
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Swelling properties of graphite oxides and graphene oxide multilayered materials
Artem
Iakunkov
* and
Alexandr V.
Talyzin
 *
*
Department of Physics, Umeå University, SE-901 87 Umeå, Sweden. E-mail: alexandr.talyzin@umu.se
First published on 10th September 2020
Abstract
Graphite oxide (GtO) and graphene oxide (GO) multilayered laminates are hydrophilic materials easily intercalated by water and other polar solvents. By definition, an increase in the volume of a material connected to the uptake of a liquid or vapour is named swelling. Swelling is a property which defines graphite oxides and graphene oxides. Less oxidized materials not capable of swelling should be named oxidized graphene. The infinite swelling of graphite oxide yields graphene oxide in aqueous dispersions. Graphene oxide sheets dispersed in a polar solvent can be re-assembled into multilayered structures and named depending on applications as films, papers or membranes. The multilayered GO materials exhibit swelling properties which are mostly similar to those of graphite oxides but not identical and in some cases surprisingly different. Swelling is a key property of GO materials in all applications which involve the sorption of water/solvents from vapours, immersion of GO into liquid water/solvents and solution based chemical reactions. These applications include sensors, sorption/removal of pollutants from waste waters, separation of liquid and gas mixtures, nanofiltration, water desalination, water-permeable protective coatings, etc. Swelling defines the distance between graphene oxide sheets in solution-immersed GO materials and the possibility for penetration of ions and molecules inside of interlayers. A high sorption capacity of GO towards many molecules and cations is defined by swelling which makes the very high surface area of GO accessible. GtO and GO swelling is a surprisingly complex phenomenon which is manifested in a variety of different ways. Swelling is strongly different for materials produced using the most common Brodie and Hummers oxidation procedures; it depends on the degree of oxidation, ad temperature and pressure conditions. The value of the GO interlayer distance is especially important in membrane applications. Diffusion of solvent molecules and ions is defined by the size of “permeation channels” provided by the swelled GO structure. According to extensive studies performed over the last decade the exact value of the inter-layer distance in swelled GO depends on the nature of solvent, temperature and pressure conditions, and the pH and concentration of solutions and exhibits pronounced aging effects. This review provides insight into the fundamental swelling properties of multilayered GO and demonstrates links to advanced applications of these materials.
1. Introduction
Graphite/graphene oxides have been the subject of tens of thousands of studies over the past decade. Initially the interest in these materials was mostly due to the possibility to convert them into graphene using thermal or chemical reduction.1,2 However, graphene oxide (GO) itself emerged over the recent years as an advanced material for many applications. For example, a lot of research was recently performed with multilayered GO laminates cited as thin films,3–5 papers6,7 or membranes8–10 depending on specific applications. GO is also used as a precursor for preparation of many other new materials and composites11–14 since it can be easily dispersed in water, reduced and functionalized in many different ways.15,16 Swelling is one of the key properties of graphite oxide (GtO) and GO multi-layered materials. For example most often the chemical functionalization of GO is done in aqueous solutions or in solutions prepared using other polar solvents. Therefore the relationship of graphite oxides and graphene oxide with water and other polar solvents is of key importance for using them in many applications.In fact, swelling of GO materials is the property which makes them so unique. Water easily penetrates between GO planes expanding the structure, while it is impossible in graphitic materials. Dispersing graphite oxide in water to produce GO is in fact swelling with infinity as an inter-layer distance. Swelling enables the expansion of the graphite oxide structure which allows the penetration of reacting ions, molecules and polymers into the inter-layers and exposes both the surfaces of each GO sheet to chemical modification reactions. A high surface area in the water-swelled state makes graphite/graphene oxides very efficient adsorbents with potential applications in removing toxic contamination and waste water treatment.17–20 Swelling of multilayered GO assemblies is also a key property enabling their membrane applications.21 Research related to using multilayered graphene oxide membranes (mostly cited simply as GO membranes) has exploded in the last 5 years demonstrating many possible applications, e.g. in separation of gases, liquids, nano-filtration and desalination.22 Expansion of the GO lattice due to swelling in polar solvents (most importantly water) enables the formation of “permeation channels” and defines the permeation properties of the membranes.18,21,23 It is clear that the progress in using graphite/graphene oxide in many applications depends on the good understanding of swelling properties and the relationship between these materials and various solvents.
Fundamental research of GtO swelling and applications has shown significant progress over the past years, but this knowledge was not always used in application driven studies. It can be partly explained by the absence of review papers which connect fundamental and applied research related to the swelling of GtO and GO. Many review papers have been published about GO until now, describing a broad range of general or highly specialized subjects, for example chemistry,15,24 electrochemistry,16 structure and preparation methods,25 reduction,26,27 biocompatibility and other biology related subjects,28,29 graphene oxide based composites,30 specific types of GO materials or applications.18,31–33 Several monographs including many review papers have been published about graphite and graphene oxides.34 However, to our knowledge no focused review paper on the swelling of graphite oxide and related materials is available.
The main purpose of this review paper is to summarize the academic research related to the swelling of GtO and GO multilayers reported in many studies and to provide links to most important applications of these materials. In fact, the history of GO swelling research accounts for over 100 years with many very interesting studies undeservingly forgotten and rarely cited. Therefore, this review will start with historical introduction followed by an overview of academic research performed in modern times (last 15 years) and demonstration of links between swelling properties and using GO as an advanced material for many applications. More detailed focus on membranes is motivated by the critical need to control the swelling of these materials for key applications.
2. Discovery of graphite oxide, and the basic information about synthesis methods and structure
Graphite oxide has been known for over 150 years and studied extensively long before it started to be considered as a “graphene-related” material. Many of the early studies of GtO are undeservingly forgotten, and others are rarely cited and not known to broader audience. As a result, many of graphite oxide properties have been re-discovered again independently in recent years while they were actually reported tens or even hundred years earlier. In fact, most of the important properties of graphite oxides were well known already by the end of the 1960s.24 As noted by one of the pioneers of GO research in modern times, A. Lerf: “most workers active in the field of GO research since 2005 are just preparing footnotes to the seminal works of Kohlschütter, Hofmann and Boehm”. Most remarkably, early observations of graphene oxide and graphene have been reported in papers authored by H.P. Boehm35,36 about 50 years before the “graphene revolution” initiated by the studies of A. Geim and K. Novoselov.37 Moreover, it should be remembered that H.P. Boehm not only coined the name “graphene”, but also officially registered it in the international registry of materials IUPAC.The difficulty in finding relevant old papers is not only limited to the absence of on-line access to old and sometimes not existing anymore journals, but also due to the fact that several other old names have been used in the early years. The name “graphite oxide” is found starting only from the 50s, earlier it was most often cited as “graphitic oxide” or “graphitic acid”. Another obstacle to learning from early papers is that many of those were published in German language and have not been translated to English until now.
The purpose of this section is to give a credit to the pioneers of graphite and graphene oxide research with references to new studies which allows us to purify knowledge verified by time. It also introduces the reader to some basic knowledge of the graphite oxide structure and properties useful as a starting point for learning about these largely exciting materials.
The discovery of graphite oxide was reported in 1859 by B.C. Brodie.38 He believed graphite to be a new chemical element. This hypothesis was obviously not correct, but Brodie's attempt to oxidize graphite had resulted in the synthesis of a new material. Repeated oxidation of graphite using a mixture of fuming nitric acid and sodium chlorate followed by washing with an excess of water yielded light yellow coloured powder consisting of transparent plates. This procedure will be named Brodie's in the following text.
Keeping in mind that no spectroscopy or diffraction methods have been known at the time of Brodie's research, his paper provides amazingly precise analysis of the material. Brodie reported the following chemical composition of the new material: 67.70% carbon, 30.37% oxygen and 1.84% hydrogen after two oxidation cycles. The C/O ratio of 2.23 reported by Brodie in 185938 is remarkably close to modern estimations provided in 2004 in the study by T. Szabo et al. where C/O = 2.22 was found for Brodie graphite oxide synthesized using the same procedure after two oxidation cycles.39
This first study of graphite oxide also reports some properties which made this material exciting for 2D chemistry and physics. In particular, Brodie reports that his new material explodes upon heating in air or in nitrogen or can be decomposed using deoxidized agents. He even performed thermal reduction of GO in a mixture of hydrocarbons with high boiling points at 270 °C. Both thermal and chemical methods of GO reduction are currently very popular to produce powdered reduced graphene oxide (rGO)2 often simply cited as “graphene”. However, already Brodie paper states that reduction is not complete and the analysis of reduced samples showed a C/O of ∼4.3.38 Complete removal of oxygen functional groups from graphene oxide without breaking the C–C bonds remains to be a challenging problem even in modern times.
The chemical properties of the new substance reported in Brodie's pioneering study included the formation of jelly in liquid ammonia and “combining with alkalies” thus providing the very first indication of swelling.38 Brodie also noted that his new material is acidic in nature, which gave the name “graphitic acid” or “Graphitsäure” in German in many followed publications.
A lot of later studies were focused on “improving” the Brodie oxidation procedure which required several oxidation cycles and largely considered as dangerous. In fact, later studies revealed that using different synthesis methods results in graphite oxides which are close relatives but not the same materials with a large variation of properties depending on the details of preparation and oxidation degree.
L. Staudenmaier (1898) used a mixture of concentrated sulphuric and nitric acids with KClO3.40 In this way the final GO could be produced in one oxidation cycle. Many other procedures for oxidation of graphite were also tested at the time41 but only one became very popular. The new method proposed in 1958 (patented one year earlier) by W.S. Hummers and R.E. Offeman was the oxidation of graphite to graphite oxide by treating graphite with a water-free mixture of concentrated sulfuric acid, sodium nitrate and potassium permanganate.42 This method was in modern times used in many modifications43–45 which involve different proportions between reagents, more precise control of temperature in the process of reaction46 or addition of various other reagents (e.g. phosphoric acid47). It should be noted that most of the synthesis “improvements” were aimed at more easy dispersion of graphite oxide in water for subsequent reduction to produce graphene.48,49
Currently all these modifications are commonly named the “Hummers” or “modified Hummers” method. Most of the studies published about “graphite oxide” during the past decade were performed using the material prepared by Hummers method and only relatively few with Brodie GO. The name “graphite oxide” is used for all materials independent of the method of preparation, including Brodie, Staudenmaier and Hummers oxidations with all variations.
Many studies have contributed to a better understanding of the graphite/graphene oxide structure over the past 100 years.50,51 However, the structure of graphite oxides remains somewhat uncertain. The “true structure” of graphene and graphite oxide was debated in tens if not hundreds of studies,52,53 very often performed using only one kind of material prepared by certain method (most often Hummers54 or Brodie55,56) with a specific degree of oxidation and different precursors. However, it also became clear that the properties of graphite oxides are significantly dependent on the method of preparation, e.g. thermal exfoliation temperatures,57,58 swelling,59–62 the mechanical strength of individual flakes,63 the sorption of polar solvents64 and the sorption capacity towards radionuclides.20
In fact, graphite/graphene oxides are a family of materials with a strong variation in the degree of oxidation and the relative amounts of various oxygen functional groups.39,65 The structure of graphite oxide depends also on the type of graphitic precursor.57,66 Moreover, the composition of GO is affected by ageing effects when stored in air.67–69 There is also a very strong difference between the swelling properties of graphite oxides produced by Brodie and Hummers oxidation as will be discussed below in this review in more detail.58,59 Therefore, it is an opinion of authors that no single “true” molecular structure can be proposed for all graphite oxides.
The detailed review of all structural studies related to graphite and graphene oxide is outside the scope of this review. Here we describe briefly only some general properties which are common for all kinds of graphite oxides and important for the understanding of swelling. Oxidation of graphite by all the abovementioned methods results in the addition of several types of functional groups on the graphene oxide planes, most importantly hydroxyls and epoxy groups, while carboxyls and carbonyls are the main functionalities on the edges of flakes.15,54 The presence of many other oxygen functional groups was also reported for different types of graphene/graphite oxides, e.g. phenols, ketones, enols etc. The oxygen functional groups are randomly distributed over the planes formed by the graphene skeleton. The non-stoichiometric composition of GO in combination with complete disorder in the positions of oxygen groups is common for all kinds of graphite/graphene oxides.39,54,70,71 Graphite and multilayered GO laminates typically show a layered structure with the interlayer distance increased to ∼6–8 Å (ref. 21 and 72) compared to ∼3.3 Å in precursor graphite.73 The expansion of the distance between graphene layers due to oxidation is one of the most important parameters of the GO structure which correlates with swelling properties. Swelling is not observed in graphitic materials as water does not penetrate between hydrophobic graphene sheets.
We consider swelling as the key property of graphite oxide or graphene oxide in stacked multilayered laminates which makes it distinct from many other kinds of oxidized graphitic materials or oxidized graphene. In the absence of swelling the materials need to be named “oxidized graphene” or “oxidized graphitic carbon”.
The simplest definition of graphite oxide is as follows: “Graphite oxide is a hydrophilic layered material prepared by oxidation of graphite and showing swelling in polar solvents”.
3. Early studies of graphite oxide swelling
In general, swelling might refer to rather different phenomena related to the sorption of water and the increase in the volume of materials. In this review we focus on intra-crystalline swelling related to the change in the crystal structure of the material caused by water or other solvents. That is distinct from intercrystalline swelling which keeps the crystal structure unchanged with the sorption of the liquid between the crystallites. Intracrystalline swelling is rather common in layered materials.74,75 It is well known that many clay minerals, e.g. montmorillonites, kaolinites, and bentonites, swell in water with expansion of the inter-layered distance due to the sorption of water between 2D layers.76–78 Clay minerals are some of the most common in nature making the swelling everyday experience in our life, e.g. when the ground road becomes muddy after the rain.It is little known and rather surprising that intracrystalline swelling was discovered first in the exotic laboratory-synthesed material graphite oxide.79,80 Rather detailed studies of the graphite oxide structure, chemistry and reduction were performed by U. Hofmann in 1930–1934.79,81,82 (Fig. 1). His study from 1934 became recently available in English translation.81 Surprisingly, it reports almost all the most important effects observed in GO due to swelling and remains one of the most complete and precise studies of swelling even in modern times. Unfortunately this study has been almost forgotten over time and many facts reported by Hofmann et al. have been re-discovered only during the past 15 years.
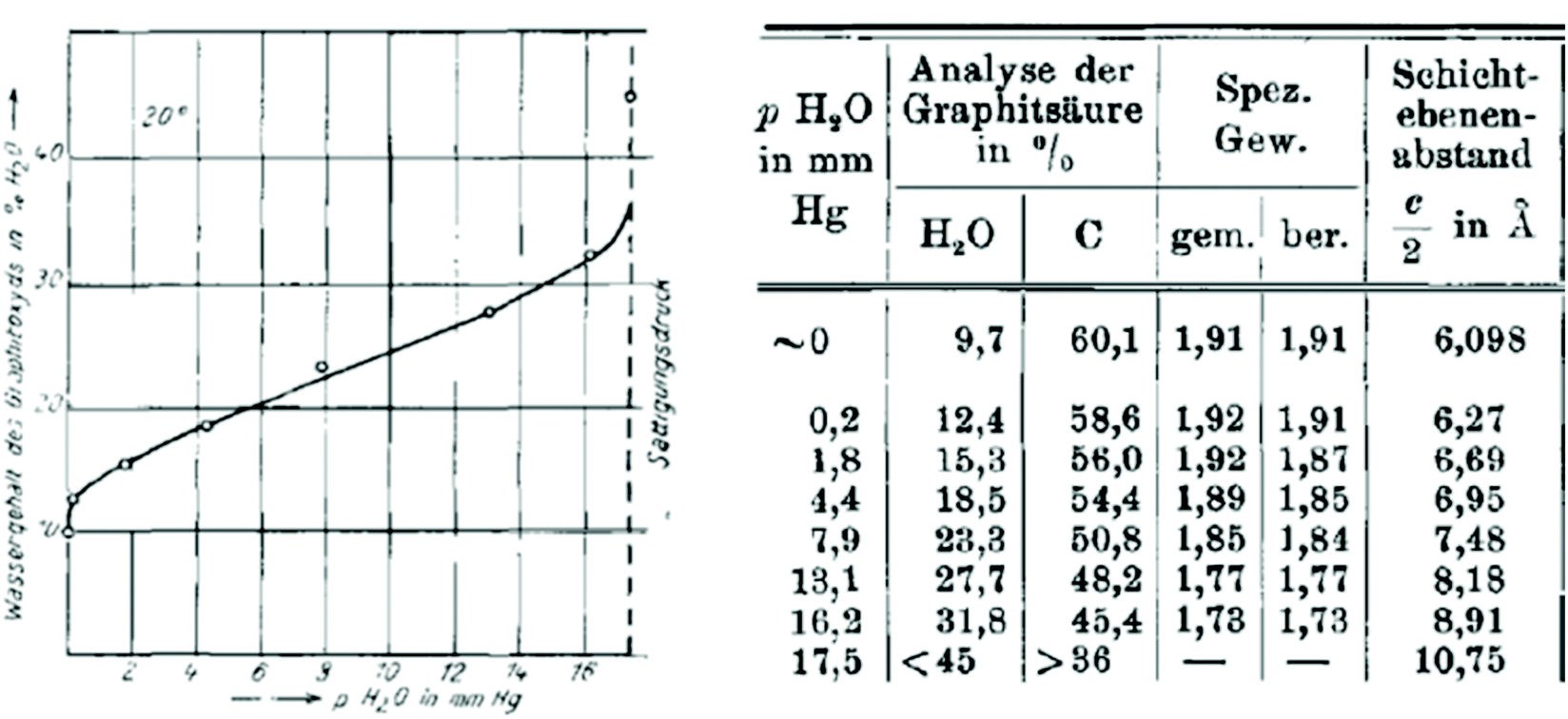 | ||
| Fig. 1 Sorption of water by graphite oxide measured in wt% and as a function of humidity (mm Hg). The table which relates the amount of absorbed water and the interlayer distance as a function of humidity.81 | ||
Swelling of graphite oxide was found in this study to occur both due to the sorption of water from vapour and in liquid water. Quantitative evaluation of water sorption as a function of air humidity was combined with XRD analysis providing remarkably precise results even considering it 90 years later.81 Graphite oxide was found to absorb water proportional to humidity (up to ∼40% by weight) with a simultaneous increase in inter-layer distance from 6.09 Å at zero humidity up to 10.75 Å. Hofmann et al. reported the heat of swelling as +19.2 cal g−1 and estimated that water molecules can form two monolayers between the neighbouring lattice planes of graphite oxide. According to the experimental results some water (∼10%) is present in the graphite oxide structure even at zero humidity and the maximal swelling in neutral water corresponds to the monomolecular film of water on both sides of graphene oxide sheets. However, the change in the interlayer distance occurs continuously as a function of water vapor pressure. This observation was rather puzzling at the moment considering that insertion of the water layer is supposed to change the inter-layer distance by minimum the size of the water molecule (∼2.5 Å). A complete water sorption isotherm of graphite oxide and the correlation between water content and inter-layer distance was recorded also by J. C. Derksen and J. R. Katz in 1934.83 This study was also first to report a remarkable pH dependence on swelling for GO in water.
Fig. 2 shows no certain values for d(001) in the pH interval 7–1383 reflecting a remarkable property of graphite oxide to disperse spontaneously producing single graphene oxide sheets. It can also be considered as ultimate swelling with infinity as a value for the interlayer distance. The formation of colloids and gels in the GtO water system was studied also in several other early studies.84 The possibility to produce dispersions with single layered graphene oxide in a basic solution is rather appreciated in modern times as a method to avoid defect-inducing mechanical treatments.85 To our knowledge the study by J. C. Derksen and J. R. Katz in 193483 is the first one to report swelling in the water solutions of various salts: nitrates, cyanides, iodides, bromides, chlorides, sulphates and chlorates. Using the same 2 M concentration of salts they demonstrated a significant difference in swelling with the range of inter-layer distance values within 10.19–14.6 Å.
 | ||
| Fig. 2 Inter-layer distance value provided by d(001) from XRD experiments as a function of pH.83 Graphite oxide spontaneously disperses into graphene oxide for the interval of pH marked as “peptisation. | ||
Unfortunately this study has been well forgotten over the years which leads in modern times to an incorrect but widely publicized assumption that multilayered GO membranes swell in all solutions exactly the same as in pure water. The “ultra-precise” filtration by the size of ions was claimed based on the incorrect assumption that swelling Is not influenced by the nature and concentration of salts in solutions.86 J. C. Derksen and J. R. Katz in 193483 reported also the first study of GO swelling in water–alcohol binary mixtures. It was also completely forgotten (the study is in German) over the years and re-discovered independently by one of the authors of this review about 80 years later.87
Hofmann et al.81 reported also rather detail study of graphite oxide thermal deoxygenation which includes the estimation of swelling in water, conductivity and the chemical analysis of the C/O composition for the GtO material annealed at different temperatures. Remarkably for the very early study of GtO thermal reduction, the data for swelling of thermally reduced GtO vs. C/O ratio remain unique even in modern days.
The C/O = 3.5 of pristine GtO in this study was relatively large for modern oxidation methods. The change in the inter-layer distance due to water swelling was 3.5 Å at ambient temperature. Annealing at 850 °C resulted in a decrease of inter-layer distance from ∼6 Å for the pristine material down to the value typical for graphite. The most important for this review part of Table 1 provides the relationship between the composition of GO and its ability of swelling. The GtO annealed at 160 °C results in C/O = 4.25 and shows a somewhat smaller interlayer distance of 5.76 Å in the dry state but keeps original swelling properties with 10 Å inter-planar distance in water. An abrupt change in swelling properties is found for samples heated at higher temperatures. The swelling is almost negligibly small for materials with a low oxidation degree C/O > 5 and completely disappears for the sample with C/O = 8.5. Since the hydrophilic properties of GO are provided by oxygen functionalities, it is expected that removing oxygen will result in more and more hydrophobic materials until graphitic carbon is produced. The Hofmann et al. study provides the first estimation for the range of C/O ratios which allow swelling in water.81 Considering the definition of graphite oxide as a material capable of swelling, the materials with C/O > ∼7 must be considered as oxidized graphite or in the case of single layers as oxidized graphene.
| Step | Temp. | %C | %H2O | %Ash | %O2 | C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O :O |
Cal per g graphitic acid | Cal per g C in graphitic acid | d(002) dry Å | d(002) wet Å | Spec. Resistance | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dry | Wet | |||||||||||
| Ω cm | ||||||||||||
| 1 | 20 | 62.8 | 9.3 | 1.8 | 24.2 | 3.5 | 4883 | 7801 | 6.14 | 11.0 | 3960 (5 × 107) | |
| 62.4 | 9.6 | 4895 | 7847 | |||||||||
| 2 | 160 | 63.9 | 8.5 | — | 24.0 | 3.6 | — | — | 5.90 | 10.7 | 251 | |
| 3 | 160 | 69.4 | 4.8 | 2.3 | 22.1 | 4.25 | 5375 | 7700 | 5.76 | 10.0 | 1.85 | 3.4 |
| 70.0 | 5.3 | 5391 | ||||||||||
| 3a | 180 | 74.6 | 4.2 | — | 18.0 | 5.53 | — | — | 4.67 | 4.97 | 2.2 | 3.6 |
| 4 | 200 | 79.6 | 2.9 | 3.1 | 15.1 | 7.1 | 6144 | 7740 | 4.41 | 4.67 | 0.39 | |
| 79.7 | 1.7 | 2.8 | 6136 | 7725 | ||||||||
| 5 | 320 | 82.1 | 2.2 | 2.9 | 12.9 | 8.5 | 6398 | 7805 | 4.05 | 4.05 | 0.20 | |
| 82.1 | 1.9 | 2.9 | 6404 | 7820 | ||||||||
| 6 | 500 | 84.6 | 3.1 | 3.3 | 9.6 | 12 | 6705 | 7870 | 3.61 | 3.61 | — | |
| 85.2 | 2.6 | 3.1 | ||||||||||
| 7 | 850 | 88.8 | 2.0 | 3.2 | 5.5 | 21 | 7047 | 7910 | 3.38 | 3.38 | 0.05 | |
| 87.6 | 3.2 | 3.2 | 7084 | 7970 | ||||||||
| Graphite | 100 | — | — | — | — | — | 7856 | 3.39 | — | 0.023 | ||
Active studies of graphite oxides and their swelling properties continued in the 1930–60s24,88 including more and more methods, e.g. the first (to our knowledge)89 electron microscopy images revealing a few layered GO flakes date back to 195288 and FTIR spectroscopy to 1955.89 The later study reported FTIR spectra recorded as a function of a water content of 0–60%. It also revealed that graphite oxide contains characteristic hydroxyl groups that may be distinguished from those of intercalated water and not easily substituted with deuterium when exposed to D2O despite the evident penetration of heavy water between GO layers.89
By the end of the 1960s the swelling of graphite oxide in water vapours was studied in much detail. The isotherm of water sorption by graphite oxide was reported by J.B. de Boer and A.B.C. van Doorn in 1958.90 They noted that 4% of water is absorbed by GO without any change in c-parameter and it changes very little until ∼10–15% of water is absorbed. A similar effect was later found for vapour sorption of ammonia by graphite oxide.91 These results indicate the presence of some “empty” space in the GO structure which can be filled with water without the expansion of the lattice. The “empty spaces” are likely related to nm size unoxidized spots covering the minor part of the GO flake surface. The studies of vapor sorption by graphite oxides can be considered as a first step towards using graphene oxide as vapour sensors in modern times. Heats of immersion and adsorption were reported for the GO–water system in 1960 by W. H. Slabaugh and C. V. Hatch.92 They were possibly the first to evaluate the surface area using water vapor sorption and the BET equation reporting it in the range of 315–350 m2 g−1.
Swelling of GO in liquid solvents was reported by the end of the 1960s for many solvents at ambient temperature with some fragmentary data for higher and lower temperatures. Particularly strong contribution to this research was made by MacEwan and co-authors. Despite the fact that several of these papers were published in the high profile journal Nature, these early papers are surprisingly little known and almost never cited.93–96 The first paper by the MacEwan group (1955) reported swelling tests for “graphitic oxide” in 19 organic solvents.93 No swelling was found in non-polar solvents (benzene, hexane + pentane). The increase of inter-layer distance by 2–4 Å detected using XRD was found for several solvents including acetone, ethylene glycol, glycerol, pyridine, small alcohols, a much larger increase in some amines (7.6 Å in naphtylamine, 9.8 Å in buthylamine) and aniline (11.5 Å).93 Remarkably, intercalation of graphite oxide with aromatic amines (including aniline) was reported again as a “first time” study in 2014.97 Several studies by MacEwan and co-authors reported the swelling of graphite oxide in a set of normal alcohols with the number of carbon atoms in the range 1–1894,95 and for aliphatic amines the number of carbon atoms was up to 20.94,98 The studies included also swelling in diamines, fatty acids and nitriles.98
The studies of graphite oxide swelling in chain alcohols performed by MacEwan et al. remain to be one of the most complete to date. It sets several questions resolved only very recently or not resolved until now.95,98Fig. 3 summarizes the data presented in two papers from this group which include swelling in liquid solvents at ambient temperature for chain alcohols starting from methanol (carbon number C = 1) to very long chains with C = 18. The data were recorded using XRD and showed an increase of inter-layer distance d(001) up to ∼50 Å. Remarkably, these very old experimental studies from 1959–1965 make obsolete some modern theoretical models which claim that octanol (C = 8) is too large molecule to penetrate between graphene oxide sheets in membrane materials.86
 | ||
| Fig. 3 Swelling of graphite oxide in 1-alcohols. The interlayer distance evaluated by XRD using d(001) vs. the number of carbon atoms in the alcohol molecule (1 for methanol, 2 for ethanol etc.). Open symbols are for data recorded in liquid solvents at ambient temperature (○- ref. 94; □- ref. 95). Ref. 95 provides also data for swelling at −50 °C (▲) recorded using frozen solid samples (except for octanol recorded in liquid) and at 70 °C (■). Inset shows the hypothetical change in orientation of alcohol molecules intercalated into the graphite oxide structure for larger alcohols.98 The data points were copied from the published figures and are not precise. | ||
The data shown in Fig. 3 demonstrate that the longer is the alcohol molecule, the larger is the swelling effect. However the increase in interlayer distance is not monotonous showing two distinct steps. The three smallest alcohol molecules intercalated into the GO structure provide almost the same layer separation by ∼10 Å. For butanol the inter-layer distance increases additionally by 5–7 Å and after that it increases almost linearly. The second less obvious step occurs around C = 10–11. One needs to note that the discrepancies between two studies of the MacEwan group (Fig. 3 for alcohols with C = 3–7) are likely to be explained by different synthesis procedures. The earlier work94 cites preliminary experiments performed in 1956, before invention of the Hummers method (1958).42 The later study from 1965 explicitly used graphite oxide prepared by the Hummers method.95
MacEwan et al. proposed a simple geometrical model (illustrated in the inset in Fig. 3) which suggests that alcohol molecules are attached to certain points on graphene oxide sheets. When the length of alcohol molecules is smaller compared to the distance between the attachment points, the orientation of molecules is parallel to GO layers (alfa-phase). When the length of alcohol molecules exceeds the average distance between hypothetical attachment points the orientation of alcohol molecules changes from parallel to stand-up (beta–phase).94,95,98 For the second step which occurs in the swelled structures of graphite oxide in alcohols with c > 10 they suggested a change in the structure of intercalated alcohol molecular layers from “liquid” to “solid” based on the details of XRD pattern analysis. The hypothetical stand-up orientation of alcohol molecules looked completely logical having only XRD data available to MacEwan et al., but did not stand when verified in later modern time studies.99 The nature of the second step in Fig. 3 observed for larger alcohols remains unexplored. However, it is interesting to note that simple geometrical models related to the change in the orientation of intercalated molecules are likely be valid for other solvents, e.g. amines and n-alkylammonium chain molecules which tend to attach to the charged points of graphene oxide sheets.100
Fig. 3 also shows a remarkable difference in the swelling of graphite oxide at three temperature points: ambient temperature, low temperature (−50°) and higher temperature of 70 °C.95 However, detailed temperature dependent studies were not performed back in the 1960s due to technical limitation. Note that the low temperature part in Fig. 3 shows mostly points for solvents which are frozen at −50 °C. The authors cited technical problems to conduct experiments in rapidly evaporating liquid solvents at low temperatures,95 thus making the studies of temperature dependent swelling in small alcohols like methanol and ethanol impossible at the moment. This gap was filled only during the past decade (see below).
Detailed studies of swelling were reported in the 1960s also for chemically modified graphite oxides in the methylated and acetated state.21,98,101
One of the most amazing achievements of graphite oxide science in the 1960s was the isolation of graphene oxide and the thermal conversion of graphene oxide to graphene. The official inventor of the term “graphene” (in the 1990s) H.P. Boehm back in 1961 listed the swelling of graphite oxide in several solvents marking as infinity the inter-layer distance in 0.01 M NaOH. He described then the complete separation of the GtO structure on individual macromolecular sheets and chemically reduced it to graphene.35,36,102 This contribution was acknowledged by the 2010 Nobel prize winner for graphene studies A. Geim in his Nobel lecture.103 Even more importantly, purely chemical separation of graphene oxide layers in the extreme swelling state is a popular approach to avoid defect inducing mechanical treatments in modern times. Moreover, some types of graphite oxides (e.g. Brodie GO) do not disperse in water even after intense sonication but spontaneously dissolve in weakly basic solutions.61
4. Early studies of “graphene oxide” membranes
Possibly the only application of GtO emerged on the industry related scale as a result of academic research in the 1960s was using graphite oxide membranes for sea water desalination but even that direction was dropped after extensive work within a government-funded program in the USA104 and almost completely forgotten. Nevertheless, the old studies of membranes are still valuable even compared to recent papers. To our knowledge the first report of using multilayered GO as a membrane dates back to 1956.88 A. Clauss and U. Hofmann reported a method to measure the water vapour pressure based on the ability of GO membranes to permeate water vapour but not air. Vacuum tight membranes were prepared using slow drying of graphene oxide dispersion on a suitable surface. Note that the term “graphene” did not exist at that moment and the membranes were named “graphitic oxide”. However, we know that the methods which were used back in the 1950–1960s provided true graphene oxide dispersions. In fact, first membrane-like foils were prepared by vacuum filtration using colloid GO dispersions as early as in 1919 but without testing any membrane properties.105 As noted in 1958 by J.B. de Boer and A.B.C. van Doorn the graphite oxide membranes are flexible and can be prepared in “any size”.90 However, the first rather detailed study of GO membranes was published by H.P. Boehm et al. in 1961.21The membranes with a thickness of 0.05 mm were found to be not permeable by gases like nitrogen and oxygen under humidity free conditions. However, rapid permeation was reported for water and “all substances which are able to penetrate between layers” following a zigzag permeation pathway shown in Fig. 4. A nearly identical picture can be found in the key studies which revived the interest in GO membranes in modern times,23 unfortunately without a reference to the early work.21 In line with a modern understanding of GO membrane permeation the study concludes that the existence of a three-dimensional pore network is unlikely in GO membranes and the mechanism of permeation is connected to swelling and diffusion of water molecules along inter-lamellar interstices. H.P. Boehm et al. tested vacuum driven vapor permeation across the membranes and estimated the velocity of water diffusion between GO layers to be 1 cm h−1. The study is also extended to the permeation of solutions and states that GO membranes are impermeable to the “substances of lower molecular weight” thus providing first insight into nanofitration properties.21 Moreover, H.P. Boehm et al. was first to measure the membrane potentials of GO foils. Since the graphene oxide sheets are negatively charged in water, cations easily permeate across the membranes while anions are repelled. This results in some charge separation on the opposite sides of the membrane immersed in aqueous electrolytes. The membrane potentials were reported by H.P. Boehm et al. for HCl, KCl, CaCl2 and BaCl2 solutions noting the significant difference between the permeation of single charged and double charged cations.21 The membrane potentials of GO were reported also in some other studies in the end of the 1960s.106 The drawbacks of the GO membranes were also noted:
● poor mechanical stability in the swollen state and extreme swelling in some solutions.
● the absence of the sieving effect for some large molecules (large alkaloid ions were found to permeate across the membrane).21
 | ||
| Fig. 4 Zigzag pathway of water permeation across the graphene oxide (graphite oxide) membrane. The size of permeation “channels” is provided by the distance between neighboring graphene oxide flakes and by the effect of swelling in water.21 | ||
The remarkable property of GO membranes to reject some salts with potential application in water desalination was a subject of a detailed four years project by the US Department of Interior finished in 1970 with reports available (but not published in per reviewed journals). The study reported excellent salt rejection properties and suggested using GO membranes enveloped by clay layers in order to improve their mechanical stability.104 However, the GO membranes were never used in industrial sea water desalination due to concurrence from other, a lot less exotic materials with better performance. Instead of GO, the first generation of desalination membranes employed by industry was based on acetylated cellulose.107,108
It can be summarized that by the end of the 1960s most of the main properties of graphite oxides were actually well known, including most general swelling properties. However, the studies had little effect on the development of practical industrial applications. Graphite and graphene oxides remained to be curiosity materials until the first half of the 2000s with many important fundamental contributions in the 1990s but rather a significant time gap in the 1970s and 1980s. For the study of the swelling properties of graphite oxide one can mark only the PhD thesis by R. Kruger with extensive studies of GO swelling and intercalation by amines, amides and several other molecules including some alcohols in the 1980s.109 Unfortunately this thesis work was not translated from German and the data were not published in peer reviewed journals.
It can only be regretted that many of the early studies were not remembered and not acknowledged in the 2000s when the interest in graphite oxides revived.
5. Swelling of graphite oxides: methods
Swelling of GO in solvent vapours and in liquid solvents is equally important to analyse as these are relevant to many applications. Swelling in liquid solvents is for example the main parameter which affects the nano-filtration properties of GO membranes.21 Swelling in vapors is important for the binary gas mixture separation properties of GO membranes which can be controlled by humidity.110 Swelling also significantly affects the mechanical properties of GO papers and membranes.6,59Most common methods for characterization of graphite oxide swelling in vapours (mostly water) and liquid have been identified already by the end of the 60s and currently widely implemented in the characterization of GtO and multilayered GO materials. Several new experimental methods were added in the recent 30 years broadening the range of possibilities. Below these methods are listed and illustrated by examples. Some common misunderstandings in using the methods are also discussed.
X-ray diffraction method
XRD is possibly the most important structural method for characterization of GO swelling as it allows to evaluate the change of interlayer distance due to the intercalation of solvent molecules. XRD was the method which was used to study GO swelling from the very early times and is extensively used in modern research using standard diffractometers and synchrotron facilities. The examples of XRD patterns recorded from graphite oxide in air and in excess of water are shown in Fig. 5a. | ||
| Fig. 5 Examples of XRD patterns recorded from graphite oxide powder (synthesized using the Brodie method) in liquid solvents: (a) synchrotron radiation (λ = 0.71170 Å) XRD recorded in transmission mode under ambient air conditions from water filled sealed glass capillaries111 (b) XRD patterns recorded using a standard diffractometer with CuKα radiation in reflection mode.112 The samples are immersed in liquid alcohols and sealed using plastic foil (the peak marked by * is from the foil). | ||
The diffraction pattern of GO powder shows very few reflections due to the absence of ordering in the packing of individual layers and the random positions of oxygen functional groups. XRD also do not provide structural information about the state of solvent molecules in GO due to complete disorder. The c-parameter of the GO structure corresponds directly to the distance between graphene oxide layers due to the turbostratic packing of layers. Typically the (001) reflection is the strongest in the XRD pattern of GO and shows an asymmetric shape. The position of the main component is considered as the easiest way to evaluate the inter-layer distance using the d(001) value. The broad and asymmetric shapes of the (010) and (110) reflections are due to non-uniform interatomic distances in slightly buckled graphene planes. XRD do not provide information about the nature or amount of oxygen functionalities in the GO structure except for the change in the inter-layer distance. Generally the stronger the oxidation, the higher the inter-layer distance. However, the difference in the d(001) between different samples needs to be treated with caution if the XRD pattern is recorded under ambient air conditions.
Water sorption causes the intercrystalline swelling of GO81,90 and the same material recorded at low humidity or e.g. in tropical climate with close to 100% humidity will exhibit XRD with a dramatic difference in the value of c-parameter. The change of the c-parameter of graphite oxide due to the increase of humidity is non-linear39,55,90 and quantitatively depends on the exact nature of the material, such as the synthesis method and oxidation degree. The change in the swelling state due to the variation of humidity is rapid according to several studies and occurs within minutes required to record an XRD pattern. Complete removal of absorbed water is very difficult even after prolonged exposure to humidity free conditions but the very carefully dried material seems to show a slower response to the increase of humidity. For example, T. Szabo et al. used 1 month of drying in a desiccator over concentrated H2SO4/silica gel to produce a dry Brodie GtO material which completely equilibrated with 50% humidity after 3 hours of exposure showing an ∼0.4 Å shift in d(001).39 Swelling of GtO in liquid water and most of other solvents is instantaneous; the maximal saturation state is achieved in 1–2 minutes.111
It can be advised to record the XRD patterns of GO under vacuum conditions (to remove humidity related lattice expansion) for standardization purpose, but that is rarely done. On the other hand, a standard test can easily be performed e.g. in liquid water thus providing information about the maximal saturation hydration state of GtO. Fig. 5 shows the XRD patterns recorded in liquid water and small alcohols. Swelling of GtO is reflected in the strong shift of (00![[small script l]](https://www.rsc.org/images/entities/i_char_e146.gif) ) reflections, while the positions of in-plane peaks are not affected. Moreover, intercalation of some solvents results in a swelling-induced ordering effect as reported in several studies87,112 The ordering effect due to swelling in alcohols is evidenced by the decrease in the width of (00) reflections compared to the solvent free state and the increased intensity of higher order peaks from this set. For example, the (002) reflection is barely visible in solvent free GO and no peaks with higher order are typically observed. Immersion of GO in e.g. alcohols and liquid amines results in a sharply higher intensity of (002) and an increased number of peaks from the (00) with up to 5–10 and even higher.99,113 Providing good sealing conditions is important for recording high quality data in solvents with high vapour pressure. The best results are achieved when experiments are performed inside of sealed capillaries (usually in transmission geometry) or under plastic foil (kapton or polyethylene) in reflection geometry (Fig. 5). One needs to observe that a strong tendency of graphite/graphene oxide flakes to align parallel to each other results in preferential orientation and the relative intensities of XRD reflections must be treated with caution as it depends on the orientation of the sample.114 Extreme caution is also needed in experiments where the XRD pattern is recorded in the solvent soaked state but in the absence of sealing. Evaporation of solvent in the process of XRD recording might result in the change of the swelling state of GO materials and inaccurate evaluation of d(001).115 Using synchrotron radiation provides an advantage of rather rapid recording of XRD images in just 1–2 minutes and a possibility to follow the changes of GO swelling under various temperature and pressure conditions with higher precision. The temperature dependent studies of GO swelling will be described in detail in the next sections. It is also very common now to use synchrotron radiation with 2D detectors which provide the images of whole diffraction rings and rather explicit information about diffuse scattering. The diffuse scattering is especially valuable as an instrument to detect the formation of gel-like phases which are observed to form in GtO immersed in some solvents.60
) reflections, while the positions of in-plane peaks are not affected. Moreover, intercalation of some solvents results in a swelling-induced ordering effect as reported in several studies87,112 The ordering effect due to swelling in alcohols is evidenced by the decrease in the width of (00) reflections compared to the solvent free state and the increased intensity of higher order peaks from this set. For example, the (002) reflection is barely visible in solvent free GO and no peaks with higher order are typically observed. Immersion of GO in e.g. alcohols and liquid amines results in a sharply higher intensity of (002) and an increased number of peaks from the (00) with up to 5–10 and even higher.99,113 Providing good sealing conditions is important for recording high quality data in solvents with high vapour pressure. The best results are achieved when experiments are performed inside of sealed capillaries (usually in transmission geometry) or under plastic foil (kapton or polyethylene) in reflection geometry (Fig. 5). One needs to observe that a strong tendency of graphite/graphene oxide flakes to align parallel to each other results in preferential orientation and the relative intensities of XRD reflections must be treated with caution as it depends on the orientation of the sample.114 Extreme caution is also needed in experiments where the XRD pattern is recorded in the solvent soaked state but in the absence of sealing. Evaporation of solvent in the process of XRD recording might result in the change of the swelling state of GO materials and inaccurate evaluation of d(001).115 Using synchrotron radiation provides an advantage of rather rapid recording of XRD images in just 1–2 minutes and a possibility to follow the changes of GO swelling under various temperature and pressure conditions with higher precision. The temperature dependent studies of GO swelling will be described in detail in the next sections. It is also very common now to use synchrotron radiation with 2D detectors which provide the images of whole diffraction rings and rather explicit information about diffuse scattering. The diffuse scattering is especially valuable as an instrument to detect the formation of gel-like phases which are observed to form in GtO immersed in some solvents.60
The same principle as in XRD can be used for the study of GtO and GO using neutron diffraction as demonstrated by several studies.116–118
Summarizing this section, the diffraction method is a powerful instrument for studies of GO swelling but it has also some limitations. For example, it provides only the estimation of the inter-layer distance based on averaging over many layers, typically hundreds or thousands. XRD does not provide quantitative evaluation for the amount of intercalated solvent. The same separation of layers can in principle be achieved by intercalation of densely packed layers or diluted layers with molecules scarcely distributed along the interlayer and serving as “pillars”. Therefore, using other complimentary methods for quantitative characterization of absorbed solvents is very important and informative.
Quantitative evaluation of solvent sorption
The ability of graphite/graphene oxide to absorb rapidly large amounts of polar solvents from vapour is useful for applications related to the removal of toxic or undesirable pollutants from air.32The simplest method to study swelling caused by sorption of water (or other polar solvent) vapours is to take measurements of weight change when humidity (or vapour pressure) is stepwise changed from one value to another.81,90,92 The weight measurement can then be taken at every step of vapour pressure after certain equilibration time (isopiestic method). The easiest test can be performed simply by exposing GO powder to solvent vapour inside of a sealed jar. Some liquid solvent is placed inside of the jar and allowed to evaporate until the saturation vapour pressure is achieved for a given temperature (often ambient). The weight change due to vapour sorption provides then the saturation value of sorption for given conditions.64,99 The dynamic isopiestic method suggests changing the vapour pressure in controlled steps and recording the weight change in every step. Saturation of sorption by some solvents occurs in GO rather rapidly which allows one to record the weight change continuously in the process of vapour pressure increase. This method was used in early studies of water sorption and in several studied performed over the past decade for sorption of several other solvents. For example, in situ weight change measurements of water sorption were performed in a sealed volume by placing inside a simple humidity sensor and a small vial with solvents.119 A quartz microbalance was also used in other studies to control the sorption of water by thin films.120 In other studies the weight change was measured only at saturation vapour pressure.64
Modern automated Dynamic Vapour Sorption systems allow one to record complete vapour sorption isotherms and to evaluate the BET surface area for a given solvent and to extract some information about the pore size. The remarkable ability of GO to swell in solvent vapours results in a very strong difference in the values of the surface area estimated using standard nitrogen BET tests, e.g. H2O BET. Nitrogen and other gases do not penetrate between individual graphene oxide sheets and adsorb on the outer surface of multilayered flakes, while water penetrates and expands inter-layers. Typical N2 BET tests of powder graphite oxides provide rather small values of the surface area on the level of few m2 g−1 while H2O BET provides for the same powder materials’ surface area values on the level of 300–600 m2 g−1 (Fig. 6).121 Considering that the theoretical surface area of graphene oxide is about 2400 m2 g−1 (somewhat smaller compared to pure graphene122), the water vapour sorption results indicate that only about ¼ of total surface is accessible for vapors. On the other hand the difference between theoretical and experimental values can be explained by the limitations of the BET model and the very small size of pores provided by GO interlayers (0.7–1.0 Å).
 | ||
| Fig. 6 Water sorption and desorption isotherms recorded using an automated DVS system for samples of (a) thermally reduced graphene oxide (rGO) (H2O BET = 113 m2 g−1) and precursor Hummers GO (H2O BET = 345 m2 g−1).121 Different shapes of isotherms reflect the hydrophobic nature of rGO with the surface area related to the highly dispersed nature of this material (N2 BET = 329 m2 g−1) and the hydrophilic nature of GO with negligibly small N2 BET surface area (<5 m2 g−1) but high water sorption due to swelling.121 | ||
The gravimetric sorption method is especially powerful when used in combination with XRD, thus providing information both about the volumetric change in the GO structure provided by expansion of inter-layers and quantitative evaluation of the absorbed solvent.81,90,123 However, the gravimetric method is not suitable for experiments with swelling in liquids.
Quantitative evaluation of the solvent adsorbed by GO in liquid solvents was successfully determined using the DSC method. The amount of solvent adsorbed by the GO structure can be determined using the known enthalpy of bulk solvent freezing/melting. The solid GO material is loaded into a sealed capsule with a known amount of liquid solvent sufficient for saturated sorption; then the sample is cooled below the freezing point of solvent and heated back to record the enthalpy of melting. The amount of bulk solvent decreases due to the intercalation into the GO structure and the part absorbed by the material does not contribute to the measured melting enthalpy. The decrease in melting enthalpy measured using DSC allows the calculation of the amount of solvent sorbed by the material.60,64,87,99 Note that the sorption is strongly temperature dependent for GO in many solvents, while the data obtained by the DSC method are valid only for the temperature point of solvent freezing. Recording temperature dependent quantitative sorption of solvents by GO in the swelled state remains to be unexplored due to the lack of suitable methods.
Methods related to the change in film thickness due to swelling
Exposure of GO to polar solvent vapors results in a significant increase of material's volume which can also be quantified for evaluation of swelling. It can possibly be done for powder materials as well, but so far this method is mostly used for GO thin films deposited on some substrates or free standing GO membranes.124 Parallel orientation of graphene oxide flakes in multilayered assemblies and thin films results in an increase of sample thickness due to swelling. This increase can be detected by optical microscopy, electron microscopy or using more advanced methods like Neutron Reflectometry (NR).125 The advantage of NR is a possibility to determine simultaneously the change in the film thickness and composition of GO intercalated by solvent vapors. Moreover, the inter-layer distance of GO laminates can be determined using reference XRD measurement, which provides the number of GO layers for a given film thickness. However, NR requires using rather advanced facilities and is very demanding for the quality of thin films.126,127Electron microscopy is usually performed under conditions of high vacuum which does not allow to do direct imaging of GO in liquid solvents. Therefore, electron microscopy of swelled structures is typically performed using frozen samples under cryogenic conditions.128
The method allows one to study solid materials formed by GO when frozen in the swelled state. However, it is not possible to compare directly the structure and composition of frozen samples with GO swelled in excess of liquid solvents. Experiments performed using XRD demonstrated that a significant part of solvent escapes from the GO structure in the process of freezing.130 Using Environmental Scanning Electron Microscopy (ESEM) the thickness of GO films was measured as a function of humidity using a specially designed setup (Fig. 7).129
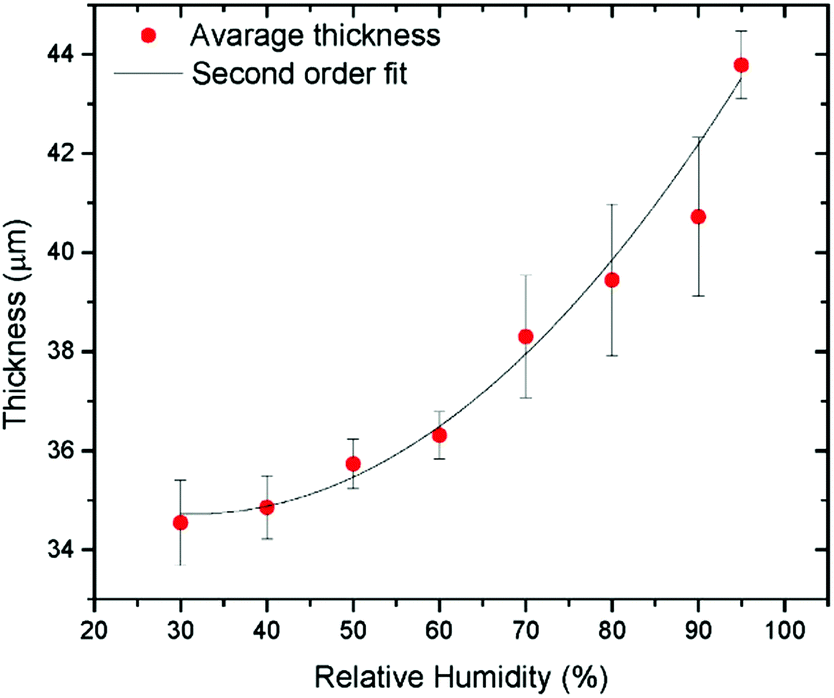 | ||
| Fig. 7 Multilayered GO film thickness measured directly using ESEM as a function of humidity.129 | ||
Some attempts to study the thickness of thin GO films using optical methods were also presented.131 However, a very high inter-layer distance of GO in water reported in this study (∼60 Å) is not independently verified and likely related to partial delamination of the film.
Most of the methods used so far to study the swelling of GO have been applied to multilayered materials. To our knowledge there is only one study where the distance between two layers of graphene oxide was directly measured using Atomic Force Microscopy (AFM) as a function of humidity. High resolution imaging in combination with height profiles on a fraction of the Ångtsrom scale is unique for the AFM method which allows one to study swelling along a single inter-layer formed by two graphene oxide sheets.132
6. Swelling of graphite oxides in water
Water is the most common solvent on the Earth and most important for applications of graphite oxides and multilayered GO laminates. For example, most often chemical modification of GO is performed in aqueous solutions.15 GO as a sorbent has great potential for waste water treatments; aqueous solutions are used in membranes for nanofiltration133–136 and sea water desalination.137 Moreover, even the deposition of GO multilayers is typically done in the water swelled state using aqueous dispersions.6 Swelling is very important for these and other applications. Therefore, research related to the hydration of graphite oxides will be reviewed in this section in more details. Swelling of GtO in water is also distinctly different compared to most of other solvents due to the small size of the water molecule and its unique chemical nature.Swelling of graphite oxides in water vapours was studied starting from the 1930s (see above) and has been re-evaluated many times over the past 90 years. In recent years the interest in the sorption of water from vapour was heated up by suggestion of application in water sensing. For example, superfast sensing abilities of graphene oxide towards water vapours were demonstrated in several studies138–140 and a simple device composed of a graphene oxide film with attached electrodes patented for mobile phone protection applications to detect liquid and vapour water.141 The change in the conductance or capacitance of graphene oxide films under conditions of varying humidity is typically used in these sensors.138,142 Graphene oxide papers were also proposed for air dehumidification.143
Sorption of water from vapour depends somewhat on the nature of graphite oxide, its oxidation degree, the method of synthesis and several other parameters. However, some sorption properties are very general for all kinds of graphite oxides. Many studies reported gravimetric water uptake and/or change in the inert-layer distance as a function of humidity.39,55,64,90,92,123,144,145
The maximal hydration at 100% humidity is typically reported to be on the level of ∼40–75 wt% depending on the type of graphite oxide. The numbers on the lower side of this interval are reported for Brodie GO (e.g. 43% in ref. 123) and on the higher side for Hummers GO (e.g. ∼75% in ref. 64). The same trend is also confirmed by the TGA method which provides data for evaporation of water from air-saturated humidity GO in the temperature interval below 100 °C. Typically Hummers GO shows a larger loss of water compared to Brodie GO for the samples studied starting from the same air humidity.58,59 However, higher water sorption is likely to be connected to stronger oxidation of GO (lower C/O ratio). For example J.B. de Boer and A.B.C. van Doorn reported water sorption isotherms for graphite oxides prepared using the Brodie procedure repeated 3 and 6 times; this corresponded to an increase of saturated water sorption from ∼65 wt% to 80 wt%,90 on the level with strongly absorbing Hummers GO. There are also indications that sulfur impurity which is usually present in Hummers GO stimulates higher water sorption. Freshly prepared multilayered GO assemblies (papers, thin films or membranes) typically show maximal water sorption very similar to the precursor graphite oxides up to ∼60–75 wt%.64,119,143,146
The sorption of water is reflected in the expansion of the GO structure along the c-direction. The increase in inter-layer distance at 100% humidity is about 4–6 Å relative to the water free state. Note that many studies reported the change in the inter-layer distance but not always relate it to specified “ambient” air humidity. The moisture-free GO typically exhibits d(001) in the range of 6–8 Å and in the saturated water vapour state it is around 11–12 Å. Once again the exact change of the inter-layer distance to swelling in vapours depends on the method of GtO preparation (e.g. Brodie vs. Hummers oxidation) and several other parameters. For example A. Lerf et al.55 studied 5 samples synthesized using the Brodie procedure but somewhat different in composition and prepared in different groups. The interlayer distance in the range of 10.5 Å–11.7 Å was reported for four relatively freshly prepared samples at the highest humidity. Some swelling was found even for the sample stored in air for about 30 years but in this case the increase of inter-layer distance by only 1 Å (up to ∼8 Å) was found.55 Once again, similar expansion of the GO lattice as a function of humidity is also found in multilayered GO laminates.23
A remarkable feature of GtO swelling in water vapours noted already in very early XRD studies back in the 1930s is the gradual change of d(001) (or more generally the c-unit cell parameter) as a function of humidity. The increase of humidity never results in step-like changes of interlayer distance related to the formation of water layers which would be easily detected by the diffraction methods. Insertion of water molecules between GO layers is expected to result in an increase of inter-layer distance related to the size of water molecules (∼2.5 Å). The gradual change of d(001), including the range of values below the size of the water molecule, is sometimes incorrectly interpreted as a true change of inter-layer distance in GO membrane “permeation channels”.124 However, it is obvious that water cannot be intercalated into GO by e.g. 1/3 of the molecule to provide a 1 Å increase for the size of the “channels”. The gradual changes in d(001) are typically explained by the effects of random interstratification. Interstratification with random stacking of differently hydrated layers is very commonly observed e.g. in hydrated clay minerals.147–149 The XRD method provides the lattice spacing value averaged over hundreds and thousands of layers. In the case of humidity dependent swelling the continuous shift in d(001) of GtO and GO laminates is typically interpreted as a change in the proportion between the numbers of differently hydrated layers.55,150,151 In the case of random stacking of differently hydrated layers only one diffraction peak is observed while the position of this reflection shifts depending on the proportion between the numbers of differently hydrated layers.
This concept was recently slightly modified to include intrastratification which is a result of the averaging of the inhomogeneous interlayer distance over the length of individual interlayers. The effect of intrastratification was introduced following the microscopic studies of humidity-dependent hydration performed using AFM on bilayered graphene oxide flakes.152
This method allows direct measurements of distance between individual graphene oxide layers and thus excludes the effects of averaging over a larger number of layers unavoidable in XRD analysis. Instead, AFM demonstrates the averaging of inter-layer distance over the length of individual interlayers when the scan is performed using relatively low resolution (Fig. 8).132
 | ||
| Fig. 8 Top panels. Left: the image of bilayered graphene oxide flakes and the height profile recorded across the smaller top flake. The height profile allows one to calculate the height difference which corresponds to the interlayer distance averaged over the whole length of scan (few hundred nm). Right top panel: the interlayer distance of bilayered GO as a function of relative humidity. Bottom panels: AFM height images of the same area on top of a double layer Hummers GO flake imaged under (a) 3% and (c) 65% RH.152 | ||
The interlayer distance in bilayered GO averaged over the lengths of a few hundred nm showed gradual changes upon variations of humidity, similarly to the change in the continuous shift of d(001) in XRD experiments. The height difference between two GO flakes gradually grows with the relative humidity (Fig. 8) from about 0.73 nm at 18% RH to more than 0.80 nm above 80% RH and increases further to 1.1 nm in liquid water. The gradual expansion of bilayer GO implies that the simple model of two flat plates separated by water monolayers very often used in the literature is oversimplified for the description of GO hydration. High resolution imaging demonstrates that hydration of the graphene oxide interlayer is inhomogeneous on the scale of a few nanometers. The topography of the GO flakes shows granular “hills and valleys” with a typical lateral size of spots in the range of 10 nm. An increase of humidity from 3% to 60% showed a number of new “hills” due to the local hydration of interlayers with height change mostly within 3–4 Å, thus corresponding roughly to the size of the water molecule.152 This suggests that hydration through water vapor is a continuous process of incorporation of water molecules into various randomly located sites within the GO interlayers. Variation of the interlayer distance over the length of the individual interlayers was proposed to be named intrastratification.152
Effects of random interstratification have been also typically used to explain the gradual changes also in the swelling state of GtO in liquid water. Expansion of the GtO lattice measured directly in liquid water has been reported in many studies over the past 100 years but mostly only at ambient temperature (Table 2). More recent studies revealed unusual anomalies in temperature and pressure dependence of water-swelled GtO.102,131
| Solvent | Brodie graphite oxide, d(001) | Hummers graphite oxide; d(001) |
|---|---|---|
| “Dry”-state | 5.5–5.9 Å,161,163–166 6.2–7.4 Å,93,97,99,111,130,162,167–182 7.8 Å183 |
6.4–6.9 Å,21,161,164 7.2–8.3 Å,58,97,127,177,184–190 8.6–9.0 Å191–194 |
| Water | ||
| Water | 7.7–11.3 Å(vapour),162,164–169 9.2–11.2 Å93,111,130,156,173,175,176,195 |
11.6–12.4 Å21,58,184,195 |
| Protic Polar solvents. Alcohols | ||
| Methanol | 9.0–10.1 Å161,174,176,183,195,196 |
13.0–14.0 Å58,127,161,181,182 |
| Ethanol | 9.2–10.0 Å161,162,174,182,183,196 |
15.0–15.5 Å21,127,161,181,182 |
| Propanol-1 | 9.2–10.0 Å,163,174,183,196 14.7 Å161 |
16.3–16.4 Å127,161 |
| Butanol-1 | 16.0 Å–17.0 Å,161,183,196 18.5 Å197 |
17.8–17.9 Å127,161 |
| Pentanol-1 | 17.0–17.2 Å,161,183,196 20.0 Å197 |
18.6–18.8 Å127,161 |
| Hexanol-1 | 19.2–20.0 Å,161,183,196 21.3 Å197 |
20.0–20.8 Å127,161 |
| Heptanol-1 | 19.0 Å196 |
22.7 Å127 |
| Octanol-1 | 8.5 Å,196 10.0 Å,183 23.1–25.0 Å99,161,175,197 |
23.8–24.8 Å127,198 |
| Nonanol-1 | 8.5 Å,196 26.7 Å197 |
26.7 Å127 |
| Dodecanol-1 | 39.2 Å197 |
|
| Tridecanol-1 | 42.9 Å197 |
|
| Tetradecanol-1 | 46.3 Å197 |
|
| Pentadecanol-1 | 46.6 Å197 |
|
| Hexadecanol-1 | 48.3 Å197 |
|
| Heptadecanol-1 | 49.3 Å197 |
|
| Octadecanol-1 | 50.4 Å197 |
|
| Ethandiol-1,2 | 9.0–9.6 Å93,161,175 |
16.6–16.8 Å21,161 |
| Propandiol-1, 3 | 9.6 Å161 |
16.6 Å161 |
| Butandiol-1,4 | 9.6 Å161 |
14.3 Å161 |
| Pentandiol-1,5 | 9.6 Å,161 14.1 Å161 |
16.8 Å161 |
| Aprotic polar solvents. Other solvents | ||
| Acetonitrile | 8.9–10.7 Å161,60,199 |
12.7 Å,161 14.1 Å177 |
| Benzonitrile | 9.2 Å161 |
9.6 Å161 |
| THF | 9.8 Å161 |
16.8 Å161 |
| Acetone | 8.9–9.2 Å93,161,175 |
9.3–9.8 Å,21,161 12.5 Å189 |
| DMSO | 9.3 Å161 |
17.7 Å,161 19.8 Å189 |
| DMF | 15.4 Å161 |
17.0 Å,161 17.7 Å189 |
| Dioxane | 9.9 Å161 |
14.2–15.5 Å21,161 |
| Pyridine | 10.8–11.8 Å93,161 |
13.7 Å161 |
| N,N-Diethylacetamide. | 16.1 Å161 |
17.1 Å161 |
| N-Methylformamide | 11.9 Å161 |
19.6 Å161 |
| N-Methylacetamide | 13.9 Å161 |
15.9 Å161 |
| N-Ethylacetamide | 13.6 Å161 |
17.3 Å161 |
| Non polar solvents. Other solvents | ||
| n-Hexane | No swelling,21 7.6–7.9 Å (“dry state” 6.3–7.0 Å)93,162 | |
| Toluene | No swelling,21 7.3–7.6 Å (6.3–7.0 Å)93,162 | |
| Nitrobenzene | 8.7–8.8 Å (5.5 Å)93,161 | 9.5 Å (6.4 Å)161 |
| Intercalation. Amines | ||
| n-Propylamine | 13.0 Å,196 16.1 Å161 |
20.3 Å161 |
| n-Butylamine | 8.3 Å–13.6 Å,170,178,200 15.0 Å,175 16.0–16.8 Å,93,196 18.1 Å,161 19.5 Å183 |
10.0 Å,190 11.8 Å,194 22.3 Å161 |
| n-Hexylamine | 21.0 Å196 |
11.8 Å193 |
| n-Octylamine | 25.0 Å,178 28.0 Å196 |
8.9 Å,179 11.7 Å,191 15.2–15.8 Å,190,192,193 28.1 Å113 |
| 1,2-Diaminoethane | 9.4 Å161 |
15.9 Å161 |
| 1,3-Diaminopropane | 9.8 Å,161 13.5 Å183 |
16.1 Å161 |
| 1,5-Diaminopentane | 13.8 Å161 |
15.0 Å161 |
| Aniline | 8.8 Å,97 17.0 Å,161 18.5 Å93 |
14.4 Å,187 18.8 Å161 |
| Cyclohexylamine | 16.5 Å161 |
19.6 Å161 |
| n-Hexadecylamine | 24.0–30.0 Å,180 44.0–49.0 Å180 |
28 Å,185 48 Å185 |
GtO immersed in liquid water shows slight expansion of the structure upon cooling due to insertion of additional water into interlayer space.153 Expansion of the GtO lattice at lower temperatures was later also observed in experiments with cooling of GO papers under fixed humidity conditions and named “pseudonegative thermal expansion”.154 An interesting effect is observed also when liquid water surrounding fully hydrated GtO gets frozen. About half of the water included in fully hydrated GtO escapes the structure when bulk water outside of GO flakes freezes (Fig. 9). The decrease of the d(001) value by ∼2.5 Å corresponds approximately to the size of the water molecule and interpreted as a loss of one water layer. However some water remains in the structure of GO even after solidification of bulk water as evidenced by the d(001) value of 8.7 Å (at 230 K), about 2.1 Å higher compared to the water free material (6.6 Å at ambient humidity).153 Therefore, temperature dependent XRD data provide evidence for two types of water intercalating GO structures at ambient temperature. The first layer of water is more strongly bound to GO sheets and has lower mobility; this water remains in the GO structure below the water freezing point. The second type of water is mobile and can be considered as liquid-like since it easily escapes from the GO structure below the freezing point and reversibly fills it again above the melting point of bulk water.153 Note also a somewhat smaller thickness of the first water layer more strongly attached to GO sheets as compared to the second liquid-like layer.
 | ||
| Fig. 9 Temperature dependence of the d(001) of Brodie graphite oxide immersed in liquid water.153 The inset shows the pressure dependence of d(001) of GO/H2O.111 Solidification of liquid water (at low temperature or high pressure) results in sharp downturn of d(001) due to partial withdrawal of water from the GO lattice. | ||
The maximum in the d(001) position observed in Fig. 9 at the point of liquid water solidification is a result of (a) the increase in hydration at lower temperature and (b) different compositions of the GO–water solvate structure in equilibrium with liquid water and solid water. Similar but a lot more pronounced maximum in the expansion of the GO lattice was found also in the pressure dependent studies of GO hydration,111 historically even earlier than in temperature dependent experiments. Brodie GO immersed in excess of water showed an increase in d(001) by ∼2 Å upon compression up to the point of water solidification (1.4 GPa), see the inset in Fig. 9. Volumetric negative compressibility is forbidden by thermodynamics. Therefore, the pressure driven expansion of the lattice can be explained only by the insertion of additional water between GO sheets. When liquid water solidifies above 1.4 GPa, part of water escapes from the GO structure. The difference in interlayer d spacing between water-immersed and water free samples was found to be about 2.5 Å at 3.5 GPa which corresponds to intercalation of one water layer which is more strongly bound to GO sheets. The maximal difference between d(001) of the hydrated state and the water free state is observed just before the water solidification point (13 Å–6.6 Å = 7.4 Å) which corresponds to insertion of three water layers. Note again that no true layered structure is observed for GtO in water and the inter-layer distance changes continuously as a function of pressure.
Fig. 9 shows that a very similar GO–H2O solvate is formed in both temperature and pressure dependent experiments in equilibrium with solid/frozen H2O. Interestingly, the maximum in interlayer d-spacing observed just below the temperature point of water solidification is found to be stronger in basic solutions and smaller in acidic solutions.155 Brodie GO pressurized in NaOH demonstrated an increase of d(001) up to 21.47 Å at 1.7 GPa compared to 6.6 Å in the dry state thus providing a structure mostly composed of water but anyway maintaining the ordered packing of GO layers. The pressure driven hydration of the GO structure was found also to be stronger in electrolyte solutions (copper acetate) and smaller in molecular solutions (sucrose).156
The pressure induced hydration first discovered in GtO111 later appeared to be a phenomenon common for materials exhibiting swelling in water. Stronger hydration at high pressure was reported for several hydrophilic layered materials with very different chemical natures, e.g. synthetic clays157 and MXenes.158 Moreover, pressure induced swelling was found also in natural clay minerals (kaolinite) providing important insight into the understanding of processes in the deep earth environment of subduction zones.159 As noted above the swelling was discovered first in rather exotic synthetic laboratory GtO materials and only later found in very common natural clay minerals.21 The history repeated itself for pressure induced hydration which was discovered first in exotic laboratory made GtO and a decade later in very common natural clay minerals.
A lot of studies were aimed in the past two decades at understanding the state of water in swelled GO structures. As noted above the water in the GtO structure is completely disordered providing no additional diffraction peaks in the XRD patterns even at temperatures below the freezing point of liquid water.153 There is extensive evidence that water adsorbed between GO sheets in the initial stages of sorption is more strongly bound while the fully hydrated structure includes also highly mobile and “liquid-like” water. The maximum isosteric heat of adsorption is less than 1.5 times that of the heat of condensation of water vapour, indicating that the process is close to physical adsorption. The early study of water sorption from vapour revealed a maximum in the heat of adsorption in the range of 0.2 to 0.4 fraction of a monolayer.92 The water molecules must first adsorb at the edges of GO flakes and then gradually separate the layers to the extent that water molecules could reach the more active sites between layers. The final lattice expansion corresponds to 2–3 monolayers of water under ambient conditions (Table 2). Note however that the term “monolayer” needs to be used with caution since the layers are not resolved by most of the experimental methods. Dynamics of water in hydrated GO was studied in detail using neutron scattering methods.55,144 These studies showed distinctly different types of water in GO (Brodie) vapour hydrated at 75% (9 Å interlayer distance) and 100% humidity (>10 Å interlayer distance). Samples equilibrated at 75% relative humidity exhibited two types of localized motions with different activation energies. Saturated hydration at 100% resulted in the detection of water with translational motion assigned to water molecules in pores between the GO particles.55,144 However, later it was suggested that some water with translational motion at 100% humidity or in liquid water is also present in between GO layers.61,153 Broadband Dielectric Spectroscopy (BDS), DSC and FTIR were used by Cerveny et al.145 to study the state of intercalated water in Brodie GO hydrated to 25% humidity. The increase of inter-layer spacing from 5.67 to 8 Å corresponded to the uptake of a water monolayer. A clear relaxation due to water molecule reorientation was found by BDS. The rotational water dynamics is dependent on the hydration level. At high water concentration (>15 wt%), water–water interactions were reported to dominate the dielectric response.145
Hydration was also studied using NMR and FTIR methods. For example the study by D. W. Lee and J. W. Seo suggested the formation of phenolic groups on the edges of GO sheets in the hydrated state.160 Moreover, a “dynamic” molecular structural model was proposed to distinguish between the properties of water free and hydrated graphene oxide.67
7. Swelling of graphite oxide in solvents other than water
Swelling of GO was studied in many solvents over the past 150 years. The best parameter which describes swelling is the value of inter-layer distance, most often evaluated using diffraction methods. Table 2 provides a summary for swelling of (most common) Brodie and Hummers GtO materials in a variety of common solvents at ambient temperature. These two types of graphite oxides exhibit also somewhat different inter-layer distances already in the “dry” state, Brodie GO showing typically the d(001) value ∼1–1.5 Å smaller compared to Hummers GO with a similar degree of oxidation. Note also that the “dry” state of graphite oxides is often reported for ambient humidity conditions. In some studies the samples were dried for a long time in a desiccator but for recording XRD patterns they were anyway exposed to ambient humidity and the recorded value might be affected by rapid sorption of water. It is more correct to report d(001) for GO recorded under dynamic vacuum conditions but these data are available only in very few studies. The difference between the vacuum dried state and structure saturated by solvent is possibly the best parameter to describe the swelling. However, the swelling state of graphite oxides in liquid solvents reported in Table 2 does not seem to depend on the air humidity (at least in many cases). The values of inter-layer spacing recorded in liquid solvents are well reproduced and easy to compare. Strictly speaking the swelling properties of GtO need to be compared only for materials with an identical oxidation degree. However, Table 2 includes data for all graphite oxides which are available in the literature, including materials with rather different degrees of oxidation.We note also that a significant part of Table 2 describes the data reported in Renate Krüger's PhD thesis from 1980161 which provides possibly the most comprehensive study of graphite oxide swelling in many solvents available by now. The data were never published in peer-reviewed journals, originally available only in German and so far known only to very few German-speaking researchers. The authors of this review were also not aware of this study until recently, otherwise it would be cited in several of our earlier publications. Only parts of data are cited in Table 2, the thesis includes also swelling of three kinds of Brodie GO. However, the difference between these three samples was not significant for most of the solvents and only one most representative Brodie GO sample was selected for Table 2.109
Analysis of Table 2 shows that swelling provides a rather broad range of interlayer distances with the highest values up to 50 Å. It also shows that swelling of Brodie and Hummers GO is significantly different for many solvents and generally stronger for Hummers GO. For some solvents the increase in inter-layer distance recorded in a liquid solvent correlates well with the size of the molecule, thus providing evidence for insertion of one solvent layer, e.g. for Brodie GtO in methanol, ethanol, acetone and acetonitrile.
On the other hand, the GtO lattice expansion in many solvents is significantly stronger compared to the solvent molecule size. In this case one needs to suggest either multilayer intercalation of solvent99 or stand-up orientation of long molecules attached to GO planes94 as will be discussed in more detail below.
Table 2 includes also solvents which are solid at ambient temperature (sugar alcohols) but in the molten state they are polar solvents. Technically the values of inter-layer distance listed in the table for sugar alcohols are not for GO immersed in liquid solvents but for the solid formed below the point of solidification.
There is a consensus that pristine (chemically not modified) GO does not swell in non-polar solvents, but few studies reported the increase of interlayer distance by values which are typically below 1 Å.93,162 A very small change in inter-layer distance should not be considered as evidence for swelling in our opinion. In some cases small shifts of (001) reflection can also be attributed to the technical details of XRD recording, e.g. due to a change in the sample height in the process of solvent evaporation.
Swelling of GO in amines is possibly a border case between swelling and functionalization. It is believed that amines are attached to graphene oxide sheets in stand-up orientation and the longer the molecules, the larger the separation of GO layers72Table 2 includes only liquid amines and in this case swelling is certainly a part of the intercalation and functionalization process.
Swelling of GO in alcohols is possibly one of the most important for practical applications. Many chemical reactions aimed at functionalization and intercalation are performed in alcohol solutions.201–203 Solvothermal reactions in alcohols under elevated conditions are also commonly used for the synthesis of various pillared GO materials204 and graphene oxide frameworks.122,205–208 Swelling is an important step in these reactions since it expands the GO lattice to allow reactive molecules and ions to access the planar surfaces of graphene oxide sheets. Most of the reactions are performed in ethanol and methanol as the most common solvents which provide an increase in the inter-layer distance by 3–8 Å depending on the type of graphite oxide. Table 2 shows that swelling of GO is progressively stronger for longer chain alcohols. Therefore, much larger molecules can possibly intercalate into the GO structure if reactions are performed in larger alcohols (up to 50 Å interlayer distance).95 This was never tested to our knowledge.
Progressively larger swelling of GtO in 1-alcohols was assigned in early studies to the stand-up orientation of larger alcohol molecules attached to graphene oxide planes.94,95,98 This was the logical assumption for the time. An alternative explanation with layered intercalation of solvent molecules parallel to the planes of GtO has emerged over the past decade. The first evidence for the multilayered structure of GO–alcohol solvates was found using pressure and temperature dependent studies of swelling. Following the discovery of the pressure induced swelling of GtO in water,111 pressure dependent studies of GtO in other polar solvents were performed. Surprisingly, instead of a gradual shift of d(001) observed for Brodie GO in water, an increase of pressure in several other GO/solvent systems resulted in a reversible step-like phase transition with an increase of inter-layer distance well correlated with the size of solvent molecules.
For Brodie GtO immersed in methanol and ethanol the increase in the interlayer distance due to the pressure induced phase transition is about 3.3 Å and 3.8 Å (respectively) in good agreement with the size of the solvent molecules (Fig. 10a). The change in the GtO structure at high pressure was interpreted as a transition from the ambient structure intercalated with one layer (1L) of methanol molecules to a high pressure two-layer (2L) intercalated structure.112 Very similar phase transitions were found later in GtO–methanol and GtO–ethanol systems at ambient pressure using a temperature dependent XRD study (Fig. 10b).209 The phase transition between 1L and 2L Brodie GO phases was also confirmed using DSC and sorption measurements.209 DSC shows a sharp exothermic anomaly at the point of the solvent insertion and an endothermic anomaly when the solvent layer is de-inserted in the heating part of the temperature cycle. The enthalpy of the transition 11.45 J g−1 was found for GO immersed in methanol.209
 | ||
| Fig. 10 Pressure (a) and temperature (b) induced phase transition from the ambient 1-layer Brodie GO–methanol structure to a 2-layer structure. (a) XRD patterns (λ = 0.7092 Å) recorded from Brodie GO immersed in excess of liquid methanol upon pressure increase.112 (b) XRD patterns recorded in the process of cooling (λ = 0.99874 Å).209 | ||
Experimental evidence for additional sorption of methanol at temperatures below the point of phase transition was provided using DSC and the isopiestic method.87,181 The composition of GtO–methanol was determined by DSC experiments as 0.55 ± 0.4 g (Meth.) g−1 (GtO) for the low temperature 2L phase and 0.28 ± 0.07 g g−1 for the ambient temperature 1L phase.181 The phase transition was observed in both Brodie GtO prepared by 1 and 2 step oxidations (C/O = 3.4 and C/O = 2.7), not affected significantly by the change in the oxidation degree of the material.59 In strong contrast to the Brodie GtO–H2O system,102,131 no change in the structure of Brodie GtO methanol and ethanol solvates was found around the point of solvent solidification.
Phase transitions between 1L and 2L solvates were reported also for Brodie GO in several other solvents: acetone,209,210 dimethylformamide (DMF), acetonitrile60,87,199 and propanol.163 The composition of several Brodie GtO solvate phases was determined using the measurements of solvent sorption at ambient temperature and near the freezing point of bulk solvent. The data confirmed the nature of phase transitions related to the insertion and removal of the solvent layer.64 There is also single observation for the absence of the phase transition in Brodie GtO in deuterium substituted propanol.211 The phase transition is observed also in a methanol–water binary solvent mixture but shifts from 284 K for 100% methanol to lower temperatures when water is added (247 K with 30% water) before it disappears for a water content over ∼35%.87 Moreover, selective sorption of methanol from methanol/water binary mixtures with the methanol fraction in the range of 20–100% was found in Brodie GO.181 This study provides a possible application for Brodie GO as a sorbent selectively removing one component from binary mixtures. Selective liquid sorption properties were also reported for the chemically modified form of GtO.100
Unlike Brodie GtO, no phase transitions related to the insertion or removal of solvent layers was so far found for Hummers GtO in any tested solvents. The difference between the swelling properties of Brodie GtO and Hummers GtO in methanol and ethanol is shown in Fig. 11 which demonstrates that these are clearly very different materials. Hummers GtO immersed in methanol and ethanol shows strong continuous “pseudonegative” thermal expansion which is explained by the insertion of an additional solvent into the GtO lattice but no step-like changes.182 Hummers GtO swelling in both alcohols is significantly stronger compared to Brodie GtO already at ambient temperature. The d(001) corresponding approximately to the insertion of two methanol or ethanol layers is found for Hummers GtO while for Brodie GtO the lattice intercalation is limited to one layer.58
 | ||
| Fig. 11 (a) Temperature dependence of the d(001) corresponding to the distance between graphene oxide layers for samples of Hummers GtO immersed in excess of methanol (blue) and ethanol (green) and Brodie graphite oxide in methanol (red). Open symbols correspond to data points recorded during sample heating back to room temperature. (b) Swelling of Hummers GtO in methanol compared to swelling of Hummers and Brodie GtO in water.182 | ||
Cooling the Hummers GtO immersed in liquid solvent results in a gradual increase of d(001) up to 19.4 and 20.6 Å at 140 K for methanol and ethanol respectively.181 The swelling of Brodie GtO and Hummers GtO in the alcohols can be described as crystalline and osmotic respectively. Crystalline swelling refers to layer by layer intercalation of solvent, while osmotic swelling is controlled by a relatively easy flow of solvent in and out of the structure regulated by osmotic effects. Osmotic swelling of Hummers GtO in ethanol and methanol182 is qualitatively similar to the swelling of both Brodie GtO and Hummers GtO in water, as shown in Fig. 3.62 It is characterized by a gradual increase of the c-lattice parameter upon cooling and an abrupt contraction of the interlayer distance at the solidification point of the liquid medium by an amount which corresponds to one solvent monolayer (Fig. 11b).
The crystalline and osmotic swelling are commonly found in clay minerals.157,212–215 Remarkably, crystalline swelling is found for Brodie GO in all so far studied solvents except for water, while for Hummers GO only osmotic swelling has so far been reported. Crystalline swelling provides an opportunity to evaluate the thickness of solvent layers confined between GO sheets. For example, the phase transition connected to the insertion and de-insertion of one solvent layer provides the thickness of one layer ∼3.3 Å for methanol, 3.8 Å for ethanol, 4.4 Å for DMF,99,209 3.5 Å for acetonitrile60 and 3.9 Å for acetone.210
The gradual shift of the d(001) peak of GO is typically assigned to random interstratification.55,150,151 As noted above (Fig. 8) swelling of GO is also inhomogeneous within a single interlayer with nanometer scale spots showing individually stronger and weaker hydration.152 The difference in the swelling properties of Brodie and Hummers GtO observed for materials with a similar degree of oxidation was assigned to strongly inhomogeneous oxidation of graphene oxide sheets produced by Hummers methods.59 In fact, Hummers GtO can be considered as a mixture (or solid solution) of many materials with different oxidation degrees on the scale of several nm, each of these showing different swelling and XRD patterns providing the lattice change averaged over all the varieties.
The temperature dependence of swelling exhibited by Hummers GO has interesting implications for several applications. For example, larger molecules can obviously be intercalated into the GO structure at lower temperatures due to stronger expansion of the lattice provided by swelling. The difference in the lattice expansion due to swelling was clearly demonstrated in the studies related to the preparation of pillared GO. Attempts to functionalize Brodie GO with tetrapod-shaped amines were not successful due to the size of molecules too large relative to the layer separation in methanol. However, for Hummers GO this worked well due to a larger inter-layer distance.122 Chemical functionalization of GO is often performed in alcohol solutions and at elevated temperatures. Higher temperatures improve the kinetics of reactions but also provide limitations to expansion of the GO lattice as demonstrated in studies on swelling.
Even stronger swelling is typical of GO in long chain molecules, most remarkably in long chain alcohols. Larger lattice expansion of GO immersed in progressively longer 1-alcohol solvents was initially related only to the length and orientation of chain-like molecules attached by one side to graphene oxide sheets. This idea was discussed in several early studies of GO swelling in alcohols.94,95,98,161 However, recent studies demonstrated a multilayered structure of Brodie GtO/1-octanol solvates with solvent molecule layers parallel to GO sheets.
Brodie GtO immersed in liquid 1-octanol was found to undergo a reversible phase transition with expansion of the inter-layer distance by 4.5 Å both upon compression (Fig. 12) and cooling.99 The phase transition can be explained only by insertion of one 1-octanol layer with orientation of molecules parallel to GO sheets. A similar transition observed at low temperatures was studied also by DSC99 providing distinct anomaly with ΔH = 25 ± 6 J g−1.
 | ||
| Fig. 12 (a) Phase transition observed in Brodie GO immersed in excess of 1-octanol at high pressures, the difference in d(001) corresponds to the thickness of one 1-octanol layer oriented parallel to GO sheets. (b) Evaporation of ethanol at ambient pressure results in layer by layer removal of 1-octanol starting from the ambient 4 layer structure to 3 layer, 2 layer and 1 layer structures. | ||
Swelling of Brodie GtO in 1-octanol under ambient conditions provides a c-parameter of ∼23.4 Å, increased by 16.7 Å compared to the material exposed to air at ambient humidity. Therefore, the structure of Brodie GtO in 1-octanol corresponds to 4 layers of solvent intercalated between GO sheets.
Direct evidence of the layered structure was provided by XRD experiments with evaporation of 1-octanol. This solvent has very low vapor pressure under ambient conditions, but using vacuum and elevated temperatures it is possible to observe layer-by-layer removal of 1-octanol (Fig. 12b). An XRD study revealed that a four layered Brodie GtO/1-octanol phase (23.4 Å) decomposed stepwise into phases with interlayer distances that correspond to one-layered (∼9.9 Å), two-layered (14.6 Å), and three-layered (19.4 Å) solvate structures. Quantitative evaluation of the octanol content in the ambient temperature Brodie GtO/1-octanol phase and low temperature phase provided values of 0.88 ± 0.05 g g−1 (0.12) and 1.10 ± 0.05 (0.15) g g−1 respectively. Assuming that the difference between these two phases correspond to the sorption/desorption of one 1-octanol layer it is 0.22 g g−1 per layer. Therefore, the ambient temperature Brodie GtO/1-octanol phase composition 0.88 g g−1 corresponds exactly to the intercalation of four layers of octanol.99
A summary of the interlayer distances reported so far for Brodie GtO in alcohols shows surprising complexity (Fig. 13). The ambient structure of Brodie GtO in small alcohols (methanol, ethanol and propanol) is one layered, three layered in butanol, pentanol and hexanol, and four layered in octanol and nonanol.99
 | ||
Fig. 13 Summary of data related to the interlayer distance of B-GO intercalated with alcohol molecules of different lengths.99 Star symbols show the data obtained in this study for B-GO in various alcohols (ref. 112 data used for methanol, ethanol, and propanol) at ambient temperature ( ) and low temperature (★). Data from ref. 93 ( ) and low temperature (★). Data from ref. 93 ( ) and ref. 95 ( ) and ref. 95 ( ) are for ambient temperature and (■) for 223 K, ref. 163 data for propanol (●) at 240 K. Reproduced from ref. 99 with permission from RSC. ) are for ambient temperature and (■) for 223 K, ref. 163 data for propanol (●) at 240 K. Reproduced from ref. 99 with permission from RSC. | ||
Phase transitions with addition of one more solvent layer are observed at low temperatures or high pressures for Brodie GO in several, but not all alcohols. It is unclear why, but no phase transitions have so far been found in e.g. pentanol and hexanol. It is very likely that multilayered intercalation will also be found for 1-alcohols with an even larger chain length. The largest interlayer distance reported for C = 18 is ∼50 Å. Assuming approximately 4.5 Å per layer this structure corresponds to intercalation of 9–10 alcohol layers. However, no detailed structural studies of GO in alcohols with C > 8 are available so far.
There is also certain lack of data for the structure of Hummers GO in alcohols. So far temperature dependent studies were performed only for Hummers GtO in methanol and ethanol, whereas the ambient structure was studied only up to alcohols with C = 9 (nonanol).69 The difference between the swelling of Brodie GtO and Hummers GtO under ambient conditions is very strong for small alcohols (e.g. methanol, ethanol, propanol) but the lattice expansion is very similar for larger alcohols (e.g. octanol, nonanol).
The data shown in Fig. 13 and 14 demonstrate that the interlayer distance of Brodie GtO and Hummers GtO can be precisely tuned using a selection of alcohol molecules and temperature/pressure conditions, which makes higher alcohols attractive solvents for the preparation of pillared structures. They also provide strong implications to the properties of multilayered GO membranes discussed in the next section.
 | ||
| Fig. 14 XRD patterns recorded from Hummers GO in liquid alcohols. Reproduced from ref. 69 with permission from RSC, temperature/pressure conditions, which makes higher alcohols attractive solvents for the preparation of pillared structures. It has also strong implications to the properties of multilayered GO membranes discussed in the next section. | ||
The swelling of graphite oxides can be strongly modified by chemical functionalization. This research field is not yet so strongly developed leaving a lot of space for future studies. Several examples of a strong change in swelling of chemically functionalized graphite oxides provided below can be useful also for the preparation of GO membranes with modified swelling and permeation properties. Already in the 1960s the swelling of two chemically modified GO was reported in much detail. Acetylation of GO using a reaction with acetic anhydride has been reported already in the 1950s.101,216 Acetylated GO (AcGO) was produced using a reaction with acetic anhydride which resulted in a material with the lattice expanded up to 10.9 Å, about 4 Å larger compared to pristine GO.101 Swelling of acetylated GO is significantly modified showing for example stronger expansion of the lattice in ketones (Table 3) However, the short report of AcGO synthesis published about 50 years ago did not provide information about the properties of precursor graphite oxide and acetylated material, e.g. no estimation of the materials’ surface area, chemical composition or spectroscopic characterization.101 The only modern study of GO acetylation performed was mostly focused on the chemical modification of AcGO.217
| Basal spacing in Å | ||
|---|---|---|
| Complexing substance | Acetylated graph. acid | Graphitic acid |
| None | 10.90 | 7.08 |
| Methanol | 11.78 | 8.04 |
| Ethanol | 12.35 | 8.63 |
| Propanol | 13.59 | 16.50 |
| 9.06 | ||
| Isopropanol | 12.18 | 14.72 |
| 8.84 | ||
| Butanol | 13.59 | 17.66 |
| 8.97 | ||
| Dimethyl ketone | 12.18 | 9.00 |
| Ethyl butyl ketone | 13.8 | 9.02 |
| Butyl propyl ketone | 14.24 | 8.7 |
| Acetic acid | 14.72 | 8.58 |
The second example of GtO chemical modification which preserves but strongly modifies swelling properties is methylation which was done by a reaction with diazomethane.98 Methylated GtO showed the inter-layer spacing expanded by ∼2.4 Å relative to precursor GO (8.9 Å in ref. 21). It was reported to swell not only in polar solvents (e.g. providing an increase up to 16.4 Å in ethanol and 18.3 Å in dioxane), but also in non-polar solvents like benzene (16.5 Å) and hexane (12.6 Å).21
However, the methylation reaction used in these studies is rather dangerous due to extreme toxicity of diazomethane and to our knowledge methylated GO has never been re-studied in modern times.
Swelling in non-polar solvents was reported also for graphite oxides after hydrophobization with n-alkylammonium cations.100 Alkylammonium chains are attached to the negative charges on graphene oxide sheets providing an organocomplex GO material with a strongly expanded lattice, e.g. 18.7 Å in the air dried state for GO–C14. The d-spacing of this complex increases in toluene up to 36.8 Å and in cyclohexane up to 41 Å.100
The hydrophobized graphite oxide was demonstrated also to selectively adsorb some solvents from binary liquid mixtures.100 Successful hydrophobization with n-alkylammonium was more recently demonstrated also for free standing GO membranes produced by vacuum filtration of graphene oxide dispersions. Swelling of these membranes in toluene resulted in an increase of c-lattice parameter up to 46–47 Å.113 However, the permeation properties of hydrophobized membranes in polar and non-polar solvents have never been studied so far to the best of our knowledge.
8. Swelling of multilayered GO materials: membranes, thin films, and papers
Graphite oxide is a multilayered material composed of graphene oxide sheets packed into a disordered structure. Graphene oxide dispersions are prepared by sonication of GtO in water or by spontaneous delamination in slightly basic solutions and deposited to form multilayered GO materials which were proposed for many applications.As cited above, the main properties of multilayered GO foils as membranes were reported back in the 1950s–60s, including the basic permeation properties, measurements of membrane potentials, and possible applications for filtration of small molecules and water desalination.21,104 The range of possible applications was significantly broadened during the last decade to include for example membranes for batteries218 and fuel cells,219–221 electrodes for supercapacitors199,222 and sorbents for removal of various contaminants.223 GO is used in these applications in the solution immersed swelled state to allow diffusion or sorption of ions.
Note that Hummers GtO is easy to disperse by sonication while it is not efficient for dispersion of Brodie GtO.224 GO dispersions can be re-assembled again into multilayered structures using a variety of methods, e.g. drop casting,23 vacuum filtration,9 spin coating125 or simple evaporation of dispersion on a suitable surface21 (Fig. 15). Multilayered GO materials are named papers,6,7,225 thin films119 or membranes8–10,23,86,226 while being essentially the same material. Note that deposition of GO flakes from dispersions occurs most commonly in water and multilayered materials are prepared under the conditions of saturated swelling. The arrangement of GO flakes in e.g. GO membranes is then strongly affected by deposition conditions and the drying procedure. The volume of the membrane shrinks by ∼30–40% in the process of water evaporation reflecting the change in the inert-layer distance from ∼12–13 Å in the hydrated state and 7–8 Å in the air dried state. Formation of lamellas composed of tens of GO layers and their significant buckling (Fig. 15) are likely connected to the shrinking of the foils in the process of drying.224
 | ||
| Fig. 15 Scheme representing the preparation of graphene oxide dispersions and using these dispersions for deposition of various multilayered GO structures. Images from ref. 113 and 234 were used with permission from RSC and AAAS. | ||
Multilayered materials are composed of the same graphene oxide sheets as precursor graphite oxides except for different packing of individual sheets. Therefore, these materials could be named simply disordered graphite oxide and the name “graphene oxide” is strictly speaking not correct. However, the name has become now very common and it does reflect some differences in the properties of graphite oxides and multilayered GO materials.
It is known that GO papers and membranes show many properties similar to graphite oxides, e.g. a very similar distance between graphene oxide sheets in the dried state and the ability to swell in polar solvents.6,9 However, recent studies revealed that this similarity is not complete. Some properties of GO membranes are easy to predict using the known properties of precursor GtO, for example the absence of swelling in non-polar solvents. However, there are also properties which are difficult to explain using only the knowledge accumulated for GtO. For example, back in the 1960 s H.P. Boehm assumed that GO membranes will be easily permeable to any polar solvents which are known to cause swelling of GtO, but he tested permeation only for water.21 More recent studies of GO membranes showed fast water permeation and decreased permeability to alcohols.10,125,133,227 It is not a trivial fact taking into account that precursor graphite oxides can be rather rapidly and reversibly intercalated by both water and ethanol with amounts corresponding to several monolayers.130,182
Therefore, we will review here the main swelling properties of GO multilayered materials and discuss the similarity/difference with the swelling properties of graphite oxides. This paper is not aimed at a detailed review of GO membrane applications and permeation properties which are described in thousands of papers at the moment and summarized in several review papers, see e.g.ref. 137, 228, 229 and 230.
Swelling of multilayered GO is related to the sorption of water or other solvents which results in an increase of the distance between individual sheets in the multilayered material. As a result it affects most of the properties of GO papers, films and membranes. For example, sorption of water under conditions of different air humidities strongly affects the mechanical properties of GO membranes and papers.7,59,231 Moreover, decreased sorption of water by Brodie GO membranes results in a mechanical strength significantly higher compared to Hummers GO membranes at any humidity.59 Sorption of water also affects the conductivity of GO very strongly, the property which is used in ultrafast humidity sensors.138,140 Humidity also affects very strongly the tribological properties of GO films.232
Possibly the most important effect of swelling is that it enables the permeation of solvents across the GO membranes. GO membranes under dry conditions are excellent gas barriers, much like graphitic foils, capable to hold vacuum and not permeable to gases.21,23,233 However, for most applications GO membranes are used under conditions of immersion in liquid solvents (most often water), e.g. for filtration86 and desalination applications104 or under exposure to water/solvent vapors for separation of binary mixtures by pervaporation.23,235 The humidity controlled size of permeation channels was also used to tune the gas separation properties of GO membranes.236
The stronger the swelling, the larger the size of “permeation channels” in the GO structure. The exact size of permeation channels is calculated differently in some studies due to different opinions related to the structure of GO.23,61,86,113 For example, the size of permeation channels in the swelled state of GO membranes was calculated assuming only not oxidized areas of graphene oxide sheets while the permeation was speculated to occur in the interconnected network of unoxidized interlayers named “graphene capillaries”.23,86,115 The size of permeation channels was calculated in these studies by taking the d(001) value of the GO membrane in the swelled state (e.g. ∼13 Å in water) and subtracting the thickness of the not oxidized graphene layer (3.4 Å) thus resulting in the size of about 9–10 Å.86,231 Other studies, including studies by the authors of this review, argued that describing hydrophilic GO membranes using hydrophobic “graphene capillaries” is not relevant. The absence of the interconnected network of graphene capillaries in GO membranes is directly confirmed from the absence of He permeation in water free membranes4 (size of “graphene channels” in water free membranes calculated as ∼3 Å allows the permeation of gases), the absence of a sizable BET surface area in dry membranes, direct electrostatic force microscopy data237 and HRTEM imaging,238 the electrical insulating nature of GO sheets, the invariance in permeation rates for laminates with different flake sizes239 and many other methods which point out that small nm sized graphene areas present on the GO surface are not interconnected until some reduction is applied.240 Therefore, our opinion is that the size of permeation channels in GO membranes needs to be considered assuming the averaged thickness of GO sheets (∼7 Å (ref. 241)). For the swelling state with the inter-layer distance of 13 Å as in the above cited example, the size of permeation channels must be then equal to ∼6 Å.61,113,242 The flow of water and other solvents needs to be considered using hydrophilic permeation channels provided by swelling of the GO structure.
Independently on discussion about the exact size of permeation channels there is no doubt that the permeation properties of GO membranes are strongly related to the inter-layer distance provided by swelling and mobility of solvents inside the interlayers.23,236,243–245 As cited above, one needs to be careful in interpreting diffraction studies which provide only the value of the inter-layer distance averaged over many layers and averaged over the whole (but strongly inhomogeneous on the nm level) GO sheets.152
The values reported for the averaged interlayer distances of GO multilayered structures in various solvents are summarized in Table 4. The data are related only to Hummers GO membranes. Brodie GO membranes were prepared but not yet used for studies of permeation.59,61
| Solvent | d(001) swelling in liquids | d(001) swelling in vapors | Thickness measurement | Computation approach |
|---|---|---|---|---|
| Dry-state | 6.4 Å,124 7.40 Å–7.6 Å,127,251,252 7.7–8.6 Å,253–268 9–10 Å269–274 |
6.0–6.5 Å,7,124,146 7.0–7.3 Å,275,276 7.9–8.0 Å126,277 |
7.9–8.0 Å131,278 |
7.0–8.0 Å279–282 10.0 Å262,283 |
| Protonic polar solvents. Water | ||||
| Water | No reflection,255,265 8.90 Å,254 10.8–11.1 Å,261,270 11.8–12.4 Å,189,262,263,266 12.7–13.1 Å,127,253,260,271–273 13.5–14.7 Å124,256–259,268 |
8.0 Å,7,276 10.0 Å,124 11.0–11.3 Å,146,277 12.0 Å,275 12.3 Å126 |
21–23 Å,254,264 62.0 Å131 |
6.5 Å,284 8.7 Å,280 10.0 Å,281 11.0 Å,282 13.5–14.0 Å279,281 |
| Protonic polar solvents. Alcohols | ||||
| Methanol | 9.1 Å,264 10.6 Å,189,266 12.0–12.5 Å,127,256 14.0 Å257 |
11.6 Å126 |
8.5 Å264 |
|
| Ethanol | 9.6–9.7 Å,127,264 11.1 Å,189,266 15.0–15.8 Å,127,246,256,267 16.5 Å,257 17.7 Å269 |
10.0 Å,126 11.5 Å277 |
10 Å264 |
7.0 Å284 |
| Propanol-1 | 16.7–17.0 Å127,257 |
|||
| Butanol-1 | 9.8 Å,256 17.9 Å,127 18.5 Å257 |
7.2 Å284 |
||
| Pentanol-1 | 18.8 Å,127 19.9 Å257 |
|||
| Hexanol-1 | 20.2 Å127 |
|||
| Heptanol-1 | 21.7 Å127 |
|||
| Octanol-1 | 10.1 Å,264 23.2 Å127 |
12 Å264 |
||
| Nonanol-1127 | 26.0 Å127 |
|||
| Aprotic polar solvents. Other solvents | ||||
| Acetone | 8.7 Å,189 9.8 Å,256 10.1 Å,264 12.9 Å267 |
10.4 Å126 |
11 Å264 |
7.2 Å284 |
| DMSO | 18.6 Å189 |
11.7 Å126 |
17 Å264 |
|
| Dioxane | 8.5 Å189 |
|||
| DMF | 13.8 Å,189 22.2 Å269 |
11.7 Å126 |
16 Å264 |
|
| NMP | 22.Å269 |
22.5 Å264 |
||
| Non polar solvents. Other solvents | ||||
| n-Hexane | 8.0–8.4 Å (”dry state” 7.7–8.0 Å)256,264,267 | 9.0 Å(7.8 Å)264 | 7.8 Å (no information about the dry state)284 | |
| Toluene | 8.2 Å (7.8 Å),264 8.8 Å(8.3 Å),267 10.1 Å (10.0 Å)269 | 8.0(7.8) Å264 |
7.3 Å (no information about the dry state)284 | |
As expected from the swelling properties of GtO (Table 2), GO membranes do not swell in non-polar solvents.246 As a result, non-polar solvents do not penetrate across GO membranes. Therefore, the membranes can be used for separation of polar solvents from non-polar solvents.246 GO membranes were also proposed as a material for preparation of protective fabrics which permeate water but serve as a barrier for toxic non-polar solvent contamination.247
Similar to GtO, GO films and membranes also very easily absorb water both from liquid and vapors. This property is used for preparation of humidity sensors which provide extremely rapid response138 and ultra-high sensitivity.33,248,249
Even after a rapid look at Table 4, it is obvious that huge variation of swelling is reported not only for re-stacked GO multilayered materials but even for solvent free “dry” GO. The permeation properties of GO membranes were also reported to be different by several orders of magnitude.250
As cited above, Hummers oxidation is used with many variations providing a large variety of materials. Therefore, we support recent calls for standardization of GO with respect to reporting certain characterization of these materials, most importantly the degree of oxidation, flake size, spectroscopy data and defect state.250 Some deviations in the numbers reported using diffraction methods can also be explained by purely technical reasons. The swelling of GO membranes in liquids is best to evaluate by XRD directly in the immersed state with sufficient for saturation excess of solvent. However, if the sample is not sealed, evaporation of solvent in the process of recording which takes usually at least a few minutes might result in more or less random underestimation of the inter-layer distance, especially for solvents with high vapor pressure.115 Some of the differences in the swelling properties of GO membranes can be related also to the details of GO layers packing in the multilayered structure (Fig. 16). Significant variations in the alignment of GO layers (buckling, wrinkles, overlaps) will be reflected in XRD patterns which average the interlayer distance over hundreds and thousands of layers. The difference in alignment of flakes can be for example due to the result of different deposition procedures. Several studies were specifically aimed at improving the alignment of GO flakes to provide a more uniform size of “permeation channels”, see e.g.ref. 133 and 135.
 | ||
| Fig. 16 Possible water-transport mechanisms through GO laminates. (A) Layer-by-layer water transport in which water flows strictly between the individual GO sheets. (B) Layer-by-layer transport, mediated by pinhole defects, in which the effective path length is significantly reduced by the pinhole defects. (C) Combination of layer-by-layer transport and transport mediated by voids and imperfect sheet stacking. Reproduced from ref. 239. | ||
The initial studies assumed the idealized structure of GO membranes composed of the flakes of identical size, a very small and regular distance between the flakes, parallel orientation of flakes and an extremely long zigzag pathway of water around the hole-free flakes (as in Fig. 16).23,86 However, real GO flakes used for preparation of membranes are different in size, irregular in the shape and not defect-free with cracks and holes present already after synthesis and greatly increased in numbers after sonication treatment typically used to prepare GO dispersions.285,286
More realistic structures with holes, point defects in GO flakes and somewhat irregular orientation of flakes were later considered as more relevant for describing real materials.280 Possibly the most adequate representation of the real GO membrane structure is a combination of Fig. 16B (with holes and cracks in individual flakes) and Fig. 16C with an irregular overlap of GO flakes.279,280 The study revealed that the permeation rate across the GO membrane is the same independent of the flake size pointing out to the holey structure shown in Fig. 16B.239
All three structures shown in Fig. 16 suggest the zigzag pathway of water molecules passing through interlayers expanded by swelling, but the length of these pathways is dramatically different.239 Therefore, calculation of the solvent flow rate in the permeation channels starting from the permeation rate across the GO membrane is very strongly affected (by 2–3 orders of magnitude) by selection of a structural model.242 Assuming realistic structural models with defects and holes in GO sheets allows one to explain the permeation rates observed in GO membranes without involving “ultra-fast” flow and exotic mechanisms discussed in some theoretical models.242 Sufficiently high water mobility of water can be achieved in hydrophilic permeation channels provided by GO sheets.287 A detailed review of the structure and permeation mechanisms for GO membranes is out of scope of this paper.
Swelling of GO membranes is clearly different from the swelling of graphite oxides. This can be illustrated by the example of GO membranes swelling in water–ethanol mixtures.224 The study reported that GO membranes are hydrated by water similarly to precursor graphite oxide powders but intercalation of alcohols is strongly hindered. Insertion of ethanol into the membrane structure at ambient temperature was found to be limited to only one monolayer (10.6 Å in Fig. 17) for Hummers (GO) in strong contrast to precursor graphite oxide powder which is intercalated with at least two ethanol monolayers (15.5 Å). The cooling experiments also demonstrated the absence of “negative thermal expansion” for Hummers GO and phase transitions connected to insertion/de-insertion of alcohols in Brodie GO membranes.224
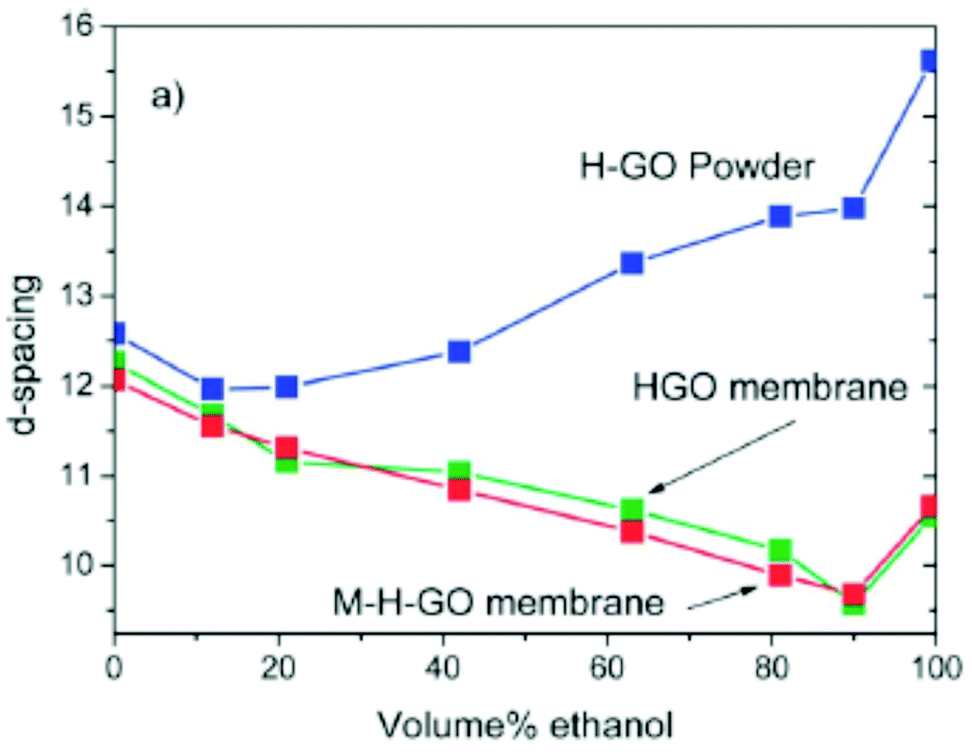 | ||
| Fig. 17 Interlayer distance, d(001), measured at 300 K for GO membranes in liquid water–ethanol binary solvents: (a) H-GO and M-H-GO membranes compared to H-GO. The M-H-GO membrane was produced in the same group which reported selective water pervaporation for water–ethanol binary mixtures.23 Reproduced from ref. 224 with permission of RSC. | ||
Quantitative estimation of vapor sorption from water–ethanol mixtures by Hummers GO films demonstrated an increase of ethanol concentration in the film relative to the composition of the source solvent125 as well as the change in the composition of several other binary mixtures as a result of sorption.119 The swelling of Hummers GO membranes in liquid solvents was also compared to the swelling of precursor Hummers GtO powder in several studies.64,113 Quantitative evaluation of water sorption using DSC and the isopiestic method revealed smaller water sorption by the GO membrane compared to precursor Hummers GtO.64 An XRD study of intercalation of several liquid solvents into the GO membrane structure, e.g. ethanol, methanol, dioxolane, acetonitrile, acetone, and chloroform showed maximum one monolayer insertion, in contrast to insertion of 2–3 layers of these solvents into the graphite oxide structure.113 However, the structure of GO membranes in some other solvents (DMSO and DMF) was found to expand similarly to precursor graphite oxide but with slower kinetics.
The membranes are found to be stable in aqueous solutions of acidic and neutral salts, but dissolve slowly in some basic solutions of certain concentrations.113 The studies concluded that GO membranes are a distinct type of material with unique solvation properties compared to parent GtO even if they are composed of the same GO flakes.113 The difference in the swelling properties of GO membranes and GtO was assigned to the regions of flake overlap which provide hindrance to the expansion of the GO lattice (Fig. 18).113
 | ||
| Fig. 18 (a) Typical shape of individual GO sheets imaged using AFM (the size of the panel is 6.4 μm). (b) Schematic geometry of overlapped graphite oxide flakes. (c) Schematic drawing of the GO membrane composed of irregularly shaped and overlapped sheets. The red circle illustrates the example of GO flake overlap with (d) the enlarged region of three overlapped GO flakes; (e) the same region under the conditions of water molecule insertion hinders larger separations of GO sheets. Reproduced from ref. 113 with permission from RSC. | ||
The swelling of GO membranes in solvents other than water shows an enormous variety in inter-layer distance. Table 4 shows the separation of GO layers in the range up to ∼50 Å depending on the nature of the solvent. The swelling depends also on the nature of dissolved molecules and ions, the concentration of solutes113 and the duration and conditions of the membrane storage.69 This variety of inter-layer distances is in conflict with the claims of universal ultra-precise ion sieving limited by the fixed diameter of permeation channels. The size of “permeation channels” was assumed in these studies to be the same as in pure water for any solvents, solutions and concentrations of solutes.23,86,115,124 For example the 1-octanol molecule was assumed as too large for permeation across GO membranes. However, Table 4 shows that 1-coctanol not only penetrates between GO sheets but also causes very strong swelling both in GtO and GO membranes (23.2 Å (ref. 127)).
Notably, some other study shows swelling in 1-octanol to be significantly smaller, 10.1 Å.264 A similarly strong difference in swelling is found in Table 4 for several other solvents. The difference in the swelling properties of GO membranes is puzzling and possibly related to several factors.
Recent studies demonstrated that a huge difference in swelling and permeation properties of at least some GO membrane materials can be explained by air-ageing over the period of several months and longer.69 Storing the membrane in air after preparation by vacuum filtration (that is in water swelled state) results in slow evaporation of water (3–4 weeks) and the increased stability of the membrane in water.224
The membranes are often dried using shelf storage for days, weeks and months but this storage time is typically not reported or taken into account. However, recent studies revealed dramatic changes in the swelling of membranes as a function of air storage time.69 Both precursor graphite oxides and freshly prepared GO membranes were found to swell equally well in the set of liquid alcohols (from methanol to 1-nonanol), Fig. 19. However, the swelling of GO membranes changed dramatically following air storage and in a rather not equal way for different solvents. Chemical modification of membranes was found to start on the surface of the multilayered structure and to penetrate slowly into the deeper regions according to the analysis of XPS data.69 The lattice expansion due to the swelling of Hummers GO in ethanol and methanol decreased significantly with time and disappeared for 1-hexanol and larger alcohol molecules after 0.6–2 years of air storage. The samples stored under ambient conditions for 5 years showed a nearly complete absence of swelling in alcohols but showed significant swelling in water. The study suggests that at least part of the difference reported in the swelling properties of GO membranes can be related simply to different times and conditions of air storage, including also our own earlier study which was performed using membranes stored for 6–8 months after preparation.224 The chemical modification of GO membranes during air storage can be considered as degradation, but also as a possibility to tune selectivity in sorption and permeation. The selectivity of GO membranes to swelling in alcohols improves due to the “degradation” (Fig. 17).
 | ||
Fig. 19 Interlayer distance (d(001)) for Hummers GtO powder (Δ) and membranes immersed in liquid alcohols: freshly prepared (1 week) sample (■), sample stored in air for 1.5 years, ( ) for 2.5 years ( ) for 2.5 years ( ), and sample stored in air for 5 years ( ), and sample stored in air for 5 years ( ). The inset shows the 2D XRD image (λ = 0.30996 Å) recorded of the Hummers GO membrane in 1-pentanol after 7.5 hours of immersion. ). The inset shows the 2D XRD image (λ = 0.30996 Å) recorded of the Hummers GO membrane in 1-pentanol after 7.5 hours of immersion. | ||
Similar observations were earlier presented for GO papers which were easily functionalized a few weeks after preparation but became less and less reactive after longer storage. One can assume that smaller swelling of GO papers does not allow penetration of molecules into the intra-lamellar structure.
A large variation of the swelling properties of GO membranes in various solvents and solutions is considered as one of the major obstacles in applications which require precise filtration parameters. Ideal nanofiltration membranes must provide exactly the same size of permeation channels independent of e.g. the concentration of solutions. This requirement is even more important for desalination applications which require size exclusion of small ions present in sea water. Therefore, a lot of research efforts were recently focused on chemical modification of GO membranes to control the inter-layer distance288–293 and to limit swelling to a degree which allows permeation but excludes small ions (e.g. Na+, K+etc.).294,295 For example, using divalent and multivalent cations was proposed as a method to bind together GO sheets.296 Intercalation of Mg2+ and Ca2+ was reported to increase the mechanical strength of GO papers.297 The control of interlayer distance in aqueous solutions was suggested using intercalation of certain cations into the GO structure.298 Another approach tested in several studies is partial reduction of GO membranes.299 Crosslinking of GO membranes using various chemical intercalation modifications was tested in several studies.300–302 Moreover, some attempts to control the swelling and permeation of GO membranes using the electrical current were presented, but the reported effect is most likely related to the cracks in the unintentionally created water impermeable rGO layer which becomes narrower or broader when the current is switched on and off.303
Another problem related to the swelling properties of GO membranes is instability in water. Freshly prepared GO multilayers are known to suffer from spontaneous delamination.304–306 When GtO synthesis methods are tuned to provide easy dispersion in water, it is not surprising that re-assembled laminates/GO membranes also easily dissolve in water or swell into a jelly-like state.307 Therefore various chemical modification methods were proposed to stabilize the membranes or GO based composite sorbents.308–314 Another method tested already back in the 1960 s is encapsulation of GO membranes into an envelope made of other materials (clays in ref. 104). Enveloping of GO membranes using CNTs was also recently proposed.315 We note also that Al3+ ions suggested as a reason for water stability of GO membranes prepared on alumina filters304 are likely to originate from small Al2O3 particles which are commonly found on the surface of membranes peeled off from the alumina filters.224
9. Conclusions
Swelling is one of the key properties of graphite oxides studied extensively over the past 150 years but still providing many surprise discoveries even in modern times. A broad variety of methods used to study GO swelling and multiple factors, which affect the swelling of GO are summarized in Fig. 20. | ||
| Fig. 20 Scheme representing the most common methods to study swelling of GO materials and a broad range of parameters which affect swelling. | ||
Fundamental studies of graphite oxide swelling in many solvents and under variation of pressure and temperature conditions provide the background for understanding multilayered graphene oxide structures. In fact, even the preparation of graphene oxide dispersions can be considered as a swelling with infinite inter-layer distance. Deposition of GO dispersions into multilayered structures occurs under conditions of saturated swelling and defines the structure of these materials after solvent evaporation. One of the most surprising findings during the last decade is a significant difference between the swelling of graphite oxides and GO multilayered foils, which is not yet well understood despite very strong research efforts. Thanks to the hydrophilic nature of GO multilayered structures are capable to absorb significant amounts of polar solvents from vapour and in liquid into the expanded inter-layers, thus making the whole surface area of individual sheets accessible for sorption of ions and molecules. The high sorption capacity of GO materials provides important applications as sorbents for waste water treatments and removal of various pollutants. Expansion of the GO lattice due to swelling enables membrane applications but also provides challenges to control the size of permeation channels which depends not only on the nature of the solvent, but also on many other parameters, like pH, nature and concentration of solutes, temperature and pressure conditions. Many applications of GO multilayers also involve the swelling and sorption of polar solvents, e.g. humidity sensors, protective semi-permeable coatings, gas and liquid mixture separation, nanofiltration, fuel cells and battery membranes and water desalination. Moreover, chemical functionalization of GO materials most often performed in solutions thus involves swelling, for preparation of various framework and pillared structures. Swelling is an efficient way to separate layers from each other providing access to functional groups located on the GO surface for chemical modification.
It can be concluded that the fundamental understanding of swelling phenomena in multilayered GO materials is strongly interconnected with many applications and requires further studies which are likely to present us with many surprises. This review is likely to be far from complete reflecting only the most important connections between the swelling of GO and applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the funding from the European Union's Horizon 2020 research and innovation program under grant agreement No. 785219 and No. 881603. The support from the Swedish Research Council grant (no. 2017-04173) is also acknowledged.Notes and references
- Y. W. Zhu, M. D. Stoller, W. W. Cai, A. Velamakanni, R. D. Piner, D. Chen and R. S. Ruoff, ACS Nano, 2010, 4, 1227–1233 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- N. A. Kotov, I. Dekany and J. H. Fendler, Adv. Mater., 1996, 8, 637–641 CrossRef CAS.
- N. Kovtyukhova, E. Buzaneva and A. Senkevich, Eur. Mat. Res., 1998, 68, 549–554 Search PubMed.
- N. I. Kovtyukhova, P. J. Ollivier, B. R. Martin, T. E. Mallouk, S. A. Chizhik, E. V. Buzaneva and A. D. Gorchinskiy, Chem. Mater., 1999, 11, 771–778 CrossRef CAS.
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. B. Dommett, G. Evmenenko, S. T. Nguyen and R. S. Ruoff, Nature, 2007, 448, 457–460 CrossRef CAS.
- N. V. Medhekar, A. Ramasubramaniam, R. S. Ruoff and V. B. Shenoy, ACS Nano, 2010, 4, 2300–2306 CrossRef CAS.
- Z. T. Luo, Y. Lu, L. A. Somers and A. T. C. Johnson, J. Am. Chem. Soc., 2009, 131, 898–899 CrossRef CAS.
- C. M. Chen, Q. H. Yang, Y. G. Yang, W. Lv, Y. F. Wen, P. X. Hou, M. Z. Wang and H. M. Cheng, Adv. Mater., 2009, 21, 3007–3011 CrossRef CAS.
- M. Krueger, S. Berg, D. Stone, E. Strelcov, D. A. Dikin, J. Kim, L. J. Cote, J. X. Huang and A. Kolmakov, ACS Nano, 2011, 5, 10047–10054 CrossRef CAS.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS.
- L. L. Xu, S. D. Jiang, B. P. Li, W. P. Hou, G. X. Li, M. A. Memon, Y. Huang and J. X. Geng, Chem. Mater., 2015, 27, 4358–4367 CrossRef CAS.
- J. H. Sun, D. L. Meng, S. D. Jiang, G. F. Wu, S. K. Yan, J. X. Geng and Y. Huang, J. Mater. Chem., 2012, 22, 18879–18886 RSC.
- J. H. Sun, L. H. Xiao, D. L. Meng, J. X. Geng and Y. Huang, Chem. Commun., 2013, 49, 5538–5540 RSC.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- M. Pumera, Electrochem. Commun., 2013, 36, 14–18 CrossRef CAS.
- X. L. Liu, R. Ma, X. X. Wang, Y. Ma, Y. P. Yang, L. Zhuang, S. Zhang, R. Jehan, J. R. Chen and X. K. Wang, Environ. Pollut., 2019, 252, 62–73 CrossRef CAS.
- J. X. Ma, D. Ping and X. F. Dong, Membranes, 2017, 7, 52 CrossRef.
- A. Y. Romanchuk, A. S. Slesarev, S. N. Kalmykov, D. V. Kosynkin and J. M. Tour, Phys. Chem. Chem. Phys., 2013, 15, 2321–2327 RSC.
- A. S. Kuzenkova, A. Y. Romanchuk, A. L. Trigub, K. I. Maslakov, A. V. Egorov, L. Amidani, C. Kittrell, K. O. Kvashnina, J. M. Tour, A. V. Talyzin and S. N. Kalmykov, Carbon, 2020, 158, 291–302 CrossRef CAS.
- H. P. Boehm, A. Clauss and U. Hofmann, J. Chim. Phys. Phys.-Chim. Biol., 1961, 58, 141–147 CrossRef CAS.
- N. Zhang, W. X. Qi, L. L. Huang, E. Jiang, J. J. Bao, X. P. Zhang, B. G. An and G. H. He, Chin. J. Chem. Eng., 2019, 27, 1348–1360 CrossRef CAS.
- R. R. Nair, H. A. Wu, P. N. Jayaram, I. V. Grigorieva and A. K. Geim, Science, 2012, 335, 442–444 CrossRef CAS.
- R. C. Croft, Q. Rev., Chem. Soc., 1960, 14, 1–45 RSC.
- S. K. Srivastava and J. Pionteck, J. Nanosci. Nanotechnol., 2015, 15, 1984–2000 CrossRef CAS.
- L. G. Guex, B. Sacchi, K. F. Peuvot, R. L. Andersson, A. M. Pourrahimi, V. Strom, S. Farris and R. T. Olsson, Nanoscale, 2017, 9, 9562–9571 RSC.
- S. Thakur and N. Karak, Carbon, 2015, 94, 224–242 CrossRef CAS.
- S. F. Kiew, L. V. Kiew, H. B. Lee, T. Imae and L. Y. Chung, J. Controlled Release, 2016, 226, 217–228 CrossRef CAS.
- B. C. Thompson, E. Murray and G. G. Wallace, Adv. Mater., 2015, 27, 7563–7582 CrossRef CAS.
- R. J. Young, I. A. Kinloch, L. Gong and K. S. Novoselov, Compos. Sci. Technol., 2012, 72, 1459–1476 CrossRef CAS.
- V. Chabot, D. Higgins, A. P. Yu, X. C. Xiao, Z. W. Chen and J. J. Zhang, Energy Environ. Sci., 2014, 7, 1564–1596 RSC.
- F. Li, X. Jiang, J. J. Zhao and S. B. Zhang, Nano Energy, 2015, 16, 488–515 CrossRef CAS.
- K. Toda, R. Furue and S. Hayami, Anal. Chim. Acta, 2015, 878, 43–53 CrossRef CAS.
- S. H. Aboutalebi, Y. H. Cho, A. M. Dimiev, S. Eigler, M. M. Gudarzi, C. M. Hayner, L. Kovbasyuk, A. Lerf, S. E. Lowe, A. Mokhir, A. V. Naumov, I. V. Pavlidis, S. Porro, I. Roppolo, F. Sharif, C. Vallés, H. W. Yoon and Y. L. Zhong, in Graphene oxide. Fundamentals and applications, ed. A. M. Dimiev and S. Eigler, Wiley, 2016 Search PubMed.
- H. P. Boehm, A. Clauss, G. Fischer and C. Hofmann, Proc. 5th Conf. on Carbon, Oxford, Pergamon, 1962, pp. 73–80.
- H. P. Boehm, Angew. Chem., Int. Ed., 2010, 49, 9332–9335 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Firsov, Nature, 2005, 438, 197–200 CrossRef CAS.
- B. C. Brodie, Philos. Trans. R. Soc. London, 1859, 149, 249–259 CrossRef.
- T. Szabo, O. Berkesi, P. Forgo, K. Josepovits, Y. Sanakis, D. Petridis and I. Dekany, Chem. Mater., 2006, 18, 2740–2749 CrossRef CAS.
- L. Staudenmaier, Ber. Dtsch. Chem. Ges., 1898, 31, 1481–1487 CrossRef CAS.
- U. Hofmann and W. Rudorff, Trans. Faraday Soc., 1938, 34, 1017–1021 RSC.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339–1339 CrossRef CAS.
- F. W. Low, C. W. Lai and S. B. Abd Hamid, J. Nanosci. Nanotechnol., 2015, 15, 6769–6773 CrossRef CAS.
- J. Chen, B. W. Yao, C. Li and G. Q. Shi, Carbon, 2013, 64, 225–229 CrossRef CAS.
- H. T. Yu, B. W. Zhang, C. K. Bulin, R. H. Li and R. G. Xing, Sci. Rep., 2016, 6, 36143 CrossRef CAS.
- C. E. Halbig, O. Martin, F. Hauke, S. Eigler and A. Hirsch, Chem. – Eur. J., 2018, 24, 13348–13354 CrossRef CAS.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS.
- Y. J. Kim, Y. H. Kahng, Y. H. Hwang, S. M. Lee, S. Y. Lee, H. R. Lee, S. H. Lee, S. H. Nam, W. B. Kim and K. Lee, Mater. Res. Express, 2017, 4, 105033 CrossRef.
- N. I. Zaaba, K. L. Foo, U. Hashim, S. J. Tan, W.-W. Liu and C. H. Voon, Procedia Eng., 2017, 184, 469–477 CrossRef CAS.
- A. Y. S. Eng, C. K. Chua and M. Pumera, Nanoscale, 2015, 7, 20256–20266 RSC.
- W. Gao, L. B. Alemany, L. J. Ci and P. M. Ajayan, Nat. Chem., 2009, 1, 403–408 CrossRef CAS.
- W. W. Cai, R. D. Piner, F. J. Stadermann, S. Park, M. A. Shaibat, Y. Ishii, D. X. Yang, A. Velamakanni, S. J. An, M. Stoller, J. H. An, D. M. Chen and R. S. Ruoff, Science, 2008, 321, 1815–1817 CrossRef CAS.
- L. B. Casablanca, M. A. Shaibat, W. W. W. Cai, S. Park, R. Piner, R. S. Ruoff and Y. Ishii, J. Am. Chem. Soc., 2010, 132, 5672–5676 CrossRef.
- A. Lerf, H. Y. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- A. Lerf, A. Buchsteiner, J. Pieper, S. Schottl, I. Dekany, T. Szabo and H. P. Boehm, J. Phys. Chem. Solids, 2006, 67, 1106–1110 CrossRef CAS.
- D. W. Lee, L. De Los Santos, J. W. Seo, L. L. Felix, A. Bustamante, J. M. Cole and C. H. W. Barnes, J. Phys. Chem. B, 2010, 114, 5723–5728 CrossRef CAS.
- H. P. Boehm and W. Scholz, Z. Anorg. Allg. Chem., 1965, 335, 74–79 CrossRef CAS.
- S. You, S. M. Luzan, T. Szabo and A. V. Talyzin, Carbon, 2013, 52, 171–180 CrossRef CAS.
- A. V. Talyzin, G. Mercier, A. Klechikov, M. Hedenstrom, D. Johnels, D. Wei, D. Cotton, A. Opitz and E. Moons, Carbon, 2017, 115, 430–440 CrossRef CAS.
- A. V. Talyzin, A. Klechikov, M. Korobov, A. T. Rebrikova, N. V. Avramenko, M. F. Gholami, N. Severin and J. P. Rabe, Nanoscale, 2015, 7, 12625–12630 RSC.
- A. V. Talyzin, T. Hausmaninger, S. J. You and T. Szabo, Nanoscale, 2014, 6, 272–281 RSC.
- S. J. You, S. Luzan, J. C. Yu, B. Sundqvist and A. V. Talyzin, J. Phys. Chem. Lett., 2012, 3, 812–817 CrossRef CAS.
- P. Feicht, R. Siegel, H. Thurn, J. W. Neubauer, M. Seuss, T. Szabo, A. V. Talyzin, C. E. Halbig, S. Eigler, D. A. Kunz, A. Fery, G. Papastavrou, J. Senker and J. Breu, Carbon, 2017, 114, 700–705 CrossRef CAS.
- M. V. Korobov, A. V. Talyzin, A. T. Rebrikova, E. A. Shilayeva, N. V. Avramenko, A. N. Gagarin and N. B. Ferapontov, Carbon, 2016, 102, 297–303 CrossRef CAS.
- K. Krishnamoorthy, M. Veerapandian, K. Yun and S. J. Kim, Carbon, 2013, 53, 38–49 CrossRef CAS.
- C. Botas, P. Alvarez, C. Blanco, R. Santamaria, M. Granda, P. Ares, F. Rodriguez-Reinoso and R. Menendez, Carbon, 2012, 50, 275–282 CrossRef CAS.
- A. M. Dimiev, L. B. Alemany and J. M. Tour, ACS Nano, 2013, 7, 576–588 CrossRef CAS.
- S. Kim, S. Zhou, Y. K. Hu, M. Acik, Y. J. Chabal, C. Berger, W. de Heer, A. Bongiorno and E. Riedo, Nat. Mater., 2012, 11, 544–549 CrossRef CAS.
- A. Iakunkov, J. H. Sun, A. Rebrikova, M. Korobov, A. Klechikov, A. Vorobiev, N. Boulanger and A. V. Talyzin, J. Mater. Chem. A, 2019, 7, 11331–11337 RSC.
- H. Y. He, T. Riedl, A. Lerf and J. Klinowski, J. Phys. Chem., 1996, 100, 19954–19958 CrossRef CAS.
- H. Y. He, J. Klinowski, M. Forster and A. Lerf, Chem. Phys. Lett., 1998, 287, 53–56 CrossRef CAS.
- A. B. Bourlinos, D. Gournis, D. Petridis, T. Szabo, A. Szeri and I. Dekany, Langmuir, 2003, 19, 6050–6055 CrossRef CAS.
- G. E. Bacon, Acta Crystallogr., 1951, 4, 558–561 CrossRef CAS.
- A. Weiss and A. Weiss, Angew. Chem., 1960, 72, 413–415 CrossRef CAS.
- M. W. Laipan, L. C. Xiang, J. F. Yu, B. R. Martin, R. L. Zhu, J. X. Zhu, H. P. He, A. Clearfield and L. Y. Sun, Prog. Mater. Sci., 2020, 109, 100631 CrossRef CAS.
- M. Mullervonmoos, G. Kahr and F. T. Madsen, Clay Miner., 1994, 29, 205–213 CrossRef.
- G. W. Brindley, K. Wiewiora and A. Wiewiora, Am. Mineral., 1969, 54, 1635–1644 CAS.
- U. Hofmann, A. Weiss, G. Koch, A. Mehler and A. Scholz, Clays Clay Miner., 1955, 4, 273–287 CrossRef.
- U. Hofmann and A. Frenzel, Ber. Dtsch. Chem. Ges., 1930, 63, 14 Search PubMed.
- U. E. J. Hofmann, Angew. Chem., 1939, 52, 708–709 CrossRef.
- U. Hofmann, A. Frenzel and E. Csalán, Justus Liebigs Ann. Chem., 1934, 510, 1–41 CrossRef CAS.
- U. F. Hofmann and A. Frenzel, Kolloid-Z., 1932, 58, 8–14 CrossRef CAS.
- J. C. Derksen and J. R. Katz, Recl. Trav. Chim. Pays-Bas, 1934, 53, 652–669 CrossRef CAS.
- H. Thiele, Kolloid-Z., 1948, 111, 15–19 CrossRef CAS.
- M. M. Gudarzi, M. H. M. Moghadam and F. Sharif, Carbon, 2013, 64, 403–415 CrossRef CAS.
- R. K. Joshi, P. Carbone, F. C. Wang, V. G. Kravets, Y. Su, I. V. Grigorieva, H. A. Wu, A. K. Geim and R. R. Nair, Science, 2014, 343, 752–754 CrossRef CAS.
- S. J. You, J. C. Yu, B. Sundqvist and A. V. Talyzin, Phys. Status Solidi B, 2012, 249, 2568–2571 CrossRef CAS.
- R. J. Beckett and R. C. Croft, J. Phys. Chem., 1952, 56, 929–941 CrossRef CAS.
- D. Hadzi and A. Novak, Trans. Faraday Soc., 1955, 51, 1614–1620 RSC.
- J. H. De Boer and A. B. C. Van Doorn, Proc. K. Ned. Akad. Wet., Ser. B: Phys. Sci., 1958, 61, 10 Search PubMed.
- W. H. Slabaugh and B. C. Seiler, J. Phys. Chem., 1962, 66, 396–401 CrossRef CAS.
- W. H. Slabaugh and C. V. Hatch, J. Chem. Eng. Data, 1960, 5, 453–455 CrossRef CAS.
- J. C. Ruiz and D. M. C. Macewan, Nature, 1955, 176, 1222–1223 CrossRef CAS.
- F. Aragon, J. C. Ruiz and D. M. C. Macewan, Nature, 1959, 183, 740–741 CrossRef CAS.
- A. R. Garcia, J. Canoruiz and D. M. C. Macewan, Nature, 1964, 203, 1063–1064 CrossRef CAS.
- D. M. C. Macewan and F. A. Delacruz, Nature, 1959, 184, 1859–1859 CrossRef.
- K. Spyrou, M. Calvaresi, E. K. Diamanti, T. Tsoufis, D. Gournis, P. Rudolf and F. Zerbetto, Adv. Funct. Mater., 2015, 25, 263–269 CrossRef CAS.
- F. A. Delacruz and D. M. C. Macewan, Kolloid Z. Z. Polym., 1965, 203, 36–42 CrossRef CAS.
- A. Klechikov, J. H. Sun, I. A. Baburin, G. Seifert, A. T. Rebrikova, N. V. Avramenko, M. V. Korobov and A. V. Talyzin, Nanoscale, 2017, 9, 6929–6936 RSC.
- I. Dekany, R. Kruger-Grasser and A. Weiss, Colloid Polym. Sci., 1998, 276, 570–576 CrossRef CAS.
- F. A. Delacruz and H. Castro, Naturwissenschaften, 1966, 53, 155–155 Search PubMed.
- H. P. Boehm, A. Clauss, G. O. Fischer and U. Hofmann, Z. Anorg. Allg. Chem., 1962, 316, 119–127 CrossRef CAS.
- A. K. Geim, Rev. Mod. Phys., 2011, 83, 851–862 CrossRef CAS.
- E. S. Bober, L. C. Flowers, P. K. Lee, D. E. Sestrich, C.-M. Wong, W. Sherman Gillam, S. Johnson and R. H. Horowitz, Research and development progress report No. 544, US Department of the Interior, reprints from the colelction of the University of Michigan Library, 1970, pp. 1–113.
- V. Kohlschütter and P. Haenni, Z. Anorg. Allg. Chem., 1919, 105, 121–144 CrossRef.
- F. Barbules, V. Petrescu and J. Paun, Rev. Roum. Chim., 1967, 12, 727–730 Search PubMed.
- A. Sagle and B. Freeman, The Future of Desalination in Texas, 2004, vol. 2, p. 137 Search PubMed.
- S. Loeb and S. Sourirajan, in Saline Water Conversion—II, American Chemical Society, 1963, vol. 38, ch. 9, pp. 117–132 Search PubMed.
- R. Kruger, PhD thesis, Ludwig-Maximilians-Universität München, 1980.
- H. W. Kim, H. W. Yoon, S. M. Yoon, B. M. Yoo, B. K. Ahn, Y. H. Cho, H. J. Shin, H. Yang, U. Paik, S. Kwon, J. Y. Choi and H. B. Park, Science, 2013, 342, 91–95 CrossRef CAS.
- A. V. Talyzin, V. L. Solozhenko, O. O. Kurakevych, T. Szabo, I. Dekany, A. Kurnosov and V. Dmitriev, Angew. Chem., Int. Ed., 2008, 47, 8268–8271 CrossRef CAS.
- A. V. Talyzin, B. Sundqvist, T. Szabo, I. Dekany and V. Dmitriev, J. Am. Chem. Soc., 2009, 131, 18445–18449 CrossRef CAS.
- A. Klechikov, J. C. Yu, D. Thomas, T. Sharifi and A. V. Talyzin, Nanoscale, 2015, 7, 15374–15384 RSC.
- P. Feicht, J. Biskupek, T. E. Gorelik, J. Renner, C. E. Halbig, M. Maranska, F. Puchtler, U. Kaiser and S. Eigler, Chem. – Eur. J., 2019, 25, 8955–8959 CrossRef CAS.
- Q. Yang, Y. Su, C. Chi, C. T. Cherian, K. Huang, V. G. Kravets, F. C. Wang, J. C. Zhang, A. Pratt, A. N. Grigorenko, F. Guinea, A. K. Geim and R. R. Nair, Nat. Mater., 2017, 16, 1198–1202 CrossRef CAS.
- J. A. Johnson, C. J. Benmore, S. Stankovich and R. S. Ruoff, Carbon, 2009, 47, 2239–2243 CrossRef CAS.
- N. S. Nazer, V. A. Yartys, T. Azib, M. Latroche, F. Cuevas, S. Forseth, P. J. S. Vie, R. V. Denys, M. H. Sorby, B. C. Hauback, L. Arnberg and P. F. Henry, J. Power Sources, 2016, 326, 93–103 CrossRef CAS.
- J. C. Schaeperkoetter, M. J. Connolly, Z. N. Buck, H. Taub, H. Kaiser and C. Wexler, ACS Omega, 2019, 4, 18668–18676 CrossRef CAS.
- A. Klechikov, J. H. Sun, A. Vorobiev and A. V. Talyzin, J. Phys. Chem. C, 2018, 122, 13106–13116 CrossRef CAS.
- Y. Yao, X. D. Chen, H. H. Guo and Z. Q. Wu, Appl. Surf. Sci., 2011, 257, 7778–7782 CrossRef CAS.
- A. Iakunkov, V. Skrypnychuk, A. Nordenstrom, E. A. Shilayeva, M. Korobov, M. Prodana, M. Enachescu, S. H. Larsson and A. V. Talyzin, Phys. Chem. Chem. Phys., 2019, 21, 17901–17912 RSC.
- J. H. Sun, F. Morales-Lara, A. Klechikov, A. V. Talyzin, I. A. Baburin, G. Seifert, F. Cardano, M. Baldrighi, M. Frasconi and S. Giordani, Carbon, 2017, 120, 145–156 CrossRef CAS.
- F. Barroso-Bujans, S. Cerveny, A. Alegria and J. Colmenero, Carbon, 2010, 48, 3277–3286 CAS.
- J. Abraham, K. S. Vasu, C. D. Williams, K. Gopinadhan, Y. Su, C. T. Cherian, J. Dix, E. Prestat, S. J. Haigh, I. V. Grigorieva, P. Carbone, A. K. Geim and R. R. Nair, Nat. Nanotechnol., 2017, 12, 546–550 CAS.
- A. Vorobiev, A. Dennison, D. Chernyshov, V. Skrypnychuk, D. Barbero and A. V. Talyzin, Nanoscale, 2014, 6, 12151–12156 RSC.
- A. Klechikov, J. Sun, A. Vorobiev and A. V. Talyzin, J. Phys. Chem. C, 2018, 122, 13106–13116 CrossRef CAS.
- A. Iakunkov, J. Sun, A. Rebrikova, M. Korobov, A. Klechikov, A. Vorobiev, N. Boulanger and A. V. Talyzin, J. Mater. Chem. A, 2019, 7, 11331–11337 RSC.
- K. Tazaki, W. S. Fyfe and M. Iwatsuki, Nature, 1988, 333, 245–247 CrossRef.
- T. Daio, T. Bayer, T. Ikuta, T. Nishiyama, K. Takahashi, Y. Takata, K. Sasaki and S. M. Lyth, Sci. Rep., 2015, 5, 11807 CrossRef.
- A. V. Talyzin, S. M. Luzan, T. Szabó, D. Chernyshev and V. Dmitriev, Carbon, 2011, 49, 1894–1899 CrossRef CAS.
- S. Zheng, Q. Tu, J. J. Urban, S. Li and B. Mi, ACS Nano, 2017, 11, 6440–6450 CrossRef CAS.
- B. Rezania, N. Severin, A. V. Talyzin and J. P. Rabe, Nano Lett., 2014, 14, 3993–3998 CrossRef CAS.
- Y. P. Tang, D. R. Paul and T. S. Chung, J. Membr. Sci., 2014, 458, 199–208 CrossRef CAS.
- N. X. Wang, S. L. Ji, G. J. Zhang, J. Li and L. Wang, Chem. Eng. J., 2012, 213, 318–329 CrossRef CAS.
- A. Akbari, P. Sheath, S. T. Martin, D. B. Shinde, M. Shaibani, P. C. Banerjee, R. Tkacz, D. Bhattacharyya and M. Majumder, Nat. Commun., 2016, 7, 10891 CrossRef CAS.
- L. Chen, J. H. Moon, X. X. Ma, L. Zhang, Q. Chen, L. N. Chen, R. Q. Peng, P. C. Si, J. K. Feng, Y. H. Li, J. Lou and L. J. Ci, Carbon, 2018, 130, 487–494 CAS.
- H. M. Hegab and L. D. Zou, J. Membr. Sci., 2015, 484, 95–106 CAS.
- S. Borini, R. White, D. Wei, M. Astley, S. Haque, E. Spigone, N. Harris, J. Kivioja and T. Ryhanen, ACS Nano, 2013, 7, 11166–11173 CrossRef CAS.
- C. Li, X. Y. Yu, W. Zhou, Y. B. Cui, J. Liu and S. C. Fan, Opt. Lett., 2018, 43, 4719–4722 CrossRef CAS.
- M. Tang, C. Zhang, J. Y. Zhang, Q. L. Zhao, Z. L. Hou and K. T. Zhan, Phys. Status Solidi A, 2020, 217, 1900869 CrossRef CAS.
- S. M. Borini, R. V. Jani, K. Kiviola, M. R. Astley and A. C. Bower, Patent US9042075B2, Nokia Technologies Oy, 2015 Search PubMed.
- S. W. Lee, B. I. Choi, J. C. Kim, S. B. Woo, Y. G. Kim, S. Kwon, J. Yoo and Y. S. Seo, Sens. Actuators, B, 2016, 237, 575–580 CrossRef CAS.
- R. L. Liu, T. Gong, K. Zhang and C. Lee, Sci. Rep., 2017, 7, 9761 CrossRef.
- A. Buchsteiner, A. Lerf and J. Pieper, J. Phys. Chem. B, 2006, 110, 22328–22338 CrossRef CAS.
- S. Cerveny, F. Barroso-Bujans, A. Alegria and J. Colmenero, J. Phys. Chem. C, 2010, 114, 2604–2612 CrossRef CAS.
- B. Lian, S. De Luca, Y. You, S. Alwarappan, M. Yoshimura, V. Sahajwalla, S. C. Smith, G. Leslie and R. K. Joshi, Chem. Sci., 2018, 9, 5106–5111 CAS.
- S. A. Solin, Annu. Rev. Mater. Sci., 1997, 27, 89–115 CrossRef CAS.
- J. L. Mcatee, Am. Mineral., 1956, 41, 627–631 CAS.
- H. Vali and H. M. Koster, Clay Miner., 1986, 21, 827–859 CrossRef CAS.
- F. Barroso-Bujans, S. Cerveny, R. Verdejo, J. J. del Val, J. M. Alberdi, A. Alegria and J. Colmenero, Carbon, 2010, 48, 1079–1087 CrossRef CAS.
- S. Ban, J. Xie, Y. J. Wang, B. Jing, B. Liu and H. J. Zhou, ACS Appl. Mater. Interfaces, 2016, 8, 321–332 CrossRef CAS.
- B. Rezania, N. Severin, A. V. Talyzin and J. P. Rabe, Nano Lett., 2014, 14, 3993–3998 CrossRef CAS.
- A. V. Talyzin, S. M. Luzan, T. Szabo, D. Chernyshev and V. Dmitriev, Carbon, 2011, 49, 1894–1899 CrossRef CAS.
- J. Zhu, C. M. Andres, J. D. Xu, A. Ramamoorthy, T. Tsotsis and N. A. Kotov, ACS Nano, 2012, 6, 8357–8365 CrossRef CAS.
- A. V. Talyzin, B. Sundqvist, T. Szabo and V. Dmitriev, J. Phys. Chem. Lett., 2011, 2, 309–313 CrossRef CAS.
- S. M. Luzan and A. V. Talyzin, J. Phys. Chem. C, 2011, 115, 24611–24614 CrossRef CAS.
- S. J. You, D. Kunz, M. Stoeter, H. Kalo, B. Putz, J. Breu and A. V. Talyzin, Angew. Chem., Int. Ed., 2013, 52, 3891–3895 CrossRef CAS.
- M. Ghidiu, S. Kota, V. Drozd and M. W. Barsoum, Sci. Adv., 2018, 4, eaao6850 CrossRef.
- H. Hwang, D. Seoung, Y. Lee, Z. X. Liu, H. P. Liermann, H. Cynn, T. Vogt, C. C. Kao and H. K. Mao, Nat. Geosci., 2017, 10, 947–953 CrossRef CAS.
- D. W. Lee and J. W. Seo, J. Phys. Chem. C, 2011, 115, 12483–12486 CrossRef CAS.
- R. Krüger, Inaugural-Dissertation, Ludwig-Maximilians-Universität München, 1980.
- A. Weiss, I. Dekany and R. Kruger-Grasser, Colloid Polym. Sci., 1998, 276, 570–576 CrossRef.
- C. Cabrillo, F. Barroso-Bujans, R. Fernandez-Perea, F. Fernandez-Alonso, D. Bowron and F. J. Bermejo, Carbon, 2016, 100, 546–555 CrossRef CAS.
- A. Buchsteiner, A. Lerf and J. Pieper, J. Phys. Chem. B, 2006, 110, 22328–22338 CrossRef CAS.
- S. Cerveny, F. Barroso-Bujans, Á. Alegría and J. Colmenero, J. Phys. Chem. C, 2010, 114, 2604–2612 CrossRef CAS.
- A. F. U. Hofmann, Ber. Dtsch. Chem. Ges., 1930, 63, 1248–1262 CrossRef.
- A. Lerf, A. Buchsteiner, J. Pieper, S. Schöttl, I. Dekany, T. Szabo and H. P. Boehm, J. Phys. Chem. Solids, 2006, 67, 1106–1110 CrossRef CAS.
- J. B. v. D. de Boer and A. B. C. van Doorn, Proc. K. Ned. Akad. Wet., Ser. B: Phys. Sci., 1958, 61, 242–252 Search PubMed.
- T. Szabó, O. Berkesi, P. Forgó, K. Josepovits, Y. Sanakis, D. Petridis and I. Dékány, Chem. Mater., 2006, 18, 2740–2749 CrossRef.
- Y. Matsuo, T. Fukunaga, T. Fukutsuka and Y. Sugie, Carbon, 2004, 42, 2117–2119 CrossRef CAS.
- Y. Matsuo, K. Tahara and Y. Sugie, Carbon, 1997, 35, 113–120 CrossRef CAS.
- S. M. Luzan and A. V. Talyzin, J. Phys. Chem. C, 2011, 115, 24611–24614 CrossRef CAS.
- A. Klechikov, S. You, L. Lackner, J. Sun, A. Iakunkov, A. Rebrikova, M. Korobov, I. Baburin, G. Seifert and A. V. Talyzin, Carbon, 2018, 140, 157–163 CrossRef CAS.
- A. V. Talyzin, B. Sundqvist, T. S. Szabó, I. Dékány and V. Dmitriev, J. Am. Chem. Soc., 2009, 131, 18445–18449 CrossRef CAS.
- C. D. Pino, A. Ramirez and J. Cano-Ruiz, Nature, 1966, 209, 906–907 CrossRef.
- S. You, J. Yu, B. Sundqvist, L. A. Belyaeva, N. V. Avramenko, M. V. Korobov and A. V. Talyzin, J. Phys. Chem. C, 2013, 117, 1963–1968 CrossRef CAS.
- A. V. Talyzin, A. Klechikov, M. Korobov, A. T. Rebrikova, N. V. Avramenko, M. F. Gholami, N. Severin and J. P. Rabe, Nanoscale, 2015, 7, 12625–12630 RSC.
- Y. Matsuo, T. Miyabe, T. Fukutsuka and Y. Sugie, Carbon, 2007, 45, 1005–1012 CrossRef CAS.
- A. B. Bourlinos, D. Gournis, D. Petridis, T. Szabó, A. Szeri and I. Dékány, Langmuir, 2003, 19, 6050–6055 CrossRef CAS.
- Y. Matsuo, K. Watanabe, T. Fukutsuka and Y. Sugie, Carbon, 2003, 41, 1545–1550 CrossRef CAS.
- S. J. You, J. C. Yu, B. Sundqvist, L. A. Belyaeva, N. V. Avramenko, M. V. Korobov and A. V. Talyzin, J. Phys. Chem. C, 2013, 117, 1963–1968 CrossRef CAS.
- S. J. You, B. Sundqvist and A. V. Talyzin, ACS Nano, 2013, 7, 1395–1399 CrossRef CAS.
- D. M. C. MacEwan, Kolloid Z. Z. Polym., 1965, 203, 36–42 CrossRef.
- D. Kim, D. W. Kim, H.-K. Lim, J. Jeon, H. Kim, H.-T. Jung and H. Lee, J. Phys. Chem. C, 2014, 118, 11142–11148 CrossRef CAS.
- Y. Matsuo, S. Higashika, K. Kimura, Y. Miyamoto, T. Fukutsuka and Y. Sugie, J. Mater. Chem., 2002, 12, 1592–1596 RSC.
- P. Liu and K. Gong, Carbon, 1999, 37, 706–707 CrossRef CAS.
- P. Xiao, M. Xiao, P. Liu and K. Gong, Carbon, 2000, 38, 626–628 CrossRef CAS.
- T. Kyotani, H. Moriyama and A. Tomita, Carbon, 1997, 35, 1185–1187 CrossRef CAS.
- A. Klechikov, J. Yu, D. Thomas, T. Sharifi and A. V. Talyzin, Nanoscale, 2015, 7, 15374–15384 RSC.
- Z. Hu and C. Liu, J. Polym. Res., 2013, 20, 39 CrossRef.
- J.-H. Jeong, M.-C. Choi, S. Nagappan, W.-K. Lee and C.-S. Ha, Polym. Int., 2018, 67, 91–99 CrossRef CAS.
- C. Nethravathi, B. Viswanath, C. Shivakumara, N. Mahadevaiah and M. Rajamathi, Carbon, 2008, 46, 1773–1781 CrossRef CAS.
- C. Nethravathi and M. Rajamathi, Carbon, 2006, 44, 2635–2641 CrossRef CAS.
- C. Nethravathi, Bhojaraj and M. Rajamathi, Carbon, 2020, 158, 97–101 CrossRef CAS.
- S. You, S. M. Luzan, T. Szabó and A. V. Talyzin, Carbon, 2013, 52, 171–180 CrossRef CAS.
- F. Aragón, J. Cano Ruiz and D. M. C. Macewan, Nature, 1959, 183, 740–741 CrossRef.
- A. R. GarcÍA, J. Cano-Ruiz and D. M. C. MacEwan, Nature, 1964, 203, 1063–1064 CrossRef.
- P. Liu, K. Gong and P. Xiao, Carbon, 1999, 37, 2073–2075 CrossRef CAS.
- J. H. Sun, A. Iakunkov, A. T. Rebrikova and A. V. Talyzin, Nanoscale, 2018, 10, 21386–21395 RSC.
- A. B. Bourlinos, D. Gournis, D. Petridis, T. Szabó, A. Szeri and I. Dékány, Langmuir, 2003, 19, 6050–6055 CrossRef CAS.
- O. Akhavan, Carbon, 2011, 49, 11–18 CrossRef CAS.
- F. M. Chen, S. B. Liu, J. M. Shen, L. Wei, A. D. Liu, M. B. Chan-Park and Y. Chen, Langmuir, 2011, 27, 9174–9181 CrossRef CAS.
- M. Mirza-Aghayan, M. Alizadeh, M. M. Tavana and R. Boukherroub, Tetrahedron Lett., 2014, 55, 6694–6697 CrossRef CAS.
- J. H. Sun, A. Klechikov, C. Moise, M. Prodana, M. Enachescu and A. V. Talyzin, Angew. Chem., Int. Ed., 2018, 57, 1034–1038 CrossRef CAS.
- A. Nordenstrom, A. Iakunkov, J. H. Sun and A. V. V. Talyzin, RSC Adv., 2020, 10, 6831–6839 RSC.
- G. Mercier, A. Klechikov, M. Hedenstrom, D. Johnels, I. A. Baburin, G. Seifert, R. Mysyk and A. V. Talyzin, J. Phys. Chem. C, 2015, 119, 27179–27191 CrossRef CAS.
- J. W. Burress, S. Gadipelli, J. Ford, J. M. Simmons, W. Zhou and T. Yildirim, Angew. Chem., Int. Ed., 2010, 49, 8902–8904 CrossRef CAS.
- G. Srinivas, J. W. Burress, J. Ford and T. Yildirim, J. Mater. Chem., 2011, 21, 11323–11329 RSC.
- S. You, S. M. Luzan, J. Yu, B. Sundqvist and A. Talyzin, J. Phys. Chem. Lett., 2012, 3, 812–817 CrossRef CAS.
- A. V. Talyzin and S. M. Luzan, J. Phys. Chem. C, 2010, 114, 7004–7006 CrossRef CAS.
- C. Cabrillo, F. Barroso-Bujans, S. Cerveny, R. Fernandez-Perea, F. Fernandez-Alonso, D. Bowron and F. J. Bermejo, Mol. Phys., 2019, 117, 3434–3444 CrossRef CAS.
- J. A. Rausellcolom and K. Norrish, J. Sci. Instrum., 1962, 39, 156–159 CrossRef.
- J. M. Cases, I. Berend, G. Besson, M. Francois, J. P. Uriot, F. Thomas and J. E. Poirier, Langmuir, 1992, 8, 2730–2739 CrossRef CAS.
- L. Mayr, S. Amschler, A. Edenharter, V. Dudko, R. Kunz, S. Rosenfeldt and J. Breu, Langmuir, 2020, 36, 3814–3820 CrossRef CAS.
- R. Kunz, S. Amschler, A. Edenharter, L. Mayr, S. Herlitz, S. Rosenfeldt and J. Breu, Clays Clay Miner., 2019, 67, 481–487 CrossRef CAS.
- J. De Boer and A. Van Doorn, Proc. K. Ned. Akad. Wetensch. B, 1958, 61160–169 Search PubMed.
- M. R. Acocella, L. D'Urso, M. Maggio, R. Avolio, M. E. Errico and G. Guerra, Langmuir, 2017, 33, 6819–6825 CrossRef CAS.
- J. Q. Huang, T. Z. Zhuang, Q. Zhang, H. J. Peng, C. M. Chen and F. Wei, ACS Nano, 2015, 9, 3002–3011 CrossRef CAS.
- K. Feng, B. B. Tang and P. Y. Wu, ACS Appl. Mater. Interfaces, 2013, 5, 1481–1488 CrossRef CAS.
- C. W. Lin and Y. S. Lu, J. Power Sources, 2013, 237, 187–194 CrossRef CAS.
- R. Sandstrom, A. Annamalai, N. Boulanger, J. Ekspong, A. Talyzin, I. Muhlbacher and T. Wagberg, Sustainable Energy Fuels, 2019, 3, 1790–1798 RSC.
- B. Xu, S. F. Yue, Z. Y. Sui, X. T. Zhang, S. S. Hou, G. P. Cao and Y. S. Yang, Energy Environ. Sci., 2011, 4, 2826–2830 RSC.
- G. X. Zhao, J. X. Li, X. M. Ren, C. L. Chen and X. K. Wang, Environ. Sci. Technol., 2011, 45, 10454–10462 CrossRef CAS.
- A. V. Talyzin, T. Hausmaninger, S. You and T. Szabo, Nanoscale, 2014, 6, 272–281 RSC.
- J. M. Zhu, L. W. Zhu, Z. F. Lu, L. Gu, S. L. Cao and X. B. Cao, J. Phys. Chem. C, 2012, 116, 23075–23082 CrossRef CAS.
- D. Pacile, J. C. Meyer, A. F. Rodriguez, M. Papagno, C. Gomez-Navarro, R. S. Sundaram, M. Burghard, K. Kern, C. Carbone and U. Kaiser, Carbon, 2011, 49, 966–972 CrossRef CAS.
- R. Liu, G. Arabale, J. Kim, K. Sun, Y. Lee, C. Ryu and C. Lee, Carbon, 2014, 77, 933–938 CrossRef CAS.
- A. Anand, B. Unnikrishnan, J. Y. Mao, H. J. Lin and C. C. Huang, Desalination, 2018, 429, 119–133 CrossRef CAS.
- N. Song, X. L. Gao, Z. Ma, X. J. Wang, Y. Wei and C. J. Gao, Desalination, 2018, 437, 59–72 CrossRef CAS.
- Y. Wei, Y. S. Zhang, X. L. Gao, Z. Ma, X. J. Wang and C. J. Gao, Carbon, 2018, 139, 964–981 CrossRef CAS.
- O. C. Compton, S. W. Cranford, K. W. Putz, Z. An, L. C. Brinson, M. J. Buehler and S. T. Nguyen, ACS Nano, 2012, 6, 2008–2019 CrossRef CAS.
- T. Arif, G. Colas and T. Filleter, ACS Appl. Mater. Interfaces, 2018, 10, 22537–22544 CrossRef CAS.
- A. Clauss and U. Hofmann, Angew. Chem., 1956, 68, 522–522 CrossRef CAS.
- S. Liu, K. Hu, M. Cerruti and F. Barthelat, Carbon, 2020, 158, 426–434 CrossRef CAS.
- K. Huang, G. P. Liu, Y. Y. Lou, Z. Y. Dong, J. Shen and W. Q. Jin, Angew. Chem., Int. Ed., 2014, 53, 6929–6932 CrossRef CAS.
- H. W. Kim, H. W. Yoon, S.-M. Yoon, B. K. Ahn, Y. H. Cho, H. J. Shin, H. Yang, U. Paik, S. Kwon, J.-Y. Choi and H. B. Park, Science, 2013, 342, 91–95 CrossRef CAS.
- D. D. Kulkarni, S. Kim, M. Chyasnavichyus, K. S. Hu, A. G. Fedorov and V. V. Tsukruk, J. Am. Chem. Soc., 2014, 136, 6546–6549 CrossRef CAS.
- K. Erickson, R. Erni, Z. Lee, N. Alem, W. Gannett and A. Zettl, Adv. Mater., 2010, 22, 4467–4472 CrossRef CAS.
- V. Saraswat, R. M. Jacobberger, J. S. Ostrander, C. L. Hummell, A. J. Way, J. Wang, M. T. Zanni and M. S. Arnold, ACS Nano, 2018, 12, 7855–7865 CrossRef CAS.
- Y. Si and E. T. Samulski, Nano Lett., 2008, 8, 1679–1682 CrossRef CAS.
- K. A. Mkhoyan, A. W. Contryman, J. Silcox, D. A. Stewart, G. Eda, C. Mattevi, S. Miller and M. Chhowalla, Nano Lett., 2009, 9, 1058–1063 CrossRef CAS.
- A. V. Talyzin, 2018, arXiv preprint arXiv:1812.03941, pp. 1–6.
- P. Z. Sun, M. Zhu, K. L. Wang, M. L. Zhong, J. Q. Wei, D. H. Wu, Z. P. Xu and H. W. Zhu, ACS Nano, 2013, 7, 428–437 CrossRef CAS.
- P. Z. Sun, F. Zheng, M. Zhu, Z. G. Song, K. L. Wang, M. L. Zhong, D. H. Wu, R. B. Little, Z. P. Xu and H. W. Zhu, ACS Nano, 2014, 8, 850–859 CrossRef CAS.
- N. A. Chumakova, A. T. Rebrikova, A. V. Talyzin, N. A. Paramonov, A. K. Vorobiev and M. V. Korobov, J. Phys. Chem. C, 2018, 122, 22750–22759 CrossRef CAS.
- L. Huang, Y. R. Li, Q. Q. Zhou, W. J. Yuan and G. Q. Shi, Adv. Mater., 2015, 27, 3797–3802 CrossRef CAS.
- R. Spitz Steinberg, M. Cruz, N. G. A. Mahfouz, Y. Qiu and R. H. Hurt, ACS Nano, 2017, 11, 5670–5679 CrossRef CAS.
- H. C. Bi, K. B. Yin, X. Xie, J. Ji, S. Wan, L. T. Sun, M. Terrones and M. S. Dresselhaus, Sci. Rep., 2013, 3, 2714 CrossRef.
- S. Pongampai, P. Pengpad, R. Meananeatra, W. Chaisriratanakul, W. Thitirungraung and R. Muanghlua, Int. Elect. Eng. Congr., 2019, 1–4 Search PubMed.
- C. A. Amadei and C. D. Vecitis, J. Phys. Chem. Lett., 2016, 7, 3791–3797 CrossRef CAS.
- Z. Jia and W. Shi, Carbon, 2016, 101, 290–295 CrossRef CAS.
- Z. Jia, Y. Wang, W. Shi and J. Wang, J. Membr. Sci., 2016, 520, 139–144 CrossRef CAS.
- F. Baskoro, C.-B. Wong, S. R. Kumar, C.-W. Chang, C.-H. Chen, D. W. Chen and S. J. Lue, J. Membr. Sci., 2018, 554, 253–263 CrossRef CAS.
- W. L. Xu, F. Zhou and M. Yu, Ind. Eng. Chem. Res., 2018, 57, 16103–16109 CrossRef CAS.
- W. Wu, J. Su, M. Jia, W. Zhong, Z. Li and W. Li, J. Mater. Chem. A, 2019, 7, 13007–13011 RSC.
- Q. Yang, Y. Su, C. Chi, C. T. Cherian, K. Huang, V. G. Kravets, F. C. Wang, J. A. Pratt, A. N. Grigorenko, F. Guinea, A. K. Geim and R. R. Nair, Nat. Mater., 2017, 16, 1198–1202 CrossRef CAS.
- R. Liu, G. Arabale, J. Kim, K. Sun, Y. Lee, C. Ryu and C. Lee, Carbon, 2014, 77, 933–938 CrossRef CAS.
- M. Zhang, Y. Mao, G. Liu, G. Liu, Y. Fan and W. Jin, Angew. Chem., Int. Ed., 2020, 59(4), 1689–1695 CrossRef CAS.
- J. Kim, S. E. Lee, S. Seo, J. Y. Woo and C.-S. Han, J. Membr. Sci., 2019, 592, 117394 CrossRef CAS.
- G. Yang, Z. Xie, M. Cran, D. Ng, C. D. Easton, M. Ding, H. Xu and S. Gray, J. Mater. Chem. A, 2019, 7, 19682–19690 RSC.
- D. W. Kim, D. Kim, B. H. Min, H. Lee and H.-T. Jung, Carbon, 2015, 88, 126–132 CrossRef CAS.
- L. Chen, G. Shi, J. Shen, B. Peng, B. Zhang, Y. Wang, F. Bian, J. Wang, D. Li, Z. Qian, G. Xu, G. Liu, J. Zeng, L. Zhang, Y. Yang, G. Zhou, M. Wu, W. Jin, J. Li and H. Fang, Nature, 2017, 550, 380–383 CrossRef CAS.
- Y. H. Cho, H. W. Kim, H. D. Lee, J. E. Shin, B. M. Yoo and H. B. Park, J. Membr. Sci., 2017, 544, 425–435 CrossRef CAS.
- S. Zheng, Q. Tu, M. Wang, J. J. Urban and B. Mi, ACS Nano, 2020, 14(5), 6013–6023 CrossRef CAS.
- K. A. Nakagawa, S. M. Kunimatsu, T. Yoshioka, T. Shintani, E. Kamio and H. Matsuyama, Membranes, 2018, 8, 130 CrossRef.
- A. V. Talyzin, T. Hausmaninger, S. You and T. Szabó, Nanoscale, 2014, 6, 272–281 RSC.
- L. Huang, Y. Li, Q. Zhou, W. Yuan and G. Shi, Adv. Mater., 2015, 27, 3797–3802 CrossRef CAS.
- K. Raidongia and J. Huang, J. Am. Chem. Soc., 2012, 134, 16528–16531 CrossRef CAS.
- S. J. Kim, D. W. Kim, K. M. Cho, K. M. Kang, J. Choi, D. Kim and H.-T. Jung, Sci. Rep., 2018, 8, 1959 CrossRef.
- J.-H. Song, H.-W. Yu, M.-H. Ham and I. S. Kim, Nano Lett., 2018, 18, 5506–5513 CrossRef CAS.
- Z. Zhao, S. Ni, X. Su, Y. Gao and X. Sun, ACS Sustainable Chem. Eng., 2019, 7(17), 14874–14882 CrossRef CAS.
- K. M. Kang, D. W. Kim, C. E. Ren, K. M. Cho, S. J. Kim, J. H. Choi, Y. T. Nam, Y. Gogotsi and H.-T. Jung, ACS Appl. Mater. Interfaces, 2017, 9, 44687–44694 CrossRef CAS.
- R. K. Joshi, P. Carbone, F. C. Wang, V. G. Kravets, Y. Su, I. V. Grigorieva, H. A. Wu, A. K. Geim and R. R. Nair, Science, 2014, 343, 752–754 CrossRef CAS.
- Z. Luo, Y. Lu, L. A. Somers and A. T. C. Johnson, J. Am. Chem. Soc., 2009, 131, 898–899 CrossRef CAS.
- B. Rezania, N. Severin, A. V. Talyzin and J. P. Rabe, Nano Lett., 2014, 14, 3993–3998 CrossRef CAS.
- J. Zhu, C. M. Andres, J. Xu, A. Ramamoorthy, T. Tsotsis and N. A. Kotov, ACS Nano, 2012, 6, 8357–8365 CrossRef CAS.
- A. Vorobiev, A. Dennison, D. Chernyshov, V. Skrypnychuk, D. Barbero and A. V. Talyzin, Nanoscale, 2014, 6, 12151–12156 RSC.
- B. Mi, S. Zheng and Q. Tu, Faraday Discuss., 2018, 209, 329–340 RSC.
- C. D. Williams, P. Carbone and F. R. Siperstein, ACS Nano, 2019, 13, 2995–3004 CrossRef CAS.
- C. D. Williams, P. Carbone and F. R. Siperstein, Nanoscale, 2018, 10, 1946–1956 RSC.
- N. Wei, X. Peng and Z. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 5877–5883 CrossRef CAS.
- R. Devanathan, D. Chase-Woods, Y. Shin and D. W. Gotthold, Sci. Rep., 2016, 6, 29484 CrossRef.
- Y. Yang, L. Mu, L. Chen, G. Shi and H. Fang, Phys. Chem. Chem. Phys., 2019, 21, 7623–7629 RSC.
- S. Jiao, K. Zhou, M. Wu, C. Li, X. Cao, L. Zhang and Z. Xu, ACS Appl. Mater. Interfaces, 2018, 10, 37014–37022 CrossRef CAS.
- A. Liscio, K. Kouroupis-Agalou, X. D. Betriu, A. Kovtun, E. Treossi, N. M. Pugno, G. De Luca, L. Giorgini and V. Palermo, 2D Mater., 2017, 4, 025017 CrossRef.
- X. D. Qi, T. N. Zhou, S. Deng, G. Y. Zong, X. L. Yao and Q. Fu, J. Mater. Sci., 2014, 49, 1785–1793 CrossRef CAS.
- F. Mouhat, F. X. Coudert and M. L. Bocquet, Nat. Commun., 2020, 11, 1566 CrossRef CAS.
- C. Cheng, G. P. Jiang, C. J. Garvey, Y. Y. Wang, G. P. Simon, J. Z. Liu and D. Li, Sci. Adv., 2016, 2, e1501272 CrossRef.
- B. Mi, S. Zheng and Q. Tu, Faraday Discuss., 2018, 209, 329–340 CAS.
- Y. H. Xi, J. Q. Hu, Z. Liu, R. Xie, X. J. Ju, W. Wang and L. Y. Chu, ACS Appl. Mater. Interfaces, 2016, 8, 15557–15566 CAS.
- A. Ghaffar, L. N. Zhang, X. Y. Zhu and B. L. Chen, Environ. Sci.: Nano, 2019, 6, 904–915 RSC.
- S. Zhao, H. T. Zhu, H. Wang, P. Rassu, Z. Wang, P. Song and D. W. Rao, J. Hazard. Mater., 2019, 366, 659–668 CrossRef CAS.
- J. Kim, S. E. Lee, S. Seo, J. Y. Woo and C. S. Han, J. Membr. Sci., 2019, 592, 117394 CrossRef CAS.
- J. Abraham, K. S. Vasu, C. D. Williams, K. Gopinadhan, Y. Su, C. T. Cherian, J. Dix, E. Prestat, S. J. Haigh, I. V. Grigorieva, P. Carbone, A. K. Geim and R. R. Nair, Nat. Nanotechnol., 2017, 12, 546–550 CrossRef CAS.
- S. Kim, R. W. Ou, Y. X. Hu, X. Y. Li, H. C. Zhang, G. P. Simon and H. T. Wang, J. Membr. Sci., 2018, 562, 47–55 CrossRef CAS.
- A. T. Li, K. Han, Y. H. Zhou, H. Q. Ye, G. G. Liu and H. H. Kung, Mater. Today Commun., 2017, 11, 139–146 CrossRef CAS.
- S. Park, K. S. Lee, G. Bozoklu, W. Cai, S. T. Nguyen and R. S. Ruoff’, ACS Nano, 2008, 2, 572–578 CrossRef CAS.
- L. Chen, G. S. Shi, J. Shen, B. Q. Peng, B. W. Zhang, Y. Z. Wang, F. G. Bian, J. J. Wang, D. Y. Li, Z. Qian, G. Xu, G. P. Liu, J. R. Zeng, L. J. Zhang, Y. Z. Yang, G. Q. Zhou, M. H. Wu, W. Q. Jin, J. Y. Li and H. P. Fang, Nature, 2017, 550, 415–418 CrossRef.
- Y. H. Xi, Z. Liu, J. Y. Ji, Y. Wang, Y. Faraj, Y. D. Zhu, R. Xie, X. J. Ju, W. Wang, X. H. Lu and L. Y. Chu, J. Membr. Sci., 2018, 550, 208–218 CrossRef CAS.
- D. K. Mahalingam, S. F. Wang and S. P. Nunes, Ind. Eng. Chem. Res., 2019, 58, 23106–23113 CrossRef CAS.
- G. K. Zhao and H. W. Zhu, Adv. Mater. Interfaces, 2020, 7, 1901535 CrossRef CAS.
- Y. X. Zhang, S. Chen, J. X. An, H. Fu, X. S. Wu, C. C. Pang and H. Gao, ACS Biomater. Sci. Eng., 2019, 5, 2732–2739 CrossRef CAS.
- K. G. Zhou, K. S. Vasu, C. T. Cherian, M. Neek-Amal, J. C. Zhang, H. Ghorbanfekr-Kalashami, K. Huang, O. P. Marshall, V. G. Kravets, J. Abraham, Y. Su, A. N. Grigorenko, A. Pratt, A. K. Geim, F. M. Peeters, K. S. Novoselov and R. R. Nair, Nature, 2018, 559, 236–241 CrossRef CAS.
- C. N. Yeh, K. Raidongia, J. J. Shao, Q. H. Yang and J. X. Huang, Nat. Chem., 2015, 7, 166–170 CrossRef CAS.
- G. P. Liu, W. Q. Jin and N. P. Xu, Chem. Soc. Rev., 2015, 44, 5016–5030 RSC.
- J. J. Zhang, Q. Q. Liu, Y. B. Ruan, S. Ling, K. Wang and H. B. Lu, Chem. Mater., 2018, 30, 1888–1897 CrossRef CAS.
- S. X. Zheng, Q. S. Tu, J. J. Urban, S. F. Li and B. X. Mi, ACS Nano, 2017, 11, 6440–6450 CrossRef CAS.
- K. H. Thebo, X. T. Qian, Q. Zhang, L. Chen, H. M. Cheng and W. C. Ren, Nat. Commun., 2018, 9, 1486 CrossRef.
- K. Delhiraja, K. Vellingiri, D. W. Boukhvalov and L. Philip, Ind. Eng. Chem. Res., 2019, 58, 2899–2913 CrossRef CAS.
- W. Z. Yu, T. Y. Yu and N. Graham, 2D Mater., 2017, 4, 311 Search PubMed.
- C. B. Wang, Z. Y. Li, J. X. Chen, Y. H. Yin and H. Wu, Appl. Surf. Sci., 2018, 427, 1092–1098 CrossRef CAS.
- C. M. Kim, S. Hong, R. Y. Li, I. S. Kim and P. Wang, ACS Sustainable Chem. Eng., 2019, 7, 7252–7259 CrossRef CAS.
- Y. T. Nam, J. Choi, K. M. Kang, D. W. Kim and H. T. Jung, ACS Appl. Mater. Interfaces, 2016, 8, 27376–27382 CrossRef CAS.
- T. T. Gao, H. B. Wu, L. Tao, L. T. Qu and C. Li, J. Mater. Chem. A, 2018, 6, 19563–19569 RSC.
- D. Tomanek and A. Kyrylchuk, Phys. Rev. Appl., 2019, 12, 024054 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |