidases (NapH1 and H3) in one pot, napyradiomycin A1 was synthesised in 22% yield. With the addition of vanadium-dependent haloperoxidase NapH4, napyradiomycin B1 was synthesised in 18% yield.
Besides the vanadium-dependent haloperoxidases, other halogenating enzymes324–327 could also provide plenty of opportunities in the enzymatic syntheses of terpenoids (and other natural products) as well as their derivatives.
Steroids
Steroids are a large group of biologically active compounds that share a common structure typically containing 17 carbon atoms fused in a tetracyclic system known as gonane (Scheme 8). This framework derives from the MEV pathway, where isoprene units are linked and the cyclisation of the triterpenoid squalene leads to basic precursors like lanosterol (in animals and fungi) and cycloartenol (in plants),328–330 but alternative biosynthetic pathways exist in bacteria and certain plants that yield the steroid core (Scheme 8).330–332 Besides being integral components of cellular membranes, steroids function as signaling molecules in metabolic pathways, cellular survival, reproduction, and disease. To precisely execute these diverse biological roles, (subtle) modifications of the four-ring scaffold are introduced by an equally diverse subset of enzymes with high stereo- and regio-selectivity that is hard to achieved by chemical synthetic methods. Hence, the over 250 known natural steroid compounds differ in functional group decoration and the oxidation state of the steroid nucleus and many of them are considered essential precursors for the manufacturing of drugs exhibiting antifungal, antimicrobial, antiviral, immune-modulating, antitumor, and anticonvulsant activities.333 Indeed, steroid-based drugs represent the second-highest marketed category of APIs – after antibiotics – with the annual global market exceeding 10 billion USD and more than 300 clinically approved natural and (semi-)synthetic steroidal compounds.334–336Traditionally, steroidal APIs have been synthesised through chemical processes that are characterised by the requirement for multiple sequential steps that offer only poor control over the stereo- and regioselectivity and very low yields.337 In the 1950s, the corticosteroid hormone cortisone was synthesised from the bile acid (BA) deoxycholic acid (DCA) over 31 steps with a yield of 0.16%. By including a fermentation step with the fungi Rhizopus arrhizus and A. niger, the number of chemical steps could be reduced to 11, markedly reducing production costs (Scheme 8).338 To date, many typical steroidal APIs are manufactured chemically but involve microbial biotransformations for the preparation of key intermediates335,337,339 or the late-stage functionalisation of steroids.340–343 The latter regularly involves stereo- and regioselective hydroxylations by P450s and will be described with focus on recombinant applications below.333,340,344,345
Although microorganisms have long been used for precursor synthesis and steroid modifications, fully microbial processes featuring efficient platform strains or recombinant microbial cell factories are scarce. Limitations are directly caused by the intrinsic properties of steroidal compounds such as low solubility in aqueous media and cellular toxicity.29 Some of these issues have been addressed by the emulsification of substrates with surfactants or the utilization of two-phase systems with organic solvents.333,339,346 Furthermore, steroid transformations in well-characterised recombinant host cells (e.g., E. coli, S. cerevisiae) regularly yield low product titres simply because steroid-modifying enzymes only poorly express or function outside their native hosts, combined with insufficient substrate uptake and unintended metabolism.75,335,347
In the following, the bio-based synthesis of steroid precursors and (recombinant) functionalization will be highlighted as well as trends that project towards the de novo synthesis of steroidal APIs and their customization for future applications.
Important substrates, in the context of microbial production of API precursors, are sterols.330,335 This sub-group of steroidal compounds, bearing a 3-hydroxyl group, include cholesterol, lanosterol, phytosterol, ergosterol, and BAs like DCA and lithocholic acid (LCA; Scheme 8). Sterols are intermediates of both anabolic and catabolic steroid pathways and are available from plant-based and animal feedstocks for biotechnological applications.335,348–352 Various strains from the genera Mycobacterium, Nocardia, and Rhodococcus were identified to transform sterols and, importantly, have been metabolically engineered to reroute fluxes towards the accumulation of value-added steroids. Examples feature the production of C-19 steroids such as 4-androstene-3,17-dione (AD), 1,4-androstadiene-3,17-dione (ADD), and testosterone in mutant strains of Mycobacterium smegmatis (M. smegmatis) starting from cholesterol. Single and multiple gene deletions including kstD and ksh, encoding 3-ketosteroid-Δ1-dehydrogenase and 3-ketosteroid-9α-dehydrogenase, accumulated AD and ADD, respectively; the heterologous overexpression of 17β-hydroxysteroid dehydrogenases (HSDHs) from different (bacterial) sources successfully converted AD into testosterone.339,353
Whereas the biotransformation of sterols into AD requires multiple reaction steps carried out by endogenous host enzymes, targeted functionalization usually involves one-step transformations by the activity of a single (engineered) enzyme. Industrially relevant steroid modifications are Baeyer–Villiger oxidations,354,355 hydrogenation and dehydrogenation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–C bonds, respectively,337,356–358 and alcohol/carbonyl group interconversions.359–362 Several wild-type organisms are used at industrial scales to perform these functionalisation reactions.333,335,363
C and C–C bonds, respectively,337,356–358 and alcohol/carbonyl group interconversions.359–362 Several wild-type organisms are used at industrial scales to perform these functionalisation reactions.333,335,363
Of particular interest are direct hydroxylations of inert C–H bonds as carried out, for example, by P450s, a superfamily of heme-containing enzymes.333,344,345,364,365 P450s can execute an impressive variety of other reactions340,366–368 and were even engineered to perform a set of ‘new-to-nature’ reactions.369–372 To direct the biological activity of steroid drugs, mainly their stereo- and regioselective hydroxylation activities are of interest.344 To name two, a hydroxyl function at position 11β is required for the anti-inflammatory activity of cortisol and prednisolone373 and the presence of two hydroxyl groups – 1α and 25α – is essential for the biological activity of vitamin D derivatives.374 Integral parts to customise hydroxylation activities have been the many well-established protein engineering techniques, exemplarily highlighted in the following.
The CYP enzyme P450BM3 from Bacillus megaterium (CYP106A2) was amongst the first bacterial steroid hydroxylases characterised375 and hydroxylates multiple pharmaceutically relevant steroids including cortisol, progesterone, and testosterone predominantly at the 15β position.376 P450BM3 is a self-sufficient CYP and, as such, does not require additional redox partner proteins for the transfer of electrons required for catalysis.364 It has been the target of numerous protein engineering studies (see section Terpenoids), not only to enhance the physiological 15β-hydroxylation activity but to invert stereoselectivities and shift regioselectivity. The group of Reetz has published thorough research on these topics, heavily employing directed evolution strategies including the combinatorial active site saturation test (CAST)377 and iterative site saturation mutagenesis (ISM).7,378,379
In two prominent examples, CASTing was used to transform the previously identified P450BM3 F87A mutant,380,381 which hydroxylates testosterone at the positions 2β and 15β with low selectivity, into biocatalysts with nearly perfect regioselectivity. Variants with the additional mutations A330W (KSA-1) and R47Y/T49F/V78L/A82M (KSA-14) catalysed the 2β- and 16β-hydroxylations of testosterone with 97% and 96% regioselectivity, respectively.382 In a subsequent study, ISM – based on information from mutability landscapes, molecular dynamics simulations, and X-ray crystallography – was used to generate P450BM3 variants with exquisite regio- and stereoselective hydroxylation activities for testosterone and four other steroids at the C16 position.383 Whereas the mutant WIFI-WC (combining the mutations R47W/S72I/A82F/F87I and Y51W/L181C from two distinct libraries after three rounds of ISM) produced 16α-hydroxy testosterone with 96% stereoselectivity, WWV-QRS (combining R47W/A82W/F87V and L181Q/T436R/M177S) produced the 16β-stereoisomer with 92% selectivity.
Most recently, the group of Wong demonstrated the crucial roles of glycine mutations in P450BM3 for different substrate binding orientations, resulting in a variant library capable of hydroxylating AD and testosterone, for example, at a wide range of positions (C1, C2, C6, C7, C15, and C16) with up to 97% selectivity.384 Very recently, Li et al. created P450BM3 mutants with 7β-hydroxylation activity towards testosterone and related steroidal compounds.385 Previously, CYP106A2 had only been described to yield the 7β-products from the steroids pregnenolone and dehydroepiandrosterone.376,386 The resulting compounds and their derivatives are considered to act as neuroprotective and anti-inflammatory agents to treat neuronal damage after stroke or trauma.387 Regarding the hydroxylation at position C7, BAs that predominantly occur in the bile of mammals,388,389 have also moved into the focus due to their clinical significance.390–392 Again, their synthesis requires tedious multi-step chemical procedures that suffer from low yields and poor control over regioselectivity.333,393,394 The synthesis of the BA ursodeoxycholic acid (UDCA) is no exception. Cholic acid (CA)351,361 or chenodeoxycholic acid (CDCA)360 were suggested as precursor molecules and the biocatalytic epimerisation of CDCA to UDCA at C7 further shortened the synthesis route and enhanced yields.395,396 However, apart from certain filamentous fungi such as Fusarium equiseti,343,397 direct hydroxylations, especially in recombinant systems, have not been described until very recently.
The CYP107D1 from Streptomyces antibioticus (OleP), which physiologically catalyses an epoxidation step in the oleandomycin biosynthesis pathway,398,399 hydroxylates testosterone at the positions 6β, 7β, 12β, and 15β.400 In contrast, BAs like LCA are hydroxylated exclusively at the 6β-position.401 Grobe et al. engineered OleP based on a semi-rational directed evolution approach and generated a triple-mutant (F84Q/S240A/V291G) with nearly perfect regioselectivity for the 7β-position.29 Hits after directed evolution were identified by a colorimetric HTP assay, which is based on the activity of a 7β-HSDH, specifically oxidizing the 7β-OH of UDCA to the corresponding ketone.359 The reaction also yields NADPH, which reduces a dye and results in an increase in absorption dependent on the concentration of UDCA.29 The assay principle offers an easy-to-implement alternative to time-consuming chromatographic methods that are currently employed to verify the success of P450 engineering approaches. Noteworthy, the heme group in CYPs has been used as ‘intrinsic chromophore’ in HTP screenings to identify potential CYP substrates (and inhibitors). Binding of a ligand causes the spin shift of the heme iron that can be detected as a signal spectrophotometrically.402–404
Although these selected examples certainly highlight the power of directed evolution to engineer CYPs to execute highly desired steroid modifications, they are typically far from industrial applications due to low yields (2% isolated yield after LCA to UDCA transformation by the best OleP mutant in E. coli co-expressing putidaredoxin and putidaredoxin reductase as redox partner proteins)29 and/or low substrate loads (1 mM testosterone for different hydroxylations in E. coli by self-sufficient P450BM3 variants).383,385 None of these studies addressed the optimisation of CYP enzyme production in vivo apart from precursor supplementation for heme production.29 Khatari et al. adjusted the stoichiometry of CYP260A1 from Sorangium cellulosum and the redox partner proteins adenoredoxin reductase (AdR) and adenoredoxin (AdX) and showed that CYP260A1 hydroxylates 11-deoxycorticosterone (11-DOC) at high ratios of the redox partners (e.g., CYP260A1:AdR:AdX = 1:3:10) mainly at the C1α-position.405 At lower ratios (CYP260A1:AdR:AdX = 1:3:5), also C1–C2-ene-11-DOC was produced in vitro. A high ratio (CYP260A1:AdR:AdX = 1:3:20) and additional recycling of NADPH mainly formed 1α-,14α-dihydroxy-11-DOC in an E. coli whole-cell biocatalyst.405,406 Besides cofactor recycling,383,407 an increase of gene copy numbers combined with genomic manipulations (integrations347,408 and deletions339,353) of target genes has been realised.335 Whereas enzyme and redox partner stoichiometry can be easily controlled in vitro, this is certainly challenging in vivo70 but should definitely be considered in future applications of CYPs. However, Khatari et al. only reached conversions up to 80% at very low substrate loads (0.2 mM of 11-DOC).405 These – to say at the least – modest performances of biocatalytic syntheses are a current and future challenge, not only of recombinant processes for steroid modifications.372,409
To date, only two recombinant processes yielding steroidal APIs have been implemented industrially. Shi et al. developed a process that converts CA to 12-oxo-CDCA, a key precursor for chemoenzymatic synthesis of UDCA, in a single step with very high productivity (68 g L−1 h−1). The responsible enzyme, a 12α-HSDH from Rhodococcus ruber, was identified using a structure-guided genome mining approach and is applied as lyophilised E. coli whole-cell powder during the process, yielding 12-oxo-CDCA.362,409
The second example is the exploitation of a metabolically engineered S. cerevisiae strain harboring an artificial biosynthetic pathway consisting of four mammalian P450s. The heterologous cascade reaction yields cortisol from glucose by mimicking human steroid biosynthesis (Scheme 8).410,411
The lack of steroid-modifying enzymes as bottleneck has been overcome with the continuous discovery of new P450s from microbial but also eukaryotic sources.362,412–417 Recently, Szaleniec et al. reviewed P450s for the degradation of cholesterol (CYP125 and CYP142 family) and steroid hydroxylations (CYP106A, CYP109, CYP154, and CYP260), as well as Rieske-type monooxygenases, 3-ketosteroid 9α-hydroxylases, and molybdenum-containing steroid C25-dehydrogenases as alternative (bacterial) steroid hydroxylases.344 The impressive successful engineering of CYPs complements the steroid hydroxylations found in nature. Together with the other useful enzymatic reactions described above, steroidal APIs have been accessed through both the application of wild-type strains and recombinant systems, yielding anti-inflammatory cortisol410,411 and prednisolone,337,373 the sex hormones testosterone and progesterone,339,353,385 derivatives thereof exhibiting neuroprotective functions,376,386,418,419 the value-added BA UDCA,29,395 as well as the biologically active (1α,25-dihydroxylated) forms of vitamin D2420,421 and D3,374,422 and many more.333,335,344,346,365,423,424 Although the optimisation of these bio-based processes has been addressed by the emerging tools from synthetic and systems biology, steroids remain ‘tough’ substrates, intermediates, and products due to their low solubility in aqueous media and cellular toxicity. However, this trend is rapidly changing since new microbial chassis for the biotransformation and functionalization of steroids including P. putida,425 different Rhodococcus sp. and related mycobacterial strains are emerging.335,426Corynebacterium glutamicum, for example, has beneficial properties for steroid biocatalysis such as efficient transport of steroidal compounds, high stress tolerance, and potentially interfering metabolic pathways are missing.427 Lastly, genetic tools have become readily available for these strains428,429 and the revolutionary CRISPR/Cas9-based recombineering tool, accelerated genomic manipulations.430,431 Hence, the development of potent microbial cell factories for the customization of steroidal APIs has never looked brighter.
Polyketides
Polyketides are a chemically rich and extremely complex class of natural products assembled by polyketide synthases from simple activated carboxylic acid building blocks.432 One way of viewing their value in nature is by observing that they are produced despite a tremendous metabolic burden, in terms of DNA and protein synthesis. Polyketide synthases can be several megadaltons in size, often several-fold larger than ribosomes.433 It is therefore not surprising that polyketides are also valuable to humans as pesticides (spinosyn A), antibiotics (erythromycin), antineoplastics (daunorubicin), immunosuppressants (FK506), antifungal (neoaureothin), antitumor (epothilone B), antiparasitic (avermectin), and cholesterol-lowering (lovastatin) drugs.434 Most of this rich diversity is produced by the modular Type I or “assembly-line” polyketide synthases.435Polyketides are an excellent example of the central theme of this review, which is to demonstrate that it is still difficult to achieve the total enzymatic synthesis of natural products without the original host organism. In this case, the reason is not merely the complexity of host metabolic pathways, but also the extreme complexity of the megasynthases themselves. Their sheer sizes make cloning and standard DNA manipulations complicated. It also makes the proteins very hard to express and fold in heterologous hosts like E. coli.436 Polyketide synthase engineering is a very promising field of study but is restricted by the same technological limitations. Furthermore, it is extremely challenging to determine the structures of complete assembly-line polyketide synthases due to their large sizes and the often-weak protein–protein interactions between modules.437–439 Overcoming the hurdles to designer polyketide synthases would be a clear sign that new trends of biocatalysis have emerged. In this section we review recent trends in PKS engineering, suggesting that there is hope despite the decades of failing to deliver on the promise of on-demand designer polyketides.
The modular Type I PKSs are commonly described as “assembly-line” complexes because each module sequentially adds a unit to the growing product so that the sequence of functional groups in the final polyketide depends on the sequence of PKS modules.434,435,437,442 Each module of an assembly-line PKS consists of at least a ketosynthase, an acyltransferase, and an acyl-carrier protein domain (Fig. 4C).443 The acyltransferase domain of a loading module first transfers an acyl group, usually from acetyl-CoA, to the phosphopantetheinyl arm of its acyl-carrier protein. The ketosynthase domain of the downstream module catalyses both translocation of the acyl group and carbon–carbon bond formation by a decarboxylative Claisen condensation of the translocated acyl group with a malonyl derivative (bound to the acyl-carrier protein as a thioester). This reaction forms a 3-ketoacyl intermediate and releases carbon dioxide, which thermodynamically drives the process.437 This intermediate may then be reduced, and further functionalised by an optional “reducing loop” composed of a keto-reductase, a dehydratase, and an enoyl-reductase.444 All these reactions are stereospecific, and the ketoreductase domains determine the stereo-configuration of the α- and β-carbon atoms of the resulting product molecules.442,444 Some modules also have methyltransferase domains and repetitions, with variation in each elongation cycle, produces polyketides of great diversity.435 Finally, a thioesterase domain cleaves the polyketide from the terminal acyl-carrier protein, often cyclizing the product to form a macrolactone.434,437,442,443
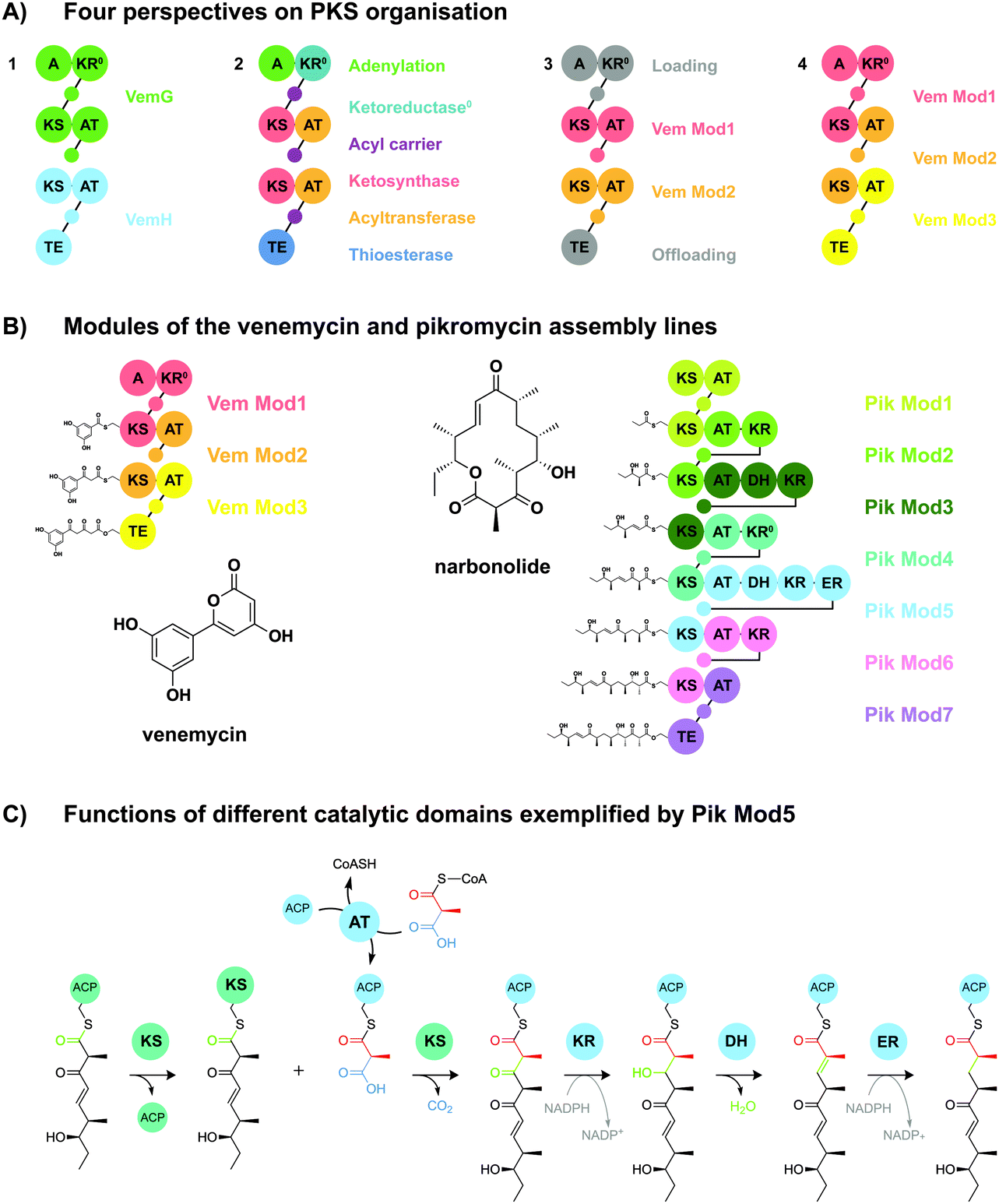 | ||
| Fig. 4 Traditional and updated assembly-line polyketide synthase module definitions. (A) Four different perspectives on the venemycin assembly-line PKS. (1) The synthase can be divided into two polypeptide chains called VemG (232 kDa) and VemH (140 kDa). (2) The PKS can also be viewed in terms of its domains. The adenylation domain (A) accepts the 3,5-dihydroxybenzoyl starter unit. The inactive ketoreductase domain (KR0) probably plays a structural role. The ketosynthase (KS), acyltransferase (AT) and acyl carrier protein (small circle) domains are the core components of PKSs. The thioesterase domain finally releases the polyketide from the synthase, often by cyclisation to form a macrolactone. (3) In the traditional view, the module boundaries are the N-terminus of the ketosynthase domain and the C-terminus of the acyl carrier protein domain. (4) In the updated definition, modules end at the C-termini of ketosynthase domains, reflecting the evolutionary co-migration of domains. (B) The venemycin and pikromycin assembly lines depicted using the new module definitions. The assembly-line steps and products are also shown. The pikromycin assembly line additionally includes dehydratase (DH) and enoylreductase (ER) domains. Note that modules can be split over different polypeptide chains. For example, Vem Mod2 is split between the VemG and VemH proteins. (C) The functions of the different catalytic domains exemplified by Pik Mod5, which has a full set of reductive domains. The domains are represented by spheres coloured by module as in (B). The process starts with a tetraketide intermediate attached to an acyl carrier protein, which is transferred to a cysteine residue on the ketosynthase domain. An acyltransferase domain loads the downstream ACP with an acyl-CoA-derived extender unit, in this case methylmalonyl-CoA. The ketosynthase catalyses decarboxylative condensation of the tetraketide intermediate and the extender unit to form an ACP-linked pentaketide intermediate. This intermediate is then subjected to reductive reactions by the ketoreductase, dehydratase, and enoylreductase domains (the carbonyl subjected to reduction is coloured green). All three reactions are optional so that other modules may stop at either the β-keto, β-hydroxy, or α,β-alkene intermediates. The resulting ACP-linked intermediate is substrate to either the next KS domain or the terminal thioesterase, which usually results in cyclisation. This figure was simplified and redrawn from the Miyazawa et al. and Smith et al.440,441 | ||
The rich chemical and structural diversity of naturally occurring polyketides can often be enhanced by structural fine-tuning.437,442 Chemical modification of natural polyketides is possible, but often inefficient, and selectivity is hard to achieve.434 However, successful modification of the biosynthetic machinery itself would enable diversely modified analogues to be produced. Therefore, scientists have been trying to reprogram the modular architecture of assembly-line PKSs since they were discovered in the 1990s.442 Not only should it be possible to produce targeted polyketide modifications, but libraries of ‘unnatural natural products' could also be created and screened for novel activities, if only we could insert, delete, or swap out individual PKS modules at will.434,445 Unfortunately, this combinatorial assembly approach is not straight forward since the chimeric assemblies are usually catalytically impaired and the production of structural analogues of medically relevant polyketides by genetic/protein engineering is still a major challenge.434,442,446,447
Interactions between domains and modules seem to be as important to the activity and fidelity of assembly-line PKSs as enzyme-substrate interactions.448 Therefore, in addition to the typical protein engineering challenges like changing substrate specificity, delicate protein–protein interactions must be maintained. Klaus et al. studied chimeras with modules from the erythromycin, rapamycin, and rifamycin PKSs. Analysis of bi-modular chimeras revealed that turnover rate correlated with efficiency of the intermodular chain translocation, which depended on interactions between the ACP and downstream KS domains. These results demonstrate that more efficient engineering of domain-domain interactions could significantly facilitate the generation of highly productive chimeric PKSs.440,448 Difficulties in engineering the protein–protein interactions necessary for modules to functionally interact seem to be largely responsible for the limited success of PKS engineering.442,448
Adding to the difficulties of designing functional protein–protein interactions is the fact that we do not know much about the overall structures of entire multi-modular PKSs.440,442,446 Assembly-line polyketide synthases are some of the largest and most complex protein structures known. Their several-megadalton sizes are probably their most striking attributes.435,437,442 Protein–protein interactions between noncovalently attached modules can be rather weak, making it hard to isolate and structurally characterise entire complexes.437–439 Interestingly, catalytic modules of assembly-line PKSs are observed in both extended and arched conformations but until very recently it was unknown whether these conformations influence catalytic activity. Khosla's group used a high-affinity antibody to lock a PKS in the extended form, which retained catalytic activity.449 Only recently did Dutta et al. and Whicher et al. use cryoelectron microscopy to determine the structure of an entire pikromycin synthase module and key stages of its catalytic cycle.437,450,451 These developments will facilitate future rational design and engineering endeavours.
The modern protein engineering approaches that work so well for predominantly monomeric/independent enzymes are hard to apply to megasynthases, where rational design is essentially impossible due to the lack of structural information. As noted in an excellent review by Khosla's team, the rich natural diversity of modular PKSs is even more astonishing considering how hard it is to engineer these proteins in the laboratory.442 It seems like understanding and mimicking natural evolutionary mechanisms is currently one of the most promising PKS engineering strategies. Natural PKS evolution depends on functional PKSs resulting from domain exchanges. Therefore, there is a growing interest in understanding the ‘natural splice points' which could accelerate rational engineering. Analysis of many PKS systems has suggested that the KS-AT linker is a natural splice site, making it an attractive target for engineering by homologous recombination.442,452,453 Peng et al. showed that hybrid aureothin and neoaureothin synthases were more active when modules were split at the KS-AT linker than when split at the traditional ACP-KS interface, confirming that the KS-AT linker is a good fusion site for module swapping.442,446,454 These findings are in line with recently updated module definitions that place the module boundaries between the KS and AT domains. Evolutionary models based on gene duplication suggest that the unit of duplication would be the KS-AT-ACP module, which is functionally required for chain elongation and matches the boundaries of single-module PKSs.442 However, Zhang et al. recently compared four very large aminopolyol-producing PKSs (each 25–30 modules long or the size of about five ribosomes). They observed that the co-migrating module consisted of AT-DH-ER-KR-ACP-KS domains rather than the traditional KS-AT-DH-ER-KR-ACP module (Fig. 4).433,455 Importantly, this new module definition has led to some promising results. The lower activities of synthases engineered using traditional boundaries seems to result from weaker interactions between acyl carrier proteins and the downstream KS units that do not co-migrate in natural evolution.440,446 Miyazawa et al. reconstituted the venemycin PKS, a short aromatic polyketide-producing assembly line, in vitro. Venemycin production was achieved by incubation of the polypeptides VemG and VemH with the substrate 3,5-dihydroxybenzoate and ATP, malonate, coenzyme A, and the malonyl-CoA ligase MatB for malonyl-CoA production. Venemycin could be isolated on the milligram scale, without the need for chromatography, from dialysis reactors which also enabled enzyme recycling.440 They performed assembly line engineering using the venemycin and pikromycin synthase modules and demonstrated that chimeric synthases designed using the updated module definitions outperformed those based on traditional module boundaries by over an order of magnitude (Fig. 5).434,440 Peng et al. used genome mining to identify nine homologous biosynthetic gene clusters encoding assembly lines for aureothin and neoaureothin, two compact and highly functionalised polyketides produced by homologous biosynthetic gene clusters. They successfully morphed the neoaureothin assembly line to produce the aureothin backbone by deletion of two modules. They found that the KS-AT linker is well suited for both insertion and deletion of modules, further supporting the alternative domain definition.446
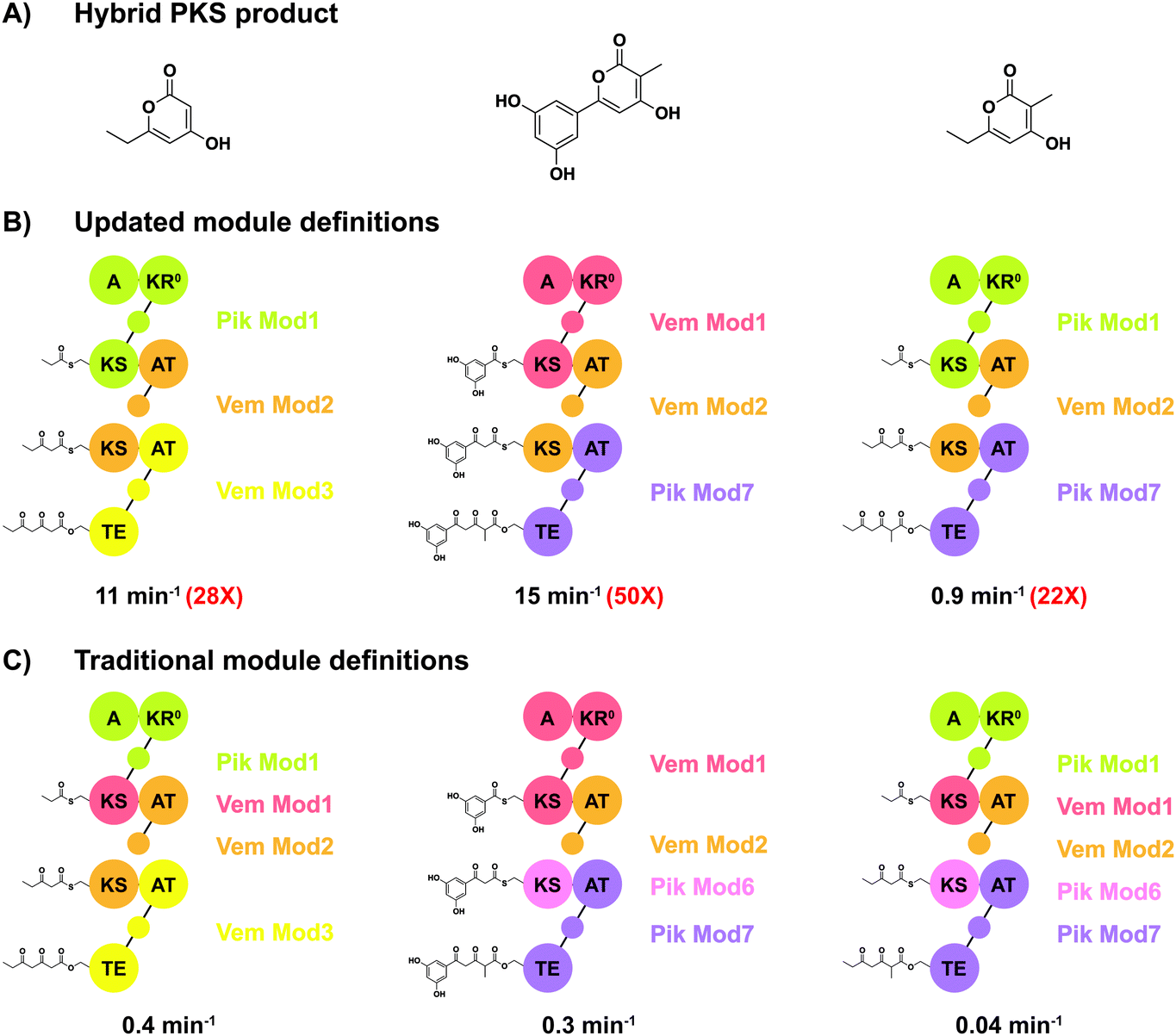 | ||
| Fig. 5 Hybrid polyketide synthases constructed using both the updated and the traditional module definitions. (A) The products of three hybrid assembly lines. (B) Three assembly lines constructed using the updated module definition are over an order of magnitude more productive (red numbers) than (C) assembly lines constructed using the traditional modules. Despite the success of assembly lines based on the new module definitions, the native VemG-VemH assembly line is still more than double as active (36 min−1). The domain abbreviations are as for Fig. 4 and again the acyl carrier proteins are represented by small circles. This figure was simplified and redrawn from Miyazawa et al.440 | ||
Despite advances in modelling natural evolutionary processes and computational prediction of optimal splice sites, a major barrier to successful engineering has been a lack of experimental data for guiding optimal selection of splice sites for generating functional chimeric PKSs.447,450,451,456,457 Experimentally accelerated molecular evolution based on homologous recombination is a recently introduced strategy for gaining valuable information on optimal splice sites for functional chimeras. Homologous recombination based on naturally occurring stretches of sequence similarity can be used for the assembly of novel chimeric PKSs, similar to natural PKS evolution.442 Chemler et al. used homologous recombination between the erythromycin (DEBS) and pikromycin coding sequences in Saccharomyces cerevisiae to generate hybrid libraries containing many functional chimeras.456 This method has the potential for generating large libraries rich in functional variants. Wlodek et al. recently described a method for adding, removing, and replacing modules, based on recombination between regions of high sequence homology within a PKS gene cluster. Rather than using yeast, they harnessed the homologous recombination machinery of a Streptomyces strain, rapidly generating diverse and highly productive assembly lines by 'accelerating' the natural evolution of modular polyketide synthases.447 They generated 17 rapamycin synthase and 9 tylactone synthase chimeras, many of which were highly active, producing titres comparable to the wild-type strain.442,447 Sherman's group described the use of PKS modules in vitro to convert a chemically synthesised thiophenyl-activated analogue of the hexaketide intermediate of tylactone biosynthesis. The intermediate was accepted by the ketosynthase of the JuvEIV PKS module and further processed by the JuvEIV and JuvEV modules to form tylactone. Macrolactonization was followed by in vivo glycosylation, in vitro P450-mediated oxidation, and chemical oxidation, resulting in the total synthesis of a range of macrolide antibiotics from the juvenimicin, M-4365, and rosamicin family.458 Analogues of tylactone intermediates accessed by homologous recombination-based genetic engineering could be valuable alternative starting points for chemoenzymatic late-stage modification, enabling structural diversification to an even larger number of macrolide antibiotics.447,458
Khosla's group recently reported the use of in vitro reconstitution to decode the orphan polyketide assembly line responsible for producing the nocardiosis-associated polyketide (NOCAP). The reconstituted PKS, in the presence of octanoyl-CoA, malonyl-CoA, NADPH, and SAM, produced octaketide and heptaketide products that could partially be structurally elucidated by MS and NMR.459 More recently, they reconstituted the entire NOCAP assembly line both in vitro and in E. coli to fully “deorphanise” the NOCAP synthase, independent of its genetically challenging and hazardous natural host.460 These approaches for studying multi-megadalton assembly lines are by far not standardised. For example, to overcome heterologous expression problems due to the exceptional size of the NOCAP PKS (3 MDa homodimer), multi-modular proteins had to be dissected into mono- or bimodular units that could be more easily expressed in E. coli.438,460 Optimisation or modifications of polyketide synthesis in the native host strain is often desirable because the gene cluster is already functionally expressed. However, as the NOCAP case demonstrates, the hosts may be hazardous or otherwise hard to culture and manipulate. One of the key advantages of E. coli is that its metabolic background is not cluttered by complex natural product biosynthesis pathways, avoiding crosstalk with heterologously introduced PKSs and facilitating the identification of novel polyketides. The metabolism and molecular biology of E. coli are also well understood, and the genetic toolkit is unrivalled.436 The E. coli genome has not only been extensively sequenced but also completely recoded.461,462 Therefore, future advances in systems level understanding could be rapidly translated into genomically-reprogrammed hosts. Despite the advantages,443,460 only a few assembly-line PKSs have been functionally reconstituted in E. coli, because E. coli is not an ideal “universal host”.436 Nontrivial engineering is necessary for biosynthesis of precursors and performing critical post-translational modifications. No assembly-line PKS has yet been completely deorphanised solely by reconstitution in E. coli. This achievement would revolutionize natural product discovery if robustly implemented.436
As in other protein engineering endeavours, the number of PKS variants interrogated is limited by the throughput of the available screening methodologies. Chromatographic analyses are time consuming but due to the complexity of the molecules, simple colorimetric or fluorometric assays are not typically applicable to PKS screening.434 While biosensors for detecting enhanced precursor (e.g., malonyl-CoA) formation have been reported,143,463 relatively few high-throughput methods for polyketide products are available. Kasey et al. recently showed that engineered variants of the promiscuous erythromycin-sensing transcription factor MphR could be used to detect related ligands like clarithromycin. This work demonstrated the potential of engineered biosensors to facilitate the directed evolution of macrolide synthases, but little work on this topic has been published recently.71,434,464 While biosensors are promising tools for HTS, they are often limited to naturally occurring biosensors. Unfortunately, biosensor engineering is challenging in itself, effectively doubling the engineering effort if an off-the-shelf biosensor is not available.465 However, for a problem as complex as PKS engineering, it might well be worth to first engineer a biosensor for screening PKS libraries. Rapid advances in both protein structure prediction466 and de novo design of bioactive protein switches165,467 might make biosensor design much simpler in the near future. Unfortunately, these advances cannot entirely solve the problem, since in many cases the structure of the target polyketide will not be known in advance, making activity-based screening indispensable. While some (e.g., antibiotic) activities are relatively easy to screen for using agar plate or microfluidic droplet-based UHTP screening technologies, others are not. In an interesting approach to screen for proapoptotic compounds, Theodorou et al. mixed bacteria in microfluidic droplets with mammalian cells, which could be assayed for apoptosis markers.468 For the foreseeable future, these complicated screening problems will have to be solved on a case-by-case basis.
Yuzawa et al. recently reported engineered strains of Streptomyces albus harbouring engineered hybrid polyketide synthases capable of converting plant biomass to methyl and ethyl ketones at titres of over 1 g L−1.469 While these product titres are too low to compete with fossil fuels, they demonstrate that engineered hybrid polyketide synthases can produce g L−1 titres, which is significant for more valuable pharmaceutical compounds. Probably the greatest challenge for the field is increasing production rates to industrially competitive values since both time and titre are relevant to the space-time yield. The ability to rationally exchange assembly line modules has long been one of the holy grails in biochemistry. While linking a loading domain to more than one extension module without losing functionality is still a major challenge, recent trends in evolution-guided engineering strategies suggest that there is hope for the future. Updated module definitions (Fig. 4 and 5), supported by high-throughput experimental recombination and characterisation of chimeras and machine learning, may well make this goal attainable.470 Advances in DNA synthesis and assembly coupled with automated and unbiased PKS characterisation is necessary to realise the full potential of machine learning in PKS research. The hurdles to designer polyketide synthases are expected to be overcome taking advantage of the newly arising tools available for biocatalysis.
De novo enzymatic total synthesis of complex drug compounds
Biocatalytic synthesis of drug compounds is a main research topic in the biopharmaceutical industry. The previous studies have mostly focused on single-step enzymatic reactions or simple multi-step enzymatic cascades to synthesise intermediates with high value. At present, more attempts are involved in the total biosynthesis of drug compounds with complex structures. Since drug molecules mostly contain non-natural structures, the design of their biosynthetic pathways has more challenges in comparison to natural products. An effective strategy is to introduce artificially designed enzymatic cascades on the basis of related natural synthetic pathways, which can reduce the complexity of pathway design. Moreover, de novo biosynthetic pathways require screening of novel enzymes or modification of existing enzymes through protein engineering to obtain biocatalysts with sufficient activity, specificity, and stability that meet the requirements of drug synthesis. Furthermore, some auxiliary enzymatic cascades are also designed to optimise the supply of cofactors and reduce the formation of by-products in the synthetic pathway. Here, we illustrate a few selected successful examples of enzymatic total synthesis of drugs and important enzymatic cascades to elaborate how novel enzymes and enzymatic cascades are involved in the de novo enzymatic synthesis of drug compounds.Islatravir
Islatravir is a translocation inhibitor against the reverse transcriptase of the human immunodeficiency virus (HIV) and thus became a potential drug candidate to treat the acquired immune deficiency syndrome (AIDS). Recently, its total enzymatic synthesis has been achieved by Merck & Co., Inc and Codexis (Scheme 9).471 The design of the enzymatic synthetic route did not follow the de novo synthesis of the nucleotide, but referred to the nucleoside salvage pathway in bacteria. Since in the salvage pathway the enzymatic reactions catalysed by purine nucleoside phosphorylase (PNP), phosphopentomutase (PPM) and deoxyribose 5-phosphate aldolase (DERA) are all reversible, the retrosynthetic route thus formed is feasible in terms of reaction kinetics and more efficient than the natural synthetic route of nucleotides. Therefore, following this retrosynthetic route, islatravir was synthesised by a three-step enzymatic cascade with 2-ethynylglyceraldehyde-3-phosphate and acetaldehyde as starting materials. Furthermore, the enzymatic synthesis of chiral 2-ethynylglyceraldehyde-3-phosphate also follows a retrosynthetic pathway, through which the target compound is obtained by sequential oxidation and phosphorylation catalysed by galactose oxidase (GOase) and pantothenate kinase (PanK) with non-chiral 2-ethynylglycerol as starting material. So far, through this novel five-step enzymatic cascade, the total synthesis of islatravir can be achieved from simple non-chiral compounds. The next challenge was to obtain the biocatalysts that meet the requirement for each step. With enzyme screening, PNP, PPM, PanK from E. coli, DERA from Shewanella halifaxensis, and GOase from Fusarium graminearum were selected as the starting biocatalyst candidates. After 2 to 12 rounds of evolution, respectively, the activity of PPM, PNP, PNK, and GOase, as well as the stereoselectivity of PanK and GOase, have been greatly improved towards the non-natural substrates and are high enough for synthetic purposes. The third step in this impressive endeavor was the optimisation of the total synthetic pathway, including cofactor regeneration, removal of product and substrate inhibition, and avoiding additional by-products produced by multi-enzymatic reactions. Several auxiliary enzymes, such as horseradish peroxidase, catalase, acetate kinase, and sucrose phosphorylase, were introduced to the main synthetic route to maintain the correct oxidation state of copper, disproportionate the hydrogen peroxide by-product, regenerate the cofactor adenosine triphosphate, and convert free phosphate to glucose-1-phosphate, respectively, and thus shift the entire equilibrium forward. Finally, 51% overall yield was achieved by this full biocatalytic route in vitro.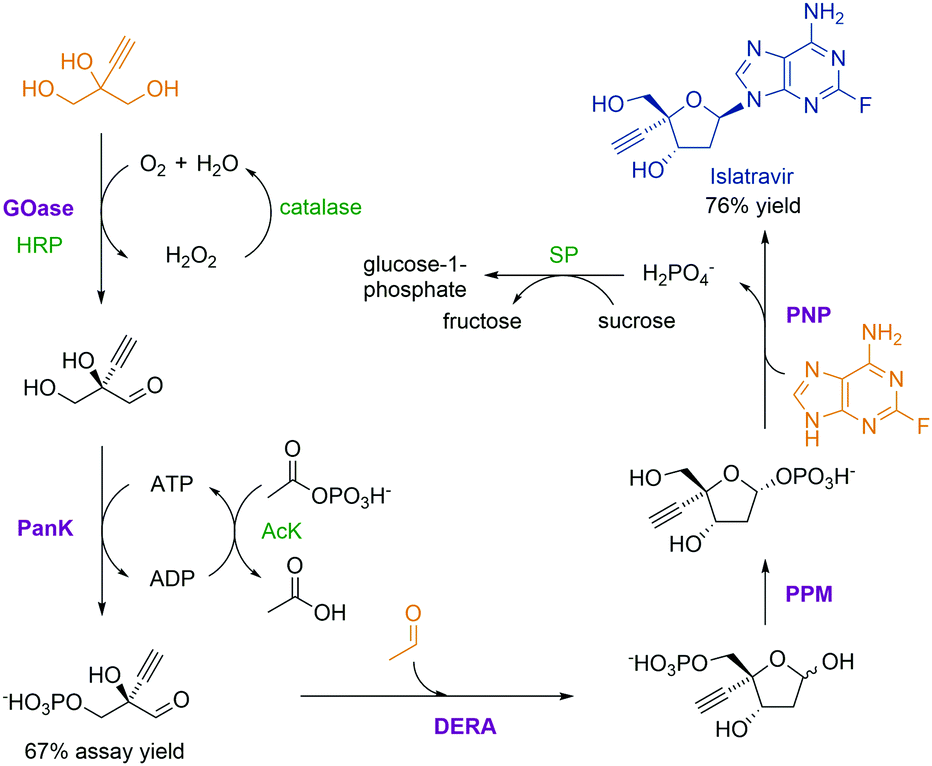 | ||
| Scheme 9 De novo enzymatic synthesis of islatravir. GOase: galactose oxidase; PanK: pantothenate kinase; DERA: deoxyribose 5-phosphate aldolase; PPM: phosphopentomutase; PNP: purine nucleoside phosphorylase; HRP: horseradish peroxidase; AcK: acetate kinase; SP: sucrose phosphorylase. Starting materials are highlighted in orange. The product is highlighted in blue. Key enzymes in synthetic routes are highlighted in purple. Auxiliary enzymes are highlighted in green. | ||
The successful total biosynthesis of islatravir is a textbook case for the design of a new synthetic route based on the salvage/degradation pathway, enzyme screening and modification assisted by protein engineering, and synthetic pathway optimisation. This will certainly inspire the total biosynthesis of other drug compounds in the pharmaceutical industry.
Cannabinoids
Cannabinoids are a class of meroterpenoids with a resorcinyl core typically decorated with a para-positioned isoprenyl, alkyl, or aralkyl side chain.472 Due to their great therapeutic potentials, cannabinoids are highly valued in the pharmaceutical and health product markets. The synthetic pathway of cannabinoids integrates the key enzymatic cascades of polyketides and terpenes biosynthesis. Briefly, fatty acids are formed into olivetolic acid (OA) or divarinic acid (DA) under catalysis by the olivetol synthase and olivetolic acid cyclase, and then prenylated by cannabigerolic acid (CBGA) synthase with GPP as donor and further cyclised by cannabinoid synthase.By integrating OA, GPP, and cannabinoid synthetic modules, Keasling and his colleagues realised the construction of a cannabinoid synthetic pathway in yeast. The obtained recombinant yeasts can synthesise cannabinoids and their non-natural derivatives up to a yield of 8 mg L−1 starting from galactose and fatty acid derivatives.473 However, the low supply of prenylation donors and the toxicity of product and intermediates to recombinant cells are still the barriers against achieving a high yield of cannabinoids by whole-cell conversion.
Therefore, Bowie and his colleagues chose a cell-free platform for the prenylation of natural products and application to cannabinoid production.474 The cell-free prenylation system contains glycolysis, acetyl-CoA, MEV, and cannabinoid modules, and involves 25 enzymes. The synthesis of the prenylation donor GPP was achieved by adding glucose from an external source and the prenylated products were synthesised by adding a variety of aromatic substrates. Constructing natural enzymatic pathways in a cell-free system often encounters problems of metabolic flow equilibrium, feedback inhibition, and low activity of enzymes towards non-natural substrates. By introducing a purge valve to allow carbon flux to continue through the glycolysis pathway without building up excess NADPH, a pyruvate dehydrogenase bypass to eliminate the inhibition by intermediates, and engineered PT (NphB) for improved activity and regioselectivity, a final yield of 1.25 g L−1 was achieved for CBGA. However, the shortcomings of this synthesis system are obvious: the 25-step enzymatic cascade is too complex, and expensive OA or DA are required as one of the starting materials.
In a recently reported cell-free system,475 the enzymatic synthesis pathway is designed into four modules: isoprenoid, aromatic polyketide, ATP regeneration, and cannabinoid modules. Isoprenoid was used as a starting material for the synthesis of GPP through a four-step enzymatic cascade (Scheme 10), which greatly shortens the original 23-step GPP synthesis pathway starting from glucose. Inexpensive acetyl-phosphate (AcP) acts as a phosphate donor for ATP regeneration, and at the same time as CoA transfer medium to generate malonyl-CoA through the catalysis of a phosphotransacetylase and a malonate decarboxylase α-subunit. This non-natural CoA transfer cascade greatly improves the efficiency of malonyl-CoA and further OA/DA synthesis. In the cannabinoid module, an efficient, water-soluble CBGA synthase replaced the natural membrane enzyme to directly prenylate OA/DA. In the end, the optimised cell-free system uses only 12 enzymes with low-cost organic acids as starting materials to produce CBGA and cannabigerovarinic acid (CBGVA) with a yield of 0.5 g L−1. This successful example demonstrates the unique advantages of cell-free systems for the enzymatic in vitro biosynthesis, including complete flexibility in pathway design, rapid design-build-test-learn cycles, precise control of all system components, and circumventing the toxicity of products and intermediates to cells.475 Meanwhile, this case also proves that careful selection of starting materials, introduction of heterologous synthetic pathways and isoenzymes, and simplification of the synthesis pathways of each module can effectively increase the supply of raw materials, improve the efficiency of coenzyme regeneration, and thus achieve product output with high yield.
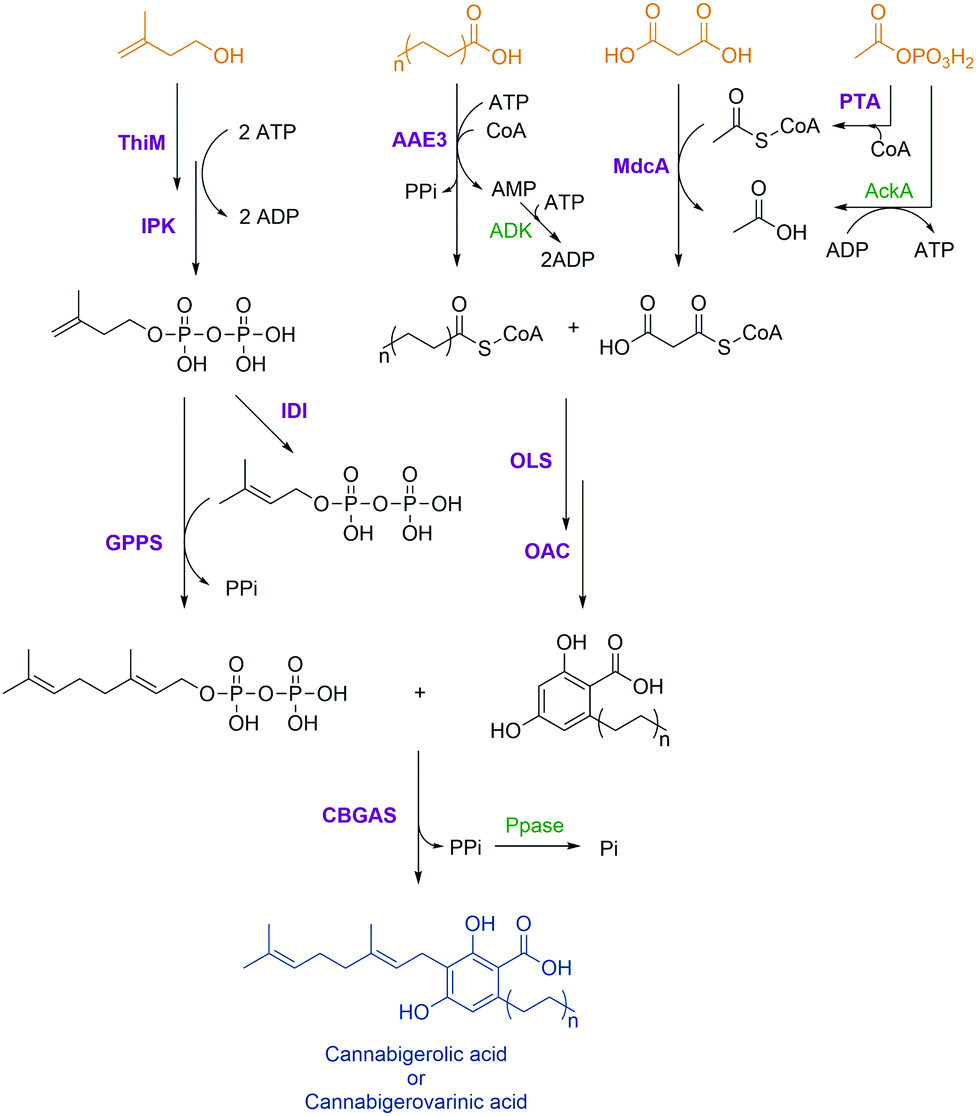 | ||
| Scheme 10 Enzymatic total synthesis of cannabinoids in a cell-free system. ThiM: hydroxyethylthiazole kinase; IPK: isopentenyl kinase; IDI: isopentyl diphosphate isomerase; GPPS: geranyl pyrophosphate synhetase; AAE3: acyl activating enzyme 3; ADK: adenylate kinase; MdcA: malonate decarboxylase α subunit; PTA: phosphotransacetylase; AckA: acetate kinase; OLS: olivetol synthase; OAC: olivetolic acid cyclase; CBGA synthase: cannabigerolic acid synthase. Starting materials are highlighted in orange. The products are highlighted in blue. Key enzymes in this synthetic route are highlighted in purple. Auxiliary enzymes are highlighted in green. | ||
Ikarugamycin
Ikarugamycin is the parent compound of bacterial polycyclic tetramate macrolactams which predominantly possess antibiotic, anti-leukemic, and anti-inflammatory properties. The chemical total synthesis of ikarugamycin normally requires around 30 steps with very low yield. However, the natural biosynthesis of ikarugamycin only involves three enzymes: IkaA, B and C. IkaA catalyses the formation of tetramic acid from acetyl-CoA, malonyl-CoA, and L-ornithine via an unusual iterative PKS/NRPS mechanism. IkaB and IkaC are reductases catalyzing the cyclisation of tetramic acid via the formation of the outer and inner bonds of the carbocycle, respectively. Gulder and his colleagues constructed a cascade of these three enzymes in vitro as an effective way for the total synthesis of ikarugamycin (Scheme 11).476 The genes of IkaA, B and C from the ikarugamycin-producing Strepomyces sp. Tü6239 were cloned for heterologous expression in E. coli. However, the soluble expression of IkaC in E. coli could not be achieved. To circumvent this problem, the IkaC homologue from Salinispora arenicola CNS-205 was identified as a replacement. Combining this with a phosphopantetheinyl transferase Sfp from Bacillus subtilis to activate the active-site serine residue of PKS/NRPS carrier proteins, the in vitro three-enzyme cascade was confirmed to achieve the synthesis of ikarugamycin with a yield of the purified product of 9%. Furthermore, acetate kinase, phosphotransacetylase and malonyl-CoA synthetase were employed as an enzymatic system for the synthesis of the expensive precursors acetyl-CoA and malonyl-CoA from acetic and malonic acid, respectively. This efficient synthetic route shows an unparalleled ingenuity and economy of natural biosynthesis. The artificially optimised precursor synthesis pathway improves the economics of in vitro enzymatic synthesis.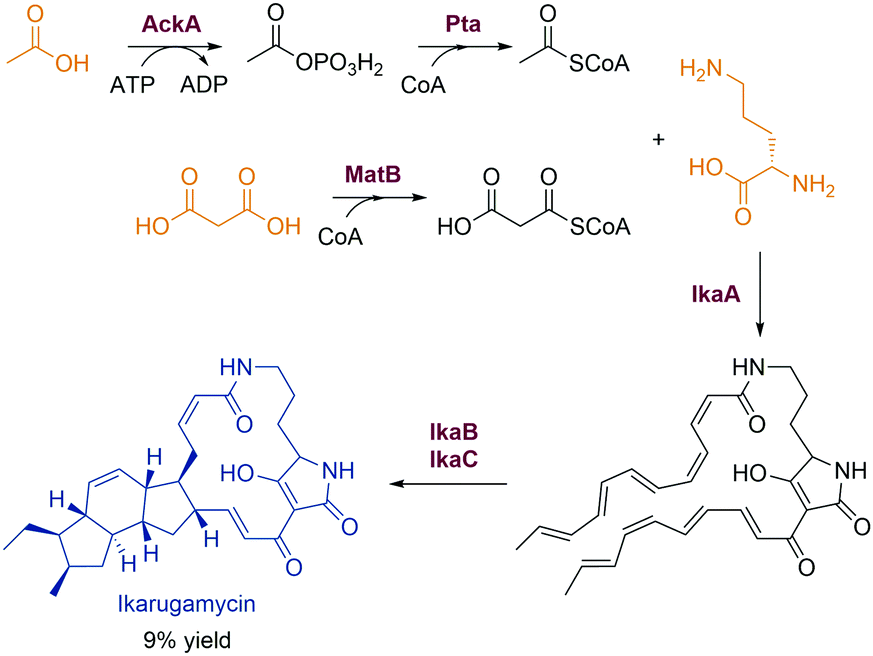 | ||
| Scheme 11 Enzymatic total synthesis of ikarugamycin. AckA: acetate kinase; Pta: phosphotransacetylase; MatB: malonyl-CoA synthetase. Starting materials are highlighted in orange. The product is highlighted in blue. Key enzymes in this synthetic route are highlighted in purple. Auxiliary enzymes are highlighted in green. | ||
Kainic acid
Kainic acid is a potent neuroexcitatory amino acid agonist that naturally occurs in some seaweeds. It was previously used as an anthelmintic drug for children, and now is mainly used for the construction of pharmacological models. Recently, Moore and his colleagues successfully revealed the natural synthesis pathway of kainic acid from the genome data of kainic acid producing algae and established a two-step synthetic cascade of kainic acid from glutamic acid (Scheme 12).477 According to the biosynthetic pathway of domoic acid, they predicted the biosynthesis of kainic acid probably starts from N-prenylation of glutamic acid, followed by an enzymatic cyclisation catalysed by α-ketoglutarate-dependent dioxygenase (KGDOX). To prove this speculation, the genome sequencing and assembling of Digenea simplex were obtained by the Oxford Nanopore Technologies (ONT) that is a novel single-molecule, long-read sequencing platform being able to generate single reads in hundreds of kilobases, which greatly facilitates resolving tandemly duplicated genes and repeats in red macroalgae genomes. Through the analysis of genome data, a kainic acid biosynthesis (kab) cluster was successfully identified containing an annotated N-prenyltransferase (N-PT) and a KGDOX, for both enzymes the activities were proven in vitro. The N-PT has restricted substrate specificity towards L-glutamic acid but modest prenyl donor promiscuity towards DMAPP and GPP. This N-PT belongs to the new family of PTs from marine algae of which the crystal structure and the catalytic mechanism have been revealed very recently.478 The KGDOX can directly convert prekainic acid to kainic acid in the presence of α-ketoglutarate (αKG), L-ascorbate and Fe3+ with 46% yield by purified enzyme and 57% yield using a recombinant E. coli. This enzymatic conversion solved the synthetic challenge of the stereocontrolled formation of the trisubstituted pyrrolidine ring and can replace the current chemical synthesis route with many steps. This impressive result shows that the use of the novel sequencing technology to unearth natural product synthesis pathways in unknown genome data has great potential. The steps of the natural synthesis pathways of natural products are sometimes not as cumbersome as imagined. A simple single step or a few steps of enzymatic reactions can also achieve the synthesis of complex compounds.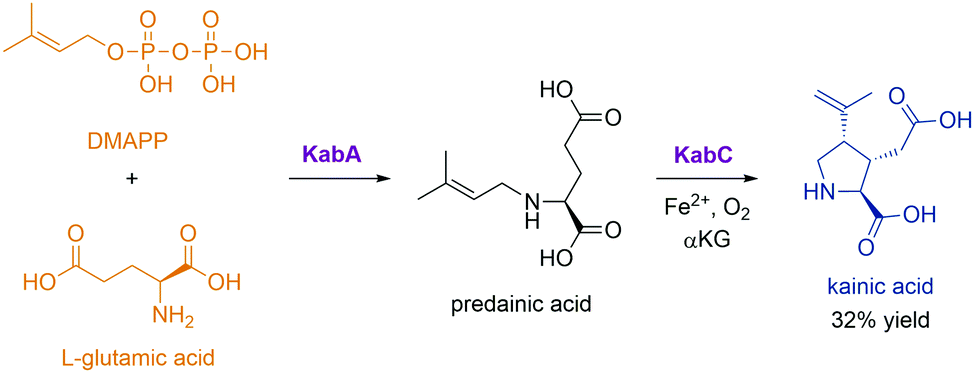 | ||
| Scheme 12 Enzymatic total synthesis of kainic acid. KabA: N-prenyltransferase; KabC: α-ketoglutarate-dependent dioxygenase; αKG: α-ketoglutarate. Starting materials are highlighted in orange. The product is highlighted in blue. Key enzymes in this synthetic route are highlighted in purple. | ||
Enterocin and wailupemycin
Enterocin and wailupemycin are polyketides with broad-spectrum antibacterial activity. In particular, enterocin can be degraded by proteases in human's gastrointestinal tract, so it is considered a safe bacteriostatic agent with high application value in the food industry. Moore and his colleagues reported the biosynthesis pathway of enterocin and wailupemycin in Streptomyces maritimus, and achieved the total enzymatic synthesis of enterocin and wailupemycin F and G in vitro.479The biosynthesis of enterocin and wailupemycin in S. maritimus uses benzoic acid and malonyl-CoA as starting materials and is catalysed by the enc II type polyketide synthase complex (Scheme 13). First, EncN catalyses an ATP-dependent activation and transfers benzoate to the acyl carrier protein EncC. Then, the benzoyl unit migrates from EncC to the ketone synthase heterodimer EncA-EncB. The resting EncC is malonated under the catalysis of malonyl-CoA:ACP transacylase (FabD), followed by a Claisen condensation reaction between benzoyl and malonyl units. This reaction process is repeated six additional times to produce an octaketide, which is then reduced by the ketoreductase EncD to obtain an intermediate common to enterocin and wailupemycin. This intermediate can be self-cyclised to generate wailupemycin D-G. However, in the presence of EncM, the linear polyketide intermediate is oxidatively converted in a Favorskii-like reaction to form the enterocin tricyclic scaffold. Finally, this scaffold is methylated by the O-MT EncK and hydroxylated by the cytochrome P450 hydroxylase EncR to produce enterocin. Interestingly, EncN has a wide substrate scope and is able to accept structural analogues of benzoic acid. As a result, 24 unnatural 5-deoxyenterocin and wailupemycin F and G analogues were successfully synthesised in vitro by adding halogenated benzoic acids, phenolic acids, and thiophene acids as starting materials.480 The use of unnatural starting materials may bring toxicity to cells in whole-cell transformation. However, an enzymatic total synthesis system in vitro does not have this disadvantage. Therefore, it is convenient to synthesise natural product derivatives by trying starting materials with different structures, which brings unlimited possibilities for expanding the activity and application of natural products.
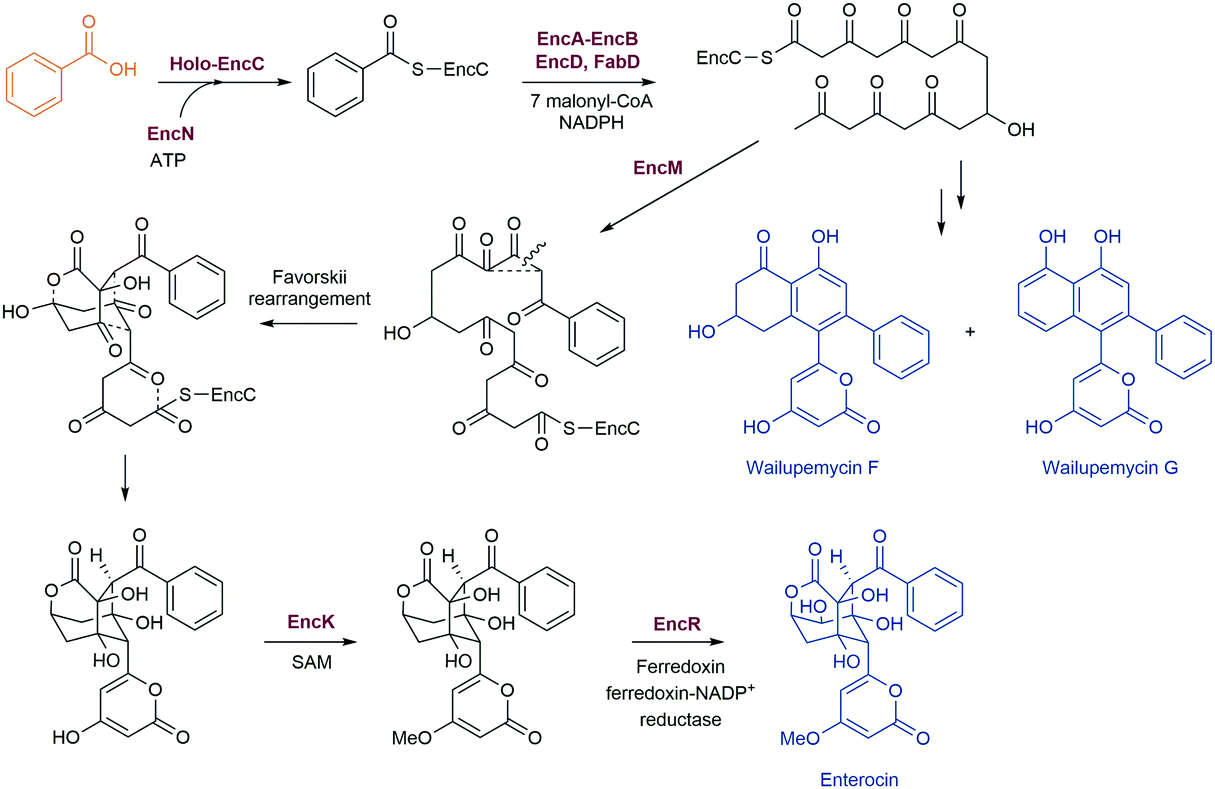 | ||
| Scheme 13 Enzymatic total synthesis of enterocin and wailupemycin. Starting materials are highlighted in orange. The products are highlighted in blue. Key enzymes in this synthetic route are highlighted in purple. | ||
Meroterpenoids
Meroterpenoids are hybrid terpenoid–polyketide natural products with many desired bioactivities such as antimicrobial, antioxidant, and anticancer. However, they are particularly challenging complex molecules for enzymatic total synthesis. The Moore group elucidated the biosynthesis of the halogenated meroterpenoids merochlorin A and B, and demonstrated the first total enzymatic synthesis of a meroterpenoid (Scheme 14).481 A type III PKS, Mcl17, catalysed the formation of 1,3,6,8-tetrahydroxynapthalene from 5 malonyl-CoA substrates. The terpenoid part of the molecule was synthesised by a unique prenyl diphosphate synthase, Mcl22, via a head-to-torso coupling of GPP with DMAPP. These two parts were connected by an aromatic prenyltransferase, Mcl23. The most surprising part came from the vanadium-dependent chloroperoxidase, Mcl24, catalyzing a cascade of naphthol chlorination and oxidative dearomatization/cyclisation steps.482 The highly complex halogenated meroterpenoids merochlorin A and B were synthesised by applying merely these four enzymes (Mcl17, Mcl22, Mcl23, and Mcl24) and simple substrates (malonyl CoA, DMAPP, GPP), showing the power of enzymes to generate complex structures.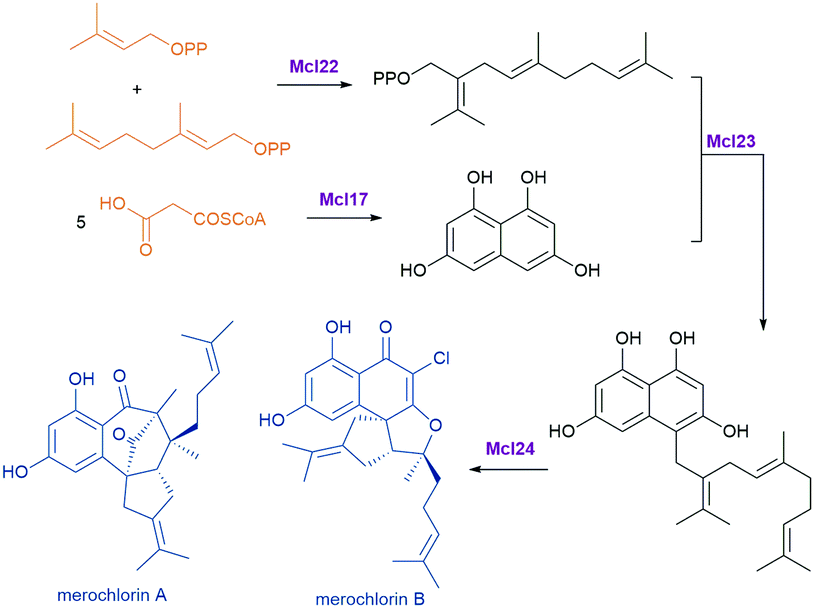 | ||
| Scheme 14 Enzymatic total synthesis of the meroterpenoids merochlorin A and B. Mcl17: type III polyketide synthase; Mcl22: prenyl diphosphate synthase; Mcl23: aromatic prenyltransferase; Mcl24: vanadium-dependent chloroperoxidase. Starting materials are highlighted in orange. The products are highlighted in blue. Key enzymes in this synthetic route are highlighted in purple. | ||
Hyoscyamine and scopolamine
Hyoscyamine and scopolamine are widely used drugs to control neuropathic pain and treat neuromuscular disorders, such as nerve agent poisoning, digestive and urinary tract spasm, and Parkinson's disease. At present, the production of these two compounds can only rely on extraction from nightshade plants, which poses a challenge to guarantee the supply. Recently, the total synthesis of hyoscyamine and scopolamine in yeast has been achieved by Smolke and Srinivasan with simple amino acids as starting materials (Scheme 15).483 The most critical step in the enzymatic synthesis of hyoscyamine and scopolamine is to use tropine and phenyllactic acid (PLA) glucoside as acceptor and donor, respectively, to generate littorine through transacylation, followed by reduction and epoxidation to produce the final products. Therefore, the whole synthesis pathway is composed of five modules: (i) putrescine module and (ii) tropine module for the synthesis of the acyl acceptor tropine, (iii) PLA glucoside module for the synthesis of the acyl donor PLA glucoside, (iv) tropane alkaloid (TA) scaffold module for transacylation, and (v) medicinal TA module for the post-modification of the littorine structure. In the putrescine module, arginine is generated through the arginine synthesis pathway with glutamic acid as starting material, which is then converted to putrescine catalysed by arginase (Car1) and ornithine decarboxylase (Spe1). In addition, spermine present in cells can be converted to putrescine by one enzyme, polyamine oxidase (Fms1). By enhancing the expression of key enzymes such as glutamate N-acetyltransferase (Arg2), Car1, Spe1 and Fms1, and introducing arginine decarboxylase from Avena sativa (AsADC) and agmatine ureohydrolase from E. coli (speB) which constitute a pathway to decarboxylate arginine, the yield of cadaverine was effectively enhanced. After entering the tropine biosynthesis module, one terminal amino group of putrescine is methylated by putrescine N-methyltransferases (AbPMT1 and DsPmT1), and the other one is deaminated into the aldehyde by an engineered N-methylputrescine oxidase (DmMPO1). The formed 4-methylaminobutanal self-cyclizes into N-methylpyrrolinium. The newly discovered key enzymes pyrrolidine ketide synthase (AbPYKS), tropinone synthase (AbCYP82M3) and cytochrome P450 reductase (AtATR1) catalyse the sequential malonylation and cyclisation of N-methylpyrrolinium to generate tropinone, in which the carbonyl group is subsequently reduced to a hydroxyl group by tropinone reductase 1 (DsTR1) to obtain tropine. In the PLA glucoside module, phenylalanine is deaminated and reduced to PLA by aromatic aminotransferases (Aro8 and Aro9) and phenylpyruvate reductase (PPR), respectively. Then a newly identified PLA UDP-glucosyltransferase (UGT84A27) achieves the glucosylation of PLA. Littorine is produced in the TA scaffold module through acyltransfer catalysed by littorine synthase (AbLS). In the last TA module, littorine in converted to hyoscyamine by littorine mutase (AbCYP80F1) and hyoscyamine dehydrogenase (HDH), and further to scopolamine by hyoscyamine 6β-hydroxylase/dioxygenase (H6H).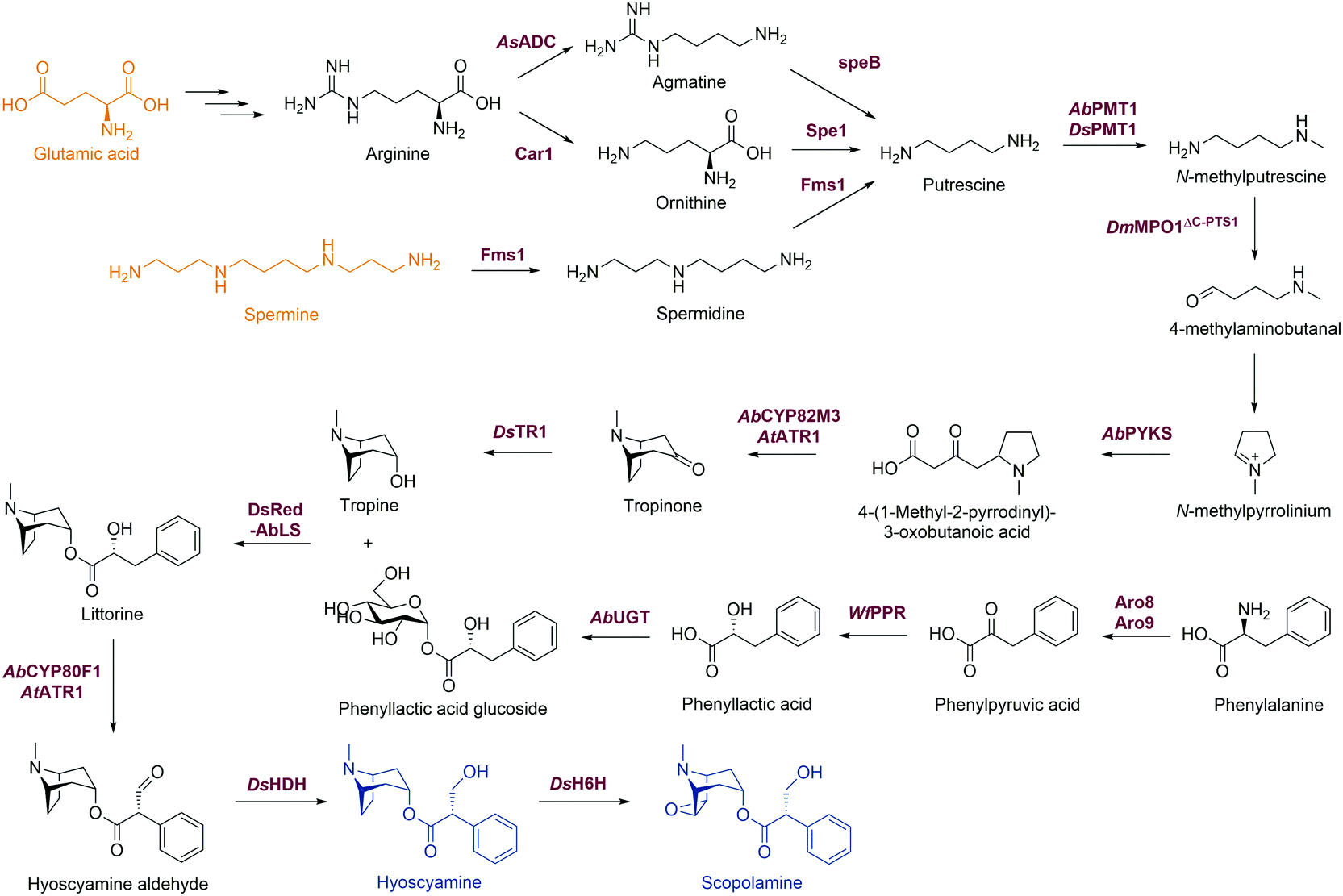 | ||
| Scheme 15 Enzymatic total synthesis of hyoscyamine and scopolamine in yeast. AsADC: arginine decarboxylase; Car1: arginase; speB: agmatine ureohydrolase; Spe1: ornithine decarboxylase; Fms1: polyamine oxidase; AbPMT1 and DsPMT1: N-methyltransferases; DmMPO1ΔC-PTS1: engineered N-methylputrescine oxidase; AbPYKS: pyrrolidine ketide synthase; AbCYP82M3: tropinone synthase; AtATR1: cytochrome P450 reductase; DsTR1: tropinone reductase 1; Aro8 and Aro9: aromatic aminotransferases; WfPPR: phenylpyruvate reductase; AbUGT: UDP-glucosyltransferase; DsRed-AbLS: littorine synthase; AbCYP82M3: tropinone synthase; AtATR1: cytochrome P450 reductase; AbCYP80F1: littorine mutase; DsHDH: hyoscyamine dehydrogenase; DsH6H: hyoscyamine 6β-hydroxylase/dioxygenase. Starting materials are highlighted in orange. The products are highlighted in blue. Key enzymes in this synthetic route are highlighted in purple. | ||
The design of the heterologous biosynthetic pathway of hyoscyamine and scopolamine basically refers to the natural plant synthesis pathway. Through the mining of plant transcriptome data, the successful identification of the key enzymes AbPYKS, AbCYP82M3, AbUGT, AbLS, AbCYP80F1, DsHDH and DsH6H was the prerequisite for constructing a complete synthetic pathway. In addition, screening orthologues and introducing microorganism-derived enzymes are able to improve the efficiency of the single-step reaction and product yield. However, the enzymes involved in the original synthesis pathway are expressed in different tissue cells in plants and various subcellular structures in plant cells. Due to lacking the corresponding differentiated cells and subcellular structures, it is a great challenge to construct such complex enzymatic cascades in most heterologous microbial cells. In this influential biosynthesis example, enzymes are expressed in the cytoplasm, mitochondria, peroxisome, vacuole, and membrane of vacuole and endoplasmic reticulum, respectively, according to their protein transmembrane structure, cofactor regeneration and electron transport chain required for activity. This strategy of ‘regionalised’ expression not only ensures the activity and function of the enzyme, but also brings convenience to the regulation of synthetic pathways. The final product titres were reported as 30 to 80 μg L−1, hence extensive further optimisation is needed to reach grams per litre. Nevertheless, this wonderful case illustrates that the integration of classic biocatalysis and synthetic biology is constantly advancing the construction of complex enzymatic synthesis pathways in engineered whole cells and improving the research achievements of enzymatic total synthesis to a higher level.
Emerging enzyme technologies: photo-biocatalysis
Although nature provides us with a huge diversity of enzyme activities that can be redesigned and combined to construct artificial pathways, the number of chemical mechanisms used by natural enzymes is limited and chemists often desire to include transformations that have not been evolved in nature yet. Mechanistically informed protein engineering created various new-to-nature enzymes that enlarged the repertoire of biocatalytic transformations.484,485 A very recent trend in the creation of novel reactivities is the development of photocatalytic proteins. Photo-biocatalysis has become a popular research field. Compared to approaches for photocatalytic co-factor and co-substrate (re)-generation or metabolic engineering of phototrophic host organisms,486–488 the new “photoenzymes” use light directly to drive mechanistically unique small molecule conversions, which are otherwise not observed in nature. As the radical-based chemistry takes place in the chiral environment of a protein's active site, the reactions are rendered enantioselective, which is an important add-on achieved by the protein.Until recently, only four enzymes were known that used light energy directly to drive small molecule conversions.488 These enzymes – including photolyases involved in DNA repair – have not been synthetically applied. The characterisation of a fatty acid decarboxylase489 found in algal lipid metabolism complements this ensemble of natural photoenzymes and now facilitates novel reaction cascades, such as the biofuel production from triglycerides (Scheme 16A).490,491 In addition, engineered variants are able to carry out the kinetic resolution of α-hydroxy carboxylic acids,492 and the unnatural amino acid phosphinothricin, with high stereoselectivity.493
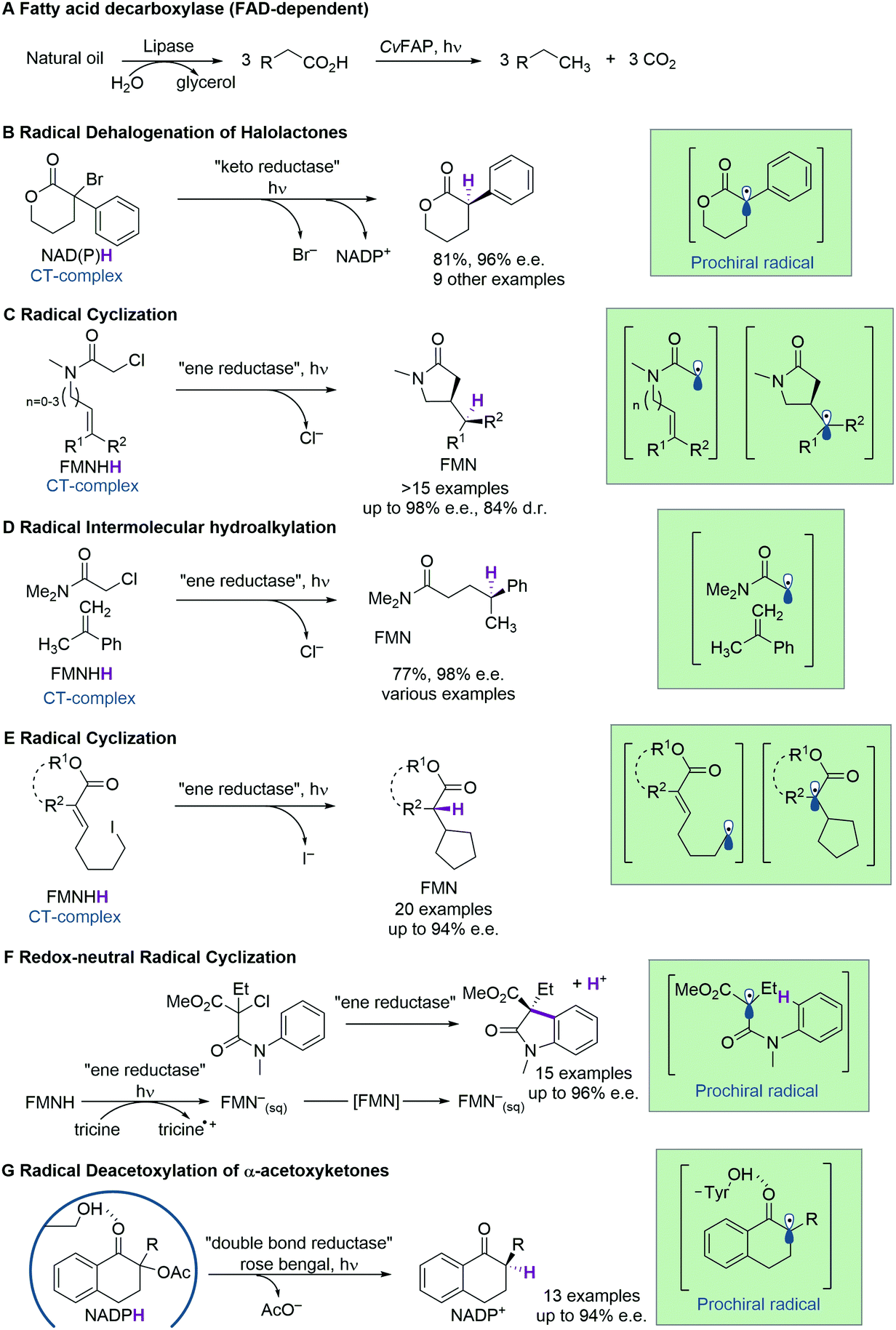 | ||
| Scheme 16 Light-induced natural and promiscuous enzymatic transformations. Fatty acid photodecarboxylase (A) is the only natural photoenzyme employed for biotransformations. The other examples (B–G) utilize the photophysical capabilities of nicotinamide and flavin cofactors to generate radical species (key intermediates are shown in the green boxes) as reactive intermediates. | ||
One approach to induce photocatalytic non-natural activities in enzymes relies on the ability of the naturally occurring nicotinamide cofactors and flavines to facilitate light-driven redox reactions that lead to radical intermediates not observed in reactions proceeding in the ground state. This possibility was explored in pioneering studies published by the Hyster lab that facilitates preparation of α-chiral lactones – a structural motif found in drug molecules such as Artemisinin and the psychotropic terpenoid Salvinorin A. Instead of constructing the chiral lactone from a chiral acid precursor, racemic α-brominated lactams are used as the starting material: a keto reductase (KRED) was shown to facilitate its enantioselective dehalogenation when irradiated with blue light (Scheme 16B).494,495 After excitation, NAD(P)H transfers a single electron to the substrate. This weakens its carbon-halogen bond and leads to loss of bromide with the subsequent formation of a prochiral radical intermediate. As the complex is still bound in the enzyme's active site, H˙ can now be delivered from NADPH+˙ in a stereoselective manner to form the chiral lactone product. Importantly, this reaction sequence is initiated while the cofactor and halogenated lactone are bound in close proximity inside the active site: only the resulting charge-transfer (CT) complex is excited at the chosen wavelength. As such a CT complex is not formed in solution, a potentially non-selective background reaction is avoided. Stereocomplementary KREDs were identified yielding the enantiomers of nine additional product analogues with up to 96% e.e. Racemic starting materials are employed, but interestingly, 85% yield clearly outperforms a classical kinetic resolution: the enzyme does not discriminate between the substrate enantiomers, and dehalogenation leads to the same central prochiral radical. However, the hydrogen atom delivery from the nicotinamide occurs only from one face and thus is highly stereoselective. This makes it possible to transform both enantiomers of the halolactone substrate to one enantiomer of the lactone product.
Based on similar principles, photocatalytic dehalogenations generated radicals suitable for cyclisation and intermolecular hydroalkylation reactions in the active sites of double bond reductases (“ene reductases”) as protein scaffolds,496–499 where a flavin mononucleotide acts as photoredox catalyst for electron and hydrogen transfer reactions. One example is the construction of β-chiral lactams, a motif found, for example, in the antiepileptic drug brivaracetam. In the conversion of α-chloroamides, the dehalogenated intermediate adds in an intramolecular addition to a double bond, and two stereocentres can be set in this way (Scheme 16C) with up to 98% e.e. and 84% d.e.499 This route differs from the conventional construction of a lactam by building and cyclizing a γ-amino acid precursor. Interestingly, this radical addition to a double bond is also possible in an intermolecular manner: in this case, a quaternary charge transfer complex suitable for excitation is formed between the protein, FMN hydroquinone, α-chloroamide, and the alkene substrate (Scheme 16D).497 Due to its modularity regarding the choice of different substrates, this hydroalkylation approach is a valuable tool for asymmetric C–C bond formation and creates a stereocentre in the γ-position of the amide function. In addition to α-halogenated ester or amide substrates, the photocatalytic approach was recently also expanded to N-alkyl-iodides, which yield unstabilised radical intermediates after the initial dehalogenation (Scheme 16E).498 All of the above-mentioned reactions require a cofactor regeneration, as reduction equivalents from FMN or NADP(H), delivered to radical intermediates during the reaction. A different mechanism is the redox-neutral cyclisation496 shown in Scheme 16F for the production of oxindoles. Here, the light-driven first step generates the flavine semiquinone form of FMN in the active site at the expense of a sacrificial tricine buffer molecule. This opens the possibility to efficiently catalyse one-electron redox reactions, which do not occur in the ground state of the ERED. The formed oxindoles feature a quaternary stereocentre and are obtained in high optical purity and up to 95% yield from the racemic starting material. Compared to the above reactions, inclusion of photoorganoredox catalysts that have strong binding to proteins can unlock further promiscuous reactions: the rose bengal radical anion, which can be formed outside of the protein by light illumination, can strongly associate with a double bond reductase and thus inject its electron into an α-acetoxyketone located in the active site (Scheme 16G).500 Here, the role of the protein is to attenuate the redox potential of the ketone substrate by a hydrogen bonding interaction to its carbonyl group. This ensures that the single electron transfer from the photocatalyst only occurs to the enzyme-bound ketone substrate and not in the bulk reaction solution, and thus facilitates the exclusive asymmetric H-atom delivery from the enzyme-bound nicotinamide cofactor. The obtained α-chiral ketones or amides can serve as valuable synthons suitable for further functionalization.
For the construction of more complex molecules, it is a plausible next step to combine photocatalytic reactions with further enzymatic conversions in a one-pot sequential fashion or even as one-pot concurrent cascade. The feasibility of this approach has been demonstrated very recently,501 and a couple of examples are available from the last two years. In most cases, the photocatalytic step generates a (reactive) intermediate that is then converted by an enzyme in the second step.487,488 Exploited photoreactions include e.g., oxyfunctionalisation of alkanes502 or alcohols503 by the water-soluble photocatalyst sodium antraquinone sulfate to yield ketones or aldehydes. Many different enzymatic reactions can then be coupled as the second step, yielding amines and cyanohydrins, amongst others. However, the one-pot two-step protocol was more efficient in terms of yield compared to the concurrent cascades. True concurrent cascades have been realised e.g., by the oxyfunctionalisation of 2-arylindoles to indol-3-ones, which can be alkylated with ketones by employing the promiscuous activities of lipases (Scheme 17A). In this way, a quaternary carbon stereocentre was constructed – a structural motif occurring in natural products such as (−)-isatisine A and (+)-hinckdentine A.504 The same Ru(bpy)3 catalyst was also used to generate a reactive but sufficiently stable radical intermediate, which could be reduced enzymatically to provide optically active pyridines, a privileged moiety in physiologically active compounds, such as the antihistamine dexchlorpheniramine:505 First, light irradiation of vinyl pyridines yields the neutral benzylic radical in the reaction solution, which then diffuses inside an enoate reductase (ERED) to get reduced to the optically active hydrocarbon (Scheme 17B). In this way, a new reactivity of EREDs was unlocked, as double bond reduction only occurs in enones, enoates, and nitroalkenes.
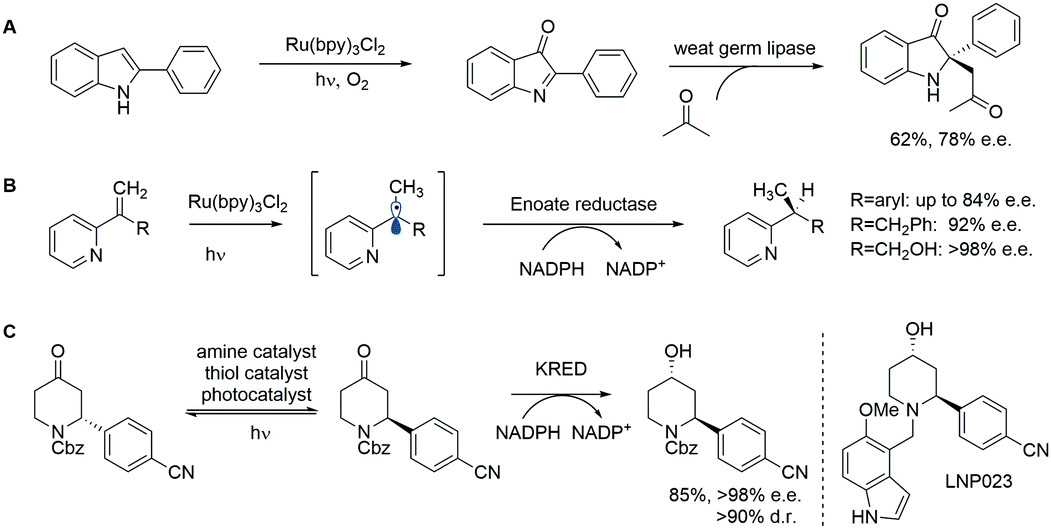 | ||
| Scheme 17 Cascade reactions combining photocatalytic and enzymatic steps. (A) Photocatalytic oxyfunctionalization generates the intermediate which is alkylated by a promiscuous lipase. (B) The photocatalyst generates a long-lived radical in the reaction solution, which is converted enzymatically. (C) A complex organo- and photocatalytic cycle allows racemization and thus to set two stereocentres in the enzymatic step. | ||
Many natural products and pharmaceuticals contain (multiple) stereocentres. If asymmetric synthesis cannot be used for setting the stereocentre, kinetic resolutions using stereoselective enzymes have been realised in many cases, but yields are often limited to 50%. A racemization step can thus substantially increase the synthesis efficiency, and photocatalysts have been shown to facilitate the desired racemization. cis/trans-photoisomerization of a CC double bond is one of the classical photocatalysed reactions and was one of the first examples coupled with an enzymatic stereoselective reduction. As the used ERED only converts the E-enantiomer, the cascade increased the efficiency of the reaction by facilitating near complete substrate conversion.501 One particular challenge is that many physiologically active molecules contain static, remote stereocentres related to a functional group, which should be targeted by the enzyme. One example is the class of 3-substituted ketones (Scheme 17C). A clever combination of an organo- and a photocatalyst facilitates racemization of the ketone enantiomers and highly selective enzymes were identified that convert preferentially one of the ketone enantiomers.506 This yielded chiral alcohols or amines featuring both excellent enantiomeric and diastereomeric ratios and could be employed for the synthesis of the drug candidate LNP023. These few exemplary cascades demonstrate the potential of combining photo- and biocatalytic steps and we anticipate a vivid exploration in this research field.
Conclusions
As chemical total synthesis is a goal of organic chemists, enzymatic total synthesis is a dream of biochemists. Since more and more achievements have resulted from in-depth studies on single-step enzyme reactions and multi-step enzymatic cascades, heterologous or in vitro total biosynthesis of natural products and complex drug compounds is becoming a reality. In this review, we emphasised that artificial intelligence, microfluidics and biosensors are greatly improving the processing speed of biological information, the mining efficiency of enzymatic reactions, and the design level of enzymatic cascades. We exemplified this progress by recent examples dealing with novel enzymes, enzymatic mechanisms and cascades discovered in the study of common natural products. These achievements gradually complement the synthetic pathways for the enzymatic total synthesis of natural products. Besides, enzymatic photocatalysis just opened a new door to construct artificial enzymatic reactions and will bring new opportunities for the streamlining and optimisation of enzymatic synthesis pathways. We also listed several impressive examples of enzymatic total synthesis in vitro, cell-free systems and recombinant whole cells to make highly complex target molecules. We hope that this review provides readers with a clear future prospect for biocatalysis: a new era of biocatalysis characterised by technological integration, intelligence and enzymatic synthesis has arrived. Clearly, the boundaries between ‘classical’ biocatalysis with a single isolated enzyme and synthetic biology using complex cascades involving a toolbox of (engineered) key enzymes are gradually disappearing.Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
This work was supported by the European Union's Horizon 2020 Research and Innovation Programme under Grant Agreement no. 814650 for the project SynBio4Flav. We also thank the Bundesministerium für Bildung und Forschung (Grant No. 01DD20002C_ZB). T. B. was supported by the Austrian Science Fund (FWF) through the Erwin Schrödinger Fellowship (grant number: J4231-B21). C. B. and S. W. were supported by a Humboldt research fellowship. M. H. received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No. 759262).Notes and references
- L. Rosenthaler, Biochem. Z., 1908, 14, 238–253 CAS.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS PubMed.
- G. E. Moore, Electronics, 1965, 38, 8 Search PubMed.
- D. M. Camacho, K. M. Collins, R. K. Powers, J. C. Costello and J. J. Collins, Cell, 2018, 173, 1581–1592 CrossRef CAS PubMed.
- J. Zou, M. Huss, A. Abid, P. Mohammadi, A. Torkamani and A. Telenti, Nat. Genet., 2019, 51, 12–18 CrossRef CAS PubMed.
- R. P. H. de Jongh, A. D. J. van Dijk, M. K. Julsing, P. J. Schaap and D. de Ridder, Trends Biotechnol., 2020, 38, 191–201 CrossRef CAS PubMed.
- G. Qu, A. Li, C. G. Acevedo-Rocha, Z. Sun and M. T. Reetz, Angew. Chem., Int. Ed., 2020, 59, 13204–13231 CrossRef CAS PubMed.
- K. K. Yang, Z. Wu and F. H. Arnold, Nat. Methods, 2019, 16, 687–694 CrossRef CAS PubMed.
- A. I. Q. Google and Collaborators, Science, 2020, 369, 1084–1089 CrossRef PubMed.
- B. Burger, P. M. Maffettone, V. V. Gusev, C. M. Aitchison, Y. Bai, X. Wang, X. Li, B. M. Alston, B. Li, R. Clowes, N. Rankin, B. Harris, R. S. Sprick and A. I. Cooper, Nature, 2020, 583, 237–241 CrossRef CAS PubMed.
- S. Gulati, V. Rouilly, X. Niu, J. Chappell, R. I. Kitney, J. B. Edel, P. S. Freemont and A. J. deMello, J. R. Soc., Interface, 2009, 6, S493–S506 CrossRef CAS PubMed.
- N. Szita, K. Polizzi, N. Jaccard and F. Baganz, Curr. Opin. Biotechnol., 2010, 21, 517–523 CrossRef CAS PubMed.
- L. Weng and J. E. Spoonamore, Micromachines, 2019, 10, 734 CrossRef PubMed.
- G. Y. Li, Y. J. Dong and M. T. Reetz, Adv. Synth. Catal., 2019, 361, 2377–2386 CAS.
- S. Mazurenko, Z. Prokop and J. Damborsky, ACS Catal., 2020, 10, 1210–1223 CrossRef CAS.
- J. Hon, S. Borko, J. Stourac, Z. Prokop, J. Zendulka, D. Bednar, T. Martinek and J. Damborsky, Nucleic Acids Res., 2020, 48, W104–W109 CrossRef CAS PubMed.
- S. Mazurenko, ChemCatChem, 2020, 12, 5590–5598 CrossRef CAS.
- M. du Lac, T. Duigou, J. Hérisson, P. Carbonell, N. Swainston, V. Zulkower, F. Shah, L. Faure, M. Mahdy, P. Soudier and J.-L. Faulon, bioRxiv, 2020 DOI:10.1101/2020.06.14.145730.
- K. F. Tipton, R. N. Armstrong, B. M. Bakker, A. Bairoch, A. Cornish-Bowden, P. J. Halling, J.-H. Hofmeyr, T. S. Leyh, C. Kettner, F. M. Raushel, J. Rohwer, D. Schomburg and C. Steinbeck, Perspect. Sci., 2014, 1, 131–137 CrossRef.
- J. Liao, M. K. Warmuth, S. Govindarajan, J. E. Ness, R. P. Wang, C. Gustafsson and J. Minshull, BMC Biotechnol., 2007, 7, 16 CrossRef PubMed.
- F. Cadet, N. Fontaine, G. Li, J. Sanchis, M. Ng Fuk Chong, R. Pandjaitan, I. Vetrivel, B. Offmann and M. T. Reetz, Sci. Rep., 2018, 8, 16757 CrossRef PubMed.
- F. Liang, X. J. Feng, M. Lowry and H. Rabitz, J. Phys. Chem. B, 2005, 109, 5842–5854 CrossRef CAS PubMed.
- S. Bartsch, R. Kourist and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2008, 47, 1508–1511 CrossRef PubMed.
- S. C. Huziel, E. Sauceda, I. Poltavsky, K.-R. Müller and A. Tkatchenko, in Machine Learning Meets Quantum Physics, ed. K. T. Schütt, S. Chmiela, O. A. von Lilienfeld, A. Tkatchenko, K. Tsuda, K.-R. Müller, Springer, Switzerland, 2020, pp. 277–307 Search PubMed.
- E. C. Alley, G. Khimulya, S. Biswas, M. AlQuraishi and G. M. Church, Nat. Methods, 2019, 16, 1315–1322 CrossRef CAS PubMed.
- A. W. Senior, R. Evans, J. Jumper, J. Kirkpatrick, L. Sifre, T. Green, C. Qin, A. Zidek, A. W. R. Nelson, A. Bridgland, H. Penedones, S. Petersen, K. Simonyan, S. Crossan, P. Kohli, D. T. Jones, D. Silver, K. Kavukcuoglu and D. Hassabis, Nature, 2020, 577, 706–710 CrossRef CAS PubMed.
- M. AlQuraishi, Cell Syst., 2019, 8, 292–301 CrossRef CAS PubMed.
- B. J. Wittmann, Y. Yue and F. H. Arnold, bioRxiv, 2020 DOI:10.1101/2020.12.04.408955.
- S. Grobe, C. P. S. Badenhorst, T. Bayer, E. Hamnevik, S. Wu, C. W. Grathwol, A. Link, S. Koban, H. Brundiek, B. Grossjohann and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 753–757 CrossRef CAS PubMed.
- Q. Tang, C. W. Grathwol, A. S. Aslan-Uzel, S. Wu, A. Link, I. V. Pavlidis, C. P. S. Badenhorst and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 1524–1527 CrossRef PubMed.
- A. Currin, N. Swainston, P. J. Day and D. B. Kell, Chem. Soc. Rev., 2015, 44, 1172–1239 RSC.
- B. Chen, S. Lim, A. Kannan, S. C. Alford, F. Sunden, D. Herschlag, I. K. Dimov, T. M. Baer and J. R. Cochran, Nat. Chem. Biol., 2016, 12, 76–81 CrossRef CAS PubMed.
- S. Lim, B. Chen, M. S. Kariolis, I. K. Dimov, T. M. Baer and J. R. Cochran, ACS Chem. Biol., 2017, 12, 336–341 CrossRef CAS PubMed.
- D. S. Tawfik and A. D. Griffiths, Nat. Biotechnol., 1998, 16, 652–656 CrossRef CAS PubMed.
- B. Kintses, C. Hein, M. F. Mohamed, M. Fischlechner, F. Courtois, C. Laine and F. Hollfelder, Chem. Biol., 2012, 19, 1001–1009 CrossRef CAS PubMed.
- J. J. Agresti, E. Antipov, A. R. Abate, K. Ahn, A. C. Rowat, J. C. Baret, M. Marquez, A. M. Klibanov, A. D. Griffiths and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4004–4009 CrossRef CAS PubMed.
- A. Sciambi and A. R. Abate, Lab Chip, 2015, 15, 47–51 RSC.
- O. Caen, S. Schutz, M. S. S. Jammalamadaka, J. Vrignon, P. Nizard, T. M. Schneider, J. C. Baret and V. Taly, Microsyst. Nanoeng., 2018, 4, 33 CrossRef PubMed.
- R. H. Cole, N. de Lange, Z. J. Gartner and A. R. Abate, Lab Chip, 2015, 15, 2754–2758 RSC.
- F. Ma, M. T. Chung, Y. Yao, R. Nidetz, L. M. Lee, A. P. Liu, Y. Feng, K. Kurabayashi and G. Y. Yang, Nat. Commun., 2018, 9, 1030 CrossRef PubMed.
- U. T. Bornscheuer, Nat. Chem. Biol., 2016, 12, 54–55 CrossRef CAS PubMed.
- P. Y. Colin, B. Kintses, F. Gielen, C. M. Miton, G. Fischer, M. F. Mohamed, M. Hyvonen, D. P. Morgavi, D. B. Janssen and F. Hollfelder, Nat. Commun., 2015, 6, 10008 CrossRef CAS PubMed.
- A. Zinchenko, S. R. Devenish, B. Kintses, P. Y. Colin, M. Fischlechner and F. Hollfelder, Anal. Chem., 2014, 86, 2526–2533 CrossRef CAS PubMed.
- F. Gielen, R. Hours, S. Emond, M. Fischlechner, U. Schell and F. Hollfelder, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E7383–E7389 CrossRef CAS PubMed.
- P. J. Zurek, P. Knyphausen, K. Neufeld, A. Pushpanath and F. Hollfelder, Nat. Commun., 2020, 11, 6023 CrossRef CAS PubMed.
- D. A. Holland-Moritz, M. K. Wismer, B. F. Mann, I. Farasat, P. Devine, E. D. Guetschow, I. Mangion, C. J. Welch, J. C. Moore, S. Sun and R. T. Kennedy, Angew. Chem., Int. Ed., 2020, 59, 4470–4477 CrossRef CAS PubMed.
- D. R. Link, S. L. Anna, D. A. Weitz and H. A. Stone, Phys. Rev. Lett., 2004, 92, 054503 CrossRef CAS PubMed.
- E. Brouzes, M. Medkova, N. Savenelli, D. Marran, M. Twardowski, J. B. Hutchison, J. M. Rothberg, D. R. Link, N. Perrimon and M. L. Samuels, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14195–14200 CrossRef CAS PubMed.
- A. R. Abate, T. Hung, P. Mary, J. J. Agresti and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19163–19166 CrossRef CAS PubMed.
- Z. Tan and J. M. Heemstra, ChemBioChem, 2018, 19, 1853–1857 CrossRef CAS PubMed.
- J. Abatemarco, M. F. Sarhan, J. M. Wagner, J. L. Lin, L. Liu, W. Hassouneh, S. F. Yuan, H. S. Alper and A. R. Abate, Nat. Commun., 2017, 8, 332 CrossRef PubMed.
- B. Kintses, L. D. van Vliet, S. R. Devenish and F. Hollfelder, Curr. Opin. Chem. Biol., 2010, 14, 548–555 CrossRef CAS PubMed.
- S. Neun, P. J. Zurek, T. S. Kaminski and F. Hollfelder, Methods Enzymol., 2020, 643, 317–343 Search PubMed.
- A. Debon, M. Pott, R. Obexer, A. P. Green, L. Friedrich, A. D. Griffiths and D. Hilvert, Nat. Catal., 2019, 2, 740–747 CrossRef CAS.
- R. Obexer, A. Godina, X. Garrabou, P. R. Mittl, D. Baker, A. D. Griffiths and D. Hilvert, Nat. Chem., 2017, 9, 50–56 CrossRef CAS PubMed.
- B. van Loo, M. Heberlein, P. Mair, A. Zinchenko, J. Schuurmann, B. D. G. Eenink, J. M. Holstein, C. Dilkaute, J. Jose, F. Hollfelder and E. Bornberg-Bauer, ACS Synth. Biol., 2019, 8, 2690–2700 CrossRef CAS PubMed.
- P. J. Zurek, R. Hours, U. Schell, A. Pushpanath and F. Hollfelder, Lab Chip, 2021, 21, 163–173 RSC.
- S. Neun, T. S. Kaminski and F. Hollfelder, Methods Enzymol., 2019, 628, 95–112 CAS.
- P. Gruner, B. Riechers, B. Semin, J. Lim, A. Johnston, K. Short and J. C. Baret, Nat. Commun., 2016, 7, 10392 CrossRef CAS PubMed.
- G. Woronoff, A. El Harrak, E. Mayot, O. Schicke, O. J. Miller, P. Soumillion, A. D. Griffiths and M. Ryckelynck, Anal. Chem., 2011, 83, 2852–2857 CrossRef CAS PubMed.
- T. Buryska, M. Vasina, F. Gielen, P. Vanacek, L. van Vliet, J. Jezek, Z. Pilat, P. Zemanek, J. Damborsky, F. Hollfelder and Z. Prokop, Anal. Chem., 2019, 91, 10008–10015 CrossRef CAS PubMed.
- M. Pan, L. Rosenfeld, M. Kim, M. Xu, E. Lin, R. Derda and S. K. Tang, ACS Appl. Mater. Interfaces, 2014, 6, 21446–21453 CrossRef CAS PubMed.
- M. Pan, F. Lyu and S. K. Tang, Anal. Chem., 2015, 87, 7938–7943 CrossRef CAS PubMed.
- L. You and F. H. Arnold, Protein Eng., 1994, 9, 77–83 CrossRef PubMed.
- X. Wang, Y. Xin, L. Ren, Z. Sun, P. Zhu, Y. Ji, C. Li, J. Xu and B. Ma, Sci. Adv., 2020, 6, eabb3521 CrossRef CAS.
- E. E. Kempa, C. A. Smith, X. Li, B. Bellina, K. Richardson, S. Pringle, J. L. Galman, N. J. Turner and P. E. Barran, Anal. Chem., 2020, 92, 12605–12612 CrossRef CAS PubMed.
- J. M. Holstein, C. Gylstorff and F. Hollfelder, ACS Synth. Biol., 2021, 10, 252–257 CrossRef CAS PubMed.
- R. M. Maceiczyk, D. Hess, F. W. Y. Chiu, S. Stavrakis and A. J. deMello, Lab Chip, 2017, 17, 3654–3663 RSC.
- M. Hosokawa, Y. Hoshino, Y. Nishikawa, T. Hirose, D. H. Yoon, T. Mori, T. Sekiguchi, S. Shoji and H. Takeyama, Biosens. Bioelectron., 2015, 67, 379–385 CrossRef CAS PubMed.
- T. Bayer, S. Milker, T. Wiesinger, F. Rudroff and M. D. Mihovilovic, Adv. Synth. Catal., 2015, 357, 1587–1618 CrossRef CAS.
- G. S. Hossain, M. Saini, R. Miyake, H. Ling and M. W. Chang, Trends Biotechnol., 2020, 38, 797–810 CrossRef CAS PubMed.
- S. Cardinale and A. P. Arkin, Biotechnol. J., 2012, 7, 856–866 CrossRef CAS PubMed.
- S. Jang, S. Jang, Y. Xiu, T. J. Kang, S.-H. Lee, M. A. G. Koffas and G. Y. Jung, ACS Synth. Biol., 2017, 6, 2077–2085 CrossRef CAS PubMed.
- A. Santos-Zavaleta, M. Sánchez-Pérez, H. Salgado, D. A. Velázquez-Ramírez, S. Gama-Castro, V. H. Tierrafría, S. J. W. Busby, P. Aquino, X. Fang, B. O. Palsson, J. E. Galagan and J. Collado-Vides, BMC Biol., 2018, 16, 91 CrossRef PubMed.
- U. Markel, K. D. Essani, V. Besirlioglu, J. Schiffels, W. R. Streit and U. Schwaneberg, Chem. Soc. Rev., 2020, 49, 233–262 RSC.
- R. Lutz and H. Bujard, Nucleic Acids Res., 1997, 25, 1203–1210 CrossRef CAS PubMed.
- A. Mullick, Y. Xu, R. Warren, M. Koutroumanis, C. Guilbault, S. Broussau, F. Malenfant, L. Bourget, L. Lamoureux, R. Lo, A. W. Caron, A. Pilotte and B. Massie, BMC Biotechnol., 2006, 6, 43 CrossRef PubMed.
- Y. J. Choi, L. Morel, T. Le François, D. Bourque, L. Bourget, D. Groleau, B. Massie and C. B. Míguez, Appl. Environ. Microbiol., 2010, 76, 5058–5066 CrossRef CAS PubMed.
- A. Gawin, S. Valla and T. Brautaset, Microbiol. Biotechnol., 2017, 10, 702–718 CrossRef CAS PubMed.
- J. A. N. Brophy and C. A. Voigt, Nat. Methods, 2014, 11, 508–520 CrossRef CAS PubMed.
- J. A. Dietrich, A. E. McKee and J. D. Keasling, Annu. Rev. Biochem., 2010, 79, 563–590 CrossRef CAS PubMed.
- D. Liu, T. Evans and F. Zhang, Metab. Eng., 2015, 31, 35–43 CrossRef CAS PubMed.
- J. A. Dietrich, D. L. Shis, A. Alikhani and J. D. Keasling, ACS Synth. Biol., 2013, 2, 47–58 CrossRef CAS PubMed.
- S. Li, T. Si, M. Wang and H. Zhao, ACS Synth. Biol., 2015, 4, 1308–1315 CrossRef CAS PubMed.
- W. C. DeLoache, Z. N. Russ, L. Narcross, A. M. Gonzales, V. J. Martin and J. E. Dueber, Nat. Chem. Biol., 2015, 11, 465–471 CrossRef CAS PubMed.
- Y. Liu, Y. Zhuang, D. Ding, Y. Xu, J. Sun and D. Zhang, ACS Synth. Biol., 2017, 6, 837–848 CrossRef CAS.
- H. H. Chou and J. D. Keasling, Nat. Commun., 2013, 4, 2595 CrossRef PubMed.
- G. Schendzielorz, M. Dippong, A. Grünberger, D. Kohlheyer, A. Yoshida, S. Binder, C. Nishiyama, M. Nishiyama, M. Bott and L. Eggeling, ACS Synth. Biol., 2014, 3, 21–29 CrossRef CAS PubMed.
- S. Raman, J. K. Rogers, N. D. Taylor and G. M. Church, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17803–17808 CrossRef CAS PubMed.
- D. C. Volke, J. Turlin, V. Mol and P. I. Nikel, Microbiol. Biotechnol., 2020, 13, 222–232 CrossRef CAS PubMed.
- M. Rienzo, S. J. Jackson, L. K. Chao, T. Leaf, T. J. Schmidt, A. H. Navidi, D. C. Nadler, M. Ohler and M. D. Leavell, Metab. Eng., 2021, 63, 102–125 CrossRef CAS PubMed.
- F. Zhang, J. M. Carothers and J. D. Keasling, Nat. Biotechnol., 2012, 30, 354–359 CrossRef CAS PubMed.
- P. Xu, L. Li, F. Zhang, G. Stephanopoulos and M. Koffas, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11299–11304 CrossRef CAS PubMed.
- S. J. Doong, A. Gupta and K. L. J. Prather, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 2964–2969 CrossRef CAS PubMed.
- S. Shi, Y. W. Choi, H. Zhao, M. H. Tan and E. L. Ang, Bioresour. Technol., 2017, 245, 1343–1351 CrossRef CAS PubMed.
- R. H. Dahl, F. Zhang, J. Alonso-Gutierrez, E. Baidoo, T. S. Batth, A. M. Redding-Johanson, C. J. Petzold, A. Mukhopadhyay, T. S. Lee, P. D. Adams and J. D. Keasling, Nat. Biotechnol., 2013, 31, 1039–1046 CrossRef CAS PubMed.
- Y. Liu, Q. Li, P. Zheng, Z. Zhang, Y. Liu, C. Sun, G. Cao, W. Zhou, X. Wang, D. Zhang, T. Zhang, J. Sun and Y. Ma, Microb. Cell Fact., 2015, 14, 121 CrossRef PubMed.
- M. Koch, A. Pandi, B. Delepine and J. L. Faulon, Data Brief, 2018, 17, 1374–1378 CrossRef PubMed.
- B. Delépine, V. Libis, P. Carbonell and J.-L. Faulon, Nucleic Acids Res., 2016, 44, W226–W231 CrossRef PubMed.
- D. Jain, IUBMB Life, 2015, 67, 556–563 CrossRef CAS PubMed.
- N. D. Taylor, A. S. Garruss, R. Moretti, S. Chan, M. A. Arbing, D. Cascio, J. K. Rogers, F. J. Isaacs, S. Kosuri, D. Baker, S. Fields, G. M. Church and S. Raman, Nat. Methods, 2016, 13, 177–183 CrossRef CAS PubMed.
- D. Xiong, S. Lu, J. Wu, C. Liang, W. Wang, W. Wang, J.-M. Jin and S.-Y. Tang, Metab. Eng., 2017, 40, 115–123 CrossRef CAS PubMed.
- M. Espinosa-Urgel, L. Serrano, J. L. Ramos and A. M. Fernández-Escamilla, Mol. Biotechnol., 2015, 57, 558–564 CrossRef CAS PubMed.
- T. Snoek, E. K. Chaberski, F. Ambri, S. Kol, S. P. Bjørn, B. Pang, J. F. Barajas, D. H. Welner, M. K. Jensen and J. D. Keasling, Nucleic Acids Res., 2019, 48, e3 CrossRef PubMed.
- S. Meinhardt, M. W. Manley, Jr., N. A. Becker, J. A. Hessman, L. J. Maher, 3rd and L. Swint-Kruse, Nucleic Acids Res., 2012, 40, 11139–11154 CrossRef CAS PubMed.
- J. Zhang, J. F. Barajas, M. Burdu, T. L. Ruegg, B. Dias and J. D. Keasling, ACS Synth. Biol., 2017, 6, 439–445 CrossRef CAS PubMed.
- B. De Paepe, G. Peters, P. Coussement, J. Maertens and M. De Mey, J. Ind. Microbiol. Biotechnol., 2017, 44, 623–645 CrossRef CAS PubMed.
- C. E. Lehning, S. Siedler, M. M. H. Ellabaan and M. O. A. Sommer, Metab. Eng., 2017, 42, 194–202 CrossRef CAS.
- G. Peters, B. De Paepe, L. De Wannemaeker, D. Duchi, J. Maertens, J. Lammertyn and M. De Mey, Biotechnol. Bioeng., 2018, 115, 1855–1865 CrossRef CAS.
- W. Cui, L. Han, J. Cheng, Z. Liu, L. Zhou, J. Guo and Z. Zhou, Microb. Cell Fact., 2016, 15, 199 CrossRef PubMed.
- Y.-H. Wang, M. McKeague, T. M. Hsu and C. D. Smolke, Cell Syst., 2016, 3, 549–562 CrossRef CAS PubMed.
- A. M. Kunjapur and K. L. J. Prather, ACS Synth. Biol., 2019, 8, 1958–1967 CrossRef CAS.
- L. K. Flachbart, S. Sokolowsky and J. Marienhagen, ACS Synth. Biol., 2019, 8, 1847–1857 CrossRef CAS.
- Y. Dabirian, X. Li, Y. Chen, F. David, J. Nielsen and V. Siewers, ACS Synth. Biol., 2019, 8, 1968–1975 CrossRef CAS PubMed.
- S. Gama-Castro, H. Salgado, A. Santos-Zavaleta, D. Ledezma-Tejeida, L. Muñiz-Rascado, J. S. García-Sotelo, K. Alquicira-Hernández, I. Martínez-Flores, L. Pannier, J. A. Castro-Mondragón, A. Medina-Rivera, H. Solano-Lira, C. Bonavides-Martínez, E. Pérez-Rueda, S. Alquicira-Hernández, L. Porrón-Sotelo, A. López-Fuentes, A. Hernández-Koutoucheva, V. D. Moral-Chávez, F. Rinaldi and J. Collado-Vides, Nucleic Acids Res., 2015, 44, D133–D143 CrossRef PubMed.
- A. Rajput, K. Kaur and M. Kumar, Nucleic Acids Res., 2015, 44, D634–D639 CrossRef PubMed.
- A. Berepiki, R. Kent, L. F. M. Machado and N. Dixon, ACS Synth. Biol., 2020, 9, 576–589 CrossRef CAS PubMed.
- A. M. Watkins, R. Rangan and R. Das, in Methods Enzymol., ed. A. E. Hargrove, Academic Press, 2019, vol. 623, pp. 177–207 Search PubMed.
- Z. Miao, R. W. Adamiak, M. Antczak, R. T. Batey, A. J. Becka, M. Biesiada, M. J. Boniecki, J. M. Bujnicki, S. J. Chen, C. Y. Cheng, F. C. Chou, A. R. Ferre-D'Amare, R. Das, W. K. Dawson, F. Ding, N. V. Dokholyan, S. Dunin-Horkawicz, C. Geniesse, K. Kappel, W. Kladwang, A. Krokhotin, G. E. Lach, F. Major, T. H. Mann, M. Magnus, K. Pachulska-Wieczorek, D. J. Patel, J. A. Piccirilli, M. Popenda, K. J. Purzycka, A. Ren, G. M. Rice, J. Santalucia, Jr., J. Sarzynska, M. Szachniuk, A. Tandon, J. J. Trausch, S. Tian, J. Wang, K. M. Weeks, B. Williams, 2nd, Y. Xiao, X. Xu, D. Zhang, T. Zok and E. Westhof, RNA, 2017, 23, 655–672 CrossRef CAS PubMed.
- S. Gong, Y. Wang, Z. Wang and W. Zhang, Int. J. Mol. Sci., 2017, 18, 2175 CrossRef PubMed.
- S. Mukherjee, S. Das Mandal, N. Gupta, M. Drory-Retwitzer, D. Barash and S. Sengupta, Bioinformatics, 2019, 35, 3541–3543 CrossRef CAS PubMed.
- I. Kalvari, E. P. Nawrocki, J. Argasinska, N. Quinones-Olvera, R. D. Finn, A. Bateman and A. I. Petrov, Curr. Protoc. Bioinform., 2018, 62, e51 CrossRef PubMed.
- M. R. Naghdi, K. Smail, J. X. Wang, F. Wade, R. R. Breaker and J. Perreault, Methods, 2017, 117, 3–13 CrossRef CAS PubMed.
- P. Calero, D. C. Volke, P. T. Lowe, C. H. Gotfredsen, D. O’Hagan and P. I. Nikel, Nat. Commun., 2020, 11, 5045 CrossRef CAS PubMed.
- C. Zhu, G. Yang, M. Ghulam, L. Li and F. Qu, Biotechnol. Adv., 2019, 37, 107432 CrossRef CAS PubMed.
- S. Jang, S. Jang, D.-K. Im, T. J. Kang, M.-K. Oh and G. Y. Jung, ACS Synth. Biol., 2019, 8, 1276–1283 CrossRef CAS PubMed.
- Z. F. Hallberg, Y. Su, R. Z. Kitto and M. C. Hammond, Ann. Rev. Biochem., 2017, 86, 515–539 CrossRef CAS PubMed.
- H. L. Pham, A. Wong, N. Chua, W. S. Teo, W. S. Yew and M. W. Chang, Nat. Commun., 2017, 8, 411 CrossRef PubMed.
- Y. Nomura and Y. Yokobayashi, J. Am. Chem. Soc., 2007, 129, 13814–13815 CrossRef CAS PubMed.
- Y. Dai, A. Furst and C. C. Liu, Trends Biotechnol., 2019, 37, 1367–1382 CrossRef CAS PubMed.
- Z. Zhang, C. Hejesen, M. B. Kjelstrup, V. Birkedal and K. V. Gothelf, J. Am. Chem. Soc., 2014, 136, 11115–11120 CrossRef CAS PubMed.
- S. Siedler, G. Schendzielorz, S. Binder, L. Eggeling, S. Bringer and M. Bott, ACS Synth. Biol., 2014, 3, 41–47 CrossRef CAS PubMed.
- R. Mahr, C. Gätgens, J. Gätgens, T. Polen, J. Kalinowski and J. Frunzke, Metab. Eng., 2015, 32, 184–194 CrossRef CAS PubMed.
- H. Yu, N. Wang, W. Huo, Y. Zhang, W. Zhang, Y. Yang, Z. Chen and Y.-X. Huo, Microb. Cell Fact., 2019, 18, 30 CrossRef PubMed.
- S. Siedler, S. G. Stahlhut, S. Malla, J. Maury and A. R. Neves, Metab. Eng., 2014, 21, 2–8 CrossRef CAS PubMed.
- S. Qian and P. C. Cirino, Curr. Opin. Chem. Eng., 2016, 14, 93–102 CrossRef.
- J. K. Rogers and G. M. Church, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2388–2393 CrossRef CAS PubMed.
- S. Siedler, N. K. Khatri, A. Zsohár, I. Kjærbølling, M. Vogt, P. Hammar, C. F. Nielsen, J. Marienhagen, M. O. A. Sommer and H. N. Joensson, ACS Synth. Biol., 2017, 6, 1860–1869 CrossRef PubMed.
- L. Li, R. Tu, G. Song, J. Cheng, W. Chen, L. Li, L. Wang and Q. Wang, ACS Synth. Biol., 2019, 8, 297–306 CrossRef PubMed.
- J. F. Juárez, B. Lecube-Azpeitia, S. L. Brown, C. D. Johnston and G. M. Church, Nat. Commun., 2018, 9, 3101 CrossRef PubMed.
- P. L. Voyvodic, A. Pandi, M. Koch, I. Conejero, E. Valjent, P. Courtet, E. Renard, J.-L. Faulon and J. Bonnet, Nat. Commun., 2019, 10, 1697 CrossRef PubMed.
- Y. Dabirian, P. Gonçalves Teixeira, J. Nielsen, V. Siewers and F. David, ACS Synth. Biol., 2019, 8, 1788–1800 CrossRef CAS PubMed.
- E. Kalkreuter, A. M. Keeler, A. A. Malico, K. S. Bingham, A. K. Gayen and G. J. Williams, ACS Synth. Biol., 2019, 8, 1391–1400 CrossRef CAS PubMed.
- W. S. Teo, K. S. Hee and M. W. Chang, Eng. Life Sci., 2013, 13, 456–463 CrossRef CAS.
- T. Lehtinen, E. Efimova, S. Santala and V. Santala, Microb. Cell Fact., 2018, 17, 19 CrossRef PubMed.
- T. Lehtinen, V. Santala and S. Santala, FEMS Microbiol. Lett., 2017, 364, fnx053 CrossRef PubMed.
- K. Mukherjee, S. Bhattacharyya and P. Peralta-Yahya, ACS Synth. Biol., 2015, 4, 1261–1269 CrossRef CAS PubMed.
- N. K. Van Wyk, H. Pretorius and I. S. Pretorius, Fermentation, 2018, 4, 54 CrossRef.
- S. K. Kim, S. H. Kim, B. Subhadra, S.-G. Woo, E. Rha, S.-W. Kim, H. Kim, D.-H. Lee and S.-G. Lee, ACS Synth. Biol., 2018, 7, 2379–2390 CrossRef CAS PubMed.
- C. Wang, M. Liwei, J. B. Park, S. H. Jeong, G. Wei, Y. Wang and S. W. Kim, Front. Microbiol., 2018, 9, 2460 CrossRef PubMed.
- M. Mazumder and D. R. McMillen, Nucleic Acids Res., 2014, 42, 9514–9522 CrossRef CAS.
- A. Chamas, H. T. M. Pham, M. Jähne, K. Hettwer, S. Uhlig, K. Simon, A. Einspanier, K. Baronian and G. Kunze, Biotechnol. Bioeng., 2017, 114, 1539–1549 CrossRef CAS PubMed.
- E. Maser and G. Xiong, J. Steroid Biochem. Mol. Biol., 2010, 121, 633–640 CrossRef CAS PubMed.
- C. Grazon, R. C. Baer, U. Kuzmanović, T. Nguyen, M. Chen, M. Zamani, M. Chern, P. Aquino, X. Zhang, S. Lecommandoux, A. Fan, M. Cabodi, C. Klapperich, M. W. Grinstaff, A. M. Dennis and J. E. Galagan, Nat. Commun., 2020, 11, 1276 CrossRef CAS PubMed.
- F. Piroozmand, F. Mohammadipanah and F. Faridbod, Synth. Syst. Biotechnol., 2020, 5, 293–303 CrossRef PubMed.
- B. De Paepe, J. Maertens, B. Vanholme and M. De Mey, ACS Synth. Biol., 2019, 8, 318–331 CrossRef CAS PubMed.
- L. Swint-Kruse and K. S. Matthews, Curr. Opin. Microbiol., 2009, 12, 129–137 CrossRef CAS PubMed.
- S. Meinhardt and L. Swint-Kruse, Proteins, 2008, 73, 941–957 CrossRef CAS PubMed.
- M. Marsafari, H. Samizadeh, B. Rabiei, A. Mehrabi, M. Koffas and P. Xu, Biotechnol. J., 2020, 15, e1900432 CrossRef PubMed.
- X.-F. Chen, X.-X. Xia, S. Y. Lee and Z.-G. Qian, Biotechnol. Bioeng., 2018, 115, 1014–1027 CrossRef CAS.
- J. K. Rogers, C. D. Guzman, N. D. Taylor, S. Raman, K. Anderson and G. M. Church, Nucleic Acids Res., 2015, 43, 7648–7660 CrossRef CAS PubMed.
- W. Wu, L. Zhang, L. Yao, X. Tan, X. Liu and X. Lu, Sci. Rep., 2015, 5, 10907 CrossRef CAS PubMed.
- A. M. Ehrenworth, T. Claiborne and P. Peralta-Yahya, Biochemistry, 2017, 56, 5471–5475 CrossRef CAS PubMed.
- M. Hicks, T. T. Bachmann and B. Wang, ChemPhysChem, 2020, 21, 132–144 CrossRef CAS PubMed.
- J. Feng, B. W. Jester, C. E. Tinberg, D. J. Mandell, M. S. Antunes, R. Chari, K. J. Morey, X. Rios, J. I. Medford, G. M. Church, S. Fields and D. Baker, eLife, 2015, 4, e10606 CrossRef PubMed.
- Y. Liu, J. Chen, D. Crisante, J. M. Jaramillo Lopez and R. Mahadevan, ACS Synth. Biol., 2020, 9, 1284–1291 CrossRef CAS PubMed.
- F. Ceroni, A. Boo, S. Furini, T. E. Gorochowski, O. Borkowski, Y. N. Ladak, A. R. Awan, C. Gilbert, G.-B. Stan and T. Ellis, Nat. Methods, 2018, 15, 387–393 CrossRef CAS PubMed.
- D. Cunningham-Bryant, J. Sun, B. Fernandez and J. G. Zalatan, ChemBioChem, 2019, 20, 1519–1523 CAS.
- Y. Lv, S. Qian, G. Du, J. Chen, J. Zhou and P. Xu, Metab. Eng., 2019, 54, 109–116 CrossRef CAS PubMed.
- M. L. Falcone Ferreyra, S. P. Rius and P. Casati, Front. Plant Sci., 2012, 3, 222 CAS.
- C. Y. Zhao, F. Wang, Y. H. Lian, H. Xiao and J. K. Zheng, Crit. Rev. Food Sci. Nutr., 2020, 60, 566–583 CrossRef CAS.
- R. P. Pandey, P. Parajuli, M. A. G. Koffas and J. K. Sohng, Biotechnol. Adv., 2016, 34, 634–662 CrossRef CAS PubMed.
- S. M. Nabavi, D. Samec, M. Tomczyk, L. Milella, D. Russo, S. Habtemariam, I. Suntar, L. Rastrelli, M. Daglia, J. Xiao, F. Giampieri, M. Battino, E. Sobarzo-Sanchez, S. F. Nabavi, B. Yousefi, P. Jeandet, S. Xu and S. Shirooie, Biotechnol. Adv., 2020, 38, 107316 CrossRef CAS PubMed.
- W. J. C. de Bruijn, M. Levisson, J. Beekwilder, W. J. H. van Berkel and J. P. Vincken, Trends Biotechnol., 2020, 38, 917–934 CrossRef CAS PubMed.
- Y. C. F. Teles, M. S. R. Souza and M. F. V. Souza, Molecules, 2018, 23, 480 CrossRef PubMed.
- A. Berim and D. R. Gang, Phytochem. Rev., 2016, 15, 363–390 CrossRef CAS.
- M. W. Hyun, Y. H. Yun, J. Y. Kim and S. H. Kim, Mycobiology, 2011, 39, 257–265 CrossRef CAS PubMed.
- J. Barros and R. A. Dixon, Trends Plant Sci., 2020, 25, 66–79 CrossRef CAS PubMed.
- S. Bartsch and U. T. Bornscheuer, Protein Eng., Des. Sel., 2010, 23, 929–933 CrossRef CAS PubMed.
- J. D. Cui, J. Q. Qiu, X. W. Fan, S. R. Jia and Z. L. Tan, Crit. Rev. Biotechnol., 2014, 34, 258–268 CrossRef CAS PubMed.
- T. Vannelli, W. W. Qi, J. Sweigard, A. A. Gatenby and F. S. Sariaslani, Metab. Eng., 2007, 9, 142–151 CrossRef CAS.
- C. N. S. Santos, M. Koffas and G. Stephanopoulos, Metab. Eng., 2011, 13, 392–400 CrossRef CAS.
- J. J. Wu, G. C. Du, J. W. Zhou and J. Chen, Metab. Eng., 2013, 16, 48–55 CrossRef PubMed.
- J. J. Wu, P. R. Liu, Y. M. Fan, H. Bao, G. C. Du, J. W. Zhou and J. Chen, J. Biotechnol., 2013, 167, 404–411 CrossRef CAS PubMed.
- S. Zhou, P. Liu, J. Chen, G. Du, H. Li and J. Zhou, Appl. Microbiol. Biotechnol., 2016, 100, 10443–10452 CrossRef CAS PubMed.
- K. T. Watts, B. N. Mijts, P. C. Lee, A. J. Manning and C. Schmidt-Dannert, Chem. Biol., 2006, 13, 1317–1326 CrossRef CAS PubMed.
- P. Cui, W. Zhong, Y. Qin, F. Tao, W. Wang and J. Zhan, Bioprocess Biosyst. Eng., 2020, 43, 1287–1298 CrossRef CAS PubMed.
- S. A. Pandith, S. Ramazan, M. I. Khan, Z. A. Reshi and M. A. Shah, Planta, 2019, 251, 15 CrossRef.
- T. Waki, R. Mameda, T. Nakano, S. Yamada, M. Terashita, K. Ito, N. Tenma, Y. Li, N. Fujino, K. Uno, S. Yamashita, Y. Aoki, K. Denessiouk, Y. Kawai, S. Sugawara, K. Saito, K. Yonekura-Sakakibara, Y. Morita, A. Hoshino, S. Takahashi and T. Nakayama, Nat. Commun., 2020, 11, 870 CrossRef CAS PubMed.
- M. N. Ngaki, G. V. Louie, R. N. Philippe, G. Manning, F. Pojer, M. E. Bowman, L. Li, E. Larsen, E. S. Wurtele and J. P. Noel, Nature, 2012, 485, 530–533 CrossRef CAS PubMed.
- M. Kaltenbach, J. R. Burke, M. Dindo, A. Pabis, F. S. Munsberg, A. Rabin, S. C. L. Kamerlin, J. P. Noel and D. S. Tawfik, Nat. Chem. Biol., 2018, 14, 548–555 CrossRef CAS PubMed.
- Y. C. Yin, X. D. Zhang, Z. Q. Gao, T. Hu and Y. Liu, Mol. Biotechnol., 2019, 61, 32–52 CrossRef CAS PubMed.
- J. R. Burke, J. J. La Clair, R. N. Philippe, A. Pabis, M. Corbella, J. M. Jez, G. A. Cortina, M. Kaltenbach, M. E. Bowman, G. V. Louie, K. B. Woods, A. T. Nelson, D. S. Tawfik, S. C. L. Kamerlin and J. P. Noel, ACS Catal., 2019, 9, 8388–8396 CrossRef CAS.
- C. Herles, A. Braune and M. Blaut, Arch. Microbiol., 2004, 181, 428–434 CrossRef CAS PubMed.
- M. Gall, M. Thomsen, C. Peters, I. V. Pavlidis, P. Jonczyk, P. P. Grunert, S. Beutel, T. Scheper, E. Gross, M. Backes, T. Geissler, J. P. Ley, J. M. Hilmer, G. Krammer, G. J. Palm, W. Hinrichs and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2014, 53, 1439–1442 CrossRef CAS PubMed.
- M. Thomsen, A. Tuukkanen, J. Dickerhoff, G. J. Palm, H. Kratzat, D. I. Svergun, K. Weisz, U. T. Bornscheuer and W. Hinrichs, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2015, 71, 907–917 CrossRef CAS PubMed.
- A. Braune, W. Engst, P. W. Elsinghorst, N. Furtmann, J. Bajorath, M. Gutschow and M. Blaut, J. Bacteriol., 2016, 198, 2965–2974 CrossRef CAS.
- A. Braune, M. Gutschow and M. Blaut, Appl. Environ. Microbiol., 2019, 85, e01233 CrossRef CAS PubMed.
- J. A. Jones, S. M. Collins, V. R. Vernacchio, D. M. Lachance and M. A. Koffas, Biotechnol. Prog., 2016, 32, 21–25 CrossRef CAS PubMed.
- B. G. Kim, S. H. Sung, Y. Chong, Y. Lim and J. H. Ahn, J. Plant Biol., 2010, 53, 321–329 CrossRef CAS.
- Y. J. Lee, B. G. Kim, Y. Park, Y. Lim, H. G. Hur and J. H. Ahn, J. Microbiol. Biotechnol., 2006, 16, 1090–1096 CAS.
- S. Darsandhari, D. Dhakal, B. Shrestha, P. Parajuli, J. H. Seo, T. S. Kim and J. K. Sohng, Enzyme Microb. Technol., 2018, 113, 29–36 CrossRef CAS PubMed.
- B. G. Kim, B. R. Jung, Y. Lee, H. G. Hur, Y. Lim and J. H. Ahn, J. Agric. Food Chem., 2006, 54, 823–828 CrossRef CAS PubMed.
- L. Wessjohann, M. Dippe, M. Tengg and M. Gruber-Khadjawi, Cascade Biocatalysis: Integrating Stereoselective and Environmentally Friendly Reactions, ed. S. Riva and W.-D. Fessner, John Wiley & Sons, New Jersey, 2014, pp. 393–426 Search PubMed.
- L. Wessjohann, A.-K. Bauer, M. Dippe, J. Ley and T. Geißler, Applied Biocatalysis: From Fundamental Science to Industrial Applications, ed. L. Hilterhaus, A. Liese, U. Kettling and G. Antranikian, John Wiley & Sons, New Jersey, 2016, pp. 121–146 Search PubMed.
- S. Mordhorst, J. Siegrist, M. Müller, M. Richter and J. N. Andexer, Angew. Chem., Int. Ed., 2017, 56, 4037–4041 CrossRef CAS PubMed.
- J. D. Finkelstein, Clin. Chem. Lab. Med., 2007, 45, 1694–1699 CAS.
- C. S. Liao and F. P. Seebeck, Nat. Catal., 2019, 2, 696–701 CrossRef CAS.
- C. Dalhoff, G. Lukinavičius, S. Klima
![[s with combining breve]](https://www.rsc.org/images/entities/char_0073_0306.gif) auskas and E. Weinhold, Nat. Chem. Biol., 2006, 2, 31–32 CrossRef CAS PubMed.
auskas and E. Weinhold, Nat. Chem. Biol., 2006, 2, 31–32 CrossRef CAS PubMed. - S. Singh, J. Zhang, T. D. Huber, M. Sunkara, K. Hurley, R. D. Goff, G. Wang, W. Zhang, C. Liu and J. Rohr, Angew. Chem., Int. Ed., 2014, 53, 3965–3969 CrossRef CAS.
- M. Thomsen, S. B. Vogensen, J. Buchardt, M. D. Burkart and R. P. Clausen, Org. Biomol. Chem., 2013, 11, 7606–7610 RSC.
- J. Micklefield, Nat. Catal., 2019, 2, 644–645 CrossRef CAS.
- R. Munakata, T. Takemura, K. Tatsumi, E. Moriyoshi, K. Yanagihara, A. Sugiyama, H. Suzuki, H. Seki, T. Muranaka, N. Kawano, K. Yoshimatsu, N. Kawahara, T. Yamaura, J. Grosjean, F. Bourgaud, A. Hehn and K. Yazaki, Commun. Biol., 2019, 2, 384 CrossRef PubMed.
- J. Yang, T. Zhou, Y. Jiang and B. Yang, Int. J. Biol. Macromol., 2020, 153, 264–275 CrossRef CAS PubMed.
- K. Zhou, X. Yu, X. Xie and S. M. Li, J. Nat. Prod., 2015, 78, 2229–2235 CrossRef CAS PubMed.
- J. Couillaud, J. Rico, A. Rubini, T. Hamrouni, E. Courvoisier-Dezord, J. L. Petit, A. Mariage, E. Darii, K. Duquesne, V. de Berardinis and G. Iacazio, ACS Omega, 2019, 4, 7838–7849 CrossRef CAS.
- U. M. Vasudevan and E. Y. Lee, Biotechnol. Adv., 2020, 41, 107550 CrossRef PubMed.
- B. Nidetzky, A. Gutmann and C. Zhong, ACS Catal., 2018, 8, 6283–6300 CrossRef CAS.
- C. S. Rha, Y. S. Jung, D. H. Seo, D. O. Kim and C. S. Park, Enzyme Microb. Technol., 2019, 129, 109361 CrossRef CAS PubMed.
- J. B. He, P. Zhao, Z. M. Hu, S. Liu, Y. Kuang, M. Zhang, B. Li, C. H. Yun, X. Qiao and M. Ye, Angew. Chem., Int. Ed., 2019, 58, 11513–11520 CrossRef CAS.
- Y. Hirade, N. Kotoku, K. Terasaka, Y. Saijo-Hamano, A. Fukumoto and H. Mizukami, FEBS Lett., 2015, 589, 1778–1786 CrossRef CAS PubMed.
- N. Sasaki, Y. Nishizaki, E. Yamada, F. Tatsuzawa, T. Nakatsuka, H. Takahashi and M. Nishihara, FEBS Lett., 2015, 589, 182–187 CrossRef CAS.
- C. Ruprecht, F. Bonisch, N. Ilmberger, T. V. Heyer, E. T. K. Haupt, W. R. Streit and U. Rabausch, Metab. Eng., 2019, 55, 212–219 CrossRef CAS PubMed.
- K. G. Vanegas, A. B. Larsen, M. Eichenberger, D. Fischer, U. H. Mortensen and M. Naesby, Microb. Cell Fact., 2018, 17, 107 CrossRef PubMed.
- T. Ito, S. Fujimoto, F. Suito, M. Shimosaka and G. Taguchi, Plant J., 2017, 91, 187–198 CrossRef CAS.
- K. Xie, X. Zhang, S. Sui, F. Ye and J. Dai, Nat. Commun., 2020, 11, 5162 CrossRef CAS.
- T. Goris, A. Perez-Valero, I. Martinez, D. Yi, L. Fernandez-Calleja, D. San Leon, U. T. Bornscheuer, P. Magadan-Corpas, F. Lombo and J. Nogales, Microbiol. Biotechnol., 2021, 14, 94–110 CrossRef CAS PubMed.
- E. Fattorusso and O. Taglialatela-Scafati, Modern alkaloids: structure, isolation, synthesis, and biology, John Wiley & Sons, New Jersey, 2008 Search PubMed.
- S. Funayama and G. A. Cordell, Alkaloids: a treasury of poisons and medicines, Elsevier, Netherlands, 2014 Search PubMed.
- S. Schlager and B. Drager, Curr. Opin. Biotechnol., 2016, 37, 155–164 CrossRef.
- E. Leonard, W. Runguphan, S. O’Connor and K. J. Prather, Nat. Chem. Biol., 2009, 5, 292–300 CrossRef CAS.
- Y. Luo, B. Z. Li, D. Liu, L. Zhang, Y. Chen, B. Jia, B. X. Zeng, H. Zhao and Y. J. Yuan, Chem. Soc. Rev., 2015, 44, 5265–5290 RSC.
- A. M. Ehrenworth and P. Peralta-Yahya, Nat. Chem. Biol., 2017, 13, 249–258 CrossRef CAS PubMed.
- A. Cravens, J. Payne and C. D. Smolke, Nat. Commun., 2019, 10, 2142 CrossRef PubMed.
- M. E. Pyne, L. Narcross and V. J. J. Martin, Plant Physiol., 2019, 179, 844–861 CrossRef CAS PubMed.
- I. Carqueijeiro, C. Langley, D. Grzech, K. Koudounas, N. Papon, S. E. O'Connor and V. Courdavault, Curr. Opin. Biotechnol., 2020, 65, 17–24 CrossRef CAS PubMed.
- A. Singh, I. M. Menéndez-Perdomo and P. J. Facchini, Phytochem. Rev., 2019, 18, 1457–1482 CrossRef.
- T. Winzer, V. Gazda, Z. He, F. Kaminski, M. Kern, T. R. Larson, Y. Li, F. Meade, R. Teodor, F. E. Vaistij, C. Walker, T. A. Bowser and I. A. Graham, Science, 2012, 336, 1704–1708 CrossRef CAS PubMed.
- S. C. Farrow, J. M. Hagel, G. A. Beaudoin, D. C. Burns and P. J. Facchini, Nat. Chem. Biol., 2015, 11, 728–732 CrossRef CAS PubMed.
- S. Galanie, K. Thodey, I. J. Trenchard, M. Filsinger Interrante and C. D. Smolke, Science, 2015, 349, 1095–1100 CrossRef CAS PubMed.
- T. Winzer, M. Kern, A. J. King, T. R. Larson, R. I. Teodor, S. L. Donninger, Y. Li, A. A. Dowle, J. Cartwright, R. Bates, D. Ashford, J. Thomas, C. Walker, T. A. Bowser and I. A. Graham, Science, 2015, 349, 309–312 CrossRef CAS PubMed.
- X. Chen, J. M. Hagel, L. Chang, J. E. Tucker, S. A. Shiigi, Y. Yelpaala, H.-Y. Chen, R. Estrada, J. Colbeck, M. Enquist-Newman, A. B. Ibáñez, G. Cottarel, G. M. Vidanes and P. J. Facchini, Nat. Chem. Biol., 2018, 14, 738–743 CrossRef CAS PubMed.
- L. Narcross, E. Fossati, L. Bourgeois, J. E. Dueber and V. J. J. Martin, Trends Biotechnol., 2016, 34, 228–241 CrossRef CAS PubMed.
- A. Nakagawa, E. Matsumura, T. Koyanagi, T. Katayama, N. Kawano, K. Yoshimatsu, K. Yamamoto, H. Kumagai, F. Sato and H. Minami, Nat. Commun., 2016, 7, 10390 CrossRef CAS.
- Y. Li, S. Li, K. Thodey, I. Trenchard, A. Cravens and C. D. Smolke, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E3922–E3931 CrossRef CAS.
- M. E. Pyne, K. Kevvai, P. S. Grewal, L. Narcross, B. Choi, L. Bourgeois, J. E. Dueber and V. J. J. Martin, Nat. Commun., 2020, 11, 3337 CrossRef CAS PubMed.
- M. D. Patil, G. Grogan and H. Yun, ChemCatChem, 2018, 10, 4783–4804 CrossRef.
- B. R. Lichman, Nat. Prod. Rep., 2021, 38, 103–129 RSC.
- R. Roddan, J. M. Ward, N. H. Keep and H. C. Hailes, Curr. Opin. Chem. Biol., 2020, 55, 69–76 CrossRef CAS PubMed.
- B. R. Lichman, J. Zhao, H. C. Hailes and J. M. Ward, Nat. Commun., 2017, 8, 14883 CrossRef CAS PubMed.
- J. R. Marshall, P. Yao, S. L. Montgomery, J. D. Finnigan, T. W. Thorpe, R. B. Palmer, J. Mangas-Sanchez, R. A. M. Duncan, R. S. Heath, K. M. Graham, D. J. Cook, S. J. Charnock and N. J. Turner, Nat. Chem., 2021, 13, 140–148 CrossRef CAS PubMed.
- R. Roddan, G. Gygli, A. Sula, D. Méndez-Sánchez, J. Pleiss, J. M. Ward, N. H. Keep and H. C. Hailes, ACS Catal., 2019, 9, 9640–9649 CrossRef CAS.
- X. Sheng and F. Himo, J. Am. Chem. Soc., 2019, 141, 11230–11238 CrossRef CAS PubMed.
- V. Erdmann, B. R. Lichman, J. Zhao, R. C. Simon, W. Kroutil, J. M. Ward, H. C. Hailes and D. Rother, Angew. Chem., Int. Ed., 2017, 56, 12503–12507 CrossRef CAS PubMed.
- J. Zhao, B. R. Lichman, J. M. Ward and H. C. Hailes, Chem. Commun., 2018, 54, 1323–1326 RSC.
- Y. Wang, N. Tappertzhofen, D. Mendez-Sanchez, M. Bawn, B. Lyu, J. M. Ward and H. C. Hailes, Angew. Chem., Int. Ed., 2019, 58, 10120–10125 CrossRef CAS PubMed.
- J. H. Schrittwieser, V. Resch, J. H. Sattler, W. D. Lienhart, K. Durchschein, A. Winkler, K. Gruber, P. Macheroux and W. Kroutil, Angew. Chem., Int. Ed., 2011, 50, 1068–1071 CrossRef CAS PubMed.
- J. H. Schrittwieser, B. Groenendaal, V. Resch, D. Ghislieri, S. Wallner, E. M. Fischereder, E. Fuchs, B. Grischek, J. H. Sattler, P. Macheroux, N. J. Turner and W. Kroutil, Angew. Chem., Int. Ed., 2014, 53, 3731–3734 CrossRef CAS PubMed.
- S. Gandomkar, E. M. Fischereder, J. H. Schrittwieser, S. Wallner, Z. Habibi, P. Macheroux and W. Kroutil, Angew. Chem., Int. Ed., 2015, 54, 15051–15054 ( Angew. Chem. , 2015 , 127 , 15265–15268 ) CrossRef CAS PubMed.
- J. Mangas-Sanchez, S. P. France, S. L. Montgomery, G. A. Aleku, H. Man, M. Sharma, J. I. Ramsden, G. Grogan and N. J. Turner, Curr. Opin. Chem. Biol., 2017, 37, 19–25 CrossRef CAS PubMed.
- M. Höhne, Nat. Catal., 2019, 2, 841–842 CrossRef.
- M. Hestericova, T. Heinisch, L. Alonso-Cotchico, J. D. Marechal, P. Vidossich and T. R. Ward, Angew. Chem., Int. Ed., 2018, 57, 1863–1868 CrossRef CAS PubMed.
- S. Wu, Y. Zhou, J. G. Rebelein, M. Kuhn, H. Mallin, J. Zhao, N. V. Igareta and T. R. Ward, J. Am. Chem. Soc., 2019, 141, 15869–15878 CrossRef CAS PubMed.
- H. Li, P. Tian, J. H. Xu and G. W. Zheng, Org. Lett., 2017, 19, 3151–3154 CrossRef CAS PubMed.
- J. M. Zhu, H. Q. Tan, L. Yang, Z. Dai, L. Zhu, H. M. Ma, Z. X. Deng, Z. H. Tian and X. D. Qu, ACS Catal., 2017, 7, 7003–7007 CrossRef CAS.
- L. Yang, J. Zhu, C. Sun, Z. Deng and X. Qu, Chem. Sci., 2020, 11, 364–371 RSC.
- P. Yao, Z. Xu, S. Yu, Q. Wu and D. Zhu, Adv. Synth. Catal., 2019, 361, 556–561 CrossRef CAS.
- V. F. Batista, J. L. Galman, D. C. G. A. Pinto, A. M. S. Silva and N. J. Turner, ACS Catal., 2018, 8, 11889–11907 CrossRef CAS.
- J. Duan, B. Li, Y. Qin, Y. Dong, J. Ren and G. Li, Bioresour. Bioprocess., 2019, 6, 37 CrossRef.
- D. Ghislieri, A. P. Green, M. Pontini, S. C. Willies, I. Rowles, A. Frank, G. Grogan and N. J. Turner, J. Am. Chem. Soc., 2013, 135, 10863–10869 CrossRef CAS.
- G. Li, P. Yao, R. Gong, J. Li, P. Liu, R. Lonsdale, Q. Wu, J. Lin, D. Zhu and M. T. Reetz, Chem. Sci., 2017, 8, 4093–4099 RSC.
- S. Y. Ju, M. X. Qian, G. Xu, L. R. Yang and J. P. Wu, Adv. Synth. Catal., 2019, 361, 3191–3199 CrossRef CAS.
- S. Ju, M. Qian, G. Xu, L. Yang and J. Wu, Adv. Synth. Catal., 2019, 361, 3191–3199 CrossRef CAS.
- V. Kohler, Y. M. Wilson, M. Durrenberger, D. Ghislieri, E. Churakova, T. Quinto, L. Knorr, D. Haussinger, F. Hollmann, N. J. Turner and T. R. Ward, Nat. Chem., 2013, 5, 93–99 CrossRef CAS PubMed.
- S. Larsson and N. Ronsted, Curr. Top. Med. Chem., 2014, 14, 274–289 CrossRef CAS PubMed.
- R. S. Nett, W. Lau and E. S. Sattely, Nature, 2020, 584, 148–153 CrossRef CAS PubMed.
- Q. Pan, N. R. Mustafa, K. Tang, Y. H. Choi and R. Verpoorte, Phytochem. Rev., 2015, 15, 221–250 CrossRef.
- L. Caputi, J. Franke, S. C. Farrow, K. Chung, R. M. E. Payne, T. D. Nguyen, T. T. Dang, I. Soares Teto Carqueijeiro, K. Koudounas, T. Duge de Bernonville, B. Ameyaw, D. M. Jones, I. J. C. Vieira, V. Courdavault and S. E. O'Connor, Science, 2018, 360, 1235–1239 CrossRef CAS PubMed.
- Y. Qu, M. Easson, R. Simionescu, J. Hajicek, A. M. K. Thamm, V. Salim and V. De Luca, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3180–3185 CrossRef CAS PubMed.
- Y. Qu, O. Safonova and V. De Luca, Plant J., 2019, 97, 257–266 CrossRef CAS PubMed.
- S. Brown, M. Clastre, V. Courdavault and S. E. O'Connor, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 3205–3210 CrossRef CAS PubMed.
- Y. Qu, M. L. Easson, J. Froese, R. Simionescu, T. Hudlicky and V. De Luca, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6224–6229 CrossRef CAS PubMed.
- W. Runguphan, X. Qu and S. E. O’Connor, Nature, 2010, 468, 461–464 CrossRef CAS.
- D. Pressnitz, E. M. Fischereder, J. Pletz, C. Kofler, L. Hammerer, K. Hiebler, H. Lechner, N. Richter, E. Eger and W. Kroutil, Angew. Chem., Int. Ed., 2018, 57, 10683–10687 CrossRef CAS PubMed.
- E. Eger, A. Simon, M. Sharma, S. Yang, W. B. Breukelaar, G. Grogan, K. N. Houk and W. Kroutil, J. Am. Chem. Soc., 2020, 142, 792–800 CrossRef CAS PubMed.
- E. Eger, J. H. Schrittwieser, D. Wetzl, H. Iding, B. Kuhn and W. Kroutil, Chem. – Eur. J., 2020, 26, 16281–16285 CrossRef CAS PubMed.
- S. Velikogne, V. Resch, C. Dertnig, J. H. Schrittwieser and W. Kroutil, ChemCatChem, 2018, 10, 3236–3246 CrossRef CAS PubMed.
- D. Ghislieri, D. Houghton, A. P. Green, S. C. Willies and N. J. Turner, ACS Catal., 2013, 3, 2869–2872 CrossRef CAS.
- J. Schrader and J. Bohlmann, Biotechnology of Isoprenoids, Springer International Publishing, 2015 Search PubMed.
- D. W. Christianson, Chem. Rev., 2017, 117, 11570–11648 CrossRef CAS PubMed.
- E. Oldfield and F.-Y. Lin, Angew. Chem., Int. Ed., 2012, 51, 1124–1137 ( Angew. Chem. , 2012 , 124 , 1150–1163 ) CrossRef CAS PubMed.
- A. Vattekkatte, S. Garms, W. Brandt and W. Boland, Org. Biomol. Chem., 2018, 16, 348–362 RSC.
- A. Frank and M. Groll, Chem. Rev., 2017, 117, 5675–5703 CrossRef CAS PubMed.
- H. Xiao, Y. Zhang and M. Wang, Trends Biotechnol., 2019, 37, 618–631 CrossRef CAS PubMed.
- E. J. N. Helfrich, G.-M. Lin, C. A. Voigt and J. Clardy, Beilstein J. Org. Chem., 2019, 15, 2889–2906 CrossRef CAS PubMed.
- M. S. Belcher, J. Mahinthakumar and J. D. Keasling, Curr. Opin. Biotechnol., 2020, 65, 88–93 CrossRef CAS PubMed.
- G. Daletos, C. Katsimpouras and G. Stephanopoulos, Trends Biotechnol., 2020, 38, 811–822 CrossRef CAS PubMed.
- M. Li, F. Hou, T. Wu, X. Jiang, F. Li, H. Liu, M. Xian and H. Zhang, Nat. Prod. Rep., 2020, 37, 80–99 RSC.
- C. Schmidt-Dannert, D. Umeno and F. H. Arnold, Nat. Biotechnol., 2000, 18, 750–753 CrossRef CAS PubMed.
- C. J. Paddon, P. J. Westfall, D. J. Pitera, K. Benjamin, K. Fisher, D. McPhee, M. D. Leavell, A. Tai, A. Main, D. Eng, D. R. Polichuk, K. H. Teoh, D. W. Reed, T. Treynor, J. Lenihan, M. Fleck, S. Bajad, G. Dang, D. Dengrove, D. Diola, G. Dorin, K. W. Ellens, S. Fickes, J. Galazzo, S. P. Gaucher, T. Geistlinger, R. Henry, M. Hepp, T. Horning, T. Iqbal, H. Jiang, L. Kizer, B. Lieu, D. Melis, N. Moss, R. Regentin, S. Secrest, H. Tsuruta, R. Vazquez, L. F. Westblade, L. Xu, M. Yu, Y. Zhang, L. Zhao, J. Lievense, P. S. Covello, J. D. Keasling, K. K. Reiling, N. S. Renninger and J. D. Newman, Nature, 2013, 496, 528–532 CrossRef CAS PubMed.
- A. L. Meadows, K. M. Hawkins, Y. Tsegaye, E. Antipov, Y. Kim, L. Raetz, R. H. Dahl, A. Tai, T. Mahatdejkul-Meadows, L. Xu, L. Zhao, M. S. Dasika, A. Murarka, J. Lenihan, D. Eng, J. S. Leng, C. L. Liu, J. W. Wenger, H. Jiang, L. Chao, P. Westfall, J. Lai, S. Ganesan, P. Jackson, R. Mans, D. Platt, C. D. Reeves, P. R. Saija, G. Wichmann, V. F. Holmes, K. Benjamin, P. W. Hill, T. S. Gardner and A. E. Tsong, Nature, 2016, 537, 694–697 CrossRef CAS.
- B. Shen, P. Zhou, X. Jiao, Z. Yao, L. Ye and H. Yu, Nat. Commun., 2020, 11, 5155 CrossRef CAS PubMed.
- P. K. Ajikumar, W. H. Xiao, K. E. Tyo, Y. Wang, F. Simeon, E. Leonard, O. Mucha, T. H. Phon, B. Pfeifer and G. Stephanopoulos, Science, 2010, 330, 70–74 CrossRef CAS PubMed.
- S. Shukal, X. Chen and C. Zhang, Metab. Eng., 2019, 55, 170–178 CrossRef CAS PubMed.
- K. M. Wilding, S.-M. Schinn, E. A. Long and B. C. Bundy, Curr. Opin. Biotechnol., 2018, 53, 115–121 CrossRef CAS.
- N. J. Claassens, S. Burgener, B. Vögeli, T. J. Erb and A. Bar-Even, Curr. Opin. Biotechnol., 2019, 60, 221–229 CrossRef CAS PubMed.
- J. U. Bowie, S. Sherkhanov, T. P. Korman, M. A. Valliere, P. H. Opgenorth and H. Liu, Trends Biotechnol., 2020, 38, 766–778 CrossRef CAS.
- R. Liu, D. Yu, Z. Deng and T. Liu, Curr. Opin. Biotechnol., 2021, 69, 1–9 CrossRef CAS.
- T. P. Korman, P. H. Opgenorth and J. U. Bowie, Nat. Commun., 2017, 8, 15526 CrossRef CAS PubMed.
- P. H. Opgenorth, T. P. Korman and J. U. Bowie, Nat. Commun., 2014, 5, 4113 CrossRef CAS PubMed.
- X. Chen, C. Zhang, R. Zou, G. Stephanopoulos and H.-P. Too, ACS Synth. Biol., 2017, 6, 1691–1700 CrossRef CAS.
- M. Dirkmann, J. Nowack and F. Schulz, ChemBioChem, 2018, 19, 2146–2151 CrossRef CAS PubMed.
- P. Rabe and J. S. Dickschat, Angew. Chem., Int. Ed., 2013, 52, 1810–1812 CrossRef CAS PubMed.
- J. S. Dickschat, Angew. Chem., Int. Ed., 2019, 58, 15964–15976 CrossRef CAS PubMed.
- P. S. Karunanithi and P. Zerbe, Front. Plant Sci., 2019, 10, 1166 CrossRef.
- D. A. Nagegowda and P. Gupta, Plant Sci., 2020, 294, 110457 CrossRef CAS.
- F. Zhou and E. Pichersky, Curr. Opin. Plant Biol., 2020, 55, 1–10 CrossRef CAS PubMed.
- J. Andersen-Ranberg, K. T. Kongstad, M. T. Nielsen, N. B. Jensen, I. Pateraki, S. S. Bach, B. Hamberger, P. Zerbe, D. Staerk, J. Bohlmann, B. L. Møller and B. Hamberger, Angew. Chem., Int. Ed., 2016, 55, 2142–2146 CrossRef CAS.
- R. Thimmappa, K. Geisler, T. Louveau, P. O'Maille and A. Osbourn, Annu. Rev. Plant Biol., 2014, 65, 225–257 CrossRef CAS PubMed.
- J. Reed, M. J. Stephenson, K. Miettinen, B. Brouwer, A. Leveau, P. Brett, R. J. M. Goss, A. Goossens, M. A. O’Connell and A. Osbourn, Metab. Eng., 2017, 42, 185–193 CrossRef CAS PubMed.
- A. C. Huang, T. Jiang, Y.-X. Liu, Y.-C. Bai, J. Reed, B. Qu, A. Goossens, H.-W. Nützmann, Y. Bai and A. Osbourn, Science, 2019, 364, eaau6389 CrossRef CAS PubMed.
- S. Diethelm, R. Teufel, L. Kaysser and B. S. Moore, Angew. Chem., Int. Ed., 2014, 53, 11023–11026 CrossRef CAS PubMed.
- S. M. K. McKinnie, Z. D. Miles, P. A. Jordan, T. Awakawa, H. P. Pepper, L. A. M. Murray, J. H. George and B. S. Moore, J. Am. Chem. Soc., 2018, 140, 17840–17845 CrossRef CAS PubMed.
- D. S. Gkotsi, J. Dhaliwal, M. M. McLachlan, K. R. Mulholand and R. J. Goss, Curr. Opin. Chem. Biol., 2018, 43, 119–126 CrossRef CAS PubMed.
- D. S. Gkotsi, H. Ludewig, S. V. Sharma, J. A. Connolly, J. Dhaliwal, Y. Wang, W. P. Unsworth, R. J. K. Taylor, M. M. W. McLachlan, S. Shanahan, J. H. Naismith and R. J. M. Goss, Nat. Chem., 2019, 11, 1091–1097 CrossRef CAS PubMed.
- H. Ludewig, S. Molyneux, S. Ferrinho, K. Guo, R. Lynch, D. S. Gkotsi and R. J. M. Goss, Curr. Opin. Struc. Biol., 2020, 65, 51–60 CrossRef CAS PubMed.
- M. Voss, S. H. Malca and R. Buller, Chem. – Eur. J., 2020, 26, 7336–7345 CrossRef CAS PubMed.
- T. Kuzuyama and H. Seto, Nat. Prod. Rep., 2003, 20, 171–183 RSC.
- G. J. Schroepfer, Jr., Ann. Rev. Biochem., 1981, 50, 585–621 CrossRef PubMed.
- J. H. Wei, X. Yin and P. V. Welander, Front. Microbiol., 2016, 7, 990 Search PubMed.
- V. S. Dubey, R. Bhalla and R. Luthra, J. Biosci., 2003, 28, 637–646 CrossRef CAS PubMed.
- H. B. Bode, B. Zeggel, B. Silakowski, S. C. Wenzel, H. Reichenbach and R. Müller, Mol. Microbiol., 2003, 47, 471–481 CrossRef CAS PubMed.
- M. V. Donova and O. V. Egorova, Appl. Microbiol. Biotechnol., 2012, 94, 1423–1447 CrossRef CAS PubMed.
- W. Y. Tong and X. Dong, Recent Pat. Biotechnol., 2009, 3, 141–153 CrossRef CAS PubMed.
- L. Fernández-Cabezón, B. Galán and J. L. García, Front. Microbiol., 2018, 9, 958 CrossRef PubMed.
- M. V. Donova, in Microbial Steroids: Methods and Protocols, ed. J.-L. Barredo and I. Herráiz, Springer New York, New York, NY, 2017, pp. 1–13 Search PubMed.
- I. Herráiz, in Microbial Steroids: Methods and Protocols, eds. J.-L. Barredo and I. Herráiz, Springer New York, New York, NY, 2017, pp. 15–27 Search PubMed.
- J. D. Carballeira, M. A. Quezada, P. Hoyos, Y. Simeó, M. J. Hernaiz, A. R. Alcantara and J. V. Sinisterra, Biotechnol. Adv., 2009, 27, 686–714 CrossRef CAS PubMed.
- L. Fernández-Cabezón, B. Galán and J. L. García, Microbiol. Biotechnol., 2017, 10, 151–161 CrossRef PubMed.
- N. D. Fessner, ChemCatChem, 2019, 11, 2226–2242 CrossRef PubMed.
- J. Wang, Y. Zhang, H. Liu, Y. Shang, L. Zhou, P. Wei, W.-B. Yin, Z. Deng, X. Qu and Q. Zhou, Nat. Commun., 2019, 10, 3378 CrossRef PubMed.
- D. H. Peterson, A. H. Nathan, P. D. Meister, S. H. Eppstein, H. C. Murray, A. Weintraub, L. M. Reineke and H. M. Leigh, Proc. Natl. Acad. Sci. U. S. A., 1953, 75, 419–421 CAS.
- H. Sawada, S. Kulprecha, N. Nilubol, T. Yoshida, S. Kinoshita and H. Taguchi, Appl. Environ. Microbiol., 1982, 44, 1249–1252 CrossRef CAS PubMed.
- M. Szaleniec, A. M. Wojtkiewicz, R. Bernhardt, T. Borowski and M. Donova, Appl. Microbiol. Biotechnol., 2018, 102, 8153–8171 CrossRef CAS PubMed.
- V. B. Urlacher and M. Girhard, Trends Biotechnol., 2019, 37, 882–897 CrossRef CAS PubMed.
- M. P. C. Marques, F. Carvalho, C. C. C. R. de Carvalho, J. M. S. Cabral and P. Fernandes, Food Bioprod. Process., 2010, 88, 12–20 CrossRef CAS.
- W. Zhang, M. Shao, Z. Rao, M. Xu, X. Zhang, T. Yang, H. Li and Z. Xu, J. Steroid Biochem. Mol. Biol., 2013, 135, 36–42 CrossRef CAS PubMed.
- R. J. Park, Steroids, 1981, 38, 383–395 CrossRef CAS PubMed.
- P. Fernandes, A. Cruz, B. Angelova, H. M. Pinheiro and J. M. S. Cabral, Enzyme Microb. Technol., 2003, 32, 688–705 CrossRef CAS.
- A. Malaviya and J. Gomes, Bioresour. Technol., 2008, 99, 6725–6737 CrossRef CAS PubMed.
- X. L. He, L. T. Wang, X. Z. Gu, J. X. Xiao and W. W. Qiu, Steroids, 2018, 140, 173–178 CrossRef CAS PubMed.
- J. Wang, X. Z. Gu, L. M. He, C. C. Li and W. W. Qiu, Steroids, 2020, 157, 108600 CrossRef CAS PubMed.
- B. Galán, I. Uhía, E. García-Fernández, I. Martínez, E. Bahíllo, J. L. de la Fuente, J. L. Barredo, L. Fernández-Cabezón and J. L. García, Microbiol. Biotechnol., 2017, 10, 138–150 CrossRef PubMed.
- T. Kołek, A. Szpineter and A. Świzdor, Steroids, 2008, 73, 1441–1445 CrossRef PubMed.
- H. Leisch, K. Morley and P. C. K. Lau, Chem. Rev., 2011, 111, 4165–4222 CrossRef CAS PubMed.
- J. K. Capyk, I. Casabon, R. Gruninger, N. C. Strynadka and L. D. Eltis, J. Biol. Chem., 2011, 286, 40717–40724 CrossRef CAS PubMed.
- S. Prakash and A. Bajaj, in Microbial Steroids: Methods and Protocols, ed. J.-L. Barredo and I. Herráiz, Springer New York, New York, NY, 2017, pp. 227–238 Search PubMed.
- S. Costa, F. Zappaterra, D. Summa, B. Semeraro and G. Fantin, Molecules, 2020, 25, 2192 CrossRef CAS PubMed.
- L. Liu, A. Aigner and R. D. Schmid, Appl. Microbiol. Biotechnol., 2011, 90, 127–135 CrossRef CAS PubMed.
- M.-M. Zheng, R.-F. Wang, C.-X. Li and J.-H. Xu, Process Biochem., 2015, 50, 598–604 CrossRef CAS.
- P. P. Giovannini, A. Grandini, D. Perrone, P. Pedrini, G. Fantin and M. Fogagnolo, Steroids, 2008, 73, 1385–1390 CrossRef CAS PubMed.
- S.-C. Shi, Z.-N. You, K. Zhou, Q. Chen, J. Pan, X.-L. Qian, J.-H. Xu and C.-X. Li, Adv. Synth. Catal., 2019, 361, 4661–4668 CrossRef CAS.
- H. N. Bhatti and R. A. Khera, Steroids, 2012, 77, 1267–1290 CrossRef CAS PubMed.
- F. Hannemann, A. Bichet, K. M. Ewen and R. Bernhardt, Biochim. Biophys. Acta, Gen. Subj., 2007, 1770, 330–344 CrossRef CAS PubMed.
- G. Di Nardo and G. Gilardi, Trends Biochem. Sci., 2020, 45, 511–525 CrossRef CAS PubMed.
- F. P. Guengerich, Chem. Res. Toxicol., 2001, 14, 611–650 Search PubMed.
- E. M. Isin and F. P. Guengerich, Biochim. Biophys. Acta, Gen. Subj., 2007, 1770, 314–329 CrossRef CAS PubMed.
- J. B. Behrendorff, W. Huang and E. M. Gillam, Biochem. J., 2015, 467, 1–15 CrossRef CAS PubMed.
- P. S. Coelho, E. M. Brustad, A. Kannan and F. H. Arnold, Science, 2013, 339, 307–310 CrossRef CAS PubMed.
- A. R. H. Narayan and D. H. Sherman, Science, 2013, 339, 283–284 CrossRef CAS PubMed.
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88–119 CrossRef CAS PubMed.
- C. K. Winkler, J. H. Schrittwieser and W. Kroutil, ACS Cent. Sci., 2021, 7, 55–71 CrossRef CAS PubMed.
- K. S. Fegan, M. T. Rae, H. O. D. Critchley and S. G. Hillier, J. Endocrinol., 2008, 196, 369–376 CAS.
- D. D. Bikle, Chem. Biol., 2014, 21, 319–329 CrossRef CAS PubMed.
- A. Berg, K. Carlström, J.-Å. Gustafsson and M. Ingelman-Sundberg, Biochem. Biophys. Res. Commun., 1975, 66, 1414–1423 CrossRef CAS PubMed.
- D. Schmitz, S. Janocha, F. M. Kiss and R. Bernhardt, Biochim. Biophys. Acta, Proteins Proteomics, 2018, 1866, 11–22 CrossRef CAS PubMed.
- M. T. Reetz, Angew. Chem., Int. Ed., 2011, 50, 138–174 CrossRef CAS PubMed.
- R. M. P. Siloto and R. J. Weselake, Biocatal. Agric. Biotechnol., 2012, 1, 181–189 CrossRef CAS.
- C. G. Acevedo-Rocha, S. Hoebenreich and M. T. Reetz, in Directed Evolution Library Creation: Methods and Protocols, ed. E. M. J. Gillam, J. N. Copp and D. Ackerley, Springer New York, New York, NY, 2014, pp. 103–128 Search PubMed.
- C. F. Oliver, S. Modi, M. J. Sutcliffe, W. U. Primrose, L.-Y. Lian and G. C. K. Roberts, Biochemistry, 1997, 36, 1567–1572 CrossRef CAS PubMed.
- C. J. C. Whitehouse, S. G. Bell and L.-L. Wong, Chem. Soc. Rev., 2012, 41, 1218–1260 RSC.
- S. Kille, F. E. Zilly, J. P. Acevedo and M. T. Reetz, Nat. Chem., 2011, 3, 738–743 CrossRef CAS PubMed.
- C. G. Acevedo-Rocha, C. G. Gamble, R. Lonsdale, A. Li, N. Nett, S. Hoebenreich, J. B. Lingnau, C. Wirtz, C. Fares, H. Hinrichs, A. Deege, A. J. Mulholland, Y. Nov, D. Leys, K. J. McLean, A. W. Munro and M. T. Reetz, ACS Catal., 2018, 8, 3395–3410 CrossRef CAS.
- W. Y. Chen, M. J. Fisher, A. Leung, Y. Cao and L. L. Wong, ACS Catal., 2020, 10, 8334–8343 CrossRef CAS.
- A. Li, C. G. Acevedo-Rocha, L. D'Amore, J. Chen, Y. Peng, M. Garcia-Borràs, C. Gao, J. Zhu, H. Rickerby, S. Osuna, J. Zhou and M. T. Reetz, Angew. Chem., Int. Ed., 2020, 59, 12499–12505 CrossRef CAS PubMed.
- N. Putkaradze, F. M. Kiss, D. Schmitz, J. Zapp, M. C. Hutter and R. Bernhardt, J. Biotechnol., 2017, 242, 101–110 CrossRef CAS PubMed.
- E. Wülfert, K. A. Pringle and E. L. Sundstrom, Pat. Appl., WO2002000224A1, 2002 Search PubMed.
- S. Kevresan, K. Kuhajda, J. Kandrac, J. P. Fawcett and M. Mikov, Eur. J. Drug Metab. Pharmacokinet., 2006, 31, 145–156 CrossRef CAS PubMed.
- A. F. Hofmann, L. R. Hagey and M. D. Krasowski, J. Lipid Res., 2010, 51, 226–246 CrossRef CAS PubMed.
- J. Y. L. Chiang, Compr. Physiol., 2013, 3, 1191–1212 Search PubMed.
- S. Fiorucci, M. Biagioli, A. Zampella and E. Distrutti, Front. Immunol., 2018, 9, 1853 CrossRef PubMed.
- W. M. Pandak and G. Kakiyama, Liver Res., 2019, 3, 88–98 CrossRef.
- H. Pellissier and M. Santelli, Org. Prep. Proced. Int., 2001, 33, 1–58 CrossRef CAS.
- X. Zhang, Y. Peng, J. Zhao, Q. Li, X. Yu, C. G. Acevedo-Rocha and A. Li, Bioresour. Bioprocess., 2020, 7, 2 CrossRef.
- F. Tonin and I. W. C. E. Arends, Beilstein J. Org. Chem., 2018, 14, 470–483 CrossRef CAS PubMed.
- F. Tonin, L. G. Otten and I. W. C. E. Arends, ChemSusChem, 2019, 12, 3192–3203 CrossRef CAS PubMed.
- S. Kulprecha, T. Ueda, T. Nihira, T. Yoshida and H. Taguchi, Appl. Environ. Microbiol., 1985, 49, 338–344 CrossRef CAS PubMed.
- L. C. Montemiglio, G. Parisi, A. Scaglione, G. Sciara, C. Savino and B. Vallone, Biochim. Biophys. Acta, Gen. Subj., 2016, 1860, 465–475 CrossRef CAS PubMed.
- G. Parisi, L. C. Montemiglio, A. Giuffrè, A. Macone, A. Scaglione, G. Cerutti, C. Exertier, C. Savino and B. Vallone, FASEB J., 2019, 33, 1787–1800 CrossRef CAS PubMed.
- H. Kabumoto, K. Miyazaki and A. Arisawa, Biosci., Biotechnol., Biochem., 2009, 73, 1922–1927 CrossRef CAS PubMed.
- S. Grobe, A. Wszołek, H. Brundiek, M. Fekete and U. T. Bornscheuer, Biotechnol. Lett., 2020, 42, 819–824 CrossRef CAS PubMed.
- J. P. von Kries, T. Warrier and L. M. Podust, Curr. Protoc. Microbiol., 2010, 16, 17.14.11–17.14.25 Search PubMed.
- S. Bleif, F. Hannemann, M. Lisurek, J. P. von Kries, J. Zapp, M. Dietzen, I. Antes and R. Bernhardt, ChemBioChem, 2011, 12, 576–582 CrossRef CAS.
- A. Luthra, I. G. Denisov and S. G. Sligar, Arch. Biochem. Biophys., 2011, 507, 26–35 CrossRef CAS.
- Y. Khatri, A. Schifrin and R. Bernhardt, FEBS Lett., 2017, 591, 1126–1140 CrossRef CAS PubMed.
- Y. Khatri, M. Ringle, M. Lisurek, J. P. von Kries, J. Zapp and R. Bernhardt, ChemBioChem, 2016, 17, 90–101 CrossRef CAS PubMed.
- S. Hoebenreich, M. Spinck and N. Nett, in Microbial Steroids: Methods and Protocols, ed. J.-L. Barredo and I. Herráiz, Springer New York, New York, NY, 2017, pp. 239–257 Search PubMed.
- F. M. Kiss, M. T. Lundemo, J. Zapp, J. M. Woodley and R. Bernhardt, Microb. Cell Fact., 2015, 14, 28 CrossRef PubMed.
- B. Hauer, ACS Catal., 2020, 10, 8418–8427 CrossRef CAS.
- F. M. Szczebara, C. Chandelier, C. Villeret, A. Masurel, S. Bourot, C. Duport, S. Blanchard, A. Groisillier, E. Testet, P. Costaglioli, G. Cauet, E. Degryse, D. Balbuena, J. Winter, T. Achstetter, R. Spagnoli, D. Pompon and B. Dumas, Nat. Biotechnol., 2003, 21, 143–149 CrossRef CAS PubMed.
- B. B. Dumas, C. Brocard-Masson, K. Assemat-Lebrun and T. Achstetter, Biotechnol. J., 2006, 1, 299–307 CrossRef CAS PubMed.
- H. Agematu, N. Matsumoto, Y. Fujii, H. Kabumoto, S. Doi, K. Machida, J. Ishikawa and A. Arisawa, Biosci. Biotechnol. Biochem., 2006, 70, 307–311 CrossRef CAS PubMed.
- A. Goyal, B.-G. Kim, K.-S. Hwang and S.-G. Lee, Biotechnol. Bioprocess Eng., 2015, 20, 431–438 CrossRef CAS.
- J. D. Rudolf, C.-Y. Chang, M. Ma and B. Shen, Nat. Prod. Rep., 2017, 34, 1141–1172 RSC.
- S. Chadha, S. T. Mehetre, R. Bansal, A. Kuo, A. Aerts, I. V. Grigoriev, I. S. Druzhinina and P. K. Mukherjee, Fungal Biol. Biotechnol., 2018, 5, 12 CrossRef PubMed.
- X. Liu, X. Zhu, H. Wang, T. Liu, J. Cheng and H. Jiang, Synth. Syst. Biotechnol., 2020, 5, 187–199 CrossRef PubMed.
- J. Shin, J. E. Kim, Y. W. Lee and H. Son, Toxins, 2018, 10, 112 CrossRef PubMed.
- C. Duport, R. Spagnoli, E. Degryse and D. Pompon, Nat. Biotechnol., 1998, 16, 186–189 CrossRef CAS PubMed.
- J. H. Weng and B. C. Chung, Steroids, 2016, 111, 54–59 CrossRef CAS PubMed.
- K. Yasuda, Y. Yogo, H. Sugimoto, H. Mano, T. Takita, M. Ohta, M. Kamakura, S. Ikushiro, K. Yasukawa, Y. Shiro and T. Sakaki, Biochem. Biophys. Res. Commun., 2017, 486, 336–341 CrossRef CAS PubMed.
- K. Hayashi, K. Yasuda, Y. Yogo, T. Takita, K. Yasukawa, M. Ohta, M. Kamakura, S. Ikushiro and T. Sakaki, Biochem. Biophys. Res. Commun., 2016, 473, 853–858 CrossRef CAS PubMed.
- L. Kattner, D. Bernardi and E. Rauch, Anticancer Res., 2015, 35, 1205–1210 CAS.
- P. Bracco, H. J. Wijma, B. Nicolai, J. A. R. Buitrago, T. Klunemann, A. Vila, P. Schrepfer, W. Blankenfeldt, D. B. Janssen and A. Schallmey, ChemBioChem, 2021, 22, 1099–1110 CrossRef CAS PubMed.
- X. Zhang, Y. Hu, W. Peng, C. Gao, Q. Xing, B. Wang and A. Li, Front. Chem., 2021, 9, 649000 CrossRef CAS PubMed.
- H. J. Ruijssenaars, E. M. Sperling, P. H. Wiegerinck, F. T. Brands, J. Wery and J. A. de Bont, J. Biotechnol., 2007, 131, 205–208 CrossRef CAS PubMed.
- A. Krivoruchko, M. Kuyukina and I. Ivshina, Catalysts, 2019, 9, 236 CrossRef.
- J. Garcia-Fernandez, B. Galan, C. Felpeto-Santero, J. L. Barredo and J. L. Garcia, Catalysts, 2017, 7, 316 CrossRef.
- A. I. Kanno, C. Goulart, H. K. Rofatto, S. C. Oliveira, L. C. C. Leite and J. McFadden, Appl. Environ. Microbiol., 2016, 82, 2240–2246 CrossRef CAS PubMed.
- R. W. Bradley, M. Buck and B. Wang, J. Mol. Biol., 2016, 428, 862–888 CrossRef CAS PubMed.
- K. C. Murphy, K. Papavinasasundaram and C. M. Sassetti, in Mycobacteria Protocols, ed. T. Parish and D. M. Roberts, Springer New York, New York, NY, 2015, pp. 177–199 Search PubMed.
- J. M. Rock, F. F. Hopkins, A. Chavez, M. Diallo, M. R. Chase, E. R. Gerrick, J. R. Pritchard, G. M. Church, E. J. Rubin, C. M. Sassetti, D. Schnappinger and S. M. Fortune, Nat. Microbiol., 2017, 2, 16274 CrossRef CAS PubMed.
- C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 4688–4716 CrossRef CAS PubMed.
- A. T. Keatinge-Clay, Angew. Chem., Int. Ed., 2017, 56, 4658–4660 CrossRef CAS PubMed.
- A. A. Malico, L. Nichols and G. J. Williams, Curr. Opin. Chem. Biol., 2020, 58, 45–53 CrossRef CAS PubMed.
- M. Grininger, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 8680–8682 CrossRef CAS PubMed.
- K. P. Yuet and C. Khosla, Metab. Eng. Commun., 2020, 10, e00106 CrossRef PubMed.
- T. Robbins, Y. C. Liu, D. E. Cane and C. Khosla, Curr. Opin. Struct. Biol., 2016, 41, 10–18 CrossRef CAS PubMed.
- B. Lowry, T. Robbins, C. H. Weng, R. V. O'Brien, D. E. Cane and C. Khosla, J. Am. Chem. Soc., 2013, 135, 16809–16812 CrossRef CAS PubMed.
- S. Y. Tsuji, D. E. Cane and C. Khosla, Biochemistry, 2001, 40, 2326–2331 CrossRef CAS PubMed.
- T. Miyazawa, M. Hirsch, Z. Zhang and A. T. Keatinge-Clay, Nat. Commun., 2020, 11, 80 CrossRef CAS PubMed.
- J. L. Smith, G. Skiniotis and D. H. Sherman, Curr. Opin. Struct. Biol., 2015, 31, 9–19 CrossRef CAS PubMed.
- A. Nivina, K. P. Yuet, J. Hsu and C. Khosla, Chem. Rev., 2019, 119, 12524–12547 CrossRef CAS PubMed.
- B. A. Pfeifer, S. J. Admiraal, H. Gramajo, D. E. Cane and C. Khosla, Science, 2001, 291, 1790–1792 CrossRef CAS PubMed.
- A. Zargar, R. Lal, L. Valencia, J. Wang, T. W. H. Backman, P. Cruz-Morales, A. Kothari, M. Werts, A. R. Wong, C. B. Bailey, A. Loubat, Y. Liu, Y. Chen, S. Chang, V. T. Benites, A. C. Hernandez, J. F. Barajas, M. G. Thompson, C. Barcelos, R. Anayah, H. G. Martin, A. Mukhopadhyay, C. J. Petzold, E. E. K. Baidoo, L. Katz and J. D. Keasling, J. Am. Chem. Soc., 2020, 142, 9896–9901 CrossRef CAS PubMed.
- B. J. Dunn and C. Khosla, J. R. Soc., Interface, 2013, 10, 20130297 CrossRef PubMed.
- H. Peng, K. Ishida, Y. Sugimoto, H. Jenke-Kodama and C. Hertweck, Nat. Commun., 2019, 10, 3918 CrossRef PubMed.
- A. Wlodek, S. G. Kendrew, N. J. Coates, A. Hold, J. Pogwizd, S. Rudder, L. S. Sheehan, S. J. Higginbotham, A. E. Stanley-Smith, T. Warneck, E. A. M. Nur, M. Radzom, C. J. Martin, L. Overvoorde, M. Samborskyy, S. Alt, D. Heine, G. T. Carter, E. I. Graziani, F. E. Koehn, L. McDonald, A. Alanine, R. M. Rodriguez Sarmiento, S. K. Chao, H. Ratni, L. Steward, I. H. Norville, M. Sarkar-Tyson, S. J. Moss, P. F. Leadlay, B. Wilkinson and M. A. Gregory, Nat. Commun., 2017, 8, 1206 CrossRef PubMed.
- M. Klaus, M. P. Ostrowski, J. Austerjost, T. Robbins, B. Lowry, D. E. Cane and C. Khosla, J. Biol. Chem., 2016, 291, 16404–16415 CrossRef CAS PubMed.
- X. Li, N. Sevillano, F. La Greca, L. Deis, Y. C. Liu, M. C. Deller, I. I. Mathews, T. Matsui, D. E. Cane, C. S. Craik and C. Khosla, J. Am. Chem. Soc., 2018, 140, 6518–6521 CrossRef CAS PubMed.
- S. Dutta, J. R. Whicher, D. A. Hansen, W. A. Hale, J. A. Chemler, G. R. Congdon, A. R. Narayan, K. Hakansson, D. H. Sherman, J. L. Smith and G. Skiniotis, Nature, 2014, 510, 512–517 CrossRef CAS PubMed.
- J. R. Whicher, S. Dutta, D. A. Hansen, W. A. Hale, J. A. Chemler, A. M. Dosey, A. R. Narayan, K. Hakansson, D. H. Sherman, J. L. Smith and G. Skiniotis, Nature, 2014, 510, 560–564 CrossRef CAS PubMed.
- C. P. Ridley, H. Y. Lee and C. Khosla, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 4595–4600 CrossRef CAS PubMed.
- J. Zucko, P. F. Long, D. Hranueli and J. Cullum, J. Ind. Microbiol. Biotechnol., 2012, 39, 1541–1547 CrossRef CAS PubMed.
- Y. Sugimoto, L. Ding, K. Ishida and C. Hertweck, Angew. Chem., Int. Ed., 2014, 53, 1560–1564 CrossRef CAS PubMed.
- L. Zhang, T. Hashimoto, B. Qin, J. Hashimoto, I. Kozone, T. Kawahara, M. Okada, T. Awakawa, T. Ito, Y. Asakawa, M. Ueki, S. Takahashi, H. Osada, T. Wakimoto, H. Ikeda, K. Shin-Ya and I. Abe, Angew. Chem., Int. Ed., 2017, 56, 1740–1745 CrossRef CAS PubMed.
- J. A. Chemler, A. Tripathi, D. A. Hansen, M. O'Neil-Johnson, R. B. Williams, C. Starks, S. R. Park and D. H. Sherman, J. Am. Chem. Soc., 2015, 137, 10603–10609 CrossRef CAS PubMed.
- S. Yuzawa, K. Deng, G. Wang, E. E. Baidoo, T. R. Northen, P. D. Adams, L. Katz and J. D. Keasling, ACS Synth. Biol., 2017, 6, 139–147 CrossRef CAS PubMed.
- A. N. Lowell, M. D. DeMars, S. T. Slocum, F. A. Yu, K. Anand, J. A. Chemler, N. Korakavi, J. K. Priessnitz, S. R. Park, A. A. Koch, P. J. Schultz and D. H. Sherman, J. Am. Chem. Soc., 2017, 139, 7913–7920 CrossRef CAS PubMed.
- J. Kuo, S. R. Lynch, C. W. Liu, X. Xiao and C. Khosla, ACS Chem. Biol., 2016, 11, 2636–2641 CrossRef CAS PubMed.
- K. P. Yuet, C. W. Liu, S. R. Lynch, J. Kuo, W. Michaels, R. B. Lee, A. E. McShane, B. L. Zhong, C. R. Fischer and C. Khosla, J. Am. Chem. Soc., 2020, 142, 5952–5957 CrossRef CAS PubMed.
- J. Fredens, K. Wang, D. de la Torre, L. F. H. Funke, W. E. Robertson, Y. Christova, T. Chia, W. H. Schmied, D. L. Dunkelmann, V. Beranek, C. Uttamapinant, A. G. Llamazares, T. S. Elliott and J. W. Chin, Nature, 2019, 569, 514–518 CrossRef CAS PubMed.
- N. Ostrov, M. Landon, M. Guell, G. Kuznetsov, J. Teramoto, N. Cervantes, M. Zhou, K. Singh, M. G. Napolitano, M. Moosburner, E. Shrock, B. W. Pruitt, N. Conway, D. B. Goodman, C. L. Gardner, G. Tyree, A. Gonzales, B. L. Wanner, J. E. Norville, M. J. Lajoie and G. M. Church, Science, 2016, 353, 819–822 CrossRef CAS PubMed.
- D. Yang, W. J. Kim, S. M. Yoo, J. H. Choi, S. H. Ha, M. H. Lee and S. Y. Lee, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9835–9844 CrossRef CAS PubMed.
- C. M. Kasey, M. Zerrad, Y. Li, T. A. Cropp and G. J. Williams, ACS Synth. Biol., 2018, 7, 227–239 CrossRef CAS PubMed.
- C. P. S. Badenhorst and U. T. Bornscheuer, Trends Biochem. Sci., 2018, 43, 180–198 CrossRef CAS PubMed.
- R. F. Service, Science, 2020, 370, 1144–1145 CrossRef CAS PubMed.
- R. A. Langan, S. E. Boyken, A. H. Ng, J. A. Samson, G. Dods, A. M. Westbrook, T. H. Nguyen, M. J. Lajoie, Z. Chen, S. Berger, V. K. Mulligan, J. E. Dueber, W. R. P. Novak, H. El-Samad and D. Baker, Nature, 2019, 572, 205–210 CrossRef CAS PubMed.
- E. Theodorou, R. Scanga, M. Twardowski, M. P. Snyder and E. Brouzes, Micromachines, 2017, 8, 230 CrossRef PubMed.
- S. Yuzawa, M. Mirsiaghi, R. Jocic, T. Fujii, F. Masson, V. T. Benites, E. E. K. Baidoo, E. Sundstrom, D. Tanjore, T. R. Pray, A. George, R. W. Davis, J. M. Gladden, B. A. Simmons, L. Katz and J. D. Keasling, Nat. Commun., 2018, 9, 4569 CrossRef PubMed.
- K. A. Bozhüyük, J. Micklefield and B. Wilkinson, Curr. Opin. Microbiol., 2019, 51, 88–96 CrossRef PubMed.
- M. A. Huffman, A. Fryszkowska, O. Alvizo, M. Borra-Garske, K. R. Campos, K. A. Canada, P. N. Devine, D. Duan, J. H. Forstater, S. T. Grosser, H. M. Halsey, G. J. Hughes, J. Jo, L. A. Joyce, J. N. Kolev, J. Liang, K. M. Maloney, B. F. Mann, N. M. Marshall, M. McLaughlin, J. C. Moore, G. S. Murphy, C. C. Nawrat, J. Nazor, S. Novick, N. R. Patel, A. Rodriguez-Granillo, S. A. Robaire, E. C. Sherer, M. D. Truppo, A. M. Whittaker, D. Verma, L. Xiao, Y. Xu and H. Yang, Science, 2019, 366, 1255–1259 CrossRef CAS PubMed.
- T. Gulck and B. L. Moller, Trends Plant Sci., 2020, 25, 985–1004 CrossRef CAS PubMed.
- X. Z. Luo, M. A. Reiter, L. d'Espaux, J. Wong, C. M. Denby, A. Lechner, Y. F. Zhang, A. T. Grzybowski, S. Harth, W. Y. Lin, H. Lee, C. H. Yu, J. Shin, K. Deng, V. T. Benites, G. Wang, E. E. K. Baidoo, Y. Chen, I. Dev, C. J. Petzold and J. D. Keasling, Nature, 2019, 567, 123–126 CrossRef CAS PubMed.
- M. A. Valliere, T. P. Korman, N. B. Woodall, G. A. Khitrov, R. E. Taylor, D. Baker and J. U. Bowie, Nat. Commun., 2019, 10, 565 CrossRef CAS PubMed.
- M. A. Valliere, T. P. Korman, M. A. Arbing and J. U. Bowie, Nat. Chem. Biol., 2020, 16, 1427–1433 CrossRef CAS PubMed.
- C. Greunke, A. Glockle, J. Antosch and T. A. M. Gulder, Angew. Chem., Int. Ed., 2017, 56, 4351–4355 CrossRef CAS PubMed.
- J. R. Chekan, S. M. K. McKinnie, M. L. Moore, S. G. Poplawski, T. P. Michael and B. S. Moore, Angew. Chem., Int. Ed., 2019, 58, 8454–8457 CrossRef CAS PubMed.
- O. R. Chekan, S. M. K. McKinnie, J. P. Noel and B. S. Moore, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12799–12805 CrossRef PubMed.
- Q. Cheng, L. Xiang, M. Izumikawa, D. Meluzzi and B. S. Moore, Nat. Chem. Biol., 2007, 3, 557–558 CrossRef CAS PubMed.
- J. A. Kalaitzis, Q. Cheng, P. M. Thomas, N. L. Kelleher and B. S. Moore, J. Nat. Prod., 2009, 72, 469–472 CrossRef CAS PubMed.
- R. Teufel, L. Kaysser, M. T. Villaume, S. Diethelm, M. K. Carbullido, P. S. Baran and B. S. Moore, Angew. Chem., Int. Ed., 2014, 53, 11019–11022 CrossRef CAS PubMed.
- Z. D. Miles, S. Diethelm, H. P. Pepper, D. M. Huang, J. H. George and B. S. Moore, Nat. Chem., 2017, 9, 1235–1242 CrossRef CAS PubMed.
- P. Srinivasan and C. D. Smolke, Nature, 2020, 585, 614–619 CAS.
- K. Chen and F. H. Arnold, Nat. Catal., 2020, 3, 203–213 CrossRef CAS.
- T. Vornholt and M. Jeschek, ChemBioChem, 2020, 21, 2241–2249 CrossRef CAS PubMed.
- S. H. Lee, D. S. Choi, S. K. Kuk and C. B. Park, Angew. Chem., Int. Ed., 2018, 57, 7958–7985 CrossRef CAS PubMed.
- F. F. Özgen, M. E. Runda and S. Schmidt, ChemBioChem, 2021, 22, 790–806 CrossRef PubMed.
- L. Schmermund, V. Jurkaš, F. F. Özgen, G. D. Barone, H. C. Büchsenschütz, C. K. Winkler, S. Schmidt, R. Kourist and W. Kroutil, ACS Catal., 2019, 9, 4115–4144 CrossRef CAS.
- D. Sorigué, B. Légeret, S. Cuiné, S. Blangy, S. Moulin, E. Billon, P. Richaud, S. Brugière, Y. Couté, D. Nurizzo, P. Müller, K. Brettel, D. Pignol, P. Arnoux, Y. Li-Beisson, G. Peltier and F. Beisson, Science, 2017, 357, 903–907 CrossRef PubMed.
- H. J. Cha, S. Y. Hwang, D. S. Lee, A. R. Kumar, Y. U. Kwon, M. Voß, E. Schuiten, U. T. Bornscheuer, F. Hollmann, D. K. Oh and J. B. Park, Angew. Chem., Int. Ed., 2020, 59, 7024–7028 CrossRef CAS PubMed.
- Y. Ma, X. Zhang, W. Zhang, P. Li, Y. Li, F. Hollmann and Y. Wang, ChemPhotoChem, 2019, 4, 39–44 CrossRef.
- J. Xu, Y. Hu, J. Fan, M. Arkin, D. Li, Y. Peng, W. Xu, X. Lin and Q. Wu, Angew. Chem., Int. Ed., 2019, 58, 8474–8478 CrossRef CAS PubMed.
- F. Cheng, H. Li, D.-Y. Wu, J.-M. Li, Y. Fan, Y.-P. Xue and Y.-G. Zheng, Green Chem., 2020, 22, 6815–6818 RSC.
- M. A. Emmanuel, N. R. Greenberg, D. G. Oblinsky and T. K. Hyster, Nature, 2016, 540, 414–417 CrossRef CAS PubMed.
- U. T. Bornscheuer, Nature, 2016, 540, 345–346 CrossRef CAS PubMed.
- M. J. Black, K. F. Biegasiewicz, A. J. Meichan, D. G. Oblinsky, B. Kudisch, G. D. Scholes and T. K. Hyster, Nat. Chem., 2019, 12, 71–75 CrossRef PubMed.
- C. G. Page, S. J. Cooper, J. S. DeHovitz, D. G. Oblinsky, K. F. Biegasiewicz, A. H. Antropow, K. W. Armbrust, J. M. Ellis, L. G. Hamann, E. J. Horn, K. M. Oberg, G. D. Scholes and T. K. Hyster, J. Am. Chem. Soc., 2021, 143, 97–102 CrossRef CAS PubMed.
- P. D. Clayman and T. K. Hyster, J. Am. Chem. Soc., 2020, 142, 15673–15677 CrossRef CAS PubMed.
- K. F. Biegasiewicz, S. J. Cooper, X. Gao, D. G. Oblinsky, J. B. Kim, S. E. Garfinkle, L. A. Joyce, B. A. Sandoval, G. D. Scholes and T. K. Hyster, Science, 2019, 364, 1166–1169 CrossRef CAS PubMed.
- K. F. Biegasiewicz, S. J. Cooper, M. A. Emmanuel, D. C. Miller and T. K. Hyster, Nat. Chem., 2018, 10, 770–775 CrossRef CAS PubMed.
- Z. C. Litman, Y. Wang, H. Zhao and J. F. Hartwig, Nature, 2018, 560, 355–359 CrossRef CAS PubMed.
- W. Zhang, E. F. Fueyo, F. Hollmann, L. L. Martin, M. Pesic, R. Wardenga, M. Höhne and S. Schmidt, Eur. J. Org. Chem., 2019, 80–84 CrossRef CAS PubMed.
- J. Gacs, W. Y. Zhang, T. Knaus, F. G. Mutti, I. W. C. E. Arends and F. Hollmann, Catalysts, 2019, 9, 305 CrossRef.
- X. Ding, C.-L. Dong, Z. Guan and Y.-H. He, Angew. Chem., Int. Ed., 2019, 58, 118–124 CrossRef CAS PubMed.
- Y. Nakano, M. J. Black, A. J. Meichan, B. A. Sandoval, M. M. Chung, K. F. Biegasiewicz, T. Zhu and T. K. Hyster, Angew. Chem., Int. Ed., 2020, 59, 10484–10488 CrossRef CAS PubMed.
- J. S. DeHovitz, Y. Y. Loh, J. A. Kautzky, K. Nagao, A. J. Meichan, M. Yamauchi, D. W. C. MacMillan and T. K. Hyster, Science, 2020, 369, 1113–1118 CrossRef CAS PubMed.
Footnote |
| † Equal contribution. |
| This journal is © The Royal Society of Chemistry 2021 |