
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aziridines and azetidines: building blocks for polyamines by anionic and cationic ring-opening polymerization
Tassilo
Gleede
a,
Louis
Reisman
 b,
Elisabeth
Rieger
a,
Pierre Canisius
Mbarushimana
b,
Paul A.
Rupar
*b and
Frederik R.
Wurm
*a
b,
Elisabeth
Rieger
a,
Pierre Canisius
Mbarushimana
b,
Paul A.
Rupar
*b and
Frederik R.
Wurm
*a
aMax-Planck-Institut für Polymerforschung, Ackermannweg 10, 55128 Mainz, Germany. E-mail: wurm@mpip-mainz.mpg.de
bDepartment of Chemistry and Biochemistry, The University of Alabama, Tuscaloosa, Alabama 35487-0336, USA. E-mail: parupar@ua.edu
First published on 13th May 2019
Abstract
Despite the difficulties associated with controlling the polymerization of ring-strained nitrogen containing monomers, the resulting polymers have many important applications, such as antibacterial and antimicrobial coatings, CO2 adsorption, chelation and materials templating, and non-viral gene transfection. This review highlights the recent advances on the polymerizations of aziridine and azetidine. It provides an overview of the different routes to produce polyamines, from aziridine and azetidine, with various structures (i.e. branched vs. linear) and degrees of control. We summarize monomer preparation for cationic, anionic and other polymerization mechanisms. This comprehensive review on the polymerization of aziridine and azetidine monomers will provide a basis for the development of future macromolecular architectures using these relatively exotic monomers.
 Tassilo Gleede | Tassilo Gleede studied Chemistry at the Johannes Gutenberg-Universität Mainz (Germany) and received his diploma degree in 2015 including a research stay at the Brookhaven National Laboratory, (New York, USA) developing positron emission radiotracers for human and plant studies in the group of Prof. Joanna Fowler. He joined the Max Planck Institute for Polymer research, Mainz, in the group of Dr F. R. Wurm for his PhD thesis. His research, supported by the Deutsche Forschungsgesellschaft (DFG), focuses on the living anionic polymerization of activated aziridines and their copolymerization behavior with other reactive monomers such as ethylene oxide. |
 Louis Reisman | Louis “Chip” Reisman III graduated with a B.S. in Chemistry from Mercer University in 2014, where he conducted research with Prof. Adam Kiefer and Prof. Caryn Seney. He recently completed his Ph.D. at the University of Alabama under the supervision of Prof. Paul Rupar investigating the anionic ring-opening polymerizations of sulfonyl-activated aziridines and azetidines. Chip will be continuing his career in chemistry by studying sustainable polymers as a postdoctoral researcher in the lab of Prof. Marc Hillmyer at the University of Minnesota. |
 Elisabeth Rieger | Elisabeth Rieger was born in 1985 in Rosenheim (Germany). She studied biomedical chemistry at the Johannes Gutenberg – Universität Mainz (Germany), including a stay at the Brookhaven National Laboratory on Long Island (USA) in the group of Prof. Joanna Fowler. She finished her PhD in the group of Dr F. R. Wurm at the Max Planck Institute for Polymer Research in Mainz in 2018. Her research focused on the sequence controlled anionic polymerization of activated aziridines monitored by real-time NMR. |
 Pierre Canisius Mbarushimana | Pierre Canisius Mbarushimana studied chemistry ay Lyon College (US) and graduated with a B.Sc. degree in 2013. After that, he pursued graduate studies in analytical chemistry at the University of Alabama and completed his PhD in the Rupar group in 2018. His research interests consist of the anionic ring-opening polymerization of activated aziridines. Currently, he holds a senior scientist position at Jordi Labs in Massachusetts. |
 Paul A. Rupar | Paul A. Rupar completed his PhD in 2009 under the supervision of Prof. Kim Baines at the University of Western Ontario. In 2010, Paul started as a NSERC Postdoctoral Researcher at the University of Bristol in the group of Prof. Ian Manners where he investigated the self-assembly of polymer nanoparticles. In 2011, Paul was a Marie Curie Postdoctoral researcher, also in the Manners group. Paul is currently an Assistant Professor in the Department of Chemistry & Biochemistry at the University of Alabama. His current research interests span from conjugated inorganic systems to the anionic polymerization of aziridines and azetidines. |
 Frederik R. Wurm | Frederik R. Wurm received his PhD in 2009 from the Johannes Gutenberg-Universität Mainz (Germany) working on nonlinear block copolymers. From 2009 to 2011 he was a postdoctoral Humboldt fellow at EPFL (Switzerland) focusing on novel bioconjugation strategies. In 2012, he joined the Max Planck Institute for Polymer Research (Germany). Frederik has published over 150 research articles and received several awards, including the Polymer Chemistry Lectureship. Frederik leads the research group “Functional Polymers” and develops new degradable and molecularly adjustable polymers. He is particularly interested in biodegradable polyphosphoesters. Additionally, his research covers areas of biopolymer modification and sequence-controlled polymerization, focusing on living anionic polymerization. |
1. Introduction
Despite being structural anlogs with comparable ring strain (Fig. 1), aziridine and oxirane have very different polymerization chemistries. Oxirane can be polymerized via a variety of mechanisms, including cationic ring-opening polymerization (CROP)1 and anionic ring-opening polymerization (AROP)2,3 to form linear poly(ethylene oxide) (PEO) with high degrees of control. In contrast, aziridine can only polymerize via CROP to produce (hyper)branched polyethylenimine (hbPEI) with little control over molecular weight, dispersity, and microstructure.4–8 The situation is similar with azetidine (Fig. 1), the nitrogen analog of oxetane, which also only forms hyperbranched poly(trimethylenimine) (hbPTMI) via CROP.9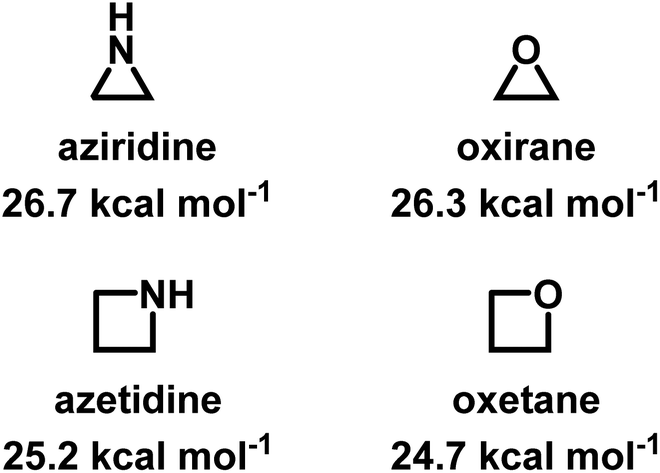 | ||
| Fig. 1 Chemical structures of aziridine, azetidine, oxirane, and oxetane and their ring-strains.10,11 | ||
Even with the challenges in controlling its polymerization, the high amine density of hbPEI lends its use in a wide range of applications including non-viral gene-transfection,7,12–44 anti-microbial and anti-viral coatings,37,45–52 CO2 capture,53–63 flocculation of negatively charged fibers in paper-making industries,64–66 metal chelation in waste water treatments,67 as additives for inkjet paper production,16 as electron injection layers in organic light-emitting diodes,68,69 and materials templating.70 As such, hbPEI is made industrially, initially under the commercial name Polymin, and today is marketed under the trade name Lupasol® by BASF.71 Aziridine is produced at a rate of ∼9000 t/a (2006),72 where, due to its toxicity, it is usually converted directly into its nontoxic intermediates and branched polymers.
The lack of control over aziridine polymerizations has significantly limited the research of linear PEI (LPEI), especially when compared to PEO and related polymers.6,73 Polyethers, especially PEO, are produced on the scale of several million tons per year and found in many everyday applications,74 while linear polyaziridines are barely used today (Fig. 2).75 This is unfortunate as the potential structural diversity of aziridines is notably greater than oxygen-containing analogs as substitution on aziridines can occur both at the carbon and nitrogen atoms of aziridine.
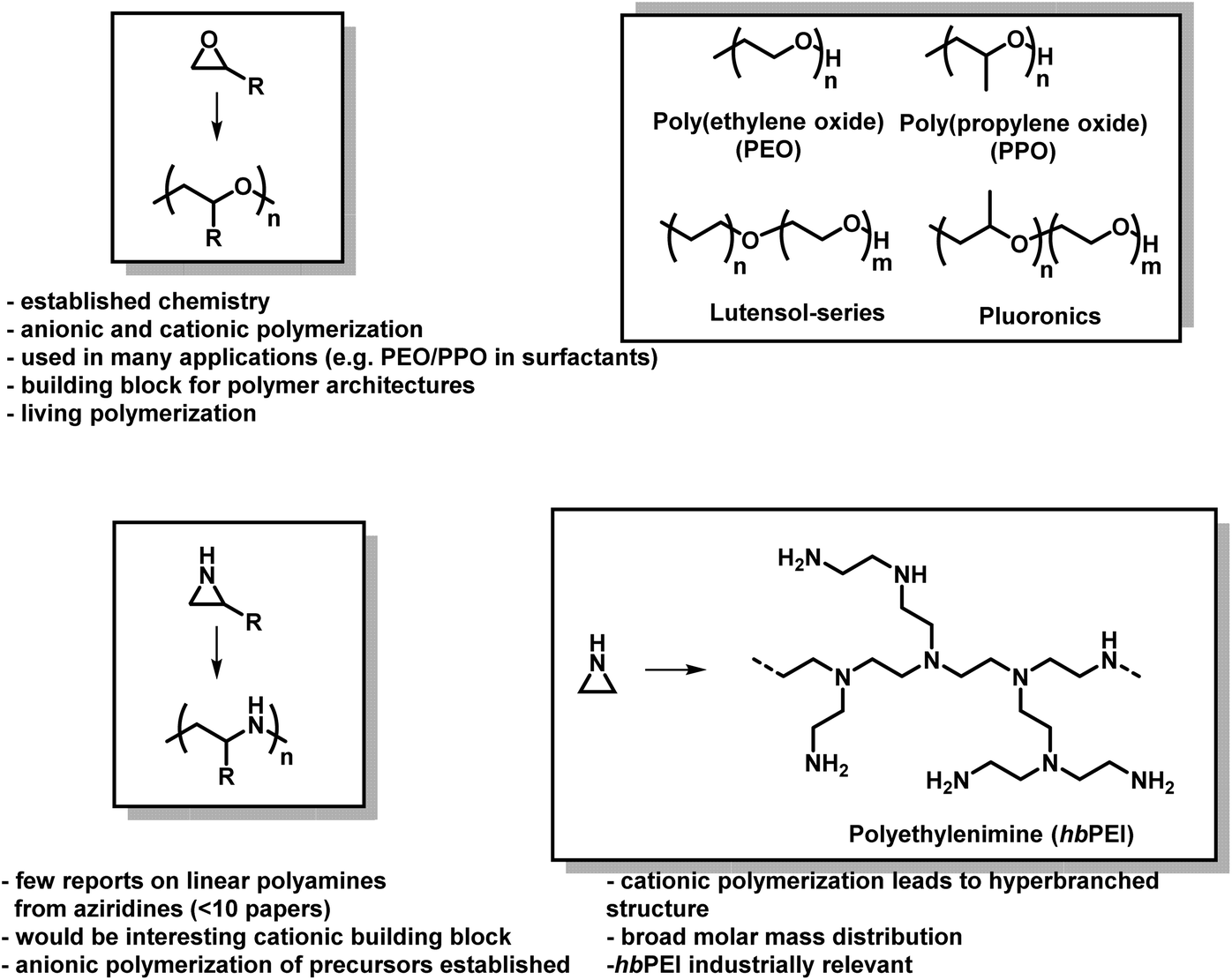 | ||
| Fig. 2 Epoxides vs. aziridines. General polymerization scheme and important facts about both material classes. | ||
Use in non-viral gene-transfection has sparked renewed academic and pharmaceutical interest in PEI, especially LPEI. LPEI is attractive compared to hbPEI as it can have a well-defined architecture with narrow molecular weight distributions. This makes it ideal for structure–property relationship studies and its incorporation into polymer–drug conjugates. However, since LPEI cannot be made from aziridine, it is instead synthesized indirectly via polyoxazolines.14 Typically, 2-oxazoline-based monomers, such as 2-ethyl-2-oxazoline, in the presence of cationic initiators, such as stannic chloride and boron trifluoride etherate, undergo a controlled CROP.76–78 Conversion of poly(2-oxazoline)s to LPEI can occur under acidic or alkaline conditions (Scheme 1).79–81 However, this route to LPEI has drawbacks in that the polymerization is difficult to control when targeting high degrees of polymerization and it is challenging to achieve quantitative removal of the acyl groups.7
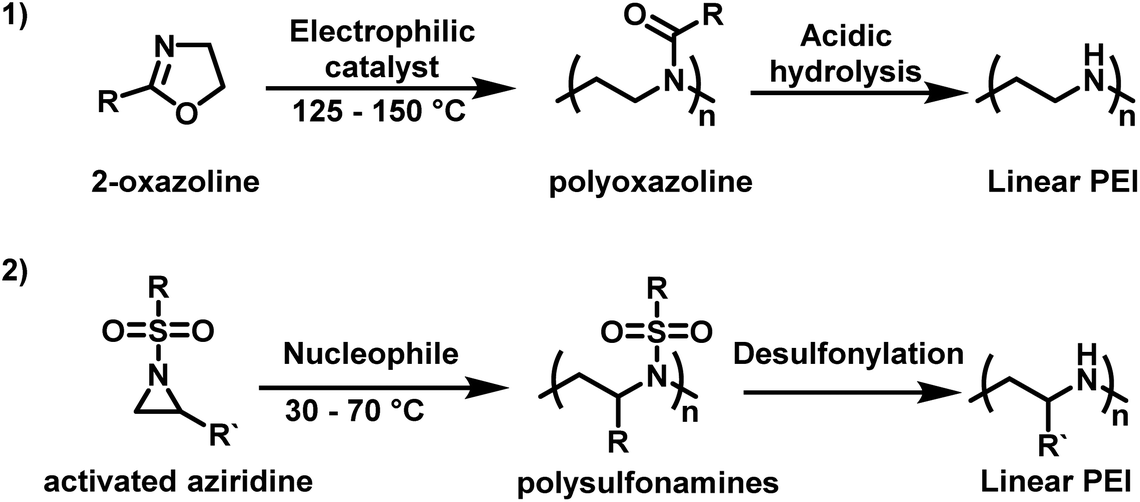 | ||
| Scheme 1 (1) Synthesis of linear polyethylenimine (LPEI) from 2-oxazoline by cationic ring-opening polymerization in comparison to (2) anionic ring-opening polymerization of sulfonyl aziridines as an alternative pathway to linear PEI derivatives.82,83 | ||
The new appreciation for the applications of LPEI and unresolved challenges associated with azirdine polymerization means that it remains an active area of research. Since there have been no recent reviews on aziridine polymerizations (Kobayashi6 was published in 1990), we feel that such a review is warranted. This is especially true given the recent breakthroughs in the AROP of activated aziridines, which have yet to be summarized in the literature.
This review gives a comprehensive overview on the synthesis of polymers based on both aziridines and azetidines. We have chosen to include azetidines in this review as their polymerization chemistry closely mirrors that of aziridines and the resulting polymers are structurally similar to that of aziridine derived polymers. We first discuss the early literature on the CROP of aziridines and azetidines. Next, we highlight recent work on the AROP and organocatalytic ring-opening polymerization (OROP) of N-sulfonyl aziridines and azetidines and strategies to produce linear polyamines from these monomers. We conclude with routes to aziridine and azetidine copolymers and highlight the possibilities for functionalization or preparation of various polyamine structures. Spaced through the review are summaries of the synthetic methods to synthesize the aziridine and azetidine monomers. Detailed analysis of the literature on gene-transfection can be found elsewhere.84 This review will also not cover oxazoline polymerization chemistry; the interested reader is encouraged to consult recent reviews.85–87
2. Cationic ring-opening polymerization of aziridines and azetidines
The differences in polymerization chemistry between nitrogen and oxygen containing strained heterocycles arise due to the reduced electronegativity of nitrogen, the presence of an acidic hydrogen atom on the nitrogen atom, and the increased nucleophilicity of the lone-pair of electrons on nitrogen. The decreased electronegativity of nitrogen prevents the nucleophilic ring-opening of aziridine and azetidine in AROP and increases the nucleophilicity of the lone pair electrons on nitrogen, leading to branching and loss of control in CROP. In addition, use of very basic nucleophiles simply deprotonates the secondary amine in aziridine and azetidine rather than inducing ring-opening. Therefore, the majority of aziridine and azetidine polymerizations proceed through a cationic mechanism.2.1 Cationic ring-opening polymerization of aziridine
The cationic polymerization of aziridine and related cyclic amines were recorded in the patent literature as early as 1937.88 However, the nature of the polymerization and the structure of the resulting polymer was first thoroughly explored by Jones and coworkers in 1944.89 Numerous studies have been performed, elaborating on this work, resulting in the mechanism, depicted in Scheme 2.5 Polymerization is initiated by electrophilic addition of an acidic catalyst to aziridine to form an aziridinium cation. An additional aziridine monomer, acting as a nucleophile, ring opens the active aziridinium ion resulting in the formation of a primary amine and a new aziridinium moiety. Subsequent aziridines attack the propagating aziridinium terminus, resulting in the linear propagation of the polymer chain. However, as the secondary amine groups in the developing polymer chain are also nucleophilic, they also ring open aziridinium species leading to branching and results in hbPEI. For hbPEI synthesized in solution, this leads to a ratio of primary![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :secondary:tertiary amines of 1:2:1 (determined by 13C NMR).90 This ratio can be varied depending upon the reaction conditions and the molar mass of the polymer (i.e. lower molar mass hbPEI has more primary amines).90
:secondary:tertiary amines of 1:2:1 (determined by 13C NMR).90 This ratio can be varied depending upon the reaction conditions and the molar mass of the polymer (i.e. lower molar mass hbPEI has more primary amines).90
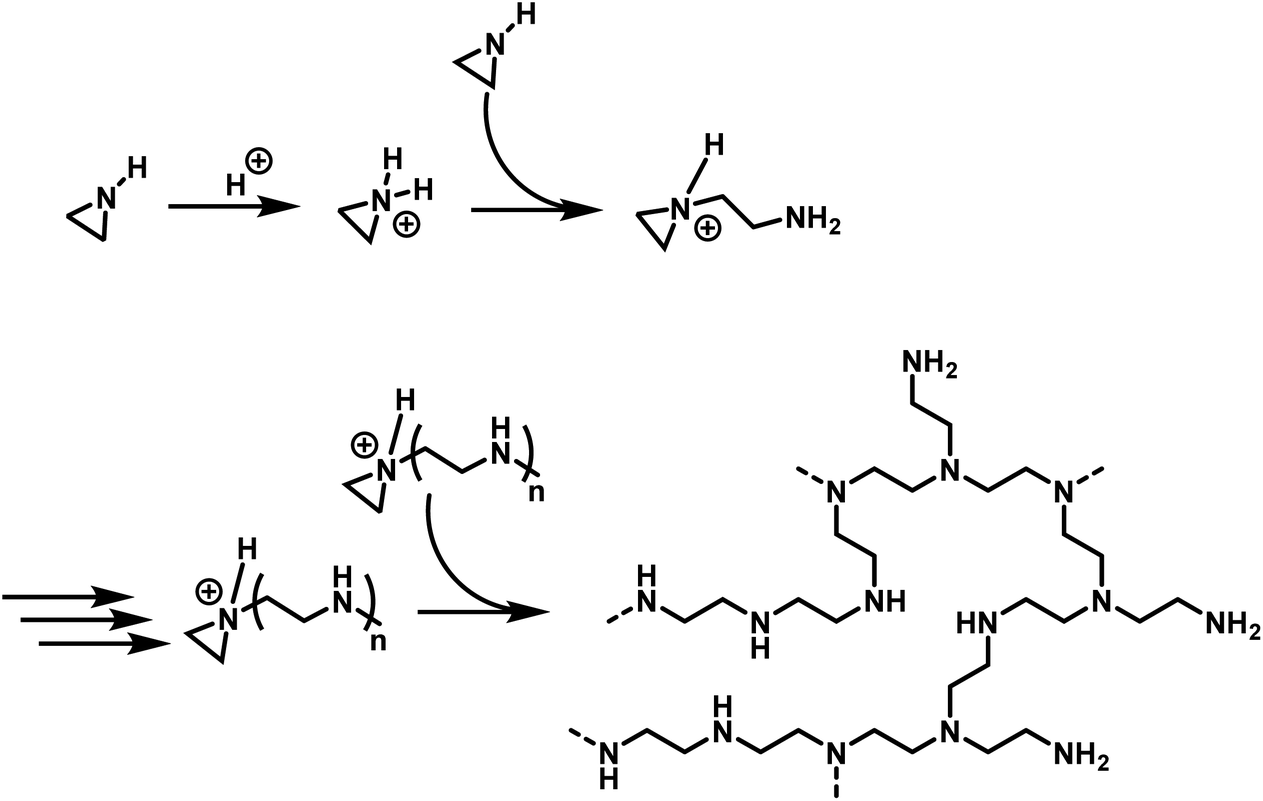 | ||
| Scheme 2 Mechanism of the cationic ring-opening polymerization of aziridine, leading to hbPEI. | ||
The polymerization of aziridine is likely more complex than that depicted in Scheme 2. Barb and coworkers91 studied the kinetics and mechanism of the polymerization of ethylenimine in the presence of different catalysts, such as p-toluenesulfonic acid, benzoic acid and others. They noted that the polymerization proceeds much like a step-growth polymerization, and that aziridine dimer is the dominate species early in the polymerization. A recent study on the polymerization of azetidine reached similar conclusion (cf. section 2.5).8 Furthermore, they noted an increase in molar masses in polymerization mixtures containing no monomeric species. This suggests that both monomer and polymer molecules are capable of activation and deactivation, thus making it more difficult to control the molecular weight and architecture of the resulting hbPEI.
2.2 Cationic ring-opening polymerization of 2-substituted aziridines
In general, the CROP of 2-substituted aziridines proceeds similarly to the parent aziridine. 2-Methylaziridine, or propylene imine, was reported to undergo polymerization initiated by BF3Et2O.92 The resulting polypropylenimine appeared as a viscous oil insoluble in water but soluble in CHCl3 and DMSO. The structure of polypropylenimine formed from the cationic polymerization of 2-methylaziridine has been determined to be highly branched, similarly to hbPEI formed from the CROP of aziridine. A photoinitiated cationic polymerization of 2-methylaziridine has also been reported.93 Regardless of the initiator used, the polymerization of 2-methylaziridine suffers from termination. This is due to nucleophilic addition of a tertiary amine within the polymer to an aziridinium, resulting in the formation of an unreactive quaternary amine.CROP of 2-phenylaziridine, initiated by methyl triflate, perchloric acid, BF3Et2O, dimethyl sulfate or hydrochloric acid, was found to form only low molecular weight polymers of ≤3000 g mol−1.94 These polymerizations did not result in full monomer consumption due to high rates of termination. Although the polymerization occurred at a much slower rate, Bakloutl and coworkers found that employing methyl triflate as initiator led to the formation of the highest molecular weight polymers of 2500–3000 g mol−1. Further investigation by studying the kinetics revealed this was due to an increase in the ratio of the propagation rate constant to the termination rate constant. It was proposed that this was due to the triflate counter ion stabilizing the aziridinium ion better than the counter ions of the other initiators.
2.3 Monomer preparation for cationic polymerization of 2-substituted aziridines
A pioneering approach to aziridines is the “β-chloroethylamine process”, which uses vicinal chloro amine hydrochloride salts and sodium hydroxide. As corrosive hydrochloric acid (HCl) is released during the reaction, which could also lead to a CROP of aziridine, this process lost industrial relevance in 1963 (Scheme 3i).72 The “Dow Process” (Scheme 3ii) was used on the industrial scale beginning in 1978, but was stopped due to the drawbacks of high corrosion rates to reactors and waste stream disposal. Per one equivalent starting material, three equivalents of ammonia are necessary, which gives the Dow Process a low atom economy.72 Today, aziridine is prepared via the “Wenker synthesis” (Scheme 3iii). This two-step process starts with the reaction of 2-aminoethanol, or other vicinal amino alcohols, with sulfuric or chlorosulfuric acid. The sulfates are treated with 5 eq. sodium hydroxide or saturated sodium carbonate solution to give the aziridine after a nucleophilic cyclization with 2-step yields of ∼90%.72 Significant waste disposal problems are avoided through high atom economy and nontoxic side products.95 For laboratory scales, sulfates can be purified by filtration and washing with excess ether. Low molecular weight aziridines such as 2-methyl aziridine can be further purified by steam distillation. If stored longer, alkali hydroxide stabilizes aziridine against spontaneous cationic polymerization. N-Substituted aziridines can also be obtained if secondary amines are used as starting material.95–97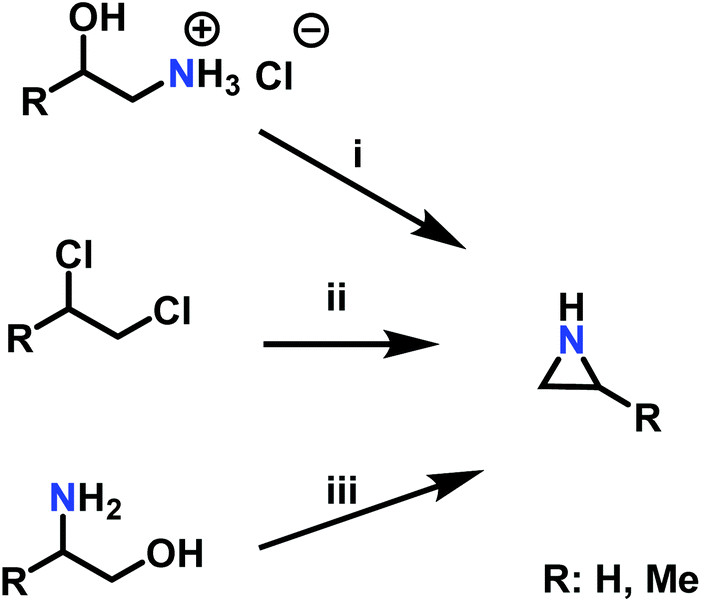 | ||
| Scheme 3 Synthesis of aziridines with and without a ring substituent (R) by i: the β-chloroethylamine process, ii: the Dow Process, iii: the Wenker synthesis. | ||
2.4 The cationic ring-opening polymerization of N-substituted aziridines
The ring-opening polymerization of N-substituted aziridines typically proceeds via a cationic mechanism. The details of the polymerization of N-substituted aziridines is like that of aziridine, with one important distinction: the possibility of an irreversible termination reaction.5 Termination occurs due to the (inter and intramolecular) nucleophilic attack of tertiary amines on aziridinium moieties, which results in the formation of unreactive, non-strained quaternary ammonium salts (Scheme 4).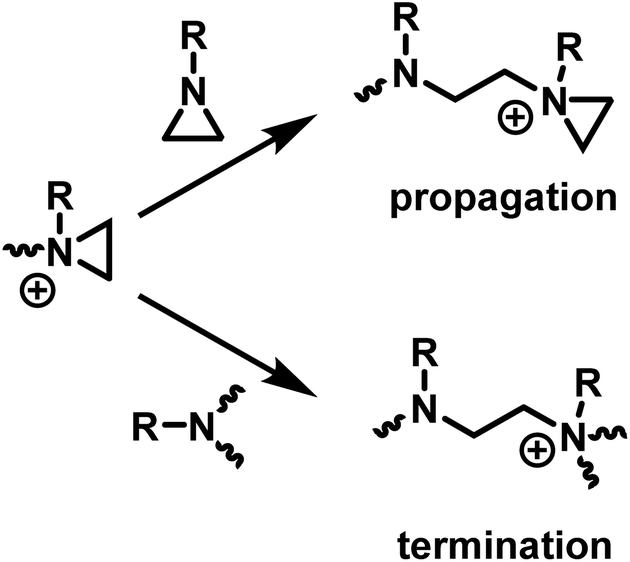 | ||
| Scheme 4 Competition between propagation and termination reactions during the CROP of N-substituted aziridines. | ||
As such, the polymerizations of simple N-substituted aziridines, often proceed only to low conversions (<55%).5 This is exemplified by the fact that in acetone the reaction of allyl bromide with 9-fold excess of N-(n-butyl)aziridine produces the piperazinium cation in a 96% yield with excess aziridine being recovered (Scheme 5).98
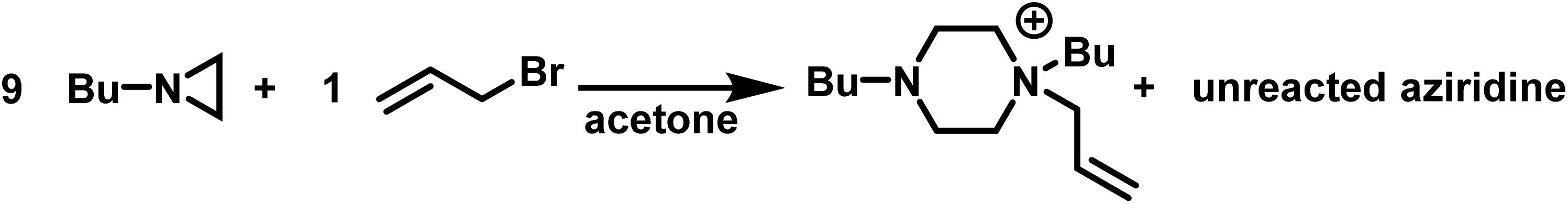 | ||
| Scheme 5 Reaction of N-(n-butyl)aziridine to form a piperazinium with no detectable polymerization. | ||
Goethals performed a series of detailed studies on the polymerization of N-alkylaziridines primarily focusing on the kinetics of the polymerization.5 A key focus of this study was to measure the propagation rate constants (kp) vs. the rates of termination (kt). Employing Et3OBF4 as a Lewis acid, due to its rapid initiation, Goethals found that alkyl substituents with low steric bulk tended to have low kp/kt ratios, which leads to the polymerizations proceeding to only low conversion. For instance, N-ethylaziridine has a kp/kt of 6 while the kp/kt for N-isopropylaziridine was 21. In contrast, N-tertbutylaziridine polymerized with essentially no termination (kp/kt ≈ ∞) and no transfer reactions, thus allowing the polymerization to be living-like. The introduction of a methyl group in the 2-position of N-alkylaziridines was found to greatly reduce the rate of termination relative to propagation. For example, the polymerization of N-benzyl aziridine stops at very low conversion due to termination (kp/kt = 85), while N-benzyl-2-methylaziridine polymerizes with almost no termination (kp/kt = 1100) (Fig. 3).
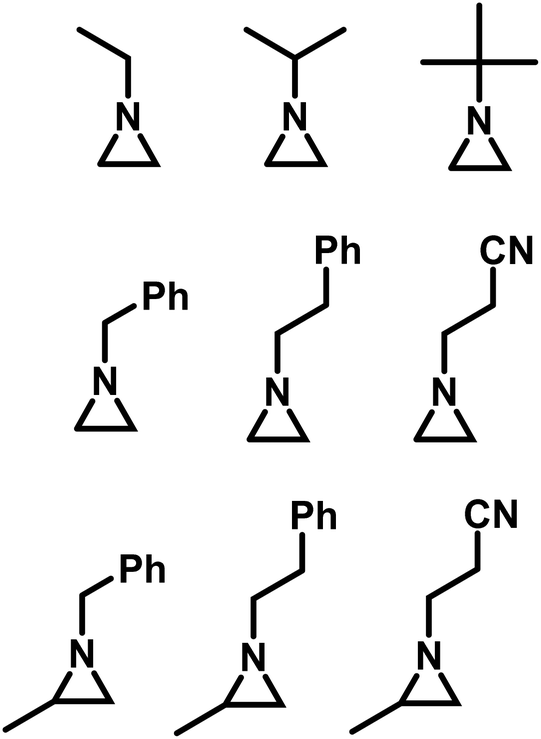 | ||
| Fig. 3 N-Alkyl aziridines that undergo CROP.5 | ||
While substitution in the 2-position greatly increases the kp/kt ratio, geminal substitution at the 2-position completely inhibits the polymerization. Specifically, N-benzyl-2,2-dimethylaziridine was only found to form N-benzyl-N-ethyl-2,2-dimethylaziridinium when reacted with Et3OBF4.5 Interestingly, N-benzyl-N-ethyl-2,2-dimethylaziridinium could initiate the polymerization of N-benzylaziridine (Scheme 6).
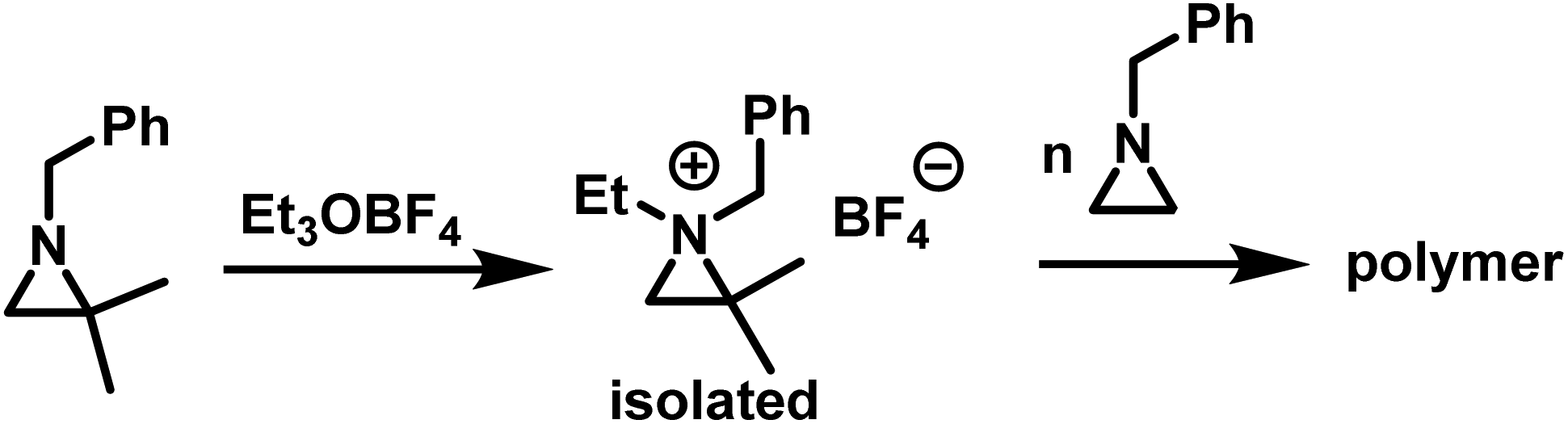 | ||
| Scheme 6 Formation of N-benzyl-N-ethyl-2,2-dimethylaziridinium and polymerization according to ref. 5. | ||
Goethals also reported on the CROP of neat N-(2-tetrahydropyranyl)aziridine initiated by Lewis acids (Scheme 7).99 This polymerization also appears to proceed without termination due to the bulky tetrahydropyranyl substituent. Hydrolysis of the polymer produced from N-(2-tetrahydropyranyl)aziridine in dilute HCl, followed by neutralization with NaOH, resulted in the formation of high molecular weight linear PEI (LPEI). Mw for the LPEI, as determined by LALLS, was as high as 19.6 kg mol−1, which is the highest molecular weight LPEI produced from an aziridine to date.
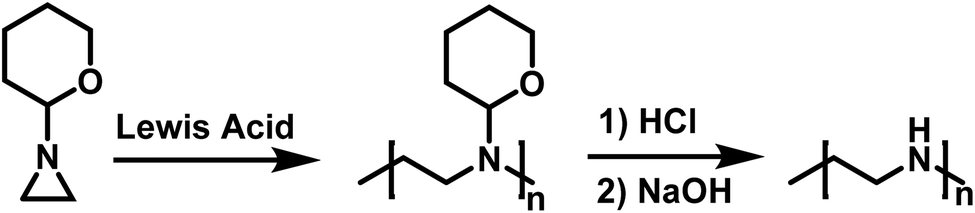 | ||
| Scheme 7 Polymerization of N-(2-tetrahydropyranyl)aziridine in route to LPEI according to ref. 99. | ||
Polymerization of N-(2-hydroxyethyl)aziridine has also been reported to occur via traditional Lewis acid catalysts and also through electroinitated polymerization.100 The resulting poly(N-(2-hydroxyethyl)aziridine) (PHEA) has been shown to be an excellent chelator of metal cations. By simple adjustment of pH, PHEA can selectively remove various metals.101 At pH = 3, PHEA can remove Cu(II) from solution with as high as 99.5% retention. With neutral pH, Co(II), Cr(III), Fe(III), Ni(II), Zn(II), and Cd(II) can be removed from solution at as high as 99.5%. These results are similar to those of hbPEI, with the exception of Cr(III) at neutral pH, when PHEA is a much stronger chelator.
CROP of aziridines have also been employed in the synthesis of copolymers. Utilizing N-(2-hydroxyethyl)aziridine, Pooley and coworkers synthesized a copolymer with 1,2,3,6-tetrahydropthalic anhydride (THPhA) (Scheme 8).102 This polymerization was accomplished in the absence of an initiator. These polymers are formed by employing a nucleophilic monomer with an electrophilic comonomer. These monomers form a zwitterion which leads to initiation and propagation in the polymerization. Pooley extended this work with N-(2-hydroxyethyl)aziridine to produce copolymers with a library of other electrophilic monomers.103–109
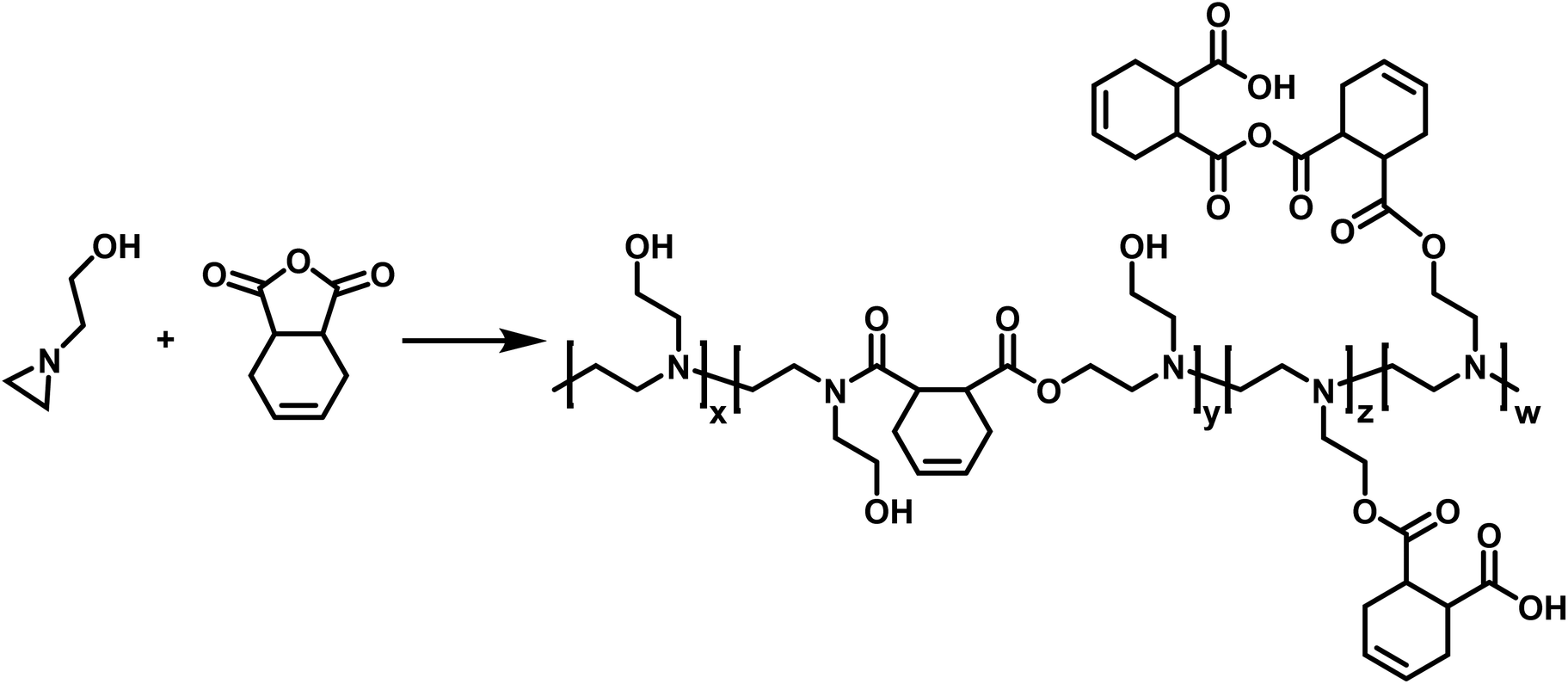 | ||
| Scheme 8 Copolymerization of N-(2-hydroxyethyl)aziridine with THPhA according to ref. 102. | ||
Although not present in the open literature, there are reports in the patent literature on the CROP of sulfonylaziridines.110 These polymerizations were performed neat by melting the monomers and were performed in the presence of Lewis acids such as AlCl3, FeCl3, and ZnCl2. Employing different monomer:catalyst feeds from 200:1 to 10000:1 polymers were obtained, but no further characterization details were given.110
2.5 Cationic ring-opening polymerization of azetidines
While the polymerization of aziridines has been studied, there are significantly fewer examples of the four-membered ring, azetidine, in the literature. Unsurprisingly, most of this work on the polymerization of azetidines focuses on the polymerization of N-substituted azetidines.4,111–115 While the first polymerization of an azetidine ring was reported by Kornfeld, in 1960, regarding the polymerization of conidine,113 Goethals provided the greatest contributions to the field due to his studies of multiple azetidines.4,5,99,115–118In 1974, Goethals reported of the polymerization of unsubstituted azetidine (Scheme 9).4 The polymerization proceeded via a cationic mechanism, similar to the CROP of aziridine, to form hyperbranched poly(trimethylenimine) (hbPTMI).5,6 This study found that after 8 h at 70 °C in methanol nearly all monomer had been consumed. Interestingly, it was found that when all the monomer had been consumed, 70% of the reaction mixture consisted of dimer. This is explained by the pKB difference of azetidine and N-methylazetidine. The pKB of azetidine is 11.29 (ref. 115) and 10.40 (ref. 119) for N-methylazetidine. Due to the differences in basicity, and the similarity in structure of the azetidine dimer to N-methylazetidine, it is expected that a proton would transfer from the protonated tertiary amine to the more basic monomer. Because of preferential formation of dimer to propagation it was hypothesized that the resulting polymer should contain many primary and tertiary amines, rather than exclusively producing secondary amines. This can be explained by two possible reaction pathways. Propagation only occurs once the tertiary amine in the dimer is protonated. The cyclic ammonium salt can then be opened by nucleophilic addition of either a primary amine or a tertiary amine. If propagation occurred by only addition of primary amines the expected polymer would contain only secondary amines. If propagation occurred by only addition of tertiary amines the expected polymer would contain equal numbers of primary and tertiary amines but no secondary amines. 1H NMR spectroscopy revealed that the PTMI produced contained 20% primary, 60% secondary, and 20% tertiary amines, suggesting that both mechanisms are occurring. However, tertiary amines may also be formed by the addition of secondary amines along the backbone of the polymer to the cyclic ammonium salt. Similarly, a tertiary amine along the backbone of the polymer could add to a cyclic ammonium salt. However, this is less probable due to the lower basicity of a tertiary amine to a secondary amine.120 Additionally, this reaction would lead to chain termination. Goethals determined in this work, by monitoring the increase in molecular weight over time, that this termination must be very slow, if occurring at all.
 | ||
| Scheme 9 The cationic ring-opening polymerization of azetidine to produce hyperbranched PTMI (hbPTMI). | ||
In 2017, Goethals’ initial work on the polymerization of unsubstituted azetidine was confirmed by Sarazen and Jones.8 They provided a report of the cationic polymerization of azetidine, which was impregnated onto a silica scaffold. These porous materials were then employed in the capturing of CO2, which is a promising preliminary application of PTMI.
2.6 Cationic ring-opening polymerization of substituted azetidines
Goethal's work was not limited to unsubstituted azetidines. He also reported the polymerization of 1,3,3-trimethylazetidine.4,115 In this work, Goethals studied the CROP of 1,3,3-trimethylazetidine. After initially screening multiple solvents and initiators, the kinetics of the polymerization were studied in nitrobenzene employing Et3OBF4 as the initiator at temperatures >60 °C.The CROP of 1,3,3-trimethylazetidine is first-order with respect to monomer concentration and the number of active chain ends remains constant throughout the polymerization with a propagation rate constant, kp, of 1.2 × 10−4 L/(mol s)−1 at 78 °C in nitrobenzene, making the polymerization notably slow. Additionally, molecular weights increased linearly with increase in the monomer to initiator ratio when studied using vapor pressure osmometry. This data, coupled with studies showing that initiation is significantly faster than propagation, suggests that the polymerization displays living character. Indeed, upon a second addition of monomer, following complete consumption of the initial monomer concentration, molecular weight increased following the same rate as the initial polymerization, confirming this hypothesis. This contrasts from other heterocyclic CROP, such as oxetanes,121 thietanes,122 and selenetanes123 in which the polymerizations either slowed or stopped at low conversions. This is attributed to the reaction of the heteroatoms in the polymer backbone to the growing chain end, producing unstrained, unreactive cations. It is suggested that the polymerization of 1,3,3-trimethylazetidine is living due to the increased basicity of 1,3,3-trimethylazetidine compared to the tertiary amines contained in the polymer backbone.120
Goethals further studied the polymerization kinetics of 1,3,3-trimethylazetidine in nitrobenzene employing Et3OBF4 as the initiator by varying the polymerization temperature and monomer to initiator ratio. In varying the temperature, an Arrhenius plot was also constructed and the activation energy of 1,3,3-trimethylazetidine was found to be 19 kcal mol−1. Additionally, in varying the monomer to initiator ratio, little deviation was found in the value of kp, suggesting the reaction is first order with respect to initiator, Et3OBF4.
3. Anionic polymerization of aziridines
Recently, a number of researchers have been interested in the newly established anionic polymerization of N-activated aziridines to prepare linear polyaziridines. In contrast to cationic polymerization, anionic polymerization uses nucleophilic initiators and propagates through an anionic chain end. Anionic polymerizations are attractive due to the high degree of control over molecular weight and dispersity of the resulting polymers compared to other polymerization methods.The first anionic polymerization of an aziridine, via an azaanion was reported by Bergman and Toste in 2005.124 When investigating the reactivity of a nucleophilic transition metal complex, Bergman, Toste, and coworkers unexpectedly observed ring-opening polymerization of enantiopure (+)-2-benzyl-N-tosylaziridine to form a poly(sulfonylazirdine).124 This molecule is activated at the ring-nitrogen by an electron-withdrawing sulfonyl group, enabling nucleophilic attack at the aziridine ring in the 3-position. The electron withdrawing effect of the sulfonyl group further stabilizes the evolving azaanion at the growing chain end by delocalization, and propagation continues via sulfonamide anions (Scheme 10). Different activating groups have also been investigated for the anionic polymerization of aziridines. Examples include diphenylphosphinyl, acetyl, and ethylcarbamoyl substituents, but exclusively the sulfonamide-aziridines were suitable for azaanionic polymerization to date.124
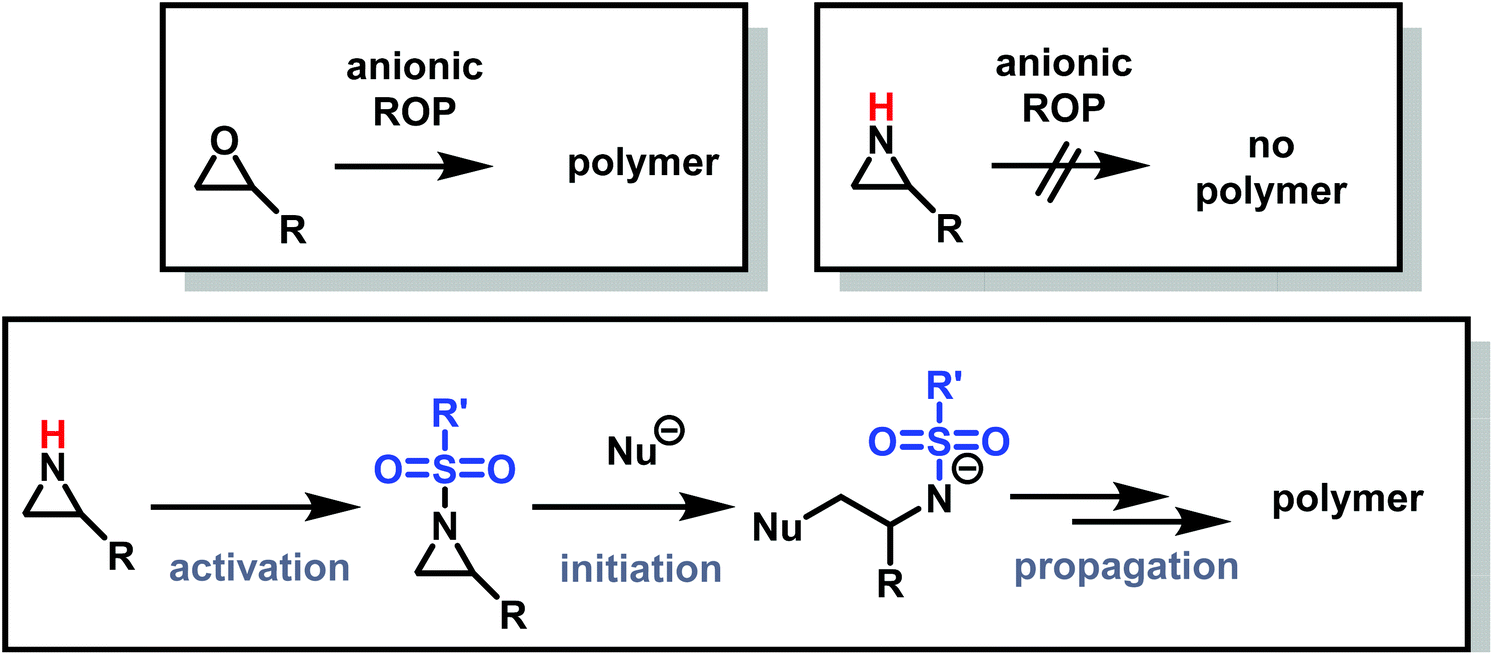 | ||
| Scheme 10 Top: Established anionic ring-opening polymerization of epoxides vs. no anionic ring-opening polymerization of aziridines. Bottom: Activation strategy of aziridines to sulfonyl-aziridines, allowing for anionic ring-opening polymerization. | ||
3.1 Preparation of sulfonyl aziridines for anionic polymerizations
Since anionic polymerization does not tolerate acidic protons, the aziridine N–H needs to be substituted with electron-withdrawing sulfonyl groups, i.e. “activation groups” (Scheme 11). In analogy to the Hinsberg reaction,125 secondary amines react with sulfonyl chlorides to produce the respective sulfonyl aziridines. Xu et al.126 presented an efficient microwave-assisted one-pot reaction of valinol, L-phenylalaninol, L-leucinol, L-alaninol and L-serine methylester, along with methyl-, phenyl-, and 4-methoxyphenyl-sulfonic chlorides to yield sulfonyl aziridines. Potential solvents for this strategy were diethyl ether, THF, acetonitrile, and dichloromethane. Bases such as alkali carbonates and hydroxides are used with DMAP as catalyst. In 30 minutes Xu et al. were able to prepare different sulfonyl aziridines with high yields of 84%–93%.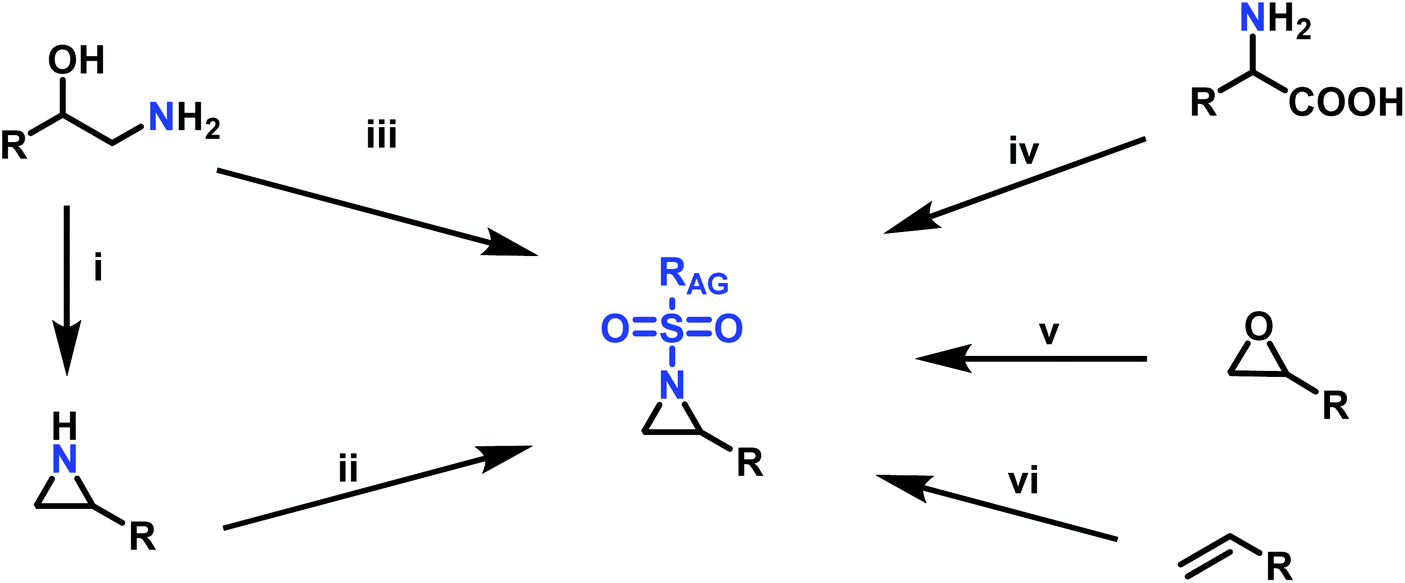 | ||
Scheme 11 Synthesis of aziridine monomers for the azaanionic polymerization: i: Wenker synthesis of aziridine: (1) vicinal amino alcohol derivate, sulfuric acid or sulfuric acid chloride (2) NaOH (aq.); ii: sulfonyl chloride, TEA, DCM; iii: sulfonic chloride, DCM, DMAP or K2CO3 (Microwave, 400 W); iv: (1) amino acid, NaOH (aq.), 0 °C, TsCl. (2) THF dry, BH3–SMe2, reflux. (3) DCM, TsCl, DMAP, Py; v: RSO2NH2, K2CO3, BnEt3N+Cl−, dioxane, 90 °C, MsCl, DCM, 0 °C to reflux; vi: (1) chloramine salts, ACN, PTAB, 12 h; (2) IPrCu(DBM), PhI![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, RSO2–NH2, chlorobenzene, r.t., 25 h; (3) Rh2(cap)4, TsNH2, NBS, K2CO3. O, RSO2–NH2, chlorobenzene, r.t., 25 h; (3) Rh2(cap)4, TsNH2, NBS, K2CO3. | ||
Amino acids have also been used to produce N-tosylaziridines in a three-step process (Scheme 12).127 This was achieved by the N-tosylation of the amino acid, followed by reduction of the carboxylic acid to yield N-tosyl-2-amino alcohols and finally O-tosylation with an in situ ring-closing. Particularly interesting is that this method does not require any purification of intermediates.
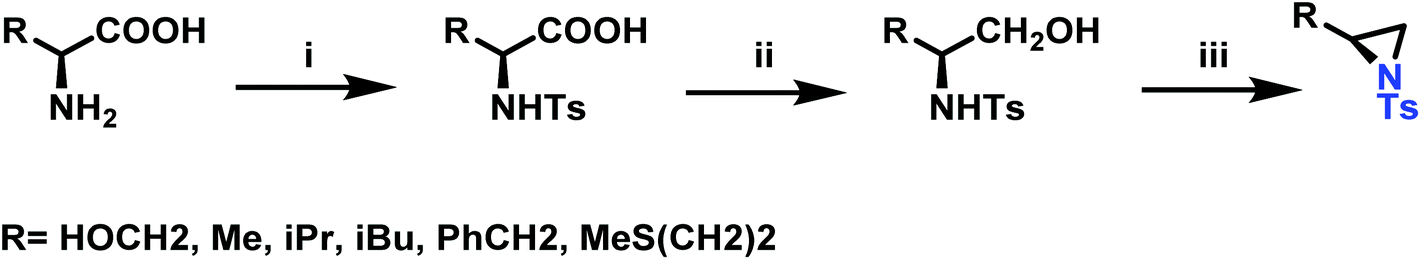 | ||
| Scheme 12 i: Preparation of N-tosyl-2-aminoacids via N-tosylation with TsCl; ii: preparation of N-tosyl-2-aminoalcohols reduction of the carbonyl group; iii: preparation of 2-substituted N-tosylaziridines. | ||
Aziridination of olefins (route (vi) in Scheme 11) was also utilized to produce sulfonyl aziridines. Sulfonyl aziridines with varying lengths of alkyl chains were produced in a single step that is tolerant to functional side groups, such as alcohols and acetals. An important advantage of this strategy is that toxic aziridine is avoided and the activated sulfonyl aziridines are obtained directly. This route employing non-functionalized alkenes to produce sulfonyl aziridines has high yields of 95% with rhodium catalysts and up to 93% with PTAB as catalyst, however, the yields vary depending on the pendant groups. Increasing the viability of this method, the catalysts are either commercially available or can be prepared with ease.128–130 Sharpless and coworkers proposed a mechanism for bromine-catalyzed aziridination (Scheme 13). In the first step, the olefin reacts with a Br+ source, given by PTAB. The brominium ion is then ring-opened by TsNCl−, to form the α-bromo-N-chloro-N-toluenesulfonamide (Step 2). Attack of the bromide anion (Br−) (or TsNCl−) on the N–Cl group of the intermediate generates the anion and a Br–X species (Step 3). Expulsion of Br− from the anion finally yields the aziridine and the regenerated Br–X species (Step 4) initiates another turn of the catalytic cycle.130 This synthesis route has been successfully used for monomer synthesis by the Wurm group by using chloramine-T and chloramine-M to synthesize MsDAz (49%) and TsDAz (47%)131 and acetal functionalized aziridine monomers (17%–30%).132
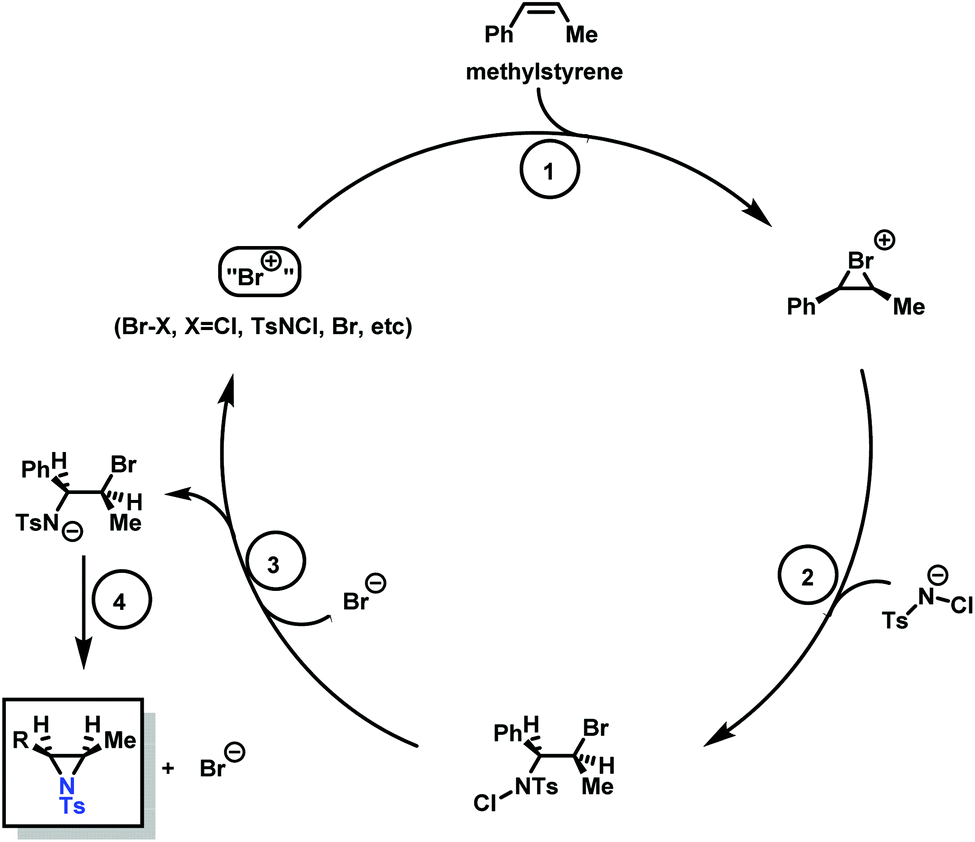 | ||
| Scheme 13 Catalytic cycle of the PTAB catalyzed aziridination of olefins, adopted from previous work of Sharpless. Copyright@1998 The American Chemical Society. Reprinted with permission from Journal of the American Chemical Society.130 | ||
Epoxides were also used as attractive starting materials for the sulfonyl aziridine synthesis (Scheme 14).124,133 2-Benzyl-1-(2,4,6-triisopropylbenzene-sulfonyl)aziridine was synthesized in two-steps: the first step was the nucleophilic ring-opening of 2-benzyloxirane with the primary sulfonamide (2,4,6-iPr3C6H2SO2NH2). This reaction requires 0.1 eq. of potassium carbonate and BnNEt3+Cl− as catalyst in dioxane (73% yield). The subsequent mesylation–cyclization of the hydroxyl-sulfonamide was achieved by the addition of mesyl chloride to activate the hydroxy group under basic conditions (86% yield). This route might be extended to other N-sulfonyl groups.134
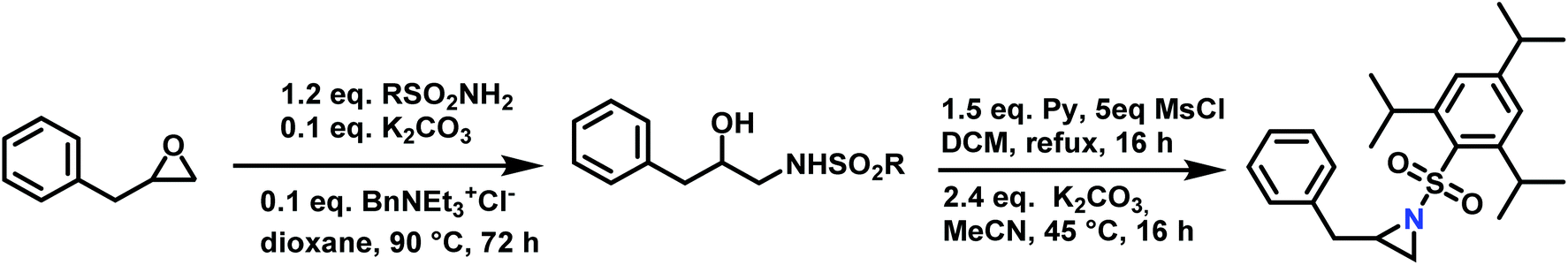 | ||
| Scheme 14 Synthesis of 2-benzyl-1-(2,4,6-triisopropylbenzenesulfonyl)aziridine from 2-benzyloxirane, R = 2,4,6-iPr3C6H2. | ||
Another route, starting from epoxides, was used by Bergman and Toste124 to synthesize 2-n-decyl-N-methanesulfonyl aziridine (MsDAz). Thomi and Wurm133 followed this procedure to synthesize 2-(oct-7-en-1-yl)-N-mesylaziridine. This procedure involves three steps; first the epoxide is ring-opened with sodium azide to give the azido-hydroxyalkane as intermediate, which is converted in the second step, by a Staudinger reaction, to the corresponding alkyl aziridine. To activate the obtained aziridine for anionic polymerization mesylchloride is used to replace the N-terminal hydrogen in the third step. Table 1 summarizes activated aziridines and azetidines which were successfully polymerized via azaanionic polymerization to date.
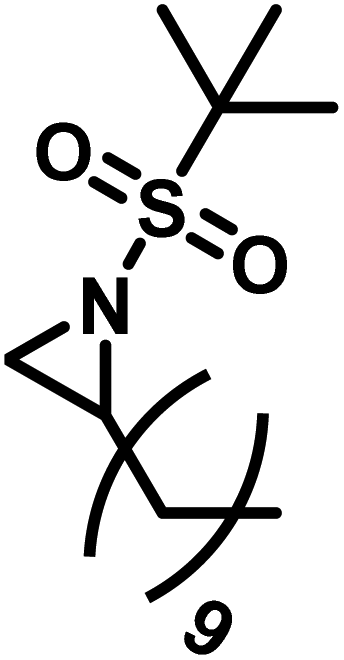
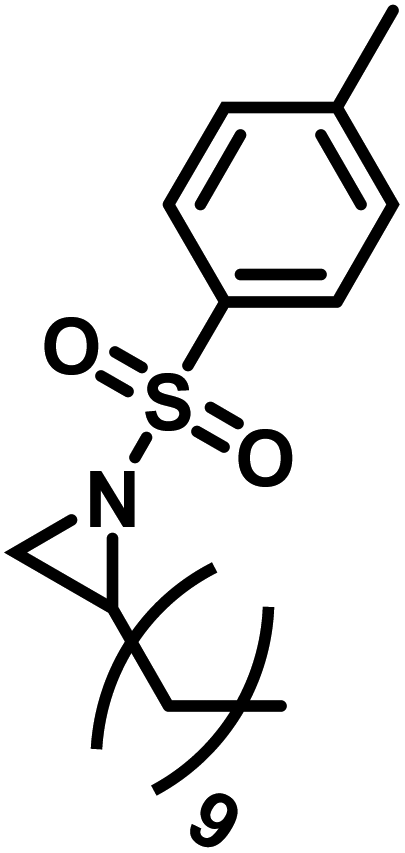
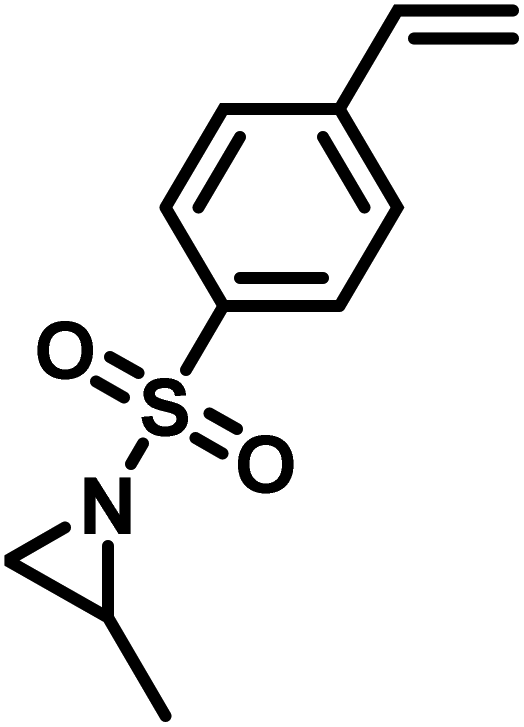
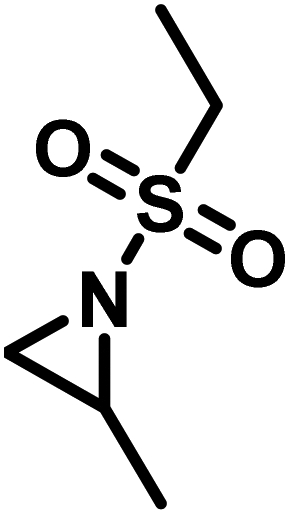
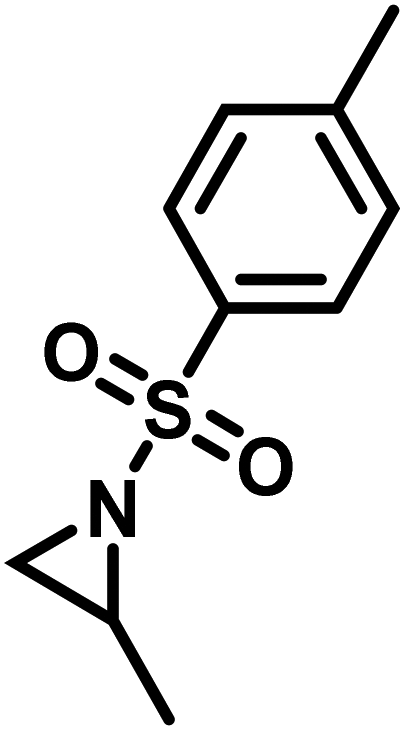
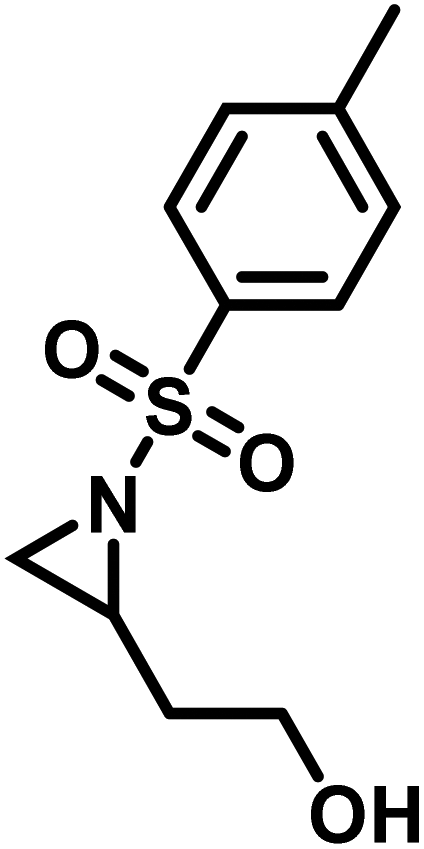
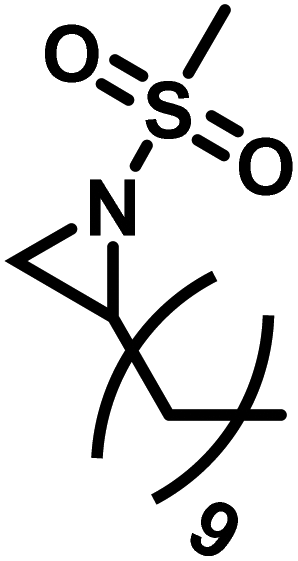
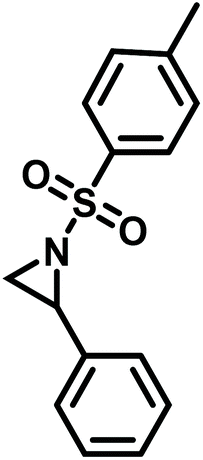
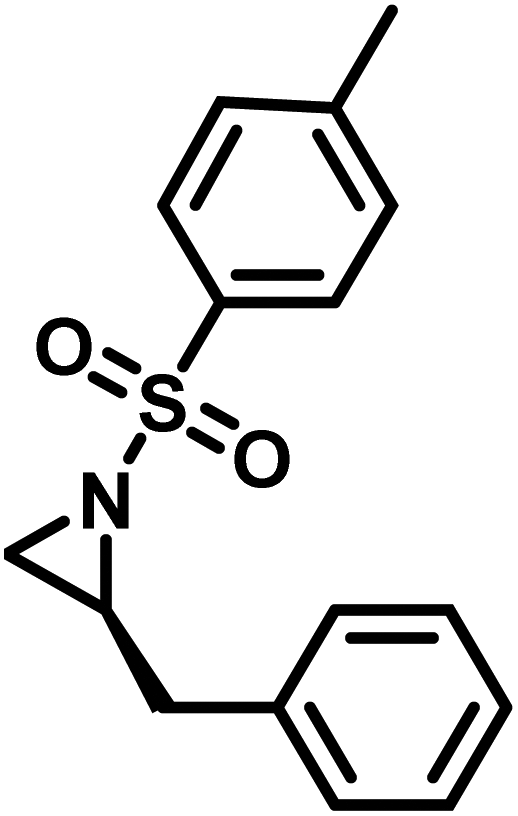
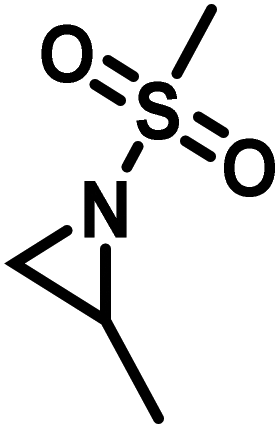
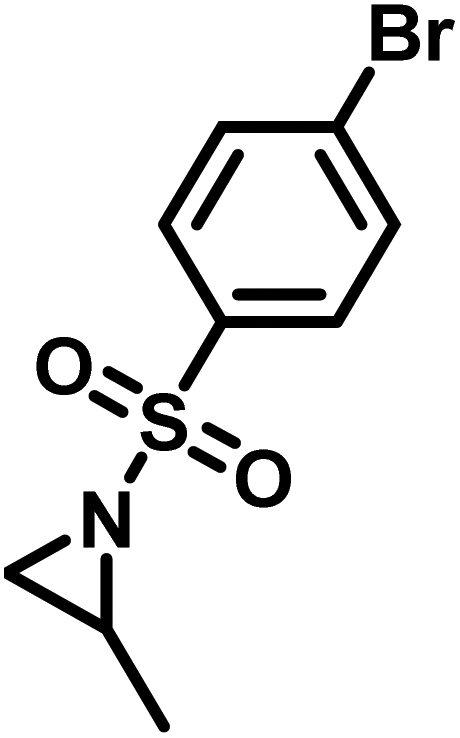
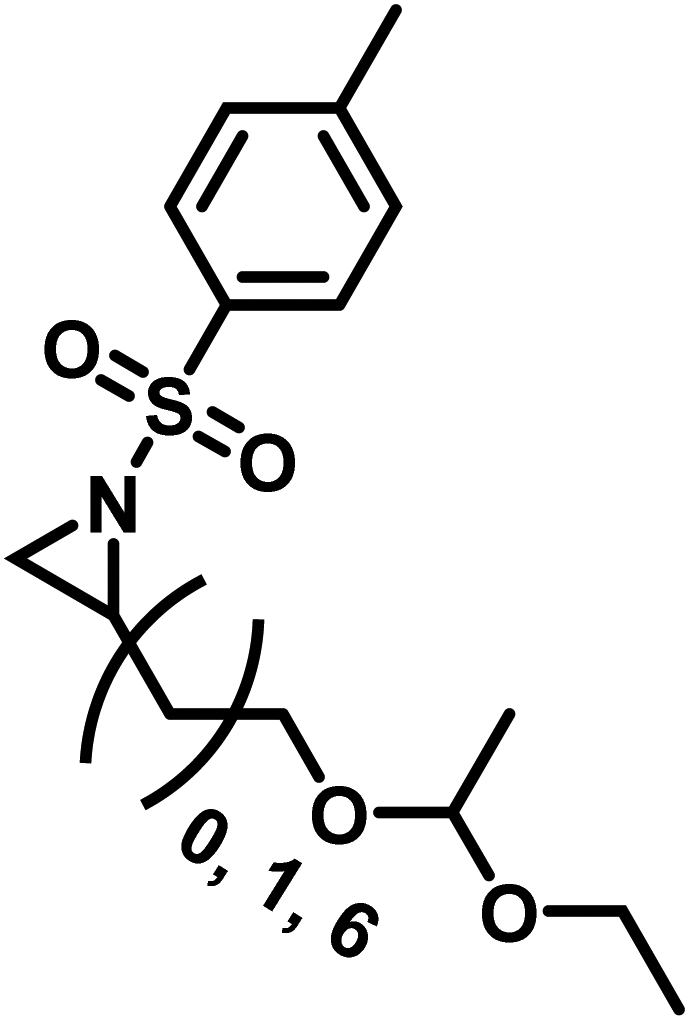
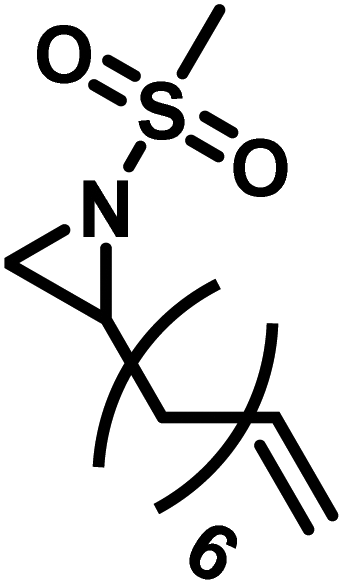
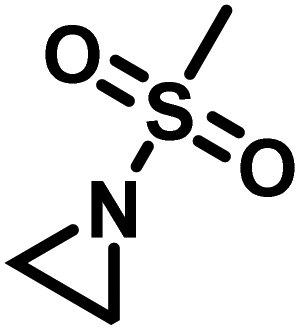
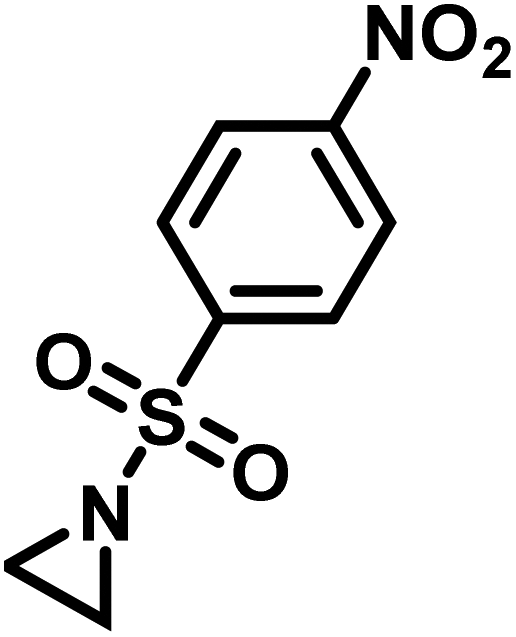
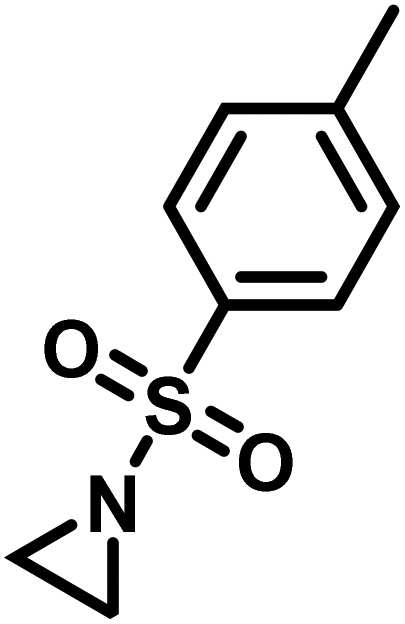
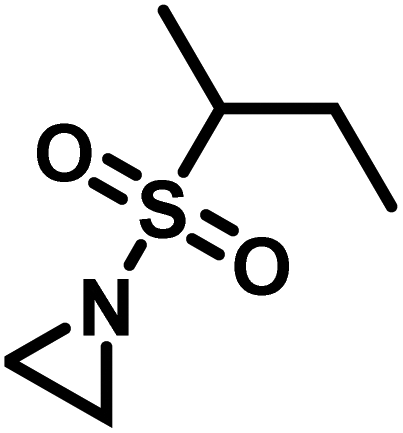
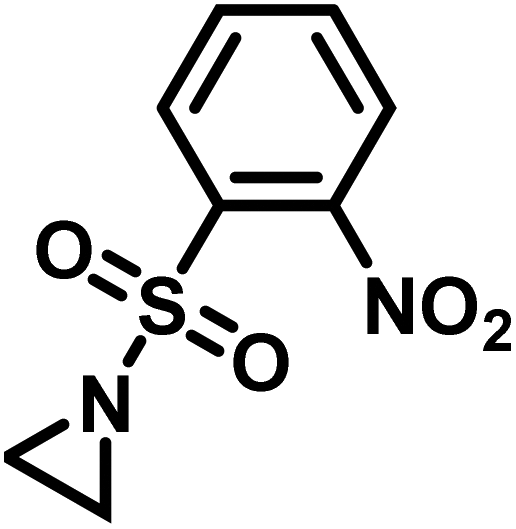
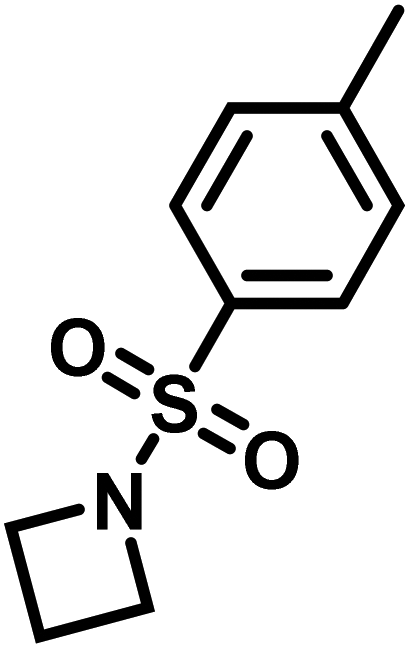
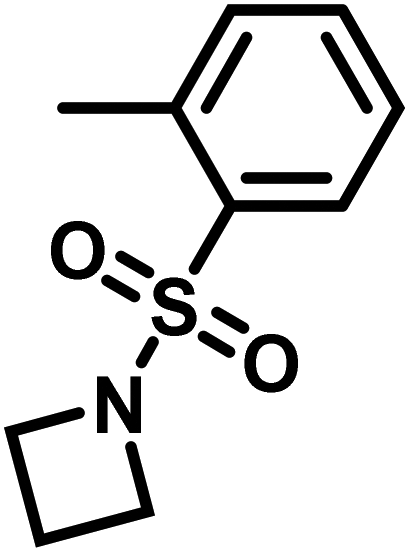
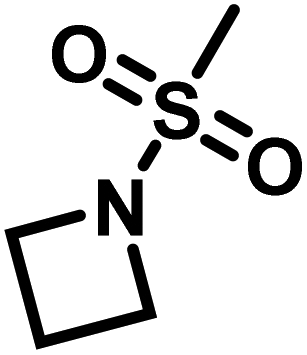
3.2 Initiators for the anionic polymerization of activated aziridines
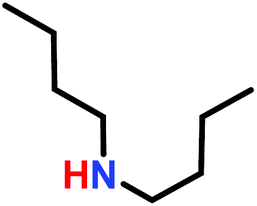
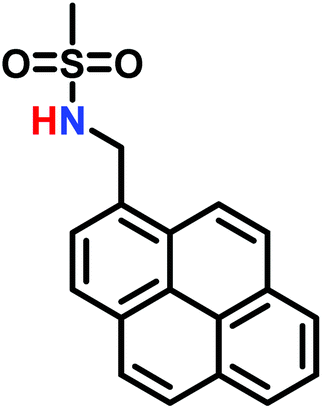
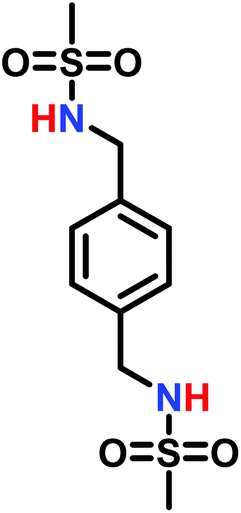
The anionic ROP of sulfonyl aziridines is typically initiated by secondary N-sulfonamide-initiators, such as the alkali salts of N-benzyl-4-methylbenzenesulfonamide,138N-pyrene-methanesulfonamide,124,132,136 or butyl lithium (Table 2).143,144 Also a bifunctional initiator N,N′-(1,4-phenylenebis(methylene))dimethane-sulfonamide was introduced in 2017.143 To date, the standard protocol uses alkali bis(trimethylsilyl)amide salts to deprotonate the sulfonamide-initiators.143 KHMDS alone was also proven to be able to ring-open sulfonyl aziridines, but bimodal molecular weight distributions were obtained.132,140 With these initiators, functional poly(sulfonylaziridine)s are available. Recently, Reisman et al.83 showed that the terminal group can be used to prepare telechelic polymers by terminating AROPs with propargyl bromide, which allows further modifications by click chemistry.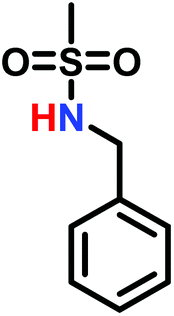


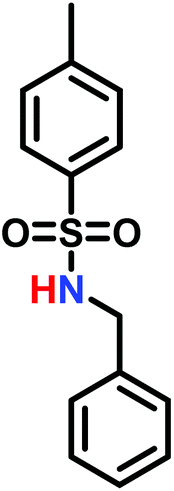
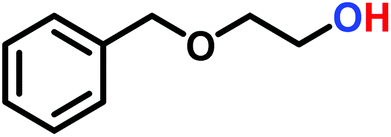
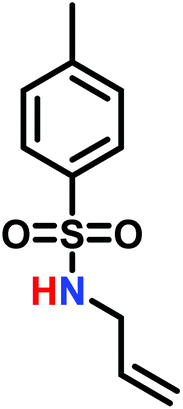
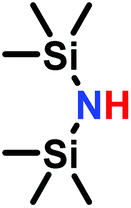
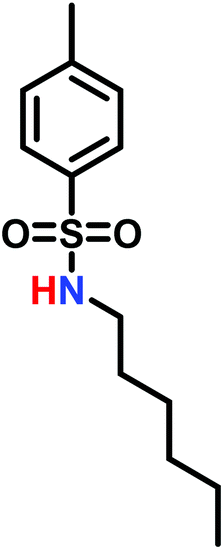
3.3 Anionic polymerization of activated aziridines
When initiated with a suitable nucleophile (Table 1), the anionic polymerization of sulfonyl aziridines follows living conditions (Scheme 15).143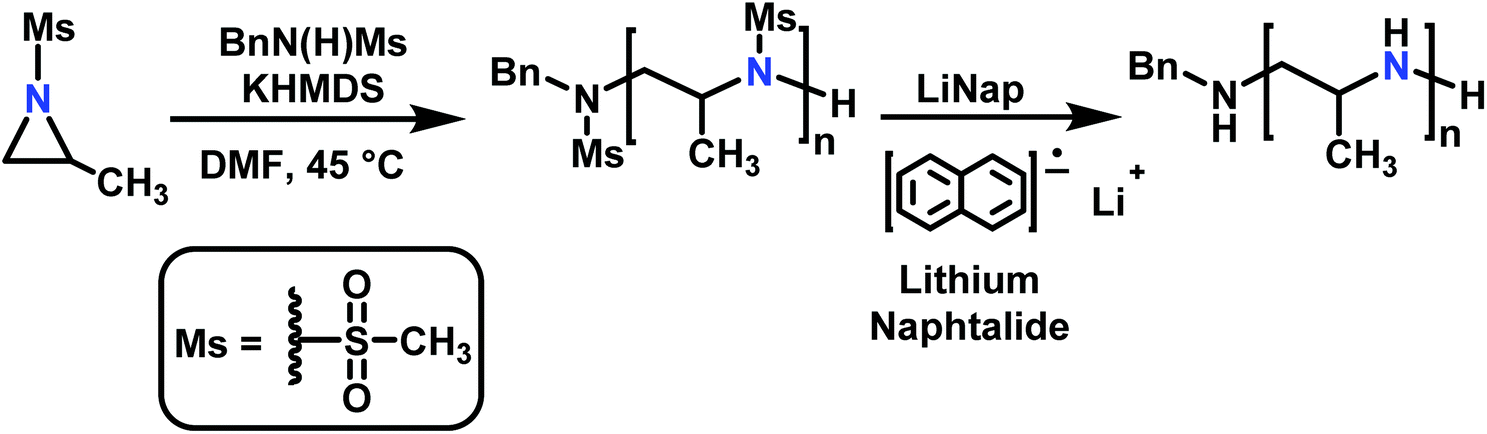 | ||
| Scheme 15 Anionic ring-opening polymerization of sulfonyl aziridines and subsequent desulfonylation (with 2-methyl-mesylaziridine as an example). | ||
The solubility of poly(sulfonylazirdine)s is highly dependent on the substituents on the sulfonyl group and the tacticity of the polymer. If (+)-2-benzyl-N-tosylaziridine was used as monomer, only insoluble oligomers were produced.124 In contrast, racemic monomers produce linear polymers with degrees of polymerization (DP) of up to 200 (with Mn = 20000 g mol−1) and narrow molecular weight distributions, Đ < 1.10.124,143 Furthermore, the polymerization follows first order kinetics with respect to monomer, suggesting a living polymerization (Fig. 4). In addition, chain extension experiments proved that the polymerization of N-sulfonylaziridines is living. The sulfonyl groups of the obtained poly(sulfonylaziridine)s can be removed after the polymerization with different strategies, e.g. using alkali metal naphthalides or acidic conditions to yield the corresponding polyamines (Scheme 15).82
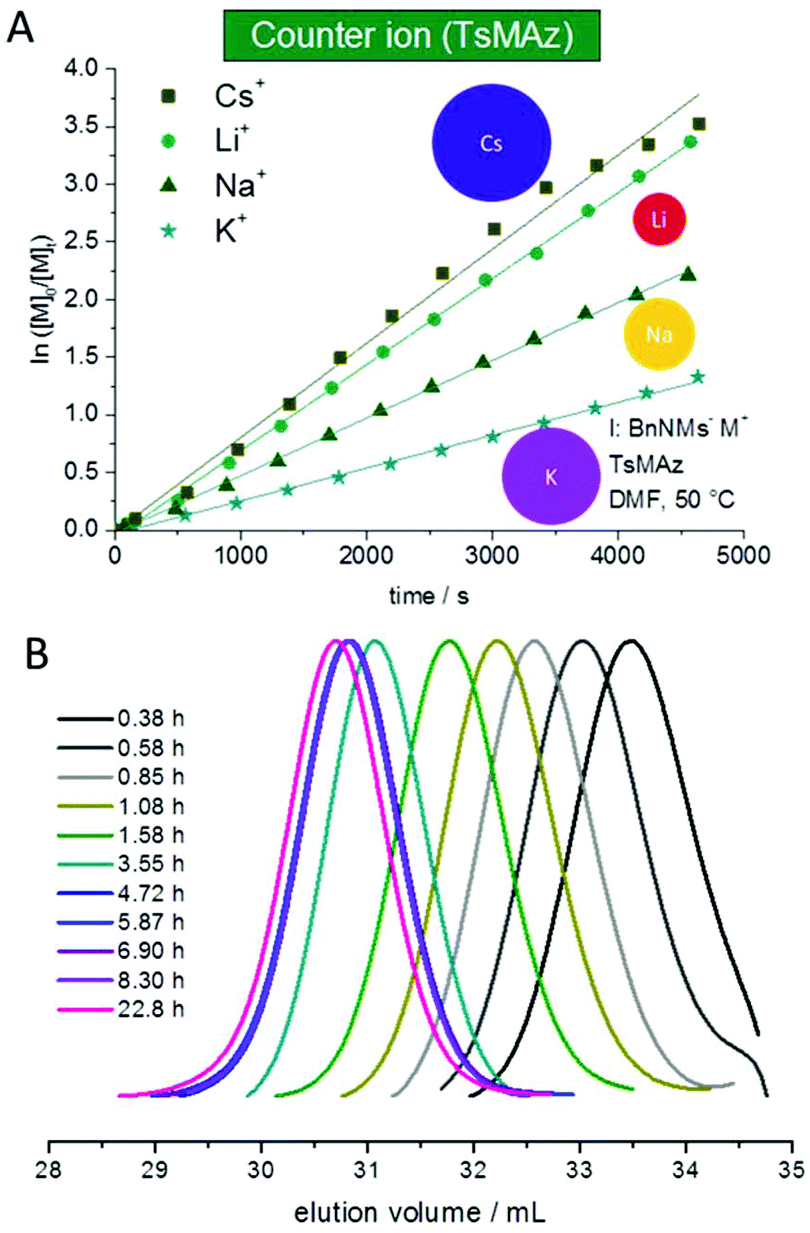 | ||
| Fig. 4 (A) Kinetic plots of ln([M]0/[M]t) vs. time for the azaanionic polymerization of TsMAz with BnNHMs (initiator) in DMF-d7 at 50 °C with different bis(trimethlsilyl)amide-salts. (B) SEC-kinetics of MsMAz, BnNKMs at 50 °C in DMF (RI-signal), reproduced from ref. 143 with permission from Royal Chemical Society, copyright 2017. | ||
The low solubility of poly(sulfonylaziridine)s was also a challenge for the polymerization of unsubstituted sulfonyl aziridines. In general, poly(sulfonylaziridine)s that lack substitution along the polymer backbone, or that have backbone substituents but are tactic, are generally insoluble in all solvents. For example, Thomi et al.144 attempted to polymerize tosylaziridine and found that only insoluble oligomers were obtained. Later, Rupar and coworkers83 were able to produce soluble polymers by copolymerizing mesylaziridine and sec-busylaziridine up to DP = 200. Such copolymers produced well-defined linear polyamines after desulfonylation by lithium metal (Scheme 16).
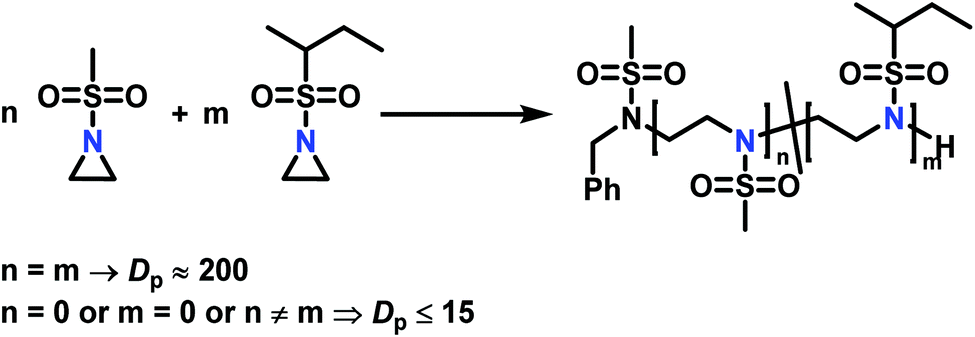 | ||
| Scheme 16 Azaanionic copolymerization of unsubstituted sulfonyl aziridines as precursors for LPEI. High degree of polymerization was only obtained when the monomers were used in a 1:1 ratio (n = m). Other ratios produced only insoluble oligomers.83 | ||
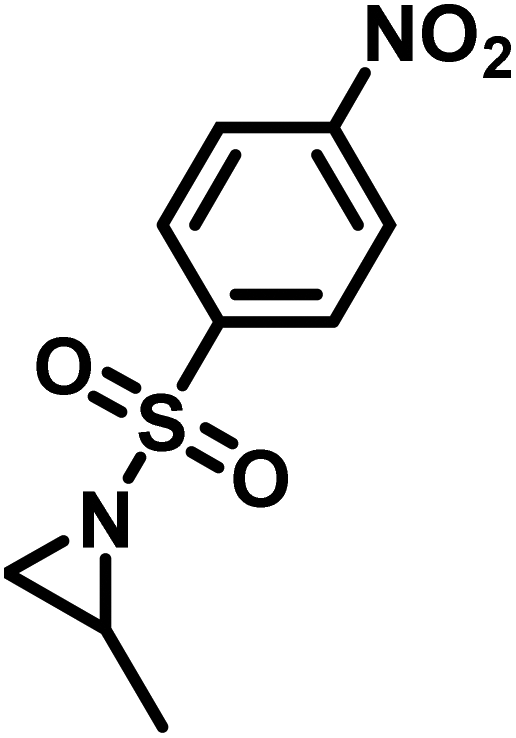
Recently, Rupar and coworkers141 reported the first example of a poly(sulfonylaziridine) homopolymer which lacked substitution on the backbone. They studied the AROP of nitrophenylsulfonyl-activated aziridine monomers, namely N-((p-nitrophenyl)sulfonyl)aziridine (pNsAz) and N-((o-nitrophenyl)sulfonyl)aziridine (oNsAz) (Scheme 17). pNsAz formed an insoluble white powder upon heating in all attempts at polymerization. With oNsAz, on the other hand, the resulting poly(oNsAz) was soluble in both DMF and DMSO at all molecular weights, making it the first example of a soluble poly(N-sulfonylaziridine) homopolymer. The obtained homopolymer was subsequently deprotected using sodium thiomethoxide in DMF at 50 °C to yield an off-white residue. Although evidence was found for the formation of LPEI from the deprotection of poly(oNsAz), satisfactory purification of the residue was not achievable. Control over the molecular weight of poly(oNsAz) was also attempted by initiating the anionic polymerization of oNsAz with BnN(Li)Ms. However, the resulting poly(oNsAz) was a mixture of the BnN(Li)Ms initiated polymer chains and hydroxyl initiated chains. This was attributed to the fact that oNsAz readily undergoes spontaneous polymerization, and thus could not be satisfactorily purified and dried.
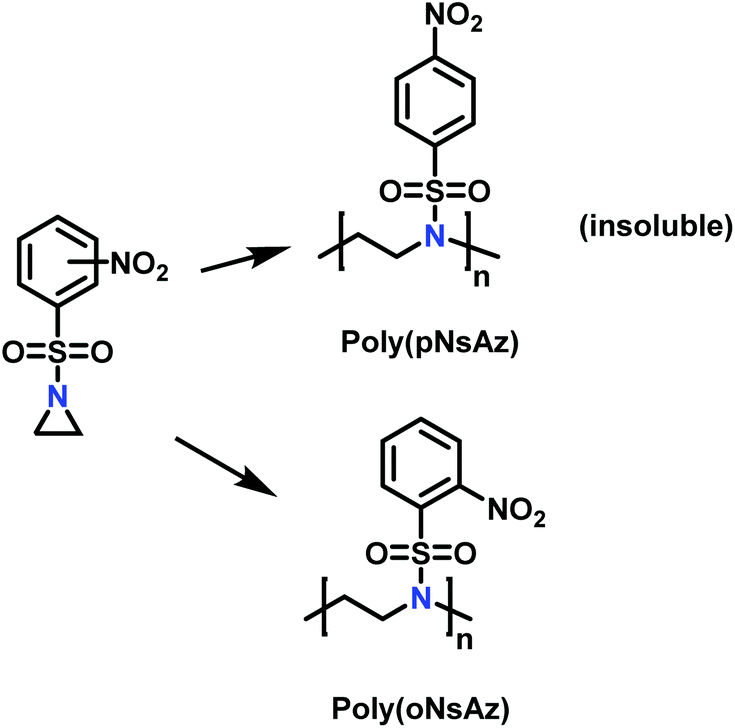 | ||
| Scheme 17 Azaaonionc polymerization of nitrophenylsulfonyl-activated aziridines.141 | ||
Copolymerizations of different sulfonyl aziridines give access to random or gradient copolymers, depending on the nature of the sulfonyl group.136 The reactivity ratios of 2-methyl tosyl aziridine (TsMAz) and 2-decyl tosyl aziridine (TsDAz) were determined via real-time 1H NMR spectroscopy and proven to be an ideal random copolymerization with r(TsMAz) = 1.08 and r(TsDAz) = 0.98 and r(TsMAz)·r(TsDAz) = 1.05. In contrast, combining monomers with different sulfonyl groups, resulted in (multi)gradient copolymers.131 Sulfonyl groups with stronger electron withdrawing effects increase the rate of polymerization, which led to gradient incorporation. Fig. 5 shows the real-time 1H NMR kinetics of a statistical terpolymerization of 2-methyl brosylaziridine (BsMAz), 2-methyl tosyl aziridine (TsMAz), and 2-methyl mesylaziridine (MsMAz), which form a copolymer with distinct domins along the polymer chain, due to the individual reactivity ratios of each monomer.131 DFT-calculations of the electrophilicity indices (ω+) support these empirically determined comonomer reactivities, with BsMAz (2.22 eV) > TsMAz (1.98 eV) > MsMAz (1.25 eV).139 Contrarily, the nucleophilicity (ω−) of the azaanion at the growing chain end changes only ca. 0.14 eV, proving that the crucial factor which determines the incorporation rate is the monomer reactivity, and not the azaanion nucleophilicity.139
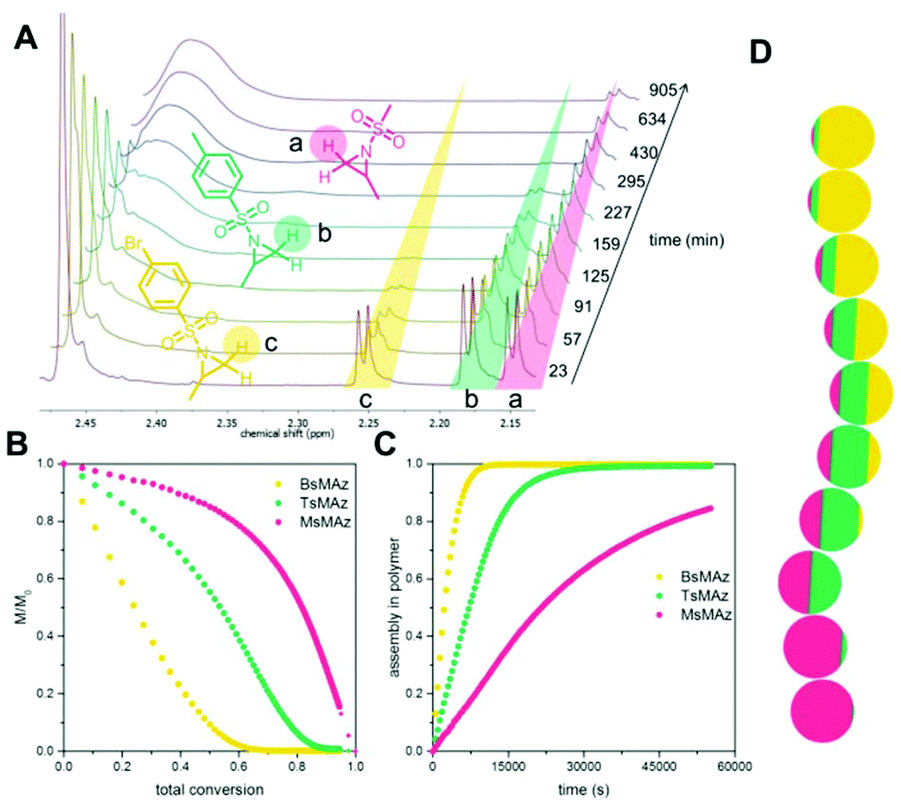 | ||
| Fig. 5 Simultaneous copolymerization of BsMAz, TsMAz, and MsMAz. (A) Real-time 1H NMR spectra of the terpolymerization of BsMAz (yellow), TsMAz (green), MsMAz (red) showing the consumption of the monomers. (B) Normalized monomer concentrations in the reaction vs. total conversion. (C) Assembly of each monomer in the polymer vs. reaction time. (D) Visualization of a single chain for poly(BsMAz-co-TsMAz-co-MsMAz) – each sphere stands for 10% conversion. (Reproduced from ref. 131 with permission from Wiley, copyright 2016). | ||
Gradient copolymers were also prepared by copolymerization of tosylated aziridines in emulsion.54 The comonomer pair TsDAz and TsMAz produce random copolymers in solution, but when separated from each other by an emulsion consisting of DMSO-droplets and cyclohexane as the continuous phase, variable gradients can be obtained by partitioning of both monomers, when the continuous phase is diluted.136,143,149 This is represented in the apparent reactivity ratios, which are rapp(TsMAz) = 4.98 and rapp(TsDAz) = 0.20 in case of a 1:20-DMSO/cyclohexane emulsion, revealing the formation of strong gradient copolymers.150–152
3.4 Functional polyaziridines prepared by anionic polymerization
Functional groups can be installed as substituents at the activating group or at the aziridine ring. 2-Oct(en)yl N-mesyl-aziridine (Fig. 6a) with an olefin functionality was homo- and copolymerized via AROP. The olefins were post-modified by a radical thiol–ene reaction with N-acetyl-L-cysteine methyl ester providing quantitative conversion.133 2-Methyl-N-(4-styrenesulfonyl) aziridine (StMAz)137 was the first bivalent aziridine derivative to undergo either anionic or radical polymerization (Fig. 6d). After anionic polymerization, thiol–ene addition of mercaptoethanol or mercaptopropionic acid to the styrenic double bond was achieved. After radical polymerization, the pending sulfonyl aziridines could be further modified by nucleophilic additions, which was described for other polymers with aziridine side groups.153–156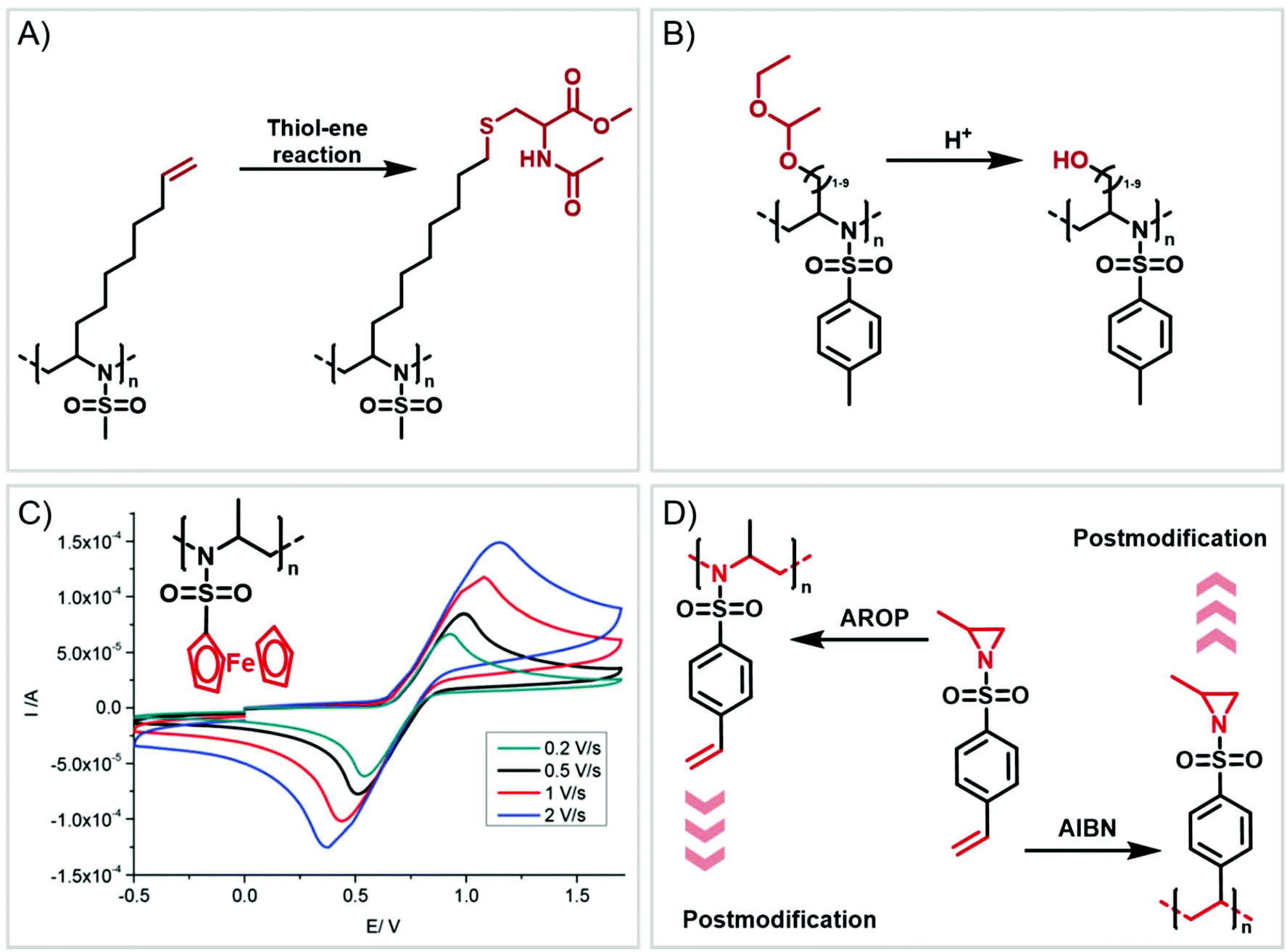 | ||
| Fig. 6 Functional polyaziridines: (A) terminal double bond can be converted via thiol–ene reaction. (B). Acetal-protected polyaziridines yield hydroxyl functionalities. (C) Ferrocene-containing polyaziridines are redox-responsive. (D) Orthogonal aziridine allows anionic and radical polymerization. | ||
Also, polyols have been prepared by the AROP of sulfonyl aziridines. In analogy to ethoxy ethyl glycidyl ether (EEGE), the well-known precursor in oxyanionic polymerization to obtain linear poly(glycerol),74,157 acetal-protected N-tosyl-activated aziridines were introduced in 2016 (Fig. 6b).132 Three different acetal-protected monomers with variable alkyl chain lengths were prepared and could be polymerized by living AROP. The hydroxyl groups were released by mild acidic hydrolysis, leaving the sulfonamides attached. Additional removal of the sulfonyl groups under reductive conditions resulted in polyamine-polyols, which might be used as chelating or transfection agents.132
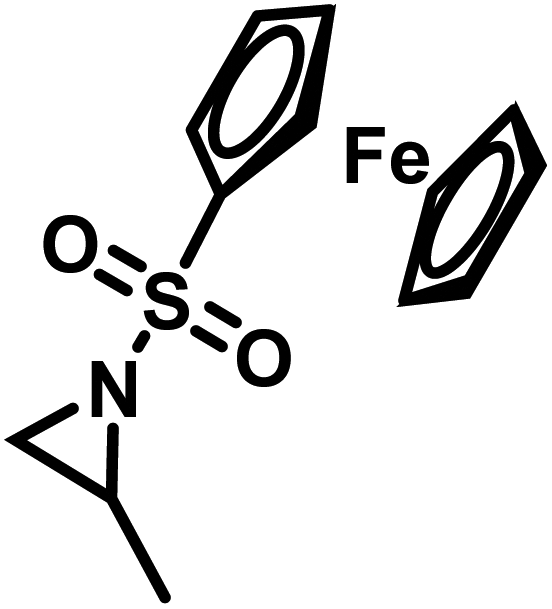
Organometallic 2-methyl-N-ferrocenylsulfonyl-aziridine was polymerized to prepare redox-responsive poly(sulfonylaziridine)s (Fig. 6c).140 Similar to other poly(sulfonylaziridine)s (see above), the homopolymerization resulted in insoluble materials. However, solid state NMR (ssNMR) and MALDI-TOF spectra supported the expected polymeric structure. Copolymerization with TsMAz or MsMAz resulted in soluble copolymers with moderate molecular weight dispersities (Đ < 1.4), and chain extension experiments proved the living nature of the polymerization. Such organometallic polymers showed reversible oxidation/reduction by cyclic voltammetry, similar to other ferrocene-containing polymers.158
4. Anionic polymerization of azetidines
Reisman et al.142 reported the first example of AROP of an azetidine monomer, N-(methanesulfonyl)azetidine (MsAzet) (Scheme 18). Unlike the three-membered ring sulfonylaziridines, the polymerization of MsAzet required high temperatures (>100 °C) in order to polymerize. The resulting polymer, p(MsAzet), proved to contain branching due to chain transfer. As evidenced by H–D exchange experiments, this chain transfer occurs through the deprotonation of methanesulfonyl groups of the polymer backbone and the monomer to form sulfamoyl methanide anions. Evidence of minimal chain transfer to DMSO that occurs through the formation of dimsyl anions was also found. More importantly, the concentration of the active chain ends was found to be constant during the polymerization of MsAzet, which indicates that the controlled and living polymerization of sulfonylazetidines can be made possible if chain transfer can be inhibited. | ||
| Scheme 18 Anionic ring-opening polymerization of N-(methanesulfonyl)azetidine.142 | ||
Recently, N-(tolylsulfonyl)azetidines were found to undergo living AROP to form linear polymers.159 These monomers do not contain protons likely to be activated under the polymerization conditions. Initial work was done by attempting to produce homopolymers from the two monomers N-(p-tolylsulfonyl)azetidine (pTsAzet) and N-(o-tolylsulfonyl)azetidine (oTsAzet) by AROP (Scheme 19). However, both resulting homopolymers precipitated from solution during polymerization at low molecular weight, similarly to sulfonylaziridine homopolymers.83,144 Drawing motivation from the literature, in which the copolymerization of two sulfonylaziridines was used to produce a soluble copolymer,83 the copolymerization of pTsAzet with oTsAzet was attempted and produced the soluble copolymer, p(pTsAzet-co-oTsAzet) (Scheme 19). Similarly to MsAzet, the polymerization showed first order kinetics with respect to the total monomer concentration and the number of active chain ends remains constant. By a series of polymerizations, it was demonstrated that the polymerization was both living and controlled and produced polymers with narrow molecular weight distributions. The sulfonyl groups of p(pTsAzet-co-oTsAzet) were removed under reductive conditions to produce the first example of LPTMI by living anionic polymerization.
 | ||
| Scheme 19 Polymerization of TsAzet monomers to produce insoluble homooligomers and a soluble copolymer.159 | ||
Additionally, due to the need for high temperatures in order to polymerize, the N-(tolylsulfonyl)azetidines could be used to produce block copolymers by living anionic polymerization in a closed-system in which all monomers are present at the time of initiation (Scheme 20, Fig. 7).159 This was accomplished by combining all monomers, pTsMAz, pTsAzet, and oTsAzet, in solution prior to initiation. Due to the differences in reactivities, pTsMAz could be polymerized selectively at lower temperatures (50 °C) while pTsAzet and oTsAzet do not polymerize. Upon total consumption of pTsMAz, the temperature was increased to 180 °C to polymerize pTsAzet and oTsAzet to produce block copolymers. This allowed for the formation of block copolymers without homopolymer impurities. In the field of high performance block copolymers, this finding is of particular importance, as small amounts of homopolymer impurities can alter the properties of block copolymer materials.
 | ||
| Scheme 20 Block copolymerization of pTsMAz with oTsAzet and pTsAzet to produce p(pTsMAz)-b-p(pTsAzet-co-oTsAzet). | ||
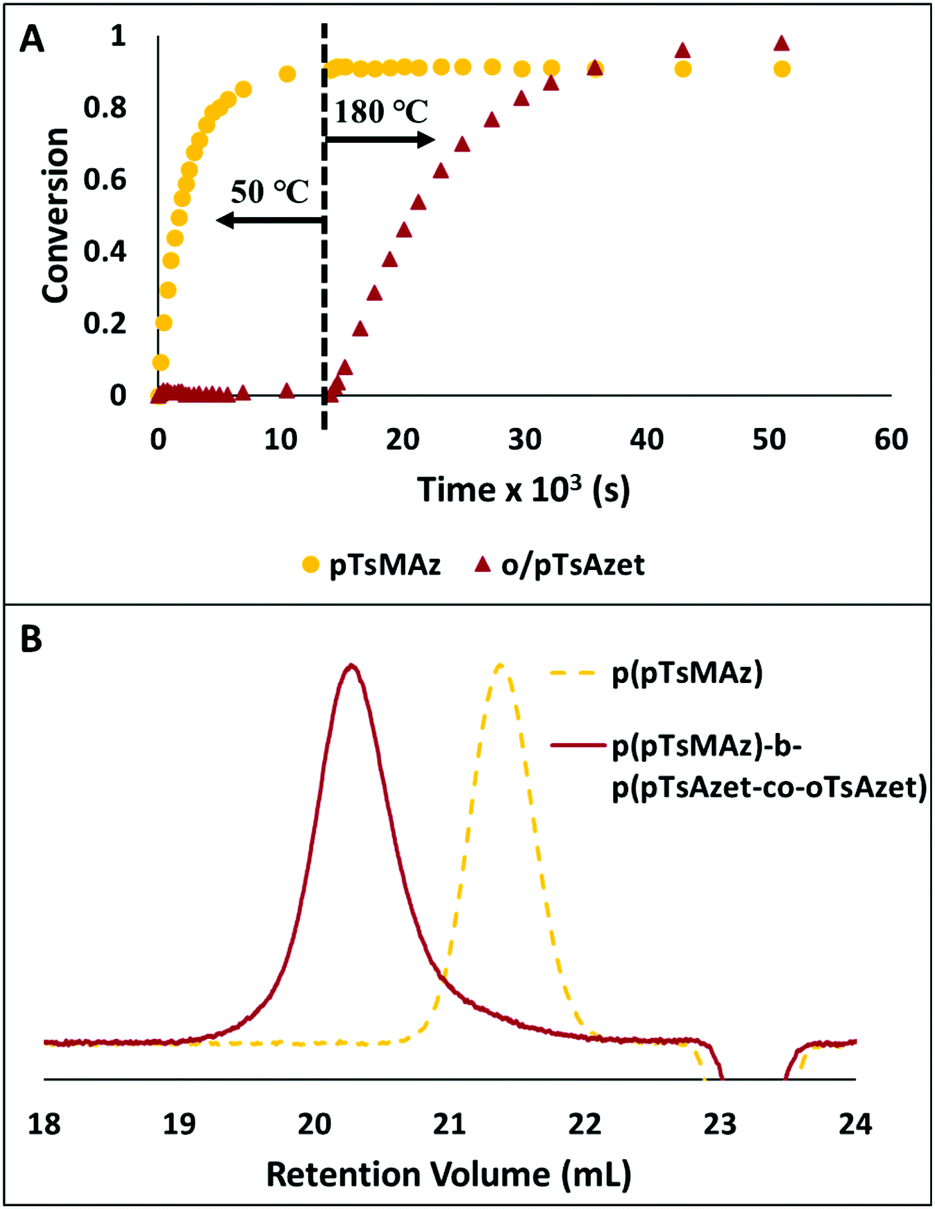 | ||
Fig. 7 (A) Plot of conversion vs. time for the block copolymerization of pTsMAz, pTsAzet, and oTsAzet in DMSOd6 to produce p(pTsMAz)20-b-p(pTsAzet-co-oTsAzet)40. The reaction is kept at 50 °C for 4 h, then heated to 180 °C for 10.25 h. The 1H NMR measured conversion of pTsMAz appears to not reach 100% due to signal overlap between the monomer and polymer resonances in 1H NMR spectra of the reaction mixture. (B) SEC trace of p(pTsMAz)20 prior to block copolymer chain extension ( ). SEC trace of p(pTsMAz)20-b-p(pTsAzet-co-oTsAzet)80 ( ). SEC trace of p(pTsMAz)20-b-p(pTsAzet-co-oTsAzet)80 ( ). Block copolymerization to produce p(pTsMAz)20-b-p(pTsAzet-co-oTsAzet)80 was performed with a [pTsMAz]:[oTsAzet]:[pTsAzet]:[I] ratio of 20:40:40:1 in NMP at 70 °C for 12 h, then 205 °C for 16 h. Reproduced from ref. 159 with permission from American Chemical Society, copyright 2018. ). Block copolymerization to produce p(pTsMAz)20-b-p(pTsAzet-co-oTsAzet)80 was performed with a [pTsMAz]:[oTsAzet]:[pTsAzet]:[I] ratio of 20:40:40:1 in NMP at 70 °C for 12 h, then 205 °C for 16 h. Reproduced from ref. 159 with permission from American Chemical Society, copyright 2018. | ||
5. Organocatalytic ring-opening polymerization (OROP) of activated aziridines
N-Heterocyclic carbenes (NHC), such as 1,3-bis(isopropyl)-4,5(dimethyl)imidazole-2-ylidene, are powerful catalysts in many types of polymerizations. Their near unlimited structural diversity, inherent high Brønsted-basicity, and nucleophilicity make NHCs powerful organocatalysts.160,161 Examples of applications of NHCs include some of the most important commercial monomers in the step-growth polymerization of terephthalaldehyde,162 the group transfer polymerization of methacrylic monomers,163 and the zwitterionic ring-opening polymerization (ZROP) of ethylene oxide (EO),164 which was discovered in 2009 by Taton and coworkers. As activated aziridines polymerize with nucleophilic bases, similar to EO, via AROP, it was of interest if organocatalytic ring-opening polymerization (OROP) can also be successfully performed with this new monomer class. The first living OROP of 2-alkyl-N-sulfonyl aziridines was presented by Carlotti, Taton and coworkers in 2016 (Scheme 21).147 The OROP of N-tosyl-2-substituedaziridines takes place in the presence of 1,3-bis(isopropyl)-4,5(dimethyl)imidazole-2-ylidene, as a sterically hindered organocatalyst, and activated secondary N-tosyl amine as the initiator. This mechanism offers a mild and metal-free route for the polymerization of activated aziridines to obtain identical polyaziridines to those from AROP, with narrow molecular weight distributions (1.04 < Đ < 1.15) and molecular weights up to 21000 g mol−1. Depending on the steric hindrance of the ring-substituent, on the monomer, the reaction time to full conversion varies between 1 and 5 days at 50 °C in THF.
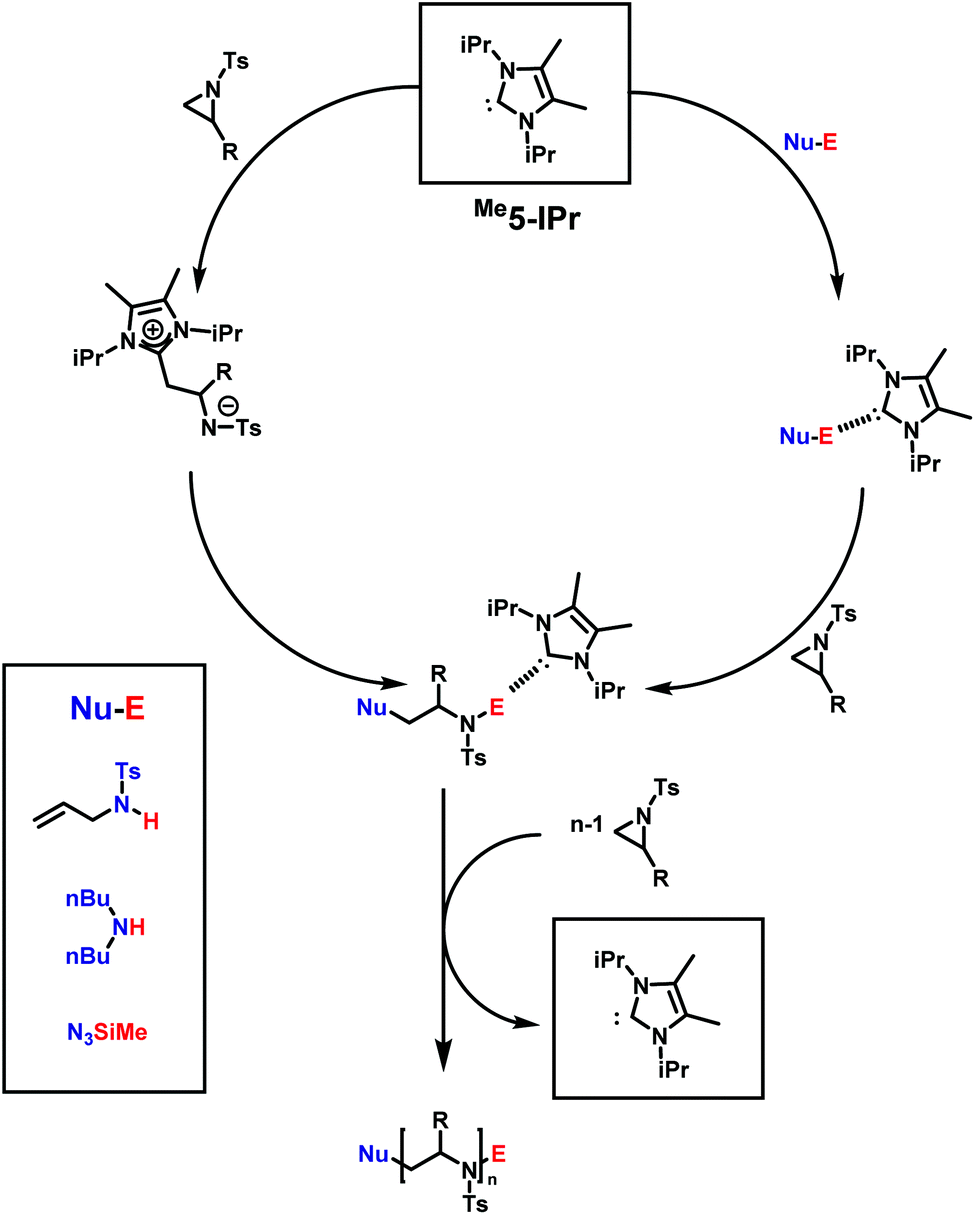 | ||
| Scheme 21 Possible mechanism for the NHC-OROP of 2-alkyl N-p-toluenesulfonyl aziridines initiated by N-allyl N-p-toluenesulfonyl amine, di-n-butylamine and trimethylsilyl azide. Reproduced from ref. 145 with permission from Elsevier, copyright 2017. | ||
Depending on the nature of the monomers, the NHCs either react as nucleophilic initiators or behave as organic catalysts by activating the initiator/active chain end. MALDI-TOF spectrometry clearly demonstrated the incorporation of the initiator (secondary N-tosyl amines) into the polymer, and a distribution with NHCs covalently bond to the polymer was not observed. The scope of practical initiators was expanded, when non-activated amines145 and unprotected aminoalcohols were investigated, which allows further post-modification of the poly(aziridine)s.
Carbene-organocatalyzed ring-opening polymerization (NHC-OROP) of activated aziridines has also been conducted with an unprotected aminoalcohol as the initiator. The NHC catalyst selectively initiated the polymerization at the secondary amine, while the alcohol group remained untouched.148 This allows for the synthesis of hydroxyl-functionalized poly(sulfonylaziridine)s which can be employed as macroinitiators for the synthesis of block copolymers. The hydroxyl group was used to initiate the ROP of lactide, catalyzed by the same carbene, to prepare PAz-b-PLLA diblock copolymers (Scheme 22).148
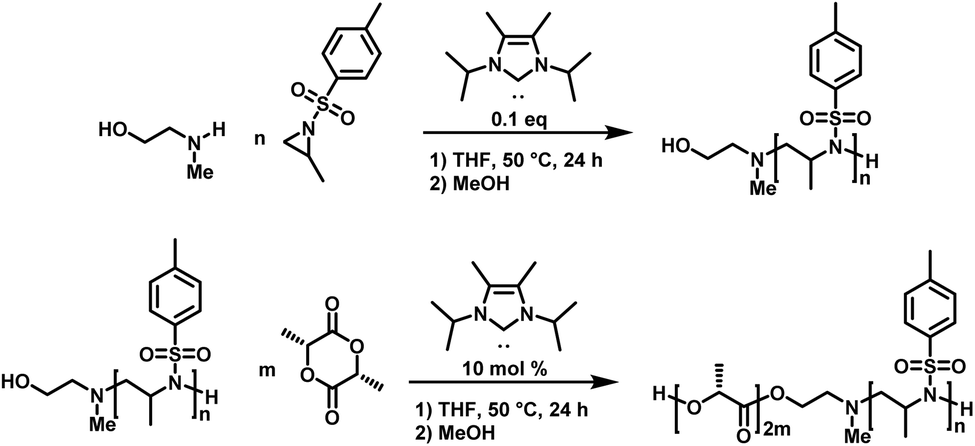 | ||
| Scheme 22 NHC-OROP of TsMAz initiated by 2-(methyl amino) ethanol, synthesis of poly(TsMAz)-b-poly(L-lactide) by sequential NHC-OROP with Me5-IPr as organocatalyst.148 | ||
Recently, another metal-free azaanionic polymerization of sulfonyl aziridines was reported,138 relying on different organic superbases, namely TMG, DBU, MTBD, TiPP and t-Bu-P4 (Scheme 23). The basicity (pKa-values of the conjugated acids) of these compounds increases in the order TMG < DBU < MTBD < TiPP < t-Bu-P4 and correlates with their increasing catalytic activity. The OROP performed best (regarding reaction time (20 min), conversion, and dispersity (Đ = 1.05)) using the most basic organic base, t-Bu-P4, but TiPP also showed satisfactory results. The remaining three bases were found to catalyze the polymerization of sulfonyl aziridines but showed higher molecular weight distributions (Đ up to 1.4). This is caused by the increased nucleophilicity of the bases leading to multiple initiators with varying rates of initiation. Overall, the strongest bases had the best catalytic activity. Moreover, the amount of catalyst could be lowered to 0.05% respect to the initiator, which indicates a very fast proton exchange, similar to oxyanionic polymerizations.74
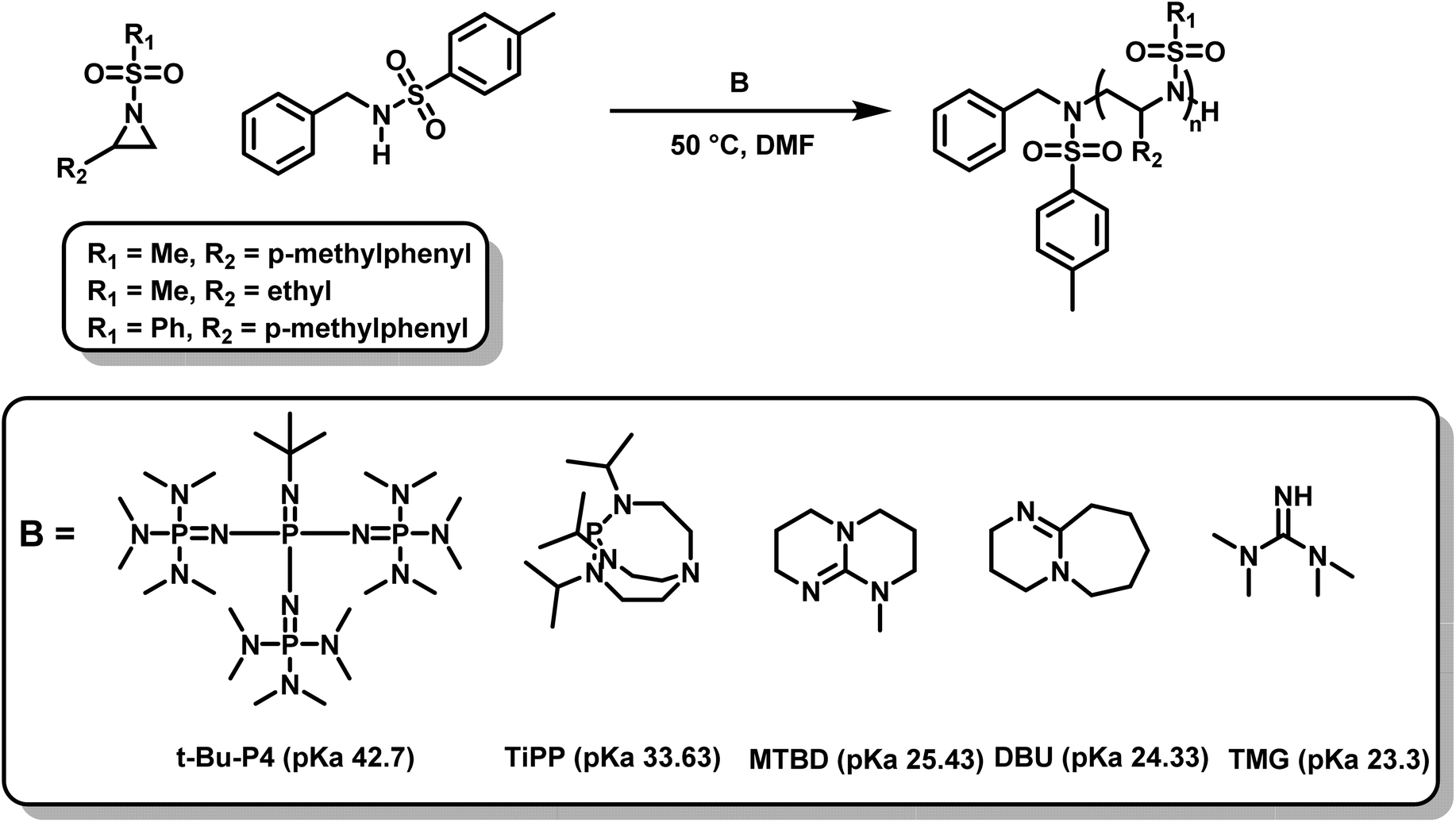 | ||
| Scheme 23 AROP of N-sulfonyl aziridines mediated by organic superbases.138 | ||
6. Desulfonylation reactions
LPEI is a linear polyamine, which is of high interest for applications in non-viral gene transfection and as polyelectrolytes.167 Today LPEI is produced from polyoxazolines following hydrolysis of the pendant amides (see above).16,26,165,166 The primary attraction to synthesizing linear PEI via the oxazoline route is due to the controlled character of the cationic polymerization of poly(2-oxazoline)s, which allows control over molar masses and dispersity.168 Generally, strongly acidic or alkaline media, and temperatures as high as 100 °C are required to transform acylated poly(2-oxazoline)s into LPEI, a process that is difficult to drive to completion. In a recent publication, Tauhardt and coworkers reported 99% hydrolysis of poly(2-oxazoline)s using 6 M HCl at 130 °C in a microwave synthesizer; the closest to complete conversion to LPEI from a poly(2-oxazoline) yet reported.169In contrast, if aziridines or azetidines are polymerized by an anionic or organocatalytic route, desulfonylation of the poly(sulfonamide)s needs to be achieved. Many published strategies exist in the literature for the reduction of low molecular weight sulfonamides to amines.170,171 According to Bergman and Toste,124 a successful desulfonylation of poly(sulfonylaziridine) was achieved by lithium napthalenide. However, no spectral analyses or molecular weight distributions of the obtained structures were given. In another approach from Wurm's group,144 tosylamides were cleaved by acidic hydrolysis with hydrobromic acid (HBr) and phenol. In spite of the harsh reaction conditions, the method was reported to be successful. Later, Wurm and coworkers were able to remove tosylamides with sodium bis(2-methoxyethoxy)aluminiumhydride (Red-Al) to ∼80% (Scheme 24).132 Rupar and coworkers were able to prepare LPEI under reductive conditions, using elemental lithium (Li), with tert-butanol (t-BuOH) in hexamethylphosphoramide (HMPA) and THF at low temperatures.83 Acidic hydrolysis under microwave irradiation, which proved to be efficient for hydrolysis of polyoxazolines,171,172 also produced desulfonylated linear polypropylenimine (LPPI, 100% desulfonylation for tosyl groups and ca. 90% for mesyl groups).82 However, chain scission could not be prevented under these harsh conditions.
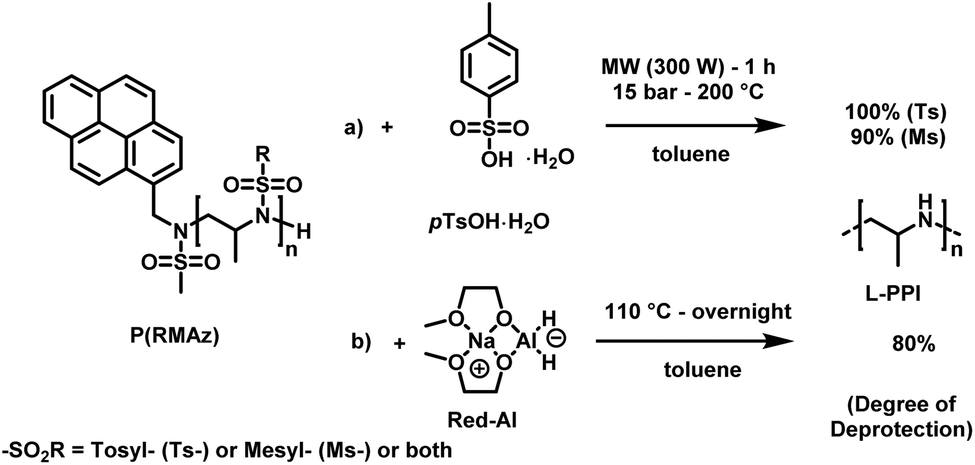 | ||
| Scheme 24 Desulfonylation methods for PAz: (a) acidic hydrolysis, using pTsOH in toluene under microwave irradiation. (b) Reductive deprotection using Red-Al in toluene.82 | ||
7. Combination of aziridines and azetidines with other polymerization techniques: copolymers and polymer architectures
7.1 Copolymers of aziridines with CO2
Ihata et al. synthesized poly(urethane-co-amine)s by copolymerization of several aziridines with CO2 (Scheme 25).173,174 The polymerizations were performed without the addition of catalyst or initiator in supercritical CO2 as the solvent, producing branched polymers of molar masses between 7000 and 15000 g mol−1. Branching occurs during the polymerization, when the secondary amines react with CO2, resulting in a carbamate and a protonated aziridine. The latter is ring-opened by nucleophilic attack, leading to branched polymers. The ratio of urethane to amine linkages in the poly(urethane-co-amine)s is affected by the CO2 pressure. By variation of the CO2 pressure from 3 to 22 MPa, copolymers with urea contents from 32 to 62% were produced. The reported yields were <35% and decreased further when the aziridine was substituted with sterically demanding side groups (i.e. 2,2 dimethylaziridine, 2-cyclohexylaziridine, etc.). The copolymers of methylaziridine and CO2 exhibited lower critical solution temperatures in water between 34 to 85 °C, which might be beneficial for the development of smart nanomaterials.
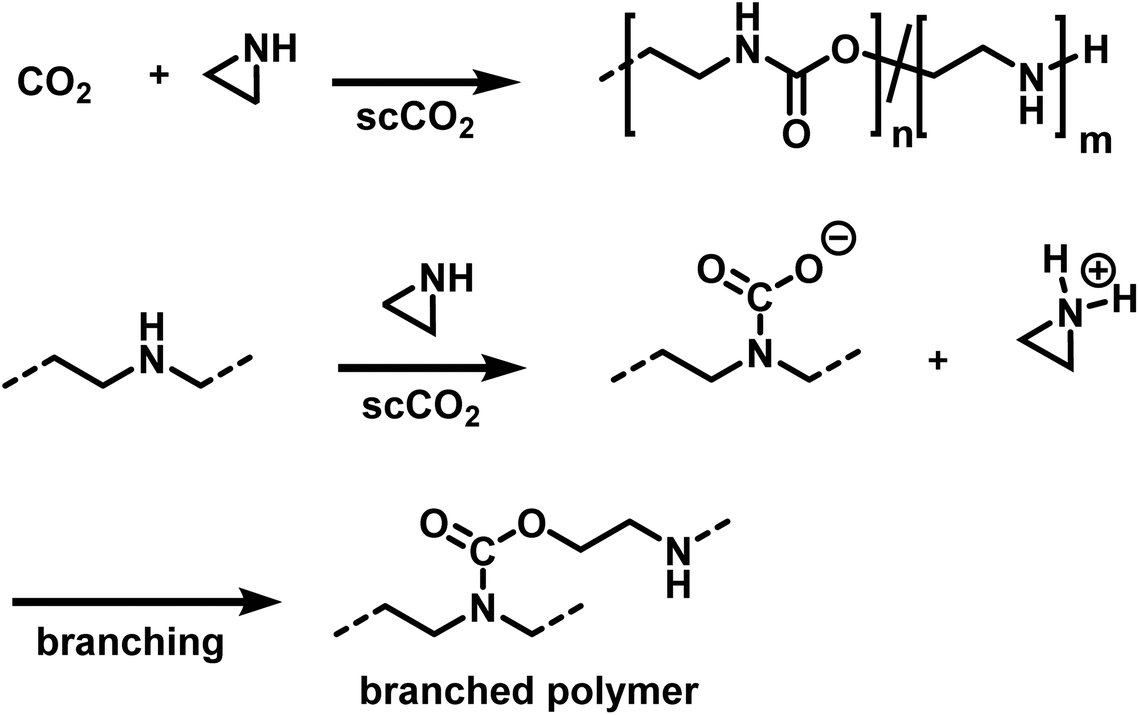 | ||
| Scheme 25 Copolymerization of aziridines and CO2 to branched poly(urethane-co-amines)s.173,174 | ||
7.2 Copolymers of aziridines with CO
Jia et al. explored the alternating copolymerization of aziridine with carbon monoxide mediated by a cobalt catalyst to prepare polyamides (Scheme 26).175 High CO pressures of up to 69 bar were necessary to obtain high polymer yields of ca. 90%. High molecular weight polyamides between 14100 and 63300 g mol−1 were synthesized. However, molar mass dispersities of up to 11.5 indicate low degrees of control over the polymerization. The authors further proposed a mechanism, in which consecutive aziridine attacks during the polymerization could lead to an amide–amine copolymer, to rationalize the broad distributions of the polymers. The amine linkages act also as nucleophiles and thereby induce branching or crosslinking.176
 | ||
| Scheme 26 Copolymerization of aziridine and carbon monoxide towards branched poly(amide-co-amines)s.175 | ||
Well-defined poly-β-peptoids can be obtained in quantitative yields with Đ = 1.11 when N-alkylated aziridines are copolymerized with carbon monoxide (Scheme 27).177N-Methyl and N-ethyl groups enhance the selectivity of the cobalt catalyst and improve the alternating copolymerization. The mechanism involves aziridine insertion into the cobalt–acyl bond, with the rate determining step being the ring-opening of the aziridine, followed by a migratory CO insertion.178 As crossover reactions, chain transfer, or combination reactions were not observed, the copolymerization of N-substituted aziridines with CO seems to follow the characteristics of a living alternating copolymerization.
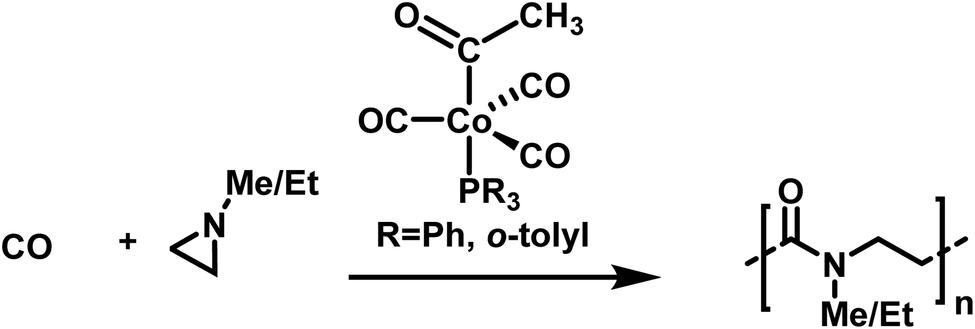 | ||
| Scheme 27 Alternating copolymerization of alkylated aziridines and carbon monoxide towards well-defined poly-β-peptoides.177 | ||
7.3 Copolymers of azetidines with CO
More recently, Jia provided the first example of a transition metal catalyzed azetidine polymerization.179 This work provided a route to poly(γ-lactams) by overcoming the difficulties associated with the ring-opening polymerization of γ-lactams.180 This was accomplished by using a cobalt catalyst to perform a carbonylative polymerization with N-n-butylazetidine and N-iso-butylazetidine. While the polymer contains only amide units for N-iso-butylazetidine, in the case of N-n-butylazetidine, it was discovered that CO is unincorporated in some instances, leaving tertiary amines along the back bone of the polymer.181 Interestingly, it was discovered that THF also participates in the reaction, leading to the formation of ester units in the polymers produced (Scheme 28).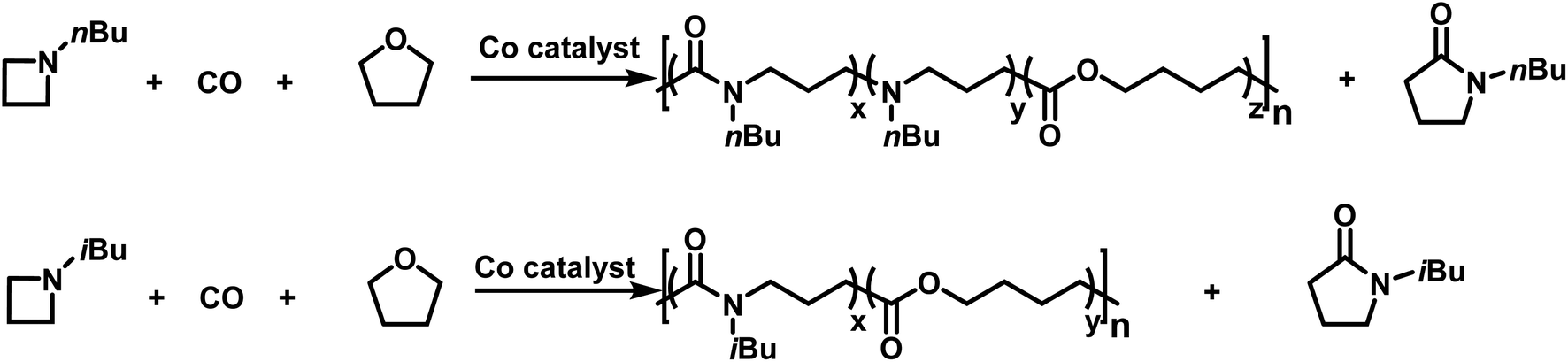 | ||
| Scheme 28 Co catalyzed carbonylative polymerization of azetidine with THF to produce poly(amide-co-ester)s.179 | ||
This was a significant finding as the incorporation of THF does not occur in the related aziridine systems.175–177,182,183 Further probing of the incorporation of THF led to the finding that increased azetidine concentration produced polymers with lower degrees of ester incorporation. This suggests that the reaction of the active chain end with THF is favored when the azetidine concentration is low. The living character, displayed by narrow molecular weight distributions and the linear increase in molecular weight with increase in conversion, coupled with in situ IR spectroscopy, suggests that the incorporation of ester units into the polymer backbone likely occurs in a gradient manner. The cobalt catalyzed carbonylative polymerization of azetidine does have a drawback in that the formation of γ-lactam also occurs. This reaction was attributed to “back-biting”,178 rather than catalyst decomposition183 due to the continued living character of the polymerizations. Jia further hypothesized that this back-biting reaction occurs at the acylazetidinium intermediate, and not the acyl-Co(CO)4 intermediate.180 This hypothesis was tested by the addition of nucleophilic I− anions to facilitate ring-opening of the acylazetidinium intermediate. The addition of LiI (2 eq. relative to Co catalyst) eliminated the γ-lactam side-product, confirming Jia's hypothesis. Curiously, it was also found that the Li counter ion also played a role in the polymerizations. This was discovered because while nBu4NI also suppressed the formation of γ-lactam, it greatly slowed the rate of polymerization. Interestingly, the addition of LiI also prevented the formation of ester linkages prior to complete consumption of azetidine. This finding allowed for the formation of block copolymers (Scheme 29). To further support the hypothesis of the Li+ cation being instrumental in the polymerization, no ester linkages were formed when nBu4NI was used as an iodide source.
 | ||
| Scheme 29 Synthesis of poly(amide-co-ester) block copolymers with LiI and sequential addition of azetidine.180 | ||
Due to the cobalt catalyzed carbonylative polymerization of azetidine having a living character, equal feeds of monomer were added over time in order to produce alternating amide and ester blocks. These polymers yielded narrow molecular weight distributions (<1.23) and produced low molecular weight polymers with similar dispersities (1.11–1.30) upon methanolysis under acidic conditions at room temperature. Complete degradation of the resulting polyamides could be further achieved by refluxing the polymers in aqueous acidic conditions. This allows for poly(amide-co-ester) block copolymers to undergo a two-stage degradation.
7.4 Combination of aziridines with other anionic polymerization techniques
In living anionic polymerization (LAP) no termination occurs, allowing the synthesis of block copolymers by sequential addition of the monomers. Thomi et al.144 prepared polystyrene-block-poly(N-tosylaziridine) by consecutive living anionic polymerization of styrene and N-tosyl aziridine (Scheme 30).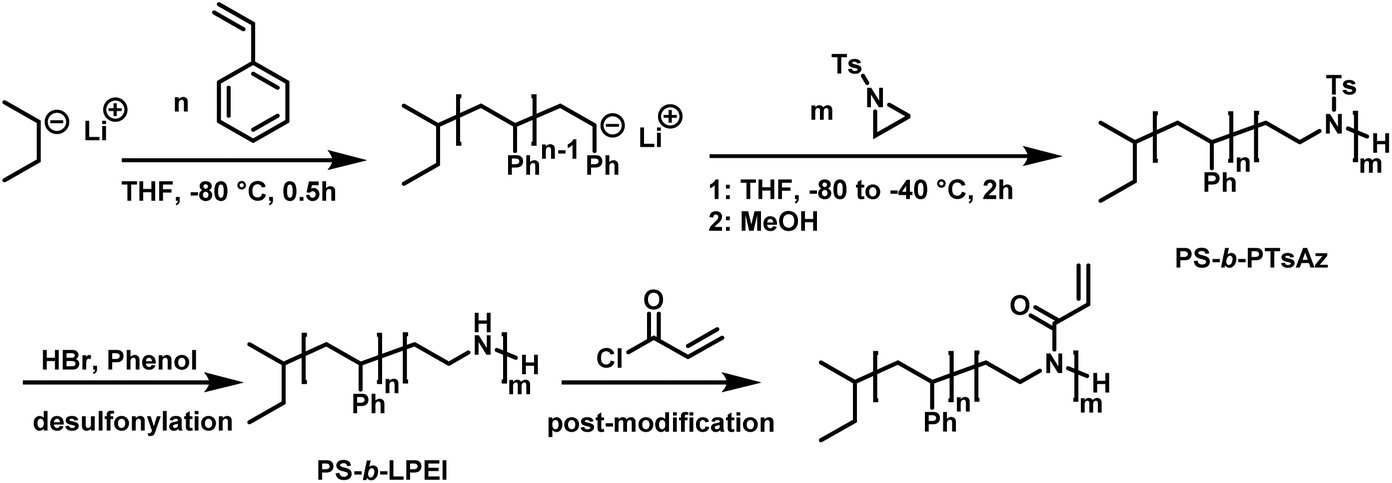 | ||
| Scheme 30 Two step synthesis of polystyrene-block-polytosylaziridine and desulfonylation to polystyrene-block-polyethylenimine.144 | ||
Quantitative transfer from the carbanions to azaanions was proven and oligomerization of the sulfonyl-activated aziridine was confirmed. Thomi et al. further demonstrated the quantitative removal of the sulfonyl groups by acidic hydrolysis with hydrobromic acid and phenol, releasing the amino groups attached to polystyrene to produce PS-b-LPEI.144 The introduced amine functionalities are a suitable platform for further efficient modifications which was shown by reaction with acryloyl chloride (Scheme 30). Short oligomers of the second block (1 < m < 5) were easily obtained, but block copolymers (up to 30 repeat units TsAz) with an increasing number of TsAz needed longer reaction times, due to the insolubility which inhibits further propagation.144
Copolymers of aziridine and ethylene oxide are interesting materials for biomedical applications or as surfactants. Attempts for the cationic ring-opening copolymerization of epoxides and N-substituted aziridines failed.184 Very recently, the anionic copolymerization of sulfonyl aziridines and ethylene oxide was achieved (Scheme 31).146 In a single step, well-defined amphiphilic block copolymers were obtained by a one-pot copolymerization. The highest difference of reactivity ratios ever reported for an anionic copolymerization (with r1 = 265 and r2 = 0.004 for 2-methyl-N-tosylaziridine/EO and r1 = 151 and r2 = 0.013 for 2-methyl-N-mesylaziridine/EO) led to the formation of block copolymers in a closed system. The amphiphilic diblock copolymers were used as a novel class of nonionic and responsive surfactants. In addition, this unique comonomer reactivity allowed fast access to multiblock copolymers: we prepared the first amphiphilic penta- or tetrablock copolymers containing aziridines in only one or two steps, respectively. These examples render the combination of epoxide and aziridine copolymerization to be a powerful strategy to sophisticated macromolecular architectures and nanostructures.
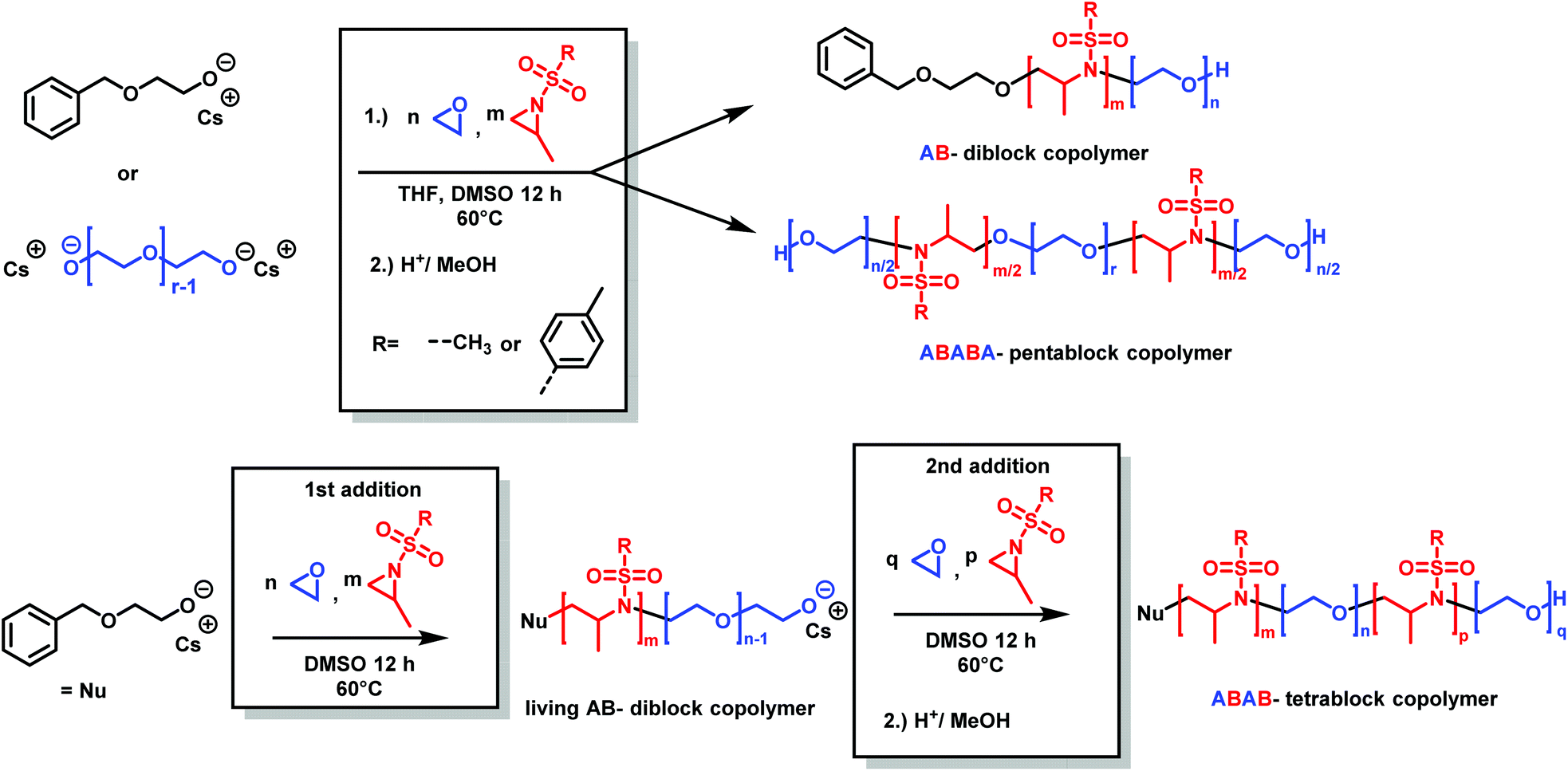 | ||
| Scheme 31 Synthesis of poly(aziridine)-b-poly(ethylene glycol) block copolymers by anionic copolymerization (2-methyl-N-tosylaziridine (TsMAz), 2-methyl-N-mesylaziridine (MsMAz), and N-tosylaziridine (TsMAz) were used in this study). Top: In a single step, either AB-diblock or ABABA-pentablock copolymers can be prepared. Bottom: Sequential addition of aziridine/EO mixture produces ABAB-tetrablock copolymers. Reproduced from ref. 146 with permission from American Chemical Society, copyright 2018. | ||
8. Polymer architectures
The aziridine building block can be used to prepare three-dimensional, polymer architectures. The typical polymer architectures for polyethylenimine-derivatives are, however, only hyperbranched (from CROP of aziridine) or linear (from CROP of oxazolines or AROP of sulfonyl aziridines). Few more complex copolymer architectures are reported to date. Current literature has studied the influence of different PEI architectures and molar masses on DNA complexation behavior, cell transfection efficiency, and toxicity.135,185–187Various multiarm star polymers with hbPEI as core were synthesized (Scheme 32). The arms of shell type star-PEIs can be obtained by different synthetic strategies. ROP allows the use of hbPEI as a macroinitiator to graft several different “shell-polymers” to the core. The most well known are: polyamide-12, ε-caprolactone, polylactide, and other polyesters. Multiarm star polymers can be afforded as well-defined nanoparticles with potential uses in nanomedicine, catalysis, and drug or gene delivery. Furthermore, star-like topologies were studied due to their unusual physical and rheological properties. These properties were found to be mainly dependant on the number of end groups, molecular weight, and the length of the arms.188
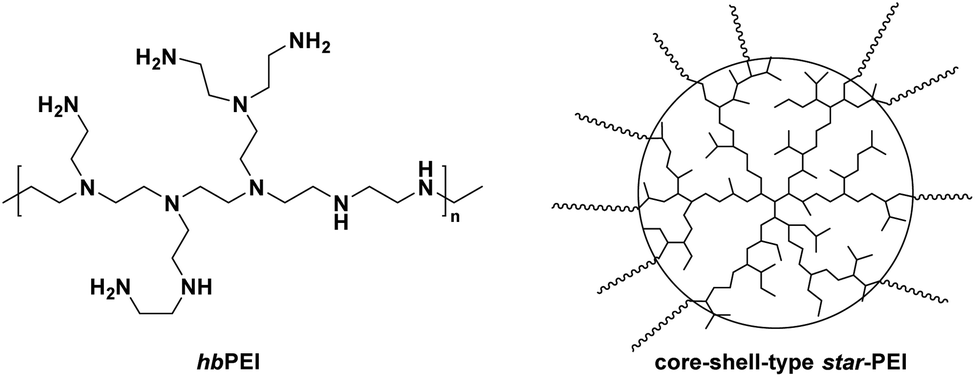 | ||
| Scheme 32 Illustration of hbPEI and core shell type star-PEI. | ||
Since the polymerization of ethyleneimine was first developed by Zomlefer and co workers89 in 1943, tuning and adjusting of the hbPEI architecture has been investigated. Comparing low molecular weight hbPEI (12 kg mol−1) (LMW-PEI) and high molecular weight hbPEI (1600 kg mol−1) (HMW-PEI) shows that the degree of branching increases with the degree of polymerization. Commercially available high molecular mass hbPEI exhibits a ratio of primary:secondary:tertiary amines close to 1:1:1, which indicates a very dense polymer structure with a branching unit on every second nitrogen.129 This ratio changes towards an excess of primary amines when decreasing the molecular weight; the amine ratio of this PEI, is mostly independent of molar masses from 8600–24300 g mol−1, is close to 3:5:2. Commercial PEI (Dp = 16) has an amine ratio of ∼4:3:3. Such LMW-PEI is usually synthesized in dilute acidic aqueous environment. The less dense structure consists on average of two linear repeating units.129,187,189 Kissel and coworkers189 demonstrated, that by varying the reaction temperature from 35 °C to 100 °C, the molecular weight of PEI can be adjusted from 24300 to 8610 g mol−1. Though certain relationships between molar mass, synthetic route, and degree of branching are known, systematic studies of these polymers regarding their properties remain challenging due to increasing dispersities with increasing Dp caused by uncontrolled crosslinking.
Cyclic PEI (c-PEI) was first synthesized by Grayson et al.190 (Scheme 33). They used propargyl p-toluene-sulfonate as initiator to polymerize ethyloxazoline under anhydrous conditions. To minimize termination and chain transfer reactions caused by aqueous impurities, the polymerization was performed in a microwave reactor. Selective termination by adding sodium azide gives α-, ω-functionalized polyoxazoline. Subsequent click-chemistry, to cyclize the polymers, followed by acidic hydrolysis, leads to c-PEI. The effect of polymer structure (i.e. linear, cyclic, branched, etc.) on DNA complexation was studied and it was determined that c-PEI polymers showed reduced toxicity and stronger complexation. This is most likely contributed to the higher charge density compared to the linear analogue. Grayson's study precisely points out the need to develop new strategies for new PEI architectures for increased understanding in terms of their properties and potential applications.
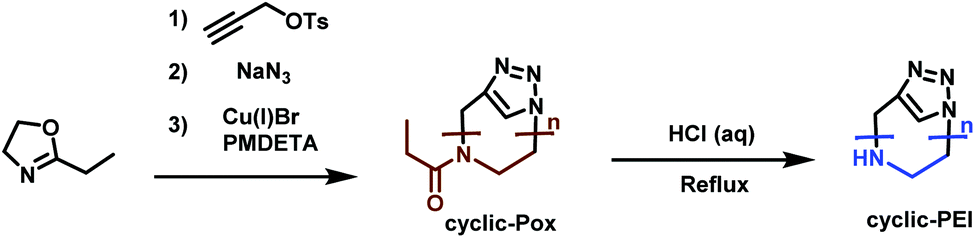 | ||
| Scheme 33 Synthesis route towards c-PEI.190 | ||
Haag and coworkers191 investigated the influence of the length of the alkylene spacer, as an efficient complexation agent for DNA needs to optimize the number of phosphate units bound. Their study investigated the complexation behavior of ethylene, propylene, and butylene spacers synthesized with different strategies (Fig. 8). Polyamines containing a butylene spacer showed stronger DNA complexation as well as decreased cytotoxicity. It was proposed, due to calculations, that the N–N distance aligns more precisely with the P–P distance of the DNA in polymers with a butylene spacer than in those with an ethylene spacer. Additionally, the increasing hydrophobicity likely also effects the biological properties.
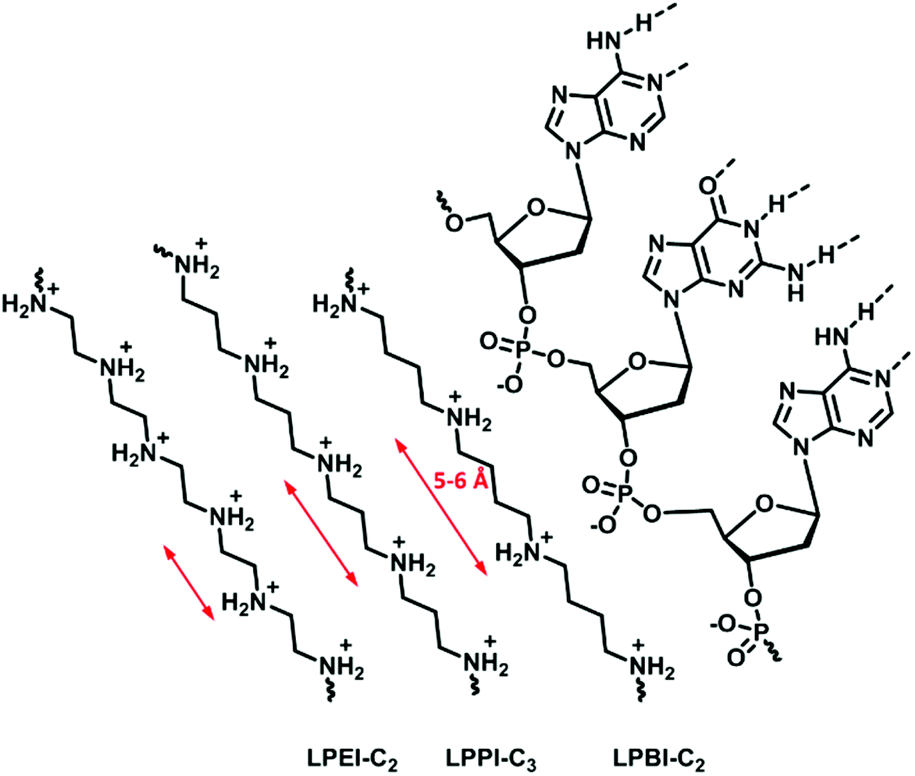 | ||
| Fig. 8 Schematic representation of the nitrogen spacing in polyamine analogs and their interaction with RNA. | ||
9. Conclusions
The polymerization of aziridines (especially ethylenimine) has had industrial relevance for several decades and polyethylenimine is used in a wide range of applications from chelators to biomedical applications. Hyperbranched PEI is available commercially at low cost, however its molecular characteristics, molar mass, and dispersity are only poorly controlled by the industrial cationic ring-opening polymerization of ethylenimine. Due to these issues, aziridine polymerizations have been sparsely studied over the course of the last couple decades. Azetidine polymers are even more exotic in both industrial and academic polymer research.However, with the development of the living anionic polymerization of sulfonyl aziridines and azetidines, the possibility to install well-defined polyamine building blocks in different macromolecular architectures has been achieved. However, to date, only very few of these polysulfonamides have been transformed into the respective polyamines, as the desulfonylation step is challenging and proceeds typically under harsh conditions, but some recent efforts have paved the way to feasible synthetic routes with control over molar mass and dispersity. While these polyamines contain high value, the polysulfonamides are also interesting, novel polymers that have been only scarcely characterized for their potential applications.
The development of the living polymerization of aziridine- and azetidine-derivatives was a milestone to develop polymer architectures with these building blocks and will open the possibility to a plethora of future applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Katharina Maisenbacher (MPIP) for assistance with graphical design. F. R. W. thanks the Deutsche Forschungsgemeinschaft (WU 750/7-1) for funding. Open Access funding provided by the Max Planck Society.References
- S. Penczek, P. Kubisa and K. Matyjaszewski, Oxiranes, Springer Berlin Heidelberg, Berlin, Heidelberg, 1985 Search PubMed.
- G. Gee, W. C. E. Higginson, P. Levesley and K. J. Taylor, J. Chem. Soc., 1959, 1338–1344, 10.1039/JR9590001338.
- G. Gee, W. C. E. Higginson and G. T. Merrall, J. Chem. Soc., 1959, 1345–1352, 10.1039/JR9590001345.
- E. H. Schacht and E. J. Goethals, Makromol. Chem., 1974, 175, 3447–3459 CrossRef CAS.
- E. J. Goethals, E. H. Schacht, P. Bruggeman and P. Bossaer, in Ring-Opening Polymerization, American Chemical Society, 1977, vol. 59, ch. 1, pp. 1–12 Search PubMed.
- S. Kobayashi, Prog. Polym. Sci., 1990, 15, 751–823 CrossRef CAS.
- M. Jäger, S. Schubert, S. Ochrimenko, D. Fischer and U. S. Schubert, Chem. Soc. Rev., 2012, 41, 4755–4767 RSC.
- M. L. Sarazen and C. W. Jones, Macromolecules, 2017, 50, 9135–9143 CrossRef CAS.
- In the literature, polytrimethylenimine (PTMI) is also called polypropylenimine (PPI). We prefer PTMI.
- S. W. Benson, F. R. Cruickshank, D. M. Golden, G. R. Haugen, H. E. O'Neal, A. S. Rodgers, R. Shaw and R. Walsh, Chem. Rev., 1969, 69, 279–324 CrossRef CAS.
- H. K. Eigenmann, D. M. Golden and S. W. Benson, J. Phys. Chem., 1973, 77, 1687–1691 CrossRef CAS.
- T. Li, L. Wu, J. Zhang, G. Xi, Y. Pang, X. Wang and T. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 31311–31320 CrossRef CAS PubMed.
- J. Pan, Z. Lyu, W. Jiang, H. Wang, Q. Liu, M. Tan, L. Yuan and H. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 14391–14398 CrossRef CAS PubMed.
- X. Ding, W. Wang, Y. Wang, X. Bao, Y. Wang, C. Wang, J. Chen, F. Zhang and J. Zhou, Mol. Pharm., 2014, 11, 3307–3321 CrossRef CAS PubMed.
- Y.-L. Lo, K.-H. Sung, C.-C. Chiu and L.-F. Wang, Mol. Pharm., 2013, 10, 664–676 CrossRef CAS PubMed.
- M. Jaeger, S. Schubert, S. Ochrimenko, D. Fischer and U. S. Schubert, Chem. Soc. Rev., 2012, 41, 4755–4767 RSC.
- S. C. De Smedt, J. Demeester and W. E. Hennink, Pharm. Res., 2000, 17, 113–126 CrossRef CAS.
- D. W. Pack, A. S. Hoffman, S. Pun and P. S. Stayton, Nat. Rev. Drug Discovery, 2005, 4, 581 CrossRef CAS PubMed.
- C. T. De Ilarduya, Y. Sun and N. Düzgüneş, Eur. J. Pharm. Sci., 2010, 40, 159–170 CrossRef PubMed.
- M. D. Giron-Gonzalez, R. Salto-Gonzalez, F. J. Lopez-Jaramillo, A. Salinas-Castillo, A. B. Jodar-Reyes, M. Ortega-Muñoz, F. Hernandez-Mateo and F. Santoyo-Gonzalez, Bioconjugate Chem., 2016, 27, 549–561 CrossRef CAS PubMed.
- M. A. Mintzer and E. E. Simanek, Chem. Rev., 2008, 109, 259–302 CrossRef PubMed.
- M. Neu, D. Fischer and T. Kissel, J. Gene Med., 2005, 7, 992–1009 CrossRef CAS PubMed.
- C. Raymond, R. Tom, S. Perret, P. Moussouami, D. L'Abbé, G. St-Laurent and Y. Durocher, Methods, 2011, 55, 44–51 CrossRef CAS PubMed.
- E. Altuntaş, K. Knop, L. Tauhardt, K. Kempe, A. C. Crecelius, M. Jäger, M. D. Hager and U. S. Schubert, J. Mass Spectrom., 2012, 47, 105–114 CrossRef PubMed.
- S. M. Moghimi, P. Symonds, J. C. Murray, A. C. Hunter, G. Debska and A. Szewczyk, Mol. Ther., 2005, 11, 990–995 CrossRef CAS PubMed.
- O. Boussif, F. Lezoualc'h, M. A. Zanta, M. D. Mergny, D. Scherman, B. Demeneix and J.-P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7297–7301 CrossRef CAS PubMed.
- M. Thomas and A. M. Klibanov, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9138–9143 CrossRef CAS PubMed.
- M. S. Shim and Y. J. Kwon, Bioconjugate Chem., 2009, 20, 488–499 CrossRef CAS PubMed.
- M.-E. Bonnet, P. Erbacher and A.-L. Bolcato-Bellemin, Pharm. Res., 2008, 25, 2972 CrossRef CAS PubMed.
- L. Wightman, R. Kircheis, V. Rössler, S. Carotta, R. Ruzicka, M. Kursa and E. Wagner, J. Gene Med., 2001, 3, 362–372 CrossRef CAS PubMed.
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin and D. G. Anderson, Nat. Rev. Genet., 2014, 15, 541 CrossRef CAS PubMed.
- T. K. Kim and J. H. Eberwine, Anal. Bioanal. Chem., 2010, 397, 3173–3178 CrossRef CAS PubMed.
- L. Xie, Y. Tan, Z. Wang, H. Liu, N. Zhang, C. Zou, X. Liu, G. Liu, J. Lu and H. Zheng, ACS Appl. Mater. Interfaces, 2016, 8, 29261–29269 CrossRef CAS PubMed.
- H. Nguyen, P. Lemieux, S. Vinogradov, C. Gebhart, N. Guerin, G. Paradis, T. Bronich, V. Alakhov and A. Kabanov, Gene Ther., 2000, 7, 126 CrossRef CAS PubMed.
- H. Petersen, P. M. Fechner, A. L. Martin, K. Kunath, S. Stolnik, C. J. Roberts, D. Fischer, M. C. Davies and T. Kissel, Bioconjugate Chem., 2002, 13, 845–854 CrossRef CAS PubMed.
- M. Breunig, U. Lungwitz, R. Liebl and A. Goepferich, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 14454–14459 CrossRef CAS PubMed.
- N. M. Milović, J. Wang, K. Lewis and A. M. Klibanov, Biotechnol. Bioeng., 2005, 90, 715–722 CrossRef PubMed.
- M. Thomas and A. M. Klibanov, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14640–14645 CrossRef CAS PubMed.
- M. Wang, P. Lu, B. Wu, J. D. Tucker, C. Cloer and Q. Lu, J. Mater. Chem., 2012, 22, 6038–6046 RSC.
- P. Xu, G. K. Quick and Y. Yeo, Biomaterials, 2009, 30, 5834–5843 CrossRef CAS PubMed.
- B.-K. Kim, D. Kim, G. Kwak, J. Y. Yhee, I.-C. Kwon, S. H. Kim and Y. Yeo, ACS Biomater. Sci. Eng., 2017, 3, 990–999 CrossRef CAS PubMed.
- Y. Zhou, F. Yu, F. Zhang, G. Chen, K. Wang, M. Sun, J. Li and D. Oupickỳ, Biomacromolecules, 2018, 19(2), 392–401 CrossRef CAS PubMed.
- F. Wang, L. Gao, L.-Y. Meng, J.-M. Xie, J.-W. Xiong and Y. Luo, ACS Appl. Mater. Interfaces, 2016, 8, 33529–33538 CrossRef CAS PubMed.
- L. Tauhardt, K. Kempe, K. Knop, E. Altuntaş, M. Jäger, S. Schubert, D. Fischer and U. S. Schubert, Macromol. Chem. Phys., 2011, 212, 1918–1924 CAS.
- V. P. Dhende, S. Samanta, D. M. Jones, I. R. Hardin and J. Locklin, ACS Appl. Mater. Interfaces, 2011, 3, 2830–2837 CrossRef CAS PubMed.
- J. Gao, E. M. White, Q. Liu and J. Locklin, ACS Appl. Mater. Interfaces, 2017, 9, 7745–7751 CrossRef CAS PubMed.
- J. Haldar, D. An, L. A. de Cienfuegos, J. Chen and A. M. Klibanov, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17667–17671 CrossRef CAS PubMed.
- A. M. Bieser and J. C. Tiller, Macromol. Biosci., 2011, 11, 526–534 CrossRef CAS PubMed.
- S. A. Koplin, S. Lin and T. Domanski, Biotechnol. Prog., 2008, 24, 1160–1165 CrossRef CAS PubMed.
- R. Kügler, O. Bouloussa and F. Rondelez, Microbiology, 2005, 151, 1341–1348 CrossRef PubMed.
- J. Lin, J. C. Tiller, S. B. Lee, K. Lewis and A. M. Klibanov, Biotechnol. Lett., 2002, 24, 801–805 CrossRef CAS.
- A. M. Bieser and J. C. Tiller, Macromol. Biosci., 2011, 11, 526–534 CrossRef CAS PubMed.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796–854 CrossRef CAS PubMed.
- J. C. Hicks, J. H. Drese, D. J. Fauth, M. L. Gray, G. Qi and C. W. Jones, J. Am. Chem. Soc., 2008, 130, 2902–2903 CrossRef CAS PubMed.
- P. Li, B. Ge, S. Zhang, S. Chen, Q. Zhang and Y. Zhao, Langmuir, 2008, 24, 6567–6574 CrossRef CAS PubMed.
- F.-Q. Liu, L. Wang, Z.-G. Huang, C.-Q. Li, W. Li, R.-X. Li and W.-H. Li, ACS Appl. Mater. Interfaces, 2014, 6, 4371–4381 CrossRef CAS PubMed.
- Q. Wang, J. Luo, Z. Zhong and A. Borgna, Energy Environ. Sci., 2011, 4, 42–55 RSC.
- X. Xu, C. Song, B. G. Miller and A. W. Scaroni, Fuel Process. Technol., 2005, 86, 1457–1472 CrossRef CAS.
- W. Chaikittisilp, R. Khunsupat, T. T. Chen and C. W. Jones, Ind. Eng. Chem. Res., 2011, 50, 14203–14210 CrossRef CAS.
- N. MacDowell, N. Florin, A. Buchard, J. Hallett, A. Galindo, G. Jackson, C. S. Adjiman, C. K. Williams, N. Shah and P. Fennell, Energy Environ. Sci., 2010, 3, 1645–1669 RSC.
- R. Sanz, G. Calleja, A. Arencibia and E. S. Sanz-Pérez, J. Mater. Chem. A, 2013, 1, 1956–1962 RSC.
- X. Xu, C. Song, J. M. Andresen, B. G. Miller and A. W. Scaroni, Energy Fuels, 2002, 16, 1463–1469 CrossRef CAS.
- S. Satyapal, T. Filburn, J. Trela and J. Strange, Energy Fuels, 2001, 15, 250–255 CrossRef CAS.
- E. Bayer, B. Y. Spivakov and K. Geckeler, Polym. Bull., 1985, 13, 307–311 CAS.
- S. Kobayashi, K. Hiroishi, M. Tokunoh and T. Saegusa, Macromolecules, 1987, 20, 1496–1500 CrossRef CAS.
- A. Von Zelewsky, L. Barbosa and C. Schläpfer, Coord. Chem. Rev., 1993, 123, 229–246 CrossRef CAS.
- B. A. Bolto, Prog. Polym. Sci., 1995, 20, 987–1041 CrossRef CAS.
- H.-C. Chen, S.-W. Lin, J.-M. Jiang, Y.-W. Su and K.-H. Wei, ACS Appl. Mater. Interfaces, 2015, 7, 6273–6281 CrossRef CAS PubMed.
- S. Stolz, Y. Zhang, U. Lemmer, G. Hernandez-Sosa and H. Aziz, ACS Appl. Mater. Interfaces, 2017, 9, 2776–2785 CrossRef CAS PubMed.
- G. F. Zou, J. Zhao, H. M. Luo, T. M. McCleskey, A. K. Burrell and Q. X. Jia, Chem. Soc. Rev., 2013, 42, 439–449 RSC.
- https://www.chempoint.com/products/basf/lupasol-polyethylenimine-adhesion-promoters/lupasol-polyethylenimine .
- U. Steuerle and R. Feuerhake, Ullmann's Encyclopedia of Industrial Chemistry, 2001 Search PubMed.
- J. B. Sweeney, Chem. Soc. Rev., 2002, 31, 247–258 RSC.
- J. Herzberger, K. Niederer, H. Pohlit, J. Seiwert, M. Worm, F. R. Wurm and H. Frey, Chem. Rev., 2016, 116, 2170–2243 CrossRef CAS PubMed.
- B. D. Monnery and R. Hoogenboom, Cationic Polymers in Regenerative Medicine, 2014, pp. 30–61 Search PubMed.
- D. A. Tomalia and D. P. Sheetz, J. Polym. Sci., Part A-1: Polym. Chem., 1966, 4, 2253–2265 CrossRef CAS.
- W. Seeliger, E. Aufderhaar, W. Diepers, R. Feinauer, R. Nehring, W. Thier and H. Hellmann, Angew. Chem., Int. Ed. Engl., 1966, 5, 875–888 CrossRef CAS PubMed.
- T. Kagiya, S. Narisawa, T. Maeda and K. Fukui, J. Polym. Sci., Part B: Polym. Lett., 1966, 4, 441–445 CrossRef CAS.
- R. Tanaka, I. Ueoka, Y. Takaki, K. Kataoka and S. Saito, Macromolecules, 1983, 16, 849–853 CrossRef CAS.
- H. M. L. Lambermont-Thijs, F. S. van der Woerdt, A. Baumgaertel, L. Bonami, F. E. Du Prez, U. S. Schubert and R. Hoogenboom, Macromolecules, 2009, 43, 927–933 CrossRef.
- T. Saegusa, A. Yamada, H. Taoda and S. Kobayashi, Macromolecules, 1978, 11, 435–436 CrossRef CAS.
- E. Rieger, T. Gleede, A. Manhart, M. Lamla and F. R. Wurm, ACS Macro Lett., 2018, 7, 598–603 CrossRef CAS.
- L. Reisman, C. P. Mbarushimana, S. J. Cassidy and P. A. Rupar, ACS Macro Lett., 2016, 5, 1137–1140 CrossRef CAS.
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin and D. G. Anderson, Nat. Rev. Genet., 2014, 15, 541 CrossRef CAS PubMed.
- P. Wilson, P. C. Ke, T. P. Davis and K. Kempe, Eur. Polym. J., 2017, 88, 486–515 CrossRef CAS.
- L. Tauhardt, K. Kempe, M. Gottschaldt and U. S. Schubert, Chem. Soc. Rev., 2013, 42, 7998–8011 RSC.
- M. A. Mees and R. Hoogenboom, Polym. Chem., 2018, 9, 4968–4978 RSC.
- J. Y. Johnson and G. W. Johnson, Great Britain Pat., GB461666 (A), 1937 Search PubMed.
- G. D. Jones, A. Langsjoen, S. M. M. C. Neumann and J. Zomlefer, J. Org. Chem., 1944, 09(2), 125–147 CrossRef CAS.
- D. R. Holycross and M. Chai, Macromolecules, 2013, 46, 6891–6897 CrossRef CAS.
- W. G. Barb, J. Chem. Soc., 1955, 2564–2577, 10.1039/JR9550002564.
- B. L. Rivas and B. Barría, Polym. Bull., 1996, 36, 157–164 CrossRef CAS.
- T. Toshikazu, Y. Z. Menceloglu and E. Takeshi, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 501–504 CrossRef.
- M. Bakloutl, R. Chaabouni, J. Sledz and F. Schué, Polym. Bull., 1989, 21, 243–250 CrossRef.
- J. Xu, X. Li and N. Chen, Synthesis, 2010, 3423–3428 CrossRef.
- J. A. Deyrup, in The Chemistry of Heterocyclic Compounds, Part 1, ed. A. Hassner, Wiley, New York, 1983, vol. 42, pp. 1–214 Search PubMed.
- E. Goethals, E. Schacht, P. Bruggeman and P. Bossaer, Cationic Polymerization of Cyclic Amines, ACS Publications, 1977, vol. 59, DOI:10.1021/bk-1977-0059.ch001.
- C. R. Dick, J. Org. Chem., 1967, 32, 72–75 CrossRef CAS.
- K. F. Weyts and E. J. Goethals, Polym. Bull., 1988, 19, 13–19 CrossRef CAS.
- J. J. Jakubowski and R. V. Subramanian, Polymer, 1980, 21, 230–232 CrossRef CAS.
- K. E. Geckeler, B. L. Rivas and R. Zhou, Angew. Makromol. Chem., 1991, 193, 195–203 CrossRef CAS.
- S. A. Pooley, G. S. Canessa and B. L. Rivas, Macromol. Chem. Phys., 1998, 199, 2293–2299 CrossRef CAS.
- B. L. Rivas, G. S. Canessa and S. A. Pooley, Makromol. Chem., Rapid Commun., 1987, 8, 365–372 CrossRef CAS.
- S. A. Pooley, G. S. Canessa, B. L. Rivas and E. Espejo, Polym. Bull., 1995, 35, 271–277 CrossRef CAS.
- S. A. Pooley, G. S. Canessa, B. L. Rivas and E. Espejo, Polym. Bull., 1996, 36, 415–422 CrossRef CAS.
- S. A. Pooley, G. S. Canessa, B. L. Rivas and E. Espejo, Polym. Bull., 1997, 39, 407–414 CrossRef CAS.
- S. A. Pooley, G. S. Canessa, B. L. Rivas and E. Espejo, Bol. Soc. Chil. Quim., 1996, 41, 261–270 CAS.
- S. A. Pooley, G. S. Canessa, B. L. Rivas and E. Espejo, Bol. Soc. Chil. Quim., 1994, 39, 305–313 CAS.
- S. A. Pooley, G. S. Canessa, B. L. Rivas and E. Espejo, Bol. Soc. Chil. Quim., 1996, 41, 71–78 CAS.
- 1967.
- S. Hashimoto and T. Yamashita, J. Macromol. Sci., Chem., 1986, 23, 295–304 CrossRef.
- S. Hashimoto, T. Yamashita and J. Hino, Polym. J., 1977, 9, 19 CrossRef CAS.
- E. R. Lavagnino, R. R. Chauvette, W. N. Cannon and E. C. Kornfeld, J. Am. Chem. Soc., 1960, 82, 2609–2613 CrossRef CAS.
- H. Oike, M. Washizuka and Y. Tezuka, Macromol. Chem. Phys., 2000, 201, 1673–1678 CrossRef CAS.
- E. H. Schacht and E. J. Goethals, Makromol. Chem., 1973, 167, 155–169 CrossRef CAS.
- E. J. Goethals and B. Dervaux, in Polymer Science: A Comprehensive Reference, ed. M. Möller, Elsevier, Amsterdam, 2012, pp. 309–330, DOI:10.1016/B978-0-444-53349-4.00108-4.
- E. J. Goethals, E. H. Schacht, Y. E. Bogaert, S. I. Ali and Y. Tezuka, Polym. J., 1980, 12, 571 CrossRef CAS.
- T. Saegusa and E. Goethals, Ring-Opening Polymerization, American Chemical Society, 1977 Search PubMed.
- A. T. Bottini and J. D. Roberts, J. Am. Chem. Soc., 1958, 80, 5203–5208 CrossRef CAS.
- H. K. Hall, J. Am. Chem. Soc., 1957, 79, 5441–5444 CrossRef CAS.
- S. Penczek and P. Kubisa, Makromol. Chem., 1969, 130, 186–209 CrossRef CAS.
- E. J. Goethals, G. G. Trossaert, P. J. Hartmann and K. Engelen, Makromol. Chem., Macromol. Symp., 1993, 73, 77–89 Search PubMed.
- E. J. Goethals, E. Schacht and D. Tack, J. Polym. Sci., Part A-1: Polym. Chem., 1972, 10, 533–539 CrossRef CAS.
- I. C. Stewart, C. C. Lee, R. G. Bergman and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 17616–17617 CrossRef CAS PubMed.
- Z. Wang, Comprehensive Organic Name Reactions and Reagents, John Wiley & Sons, Inc., 2009, vol. 3, ISBN: 978-0-471-70450-8 Search PubMed.
- H. Xu, H. Tian, L. Zheng, Q. Liu, L. Wang and S. Zhang, Tetrahedron Lett., 2011, 52, 2873–2875 CrossRef CAS.
- M. B. Berry and D. Craig, Synlett, 1992, 41–44 CrossRef CAS.
- A. J. Catino, J. M. Nichols, R. E. Forslund and M. P. Doyle, Org. Lett., 2005, 7, 2787–2790 CrossRef CAS PubMed.
- A. V. Gontcharov, H. Liu and K. B. Sharpless, Org. Lett., 1999, 1, 783–786 CrossRef CAS.
- J. U. Jeong, B. Tao, I. Sagasser, H. Henniges and K. B. Sharpless, J. Am. Chem. Soc., 1998, 120, 6844–6845 CrossRef CAS.
- E. Rieger, A. Alkan, A. Manhart, M. Wagner and F. R. Wurm, Macromol. Rapid Commun., 2016, 37, 833–839 CrossRef CAS PubMed.
- E. Rieger, A. Manhart and F. R. Wurm, ACS Macro Lett., 2016, 5, 195–198 CrossRef CAS.
- L. Thomi and F. R. Wurm, Macromol. Symp., 2015, 349, 51–56 CrossRef CAS.
- P. O'Brien and J. Huang, Synthesis, 2006, 425–434, DOI:10.1055/s-2006-926282.
- Y. Zhao, F. Sakai, L. Su, Y. Liu, K. Wei, G. Chen and M. Jiang, Adv. Mater., 2013, 25, 5215–5256 CrossRef CAS PubMed.
- E. Rieger, J. Blankenburg, E. Grune, M. Wagner, K. Landfester and F. R. Wurm, Angew. Chem., Int. Ed., 2018, 57, 2483–2487 CrossRef CAS PubMed.
- T. Gleede, E. Rieger, T. Homann-Müller and F. R. Wurm, Macromol. Chem. Phys., 2017, 1700145, DOI:10.1002/macp.201700145.
- X. Wang, Y. Liu, Z. Li, H. Wang, H. Gebru, S. Chen, H. Zhu, F. Wei and K. Guo, ACS Macro Lett., 2017, 6, 1331–1336 CrossRef CAS.
- T. Gleede, E. Rieger, L. Liu, C. Bakkali-Hassani, M. Wagner, S. Carlotti, D. Taton, D. Andrienko and F. R. Wurm, Macromolecules, 2018, 51, 5713–5719 CrossRef CAS.
- T. Homann-Müller, E. Rieger, A. Alkan and F. R. Wurm, Polym. Chem., 2016, 7, 5501–5506 RSC.
- P. C. Mbarushimana, Q. Liang, J. M. Allred and P. A. Rupar, Macromolecules, 2018, 51(3), 977–983 CrossRef CAS.
- L. Reisman, E. A. Rowe, Q. Liang and P. A. Rupar, Polym. Chem., 2018, 9, 1618–1625 RSC.
- E. Rieger, T. Gleede, K. Weber, A. Manhart, M. Wagner and F. R. Wurm, Polym. Chem., 2017, 8, 2824–2832 RSC.
- L. Thomi and F. R. Wurm, Macromol. Rapid Commun., 2014, 35, 585–589 CrossRef CAS PubMed.
- C. Bakkali-Hassani, E. Rieger, J. Vignolle, F. R. Wurm, S. Carlotti and D. Taton, Eur. Polym. J., 2017, 95, 746–755 CrossRef CAS.
- T. Gleede, E. Rieger, J. Blankenburg, K. Klein and F. R. Wurm, J. Am. Chem. Soc., 2018, 140, 13407–13412 CrossRef CAS PubMed.
- C. Bakkali-Hassani, E. Rieger, J. Vignolle, F. R. Wurm, S. Carlotti and D. Taton, Chem. Commun., 2016, 52, 9719–9722 RSC.
- C. Bakkali-Hassani, C. Coutouly, T. Gleede, J. Vignolle, F. R. Wurm, S. Carlotti and D. Taton, Macromolecules, 2018, 51, 2533–2541 CrossRef CAS.
- E. Rieger, T. Gleede, K. Weber, A. Manhart, M. Wagner and F. R. Wurm, Polym. Chem., 2017, 8, 2824–2832 RSC.
- V. Jaacks, Angew. Chem., 1967, 79, 419 CrossRef.
- F. T. Wall, J. Am. Chem. Soc., 1941, 63, 1862–1866 CrossRef CAS.
- J. Blankenburg, M. Wagner and H. Frey, Macromolecules, 2017, 50, 8885–8893 CrossRef CAS.
- D. C. McLeod and N. V. Tsarevsky, Macromol. Rapid Commun., 2016, 37, 1694–1700 CrossRef CAS PubMed.
- H.-J. Jang, J. T. Lee and H. J. Yoon, Polym. Chem., 2015, 6, 3387–3391 RSC.
- H. K. Moon, S. Kang and H. J. Yoon, Polym. Chem., 2017, 8, 2287–2291 RSC.
- T. Suzuki, J.-i. Kusakabe, K. Kitazawa, T. Nakagawa, S. Kawauchi and T. Ishizone, Macromolecules, 2010, 43, 107–116 CrossRef CAS.
- A. Thomas, S. S. Muller and H. Frey, Biomacromolecules, 2014, 15, 1935–1954 CrossRef CAS PubMed.
- A. Alkan, R. Klein, S. I. Shylin, U. Kemmer-Jonas, H. Frey and F. R. Wurm, Polym. Chem., 2015, 6, 7112–7118 RSC.
- L. Reisman, E. A. Rowe, E. M. Jackson, C. Thomas, T. Simone and P. A. Rupar, J. Am. Chem. Soc., 2018, 140, 15626–15630 CrossRef CAS PubMed.
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS.
- S. Naumann and A. P. Dove, Polym. Chem., 2015, 6, 3185–3200 RSC.
- J. Pinaud, K. Vijayakrishna, D. Taton and Y. Gnanou, Macromolecules, 2009, 42, 4932–4936 CrossRef CAS.
- J. Raynaud, Y. Gnanou and D. Taton, Macromolecules, 2009, 42, 5996–6005 CrossRef CAS.
- J. Raynaud, C. Absalon, Y. Gnanou and D. Taton, J. Am. Chem. Soc., 2009, 131, 3201–3209 CrossRef CAS PubMed.
- I. Perevyazko, A. S. Gubarev, L. Tauhardt, A. Dobrodumov, G. M. Pavlov and U. S. Schubert, Polym. Chem., 2017, 8, 7169–7179 RSC.
- A. Akinc, M. Thomas, A. M. Klibanov and R. Langer, J. Gene Med., 2005, 7, 657–663 CrossRef CAS PubMed.
- S. Taranejoo, J. Liu, P. Verma and K. Hourigan, J. Appl. Polym. Sci., 2015, 132, 42096 CrossRef.
- K. Aoi and M. Okada, Prog. Polym. Sci., 1996, 21, 151–208 CrossRef CAS.
- L. Tauhardt, K. Kempe, K. Knop, E. Altuntaş, M. Jäger, S. Schubert, D. Fischer and U. S. Schubert, Macromol. Chem. Phys., 2011, 212, 1918–1924 CAS.
- D. A. Alonso and P. G. Andersson, J. Org. Chem., 1998, 63, 9455–9461 CrossRef CAS.
- H. Senboku, K. Nakahara, T. Fukuhara and S. Hara, Tetrahedron Lett., 2010, 51, 435–438 CrossRef CAS.
- I. V. Kubrakova, A. A. Formanovsky and I. V. Mikhura, Mendeleev Commun., 1999, 9, 65–66 CrossRef.
- O. Ihata, Y. Kayaki and T. Ikariya, Macromolecules, 2005, 38, 6429–6434 CrossRef CAS.
- O. Ihata, Y. Kayaki and T. Ikariya, Angew. Chem., Int. Ed., 2004, 43, 717–719 CrossRef CAS PubMed.
- L. Jia, E. Ding and W. R. Anderson, Chem. Commun., 2001, 1436–1437, 10.1039/B103899K.
- J. Zhao, E. Ding, A. M. Allgeier and L. Jia, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 376–385 CrossRef CAS.
- L. Jia, H. Sun, J. T. Shay, A. M. Allgeier and S. D. Hanton, J. Am. Chem. Soc., 2002, 124, 7282–7283 CrossRef CAS PubMed.
- D. J. Darensbourg, A. L. Phelps, N. L. Gall and L. Jia, J. Am. Chem. Soc., 2004, 126, 13808–13815 CrossRef CAS PubMed.
- G. Liu and L. Jia, Angew. Chem., Int. Ed., 2006, 45, 129–131 CrossRef CAS PubMed.
- H. Sekiguchi, P. Tsourkas, F. Carriere and R. Audebert, Eur. Polym. J., 1974, 10, 1185–1193 CrossRef CAS.
- Due to this finding, N-isopropylazetidine was used throughout the study.
- G. Liu and L. Jia, J. Am. Chem. Soc., 2004, 126, 14716–14717 CrossRef CAS PubMed.
- H. Xu, N. LeGall, L. Jia, W. W. Brennessel and B. E. Kucera, J. Organomet. Chem., 2005, 690, 5150–5158 CrossRef CAS.
- C. G. Overberger and M. Tobkes, J. Polym. Sci., 1964, 2, 2181–2187 Search PubMed.
- A. Hall, L. Parhamifar, M. K. Lange, K. D. Meyle, M. Sanderhoff, H. Andersen, M. Roursgaard, A. K. Larsen, P. B. Jensen, C. Christensen, J. Bartek and S. M. Moghimi, Biochim. Biophys. Acta, Bioenerg., 2015, 1847, 328–342 CrossRef CAS PubMed.
- B. R. Twaites, C. de las Heras Alarcón, D. Cunliffe, M. Lavigne, S. Pennadam, J. R. Smith, D. C. Górecki and C. Alexander, J. Controlled Release, 2004, 97, 551–566 CrossRef CAS PubMed.
- D. Fischer, T. Bieber, Y. Li, H.-P. Elsässer and T. Kissel, Pharm. Res., 1999, 16, 1273–1279 CrossRef CAS.
- B. W. Tylkowski, K. Wieszczycka and R. Jastrzab, Polymer Engineering, De Gruyter, 2017, ISBN 978-3-11-046828-1 Search PubMed.
- A. von Harpe, H. Petersen, Y. Li and T. Kissel, J. Controlled Release, 2000, 69, 309–322 CrossRef CAS PubMed.
- M. A. Cortez, W. T. Godbey, Y. Fang, M. E. Payne, B. J. Cafferty, K. A. Kosakowska and S. M. Grayson, J. Am. Chem. Soc., 2015, 137, 6541–6549 CrossRef CAS PubMed.
- W. Fischer, B. Brissault, S. Prévost, M. Kopaczynska, I. Andreou, A. Janosch, M. Gradzielski and R. Haag, Macromol. Biosci., 2010, 10, 1073–1083 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |