
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReductive dehydrocoupling of diphenyltin dihydride with LiAlH4: selective synthesis and structures of the first bicyclo[2.2.1]heptastannane-1,4-diide and bicyclo[2.2.2]octastannane-1,4-diide†
Beate G.
Steller
 a,
Roland C.
Fischer
*a,
Michaela
Flock
a,
Michael S.
Hill
b,
David J.
Liptrot
*b,
Claire L.
McMullin
b,
Nasir A.
Rajabi
b,
Kathrin
Tiefling
a and
Andrew S. S.
Wilson
b
a,
Roland C.
Fischer
*a,
Michaela
Flock
a,
Michael S.
Hill
b,
David J.
Liptrot
*b,
Claire L.
McMullin
b,
Nasir A.
Rajabi
b,
Kathrin
Tiefling
a and
Andrew S. S.
Wilson
b
a6330 Institute of Inorganic Chemistry, Graz University of Technology, Stremayrgasse 9/V, Austria. E-mail: roland.fischer@tugraz.at
bDepartment of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: D.J.Liptrot@bath.ac.uk
First published on 29th November 2019
Abstract
The reaction of diphenyltin dihydride with LiAlH4 gives access to a set of charged tin cages as their lithium salts. Variation in the ratio of reactants provides a perstannabicyclooctane dianion and a perstannanorbornane as the di- and monoanions. These compounds can be synthesised selectively by careful stoichiometric control and have been characterised by single crystal X-ray diffractometry, NMR and UV-vis spectroscopy. Computational exploration of the electronic structures of these compounds was undertaken and, in agreement with structural and spectroscopic features, indicated significant σ-delocalisation in the tin skeletons.
The chemistry of anionic oligo-tin cage and cluster compounds is dominated by a plethora of Zintl phases, discrete Zintl ions,1 [Snn]x−, or their derivatisation products, [RmSnn]x−.2 Derivatisation reactions of the Zintl ion Sn94− have led to the trianionic compounds RSn93−.3 Reaction of thermolabile tin(I) halides with [Si(SiMe3)3]− resulted in the isolation of neutral Sn10[Si(SiMe3)3]6,4 the monoanion Sn10[Si(SiMe3)3]5−,5 and the dianions Sn9[Si(SiMe3)3]42− and Sn10[Si(SiMe3)3]42−.6,7 Moreover, the anionic cluster Sn9R7(NHC)− (R = CH(SiMe3)2, NHC = 1,2,3,4-tetramethylimidazol-2-ylidene) was isolated from the reaction of the trihydride RSnH3 with the corresponding NHC.8 Alongside these, several neutral metalloid clusters of the form SnnRm (n > m) have been obtained from reductive or dehydrogenative coupling methods.9 Closely related to metalloid compounds,10a albeit with a superstoichometric ratio of substituents to tin, are the elementoid10b [1.1.1]pentastannapropellanes, Sn5R6,11 tetracyclic Sn7R812 and the hexastannabenzene isomer, Sn6R6.13 The aforementioned compounds share a non-classical, i.e. 3D-delocalised, bonding situation. This confers a narrow energy gap between frontier orbitals which facilitates interesting addition chemistry and redox behaviour.14 In contrast to the well represented class of simple monocyclic rings, (R2Sn)n,15 purely σ-bonded tin cages, (RSn)n, are sparse. Examples include tricyclo- and pentacycloprismanes of the stoichiometry, R6Sn6 and R10Sn10,8,16 cubanes, R8Sn8,17 and a tetrahedral cage molecule with edge-bridging methandiyl substituents.18 Anionic, covalent, oligotin cages include Sn8R62− (R = Si-t-Bu3)16c and Sn5(CH3)R6− and the radical anion Sn5R6− (R = 2,6-Et2-C6H3). The latter species originate from the addition of methyl lithium to, and the electrochemical one-electron reduction of, a [1.1.1]pentastannapropellane.14b Organotindihydrides are common starting materials in the synthesis of oligotin compounds. Their dehydrogenative coupling has also been widely applied in the synthesis of linear tin polymers and cyclic oligomers (R2Sn)n,19 whilst the reaction of a tin dihydride with sodium in liquid ammonia led to the formation of Ph2SnHNa and NaPh2SnSnPh2Na.20
Diphenyltin dihydride can be synthesised by the reaction of LiAlH4 with diphenyltin dichloride.21 During our repetition of this literature method, we noted the reaction mixture would often acquire a deep red or yellow colour upon the use of large excesses of LiAlH4. This resulted in a significant decrease in yield of the desired diphenyltin dihydride. Thus, in order to elucidate the nature of possible over-reaction products, analytically pure Ph2SnH2 was reacted with LiAlH4.22
In an initial reaction, an equimolar mixture of the two reagents in THF was observed to provide a persistent bubbling and the formation of a dark red solution, as well as a flocculent white precipitate, identified as AlH3 by IR spectroscopy. Addition of 12-crown-4 (hereafter, 12-Cr-4) and storage at −30 °C yielded a small amount of an intensely yellow material that was thought to be a single, crystalline product. X-ray diffraction analysis, however, indicated this material to be a disordered mixture of two oligostannane dianions (1 and 2). In order to access analytically pure samples of 1 and 2, the ratio of LiAlH4 to Ph2SnH2 was optimised.
The reaction of 7 equivalents of diphenyltin dihydride with two equivalent of LiAlH4 in THF or DME gave access to crystalline, analytically pure [Ph10Sn7]2−[Li(12-Cr-4)2]+2, 1, in moderate yields after the addition of 12-Cr-4 and storage at −30 °C. Similarly, [Ph12Sn8]2−[Li(12-Cr-4)2]+2, 2, was obtained by applying analogous conditions albeit with a 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio of Ph2SnH2:LiAlH4. These optimised conditions yielded material suitable for X-ray crystallography as orange and red crystals, respectively. Higher tin to alumane ratios of 8:1 and above yielded the bicyclo[2.2.2]heptastanna-1-ide [Ph13Sn8]−[Li(12-Cr-4)2]+, 3. 3 was characterised by NMR and UV-vis spectroscopy as well as X-ray crystallography.23 A Ph2SnH2:LiAlH4 ratio of 1:0.75 and 1:2 led to the formation of elemental tin and compounds 4 and 5 which contain discrete [PhAlH3]− and [Ph3AlH]− anions and charge separated [Li·(12-Cr-4)2]+ or [Li·(DME)(12-Cr-4)]+ counterions. The formation of 4 and 5 demonstrates net phenyl group transfer from tin to aluminium which provides the tertiary tin bridgeheads in 1–3 with less than the original two phenyl groups per tin. NMR spectroscopic interrogation of the reaction mixtures indicated the initial formation of benzene and of the anion [HPh2Sn]− (119Sn: −180.5 ppm, 1J1H–119Sn = 152 Hz),20 which undergoes subsequent dehydrogenative coupling with excess Ph2SnH2 to cause the intermediate formation of various unidentified oligostannyl anions. Isotopic labelling experiments showed the predominant formation of H2 in the reaction of Ph2SnH2 with LiAlD4, while treatment of Ph2SnD2 with LiAlH4 gave D2 (see ESI† page S31).
:2 ratio of Ph2SnH2:LiAlH4. These optimised conditions yielded material suitable for X-ray crystallography as orange and red crystals, respectively. Higher tin to alumane ratios of 8:1 and above yielded the bicyclo[2.2.2]heptastanna-1-ide [Ph13Sn8]−[Li(12-Cr-4)2]+, 3. 3 was characterised by NMR and UV-vis spectroscopy as well as X-ray crystallography.23 A Ph2SnH2:LiAlH4 ratio of 1:0.75 and 1:2 led to the formation of elemental tin and compounds 4 and 5 which contain discrete [PhAlH3]− and [Ph3AlH]− anions and charge separated [Li·(12-Cr-4)2]+ or [Li·(DME)(12-Cr-4)]+ counterions. The formation of 4 and 5 demonstrates net phenyl group transfer from tin to aluminium which provides the tertiary tin bridgeheads in 1–3 with less than the original two phenyl groups per tin. NMR spectroscopic interrogation of the reaction mixtures indicated the initial formation of benzene and of the anion [HPh2Sn]− (119Sn: −180.5 ppm, 1J1H–119Sn = 152 Hz),20 which undergoes subsequent dehydrogenative coupling with excess Ph2SnH2 to cause the intermediate formation of various unidentified oligostannyl anions. Isotopic labelling experiments showed the predominant formation of H2 in the reaction of Ph2SnH2 with LiAlD4, while treatment of Ph2SnD2 with LiAlH4 gave D2 (see ESI† page S31).
1 and 2 constitute the first reports of a bicyclo[2.2.1]heptastannane-1,4-diide and bicyclo[2.2.2]octastannane-1,4-diide, respectively, and both crystallise in the presence of 2 [Li·(12-Cr-4)2]+ counterions as charge separated structures. In the solid state structure of the bicyclo[2.2.1]heptastannane-1,4-diide 1 (Fig. 1) the anionic bridgeheads Sn1 and Sn7 are separated by an intramolecular distance of 5.04 Å. The angles around Sn1 and Sn7 sum to 265.8° (Sn1) and 264.6° (Sn7), suggesting high p-orbital contribution in bonding orbitals at Sn1 and Sn7 and implying significant s-orbital character for the lone pair at these anionic tin atoms.
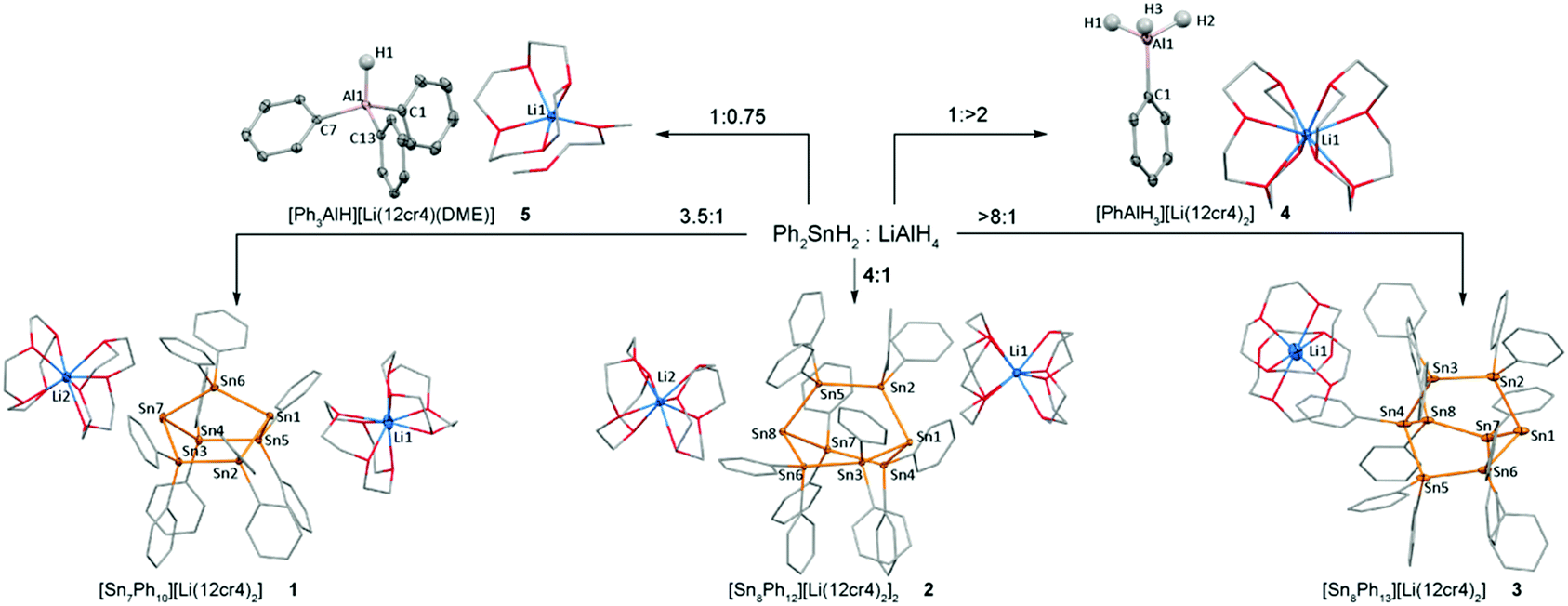 | ||
| Fig. 1 Formation of anionic oligotin cages 1–3 and phenyl aluminium hydrides 4 and 5. Metal atoms are drawn at 30% probability level, only hydrogen atoms attached to Al are shown, all other H atoms are omitted for clarity. For full geometric parameters, see ESI† pages S-7–S-9. Selected bond angles (°) 1: Sn2–Sn1–Sn5 94.37(2); Sn2–Sn1–Sn6 86.75(1); Sn5–Sn1–Sn6 84.67(1); Sn3–Sn7–Sn4 93.52(2); Sn3–Sn7–Sn6 85.41(1); Sn4–Sn7–Sn6 85.69(1). 2: Sn2–Sn1–Sn3 91.29(2); Sn2–Sn1–Sn4 91.41(2); Sn3–Sn1–Sn4 93.35(2); Sn5–Sn8–Sn6 90.95(2); Sn5–Sn8–Sn7 89.00(2); Sn6–Sn8–Sn7 94.13(2). 3: Sn2–Sn1–Sn6 96.92(8); Sn2–Sn1–Sn7 95.47(8); Sn6–Sn1–Sn7 95.30(8); Sn3–Sn4–Sn5 106.93(8); Sn3–Sn4–Sn8 105.22(8); Sn5–Sn4–Sn8 106.47(8). | ||
The bicyclo[2.2.2]octastanna-1,4-diide 2 (Fig. 1), features a less constrained geometry in the solid state with angles around the bridgehead atoms Sn1 and Sn8 just above 90° (sum of angles around Sn1 = 276.05° and Sn8 = 274.08°). The monoanion 3 displays two distinctly different environments for the bridgehead tin atoms with a highly pyramidalised anionic tin atom Sn1 (sum of angles 287.69°) and a more tetrahedral geometry at the phenyl substituted, formally neutral bridgehead Sn4 (sum of Sn–Sn–Sn angles 318.62°). The bridgehead atoms, Sn1 and Sn4, are separated by 5.27 Å in 3, which is ca. 0.67 Å less than in 2. In contrast to 1, compounds 2 and 3 are both twisted in the solid state (see ESI† Fig. S2.2, page S-7 and S2.3, page S-8). Compounds 4 and 5 (Fig. 1) containing discrete [PhAlH3]− and [Ph3AlH]− anions and charge separated [Li·(12-Cr-4)2]+ or [Li·(DME)(12-Cr-4)]+ counterions, respectively, display tetrahedral aluminium atoms with Al–C distances slightly greater than 2.0 Å.
The 119Sn{1H} NMR spectra of 1, 2 and 3 indicate that these cages persist in solution, as evidenced by chemical shifts and the observed 119Sn/117Sn couplings (Fig. 2, ESI† Section 4 pages S-14–S-26). 1 displays three resonances at 200.3 (Ph2Sn), 35.7 (Ph2SnSnPh2) and −857.3 (Sn−) ppm for which the coupling pattern is in full agreement with the bicyclo[2.2.1]heptastanna–1,3–diide framework in solution. 1J119Sn–119/117Sn coupling constants originating from the anionic bridgehead atoms are large with values of 4640/4430 Hz for the interaction with the monotin–bridge. In the case of coupling between the bridgehead tin atoms and the ditin bridges, values of 5990/5710 Hz for the 1J119Sn–119/117Sn coupling and 1130/1080 Hz for the 2J119Sn–119/117Sn coupling are observed. The relatively large 1J values compare to a smaller 1J119Sn–117Sn coupling constant of 3940 Hz for chemically equivalent tin atoms within the same ditin bridge.
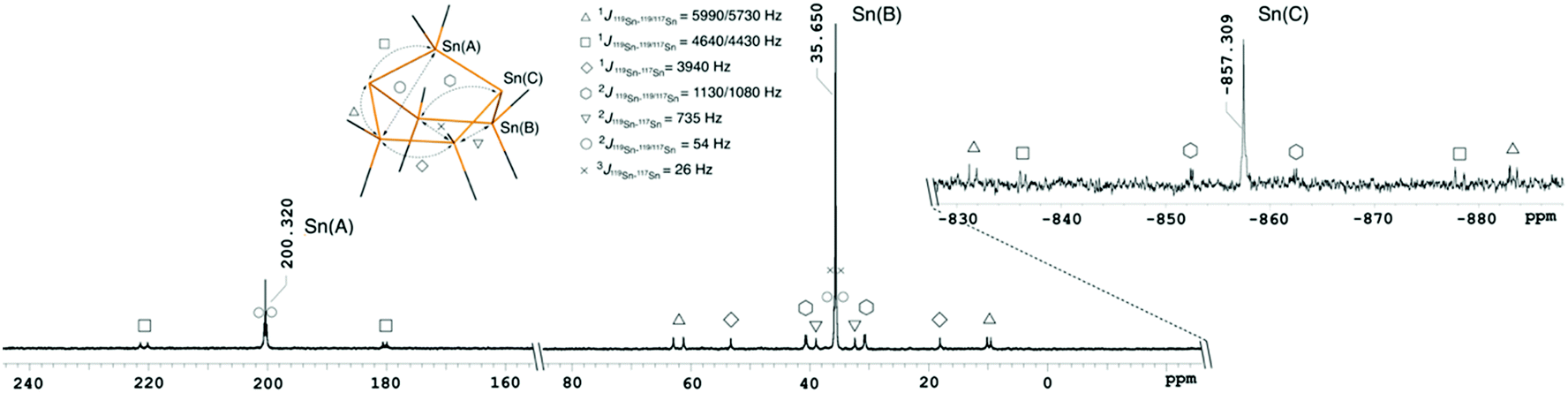 | ||
| Fig. 2 119Sn{1H} NMR spectrum of 1 in d3-acetonitrile showing Sn–Sn couplings indicative of the solution integrity of these compounds. | ||
Owing to its higher molecular symmetry, only two resonances are observed in the 119Sn{1H} NMR spectrum of 2, with peaks at −316.8 (Ph2SnSnPh2) and −585.0 (Sn−) ppm. The 1J and the 2J119Sn–119/117Sn coupling constants between the bridgehead tin atoms and the Ph2SnSnPh2 bridges are 5020/4800 Hz and 807/770 Hz, respectively. Again, a smaller 1J119Sn–117Sn coupling constant of 3610 Hz is observed for neighbouring diphenyltin fragments. The monoanion 3 displays four distinct signals in the 119Sn{1H} NMR spectrum at −183.1 (Ph2SnSn−), −238.6 (Ph2SnSnPh), −470.8 (SnPh) and −757.9 (Sn−) ppm. Notably, the coupling constant of the tricoordinate, anionic tin atom in 3 is larger (1J119Sn–119/117Sn = 6410/6090 Hz) in comparison to the signal of the neutral tin bridgehead, which is only 1150/1090 Hz.
Experimentally observed NMR chemical shifts were replicated by DFT calculations. To support the bonding model proposed based on the structural analysis of 1–3, the anions in these compounds were interrogated by DFT calculations employing different methods and basis sets. (For computational details and references see ESI† page S-84). The highest occupied molecular orbitals of 1–3 are shown in Fig. 3. These orbitals are in each case associated with the lone pairs on the anionic bridgehead tin atom(s) with contribution of the tin skeleton and show significant s-character.
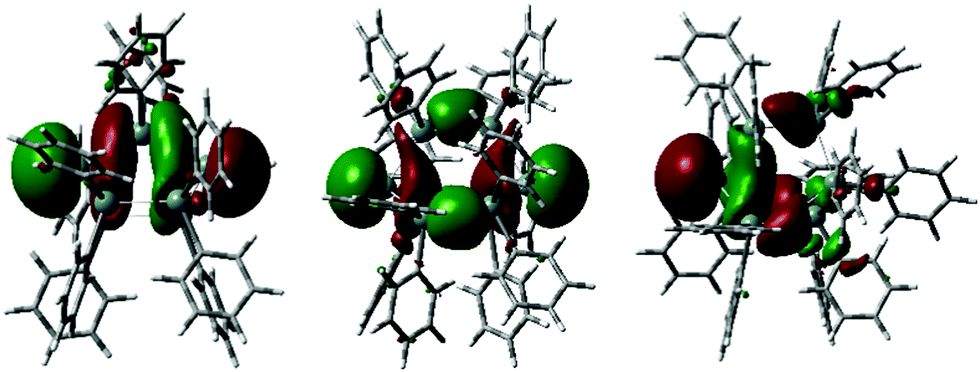 | ||
| Fig. 3 Highest occupied molecular orbitals of 1–3, for full computational details see ESI† Section 5, pages S-34–S-83. | ||
This supports the conclusion of significant p-orbital involvement in the Sn–Sn bonding of the bridgehead tin atoms inferred from their experimental solid state structures. The frontier orbitals of 1–3 (Fig. 3 and ESI† pages S-37–S-40) display distinct groupings into sets of orbitals with σ-, σ*- and π*-character interpreted as the onset of band-like behaviour. The HOMO to HOMO−8 (ΔEHOMO–HOMO−8 = 1.78 eV) for 2 and HOMO to HOMO−7 orbitals for 1 (ΔEHOMO–HOMO−7 = 2.03 eV) and 3 (ΔEHOMO–HOMO−7 = 1.66 eV) essentially represent the σ-bonded tin cores with only minor orbital contributions from the phenyl substituents. Within HOMO–LUMO energy gaps of only 2.33 eV (1), 2.15 eV (2) and 2.47 eV (3), the character of the frontier orbitals changes to phenyl based π* character.
The LUMO to LUMO+n orbitals (n = 20 in 1, n = 23 in 2, n = 25 in 3) are similar in orbital energy and all localised on the phenyl substituents. This manifold of π* orbitals is then followed by a set of orbitals with predominant σ* character of the tin framework, which span energy ranges of 0.78 eV (1), 0.81 eV (2) and 1.34 eV (3). These closely spaced MOs are reflected in the electronic absorptions displayed by 1–3. The UV-visible spectra of these species do not display distinct absorption maxima and are instead broad and tailing, suggestive of weak but extensive absorptions. Nevertheless, the visibly observed colours for crystals of 1 (orange-yellow), 2 (orange-red) and 3 (bright yellow) are consistent with the expected effects of cage size and charge upon σ-delocalisation24 inferred from the DFT calculations.
In summary, the reaction of Ph2SnH2 with LiAlH4 provides facile access to the charge-separated species, 1, 2 and 3, comprising an unprecedented set of structural motifs in anionic oligostannane cages. 1 and 2 constitute new dianionic covalent tin cages and we propose that they will provide convenient synthons in further transformations because of their charged nature and solution integrity. The electronic structure of 1–3 was interrogated computationally and support the presence of σ-delocalisation.
We wish to acknowledge the European COST Network for Smart Inorganic Polymers for providing funding to allow collaboration on this work. B. G. Steller gratefully acknowledges the Austrian Academy of Sciences for supporting this work with the DOC Fellowship. We thank the EPSRC (UK) for the support of a DTP studentship for A. S. S. Wilson and grant EP/R020752/1. Some DFT calculations were carried out using the Balena High Performance Computing (HPC) Service at the University of Bath.
Conflicts of interest
The authors declare no conflict of interest.Notes and references
- (a) S. C. Sevov and J. M. Goicoechea, Organometallics, 2006, 25, 5678–5692 CrossRef CAS; (b) T. F. Fässler, Coord. Chem. Rev., 2001, 215, 347–377 CrossRef.
- (a) A. Schnepf, Chem. Soc. Rev., 2007, 36, 745–758 RSC; (b) A. Schnepf, Struct. Bonding, 2016, 193–223 Search PubMed.
- (a) F. S. Kocak, D. O. Downing, P. Zavalij, Y. F. Lam, A. N. Vedernikov and B. Eichhorn, J. Am. Chem. Soc., 2012, 134, 9733–9740 CrossRef CAS PubMed; (b) F. S. Kocak, P. Y. Zavalij, Y.-F. Lam and B. W. Eichhorn, Chem. Commun., 2009, 4197–4199 RSC; (c) F. S. Kocak, D. O. Downing, P. Zavalij, Y. F. Lam, A. N. Vedernikov and B. Eichhorn, J. Am. Chem. Soc., 2012, 134, 9733–9740 CrossRef CAS PubMed; (d) D. J. Chapman and S. C. Sevov, Inorg. Chem., 2008, 47, 6009–6013 CrossRef CAS PubMed; (e) C. B. Benda, M. Waibel and T. F. Fässler, Angew. Chem., Int. Ed., 2015, 54, 522–526 CAS.
- (a) C. Schrenk, A. Kubas, K. Fink and A. Schnepf, Angew. Chem., Int. Ed., 2011, 50, 7273–7277 CrossRef CAS PubMed; (b) C. Schrenk, I. Schellenberg, R. Pöttgen and A. Schnepf, Dalton Trans., 2010, 39, 1872–1876 RSC; (c) C. Schrenk and A. Schnepf, Chem. Commun., 2010, 46, 6756–6758 RSC.
- (a) C. Schrenk, J. Helmlinger and A. Schnepf, Z. Anorg. Allg. Chem., 2012, 638, 589–593 CrossRef CAS; (b) C. Schrenk, B. Gerke, R. Pöttgen, A. Clayborne and A. Schnepf, Chem. – Eur. J., 2015, 21, 8222–8228 CrossRef CAS PubMed.
- C. Schrenk, F. Winter, R. Pöttgen and A. Schnepf, Inorg. Chem., 2012, 51, 8583–8588 CrossRef CAS PubMed.
- C. Schrenk, F. Winter, R. Pöttgen and A. Schnepf, Chem. – Eur. J., 2015, 21, 2992–2997 CrossRef CAS PubMed.
- J.-J. Maudrich, C. P. Sindlinger, F. S. W. Aicher, K. Eichele, H. Schubert and L. Wesemann, Chem. – Eur. J., 2017, 23, 2192–2200 CrossRef CAS PubMed.
- (a) E. Rivard, J. Steiner, J. C. Fettinger, J. R. Giuliani, M. P. Augustine and P. P. Power, Chem. Commun., 2007, 4919–4921 RSC; (b) G. Prabusankar, A. Kempter, C. Gemel, M. K. Schröter and R. A. Fischer, Angew. Chem., Int. Ed., 2008, 47, 7234–7237 CrossRef CAS PubMed; (c) B. E. Eichler and P. P. Power, Angew. Chem., Int. Ed., 2001, 40, 796–797 CrossRef CAS; (d) A. F. Richards, B. E. Eichler, M. Brynda, M. M. Olmstead and P. P. Power, Angew. Chem., Int. Ed., 2005, 44, 2546–2549 CrossRef CAS PubMed; (e) J. Wiederkehr, C. Wölper and S. Schulz, Chem. Commun., 2016, 52, 12282–12285 RSC; (f) M. Brynda, R. Herber, P. B. Hitchcock, M. F. Lappert, I. Nowik, P. P. Power, A. V. Protchenko, A. Růžička and J. Steiner, Angew. Chem., Int. Ed., 2006, 45, 4333–4337 CrossRef CAS PubMed; (g) C. P. Sindlinger, A. Stasch, H. F. Bettinger and L. Wesemann, Chem. Sci., 2015, 6, 4737–4751 RSC; (h) C. P. Sindlinger, W. Grahneis, F. S. W. Aicher and L. Wesemann, Chem. – Eur. J., 2016, 22, 7554–7566 CrossRef CAS PubMed; (i) C. P. Sindlinger, F. S. W. Aicher, H. Schubert and L. Wesemann, Angew. Chem., Int. Ed., 2017, 56, 2198–2202 CrossRef CAS PubMed; (j) L. R. Sita, Acc. Chem. Res., 1994, 27, 191–197 CrossRef CAS.
- (a) A. Schnepf and H. Schnöckel, Angew. Chem., Int. Ed., 2002, 41, 3532–3554 CrossRef CAS; (b) A. Schnepf and H. Schnöckel, Nanostructural Element Modifications: Synthesis and Structure of Elementoid Gallium Clusters, in ACS Symposium Series 822, Group 13 Chemistry, ed. P. J. Shapiro and D. A. Atwood, American Chemical Society, Washington DC, 2002 Search PubMed.
- (a) L. R. Sita and R. D. Bickerstaff, J. Am. Chem. Soc., 1989, 111, 6454–6456 CrossRef CAS; (b) C. Drost, M. Hildebrand and P. Lönnecke, Main Group Met. Chem., 2002, 25, 93–98 CAS.
- L. R. Sita and I. Kinoshita, J. Am. Chem. Soc., 1992, 114, 7024–7029 CrossRef CAS.
- C. P. Sindlinger and L. Wesemann, Chem. Sci., 2014, 5, 2739–2746 RSC.
- (a) L. R. Sita and I. Kinoshita, J. Am. Chem. Soc., 1991, 113, 5070–5072 CrossRef CAS; (b) L. R. Sita and I. Kinoshita, J. Am. Chem. Soc., 1990, 112, 8839–8843 CrossRef CAS; (c) D. Nied, E. Matern, H. Berberich, M. Neumaier and F. Breher, Organometallics, 2010, 19, 6018–6037 Search PubMed; (d) P. Vasko, S. Wang, H. M. Tuononen and P. P. Power, Angew. Chem., Int. Ed., 2015, 54, 3802–3805 CrossRef CAS PubMed; (e) D. Nied and F. Breher, Chem. Soc. Rev., 2011, 40, 3455–3466 RSC.
- C. Marschner and J. Hlina, Catenated Compounds – Group 14 (Ge, Sn, Pb), in Comprehensive Inorganic Chemistry II, ed. J. Reedijk and K. Poeppelmeier, Elsevier, Oxford, 2013, vol. 1 Search PubMed.
- (a) S. Masamune and L. R. Sita, J. Am. Chem. Soc., 1985, 107, 6390–6391 CrossRef CAS; (b) L. R. Sita and I. Kinoshita, J. Am. Chem. Soc., 1991, 113, 1856–1857 CrossRef CAS; (c) N. Wiberg, H.-W. Lerner, H. Nöth and W. Ponikwar, Angew. Chem., Int. Ed., 1999, 38, 1103–1105 CrossRef CAS.
- (a) L. R. Sita and I. Kinoshita, Organometallics, 1990, 9, 2865–2867 CrossRef CAS; (b) N. Wiberg, H. W. Lerner, S. Wagner, H. Nöth and T. Seifert, Z. Naturforsch. B, 1999, 54, 877–880 CAS.
- M. Wagner, M. Lutter, B. Zobel, W. Hiller, M. H. Prosenc and K. Jurkschat, Chem. Commun., 2015, 51, 153–156 RSC.
- (a) T. Imori, V. Lu, H. Cai and T. D. Tilley, J. Am. Chem. Soc., 1995, 117, 9931–9940 CrossRef CAS; (b) P. Braunstein, J. Durand, X. Morise, A. Tiripicchio and F. Ugozzoli, Organometallics, 2000, 19, 444–450 CrossRef CAS; (c) P. Braunstein and X. Morise, Chem. Rev., 2000, 100, 3541–3552 CrossRef CAS PubMed; (d) N. R. Neale and T. D. Tilley, J. Am. Chem. Soc., 2002, 124, 3802–3803 CrossRef CAS PubMed; (e) F. Choffat, P. Smith and W. Caseri, J. Mater. Chem., 2005, 15, 1789–1792 RSC; (f) F. Choffat, S. Käser, P. Wolfer, D. Schmid, R. Mezzenga, P. Smith and W. Caseri, Macromolecules, 2007, 40, 7878–7889 CrossRef CAS.
- M. Trummer and W. Caseri, Organometallics, 2010, 29, 3862–3867 CrossRef CAS.
- (a) A. E. Finholt, A. C. Bond, Jr., K. E. Wilzbach and H. I. Schlesinger, J. Am. Chem. Soc., 1947, 69, 2692–2696 CrossRef CAS; (b) N. Braia, Y. Saihi, A. E. Azzouzi and F. Ferkous, Asian J. Chem., 2009, 21, 4628–4634 CAS.
- The use of analytically pure LiAlH4 (recryst. from diethyl ether) is crucial for the successful control of the stoichiometry.
- The anionic part in 3 was well resolved and is fully consistent with NMR data. The X–ray structure determination of the cationic part and residual solvent molecules, however, was hampered by twinning and disorder. See ESI† pages S-4 and S-5.
- (a) M. S. Hill, Homocatenation of Metal and Metalloid Main Group Elements, in Metal-Metal Bonding: Structure and Bonding, ed. G. Parkin, Springer, Berlin, 2010, vol. 136 Search PubMed; (b) M. Jovanovic, D. Antic, D. Rooklin, A. Bande and J. Michl, Chem. – Asian J., 2017, 12, 1250–1263 CrossRef CAS PubMed; (c) M. Jovanovic and J. Michl, J. Am. Chem. Soc., 2018, 140, 11158–11160 CrossRef CAS PubMed; (d) M. Jovanovic and J. Michl, J. Am. Chem. Soc., 2019, 141, 13101–13113 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, spectroscopic data, refinement details for single crystal X-ray crystallography. CCDC 1561478–1561482. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc07976a |
| This journal is © The Royal Society of Chemistry 2020 |