
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConcise asymmetric total synthesis of (−)-patchouli alcohol†
Guang-Qiang
Xu
,
Guo-Qiang
Lin
and
Bing-Feng
Sun
*
CAS Key Laboratory of Synthetic Chemistry of Natural Substances, Shanghai Institute of Organic Chemistry, 345 Lingling Road, Shanghai 200032, China. E-mail: bfsun@sioc.ac.cn
First published on 7th July 2017
Abstract
The asymmetric total synthesis of (−)-patchouli alcohol was accomplished in a concise manner. Key reactions include a highly diastereo- and enantioselective formal organocatalytic [4 + 2] cycloaddition reaction, a radical denitration reaction, and an oxidative carboxylation reaction. The formal synthesis of norpatchoulenol was achieved as well.
The sesquiterpene patchouli alcohol (1, Fig. 1), the major component of patchouli oil, has been known for one and a half centuries since Gal and co-workers reported its isolation from Pogostemon cablin (Blanco) Benth in 1869.1 The molecular structure of patchouli alcohol (1), however, had been unknown until its establishment in 1963 through single crystal X-ray diffraction analysis.2 It possesses a compact tricyclo[5.3.1.03,8]undecane carbon skeleton decorated with three contiguous quaternary centers, closely akin to norpatchoulenol3 (2) and seychellene4 (3) (Fig. 1). Biologically, patchouli alcohol exhibits various activities including calcium ion antagonism,5 antibacterial,6 anti-inflammatory,7 and anti-tumor8 activities. The combination of the synthetic challenge and the biological interest makes patchouli alcohol a highly attractive target for synthetic chemists in the past three decades.9 In this paper, we report a concise asymmetric total synthesis of patchouli alcohol based on a new synthetic strategy.
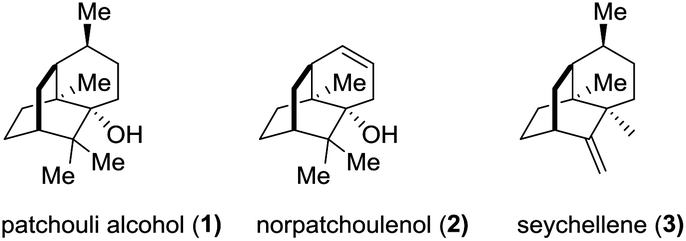 | ||
| Fig. 1 Patchouli alcohol (1), norpatchoulenol (2), and seychellene (3). | ||
The retrosynthetic analysis of 1 is depicted in Scheme 1. In the light of Bertrand's procedure,9g an intramolecular reductive radical coupling of 4 was envisioned to accomplish the target molecule. The MOM-protected carbinol in 4 could be traced back to a carboxylic acid group and further to a furan ring, thereby defining 59j and 6 as successive precursors. By incorporating a nitro group into the framework, the key intermediate 7 was envisaged and a denitration reaction could convert 7 to 6. Based on the formal organocatalytic [4 + 2] cycloaddition reaction developed by us and later successfully applied in natural product synthesis,10 compound 7 would be assembled from the reaction of 8 and 9.
 | ||
| Scheme 1 Retrosynthetic analysis of patchouli alcohol (1). | ||
The synthetic journey commenced with the execution of the critical [4 + 2] reaction of 8 and 9 (Scheme 2). The one-pot reaction of 8 and 9 was first catalyzed by 5 mol% 10 before being treated with 0.3 equivalents of DBU at room temperature, providing 7 in 61% yield and 94% ee with a diastereomeric ratio of 20/1 (Scheme 2).
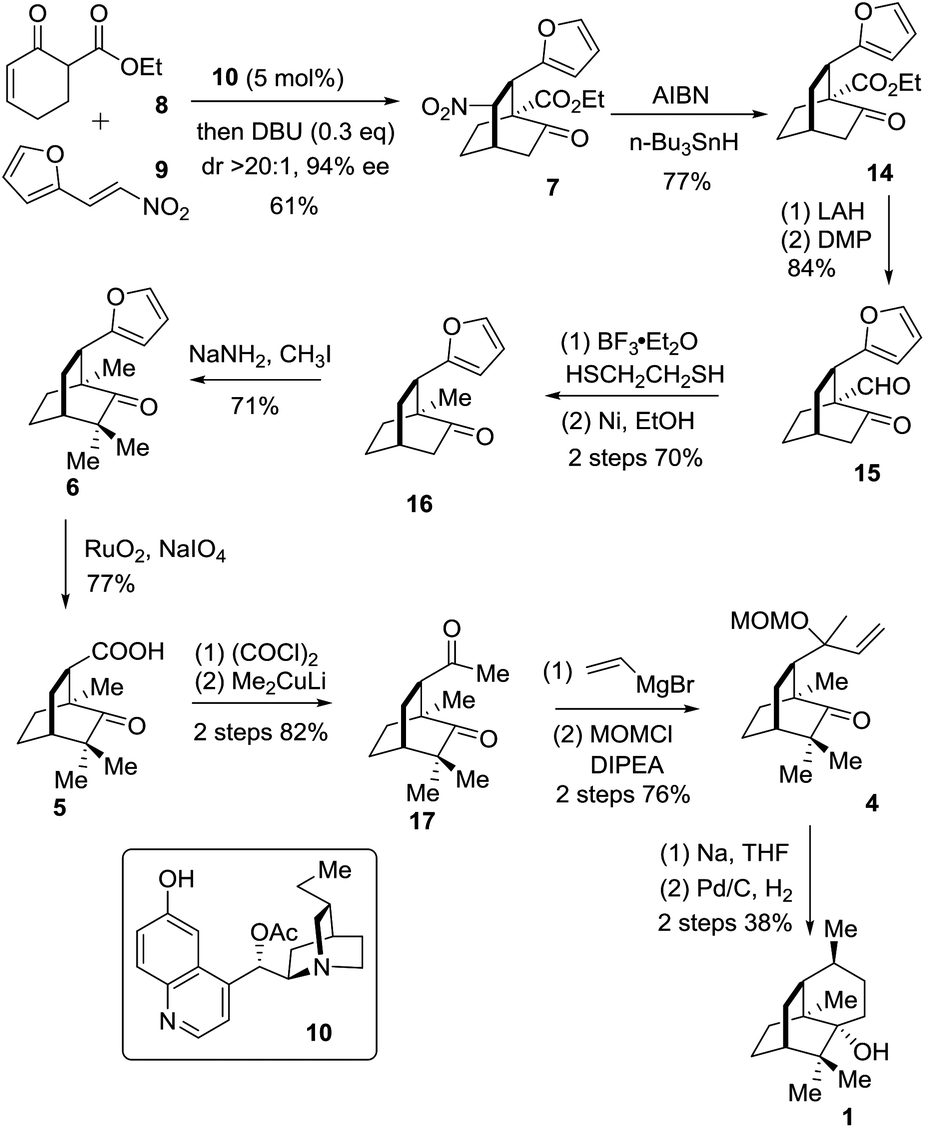 | ||
| Scheme 2 Total synthesis of patchouli alcohol (1). | ||
Mechanistically, this [4 + 2] reaction proceeded in two steps (Scheme 3). In the first step, 7 may form a complex with 10via hydrogen bonding interactions, thereby discriminating the two faces of the reactive enol. The nitroolefin approaches the enol selectively from the less hindered α-face with the substituents of the two bond-forming carbons positioned in a staggered arrangement (11), delivering 12. In the second step, when DBU is used as the base, 13 might be operative to minimize the steric repulsion between the nitro and the phenyl groups, leading to the formation of 7 as the major product. Remarkably, this reaction could be readily scaled up to a ten-gram scale.
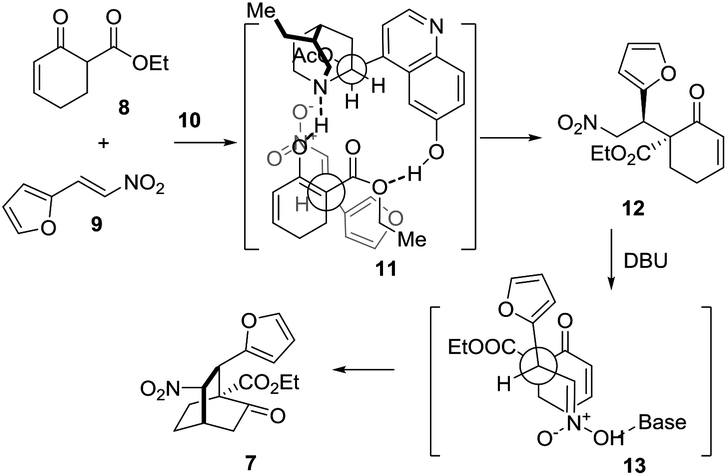 | ||
| Scheme 3 Proposed stereocontrol model for the formal [4 + 2] cycloaddition reaction of 8 and 9 catalyzed by 10. | ||
The strategic incorporation of the nitro group into the skeleton enabled us to assemble the bicyclic framework efficiently in a highly enantioselective manner. The key denitration was next investigated. As listed in Table 1, the conversion of 5 to 8 was examined with various solvents and temperatures. The optimal set of conditions involved AIBN/n-Bu3SnH in mesitylene at 150 °C that provided 14 in 77% yield after a reaction time of less than half an hour (entry 4).
| Entry | Conditions | Eq. (n-Bu3SnH) | Yield (%) |
|---|---|---|---|
| 1 | Toluene, 110 °C, 10 h | 2.5 | No reaction |
| 2 | Xylenes, 120 °C, 10 h | 2.5 | No reaction |
| 3 | Mesitylene, 150 °C, 10 h | 2.5 | 50 |
| 4 | Mesitylene, 150 °C, 25 min | 5 | 77 |
With the chiral [2.2.2] bicyclic skeleton 14 in hand, we proceeded to the next synthetic stage (Scheme 2). Reduction of the keto ester 8 with LAH followed by oxidation with DMP afforded the keto aldehyde 15 in 84% overall yield. Compound 15 was deoxygenated to give 16 in an overall yield of 70% via a two-step sequence involving thioacetalization with ethanedithiol and the succeeding desulfurization with RANEY® nickel. The geminal dimethyl groups were next installed by treating 16 with NaNH2 and CH3I to give 6 in 71% yield. The oxidation of 6 with NaIO4 in the presence of a catalytic amount of RuO2·H2O (1 mol%) in CCl4/MeCN/H2O underwent smoothly to afford the carboxylic acid 5 in a yield of 77%. Compound 5 was converted into 17 in 82% overall yield via the addition of lithium dimethylcuprate to the preformed acid chloride.9j The Grignard addition of vinylmagnesium bromide occurred on the less hindered carbonyl group in 17 providing the tertiary allyl alcohol in a chemoselective manner. Exposure of the resultant tertiary allyl alcohol to methoxymethyl chloride (MOMCl) and diisopropylethylamine (DIPEA) in CH2Cl2 at 0 °C for 24 hours gave 4 in 76% overall yield.11 The eventual single electron mediated 6-endo-trig cyclization reaction using Bertrand's conditions successfully delivered the annulated product which upon a catalytic hydrogenation reaction on Pd/C engendered 1 in 38% overall yield. The specific optical rotation of the synthetic sample ([α]24D −95.0 (c = 0.4, CHCl3)) matched that of the natural compound ([α]20D −94.6 (c = 1.21, CHCl3)).9e
An alternative strategy involving SmI2 mediated radical cyclization was attempted (Scheme 4). Thus, compound 5 was transformed into the vinyl ketone 18 through a Stille coupling reaction. To our disappointment, treatment of 18 with SmI2 did not generate the anticipated intramolecular reductive cyclization product 19 (Scheme 4). Instead, the dimerization product was identified by LRMS and HRMS techniques. Considering that the lower carbonyl group was trapped by two neighboring quaternary carbon centers, the grave steric hindrance might account for the deviation of the cyclization reaction.
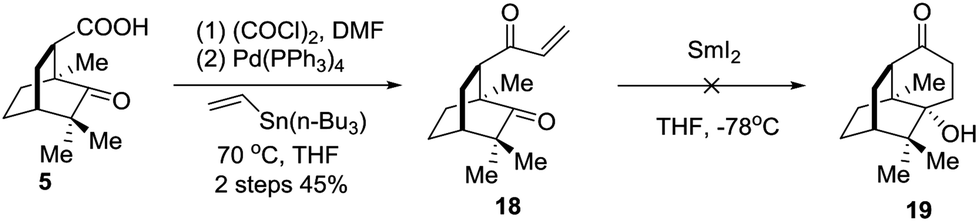 | ||
| Scheme 4 Synthesis of 18. | ||
Eventually, compound 5 was employed to synthesize 20via a redox procedure in a yield of 78% (Scheme 5). Compound 20 was a known synthetic precursor to norpatchoulenol (2).12
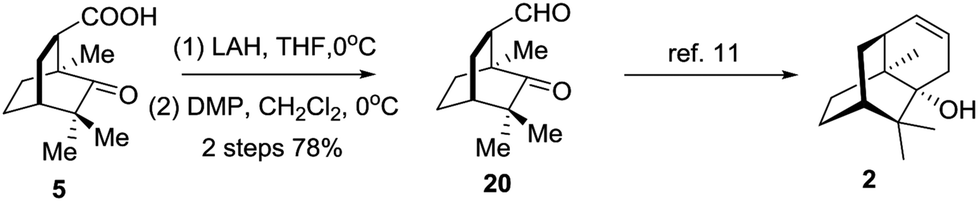 | ||
| Scheme 5 Formal synthesis of (−)-norpatchoulenol. | ||
Conclusions
In summary, we have realized the asymmetric total synthesis of (−)-patchouli alcohol with a new strategy. Key features include a highly enantioselective and diastereoselective organocatalytic [4 + 2] approach to the [2.2.2] bicyclic core, an efficient n-Bu3SnH mediated reductive denitration and an oxidative carboxylation reaction. The total synthesis spans 14 steps in a good overall yield from simple starting materials. The newly developed synthetic route may readily lend itself to the total synthesis of relevant natural products. Endeavours along this line are underway and will be reported in due course.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (grant no. 21290180, 21472210, 21672243) and the Youth Innovation Promotion Association CAS (2012202).Notes and references
- (a) H. Gal, Ann. Chem., 1869, 150, 374 CrossRef; (b) J. C. R. de Montgolfier, Acad. Sci., Paris, C. R., 1877, 84, 88 Search PubMed.
- M. Dobler, J. D. Dunitz, B. Gubler, H. P. Weber, G. Buchi and J. O. Padilla, Proc. Chem. Soc., 1963, 383 CAS.
- G. Wolff and G. Ourisson, Tetrahedron, 1969, 25, 4903 CrossRef CAS.
- P. Teisseire, P. Maupetti and B. Corbier, Recherches, 1974, 19, 8 CAS.
- (a) K. Ichikawa, T. Kinoshita and U. Sankawa, Chem. Pharm. Bull., 1989, 37, 345 CrossRef CAS PubMed; (b) D. W. Brann, K. Dhandapani and C. Wakake, Steroids, 2007, 72, 381 CrossRef CAS PubMed.
- (a) N. J. Kongkathip, Nat. Sci., 2009, 43, 519 CAS; (b) K. Osawa, T. Matsumoto and T. Maruyama, Bull. Tokyo Dent. Coll., 1990, 31, 17 CAS; (c) F. Wan, P. Fu and P. Cheng, Chin. J. Integr. Med., 2014, 1 Search PubMed.
- (a) Y. C. Li and J. N. Chen, Exp. Ther. Med., 2011, 545 Search PubMed; (b) H. T. Wang, Z. Z. Wang and Z. C. Wang, Biomed. Pharmacother., 2016, 83, 930 CrossRef CAS PubMed; (c) J. H. Xie, Y. Xu and Z. R. Su, Int. Immunopharmacol., 2016, 35, 43 CrossRef CAS PubMed.
- J. B. Jeong, J. Choi and S. H. Lee, Int. Immunopharmacol., 2013, 16, 184 CrossRef CAS PubMed.
- (a) G. Buchi and W. D. Macleod, J. Am. Chem. Soc., 1962, 84, 3205 CrossRef CAS; (b) G. Buchi, J. Am. Chem. Soc., 1964, 86, 4438 CrossRef; (c) S. Danishefsky and D. Dumas, Chem. Commun., 1968, 1287 RSC; (d) R. N. Mirrington and K. J. Schmalzl, J. Org. Chem., 1972, 37, 2871 CrossRef CAS; (e) F. Naf and G. Ohloff, Helv. Chim. Acta, 1974, 57, 1868 CrossRef; (f) K. Yamada and Y. Kyotani, Tetrahedron, 1979, 35, 293 CrossRef CAS; (g) M. Bertrand, P. Teisseire and G. Pelerin, Tetrahedron Lett., 1980, 21, 2055 CrossRef CAS; (h) R. B. Zhao and Y. L. Wu, Chin. J. Chem., 1991, 9, 377 CrossRef CAS; (i) P. Fludzinski, T. V. Magee and G. Stork, Tetrahedron Lett., 1995, 36, 7607 CrossRef; (j) G. S. R. Subba Rao and K. P. Kaliappan, J. Chem. Soc., Perkin Trans. 1, 1997, 1385 Search PubMed; (k) A. Srikrishna and G. Satyanarayana, Tetrahedron: Asymmetry, 2005, 16, 3992 CrossRef CAS.
- (a) Y. Z. Li, J. Wang, W. B. Sun, Y. F. Shan, B. F. Sun, G. Q. Lin and J. P. Zou, Org. Chem. Front., 2015, 2, 274 RSC; (b) J. Wang, W. B. Sun, Y. Z. Li, X. Wang, B. F. Sun, G. Q. Lin and J. P. Zou, Org. Chem. Front., 2015, 2, 674 RSC.
- Compound 4 is a 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric mixture isomeric at the allylic carbon center. In the ESI† the 1H and 13C NMR data were read from the spectra of the mixture.
:1 diastereomeric mixture isomeric at the allylic carbon center. In the ESI† the 1H and 13C NMR data were read from the spectra of the mixture. - M. Bertrand and P. Teisseire, Tetrahedron Lett., 1980, 21, 2051 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data, and copies of 1H, 13C and 2D NMR spectra. See DOI: 10.1039/c7qo00459a |
| This journal is © the Partner Organisations 2017 |