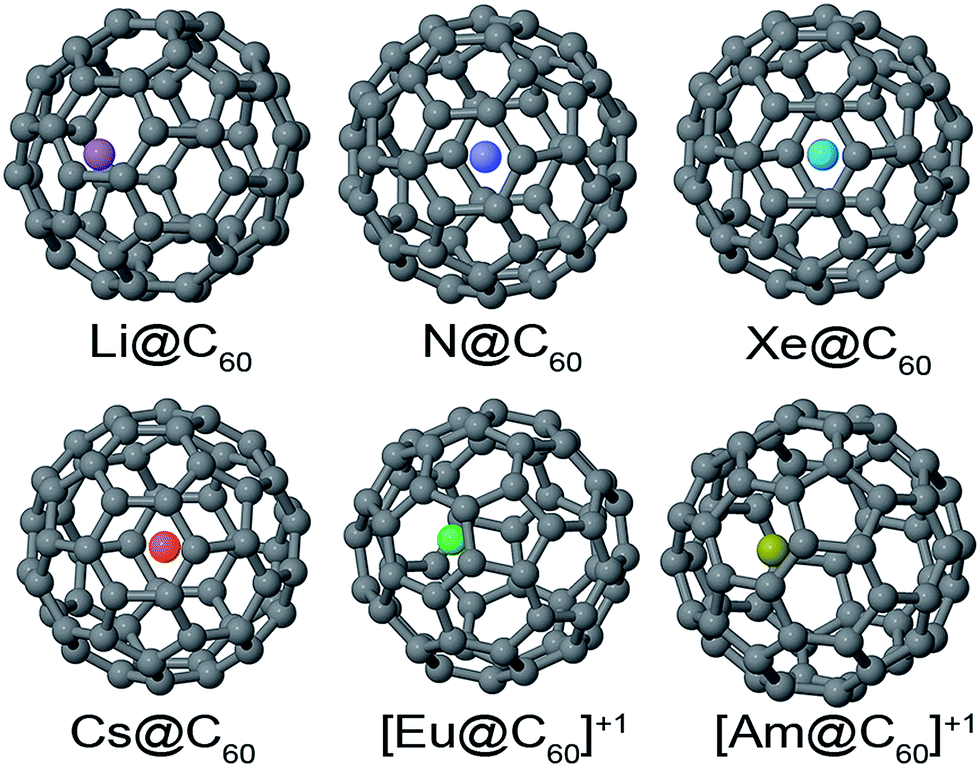
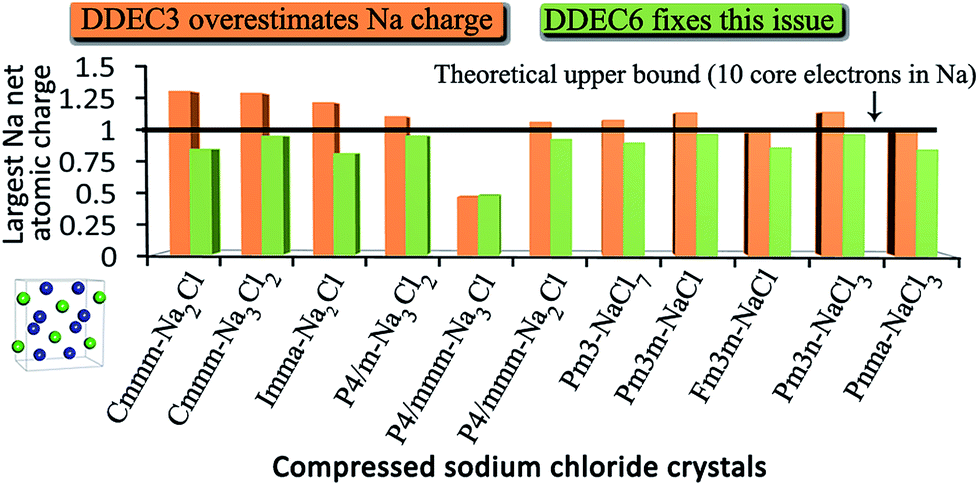
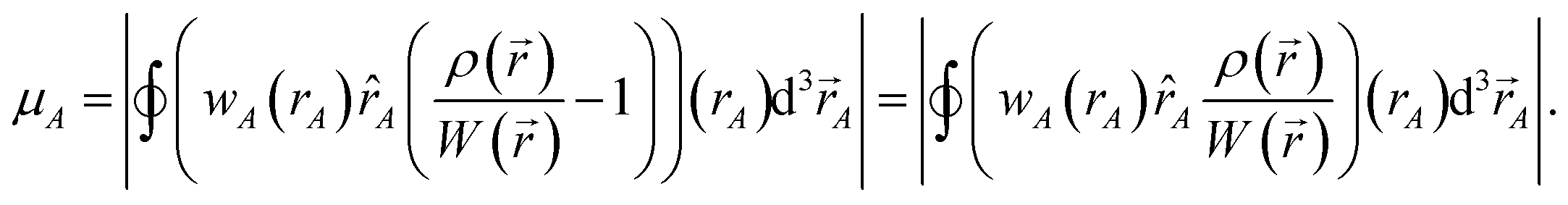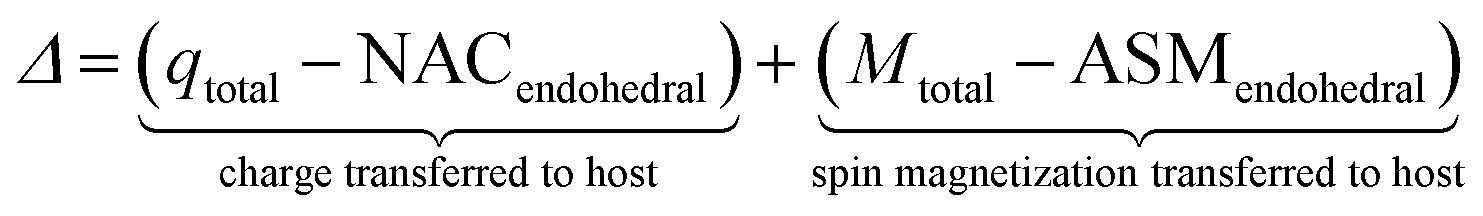DOI:
10.1039/C6RA04656H
(Paper)
RSC Adv., 2016,
6, 47771-47801
Introducing DDEC6 atomic population analysis: part 1. Charge partitioning theory and methodology†
Received
22nd February 2016
, Accepted 11th April 2016
First published on 13th April 2016
Abstract
Net atomic charges (NACs) are widely used in all chemical sciences to concisely summarize key information about the partitioning of electrons among atoms in materials. The objective of this article is to develop an atomic population analysis method that is suitable to be used as a default method in quantum chemistry programs irrespective of the kind of basis sets employed. To address this challenge, we introduce a new atoms-in-materials method with the following nine properties: (1) exactly one electron distribution is assigned to each atom, (2) core electrons are assigned to the correct host atom, (3) NACs are formally independent of the basis set type because they are functionals of the total electron distribution, (4) the assigned atomic electron distributions give an efficiently converging polyatomic multipole expansion, (5) the assigned NACs usually follow Pauling scale electronegativity trends, (6) NACs for a particular element have good transferability among different conformations that are equivalently bonded, (7) the assigned NACs are chemically consistent with the assigned atomic spin moments, (8) the method has predictably rapid and robust convergence to a unique solution, and (9) the computational cost of charge partitioning scales linearly with increasing system size. We study numerous materials as examples: (a) a series of endohedral C60 complexes, (b) high-pressure compressed sodium chloride crystals with unusual stoichiometries, (c) metal–organic frameworks, (d) large and small molecules, (e) organometallic complexes, (f) various solids, and (g) solid surfaces. Due to non-nuclear attractors, Bader's quantum chemical topology could not assign NACs for some of these materials. We show for the first time that the Iterative Hirshfeld and DDEC3 methods do not always converge to a unique solution independent of the initial guess, and this sometimes causes those methods to assign dramatically different NACs on symmetry-equivalent atoms. By using a fixed number of charge partitioning steps with well-defined reference ion charges, the DDEC6 method avoids this problem by always converging to a unique solution. These characteristics make the DDEC6 method ideally suited for use as a default charge assignment method in quantum chemistry programs.
1. Introduction
Net atomic charges (NACs) are a ubiquitous concept in all chemical sciences. It is difficult to imagine chemistry being learned at either an introductory or advanced level without some reference to NACs.1 For example, the pH scale measuring hydrogen ion activities in solution embodies the concept of hydrogen atoms carrying positive NACs. In biochemistry, many cellular functions depend on the transport of charged atoms such as K+, Na+, Ca2+, and Cl− across cell membranes.2 Experiments measuring the water molecule's dipole moment imply a negative NAC on its oxygen atom and a positive NAC on each of its two hydrogen atoms.3 NACs also play an important role in solid state physics, where oxygen atoms in solid oxides carry negative NACs to enable oxygen ion transport.4 Zwitterions, which are widely encountered in amino acids, illustrate that important chemical behaviors depend not only on the overall molecular charge but also on the net charges of local regions within a molecule.5
Our overall objective is to develop an atomic population analysis method with characteristics suitable for use as a default method in popular quantum chemistry programs. This requires the method to maximize broad applicability and minimize failure. The algorithm should converge rapidly and reliably to a unique solution with low dependence on the basis set choice. The assigned NACs, atomic spin moments (ASMs), and other atoms-in-materials (AIM) properties should be chemically consistent, accurately describe electron transfer directions, and correlate to experimental data for reference materials.
Existing atomic population analysis methods are not well-suited for use as a default method in quantum chemistry programs: (1) Mulliken6 and Davidson-Löwdin7 population analyses do not have any mathematical limits as the basis set is systematically improved toward completeness, and they are not directly applicable to plane-wave basis sets.8 (2) Bader's quantum chemical topology (QCT) has many theoretically desirable properties, but it can lead to non-nuclear attractors that produce undefined NACs.9 (3) Electrostatic potential fitting methods (e.g., ESP,10 Chelp,11 Chelpg,12 REPEAT13) do not have good conformational transferability and assign unreasonable charge values to some buried atoms.13–17 Including constraints (e.g., RESP13,14 methods) improves this, but the form of these constraints is flexible leading to numerous possible charge values. Simultaneously fitting across multiple conformations is an another possible solution, but this requires computing electron distributions for many different system geometries.17 Also, nonporous systems do not possess a surface outside which to fit the electrostatic potential. (4) The original Hirshfeld (HD)18 method, which is based on neutral reference atom densities, usually underestimates NAC magnitudes.19–22 While the Iterative Hirshfeld (IH) method improves the NAC magnitudes,21 it exhibits the bifurcation problem described in Section 2.4. (5) The Charge Model 5 (CM5) was parameterized to give NACs that approximately reproduce static molecular dipole moments.20,23 The CM5 charges include an empirical correction to the HD NACs.20,23 The main limitation of CM5 is that it only partitions the integrated number of electrons for each atom, and thus cannot be used to compute AIM properties such as atomic multipoles, bond orders, etc. (6) Because Atomic Polar Tensor (APT)24 and Born effective charges25 require computing system response properties (via perturbation theory or atomic displacements), they are not well-suited for use as a default atomic population analysis method. (7) The Natural Population Analysis (NPA),8 Natural Bond Orbital (NBO),26 Adaptive Natural Density Partitioning (ANDP),27 Intrinsic Atomic Orbital,28 Intrinsic Bond Orbital,28 and several related approaches29 provide chemically meaningful localized atomic and bonding orbitals. These approaches have a number of advantages, including the recovery of heuristic chemical bond forming concepts. In the near future, modifications of some of these methods might emerge with sufficiently wide applicability and convergence robustness to be used as default methods in quantum chemistry programs. Recently, some of these methods have been tested on a limited number of periodic or semi-periodic materials,29–32 but not yet on enough materials to draw definite conclusions about their suitability for being used as a default method for analyzing dense solids. Because these methods require generating localized orbitals, using plane-waves or other delocalized basis functions poses a challenge for their application.30
In this article we present a new atomic population analysis method, called DDEC6, that is a refinement of the Density Derived Electrostatic and Chemical (DDEC) approach. This method is an explicit functional of the electron and spin distributions with no explicit basis set dependence. We developed the DDEC6 charge partitioning algorithm using a scientific engineering design approach that resembles the process used to build airplanes. Similar to constructing an AIM partitioning, there is more than one conceivable way to build an airplane. One could make an airplane longer or shorter, for example. Yet, it is not accurate to say airplane design (or AIM partitioning design) is an arbitrary process, because the scientific method is utilized. Like airplanes, our AIM partitioning method has been scientifically engineered to meet chosen performance goals. This involved a process of constructing and testing prototypes to refine the design until all performance goals were achieved. When a new airplane is designed, prototypes are built and tested in wind tunnels to determine which shapes achieve appropriate lift, minimize drag, and respond favorably to air turbulence. Not only should an airplane fly, but it should take off and land smoothly, have good fuel efficiency, and so forth. All of these aspects are tested when developing a new airplane design. Our process for building an AIM partitioning method is similar. Specifically, we built and tested prototypes to improve the control, efficiency, accuracy, and robustness. We made extensive comparisons to experimental data during this development process.33
This scientific engineering design approach requires choosing performance goals. Table 1 lists nine desirable features we chose for assigning NACs. The first criterion is to assign exactly one electron distribution per atom in the material. This criterion is fulfilled by many but not all charge assignment methods. For example, Bader's QCT yields non-atomic electron distributions in materials with non-nuclear attractors.9,34,35 The second criterion is to assign core electrons to their host atom. This criterion is not appropriate for APT and Born effective charges that quantify the system's response to nuclear displacements. Methods that directly fit the electrostatic potential without regard for atomic chemical states also do not satisfy this criterion. Since a goal of AIM methods is to describe atomic chemical states, they should preferably assign core electrons to the host atom. The third criterion is to assign NACs as functionals of {ρ(![[r with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0072_20d1.gif) )}. The main purposes of our NACs are to convey information about charge transfer between atoms and to approximately reproduce the electrostatic potential surrounding a material. Since charge transfer between atoms cannot occur without effecting ρ() and ρ() determines the electrostatic field surrounding the material, it makes sense to construct the NACs as functionals of {ρ()}. The fourth criterion is to assign atomic electron distributions to give an efficiently converging polyatomic multipole expansion. Polyatomic multipole expansions including multipolar and charge penetration terms of arbitrarily high order provide a formally exact representation of the electrostatic potential.36–41 In practice, this expansion is normally truncated at some finite order; therefore, we wish to reproduce the electrostatic potential with good accuracy using the leading terms of the polyatomic multipole expansion. The fifth criterion is the assigned NACs should usually follow Pauling scale electronegativity trends. The Pauling scale electronegativity was parameterized to describe typical electron transfer directions in chemical bonds, where higher electronegativity elements typically take electrons from lower electronegativity elements.42,43 The sixth criterion is that NACs for a particular element have good transferability among different conformations that are equivalently bonded. We choose this criterion, because one of our goals is to assign NACs with good conformational transferability that are well-suited to construct flexible force-fields for classical atomistic simulations of materials. The seventh criterion is that the assigned NACs should be chemically consistent with the assigned ASMs. We will have more to say about this seventh criterion in Section 5.4. The eighth criterion is that the AIM distributions should have predictably rapid and robust convergence to a unique solution. The ninth criterion is that the computation cost of charge partitioning should ideally scale linearly with increasing system size. This criterion is desirable to have the method's computational cost remain competitive as the number of atoms in the unit cell increases.
)}. The main purposes of our NACs are to convey information about charge transfer between atoms and to approximately reproduce the electrostatic potential surrounding a material. Since charge transfer between atoms cannot occur without effecting ρ() and ρ() determines the electrostatic field surrounding the material, it makes sense to construct the NACs as functionals of {ρ()}. The fourth criterion is to assign atomic electron distributions to give an efficiently converging polyatomic multipole expansion. Polyatomic multipole expansions including multipolar and charge penetration terms of arbitrarily high order provide a formally exact representation of the electrostatic potential.36–41 In practice, this expansion is normally truncated at some finite order; therefore, we wish to reproduce the electrostatic potential with good accuracy using the leading terms of the polyatomic multipole expansion. The fifth criterion is the assigned NACs should usually follow Pauling scale electronegativity trends. The Pauling scale electronegativity was parameterized to describe typical electron transfer directions in chemical bonds, where higher electronegativity elements typically take electrons from lower electronegativity elements.42,43 The sixth criterion is that NACs for a particular element have good transferability among different conformations that are equivalently bonded. We choose this criterion, because one of our goals is to assign NACs with good conformational transferability that are well-suited to construct flexible force-fields for classical atomistic simulations of materials. The seventh criterion is that the assigned NACs should be chemically consistent with the assigned ASMs. We will have more to say about this seventh criterion in Section 5.4. The eighth criterion is that the AIM distributions should have predictably rapid and robust convergence to a unique solution. The ninth criterion is that the computation cost of charge partitioning should ideally scale linearly with increasing system size. This criterion is desirable to have the method's computational cost remain competitive as the number of atoms in the unit cell increases.
Table 1 Nine desirable features (performance goals) we have chosen for assigning NACs
| (1). Exactly one assigned electron distribution per atom |
| (2). Core electrons remain assigned to the host atom |
| (3). NACs are functionals of the total electron density distribution |
| (4). Assigned atomic electron distributions give an efficiently converging polyatomic multipole expansion |
| (5). NACs usually follow Pauling scale electronegativity trends |
| (6). NACs for a particular element have good transferability among different conformations that are equivalently bonded |
| (7). The assigned NACs are chemically consistent with the assigned ASMs |
| (8). Predictably rapid and robust convergence to a unique solution |
| (9). Computational cost of charge partitioning scales linearly with increasing system size |
As a point of clarification, we intend that these criteria should be satisfied across a broad range of materials encompassing molecules, ions, nanostructures, solid surfaces, porous solids, nonporous solids, and other complex materials. Notably, developing a reliable method for charge partitioning in dense periodic solids is not simply the task of adding periodic boundary conditions to a charge partitioning method initially developed for small molecules. Small molecules are comprised mainly of surface atoms with few buried atoms. In contrast, dense solids are comprised mainly of buried atoms with few surface atoms. Therefore, charge assignment methods that work well for surface atoms but poorly for buried atoms are problematic for bulk solids. Currently, the most commonly used charge partitioning method for dense solids is Bader's QCT.44 Because two charge partitioning methods that give practically equivalent results for molecules with lots of surface atoms sometimes produce spectacularly different results when applied to dense solids,19 correlations between NAC methods for molecular test sets should not be extrapolated to dense solids. In summary, charge partitioning in dense solids is an intrinsically more difficult problem than charge partitioning in small molecules.
The remainder of this article is organized as following. Section 2 presents an overview of charge partitioning operators, atoms-in-materials methods, the DDEC approach, and the bifurcation or ‘runaway charges’ problem. Section 3 presents the theory and equations defining the DDEC6 method. Section 4 summarizes computational details for the quantum chemistry calculations, electrostatic potential expansion, and Ewald summation. Section 5 contains the performance test results: 5.1 Convergence speed, 5.2 Atomic dipole magnitudes, 5.3 Conformational transferability and accuracy for reproducing the electrostatic potential, 5.4 Quantifying the consistency between assigned NACs and ASMs, 5.5 Retaining core electrons on the host atom and assigning exactly one electron distribution per atom, and 5.6 NACs usually follow electronegativity trends. Section 6 contains our conclusions.
2. Theory of atoms in materials
2.1 Charge partitioning operators
Chemical systems are comprised of atomic nuclei surrounded by an electron cloud. This electron cloud can be computed using quantum chemistry calculations. Throughout this article we use the Born–Oppenheimer approximation, in which the electron cloud is assumed to equilibrate rapidly with respect to the nuclear motions. The NAC for atom A (qA) equals its nuclear charge (zA) minus the number of electrons assigned to it (NA):
Herein we use the same notation as previously, except we use (L1, L2, L3) instead of (k1, k2, k3) to specify a translated image of atom A: “Following Manz and Sholl,45 we begin by defining a material as a set of atoms {A} located at positions {![[R with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0052_20d1.gif) A}, in a reference unit cell, U. For a nonperiodic system (e.g., a molecule), U is any parallelpiped enclosing the entire electron distribution. The reference unit cell has L1 = L2 = L3 = 0, and summation over A means summation over all atoms in this unit cell. For a periodic direction, Li ranges over all integers with the associated lattice vector
A}, in a reference unit cell, U. For a nonperiodic system (e.g., a molecule), U is any parallelpiped enclosing the entire electron distribution. The reference unit cell has L1 = L2 = L3 = 0, and summation over A means summation over all atoms in this unit cell. For a periodic direction, Li ranges over all integers with the associated lattice vector ![[v with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0076_20d1.gif) i. For a nonperiodic direction, Li = 0 and i is the corresponding edge of U. Using this notation, the vector and distance relative to atom A are given by
i. For a nonperiodic direction, Li = 0 and i is the corresponding edge of U. Using this notation, the vector and distance relative to atom A are given by
| | |
A = − L11 − L22 − L33 − A.
| (2) |
and
rA = |
A|.”
19 In this article, we are only interested in studying time-independent states of chemical systems. For such systems, a time-independent electron distribution
can be theoretically computed or experimentally measured
46,47 where
Ψel is the system's multi-electronic wavefunction within the Born–Oppenheimer approximation and
![[small rho, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e0b7.gif)
(
) is the electron density operator.
Before considering specifically how to partition the electron density operator among atoms in a material at each spatial position, we first consider various schemes that partition only the integrated number of electrons. Electrostatic potential fitting (ESP,10 Chelp,11 Chelpg,12 REPEAT13) methods optimize NACs by minimizing the root-mean-squared-error (RMSE) over a chosen set of grid points located outside the material's van der Waals surface. The APT charge quantifies the change in dipole moment due to the displacement of a nucleus.24 APT and related dipole-change-derived NACs are useful for representing infrared (IR) spectra intensities.48 In periodic materials, Born effective and related charge methods quantify the change in electric polarization due to the displacement of a nucleus and its periodic images.25 A key limitation of electrostatic potential fitting, APT, and Born effective charges is that they are not designed to retain core electrons. For example, the Born effective charge of Ti in the cubic phases of BaTiO3, CaTiO3, SrTiO3, and PbTiO3 ranges from 6.7 to 7.2, which exceeds the nominal number of 4 valence electrons for a free Ti atom.49 CM5, which uses HD charges as input, was parameterized to give NACs that approximately reproduce static molecular dipole moments.20 The CM5 NACs do a much better (but not perfect) job of retaining core electrons on the host atom.20,23 The Voronoi deformation density (VDD) method assigns NACs according to the integral of the deformation density (i.e., the difference between ρ() and a sum of spherically symmetric neutral reference atoms) over the Voronoi cell enclosing each atom.50
AIM methods partition the electron distribution at each spatial position subject to the constraints
| |
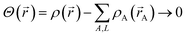 | (4) |
| | |
ρA(A) ≥ 0
| (5) |
where
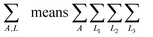
denoting summation over all atoms in the material, and
ρA(
A) is the electron distribution assigned to atom
A. Because
ρ(
) ≥ 0, constraint
(4) allows
to be interpreted as the probability of assigning an electron at position
to atom
A. AIM methods include HD,
18 IH and related charge partitioning methods,
21,51–54 Iterated Stockhold Atoms (ISA),
55,56 DDEC,
19,45 radical Voronoi tessellation,
57,58 etc. There has been some debate on how to best define the atomic probability factors, {
fA(
A)}. The ISA method optimizes the set of atomic electron density distributions {
ρA(
A)} to resemble their spherical averages {
ρavgA(
rA)}.
55,56 The HD
18 and IH
21 methods optimize {
ρA(
A)} to resemble a set of spherical reference atoms {
ρrefA(
rA)}. Nalewajski and Parr
59 and Parr
et al.60 argued for the HD definition based on information theory and philosophical considerations with the {
fA(
A)} considered as noumenons. Matta and Bader argued for a definition based on Virial compartments describing experimentally observed additive property relationships.
61 Bader's quantum chemical topology (QCT) partitions the electron cloud into non-overlapping compartments that satisfy the Virial theorem because they have zero-flux surfaces: ∇
ρ·d
![[n with combining circumflex]](https://www.rsc.org/images/entities/i_char_006e_0302.gif)
= 0 where d
is the differential surface normal unit vector.
62–64 Because non-atomic Bader compartments exist in materials with non-nuclear attractors,
9 Bader's QCT is not strictly a partition into atomic electron distributions. However, Bader's QCT has historically been categorized with AIM methods, because it pioneered the theoretical development of the AIM concept.
62–65 For the study of electrides, a non-nuclear attractor describing the electron ion would normally be considered an advantage.
34
2.2 Fundamentals of vectorized charge partitioning
We use the term vectorized charge partitioning to denote the class of AIM charge partitioning methods for which the relative probability of assigning electrons at position A to atom A can be represented in terms of some spherically symmetric atomic weighting factor, wA(rA):| | |
ρA(A)/ρ() = wA(rA)/W()
| (7) |
where| |
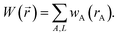 | (8) |
We call this vectorized charge partitioning, because for each atom wA(rA) forms a one-dimensional array of wA values corresponding to a series of rA values. The whole quest to define the charge partitioning method thus reduces the problem of finding a three-dimensional array of fA(A) values for each atom to that of finding a one-dimensional array of wA(rA) values for each atom. This reduction in parameter space from three to one degrees of freedom per atom makes vectorized charge partitioning computationally efficient, because one-dimensional rather than three-dimensional arrays need to be computed and stored for each atom.
A key use of NACs is to construct point-charge models to regenerate the electrostatic potential in classical molecular dynamics and Monte Carlo simulations.66 From Gauss's Law of Electrostatics it directly follows that the electrostatic potential exerted outside a spherically symmetric charge distribution is identical to an equivalent point charge placed at the sphere's center. Hence, it is wise to assign approximately spherically symmetric atomic electron distributions
| | |
ρA(A) ≈ ρavgA(rA)
| (9) |
so that a point-charge model comprised of the NACs will approximately reproduce the electrostatic potential surrounding the material. This can be accomplished by making
| | |
W() ≈ ρ()
| (10) |
for then
eqn (7) simplifies to
| | |
ρA(A) ≈ wA(rA).
| (11) |
If
eqn (10) is true everywhere in the system, then
eqn (9) and
(11) are also true everywhere in the system. This will make a point-charge model constructed from the NACs approximately reproduce the electrostatic potential surrounding the material. Thus, satisfying
eqn (10) is a key objective.
The differential path action dS allows us to study convergence properties of vectorized charge partitioning methods:19
| |
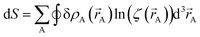 | (12) |
where
| |
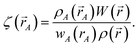 | (13) |
Stationary points of the path action
S = ∫d
S occur where
| |
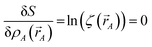 | (14) |
for every atom, which yields
eqn (7) as the only solution(s).
19 Due to its path dependence,
S is not a functional of {
ρA(
A)}. The path action
S is a kind of mapping that takes a path in {
ρA(
A)} optimization space as its input and returns a real number as its result. In practice, only the differential path action d
S needs to be considered.
A second key purpose of NACs is to represent the chemical states of atoms in materials. This requires the assigned {ρA(A)} to have atomic-like properties. The spherically averaged electron distributions of isolated atoms decay approximately exponentially with increasing rA ≥ 2 Å:
| |
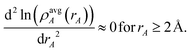 | (15) |
To maximize the transferability of atomic chemical properties between isolated atoms, molecules, porous solids, solid surfaces, non-porous solids, and nano-structures, the atomic electron distributions in materials should be assigned to approximately follow
eqn (15).
Another key consideration is the number of electrons assigned to each atom should resemble the number of electrons contained in the volume of space dominated by that atom. The volume of space dominated by atom A is defined as the spatial region for which ρA(A) > ρB(B) for every B ≠ A. If the wA(rA) for anions are too diffuse in ionic crystals, this might cause too many electrons in the volume of space dominated by the cations to be mistakenly assigned to the anions. As shown in Section 5.5, this can lead to situations where the total number of electrons assigned to the cations is even lower than their number of core electrons. To avoid this mistake, some care should be given to quantify how many electrons are in the volume of space dominated by each atom. The number of electrons assigned to each atom should then be optimized to resemble this value, subject to additional optimization criteria.
To maximize chemical transferability, it is desirable to have each atom in a material resemble a reference ion of the same element having similar (but not necessarily identical) net charge. For example, a Na1+ ion in a material should resemble a Na reference ion having a charge of approximately +1. Therefore, we use reference ions to construct part of the atomic weighting factors, {wA(rA)}.
The HD method uses neutral atoms as the reference states:18
| | |
wHDA(rA) = ρrefA(rA, qrefA = 0).
| (16) |
The extremely poor performance of the HD method can be explained by the fact that in partially or totally ionic systems
ρ(
) does not approximately equal the sum of neutral atom densities:
| |
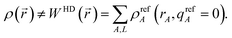 | (17) |
Thus,
eqn (9)–(11) are not consistently satisfied by the HD method. Consequently, the HD NACs usually give a poor representation of the electrostatic potential surrounding a material. The IH method improves upon the HD method by using self-consistently charged reference states:
21| | |
wIHA(rA) = ρrefA(rA, qrefA = qA).
| (18) |
While the IH method offers a clear improvement over the HD method, the performance of the IH method is still not optimal. Specifically, the IH method does not accurately account for the relative contraction or expansion of each ionic state due to its local environment. For example, an atomic anion in an ionic crystal is usually more contracted than the corresponding isolated atomic anion, because the cations in the ionic crystal provide charge balance and electrostatic screening that reduces electrostatic repulsion between excess electrons in the bound atomic anion. While it is possible to use charge-compensated reference ions in the IH method,53,67 the overall accuracy of constructing W() ≈ ρ() is still limited in the IH method by using a single set of reference ions that do not respond to their local environment. This problem is overcome in the DDEC3 and DDEC6 methods by conditioning the reference ion densities to match the specific material. This conditioning describes the contraction or expansion of reference ions in response to their local environment while still only requiring a single reference ion library as input.19
Eqn (10) will be fulfilled across the widest variety of systems if {wA(rA)} are themselves derived from partitions of ρ(). The ISA method is an early example of a charge partitioning scheme in which {wA(rA)} are derived from a partition of ρ().55,56 In the ISA method,
| | |
wISAA(rA) = ρavgA(rA).
| (19) |
Although the ISA method clearly fulfills
eqn (9)–(11), the ISA NACs have poor conformational transferability and are chemically inaccurate for many materials (especially, non-porous materials).
19,45,68–70 In Section 3 below, we construct a new charge partitioning scheme, called DDEC6, that combines electron localization, reference ion weighting, and spherical averaging to create an accurate and robust charge partitioning method.
2.3 The Density Derived Electrostatic and Chemical (DDEC) approach
The DDEC approach optimizes {wA(rA)} to simultaneously resemble reference ion states and {ρavgA(rA)}.19,45 Making {wA(rA)} resemble reference ion states maximizes the chemical transferability of the assigned {ρA(A)} and NACs, while making {wA(rA)} resemble {ρavgA(rA)} causes the NACs to approximately reproduce the electrostatic potential surrounding the material.19,45
Three variants of the DDEC method have been previously published.19,45 In the DDEC/c1 and DDEC/c2 methods, the atomic weighting factors are defined by
| | |
wDDEC/c1,c2A(rA) = (ρrefA(rA, qrefA = qA))χ(ρavgA(rA))1−χ
| (20) |
with
χDDEC/c1 =
χDDEC/c2 = 1/10.
45 During the iterative updates, the reference ion charges are updated to match the AIM charges, as done for the IH method. The reference ions, {
ρrefA(
rA,
qrefA)}, are computed using charge compensation and dielectric screening.
45 In the DDEC/c1 method, charge compensation and dielectric screening were modeled by computing the reference ion densities for atoms placed in a periodic array with a uniform compensating background charge.
45 In the DDEC/c2 method, charge compensation and dielectric screening were modeled by computing the reference ion densities for atoms enclosed by a spherical charge compensation shell.
45 For anions, the shell radius and compensating charge are carefully selected to minimize the system's total energy.
45 For cations of charge +
q, the compensating charge is −
q and the shell radius is the average radius of the outermost
q occupied Kohn–Sham orbitals of the isolated neutral atom.
45 We recently reported a complete set of these charge-compensated reference ions for all elements atomic number 1 to 109.
71 In the DDEC3 method, these reference ion densities are smoothed to ensure the following.
19 (1) Each smoothed reference ion density,
![[small rho, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d1.gif) refA
refA(
rA,
qrefA), decreases monotonically with increasing
rA.
19 (2) For a particular
rA value,
refA(
rA,
qrefA) decreases with increasing
qrefA.
19 (3) The difference
refA(
rA,
qrefA) −
refA(
rA, (
qrefA + 1)) decreases monotonically with increasing
rA.
19 The DDEC6 method introduced in this article uses the same smoothed reference ion library as the DDEC3 method.
In this article, the term charge partitioning functional means a functional F whose stationary points yield the corresponding atomic charge distributions. A point is a stationary point if and only if the full derivative of F is zero: dF = 0. This requires all of the first-order partial and variational derivatives of F with respect to the independent variables and functions, respectively, to be zero. Nalewajski and Parr showed the HD method minimizes the charge partitioning functional
| |
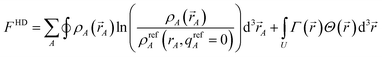 | (21) |
where
Γ(
) is a Lagrange multiplier enforcing constraint
(4).
59 Later, Bultinck
et al. showed the ISA method minimizes a similar charge partitioning functional where
ρavgA(
rA) appears instead of
ρrefA(
rA,
qrefA = 0):
70| |
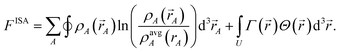 | (22) |
A functional or path action is convex if and only if its curvature is non-negative. Smooth convex functionals and path actions have a unique minimum. Both the ISA and non-iterative Hirshfeld charge partitioning functionals are convex and possess a single minimum leading to unique solution.70
The DDEC/c1,c2 methods minimize the partial derivative of
| |
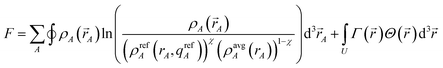 | (23) |
with respect to {
ρA(
A)} while holding the {
qrefA} constant if {
qrefA} = {
qA}.
45 Most importantly,
eqn (23) is emphatically
not a charge partitioning functional for the DDEC/c1,c2 methods. Specifically, minimizing
F does not automatically enforce the constraint {
qrefA} = {
qA}. This constraint can be enforced by simply replacing {
qrefA} with {
qA} in
eqn (23), but minimizing the resulting functional does
not yield the weighting factors shown in
eqn (20). Instead of directly replacing {
qrefA} with {
qA} in
eqn (23), the constraint {
qrefA} = {
qA} could be enforced using the method of Lagrange multipliers to yield a completely equivalent result that once again does not reproduce
eqn (20). Therefore, the object to be minimized for the DDEC/c1,c2 methods is not the functional shown in
eqn (23), but rather Manz's path action
S described in the previous section. This can be a potential source of confusion, because the first paper on the DDEC/c1,c2 method predated the introduction of the path action
S and relied on partial (not proper) minimization of
eqn (23).
45 With the introduction of the path action
S, this earlier approach that was not completely variational should be abandoned.
The same problem has also occurred in early literature on the IH method that predated Manz's path action S. Specifically, setting χ = 1 in eqn (23) does emphatically not yield a charge partitioning functional for the IH method, because it fails to properly impose the constraint {qrefA} = {qA} employed in the IH method. Enforcing {qrefA} = {qA} via a direct substitution in eqn (23) or through the addition of Lagrange multipliers to eqn (23) does not yield the IH weighting factors, but rather yields a new AIM method whose performance was not as good as the IH method.54 The object to be minimized for the IH method is the path action S. Interestingly, a proposed proof72 that the IH method always converges to a unique minimum (independent of the starting conditions) is invalid, because it was based on partial (not proper) minimization of eqn (23) rather than using the correct object to be minimized. In the following section, we present specific examples of materials for which the converged IH NACs depend on the initial guess, thus proving the IH optimization landscape is sometimes non-convex.
The DDEC3 method was introduced to improve the accuracy of charge partitioning in dense solids containing short bond lengths.19 For these materials, the DDEC/c1,c2 methods yielded extremely poor results.19 The DDEC3 method improved the accuracy by introducing reference ion conditioning (see Section 3.1) and a constraint forcing the wA(rA) tails of buried atoms to decay at least as fast as exp(−1.75 rA).19 The DDEC3 method starts with the same reference ion library as the DDEC/c2 method. DDEC3 smooths these reference ions to improve the optimization landscape curvature.19 Finally, the reference ion weighting, χDDEC3 = 3/14, was set to a theoretically derived value that achieves a balance for all materials.19
2.4 The bifurcation or ‘runaway charges’ problem
The ‘runaway charges’ problem was first noted for the DDEC/c2 method in the paper by Manz and Sholl introducing the DDEC3 method.19 The DDEC3 method was introduced to correct this problem for dense materials by introducing reference ion conditioning and constraints preventing the tails of buried atoms from becoming too diffuse.19 The DDEC3 method dramatically improved over DDEC/c2, but stopped short of a provably convex optimization functional.19 Among the hundreds of materials studied to date, we have only noticed one system for which the ‘runaway charges’ problem still occurs for the DDEC3 method: the H2 triplet state for a constrained bond length of 50 pm. The discovery of one system with this problem indicates other systems with this same problem probably exist. The two H atoms in H2 are symmetrically equivalent in the CISD/aug-cc-pvqz wavefunction and electron distribution. As shown in Fig. 1, during DDEC3 partitioning the NACs diverged from the initial values (HD charges) of +3.3 × 10−4 and −3.3 × 10−4 to final converged values of +0.50 and −0.50 after 37 iterations. This indicates the optimization landscape contains a bifurcation instability that leads to symmetry breaking. Only tiny initial differences in the input density grid files determine which of the two H atoms will head towards a NAC of +0.50 while the remaining one heads towards −0.50. The same type of bifurcation instability also occurs for the IH method in some dense solids. As shown in Fig. 1, during IH partitioning the NACs of symmetry-equivalent atoms bifurcate from the initial values (HD charges) of +4.8 × 10−4 and −4.8 × 10−4 to +0.97 and −0.97 for the Mn crystal and from +4.3 × 10−3, −5.4 × 10−3, −3.5 × 10−3, and +4.6 × 10−3 to −0.36, −0.39, −0.38, and +1.1 for the Rh crystal before the VASP program gave up after 150 charge cycles. (This IH analysis was performed in VASP 5.3.5 using the PBE73 functional.) Again, tiny differences in the initial conditions determines which of the symmetry-equivalent atoms will head towards the large positive NACs and which will head towards the negative NACs.
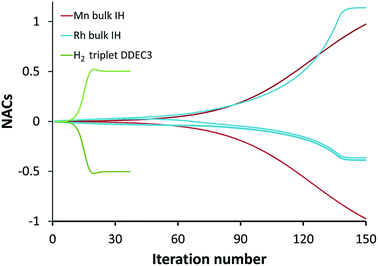 |
| | Fig. 1 Bifurcation (i.e., spontaneous symmetry breaking) during DDEC3 and IH charge partitioning. This is called the ‘runaway charges’ problem. | |
To understand why previous DDEC and IH methods sometimes yield bifurcation, we now compute their optimization landscape curvature. The path action's curvature is given by19
| |
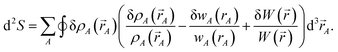 | (24) |
“If the curvature is positive everywhere,
i.e., d
2S > 0, then the action has only one stationary point, and this stationary point is its global minimum.”
19 The solution lies within the search space satisfying constraint (4). Restricting {
ρA(
A)} to this valid search space,
| |
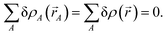 | (25) |
Let
![[S with combining macron]](https://www.rsc.org/images/entities/i_char_0053_0304.gif)
be the path action
S restricted to paths lying inside this valid search space. Combining
eqn (24) and
(25) yields the optimization landscape curvature:
| |
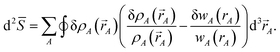 | (26) |
Substituting wmodelA = (ρsome_refA(rA, qrefA))χ(ρavgA(rA))1−χ as a DDEC-style weighting factor yields the curvature
| | |
d2model = (1 − χ)d2ISA + χd2ref
| (27) |
where
| |
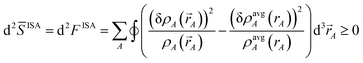 | (28) |
| |
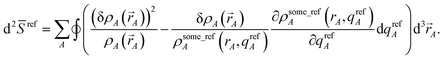 | (29) |
Bultinck
et al. previously proved the ISA curvature (
eqn (28)) is non-negative.
70
We now consider several special cases. Case 1: The ISA method, which corresponds to χ = 0. This case gives d2ISA ≥ 0 (i.e., positive semi-definite curvature) indicating a convex optimization landscape with a unique solution. However, the optimization landscape can be approximately flat (d2ISA → 0) in regions. Case 2: The non-iterative Hirshfeld method, which corresponds to χ = 1 and dqrefA = 0. This case gives
| |
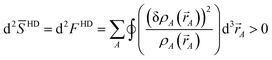 | (30) |
for any |δ
ρA(
A)| > 0. This positive definite curvature indicates a convex optimization landscape with a unique solution. Because the curvature is positive definite, the optimization landscape is not approximately flat anywhere. Case 3: The IH method, which corresponds to
χ = 1 and
| | |
dqrefA = dqA = −∮δρA(A)d3A.
| (31) |
Since {d
qrefA} can be nonzero, the second term appearing in
eqn (29) might make d
2ref negative. Consequently, we cannot guarantee that d
2ref is non-negative. From
eqn (29), the convexness or non-convexness of the IH method for a specific material depends on the particulars of the reference ion set used. Thus, under some conditions the IH method may have a negative optimization curvature. This yields the bifurcation or ‘runaway charges’ problem shown in
Fig. 1 and
Table 2. Case 4: The DDEC/c2 method, which corresponds to
χ = 1/10 and d
qrefA as defined in
eqn (31). This case has similar characteristics to Case 3 and yields the bifurcation or ‘runaway charges’ problem for the same reason. Case 5: The DDEC3 method improved over the DDEC/c2 method by increasing the d
2ref curvature term in
eqn (29),
19 but it stopped short of a provably convex optimization landscape. Thus, under rare circumstances (
e.g., H
2 triplet with 50 pm constrained bond length in
Fig. 1) it leads to the bifurcation or ‘runaway charges’ problem.
Table 2 Effect of initial guess on the converged NACs for H2 triplet with a 50 pm constrained bond length (CISD/aug-cc-pvqz electron distribution). Because the DDEC3 and IH results depend on the initial guess, they do not have a convex optimization functional or a unique solution. Because the DDEC6 NACs follow a fixed protocol of seven charge partitioning steps, they do not require any form of initial guess and converged to the unique solution (−0.0209,0.0209). Also, the convex charge partitioning functional converges to a unique solution independent of the starting guess
| Run # |
Initial guess |
DDEC3 NACs |
IH NACs |
Convex functional NACs |
| 1 |
(0,0) |
(−0.5025,0.5025) |
(−0.5978,0.5978) |
(−0.0093,0.0093) |
| 2 |
(0.5,−0.5) |
(0.5014,−0.5014) |
(0.5950,−0.5950) |
(−0.0093,0.0093) |
| 3 |
(−0.5,0.5) |
(−0.5025,0.5025) |
(−0.5978,0.5978) |
(−0.0093,0.0093) |
In the following sections, we introduce a new charge partitioning method called DDEC6 that alleviates the ‘runaway charges’ problem. Examining eqn (29), the iterative update of qrefA by requiring the self-consistency condition qrefA = qA yields the possibility of negative optimization landscape curvature and hence bifurcation. The DDEC6 method replaces the self-consistency requirement qrefA = qA with a new strategy for computing qrefA. Within a magnified integration tolerance, the DDEC6 NACs were symmetrically equivalent: ±2.1 × 10−2 (H2 triplet), ±1.8 × 10−5 (Mn solid), and −2.8 × 10−5, +1.5 × 10−4, −7.4 × 10−5, and −4.7 × 10−5 (Rh solid).
The DDEC6 method cannot lead to ‘runaway charges’ in any material, because it involves a prescribed sequence of exactly seven charge partitioning steps. For purposes of illustration, assume the initial symmetry breaking present in the input grid files is some small positive amount ε. For purposes of illustration, let us further assume that during each subsequent charge partitioning step the amount of symmetry breaking is multiplied by some finite factor K. After X charge partitioning steps, the amount of symmetry breaking will thus be KXε. If X is small or if |K| ≤ 1, the amount of symmetry breaking will be a modest multiple of ε. In the DDEC6 method, X = 7, so that even when |K| > 1 the magnitude of symmetry breaking can be contained as long as ε is small. By making the input density grids more precise, the value of ε and hence the final DDEC6 symmetry breaking (∼K7ε) can be made as small as desired. On the other hand, if X is large and |K| > 1, the symmetry breaking will be profoundly severe (i.e., ‘runaway charges’). For the DDEC/c2, DDEC3, and IH methods, X may be arbitrarily large leading to ‘runaway charges’ when |K| > 1. Since X is arbitrarily large for these methods, the value of KXε cannot be contained for |K| > 1 even if ε is made a smaller positive number. Thus, improving the input density grid precision does not necessarily alleviate the ‘runaway charges’ problem for the DDEC/c2, DDEC3, and IH methods.
We also tested a second strategy that solves the bifurcation or ‘runaway charges’ problem by constructing the self-consistent convex charge partitioning functional:
| |
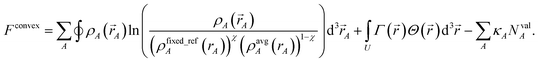 | (32) |
κA is a Lagrange multiplier that enforces the constraint
| | |
NvalA = ∮ρA(A) − NcoreA ≥ 0.
| (33) |
{
ρfixed_refA(
rA)} is a set of fixed reference densities that is not updated during the self-consistent cycles. Minimizing
Fconvex| |
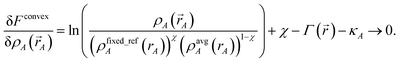 | (34) |
gives the solution
| | |
wconvexA(rA) = eκA(ρfixed_refA(rA))χ(ρavgA(rA))1−χ.
| (35) |
The curvature is
| |
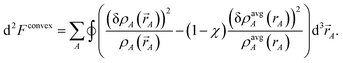 | (36) |
Inserting
eqn (28) into
(36) yields
| |
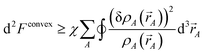 | (37) |
which is positive definite if
χ > 0. Thus, the functional is convex with a unique minimum. Through computational tests, we found nearly optimal results are obtained by setting
χconvex = 1/2. We computed {
ρfixed_refA(
rA)} by applying the upper and lower bound exponential decay constraints while minimizing a distance measure between
ρfixed_refA(
rA) and
ρcondA(
rA)〈
ρ(
)/
ρcond(
)〉
rA. (See Section S3 of the ESI
† for computational details of the Convex functional.) As shown by results in Section 3.1, the overall performance of this method is slightly inferior to the DDEC6 method.
We performed computational tests proving the bifurcation or ‘runaway charges’ problem is associated with non-unique minima on the optimization landscape. Table 2 summarizes computational results for the H2 triplet system. For consistency, all four methods compared used the same density grids, same reference ion library,71 and same integration method. (This integration method is explained in Section S2 of the ESI.†) The pair of numbers (q1,q2) denote the charges associated with the first and second H atoms, respectively. The initial guess for the IH and DDEC3 methods is the starting reference ion charge. For the DDEC3 method, the conditioned reference densities and the wA(rA) were initialized to equal the starting reference ion densities. For the Convex functional (eqn (32)), ρavgA(rA) was initialized to equal a reference ion density having the charge listed as the initial guess. The DDEC3 and IH methods exhibited pronounced bifurcation, with the converged NACs highly dependent on the initial guess. In contrast, the Convex functional exhibited the unique solution (−0.0093,0.0093) independent of the initial guess. Because the DDEC6 NACs follow a fixed protocol of seven charge partitioning steps, they do not require any form of initial guess and converged to the unique solution (−0.0209,0.0209). These results prove the DDEC3 and IH methods do not have a convex optimization functional for some materials.
3. The DDEC6 method
DDEC6 partitioning is always performed using an all-electron density. For quantum chemistry calculations performed using effective core potentials (also known as pseudopotentials), the core electrons replaced by the effective core potentials are added back in prior to DDEC6 partitioning. Also, throughout all stages of DDEC6 partitioning, zero tolerances are used to avoid division by zero errors. Specifically, if-then loops were included to smoothly handle attempted divisions by numbers with magnitudes smaller than a chosen zero tolerance (e.g., 10−10). Here, we present the DDEC6 steps and equations in a way that is independent of the integration grids employed. Specific integration procedures are described in Section S2 of the ESI.†
3.1 Conditioning steps and the equivalent reference ion weighting χequiv
The previous section demonstrated that using a fixed number of charge partitioning steps (e.g., X = 7) can alleviate the ‘runaway charges’ problem. In this section, we show how to construct schemes having a fixed number of charge partitioning steps that achieve the DDEC goal of simultaneously optimizing {ρA(A)} to resemble the smoothed reference ion densities {refA(rA, qrefA)} and the spherical average densities {ρavgA(rA)}.
First, we review the meaning of ‘conditioning’. Conditioning refers to the process in which some set of weighting functions—let us call them by the generic name {ρgenericA(rA)}—are used in a stockholder partitioning to obtain a new set of spherically averaged weighting factors, {ρconditionedA(rA)}:
| | |
ρconditionedA(rA) = ρgenericA(rA)〈ρ()/ρgeneric()〉rA
| (38) |
| |
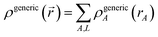 | (39) |
where 〈〉
rA denotes spherical averaging. We could perform another conditioning step by reinserting
ρgenericA(
rA) =
ρconditionedA(
rA) into the right-hand sides of
eqn (38) and
(39). This process could be repeated until some desired number,
c, of conditioning steps have been performed. Starting with the smoothed reference ions,
refA(
rA,
qrefA), a single conditioning step produces
| | |
YavgA(rA) = refA(rA, qrefA)〈ρ()/ref()〉rA
| (40) |
| |
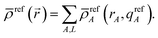 | (41) |
A second conditioning step produces
| | |
ρdouble_condA(rA) = refA(rA, qrefA)〈ρ()/ref()〉rA〈ρ()/Yavg()〉rA
| (42) |
| |
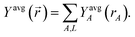 | (43) |
After
c conditioning steps,
| |
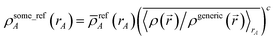 | (44) |
where

is a geometric average of all of the 〈
ρ(
)/
ρgeneric(
)〉
rA style terms.
All of the previous DDEC schemes had
| | |
wA(rA) ≈ (ρsome_refA(rA))χ(ρavgA(rA))1−χ.
| (45) |
Inserting
eqn (45) into
(7), taking the spherical average of both sides, and rearranging yields
| |
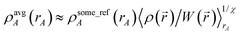 | (46) |
In general, {
ρsome_refA(
rA)} may be produced by conditioning the smoothed reference ions
refA(
rA,
qrefA) a total of c times. Substituting
eqn (44) into
(46) yields
| |
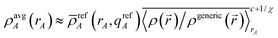 | (47) |
where the overbar denotes the (weighted) geometric average of 〈
ρ(
)/
ρgeneric(
)〉
rA style terms (such as those appearing in
eqn (40),
(42) and
(46)). Comparing
eqn (46) and
(47), the equivalent amount of reference ion weighting on a conditioning adjusted basis is therefore
| |
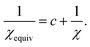 | (48) |
We now discuss specific examples. Case 1: DDEC/c1 and DDEC/c2 methods used no reference ion conditioning (i.e., c = 0) with χ = 1/10 to give χDDEC2equiv = 1/10.45 Case 2: The HD and IH methods use no reference ion conditioning (i.e., c = 0) with χ = 1 to give χHD/IHequiv = 1. Case 3: The DDEC3 method uses one conditioning step (i.e., c = 1) with χ = 3/14 to give χDDEC3equiv = 3/17. Case 4: The ISA method has two completely equivalent representations: either an infinite number of conditioning steps (i.e., c = ∞) or χ = 0 to yield χISAequiv = 0. (Exactly the same {ρISAA(A)} are obtained in either representation. In practice, the two representations are not distinguishable and have the same computational algorithm.)
When developing the DDEC3 method, Manz and Sholl showed that one conditioning step (i.e., c = 1) combined with χ = 3/14 yields an appropriate balance between reference ion weighting and spherical averaging independent of the material.19 A scheme containing a fixed number of charge partitioning steps can be constructed to yield a similar χequiv value. Specifically, if refA(rA, qrefA) are conditioned four times to yield {wDDEC6A(rA)} (i.e., c = 4 and χ = 1) and hence five times to yield the final DDEC6 {ρavgA(rA)}, this corresponds to χDDEC6equiv = 1/5. χDDEC3equiv = 3/17 lies between this value of 1/5 and the value of 1/6 that would be obtained from a total of six conditioning steps. We chose five conditioning steps, because this is more conservative. Increasing the number of conditioning steps leads to a slight decrease in the atomic multipoles at the expense of losing some of the chemical transferability. Yet even with five conditioning steps, we achieved DDEC6 atomic multipoles approximately 2–5% lower in magnitude on average (see Section 5.2) than the DDEC3 atomic multipoles. As explained in subsequent sections, this is due to using an accurate fixed reference ion charge and a weighted spherical average during some of the conditioning steps.
We now return to the bifurcation or ‘runaway charges’ problem demonstrated in the previous section. The first possible solution is to use a fixed number of charge partitioning steps. In other words, to use c = 4 and χ = 1 for a total of five conditioning steps: χDDEC6equiv = 1/5. The second possible solution is to use 0 < χ < 1 where {ρA(A)} are recovered by minimizing a provably convex optimization functional or path action. For example, Fconvex shown in eqn (32). Two conditioning steps (i.e., c = 2) are involved in computing {ρfixed_refA(rA)}. Using χconvex = 1/2 yields χconvexequiv = 1/4. We programmed both of these strategies and tested them for all systems described in this article. (Our computational method for computing the Convex functional NACs is described in Section S3 of the ESI.†) Both strategies used identical {qrefA} values. Both strategies alleviated the bifurcation or ‘runaway charges’ problem. While both approaches yielded reasonable results, the first approach proved superior to the second. The first approach allowed using a weighted spherical average in place of a simple spherical average during some of the conditioning steps. The second approach required a simple spherical average during the self-consistency iterations to prove convexness of the functional (see eqn (36)). The first approach was more computationally efficient and converged in 7 charge cycles compared to 11–14 charge cycles for the second approach. As shown in Tables 3 and 4, the first approach yielded slightly lower atomic multipole moments on average, reproduced the electrostatic potential slightly more accurately on average, and described electron transfer trends in dense solids slightly more accurately on average. Therefore, in the end we decided to go with the fixed number of charge partitioning steps (approach 1: DDEC6) as opposed to minimizing Fconvex (approach 2).
Table 3 Performance of different DDEC algorithms compared to DDEC6. Bold numbers indicate a result better than DDEC6. Italic numbers indicate a result worse than DDEC6. Numbers neither bold nor italic are within ±0.01 with respect to DDEC6 and are considered to be the same as the DDEC6 values
| |
DDEC6 (7 steps) |
DDEC3 |
4 charge partitioning steps |
10 charge partitioning steps |
Simple spherical average |
Convex functional |
| R2 for Ti solids |
0.704 |
0.360 |
0.739 |
0.671 |
0.704 |
0.718 |
| Slope [R2] for atomic dipoles (surface atoms) |
1.000 [1.000] |
1.046 [0.926] |
1.119 [0.825] |
0.956 [0.980] |
1.041 [0.955] |
1.061 [0.944] |
| Slope [R2] for atomic dipoles (solids) |
1.000 [1.000] |
1.022 [0.950] |
1.122 [0.950] |
0.943 [0.987] |
0.995 [0.995] |
1.011 [0.987] |
| Mg NAC (for MgO) |
1.465 |
2.012 |
1.312 |
1.508 |
1.473 |
1.436 |
| Ru NAC (for Li3RuO2) |
−0.083 |
−0.172 |
0.193 |
−0.149 |
−0.195 |
−0.088 |
| P NAC (for H2PO4−) |
1.622 |
1.800 |
1.656 |
1.586 |
1.682 |
1.673 |
| Li NAC (for Li2O) |
0.902 |
0.984 |
0.864 |
0.914 |
0.927 |
0.912 |
| H NAC (for water) |
0.395 |
0.417 |
0.418 |
0.381 |
0.410 |
0.414 |
| Cl NAC (Imma Na2Cl) |
−1.628 |
−2.439 |
−1.972 |
−1.492 |
−1.679 |
−1.809 |
| H NAC (H2 triplet) |
±0.021 |
±0.503 |
±0.004 |
±0.040 |
±0.019 |
±0.009 |
| Li3 dipole error |
0.645 |
0.900 |
0.420 |
0.666 |
0.771 |
0.635 |
| Minutes (NaCl 54 atoms) |
6.6 |
34.6 |
6.0 |
7.6 |
6.6 |
7.8 |
| Score for the method |
0 |
−12 |
0 |
0 |
−7 |
−5 |
Table 4 Comparison of accuracy for representing the electrostatic potential across multiple conformations of carboxylic acids, Li2O molecule, and Zn-nicotinate MOF. The first column lists the source of the NACs used to model the electrostatic potential across the various conformations. The reported values are the RMSE in kcal mol−1 averaged across all conformations. For each comparison, the best value is shown in boldface type
| NACs |
Carboxylic acids |
Li2O molecule |
Zn-nicotinate MOF |
| DDEC6 |
Convex functional |
DDEC6 |
Convex functional |
DDEC6 |
Convex functional |
| Conformation specific |
1.06 |
0.98 |
5.76 |
5.80 |
2.99 |
3.31 |
| Conformation averaged |
1.32 |
1.38 |
6.48 |
6.56 |
3.13 |
3.49 |
| Low energy conformation |
1.42 |
1.52 |
7.17 |
7.36 |
3.39 |
3.84 |
Table 3 summarizes computational tests for six different DDEC algorithms. The column labeled ‘Simple spherical average’ uses an algorithm identical to the DDEC6 method, except ρavgA(rA) is used in place of ρwavgA(rA). The performance of this algorithm is substantially worse than DDEC6 but still an improvement over DDEC3. The column labeled ‘Convex functional’ uses the charge partitioning functional of eqn (32). The Convex functional also did not perform as well on average as DDEC6. The columns labeled ‘4 charge partitioning steps’ and ‘10 charge partitioning steps’ use an algorithm identical to the DDEC6 method but stop after 4 and 10 charge partitioning steps, respectively, instead of the 7 charge partitioning steps used in the DDEC6 method. The overall performance of the ‘4 charge partitioning steps’ and ‘10 charge partitioning steps’ algorithms were similar to that of the DDEC6 method. Specifically, results for about half of the materials are improved upon going from 7 to 4 charge partitioning steps, while results for the other half of the materials are improved upon going from 7 to 10 charge partitioning steps. We chose 7 charge partitioning steps for the DDEC6 method, because it offers a suitable compromise.
The overall performance of each method was scored based on twelve computational tests:
1. Squared correlation coefficient (R2) between computed NACs and the X-ray photoelectron spectroscopy (XPS) core-electron binding energy shifts for the Ti-containing solids Ti, TiB2, TiO, TiN, BaTiO3, TiCl4, PbTiO3, CaTiO3, SrTiO3, and TiO2.33 R2 closer to one indicates a stronger correlation.
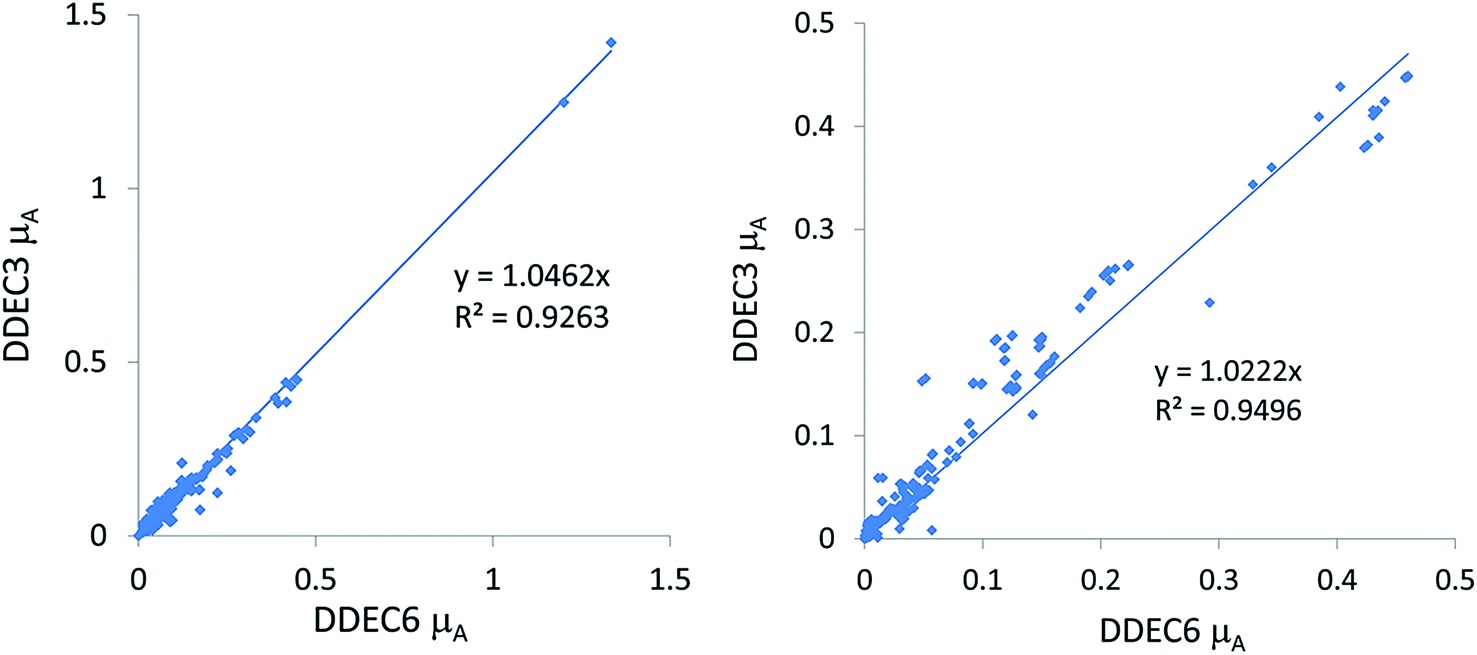
2. The slope for a plot analogous to the left panel of the plot in Section 5.2 where each charge model is compared to DDEC6. A smaller slope (considered better) indicates smaller atomic dipole magnitudes (μA) for the test set containing mostly surface atoms.
3. The slope for a plot analogous to the right panel of the plot in Section 5.2 where each charge model is compared to DDEC6. A smaller slope (considered better) indicates smaller atomic dipole magnitudes (μA) for the test set containing dense materials.
4. The NAC of Mg in crystalline MgO.33 A NAC closer to ∼1.7 was considered better.
5. The NAC of Ru in crystalline Li3RuO2.33 A NAC closer to ∼0.1 was considered better.
6. The NAC of P in the H2PO4− molecular ion.33 A NAC closer to ∼1.5 is considered better.
7. The NAC of Li in linear Li2O.33 A NAC closer to ∼0.87 is considered better.
8. The NAC of H in the H2O molecule.33 A NAC closer to ∼0.37 is considered better.
9. The NAC of Cl in the high-pressure Imma Na2Cl crystal. A NAC closer to −1.35 is considered better.
10. The NAC of H in the H2 molecule (triplet state, 50 pm constrained bond length, CISD/aug-cc-pvqz). A NAC closer to zero is better.
11. The Li3 molecule dipole error (atomic units) quantified as μA for the NAC model minus μA for the DFT-computed electron distribution (PBE + D3 (ref. 74)/aug-cc-pvtz optimized geometry and electron distribution). An error closer to zero is better.
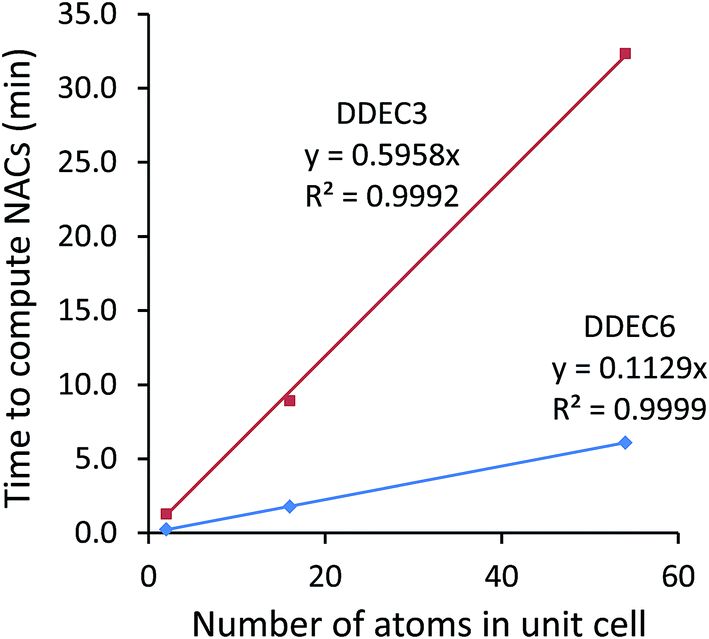
12. The time in minutes required to perform charge analysis on the NaCl crystal supercell containing 54 atoms running on a single processor core in Intel Xeon E5-2680v3 on the Comet supercomputing cluster at SDSC. This is the total wall time from program start to program finish, including the input file reading, core electron partitioning, valence electron partitioning, computation of multipole moments, output file printing, etc.
The target values for tests 4, 5, and 9 (dense solids) are charges that approximate the number of electrons in the volume dominated by each atom and were chosen to approximate Bader charges.33 The target values for tests 6, 7, and 8 (molecules) are charges that will more closely reproduce the electrostatic potential and were chosen to approximate ESP charges.
NACs are often used to construct force-fields for classical molecular dynamics and Monte Carlo simulations. For these applications, the NACs should approximately reproduce the molecular electrostatic potential (MEP). Conformational transferability is important for the construction of flexible force-fields. Table 4 compares the accuracy of the DDEC6 and Convex functional methods for reproducing the electrostatic potential across various conformations of carboxylic acids, Li2O molecule, and Zn-nicotinate metal–organic framework. Results listed under the ‘carboxylic acids’ column in Table 4 are the averages for the previously published conformations of five carboxylic acids.19,33 The Li2O conformations span angles from 90° to 180° in 10° increments.33 The Zn-nicotinate conformations are described in Section 5.3. When NACs were optimized specifically for each molecular conformation, V() was described most accurately using the Convex functional for the carboxylic acids and most accurately by the DDEC6 method for the Li2O molecule and Zn-nicotinate MOF. When the conformation averaged and low-energy conformation NACs were used to construct point-charge models for every system conformation, the DDEC6 method reproduced V() most accurately for all three types of materials. This shows the DDEC6 NACs are more accurate than the Convex functional NACs for reproducing V() across multiple system conformations. In summary, the DDEC6 NACs have better overall performance than the Convex functional NACs.
Finally, we performed additional computational tests proving the equivalence relations described in this section. Specifically, we also analyzed nearly all systems in this article using c = 1 and χ = 1/3 (yielding χequiv = 1/4) and obtained similar (but not strictly identical) results to the c = 2 and χ = 1/2 case discussed above. The computational cost was increased to ∼24 charge cycles for convergence.
3.2 Determining the DDEC6 reference ion charge value (charge cycles 1 and 2)
The reference ion charge is the most significant difference between the DDEC6 and DDEC3 methods. In the DDEC3 method, the reference ion charge is the same as the AIM NAC: qrefA = qA. While setting qrefA = qA has some theoretical appeal, it also comes with an important disadvantage. In dense materials, the diffuse nature of anions can cause the number of electrons assigned to an anion to be much greater than the number of electrons in the volume dominated by the anion. This causes several related problems. First, NACs assigned with qrefA = qA may fail to assign core electrons to the proper atom in some materials (see Section 5.5). Second, they do not properly describe electron transfer trends in some materials.33 Third, the correlation between NACs and core electron binding energy shifts will be weakened due to the overly delocalized assignment of electrons to the anions (see Ti-containing compounds in Table 3). Fourth, the accuracy of reproducing the electrostatic potential may be slightly degraded in some materials for which the anion charges are overestimated in magnitude.33
There are two competing philosophies of electrons belonging to an atom: localized and stockholder atomic charges. For localized atomic charges, electrons in the volume dominated by an atom are assigned almost entirely to that atom. Non-overlapping compartments, such as those encountered in Bader's QCT and Voronoi cell partitioning, are the most extreme limit of localized charge partitioning.50,57,58,62,63 Localized NACs (e.g., Bader or Voronoi cell NACs) convey useful information about charge transfer trends and core-electron binding energy shifts, but they are not well-suited to reproducing the electrostatic potential surrounding a material.19,33,45,75,76 On the other hand, stockholder-type charge partitioning schemes assign overlapping atomic electron distributions.18 DDEC3 NACs, which do not incorporate localized atomic charge information, are usually more accurate than Bader NACs at reproducing V() but suffer the problems mentioned in the previous paragraph as shown in Section 5.5 and ref. 33.
To achieve the best of both worlds, the DDEC6 method uses a fixed reference ion charge consisting of a weighted average of localized and stockholder charges. Specifically, the DDEC6 reference ion charge value, qrefA, is set using the following scheme:
| |
qs,refA = (1 − ![[small script l]](https://www.rsc.org/images/entities/i_char_e146.gif) )qs,StockA + qs,LocA, s = {1,2} )qs,StockA + qs,LocA, s = {1,2}
| (50) |
| | |
qi,Stock/LocA = zA − Ni,Stock/LocA
| (51) |
| |
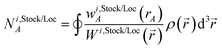 | (52) |
| |
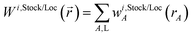 | (53) |
| | |
w1,StockA(rA) = refA(rA, 0)
| (54) |
| | |
w1,LocA(rA) = (refA(rA, 0))4
| (55) |
| | |
w2,StockA(rA) = refA(rA, q1,refA)
| (56) |
| | |
w2,LocA(rA) = (refA(rA, q1,refA))4.
| (57) |
N1,LocA and N2,LocA are measures of the number of electrons in the volume dominated by each atom. N1,LocA is computed using the neutral atom reference densities (eqn (55)). N2,LocA is computed using charged reference ions (eqn (57)). The fourth power appearing in eqn (55) and (57) makes ws,LocA(rA) {s = 1,2} change continuously while also becoming negligible in the volume of space for which refA(rA) < refB(rB). When using an exponent of 4, the ratio refA(rA)/refB(rB) = 2 corresponds to assigning 16 times higher density to atom A compared to atom B at this grid point. This provides an appropriate balance between transition sharpness and smoothness. Using an exponent less (more) than 4 would cause the density transition between atoms to be more gradual (sharper). An arbitrarily high exponent would correspond to the limit of non-overlapping atoms. Eqn (50) updates the reference ion charges by adding a fraction of this localized charge to a fraction (1 − ) of the stockholder charge based on minimizing the information distance to the (prior) reference ion densities. The final reference ion charge (eqn (49)) determined in this manner is a compromise between counting electrons in the volume dominated by each atom and counting electrons in proportion to the (prior) reference ion densities.
The superscript numerals 1 and 2 refer to the charge cycle in which that quantity is computed. The atomic weighing factors for the first charge cycle are computed using neutral reference atoms (eqn (54) and (55)). The atomic weighting factors for the second charge cycle are computed using eqn (56) and (57) based on the results of the first charge cycle. Each of the first two charge cycles first computes Wi,Stock() and Wi,Loc() by a loop over atoms and grid points to compute the sum in eqn (53). Each of the first two charge cycles then performs another loop over atoms and grid points to compute Ni,StockA and Ni,LocA via eqn (52). The stockholder, localized, and updated reference charges are then computed via eqn (50) and (51). The first charge cycle yields the HD NACs: qHDA = q1,StockA. (Even though the CM5 NACs are not used in DDEC6 charge partitioning, they were computed at this stage using the HD NACs and the CM5 definition of Marenich et al.20) The second charge cycle yields the DDEC6 reference ion charges (eqn (49)).
The fraction of localized atomic charge (qs,LocA) used to compute these fixed reference ion charges was decided through a scientific engineering design approach in which we tested dozens of alternatives. (See Section S1 of the ESI† for a summary of additional alternatives explored.) Table 5 compares results for = 0 to 1 in 1/6 increments. The accuracy was quantified using five tests:
Table 5 Effect of changing the fraction of localized to stockholder charge when computing the fixed reference ion charge value. See the main text for a description of the five tests. The best value for each row is shown in boldface type
| |
Fraction of localized charge in reference ion charge |
| 0 |
1/6 |
1/3 |
1/2 |
2/3 |
5/6 |
1 |
| R2 Ti compounds |
0.672 |
0.686 |
0.696 |
0.703 |
0.704 |
0.708 |
0.709 |
| ΔCo NAC |
0.126 |
0.116 |
0.097 |
0.081 |
0.052 |
0.023 |
0.007 |
| Mg NAC in MgO |
1.166 |
1.229 |
1.304 |
1.384 |
1.465 |
1.542 |
1.613 |
| RMSE H2O |
2.150 |
1.768 |
1.443 |
1.222 |
1.164 |
1.295 |
1.565 |
| RMSE H2PO4− |
1.397 |
1.284 |
1.298 |
1.430 |
1.647 |
1.920 |
2.219 |
1. Squared correlation coefficient (R2) between computed NACs and the X-ray photoelectron spectroscopy (XPS) core-electron binding energy shifts for the Ti-containing solids Ti, TiB2, TiO, TiN, BaTiO3, TiCl4, PbTiO3, CaTiO3, SrTiO3, and TiO2.33 R2 closer to one indicates a stronger correlation.
2. The NAC of the Co atom in solid CoO2 minus that in solid LiCoO2. Simple chemical arguments and electron density difference maps indicate this results should be significantly positive.23
3. The NAC of Mg in crystalline MgO.33 A NAC closer to ∼1.7 was considered better.
4. The root-mean-squared error (RMSE) in kcal mol−1 for reproducing the electrostatic potential surrounding the H2O molecule. A lower RMSE is better.
5. The root-mean-squared error (RMSE) in kcal mol−1 for reproducing the electrostatic potential surrounding the H2PO4− molecular ion. A lower RMSE is better.
As shown in Table 5, some of these tests benefited from small values of while others benefited from large values of . The minimum RMSE for H2O occurs near = 2/3. Two of the remaining four tests exhibited optimal performance for > 2/3, while the remaining two tests exhibited optimal performance for < 2/3. Therefore, we have chosen
| | |
= 2/3
| (58) |
as a suitable compromise to define the DDEC6 method.
3.3 Computing the conditioned reference ion density (charge cycle 3)
The next step (i.e., third charge cycle) is to compute the conditioned reference ion densities, ρcondA(rA). This conditioning matches the reference ion densities to the specific material of interest. First, a loop of over grid points and atoms is performed to compute ref() via eqn (41). Then another loop over grid points and atoms is performed to compute YavgA(rA) via eqn (40). In the DDEC6 method, each conditioned reference density, ρcondA(rA), is constrained to monotonically decrease with increasing rA| |
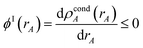 | (59) |
and to integrate to the number of electrons in the reference ion:| |
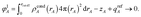 | (60) |
These constraints were introduced for theoretical appeal to ensure expected behavior. In tests we performed, these constraints had only a small effect on the NACs. They are not present in the DDEC3 method.
{ρcondA(rA)} is found by minimizing the functional
| |
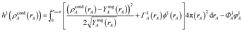 | (61) |
where
ΓIA(
rA) and
ΦIA are Lagrange multipliers enforcing constraints
(59) and
(60), respectively. The minimum of
hI is found by setting
| |
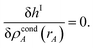 | (62) |
Inserting eqn (61) into (62) and using integration by parts to simplify gives the solution
| |
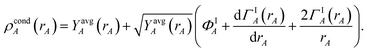 | (63) |
Because
| |
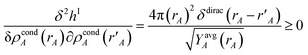 | (64) |
it directly follows that
hI(
ρcondA(
rA)) is a convex functional. Therefore, {
ρcondA(
rA)} is uniquely determined for a given {
YavgA(
rA)} input.
Section S4.1 of the ESI† describes the iterative algorithm we used to compute {ρcondA(rA)}. This description includes an algorithm flow diagram in Fig. S1 of the ESI.† Because our algorithm cuts the size of the search domain by better than half in each reshaping cycle, it is mathematically guaranteed to always converge to the correct solution in a few reshaping cycles.
After computing {ρcondA(rA)}, a loop over grid points and atoms is performed to compute
| |
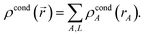 | (65) |
Then another loop over grid points and atoms is performed to compute
| |
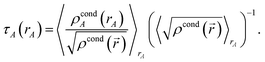 | (66) |
3.4 Updating {wA(rA)} (charge partitioning steps 4 to 7)
First, we provide a brief overview of charge partitioning steps 4 to 7. The fourth through seventh charge partitioning steps compute the weighted spherical average and enforce the exponential tail constraints. Because it is advisable to enforce the NvalA ≥ 0 constraint only after the exponential tail constraints have already been enforced, the NvalA ≥ 0 constraint is enforced beginning with the fifth charge partitioning step. κA is the Lagrange multiplier that enforces NvalA ≥ 0. We now explain the distinction between the terms ‘charge partitioning step’ and ‘charge cycle’. Here, the term ‘charge cycle’ refers to a sequence of loops that involves computing W() from {wA(rA)}, followed by a stockholder partition (eqn (68)) to compute {NA}, and finally the update of {wA(rA)}. Charge cycles that update {κA} but not any other contributions to {wA(rA)} are part of the same ‘charge partitioning step’. Thus, in general, each charge cycle follows one of two paths: (a) if the value of update_kappa is FALSE, the charge partitioning cycle follows path 6a in which the exponential tail constraints are applied, and (b) if the value of update_kappa is TRUE, the charge partitioning cycle follows path 6b in which {κA} is updated. The first charge cycle in a charge partitioning step follows path 6a and the subsequent charge cycles (if any) in a charge partitioning step follow path 6b. Fig. S2 in the ESI† is a flow diagram summarizing the DDEC6 method.
Now, we provide the detailed sequence for charge partitioning steps 4 to 7. The fourth charge cycle uses:
| | |
wA(rA) = ρcondA(rA).
| (67) |
Initialize the following variables: update_kappa = FALSE, completed_steps = 3, charge_cycle = 4, and {
κA} = {0.0}. The fourth and later charge cycles use the following sequence of steps:
1. In the first loop over grid points and atoms, the sum in eqn (8) is computed at each grid point.
2. In the second loop over grid points and atoms, the following quantities are computed:
| | |
ρA(A) = wA(rA)ρ()/W()
| (68) |
| | |
ρavgA(rA) = 〈ρA(A)〉rA
| (69) |
| |
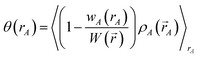 | (70) |
| |
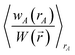 | (71) |
| | |
NA = ∮ρA(A)d3A.
| (72) |
All of these quantities except {
ρA(
A)} are stored.
3. A weighted spherical average density, ρwavgA(rA), is then computed:
| |
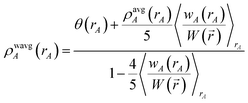 | (73) |
Eqn (73) has the form of a weighted spherical average density
| |
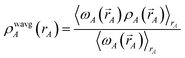 | (74) |
where
ρA(
A) is weighted at each grid point proportional to
| |
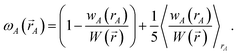 | (75) |
Examining
eqn (75), the relative weight assigned to each point is bounded by
| | |
0.1 ≲ ωA(A) < 1.2.
| (76) |
The lower bound of ∼0.1 occurs, because it is not possible to have 〈
wA(
rA)/
W(
)〉
rA = 0 if
wA(
rA)/
W(
) ≈ 1 for any grid point on the same radial shell. For a specific
rA, positions with larger
wA(
rA)/
W(
) receive smaller
ωA(
A), and positions with smaller
wA(
rA)/
W(
) receive larger
ωA(
A). Thus,
ρwavgA(
rA) weights portions of
ρA(
A) that overlap other atoms more heavily than those portions that do not overlap other atoms. At this stage, we also compute:
| |
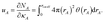 | (77) |
4. On the fourth charge cycle, update_kappa is not altered. On the fifth and subsequent charge cycles, update_kappa is set to TRUE if NvalA < −10−5 electrons (e) for any atom. If update_kappa is TRUE and the NA changes between successive charge cycles were less than 10−5 electrons for each atom two consecutive times in a row then update_kappa is reset to FALSE and {κA} is reset to {0.0}. Otherwise, the current value of update_kappa is not altered. This sequence of steps has the effect of: (a) setting update_kappa to TRUE when the number of valence electrons is negative for any atom on the fifth and later charge cycles, (b) setting update_kappa to FALSE when {κA} are converged for a particular charge partitioning step, and (c) starting each charge partitioning step with the initial guess {κA} = {0.0}.
5. If the value of update_kappa is FALSE (i.e., {κA} are converged for that charge partitioning step), then completed_steps is incremented by +1. If completed_steps = 7, the iterative charge cycles are finished and exit at this point.
6a. If the value of update_kappa is FALSE, then {wA(rA)} is updated to impose the constraints
| |
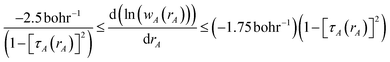 | (78) |
which are illustrated in
Fig. 2.
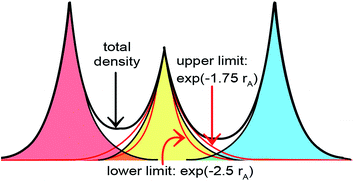 |
| | Fig. 2 Illustration of exponential decay constraints applied to wA(rA) in the buried atom tails. The red curves are not drawn to scale. | |
(i) Constraint preventing buried tails from becoming too diffuse: analogous to the DDEC3 method,19 GA(rA) is computed to make sure the tails of buried atoms do not become too diffuse. In the DDEC6 method, we set
instead of the DDEC3 expression for
σA(
rA). The constraints
| |
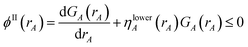 | (80) |
| | |
ηlowerA(rA) = (1.75 bohr−1)(1 − (τA(rA))2)
| (81) |
| |
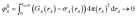 | (82) |
are imposed by minimizing the following optimization functional
| |
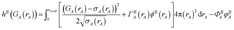 | (83) |
where
ΓIIA(
rA) and
ΦIIA are Lagrange multipliers enforcing constraints
(80) and
(82), respectively.
19 hII(
GA(
rA)) is a convex functional with the unique minimum:
19| |
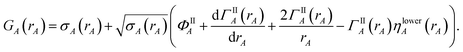 | (84) |
Section S4.3 of the ESI
† describes the iterative algorithm we used to compute {
GA(
rA)}. This description includes an algorithm flow diagram in Fig. S3 of the ESI.
† Because our algorithm cuts the size of the search domain by better than half in each reshaping cycle, it is mathematically guaranteed to always converge to the correct solution in a few reshaping cycles.
(ii) Constraint preventing buried tails from becoming too contracted: HA(rA) is then computed to make sure the tails of buried atoms do not become too contracted. Specifically, the constraints
| |
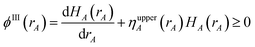 | (85) |
| |
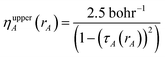 | (86) |
| |
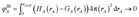 | (87) |
are imposed by minimizing the following optimization functional
| |
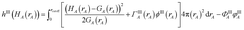 | (88) |
where
ΓIIIA(
rA) and
ΦIIIA are Lagrange multipliers enforcing constraints
(85) and
(87), respectively.
hIII(
HA(
rA)) is a convex functional:
| |
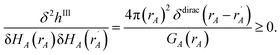 | (89) |
The unique minimum is
| |
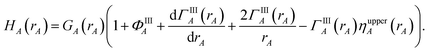 | (90) |
In practice,
HA(
rA) can be easily found by starting with the initial estimate
HestA(
rA) =
GA(
rA) and enforcing constraint
(85) by recursively setting
| | |
HestA(rA) = max(HestA(rA), HestA(rA − ΔrA)exp(−ηupperA(rA)ΔrA))
| (91) |
beginning with the second radial shell and continuing outward until the last radial shell. Finally,
HA(
rA) is normalized to satisfy constraint
(87):
| |
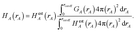 | (92) |
Comments on these exponential tail constraints: Constraint (80) preventing the tails of buried atoms from becoming too diffuse is the same for the DDEC3 and DDEC6 methods. The DDEC6 method adds constraint (85) to prevent the tails of buried atoms from becoming too contracted. The limiting exponent of 2.5 bohr−1 corresponds to wA(rA) in the buried tail being cut in half for an rA increase of 0.277 bohr (0.147 Å). There is no reason for wA(rA) to decrease more rapidly than this in the buried tail. The integrals of the optimization functionals hI,II,III in eqn (61), (83) and (88) have similar forms, except that a square root appears in the denominator of the first integral in hI,II but not in hIII. This exponent in the denominator of the first integral of the optimization functional is called the reshaping exponent.19 A reshaping exponent ξ = 1/2 is used in the optimization functional hII (eqn (83)) that enforces constraint (80) preventing tails of buried atoms from becoming too diffuse.19 A reshaping exponent ξ = 1/2 is also used in the optimization functional hI (eqn (61)) that reshapes the conditioned reference ion densities. The value ξ = 1/2 is appropriate for these cases, because it shifts electron density from the tail region into the intermediate region where atoms interface.19 A reshaping exponent ξ = 1 is used in the optimization functional hIII (eqn (88)) that enforces constraint (85) preventing tails of buried atoms from becoming too contracted. Eqn (91) implementing constraint (85) adds electron density to the tail region thereby requiring density to be removed during renormalization. Removing electron density during renormalization with ξ < 1 is ill-behaved, because this will completely deplete the electron density in the buried tail region due to wA(rA)ξ/wA(rA) ≫ 1 when wA(rA) ≪ 1. Using ξ = 1 avoids this problem.
6b. If the value of update_kappa is TRUE, then {κA} is updated by setting
| | |
κA = max(0, (κoldA − NvalA/uA))
| (93) |
where
κoldA is the value of
κA before the update is applied. The {
wA(
rA)} is then updated
via eqn (96). For an individual charge partitioning step, this process corresponds to minimizing the functional
| |
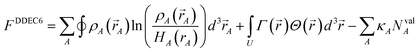 | (94) |
where
Γ(
) and {
κA} are Lagrange multipliers enforcing constraints
(4) and
(33), respectively. Within an individual charge partitioning step, the {
HA(
rA)} remain constant during the update of {
κA}. Consequently, the curvature is given by
| |
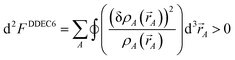 | (95) |
for any |δ
ρA(
A)| > 0. This positive definite curvature indicates a convex optimization landscape with a unique solution. For an atom without any overlaps (
i.e., isolated atomic ion limit),
uA = 0 and the converged
ρA(
A) is independent of
κA. Therefore, when
uA is negligible (
e.g.,
uA < 10
−7) we set
κA = 0.0 to avoid division by zero in
eqn (93).
7. The updated atomic weighting factor is
The charge_cycle is incremented by +1, and the calculation returns to # 1 above to start the next charge cycle.
3.5 Summary of changes between DDEC3 and DDEC6 methods
Table 6 summarizes the five differences between the DDEC3 and DDEC6 methods. First, the DDEC6 method uses fixed reference ion charge values rather than self-consistently updating them as in the DDEC3 method. Second, the DDEC6 method uses c = 4 and χ = 1 to yield a fixed number of charge partitioning steps with χDDEC6equiv = 1/5. In contrast, the DDEC3 method uses a self-consistent iterative scheme with c = 1 and χDDEC3 = 3/14 to yield χDDEC3equiv = 3/17. Third, the DDEC6 method uses a weighted spherical average in place of the simple spherical average used in the DDEC3 method. Fourth, the DDEC6 method incorporates constraints to make the conditioned reference ion densities monotonically decreasing and to integrate to NrefA. Fifth, the DDEC6 method adds a constraint to ensure the buried tails of wA(rA) do not decay too quickly. Both the DDEC3 and DDEC6 methods include the same constraint to ensure the buried tails of wA(rA) do not decay too slowly. These constraints make the buried tail of wA(rA) decay exponentially with increasing rA.
Table 6 List of differences between DDEC3 and DDEC6 methods
| |
|
DDEC3 |
DDEC6 |
| 1 |
Reference ion charge |
Iteratively set to AIM charge: qrefA = qA |
Non-iteratively set based on a special partitioning: qrefA = q2,refA |
| 2 |
How spherical averaging is incorporated |
c = 1, χ = 3/14, χDDEC3equiv = 3/17 |
c = 4, χ = 1, χDDEC6equiv = 1/5 |
| 3 |
Type of spherical average used |
Simple |
Weighted |
| 4 |
Constraints applied to each conditioned reference ion density |
— |
Monotonically decreasing with increasing rA and integrates to NrefA = zA − qrefA |
| 5 |
Tail constraints applied to wA(rA) during 4th and later charge cycles |
Tails of buried atoms decay no slower than exp(−1.75 rA) |
Tails of buried atoms decay no slower than exp(−1.75 rA) and no faster than exp(−2.5 rA) |
4. Computational details
4.1 Quantum chemistry calculations
We performed periodic quantum chemistry calculations using VASP77,78 software. Our VASP calculations used the projector augmented wave (PAW) method79,80 to perform all-electron frozen-core calculations including scalar relativistic effects with a plane-wave basis set cutoff energy of 400 eV. Calculations specifying “2 frozen Na core electrons” or “10 frozen Na core electrons” used PAWs for the Na atom including 2 or 10 frozen core electrons, respectively. For all systems, the number of k-points times the unit cell volume exceeded 4000 Å3. This is enough k-points to converge relevant properties including geometries and AIM properties (NACs, ASMs, etc.). Except for the solid surfaces, geometry optimizations relaxed both the unit cell vectors and ionic positions. The solid surface calculations used the DFT-optimized bulk lattice vectors and relaxed the ionic positions. Where noted, experimental crystal structures or other geometries from the published literature were used. A Prec = Accurate (∼0.14 bohr) electron density grid spacing was used. Bader NACs were computed using the program of Henkelman and coworkers.44
We performed non-periodic quantum chemistry calculations using GAUSSIAN 09 (ref. 81) software. ESP NACs were computed in GAUSSIAN 09 using the Merz–Singh–Kollman scheme.10,82
Some materials mentioned in the developmental tests of this article are studied in more detail in the sequel article. These include H2O molecule, H2PO4− molecular ion, formamide, Ti-containing solids, various Li2O molecular conformations, B-DNA decamer, MgO solid, LiCoO2 solid, CoO2 solid, Li3RuO2 solid, Fe2O3 solid, five carboxylic acids in various conformations, natrolite, Mo2C surface slab with K adatom, NaF surface slab, SrTiO3 surface slab, Pd crystal and alloys with interstitial H atom, B4N4 cluster, BN nanotube, BN sheet, Fe4O12N4C40H52 single molecule magnet with non-collinear magnetism, Mn12-acetate single molecule magnet, and the metal–organic frameworks ZIF-8, ZIF-90, IRMOF-1, and large-pore MIL53(Al). The computational details (e.g., basis sets and exchange–correlation functional), geometries, and full list of DDEC6 NACs for these materials are presented in the sequel article.33
4.2 Electrostatic potential expansion: atomic multipole moments and charge penetration terms
In this section, we review the basic principles of expanding the electrostatic potential, V(), into atomic contributions. The electrostatic potential is important for understanding interactions in all chemical systems.83–85 AIM methods provide a formally exact expansion of V() by partitioning the total electron distribution ρ() into atomic electron distributions, {ρA(A)}:| |
 | (97) |
| |
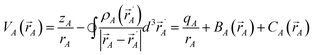 | (98) |
where BA(A) and CA(A) are terms due to atomic multipoles (AMs) and penetration of the atom's electron cloud, respectively.39,45,84,86,87 Outside ρA(A), the charge penetration term CA(A) vanishes reducing VA(A) to a multipole expansion.88 Although eqn (98) is formally exact for all AIM methods, in practice BA(A) and CA(A) are truncated at some finite orders leading to approximate V() expansions.86,87,89 Therefore, AIM methods providing more rapidly converging V() expansions are more convenient for constructing force-fields. For constructing flexible force-fields, the AIM NACs should preferably have good conformational transferability. Alternative expansions of V() and system multipole moments based on distributed multipole analysis and Gaussian density functions are given in the related literature.36–38,40,41
For each system, we computed atomic multipoles up to quadrupole order using well-known formulas. Atomic dipoles,
| |
μA = |![[small mu, Greek, vector]](https://www.rsc.org/images/entities/i_char_e0e9.gif) A| A|
| (100) |
are the leading component of
BA(
A). Atomic quadrupoles have the form
| | |
QT = −∮TAρA(A)d3A
| (101) |
where
T is a second degree polynomial. The five linearly independent quadrupole components can be expressed as (i)
TA = (
X −
XA)(
Y −
YA) for
Qxy, (ii)
TA = (
X −
XA)(
Z −
ZA) for
Qxz, (iii)
TA = (
Y −
YA)(
Z −
ZA) for
Qyz, (iv)
TA = (
X −
XA)
2 − (
Y −
YA)
2 for
Qx2 − y2, and (v)
TA = 3(
Z −
ZA)
2 − (
rA)
2 for
Q3z2−r2, where
A = (
XA,
YA,
ZA) is the nuclear position. The traceless atomic quadrupole tensor,
, is defined by
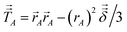
, where
is the identity tensor. The three eigenvalues of
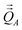
are independent of molecular orientation and coordinate system, and they sum to zero. For a spherical atom, all three quadrupole eigenvalues are zero. However, three zero quadrupole eigenvalues does not mean the atom is necessarily spherical, because such an atom could have non-zero dipole or higher order multipole (
e.g., octapole) moments that indicate deviation from spherical symmetry. An oblate spheroidal density has one negative and two equal positive quadrupole eigenvalues. A prolate spheroidal density has one positive and two equal negative quadrupole eigenvalues. An ellipsoidal density can have three unequal quadrupole eigenvalues.
For finite clusters, molecules, and ions, the total dipole moment, μ = ||, is given by
| |
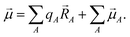 | (102) |
Therefore, a charge model including both NACs and atomic dipoles reproduces
exactly to within a grid integration tolerance. A point-charge only model includes the first term in
eqn (102) but neglects the atomic dipole terms. Unless a point-charge only model is explicitly defined to reproduce
(or higher order multipole) exactly, it will generally reproduce
(or higher order multipole) only approximately except in cases where
(or higher order multipole) is zero by symmetry.
90 (The general idea to constrain atom-centered point charges to exactly reproduce the molecular dipole moment is impossible for planar molecules placed in an electric field perpendicular to the molecule's plane. For this reason, we abandon the idea to constrain atom-centered point charges to exactly reproduce the system's dipole moment.) The total
pth order multipole of a finite cluster, molecule, or ion can be expressed as a sum over NACs and atomic multipoles up to
pth order.
86,91
Spherical cloud penetration, CavgA(rA), is the leading term in CA(A):
| | |
CA(A) = CavgA(rA) + Cnon-sphericalA(A)
| (103) |
| |
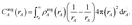 | (104) |
Eqn (104) is the well-known result arising from basic electrostatic principles. The tail of
ρavgA(
rA) decays approximately exponentially with increasing
rA:
| | |
ρavgA(rA) ≈ eב−אrA for rmin_fit ≤ rA ≤ rcutoff
| (105) |
Inserting
(105) into
(104) yields,
| |
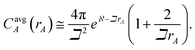 | (106) |
To determine the parameters
א and
ב for each atom, the C
HARGEMOL program performed a linear least squares fit of
ב −
אrA to ln(
ρavgA(
rA)) over the range
rmin_fit = 2 Å to
rcutoff = 5 Å.
19 The
R-squared correlation coefficient for this linear regression was usually >0.99 indicating a nearly exact fit. Previous studies have used different variations of exponential or Gaussian decaying densities (sometimes multiplied by polynomials in
rA) or exponential damping of the 1/
r or multipole potential to provide approximations of charge penetration energies.
41,92–96
A charge model's accuracy for reproducing V() can be quantified by the root mean-squared error (RMSE) in the electrostatic potential quantified over a chosen a set of grid points.11–13,15,19,97,98 The specific method we used to compute RMSE is detailed in previous work and includes a constant potential adjustment to equalize the average electrostatic potentials (over the chosen set of grid points) of the charge model and full electron distribution.13,19 The charge model may include point charges, dipoles and/or higher order multipoles, spherical cloud penetration, and/or aspherical cloud penetration terms. The relative root mean squared error (RRMSE) is the RMSE for the charge model divided by the RMSE of a null charge model having {VA(A)} = 0.10,13,15,16,82 The RMSE and RRMSE were computed over a uniform grid of points lying between surfaces defined by γinner and γouter times the van der Waals (vdW) radii. We set (γinner,γouter) = (1.4,2.0) for nonperiodic materials and (γinner,γouter) = (1.3,20.0) for periodic materials.15,16,82 For three-dimensional materials containing only nanopores (e.g., MOFs, zeolites), γouter = 20.0 is sufficiently large that the RMSE grid points span the entire pores. We used the same UFF vdW radii as Campaña et al.13 (REPEAT program) which are listed in the ESI† of Watanabe et al.15
4.3 Ewald summation
In the periodic materials, the Ewald summation method of Smith, including NACs and (optionally) atomic dipoles, was used to compute electrostatic potentials for RMSE calculations.99 This Ewald summation separates the Coulomb potential into a short-range portion summed in real space and a long-range portion summed in reciprocal space:| | |
V() = Vshort-range() + Vlong-range()
| (107) |
| |
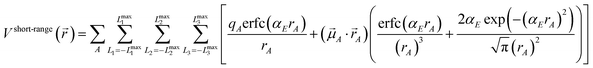 | (108) |
We set the Ewald summation convergence parameter to αE = π/(10 Å). Enough real space replications of the unit cell were included such that every point in the reference unit cell was surrounded by at least 3/αE real space distance in all directions:| |
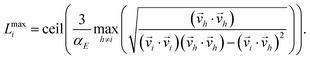 | (109) |
This corresponds to an erfc(αErA)/rA cutoff of (αE/3)erfc(3) = 6.9 × 10−7 bohr−1. The reciprocal lattice vectors are defined by| |
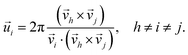 | (110) |
The reciprocal space summation encompassed integer multiples bi of the corresponding reciprocal lattice vectors| |
k2 = ![[k with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_006b_20d1.gif) · ·
| (112) |
to yield the long-range portion of the electrostatic potential| |
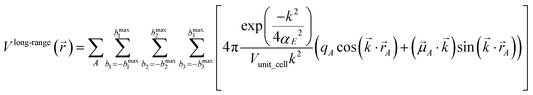 | (113) |
where A in eqn (113) is computed using (L1, L2, L3) = (0, 0, 0). The term (b1, b2, b3) = (0, 0, 0) is excluded from the sum in eqn (113). Vunit_cell is the unit cell volume. Our reciprocal space cutoff| |
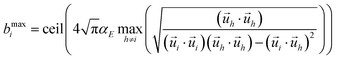 | (114) |
includes at least all reciprocal space vectors having 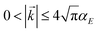 . Noting that each term in the reciprocal space term includes exp(−k2/(4αE2)) as a multiplier, our reciprocal space cutoff corresponds to exp(−k2/(4αE2)) ≈ exp(−4π) ≈ 3.5 × 10−6. Because it is a short-range effect, spherical charge penetration can be included entirely in the real space summation using the analytic potential of eqn (106). While the spherical cloud penetration effect is small over grid points used to compute RMSE, it becomes increasingly important for smaller rA values.
. Noting that each term in the reciprocal space term includes exp(−k2/(4αE2)) as a multiplier, our reciprocal space cutoff corresponds to exp(−k2/(4αE2)) ≈ exp(−4π) ≈ 3.5 × 10−6. Because it is a short-range effect, spherical charge penetration can be included entirely in the real space summation using the analytic potential of eqn (106). While the spherical cloud penetration effect is small over grid points used to compute RMSE, it becomes increasingly important for smaller rA values.
5. Performance tests
5.1 Convergence speed
DDEC charge and spin partitioning use a cutoff radius (e.g., 5 Å) to achieve a computational cost that scales linearly with increasing number of atoms in the unit cell after the initial electron and spin density grids have been generated.19,100 When combined with a linearly scaling quantum chemistry program (e.g., ONETEP), this provides computationally efficient charge analysis even for systems containing thousands of atoms in the unit cell.101,102 Fig. 3 plots the wall time for computing DDEC3 and DDEC6 NACs, atomic multipoles, and electron cloud decay exponents for the NaCl crystal (ambient pressure) as a function of the number of atoms in the unit cell. The unit cells containing 16 and 54 atoms were constructed by forming 2 × 2 × 2 and 3 × 3 × 3 supercells, respectively, that were used as input for computing the density grid files in VASP. This calculation utilized a volume of 2 × 10−3 bohr3 per grid point. The wall time in Fig. 3 begins when the CHARGEMOL program is first entered prior to reading the VASP density grid files and continues until the moment after the computed NACs, atomic multipoles, and electron cloud decay exponents have been written to the net_atomic_charges.xyz file. As expected, Fig. 3 shows the required wall time depends linearly on the number of atoms in the unit cell. Even though the computation was run on a single processor core, only six minutes were required for DDEC6 charge analysis of the unit cell containing 54 atoms. This was only one-fifth of the time required for DDEC3 charge analysis of the same material. Much larger times are required for DDEC3 or DDEC6 calculations reading in Gaussian basis set coefficients (e.g., GAUSSIAN 09 generated .wfx files), because in such cases the density grids must be explicitly computed within the CHARGEMOL program.
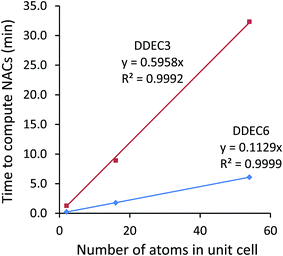 |
| | Fig. 3 The computational cost of DDEC3 and DDEC6 charge partitioning scales linearly with increasing system size. Computation for NaCl crystal (ambient pressure) performed with serial Fortran CHARGEMOL program executed on a single processor core in Intel Xeon E5-2680v3 on the Comet supercomputing cluster at the San Diego Supercomputing Center. | |
The main difference in computational cost between DDEC3 and DDEC6 arises from the number of charge cycles required for convergence. In fact, the electron partitioning scheme is the only computational difference between DDEC3 and DDEC6. As shown in Table 7, more charge cycles are required for DDEC3 convergence than for DDEC6 convergence. For all materials we studied, fewer than 200 DDEC3 charge cycles were required.19 For all materials, seven DDEC6 charge partitioning steps are required. More than one DDEC6 charge cycle per charge partitioning step is required only when the NA ≥ NcoreA constraint is binding, because in this case {κA} must be iteratively computed. For Cs@C60 with 54 simulated frozen core electrons, 18 DDEC6 charge cycles were required to complete the seven charge partitioning steps. All other materials we studied converged in seven DDEC6 charge cycles. It is gratifying that the extra accuracy of DDEC6 compared to DDEC3 comes with a reduced computational cost.
Table 7 Number of charge cycles to convergence for selected systems
| |
DDEC3 |
DDEC6 |
| For unit cells containing 2, 16, and 54 atoms. From ref. 19. Using 10 frozen Na core electrons. Using 2 frozen Na core electrons. Using 46 frozen Cs core electrons. Using 54 simulated frozen Cs core electrons by treating 8 of the 9 PAW valence electrons as core. |
| NaCl crystal (ambient pressure) |
107–109a |
7a |
| TiO solid |
130 |
7 |
| Mo2C slab with K adatom |
87b |
7 |
| Fe2O3 solid |
173b |
7 |
| Zn nicotinate MOF |
75 |
7 |
| Water molecule |
37 |
7 |
| DNA decamer |
83 |
7 |
| Linear Li2O molecule |
52 |
7 |
| Na3Cl (P4mmm crystal) |
62,c 75d |
7c,d |
| Fe4O12N4C40H52 noncollinear single molecule magnet |
90b |
7 |
| Cs@C60 |
51e |
7e (18f) |
5.2 Atomic dipole magnitudes
To produce an efficiently converging atom-centered polyatomic multipole expansion, the atomic dipoles and higher atomic multipoles should not be too large in magnitude. Fig. 4 compares DDEC3 to DDEC6 atomic dipole magnitudes in atomic units. The left panel contains the following materials comprised almost entirely of surface atoms: (a) B4N4 cluster, (b) BN nanotube, (c) h-BN sheet, (d) formamide (PW91 exchange–correlation functional with 6-311++G** and planewave basis sets), (e) the metal–organic frameworks IRMOF-1 (DFT-optimized and X-ray diffraction geometries), large-pore MIL-53(Al), ZIF-90, ZIF-8, Zn-nicotinate (PW91 optimized geometry), and CuBTC, (f) ZrN4C52H72 organometallic complex, (g) ZrO4N4C52H72 organometallic complex, (h) [GdI]2+ (SDD and planewave basis sets), (i) the MgI, MoI, SnI, TeI, and TiI molecules using both SDD and planewave basis sets, (j) [Cr(CN)6]3−, (k) the ozone singlet and triplet spin states using the PW91, B3LYP, CCSD, SAC-CI, and CAS-SCF exchange–correlation theories, (l) ozone +1 cation doublet (PW91, B3LYP, and CCSD methods), (m) the Fe4O12N4C40H52 noncollinear single molecule magnet, and (n) [Cu2N10C36H52]2+ spin triplet. The right panel contains the following dense materials: TiCl4 crystal, SrTiO3 surface slab, Pnma NaCl3 crystal (2 frozen Na core electrons), P4mmm Na2Cl crystal (2 frozen Na core electrons), natrolite, NaF surface slab, Mo2C surface with K adatom, Cmmm Na2Cl crystal (2 frozen Na core electrons), Pd crystal with interstitial H atom, Pd3Hf crystal with interstitial H atom, Pd3In crystal with interstitial H atom, Pd3V crystal with interstitial H atom. The slopes of the best fit lines constrained to have an intercept of (0,0) were 1.0462 (1.0222) with R-squared correlation coefficient = 0.9263 (0.9496) for the surface atom materials (dense materials). This shows the atomic dipole magnitudes are about 2–5% larger in magnitude for DDEC3 compared to DDEC6. These small atomic multipoles allow DDEC6 NACs to approximately reproduce V() surrounding a material.
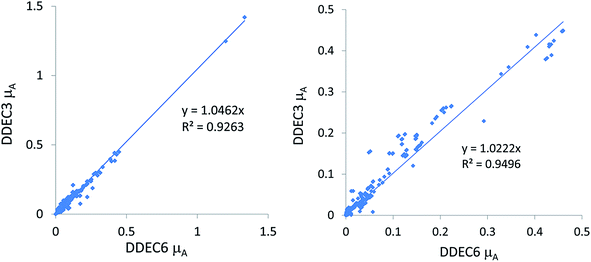 |
| | Fig. 4 Comparison of DDEC3 to DDEC6 atomic dipole magnitudes. Left: Materials comprised almost entirely of surface atoms. Right: Dense materials comprised mainly of buried atoms. | |
Why are the DDEC6 atomic dipole magnitudes slightly smaller on average than those for the DDEC3 method? The atomic dipole magnitude, μA, can be written as
| |
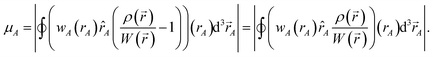 | (115) |
One strategy to slightly reduce
μA is to make the assigned {
ρA(
A)} slightly less diffuse without significantly altering
ρ(
)/
W(
). Because the integral contributions in
eqn (115) are proportional to
rA, minimizing the contributions for large
rA values will decrease
μA. The DDEC6 anions are typically less diffuse than the DDEC3 anions, because DDEC6 uses a partially localized reference ion charge (
qrefA =
q2,refA) instead of the AIM charge used as reference (
qrefA =
qA) in DDEC3. A second strategy for minimizing
μA is to make
ρ(
)/
W(
) as close to 1 as feasible in the bonding regions where atoms overlap. The integral contributions in
eqn (115) do not depend on
wA(
rA) in regions where atoms do not overlap significantly, because
wA(
rA) ≈
W(
) in those regions. The weighted spherical average,
ρwavgA(
rA), weights more heavily the regions where atom overlaps are substantial. Thus,
ρwavgA(
rA) makes
ρ(
)/
W(
) as close to 1 as feasible specifically within the regions where atom overlaps are substantial. This is precisely those regions where integral contributions to
μA can be suppressed. For this reason, using
ρwavgA(
rA) substantially outperforms the simple spherical average,
ρavgA(
rA), for the purpose of minimizing
μA. This reduction in
μA also causes the NACs to more accurately reproduce
V(
) surrounding the material.
5.3 Conformational transferability and accuracy for reproducing the electrostatic potential
For some applications, the preferred strategy is to use NACs from quantum chemistry calculations to build an electrostatic model in flexible force-fields for classical molecular dynamics and Monte Carlo simulations. Gas adsorption and diffusion in porous crystalline materials is a common example.66 The simulations of large biomolecules is another common example.103 For these applications, flexibility of the material may play a key role.104 Thus, it is important for the NACs to simultaneously have good conformational transferability and approximately reproduce the electrostatic potential around the material. This is a challenging criterion, because NACs directly fit to the electrostatic potential (without additional fitting criteria) often have poor conformational transferability.14,17
Fig. 5 shows the Zn-nicotinate metal–organic framework (MOF). This structure is comprised of one-dimensional pore channels having an approximately square cross-section. The electrostatic potential is most positive near the atomic nuclei and becomes most negative near the pore centers. In Fig. 5, a contour of electrostatic potential isovalue is displayed as a green surface.
 |
| | Fig. 5 The geometry-optimized Zn-nicotinate MOF with one-dimensional pore channels. The lines mark the unit cell boundaries. The pore cross-sections are approximately square. The green surface corresponds to an electrostatic potential isovalue. The electrostatic potential becomes more negative closer to the pore centers and more positive closer to the atomic nuclei. Atom colors: C (gray), N (blue), O (red), Zn (orange), H (pink). | |
To assess the effects of framework flexibility on the Zn-nicotinate MOF, we performed an ab initio molecular dynamics (AIMD) calculation in VASP. This AIMD simulation used a planewave cutoff energy of 400 eV, the PAW method, and PBE functional for 1200 femtoseconds (fs) with a time step of 1 fs. (Electronic energies were converged to 10−4 eV. A normal precision grid was used, which had a 0.36 (0.18) bohr uniform grid spacing along each lattice direction for the orbital (electron density) fast Fourier transforms.) A canonical ensemble at T = 300 K was simulated using a Nosé thermostat.105 The Nosé thermal coupling parameter105 was adjusted until the period of thermal oscillations was approximately 16 fs (i.e., 16 time steps). This gave reasonable temperature fluctuations that preserved the MOF's chemical integrity. A period of ∼250 fs was allowed for thermal equilibration. Twenty-one conformations were used for the subsequent charge analysis: the DFT-optimized minimum energy geometry and 20 AIMD conformations corresponding to time steps 250, 300, 350, … 1200. For each of these conformations, the valence and total all-electron densities and electrostatic potential were generated in VASP using single-point (fixed-geometry) calculations with a PREC = Accurate (∼0.14 bohr) grid.
Electrostatic potential fitting NACs were calculated using the REPEAT method and associated software code by Campaña, Mussard, and Woo.13 For the REPEAT method, NACs were fit outside surfaces defined by γR = 1.0 and 1.3 times the atomic vdW radii. As previously noted, REPEAT NACs are highly sensitive to the particular value of this vdW multiplier γR.13,15,16 Campaña et al.13 recommended the value γR = 1.0. Chen et al. recommended the value γR = 1.3.16
Table 8 summarizes electrostatic potential RMSE and RRMSE values averaged over all 21 system conformations. These were computed on a uniform grid defined by an inner vdW multiplier of 1.3 and an outer vdW multiplier limited only by the pore size. When using the conformation averaged NACs and the conformation specific NACs, the REPEAT method produced a more accurate representation of the electrostatic potential than the DDEC6 NACs. By definition, electrostatic potential fitting methods (such as REPEAT) should produce a more accurate representation of the electrostatic potential than other types of atom-centered point charge models when using the conformation specific NACs. Including atomic dipoles in the conformation specific NACs dramatically lowered the DDEC6 RMSE from 2.99 to 0.55 kcal mol−1, which was even better than the REPEAT values for both γR = 1.0 and 1.3. This means that for reproducing the electrostatic potential of a rigid framework, the DDEC6 method including atomic dipoles sometimes outperforms the REPEAT NACs. As shown in Table 8, including the spherical charge penetration term had negligible effect. When using the low energy conformation NACs, REPEAT with γR = 1.0 yielded the lowest RMSE and RRMSE values. For the low energy conformation NACs, the DDEC6 RMSE and RRMSE values were between the REPEAT values using γR = 1.0 and 1.3.
Table 8 Average RMSE (kcal mol−1) and RRMSE of Zn-nicotinate metal–organic framework at 21 different structural conformations. The values in parentheses include spherical charge penetration effects
| |
DDEC6 |
REPEAT (γR = 1.0) |
REPEAT (γR = 1.3) |
| M (M + SCP) |
D (D + SCP) |
| Not computed, because the variation in the molecular conformation affects the orientation of the atomic dipoles. |
| Using the conformation averaged NACs |
| RMSE |
3.13 |
a |
1.05 |
1.01 |
| RRMSE |
0.49 |
a |
0.22 |
0.20 |
| Using the conformation specific NACs |
| RMSE |
2.99 (3.00) |
0.55 (0.47) |
0.65 |
0.60 |
| RRMSE |
0.47 (0.47) |
0.10 (0.09) |
0.11 |
0.10 |
| Using the low energy conformation NACs |
| RMSE |
3.39 (3.38) |
a |
2.34 |
3.56 |
| RRMSE |
0.53 (0.53) |
a |
0.40 |
0.66 |
Table 9 summarizes information about the conformational transferability of the NACs. The DDEC6 NACs had excellent conformational transferability with a mean unsigned deviation (MUD) ≤∼0.01 for each atom type. Moreover, the max and min DDEC6 NACs for each atom type differed by <0.1 e. The REPEAT method had better conformational transferability with γR = 1.0 than with γR = 1.3. For γR = 1.0, three of the atom types exhibited fluctuations > 0.5 e as measured by the difference between max and min NACs. For γR = 1.3, the Zn NAC varied from −0.49 to 1.38 across the different conformations, and two of the other atom types also exhibited fluctuations >1 e. With the exception of atom type O(1), all of the min and max values of the DDEC6 NACs were between the min and max values of the REPEAT NACs using γR = 1.3. With the exception of atom types C(6), O(1), and O(2), all of the min and max values of the DDEC6 NACs were between the min and max values of the REPEAT NACs using γR = 1.0.
Table 9 Average, maximum, minimum, and mean unsigned deviation of NACs for each atom type in Zn-nicotinate using DDEC6 and REPEAT methods
| Atom type |
DDEC6 |
REPEAT (γR = 1.0) |
REPEAT (γR = 1.3) |
| Avg |
Max |
Min |
MUD |
Avg |
Max |
Min |
MUD |
Avg |
Max |
Min |
MUD |
| C(1) |
−0.12 |
−0.10 |
−0.14 |
0.007 |
−0.16 |
0.08 |
−0.36 |
0.09 |
−0.14 |
0.34 |
−0.50 |
0.18 |
| C(2) |
−0.06 |
−0.05 |
−0.08 |
0.007 |
−0.06 |
0.17 |
−0.23 |
0.07 |
0.03 |
0.56 |
−0.20 |
0.11 |
| C(3) |
−0.02 |
0.00 |
−0.04 |
0.007 |
0.05 |
0.25 |
−0.14 |
0.07 |
−0.04 |
0.32 |
−0.42 |
0.13 |
| C(4) |
0.08 |
0.10 |
0.05 |
0.012 |
0.14 |
0.33 |
−0.19 |
0.09 |
0.03 |
0.40 |
−0.42 |
0.17 |
| C(5) |
0.09 |
0.14 |
0.06 |
0.011 |
0.19 |
0.50 |
−0.05 |
0.11 |
0.20 |
0.77 |
−0.33 |
0.21 |
| C(6) |
0.56 |
0.58 |
0.52 |
0.013 |
0.37 |
0.51 |
0.20 |
0.06 |
0.26 |
0.59 |
0.04 |
0.13 |
| H(1) |
0.09 |
0.10 |
0.07 |
0.006 |
0.04 |
0.16 |
−0.03 |
0.03 |
0.10 |
0.26 |
−0.04 |
0.07 |
| H(2) |
0.10 |
0.11 |
0.08 |
0.004 |
0.03 |
0.11 |
−0.03 |
0.03 |
0.00 |
0.22 |
−0.37 |
0.08 |
| H(3) |
0.12 |
0.13 |
0.11 |
0.006 |
0.09 |
0.20 |
−0.04 |
0.04 |
0.05 |
0.28 |
−0.18 |
0.09 |
| H(4) |
0.12 |
0.14 |
0.11 |
0.004 |
0.08 |
0.18 |
0.00 |
0.03 |
0.12 |
0.30 |
−0.07 |
0.07 |
| N |
−0.23 |
−0.21 |
−0.25 |
0.007 |
−0.37 |
−0.10 |
−0.63 |
0.11 |
−0.23 |
0.47 |
−0.68 |
0.24 |
| O(1) |
−0.56 |
−0.53 |
−0.58 |
0.011 |
−0.39 |
−0.28 |
−0.49 |
0.04 |
−0.23 |
0.05 |
−0.51 |
0.13 |
| O(2) |
−0.53 |
−0.50 |
−0.56 |
0.012 |
−0.41 |
−0.32 |
−0.51 |
0.03 |
−0.37 |
−0.21 |
−0.65 |
0.07 |
| Zn |
0.74 |
0.76 |
0.70 |
0.010 |
0.80 |
0.91 |
0.54 |
0.08 |
0.47 |
1.38 |
−0.49 |
0.33 |
What are the implications of these results for developing force-fields to reproduce the electrostatic potential surrounding materials? If the goal is to reproduce the electrostatic potential as accurately as possible surrounding a rigid material using an atom-centered point-charge model without regard for the chemical meaning of those NACs, then methods such as ESP,10 Chelp,11 or Chelpg12 for molecular systems or REPEAT13 for periodic materials or the Wolf-summation technique of Chen et al.16 are preferable, because these methods minimize RMSE without regard for the chemical meaning of the NACs. If the goal is to produce chemically meaningful NACs that reproduce the electrostatic potential as accurately as possible surrounding a rigid material, the DDEC6 method is preferable with or without including atomic dipoles, because this method assigns atomic electron distributions to resemble real atoms and reproduce the electrostatic potential. For constructing flexible, non-reactive force-fields, NACs based on the low-energy structure or an average across multiple system conformations can be used. Depending on the material and computational details, either the DDEC6, REPEAT13 (especially its extension to simultaneously fit multiple conformations17), ESP,10 Chelp,11 Chelpg,12 or Wolf-summation technique16 may yield the more accurate flexible force-field NACs.
5.4 Quantifying the consistency between assigned NACs and ASMs
An AIM method should preferably yield chemically consistent NACs and ASMs. For a special type of system, the consistency between assigned NACs and ASMs can be quantitatively measured. Consider a single neutral atom or a +1 atomic cation having only one easily removable electron. For convenience, we refer to these atoms or atomic ions as containing only one labile electron. Next, consider an uncharged host system containing only deeply bound electrons that are paired. If we combine the atom or atomic ion having one labile electron with the host system having paired electrons to form a weakly or ionically bound endohedral complex, a portion of the labile electron's density may be transferred to the host system's atoms. Since there is only one labile electron in a background of strongly held effectively paired electrons (in the endohedral and host system atoms) and (optionally) strongly held like-spin unpaired electrons (in the endohedral atom), the labile electron's spin cannot be locally cancelled by any other electrons in the system. In this case, the amount of electron density transferred from the endohedral atom to the host system should equal the amount of spin magnetization density transferred from the endohedral atom to the host system. This leads to the following quantification of consistency| |
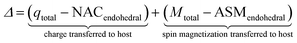 | (116) |
where qtotal ≥ 0 is the total system charge (in atomic units), Mtotal ≥ 0 is the system's total spin magnetic moment (in atomic units), and NACendohedral and ASMendohedral are the assigned NAC and ASM of the endohedral atom in the endohedral complex. In the ideal case, Δ → 0, because the single labile electron should transfer equal amounts of spin magnetization and negative charge to the host. This condition should also be fulfilled in situations where a weakly bound endohedral atom has approximately zero labile electrons. It is not necessarily fulfilled in cases where the number of labile electrons exceeds one, because in such cases the multiple labile electrons might be transferred into orbitals of opposing spins leading to physically different amounts of transferred spin magnetization and transferred negative charge. Nor should it be fulfilled in cases where a strong covalent bond forms between the endohedral atom and the host.
As specific examples, we consider the Li@C60, N@C60, Cs@C60, Xe@C60, [Eu@C60]1+, and [Am@C60]1+ endohedral fullerenes. The C60 host has only deeply held paired electrons.106 (Experiments show C60 has a first ionization energy of 6.4–7.9 eV, an electron affinity of approx. 2.6–2.8 eV, and a first optical transition of approx. 3.2 eV.107–110) These complexes were chosen as examples, because they span a wide range from light to heavy elements having zero to one labile electrons. The Li and Cs elements have a nominal s1 valence configuration; this outer s-electron is donated to the Li@C60 and Cs@C60 systems as the labile electron. The Eu1+ and Am1+ elements have a nominal f7s1 valence configuration; the half-filled f-shell is tightly held while the outer s-electron is donated to the [Eu@C60]1+ and [Am@C60]1+ systems as the labile electron. (Spectroscopic experiments show Eu in Eu@C60 is in the +II oxidation state, meaning the seven f electrons remain bound to the Eu atom.111,112) Because Xe is a noble gas element, the Xe@C60 system has no labile electrons. Spectroscopic experiments show that in N@C60, the endohedral N atom has a quartet spin state analogous to the isolated N atom.113 This can be explained by the high electronegativity of the N atom, which retains its three unpaired electrons. Thus, we consider none of the electrons in N@C60 to be labile.
We optimized the geometries and electron distributions of these endohedral fullerenes in VASP using the PBE functional with the PAW method and a 400 eV plane-wave cutoff. A 20 Å × 20 Å × 20 Å cubic unit cell was used. The positions of all atoms in the system were optimized until the forces on every atom were negligible. Due to the almost spherical nature of the C60 enclosure, only the equilibrium displacement of the endohedral atom from the cage's center should be considered significant. Therefore, we did not attempt multiple initial geometries with different angular variations in the endohedral atom's position. Fig. 6 displays the optimized geometries. In Table 10, the optimized offset is the distance of the endohedral atom from the center of the C60 group. (The center of the C60 group was computed by averaging the (x, y, z) coordinates of the carbon atoms.) The N, Xe, and Cs atoms were located at the center of the C60 group (within a computational tolerance). The central position of the N atom and quartet spin state agree with electron paramagnetic resonance (EPR) and electron-nuclear double resonance (ENDOR) experiments.113 As reviewed by Popov et al., a wide variety of spectroscopic experiments show the noble gas atom in Ng@C60 complexes (Ng = He, Ne, Ar, Kr, or Xe) resides at the cage's central position.114 The Cs@C60 geometry optimization converged to an energy minimum having a centrally located Cs atom, even though the calculation was started using a non-zero offset of 0.46 Å. This shows the central Cs position is at least a local (and perhaps global) energy minimum. In the optimized structures, the Li, Eu, and Am ions were displaced by >1 Å from the center. Our calculated offset for Li@C60 is in good agreement with previous computational studies.115,116 Prior calculations on neutral Eu@C60 and Am@C60 complexes also indicate an off-center position.117–119
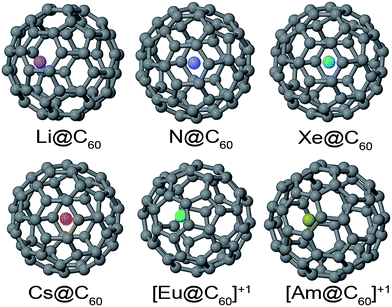 |
| | Fig. 6 Endohedral complexes used to test the charge and spin transfer consistency. The N, Xe, and Cs atoms are approximately centered in the C60 cage. The Li, Eu, and Am atoms attract to one side of the C60 cage. The sizes of the endohedral atoms are not drawn to scale. | |
Table 10 Computed NACs and ASMs for the enclosed atom in endohedral doped bucky-balls
| System |
Optimized offset (Å) |
Total unpaired electrons |
HD |
CM5 |
Bader |
DDEC6 |
| NAC |
ASM |
NAC |
NAC |
ASM |
NAC |
ASM |
| Using 46 frozen Cs core electrons. Using 54 simulated frozen Cs core electrons by treating 8 of the 9 PAW valence electrons as core. |
| Li@C60 |
1.52 |
1 |
0.323 |
0.002 |
0.566 |
0.899 |
0.000 |
0.903 |
−0.001 |
| N@C60 |
0.06 |
3 |
0.137 |
2.816 |
0.118 |
0.013 |
2.886 |
0.142 |
2.854 |
| Xe@C60 |
0.01 |
0 |
0.293 |
0.000 |
0.304 |
0.092 |
0.000 |
0.316 |
0.000 |
| Cs@C60 |
0.02 |
1 |
0.404 |
0.002 |
1.468 |
0.917 |
−0.001 |
1.057a (1.000b) |
−0.002a (−0.002b) |
| [Eu@C60]1+ |
1.10 |
8 |
0.542 |
7.501 |
1.050 |
1.566 |
6.879 |
1.368 |
7.515 |
| [Am@C60]1+ |
1.19 |
8 |
0.608 |
7.355 |
1.049 |
1.579 |
6.545 |
1.318 |
7.390 |
| Mean absolute inconsistency: |
0.601 |
0.386 |
0.302 |
0.147 |
In Table 10, the mean absolute inconsistency between the assigned NACs and ASMs is quantified as the average of the absolute value of Δ for the six materials. Because CM5 is a correction to the HD NACs, the CM5 method utilized the HD ASMs. Among the four methods, the HD NACs and ASMs were the most inconsistent with an average inconsistency of 0.6 electrons. The DDEC6 NACs and ASMs were the most consistent with an average inconsistency of 0.15 electrons. The CM5 and Bader methods had intermediate performance. Because the HD and DDEC6 ASMs were nearly the same, the poor performance of the HD method must have been due to its inaccurate NACs.
Examining the DDEC6 results in Table 10, ∼1 electron was transferred from the Li and Cs atoms, leaving a Li1+ or Cs1+ cation in the center having negligible unpaired spin. The CM5 NAC of 1.468 for the Cs atom seems too high, because this implies removal of some of its outer core electrons. The HD, CM5, and DDEC6 methods all gave ∼0.3 electrons transferred in the Xe system, but the Bader method gave ∼0.1 transferred electrons in this material. All four methods gave the least amount of electron transfer for the N system compared to other systems. In the Eu and Am cationic systems, ∼0.4 electrons were transferred from the endohedral metal atom to the C60 host to give a NAC of ∼1.4 and an ASM of ∼7.4 for the endohedral metal atom. Overall, these results demonstrate reasonable consistency between the assigned DDEC6 NACs and ASMs.
5.5 Retaining core electrons on the host atom and assigning exactly one electron distribution per atom
We were first motivated to improve upon the DDEC3 method by a series of calculations on sodium chloride crystals having unusual stoichiometries. Specifically, we computed NACs for the ten high-pressure crystal structures reported by Zhang et al.120 and the ambient-pressure NaCl structure shown in Fig. 7. We generated the electron density in VASP using the PBE functional and (a) the experimental X-ray diffraction geometries120 for the ten high-pressure crystals and (b) the PBE-optimized geometry for the ambient-pressure Fm3m-NaCl. As shown in Table 11 and Fig. 8, the DDEC3 NAC for at least one Na atom is larger than +1.0 for the following cases: (a) 1.311 for Na(3) atoms in Cmmm-Na2Cl crystal at 180 GPa, (b) 1.235 for Na(1) and 1.295 for Na(2) atoms in Cmmm-Na3Cl2 crystal at 280 GPa, (c) 1.219 for Na atoms in Imma-Na2Cl crystal at 300 GPa, (d) 1.035 for Na(1) and 1.108 for Na(2) atoms in P4/m-Na3Cl2 crystal at 140 GPa, (e) 1.063 for Na(1) atoms in P4/mmm-Na2Cl crystal at 120 GPa, (f) 1.078 for Na(1) atoms in Pm3-NaCl7 crystal at 200 GPa, (g) 1.136 for Na atoms in Pm3m-NaCl crystal at 140 GPa, (h) 1.140 for Na atoms in Pm3n-NaCl3 crystal at 200 GPa, and (i) 1.011 for Na atoms in Pnma-NaCl3 crystal at 40 GPa. Because a neutral sodium atom has one electron in its valence shell, an AIM-based NAC for a sodium atom in sodium-containing solids should ideally be ≤+1.0. (Non-AIM-based NACs such as APT, Born effective, and ESP charges are not expected to have this property.) A Na NAC greater than +1.0 would indicate that some electrons from the closed [1s22s2p6] shells are donated to other atoms, but such a donation should be energetically unfavorable under chemically relevant conditions due to the high ionization energy of closed shell configurations. (For comparison, the first ionization energy of a Ne atom having 1s22s2p6 electron configuration is 21.56 eV.121) Based on these results, we concluded that the DDEC3 method overestimates atomic charge magnitudes in some materials. If ten Na core electrons are frozen, the DDEC3 NACs for the Na atoms are constrained to be ≤+1.0 as shown in Table 11, but this is not a satisfactory solution because we want NACs to be approximately independent of the number of frozen core electrons.
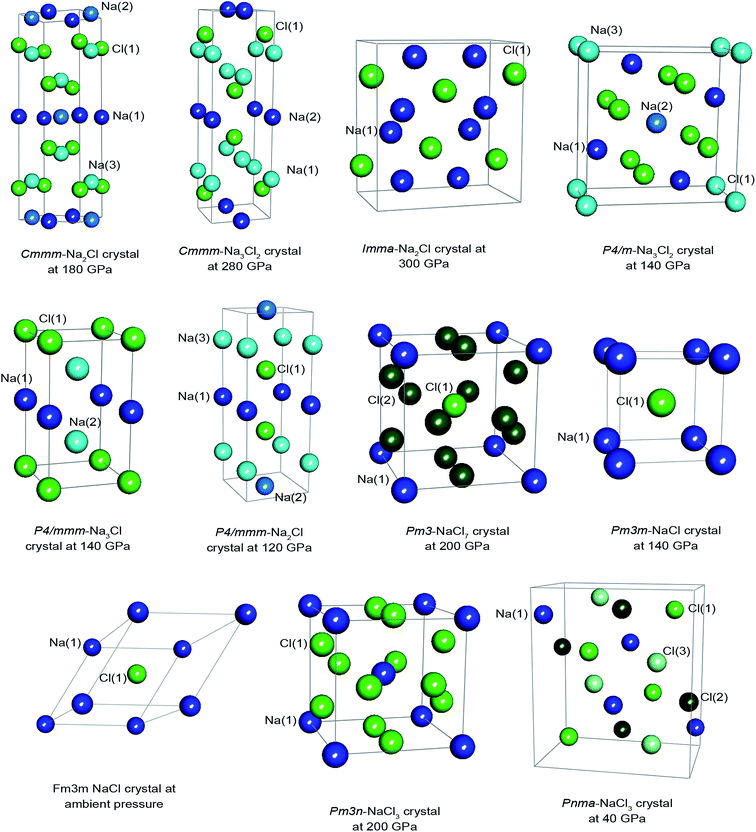 |
| | Fig. 7 Sodium chloride crystal structures. The lines mark the unit cell boundaries. | |
Table 11 DDEC and Bader net atomic charges of sodium chloride crystals
| Atom type |
Number of atoms |
DDEC3a |
DDEC6a |
Badera |
| Values listed for 2 frozen Na core electrons; values in parentheses for 10 frozen Na core electrons. Similar Bader NACs were reported previously in ref. 122. Similar Bader NACs were reported previously in ref. 120. Na charge of 1.05 computed with IH/R3 all-electron reported previously in ref. 67. Bader NACs cannot be reported because Bader analysis yields more compartments than atoms (see Table 12). For P4/mmm-Na3Cl crystal at 140 GPa and P4/mmm-Na2Cl crystal at 120 GPa, the Bader NACs for two Na frozen core electrons are listed but non-nuclear attractors occur for ten Na frozen core electrons. |
| Cmmm-Na2Cl crystal at 180 GPa |
| Na(1) |
2 |
0.392 (0.319) |
0.316 (0.334) |
e |
| Na(2) |
2 |
0.566 (0.592) |
0.547 (0.563) |
e |
| Na(3) |
4 |
1.311 (1.000) |
0.849 (0.842) |
e |
| Cl(1) |
4 |
−1.790 (−1.455) |
−1.281 (−1.290) |
e |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| Cmmm-Na3Cl2 crystal at 280 GPa |
| Na(1) |
2 |
1.235 (1.000) |
0.954 (0.891) |
0.780 (0.560) |
| Na(2) |
4 |
1.295 (1.000) |
0.871 (0.866) |
0.643 (0.291) |
| Cl(1) |
4 |
−1.912 (−1.500) |
−1.348 (−1.311) |
−1.033 (−0.571) |
|
| Imma-Na2Cl crystal at 300 GPab |
| Na(1) |
8 |
1.219 (1.000) |
0.814 (0.785) |
0.676 (0.317) |
| Cl(1) |
4 |
−2.439 (−2.000) |
−1.628 (−1.570) |
−1.351 (−0.633) |
|
| P4/m-Na3Cl2 crystal at 140 GPa |
| Na(1) |
4 |
1.035 (1.000) |
0.808 (0.777) |
e |
| Na(2) |
1 |
1.108 (1.000) |
0.956 (0.902) |
e |
| Na(3) |
1 |
−0.461 (−0.396) |
−0.310 (−0.226) |
e |
| Cl(1) |
4 |
−1.197 (−1.151) |
−0.969 (−0.946) |
e |
|
| P4/mmm-Na3Cl crystal at 140 GPa |
| Na(1) |
1 |
−0.237 (0.184) |
−0.246 (−0.202) |
0.06e |
| Na(2) |
2 |
0.465 (0.466) |
0.477 (0.480) |
0.531e |
| Cl(1) |
1 |
−0.693 (−0.749) |
−0.709 (−0.758) |
−1.122e |
|
| P4/mmm-Na2Cl crystal at 120 GPa |
| Na(1) |
1 |
1.063 (1.000) |
0.927 (0.889) |
0.756e |
| Na(2) |
1 |
−0.259 (−0.187) |
−0.242 (−0.201) |
0.041e |
| Na(3) |
2 |
0.541 (0.503) |
0.487 (0.486) |
0.511e |
| Cl(1) |
2 |
−0.943 (−0.910) |
−0.830 (−0.830) |
−0.909e |
|
| Pm3-NaCl7 crystal at 200 GPac |
| Na(1) |
1 |
1.078 (1.000) |
0.899 (0.874) |
0.883 (0.652) |
| Cl(1) |
1 |
0.297 (0.260) |
0.202 (0.196) |
0.090 (0.088) |
| Cl(2) |
6 |
−0.229 (−0.210) |
−0.184 (−0.178) |
−0.162 (−0.123) |
|
| Pm3m-NaCl crystal at 140 GPa |
| Na(1) |
1 |
1.136 (1.000) |
0.966 (0.916) |
0.862 (0.673) |
| Cl(1) |
1 |
−1.136 (−1.000) |
−0.966 (−0.916) |
−0.862 (−0.673) |
|
| NaCl crystal at ambient pressured |
| Na(1) |
1 |
0.981 (0.978) |
0.859 (0.848) |
0.840 (0.829) |
| Cl(1) |
1 |
−0.981 (−0.978) |
−0.859 (−0.848) |
−0.840 (−0.829) |
|
| Pm3n-NaCl3 crystal at 200 GPac |
| Na(1) |
2 |
1.140 (1.000) |
0.962 (0.909) |
0.913 (0.653) |
| Cl(1) |
6 |
−0.380 (−0.333) |
−0.321 (−0.303) |
−0.304 (−0.218) |
|
| Pnma-NaCl3 crystal at 40 GPac |
| Na(1) |
4 |
1.011 (1.000) |
0.842 (0.853) |
0.815 (0.770) |
| Cl(1) |
4 |
−0.718 (−0.709) |
−0.590 (−0.597) |
−0.530 (−0.501) |
| Cl(2) |
4 |
0.105 (0.101) |
0.054 (0.055) |
−0.030 (−0.028) |
| Cl(3) |
4 |
−0.398 (−0.392) |
−0.307 (−0.311) |
−0.255 (−0.242) |
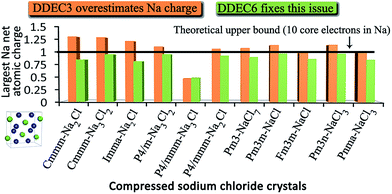 |
| | Fig. 8 Largest magnitude Na atomic charges in compressed sodium chloride crystals. These were computed using 2 frozen Na core electrons. Based on chemical arguments, at least 10 electrons should be assigned to each Na atom. The DDEC3 method gives many Na atom charges >1, which indicates some electrons are not assigned to the correct atom. The DDEC6 method fixes this problem. | |
This observation led us to explore numerous potential modifications to the DDEC method, which after testing dozens of potential modifications culminated in the DDEC6 method. As shown in Table 11 and Fig. 8, the DDEC6 NACs have the expected behavior being ≤+1.0 for each of the Na atoms. Moreover, the DDEC6 NACs were nearly insensitive to whether 2 or 10 frozen Na core electrons were used.
Bader's quantum chemical topology62–64 cannot be used to compute NACs for some of these materials, because it assigns compartments not belonging to any atom (or to multiple atoms simultaneously) in the following cases: (a) Cmmm-Na2Cl crystal at 180 GPa irrespective of the number of frozen Na core electrons, (b) P4/m-Na3Cl2 crystal at 140 GPa irrespective of the number of frozen Na core electrons, (c) P4/mmm-Na3Cl crystal at 140 GPa when using 10 frozen Na core electrons, and (d) P4/mmm-Na2Cl crystal at 120 GPa when using 10 frozen Na core electrons. Bader compartments for these four materials are detailed in Table 12. As it should be, the assignment of these Bader compartments was based on the full (i.e., valence + (frozen) core) electron density, not simply the valence density or the valence pseudodensity. At first, one might propose each non-nuclear attractor could be assigned to one of the nearby atoms, but this is not satisfactory because in some cases such an assignment cannot be made without destroying the crystal symmetry. For example, the P4/mmm-Na2Cl crystal at 120 GPa (modeled with 10 frozen Na core electrons) contains one non-nuclear attractor whose closest atoms are the two equivalent Na(3) atoms; therefore, it is impossible to assign this non-nuclear attractor to one of the closest atoms without breaking the crystal symmetry. Alternatively, one could propose to divide the electron density and/or volume of each non-nuclear attractor amongst the nearby atoms in a way that preserves the system's symmetry, but it is not presently clear whether this could be done in a way that preserves most of the important properties of the Virial compartments. Specifically, each of the Bader compartments satisfies the Virial theorem and behaves as an open quantum system, but divided pieces of such compartments may not.62,64 It might be possible that divisions of a non-nuclear attractor could be made that satisfy the net zero flux condition (and Virial theorem) over each division volume but not the local zero flux condition in the bounding surfaces, but it is not presently clear whether such a partitioning would always have a unique definition even if constrained to preserve the system's symmetry.
Table 12 Bader compartment populations for crystals with non-nuclear attractors
| Compartment type |
Number of compartments |
Enclosed atom |
Number of electronsa |
| Values listed for 2 frozen Na core electrons; values in parentheses for 10 frozen Na core electrons. |
| Population for Cmmm-Na2Cl crystal at 180 GPa |
| 1 |
2 |
Na(1) |
10.416 (10.425) |
| 2 |
2 |
Na(2) |
10.326 (10.437) |
| 3 |
4 |
Na(3) |
10.260 (10.637) |
| 4 |
4 |
Cl(1) |
18.116 (17.623) |
| 5 |
4 |
None |
0.253 (0.308) |
|
| Population for P4/m-Na3Cl2 crystal at 140 GPa |
| 1 |
4 |
Na(1) |
10.222 (10.392) |
| 2 |
1 |
Na(2) |
10.197 (10.340) |
| 3 |
1 |
Na(3) |
10.616 (10.543) |
| 4 |
4 |
Cl(1) |
17.955 (17.752) |
| 5 |
1 |
None |
0.456 (0.544) |
|
| Population for P4/mmm-Na3Cl crystal at 140 GPa |
| 1 |
1 |
Na(1) |
10.940 (10.655) |
| 2 |
2 |
Na(2) |
10.469 (10.507) |
| 3 |
1 |
Cl(1) |
18.122 (17.871) |
| 4 |
4 |
None |
(0.115) |
|
| Population for P4/mmm-Na2Cl crystal at 120 GPa |
| 1 |
1 |
Na(1) |
10.244 (10.326) |
| 2 |
1 |
Na(2) |
10.959 (10.655) |
| 3 |
2 |
Na(3) |
10.489 (10.486) |
| 4 |
2 |
Cl(1) |
17.909 (17.776) |
| 5 |
1 |
None |
(0.495) |
In materials for which there is a one-to-one correspondence between Bader compartments and atoms (i.e., no non-nuclear attractors), the Bader NACs are computed by integrating the number of electrons over each compartment. In such cases, the Bader method often yields reasonable NACs for dense ionic solids. Examining Table 11, the DDEC6 and Bader NACs using 2 frozen Na core electrons exhibited similar trends for all of the sodium chloride crystals where the Bader NACs were defined. Of particular interest, the Cl NAC was significantly more negative than −1.0 for some of the materials. The Bader NACs were more sensitive than the DDEC6 NACs to whether 2 or 10 frozen Na core electrons were used. For example, in Imma-Na2Cl crystal at 300 GPa the DDEC6 NAC for the Cl atom was −1.628 (2 frozen Na core electrons) and −1.570 (10 frozen Na core electrons) compared to the Bader Cl NAC of −1.351 (2 frozen Na core electrons) and −0.633 (10 frozen Na core electrons). The reason for this larger sensitivity of the Bader NACs on the number of frozen core electrons is that according to an integration routine now used in popular Bader analysis programs the frozen core electrons are assigned wholly to the host atom while non-frozen electrons crossing into neighboring Bader compartments are divided amongst several atoms.44 This artifact could be removed by partitioning all electrons (i.e., both frozen and non-frozen) according to their density in each of the Bader compartments, yet even so the sensitivity of the number of Bader compartments on the number of frozen core electrons (e.g., P4/mmm-Na3Cl crystal at 140 GPa and P4/mmm-Na2Cl crystal at 120 GPa) would persist. Alternatively, one could choose a small number of frozen core electrons to ensure the amount of frozen core electron density spilling into neighboring compartments is negligible. Consequently, Bader NACs with 2 frozen Na core electrons are more reliable than those with 10 frozen Na core electrons.
Interestingly, a recent computational study has predicted the existence of even more high-pressure sodium chloride phases with unusual stoichiometries.122 That study also performed Bader analysis on several high-pressure sodium chloride crystals, but all but one of those computations were performed for different pressures or different phases than the Bader charge results presented here.122 The non-nuclear attractor for P4/m-Na3Cl2 (at 125 GPa) and Cmmm-Na2Cl (at 200 GPa) was noted in that study.122
5.6 NACs usually follow electronegativity trends
A method for computing NACs should accurately describe electron transfer directions. Table 13 lists the Spearman rank coefficient between DDEC6 NACs and Pauling scale electronegativity for 14 materials containing four or more different elements. The Spearman rank coefficient quantifies how well two variables can be related by a monotonic function. A Spearman rank coefficient of +1.0 (−1.0) indicates the two variables are related by a perfectly monotonically increasing (decreasing) function. A Spearman rank coefficient of 0.0 indicates the two variables are not correlated at all. For each material, the average DDEC6 NAC was computed for each element. For each material, the elements were ranked from highest to lowest electronegativity. For example, in B-DNA the elements ranked according to electronegativity were 1. O, 2. N, 3. C, 4. P, 5. H, 6. Na. A linear least-squares regression between the average NACs and the whole number electronegativity ranks was then performed. The Spearman rank coefficient equals Pearson's correlation coefficient R for this linear regression, where 0 ≤ R2 ≤ 1 is the squared correlation coefficient.
Table 13 Spearman rank coefficient quantifying the ordering relationship between average DDEC6 NACs for each element and the Pauling scale electronegativities
| Material |
Elements |
Spearman rank coefficient |
| Formamide geometry optimized with B3LYP/6-311++G**. IRMOF X-ray structure. Mn12-acetate single molecule magnet geometry optimized with PBE/LANL2DZ. Zn nicotinate geometry optimized with PBE/planewave. |
| B-DNA |
C, H, N, Na, O, P |
0.94 |
| Cu2 pyridine complex2+ |
C, Cu, H, N |
1.00 |
| CuBTC |
C, Cu, H, O |
0.80 |
| Fe4O12N4C40H52 noncollinear SMM |
C, Fe, H, N, O |
1.00 |
| Formamidea |
C, H, N, O |
0.60 |
| IRMOFb |
C, H, O, Zn |
1.00 |
| lp-MIL-53 |
Al, C, H, O |
1.00 |
| Mn12-acetate SMMc |
C, H, Mn, O |
1.00 |
| Natrolite |
Al, H, Na, O, Si |
0.60 |
| ZIF8 |
C, H, N, Zn |
1.00 |
| ZIF90 |
C, H, N, O, Zn |
0.90 |
| Zn nicotinated |
C, H, N, O, Zn |
1.00 |
| Zr bisperoxy complex |
C, H, N, O, Zr |
1.00 |
| Zr puckered bare complex |
C, H, N, Zr |
1.00 |
Nine of the 14 materials had a Spearman rank coefficient of 1.00, indicating the average DDEC6 NACs followed exactly the same order as the element electronegativities. The remaining five materials had Spearman rank coefficients between 0.60 and 0.94, indicating the average DDEC6 NACs followed approximately but not exactly the same order as the element electronegativities. These results show DDEC6 NACs usually (but not always) follow Pauling scale electronegativity trends. The exceptions are not to be regarded as a deficiency of either the DDEC6 NACs or the Pauling scale electronegativities, because element electronegativities can only describe the usual direction of electron transfer. The specific direction of electron transfer is affected by the chemical environment. For example, while electrons are usually transferred from carbon to the more electronegative oxygen, experiments show carbon monoxide is an exception with electron transfer from oxygen towards carbon.123 Boron monofluoride is another exception with electrons transferred from fluorine towards boron.124 Furthermore, multivalent cations can sometimes acquire a positive NAC greater than that of less electronegative monovalent cations, because the multivalent cations may acquire a NAC greater than +1. For example, the multivalent P, Al, and Si atoms in B-DNA and natrolite acquired higher NACs than the monovalent Na atoms.
6. Conclusions
The main utility of net atomic charges (NACs) is they concisely convey important information about the electron distribution in materials. Due to the continuous nature of the electron cloud in a material, there is some flexibility in how to partition the total electron distribution among atoms-in-materials.125 In this article, we introduced a new method, called DDEC6, for defining atoms-in-materials and computing NACs in periodic and non-periodic materials. The DDEC6 NACs are well-suited both for understanding charge-transfer in materials and for constructing flexible force-fields for classical atomistic simulations of materials. Our method can be applied with equal validity to small and large molecules, ions, porous and non-porous solids, solid surfaces, nanostructures, and magnetic and non-magnetic materials irrespective of the basis set type used. This broad applicability makes it ideally suited for use as a default atomic population analysis method in quantum chemistry programs. Actually incorporating DDEC6 into popular quantum chemistry programs will require additional work. For example, it might be desirable to implement DDEC6 on the same integration grid already used in the respective quantum chemistry program.
We used a scientific engineering design approach to achieve nine performance goals: (1) the total electron distribution is partitioned among the atoms by assigning exactly one electron distribution to each atom, (2) core electrons remain assigned to the host atom, (3) NACs are formally independent of the basis set type because they are functionals of the total electron distribution, (4) the assigned atomic electron distributions give an efficiently converging polyatomic multipole expansion, (5) the assigned NACs usually follow Pauling scale electronegativity trends, (6) NACs for a particular element have good transferability among different conformations that are equivalently bonded, (7) the assigned NACs are chemically consistent with the assigned ASMs, (8) the method has predictably rapid and robust convergence to a unique solution, and (9) the computational cost of charge partitioning scales linearly with increasing system size.
The DDEC6 method makes five modifications relative to the DDEC3 method: (a) a fixed reference ion charge is used based on a weighted average of the electron population in a localized atomic compartment and a stockholder partition, (b) the conditioned reference ions are constrained to decay monotonically with increasing rA and to integrate to the correct number of electrons (NrefA = zA − qrefA), (c) four conditioning steps instead of (ρsome_refA)χ(ρavgA)1−χ are used to construct wA(rA), (d) a weighted spherical average improves the effect of spherical averaging during charge partitioning, and (e) the atomic weighting factor wA(rA) is constrained to decay no faster than exp(−2.5 rA) in an atom's buried tail.
We now summarize key computational results. We showed for the first time that the DDEC3 and IH methods sometimes converge to non-unique solutions that depend on the starting guess, because their optimization landscapes are sometimes non-convex. For example, the DDEC3 and IH methods assigned NACs that severely broke the molecular symmetry for the H2 triplet molecule with constrained 50 pm bond length. The DDEC6 method removes this problem by using a fixed reference ion charge with a total of seven charge partitioning steps. For a series of high-pressure sodium chloride crystals with unusual stoichiometries, we found the DDEC3 method sometimes gives NACs in excess of +1.0 for the Na atoms and Bader's quantum chemical topology sometimes yields non-nuclear attractors while the DDEC6 method exhibits neither of these problems. For six endohedral fullerenes containing zero to one labile electrons, the consistency between assigned NACs and ASMs was quantified for the Hirshfeld, CM5, Bader, and DDEC6 methods. Among these four methods, the DDEC6 method gave the most consistent agreement between assigned NACs and ASMs. A study of various conformations of the Zn-nicotinate MOF showed the DDEC6 NACs have excellent conformational transferability and are ideally suited for constructing flexible force-fields to approximately reproduce the electrostatic potential across various system conformations. Computational tests for various molecular and solid materials showed the DDEC6 atomic dipole magnitudes are slightly smaller on average than the DDEC3 ones. This is desirable to produce an efficiently converging atom-centered polyatomic multipole expansion for reproducing the electrostatic potential surrounding the material. The DDEC6 NACs also follow Pauling scale electronegativity trends on average, but of course the specific electron transfer direction between two elements is affected by their chemical environment. Due to the fewer number of required charge partitioning steps, DDEC6 charge partitioning converges several times faster than DDEC3 charge partitioning.
In summary, DDEC6 offers substantially improved accuracy and lower computational cost than DDEC3. We therefore recommend the DDEC3 method be replaced with the DDEC6 method. We performed additional computational tests of the DDEC6 method across the wider set of materials described in the sequel article.33 All of those results agree with the findings presented here.
Acknowledgements
Supercomputing resources were provided by the Extreme Science and Engineering Discovery Environment (XSEDE). XSEDE is funded by NSF grant ACI-1053575. XSEDE project grant TG-CTS100027 provided allocations on the Trestles and Comet clusters at the San Diego Supercomputing Center (SDSC) and the Stampede cluster at the Texas Advanced Computing Center (TACC).
References
- A. J. Shusterman and L. M. Hoistad, Chem. Educ., 2001, 6, 36–40 CrossRef CAS.
- E. Gouaux and R. MacKinnon, Science, 2005, 310, 1461–1465 CrossRef CAS PubMed.
- S. A. Clough, Y. Beers, G. P. Klein and L. S. Rothman, J. Chem. Phys., 1973, 59, 2254–2259 CrossRef CAS.
- S. J. Skinner and J. A. Kilner, Mater. Today, 2003, 6, 30–37 CrossRef CAS.
- W. D. Price, R. A. Jockusch and E. R. Williams, J. Am. Chem. Soc., 1997, 119, 11988–11989 CrossRef CAS PubMed.
- R. S. Mulliken, J. Chem. Phys., 1955, 23, 1833–1840 CrossRef CAS.
- G. Bruhn, E. R. Davidson, I. Mayer and A. E. Clark, Int. J. Quantum Chem., 2006, 106, 2065–2072 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- W. L. Cao, C. Gatti, P. J. MacDougall and R. F. W. Bader, Chem. Phys. Lett., 1987, 141, 380–385 CrossRef CAS.
- B. H. Besler, K. M. Merz and P. A. Kollman, J. Comput. Chem., 1990, 11, 431–439 CrossRef CAS.
- L. E. Chirlian and M. M. Francl, J. Comput. Chem., 1987, 8, 894–905 CrossRef CAS.
- C. M. Breneman and K. B. Wiberg, J. Comput. Chem., 1990, 11, 361–373 CrossRef CAS.
- C. Campaña, B. Mussard and T. K. Woo, J. Chem. Theory Comput., 2009, 5, 2866–2878 CrossRef PubMed.
- C. I. Bayly, P. Cieplak, W. D. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
- T. Watanabe, T. A. Manz and D. S. Sholl, J. Phys. Chem. C, 2011, 115, 4824–4836 CAS.
- D. L. Chen, A. C. Stern, B. Space and J. K. Johnson, J. Phys. Chem. A, 2010, 114, 10225–10233 CrossRef CAS PubMed.
- A. Gabrieli, M. Sant, P. Demontis and G. B. Suffritti, J. Chem. Theory Comput., 2015, 11, 3829–3843 CrossRef CAS PubMed.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129–138 CrossRef CAS.
- T. A. Manz and D. S. Sholl, J. Chem. Theory Comput., 2012, 8, 2844–2867 CrossRef CAS PubMed.
- A. V. Marenich, S. V. Jerome, C. J. Cramer and D. G. Truhlar, J. Chem. Theory Comput., 2012, 8, 527–541 CrossRef CAS PubMed.
- P. Bultinck, C. Van Alsenoy, P. W. Ayers and R. Carbo-Dorca, J. Chem. Phys., 2007, 126, 144111 CrossRef PubMed.
- E. R. Davidson and S. Chakravorty, Theor. Chim. Acta, 1992, 83, 319–330 CrossRef CAS.
- B. Wang, S. H. L. Li and D. G. Truhlar, J. Chem. Theory Comput., 2014, 10, 5640–5650 CrossRef CAS PubMed.
- J. Cioslowski, J. Am. Chem. Soc., 1989, 111, 8333–8336 CrossRef CAS.
- P. Ghosez, J. P. Michenaud and X. Gonze, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 6224–6240 CrossRef CAS.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- D. Y. Zubarev and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2008, 10, 5207–5217 RSC.
- G. Knizia, J. Chem. Theory Comput., 2013, 9, 4834–4843 CrossRef CAS PubMed.
- T. Janowski, J. Chem. Theory Comput., 2014, 10, 3085–3091 CrossRef CAS PubMed.
- B. D. Dunnington and J. R. Schmidt, J. Chem. Theory Comput., 2012, 8, 1902–1911 CrossRef CAS PubMed.
- T. R. Galeev, B. D. Dunnington, J. R. Schmidt and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2013, 15, 5022–5029 RSC.
- L. P. Lee, D. J. Cole, M. C. Payne and C. K. Skylaris, J. Comput. Chem., 2013, 34, 429–444 CrossRef CAS PubMed.
- N. Gabaldon Limas and T. A. Manz, Introducing DDEC6 Atomic Population Analysis: Part 2. Computed Results for a Wide Range of Periodic and Nonperiodic Materials, RSC Adv., 2016, 6 10.1039/C6RA05507A.
- S. G. Dale, A. Otero-de-la-Roza and E. R. Johnson, Phys. Chem. Chem. Phys., 2014, 16, 14584–14593 RSC.
- V. Luana, P. Mori-Sanchez, A. Costales, M. A. Blanco and A. M. Pendas, J. Chem. Phys., 2003, 119, 6341–6350 CrossRef CAS.
- T. J. Giese and D. M. York, J. Chem. Phys., 2008, 128, 064104 CrossRef PubMed.
- A. J. Stone, Chem. Phys. Lett., 1981, 83, 233–239 CrossRef CAS.
- R. J. Wheatley, Mol. Phys., 1993, 79, 597–610 CrossRef CAS.
- S. Cardamone, T. J. Hughes and P. L. A. Popelier, Phys. Chem. Chem. Phys., 2014, 16, 10367–10387 RSC.
- A. J. Stone, J. Chem. Theory Comput., 2005, 1, 1128–1132 CrossRef CAS PubMed.
- D. M. Elking, G. A. Cisneros, J. P. Piquemal, T. A. Darden and L. G. Pedersen, J. Chem. Theory Comput., 2010, 6, 190–202 CrossRef CAS PubMed.
- Electronegativity, in CRC Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press, Boca Raton, Florida, 88th edn, 2007–2008, p. 9.81 Search PubMed.
- L. Pauling, J. Am. Chem. Soc., 1932, 54, 3570–3582 CrossRef CAS.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- T. A. Manz and D. S. Sholl, J. Chem. Theory Comput., 2010, 6, 2455–2468 CrossRef CAS PubMed.
- E. D. Stevens, J. Rys and P. Coppens, J. Am. Chem. Soc., 1978, 100, 2324–2328 CrossRef CAS.
- J. L. Staudenmann, P. Coppens and J. Muller, Solid State Commun., 1976, 19, 29–33 CrossRef CAS.
- A. Milani, M. Tommasini and C. Castiglioni, Theor. Chem. Acc., 2012, 131, 1139 CrossRef.
- W. Zhong, R. D. King-Smith and D. Vanderbilt, Phys. Rev. Lett., 1994, 72, 3618–3621 CrossRef CAS PubMed.
- C. F. Guerra, J. W. Handgraaf, E. J. Baerends and F. M. Bickelhaupt, J. Comput. Chem., 2004, 25, 189–210 CrossRef CAS PubMed.
- D. Geldof, A. Krishtal, F. Blockhuys and C. Van Alsenoy, J. Chem. Theory Comput., 2011, 7, 1328–1335 CrossRef CAS PubMed.
- T. Verstraelen, P. W. Ayers, V. Van Speybroeck and M. Waroquier, J. Chem. Theory Comput., 2013, 9, 2221–2225 CrossRef CAS PubMed.
- D. E. P. Vanpoucke, P. Bultinck and I. Van Driessche, J. Comput. Chem., 2013, 34, 405–417 CrossRef CAS PubMed.
- D. Ghillemijn, P. Bultinck, D. Van Neck and P. W. Ayers, J. Comput. Chem., 2011, 32, 1561–1567 CrossRef CAS PubMed.
- T. C. Lillestolen and R. J. Wheatley, Chem. Commun., 2008, 5909–5911 RSC.
- T. C. Lillestolen and R. J. Wheatley, J. Chem. Phys., 2009, 131, 144101 CrossRef PubMed.
- B. J. Gellatly and J. L. Finney, J. Mol. Biol., 1982, 161, 305–322 CrossRef CAS PubMed.
- M. Thomas, M. Brehm and B. Kirchner, Phys. Chem. Chem. Phys., 2015, 17, 3207–3213 RSC.
- R. F. Nalewajski and R. G. Parr, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 8879–8882 CrossRef CAS.
- R. G. Parr, P. W. Ayers and R. F. Nalewajski, J. Phys. Chem. A, 2005, 109, 3957–3959 CrossRef CAS PubMed.
- C. F. Matta and R. F. W. Bader, J. Phys. Chem. A, 2006, 110, 6365–6371 CrossRef CAS PubMed.
- R. F. W. Bader, Acc. Chem. Res., 1975, 8, 34–40 CrossRef CAS.
- R. F. W. Bader, P. J. MacDougal and C. D. H. Lau, J. Am. Chem. Soc., 1984, 106, 1594–1605 CrossRef CAS.
- R. F. W. Bader, J. Phys. Chem. A, 2007, 111, 7966–7972 CrossRef CAS PubMed.
- R. F. W. Bader, T. A. Keith, K. M. Gough and K. E. Laidig, Mol. Phys., 1992, 75, 1167–1189 CrossRef CAS.
- Q. Yang, D. Liu, C. Zhong and J.-R. Li, Chem. Rev., 2013, 113, 8261–8323 CrossRef CAS PubMed.
- D. E. P. Vanpoucke, I. Van Driessche and P. Bultinck, J. Comput. Chem., 2013, 34, 422–427 CrossRef CAS PubMed.
- T. Verstraelen, E. Pauwels, F. De Proft, V. Van Speybroeck, P. Geerlings and M. Waroquier, J. Chem. Theory Comput., 2012, 8, 661–676 CrossRef CAS PubMed.
- T. Verstraelen, P. W. Ayers, V. Van Speybroeck and M. Waroquier, Chem. Phys. Lett., 2012, 545, 138–143 CrossRef CAS.
- P. Bultinck, D. L. Cooper and D. Van Neck, Phys. Chem. Chem. Phys., 2009, 11, 3424–3429 RSC.
- T. A. Manz and N. Gabaldon Limas, A Complete Set of Reference Ion and Core Densities for the Density-Derived Electrostatic and Chemical Methods, 2016, submitted for publication Search PubMed.
- P. Bultinck, P. W. Ayers, S. Fias, K. Tiels and C. Van Alsenoy, Chem. Phys. Lett., 2007, 444, 205–208 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- M. J. Guittet, J. P. Crocombette and M. Gautier-Soyer, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 125117 CrossRef.
- K. B. Wiberg and P. R. Rablen, J. Comput. Chem., 1993, 14, 1504–1518 CrossRef CAS.
- J. Hafner, J. Comput. Chem., 2008, 29, 2044–2078 CrossRef CAS PubMed.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. J. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- U. C. Singh and P. A. Kollman, J. Comput. Chem., 1984, 5, 129–145 CrossRef CAS.
- P. Politzer, P. R. Laurence and K. Jayasuriya, Environ. Health Perspect., 1985, 61, 191–202 CrossRef CAS PubMed.
- P. Politzer and J. S. Murray, Theor. Chem. Acc., 2002, 108, 134–142 CrossRef CAS.
- E. Scrocco and J. Tomasi, in Advances in Quantum Chemistry, ed. P. Lowdin, Academic Press, New York, 1978, vol. 11, pp. 115–193 Search PubMed.
- D. S. Kosov and P. L. A. Popelier, J. Phys. Chem. A, 2000, 104, 7339–7345 CrossRef CAS.
- D. M. Elking, L. Perera and L. G. Pedersen, Comput. Phys. Commun., 2012, 183, 390–397 CrossRef CAS PubMed.
- A. Volkov, T. Koritsanszky and P. Coppens, Chem. Phys. Lett., 2004, 391, 170–175 CrossRef CAS.
- M. A. Spackman, Chem. Phys. Lett., 2006, 418, 158–162 CrossRef CAS.
- J. M. Gruschus and A. Kuki, J. Comput. Chem., 1990, 11, 978–993 CrossRef CAS.
- K. E. Laidig, J. Phys. Chem., 1993, 97, 12760–12767 CrossRef CAS.
- B. Wang and D. G. Truhlar, J. Chem. Theory Comput., 2010, 6, 3330–3342 CrossRef CAS PubMed.
- D. Das, K. P. Eurenius, E. M. Billings, P. Sherwood, D. C. Chatfield, M. Hodoscek and B. R. Brooks, J. Chem. Phys., 2002, 117, 10534–10547 CrossRef CAS.
- G. A. Cisneros, S. N. I. Tholander, O. Parisel, T. A. Darden, D. Elking, L. Perera and J. P. Piquemal, Int. J. Quantum Chem., 2008, 108, 1905–1912 CrossRef CAS PubMed.
- P. N. Day, J. H. Jensen, M. S. Gordon, S. P. Webb, W. J. Stevens, M. Krauss, D. Garmer, H. Basch and D. Cohen, J. Chem. Phys., 1996, 105, 1968–1986 CrossRef CAS.
- M. A. Freitag, M. S. Gordon, J. H. Jensen and W. J. Stevens, J. Chem. Phys., 2000, 112, 7300–7306 CrossRef CAS.
- S. Van Damme, P. Bultinck and S. Fias, J. Chem. Theory Comput., 2009, 5, 334–340 CrossRef CAS PubMed.
- S. R. Cox and D. E. Williams, J. Comput. Chem., 1981, 2, 304–323 CrossRef CAS.
- W. Smith, CCCP5 Newsletter, 1998, vol. 46, pp. 18–30 Search PubMed.
- T. A. Manz and D. S. Sholl, J. Chem. Theory Comput., 2011, 7, 4146–4164 CrossRef CAS PubMed.
- L. P. Lee, D. J. Cole, C.-K. Skylaris, W. L. Jorgensen and M. C. Payne, J. Chem. Theory Comput., 2013, 9, 2981–2991 CrossRef CAS PubMed.
- L. P. Lee, N. G. Limas, D. J. Cole, M. C. Payne, C.-K. Skylaris and T. A. Manz, J. Chem. Theory Comput., 2014, 10, 5377–5390 CrossRef CAS PubMed.
- N. Foloppe and A. D. MacKerell, J. Comput. Chem., 2000, 21, 86–104 CrossRef CAS.
- F.-X. Coudert, Chem. Mater., 2015, 27, 1905–1916 CrossRef CAS.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- S. Guha and K. Nakamoto, Coord. Chem. Rev., 2005, 249, 1111–1132 CrossRef CAS.
- A. Rosen and B. Wastberg, J. Chem. Phys., 1989, 90, 2525–2526 CrossRef CAS.
- D. M. Cox, D. J. Trevor, K. C. Reichmann and A. Kaldor, J. Am. Chem. Soc., 1986, 108, 2457–2458 CrossRef CAS PubMed.
- J. R. Heath, R. F. Curl and R. E. Smalley, J. Chem. Phys., 1987, 87, 4236–4238 CrossRef CAS.
- S. H. Yang, C. L. Pettiette, J. Conceicao, O. Cheshnovsky and R. E. Smalley, Chem. Phys. Lett., 1987, 139, 233–238 CrossRef CAS.
- L. Moro, R. S. Ruoff, C. H. Becker, D. C. Lorents and R. Malhotra, J. Phys. Chem., 1993, 97, 6801–6805 CrossRef CAS.
- T. Inoue, Y. Kubozono, S. Kashino, Y. Takabayashi, K. Fujitaka, M. Hida, M. Inoue, T. Kanbara, S. Emura and T. Uruga, Chem. Phys. Lett., 2000, 316, 381–386 CrossRef CAS.
- N. Weiden, H. Kass and K. P. Dinse, J. Phys. Chem. B, 1999, 103, 9826–9830 CrossRef CAS.
- A. A. Popov, S. F. Yang and L. Dunsch, Chem. Rev., 2013, 113, 5989–6113 CrossRef CAS PubMed.
- T. Aree, T. Kerdcharoen and S. Hannongbua, Chem. Phys. Lett., 1998, 285, 221–225 CrossRef CAS.
- D. Tomanek and Y. S. Li, Chem. Phys. Lett., 1995, 243, 42–44 CrossRef CAS.
- M. V. Ryzhkov, A. L. Ivanovskii and B. Delley, Nanosyst.: Phys., Chem., Math., 2014, 5, 494–508 Search PubMed.
- S. Suzuki, M. Kushida, S. Amamiya, S. Okada and K. Nakao, Chem. Phys. Lett., 2000, 327, 291–298 CrossRef CAS.
- A. V. Krisilov, I. V. Nechaev, A. L. Kotova, K. S. Shikhaliev, V. E. Chernov and B. A. Zon, Comput. Theor. Chem., 2015, 1054, 100–108 CrossRef CAS.
- W. W. Zhang, A. R. Oganov, A. F. Goncharov, Q. Zhu, S. E. Boulfelfel, A. O. Lyakhov, E. Stavrou, M. Somayazulu, V. B. Prakapenka and Z. Konopkova, Science, 2013, 342, 1502–1505 CrossRef CAS PubMed.
- Ionization Energies of Atoms and Atomic Ions, in CRC Handbook of Chemistry
and Physics, ed. D. R. Lide, CRC Press, Boca Raton, Florida, 88th edn, 2007–2008, p. 10.203 Search PubMed.
- G. Saleh and A. R. Oganov, Phys. Chem. Chem. Phys., 2016, 18, 2840–2849 RSC.
- W. L. Meerts, F. H. Deleeuw and A. Dymanus, Chem. Phys., 1977, 22, 319–324 CrossRef CAS.
- F. Fantuzzi, T. M. Cardozo and M. A. C. Nascimento, J. Phys. Chem. A, 2015, 119, 5335–5343 CrossRef CAS PubMed.
- J. Meister and W. H. E. Schwarz, J. Phys. Chem., 1994, 98, 8245–8252 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Summary of alternative charge partitioning algorithms considered; summary of integration routines; iterative algorithm for computing the Convex functional NACs; flow diagrams for the DDEC6 method; and .xyz files (which can be read using any text editor or the free Jmol visualization program downloadable from http://jmol.sourceforge.net) containing geometries, net atomic charges, atomic dipoles and quadrupoles, fitted tail decay exponents, and ASMs. See DOI: 10.1039/c6ra04656h |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
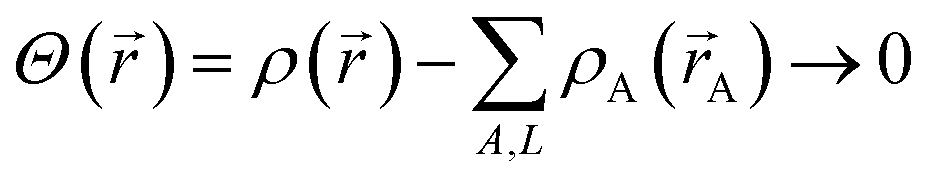
 denoting summation over all atoms in the material, and ρA(
denoting summation over all atoms in the material, and ρA(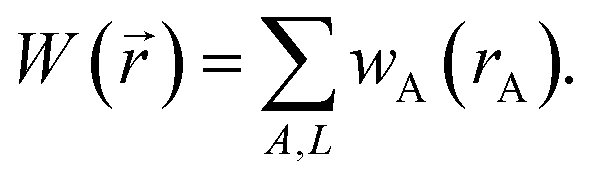
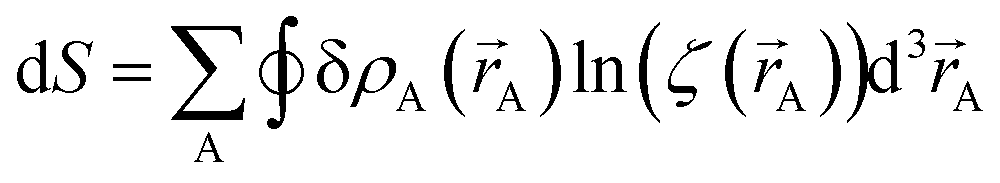
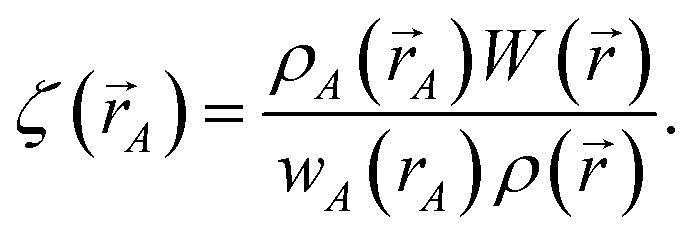
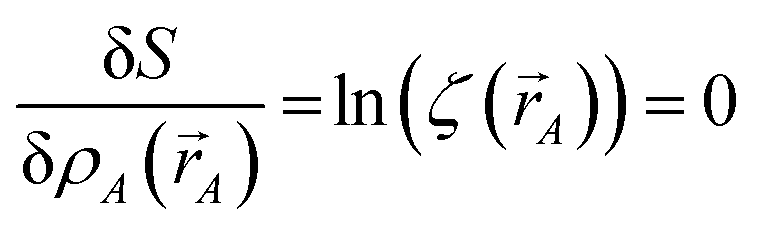
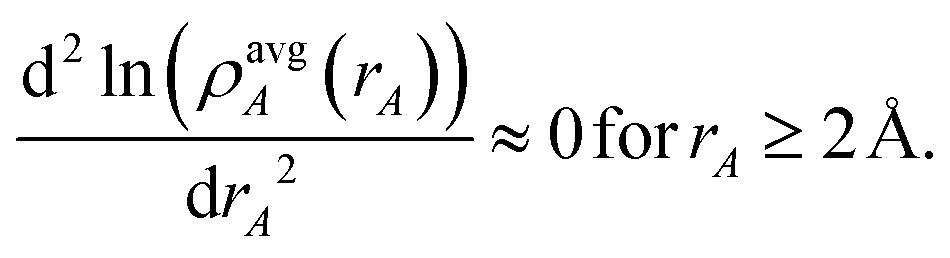
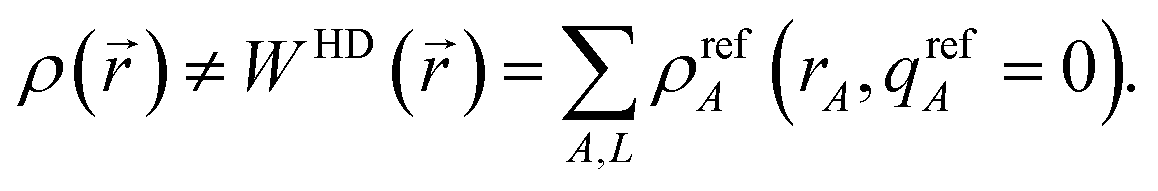
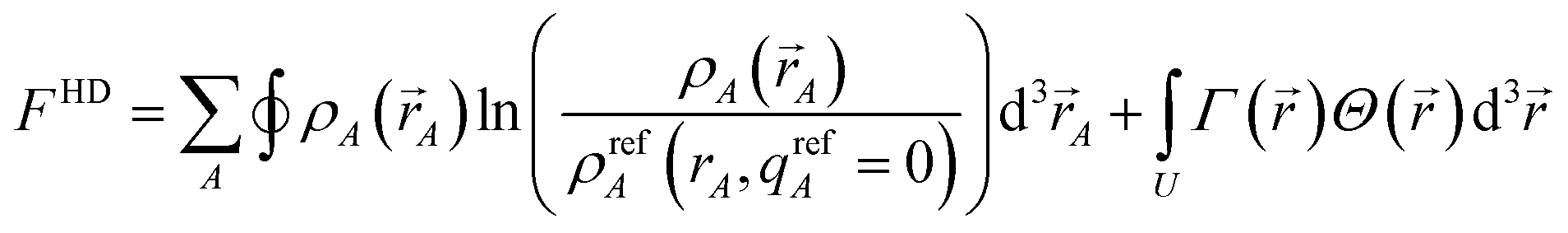
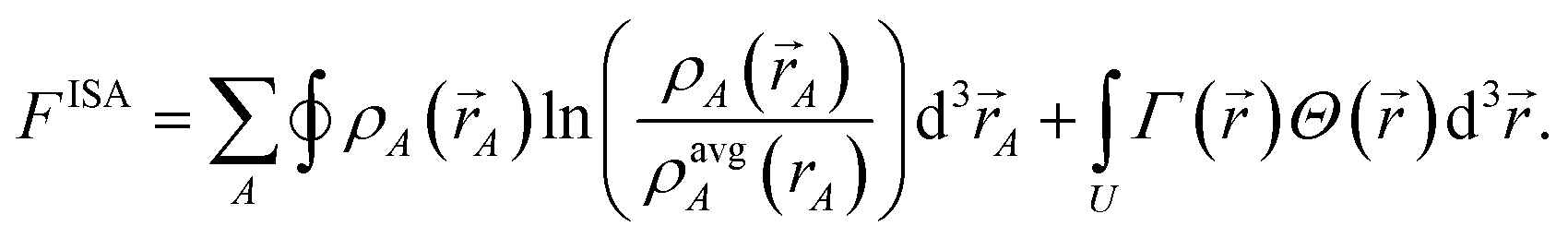
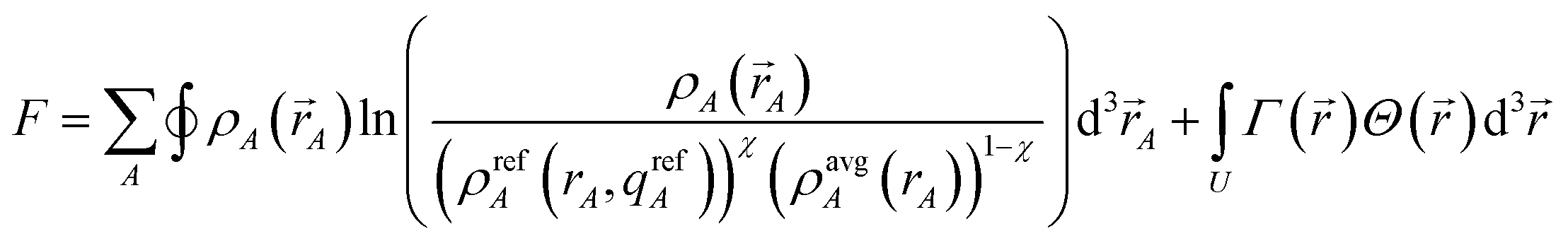
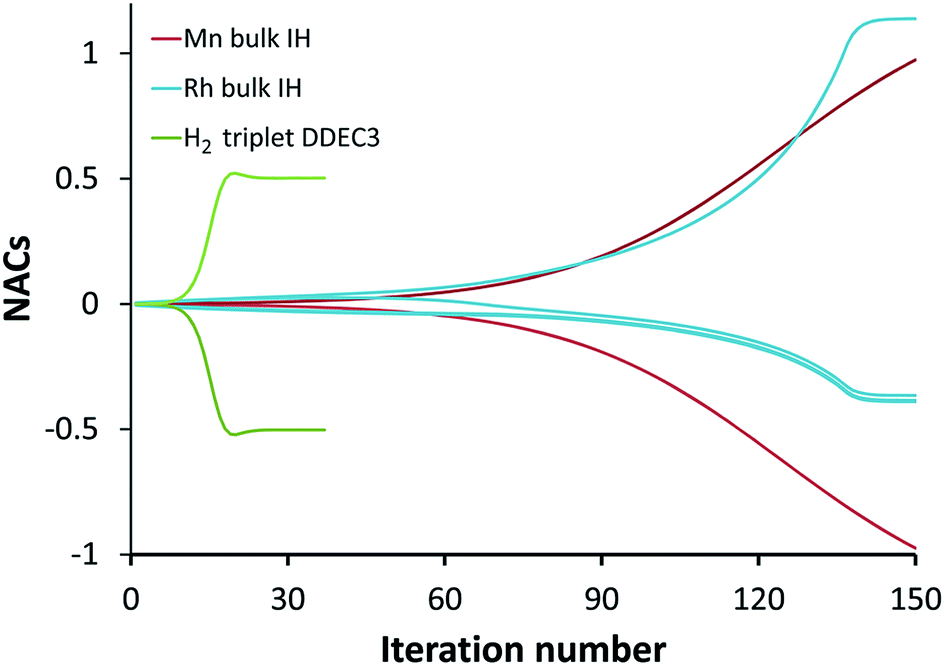
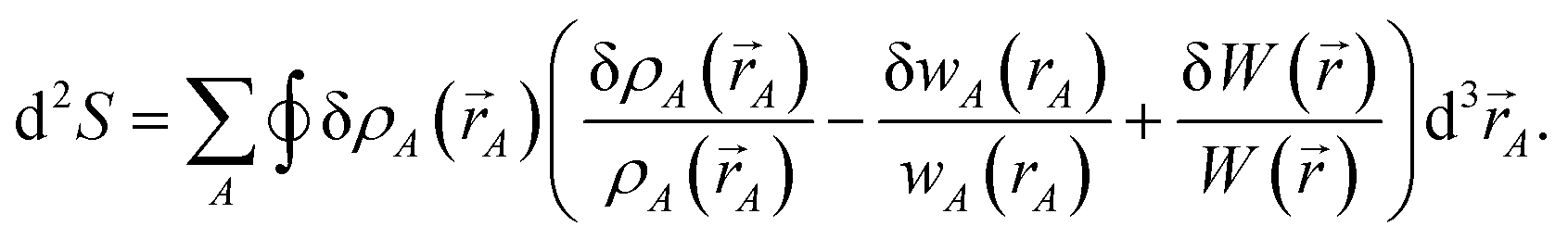
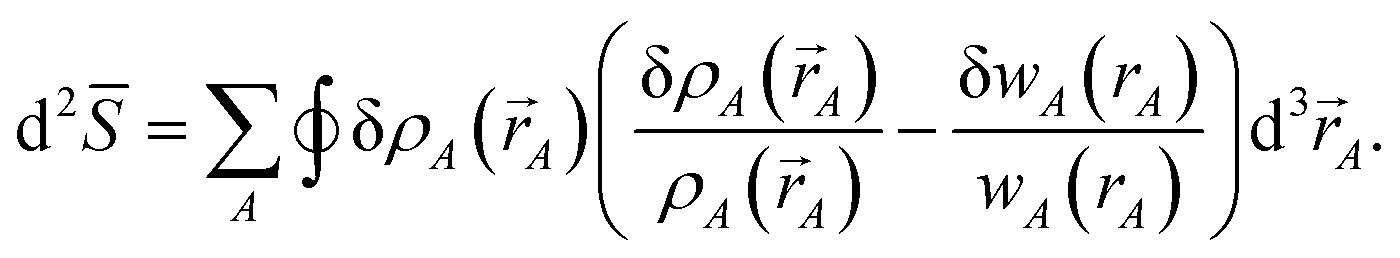
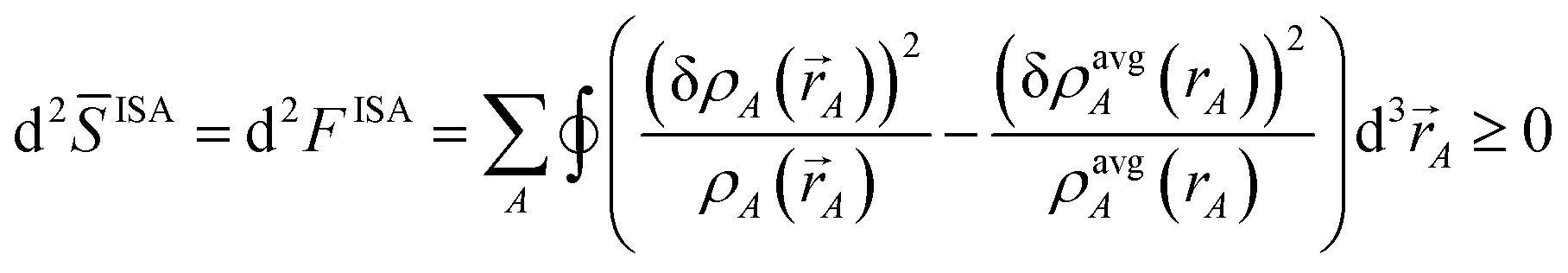
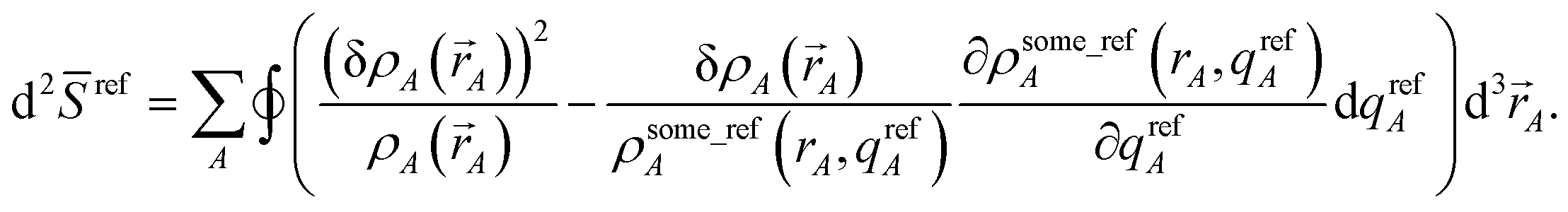
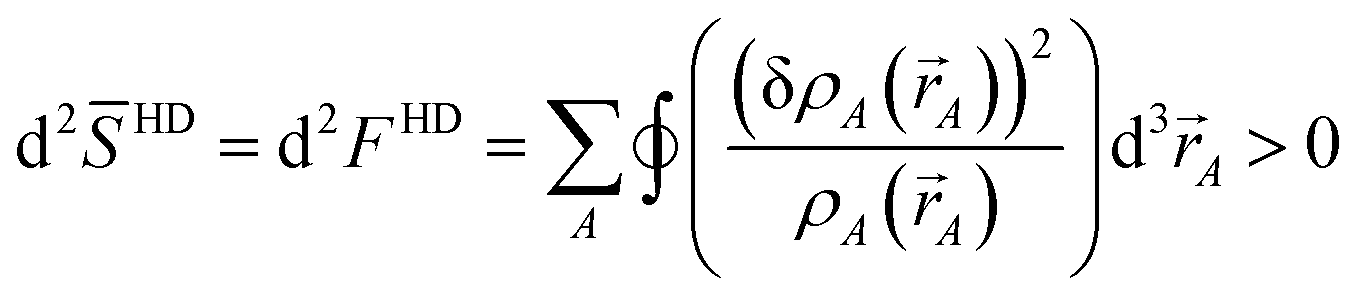
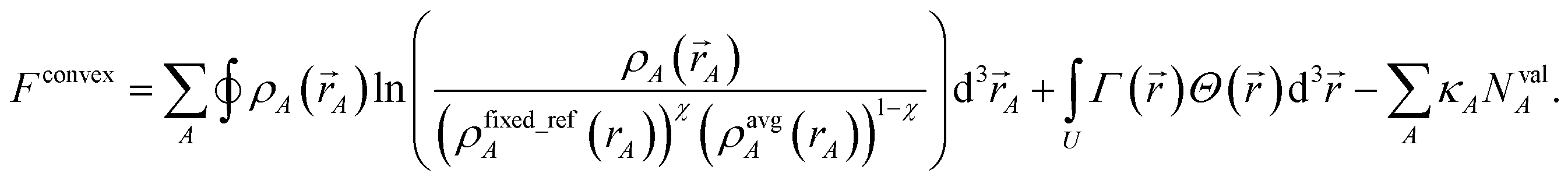
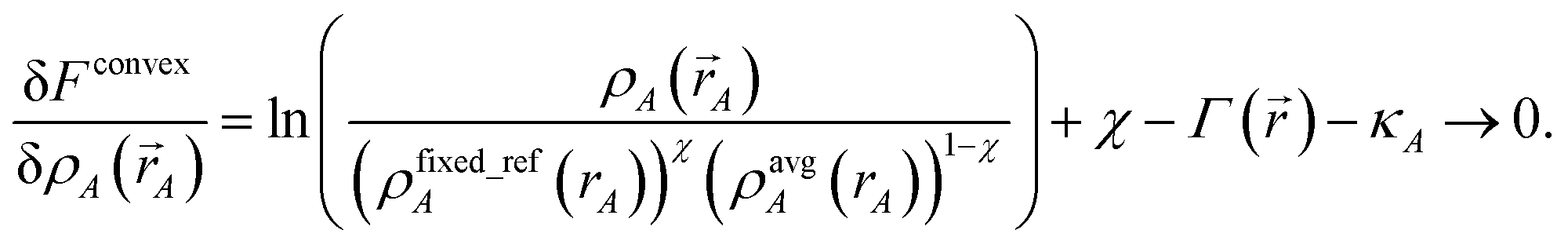
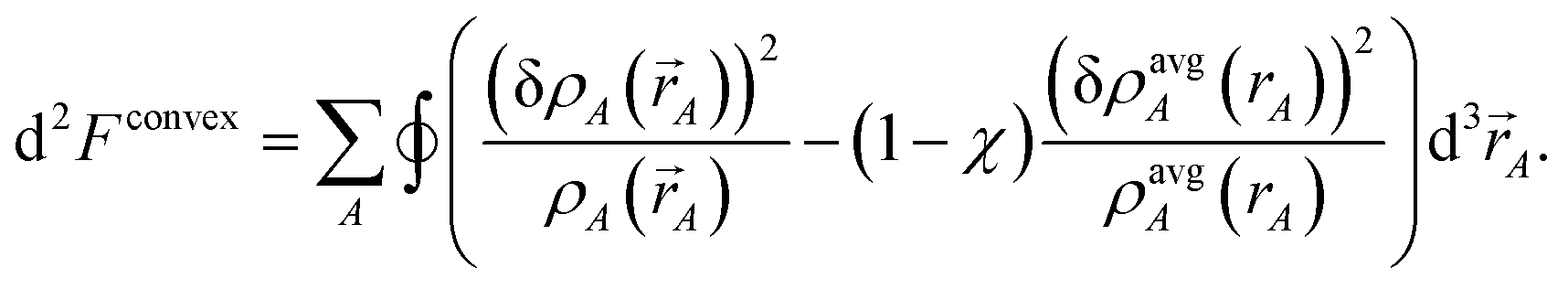
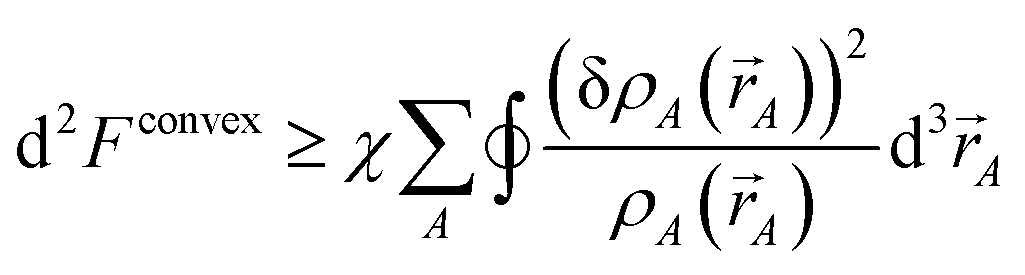
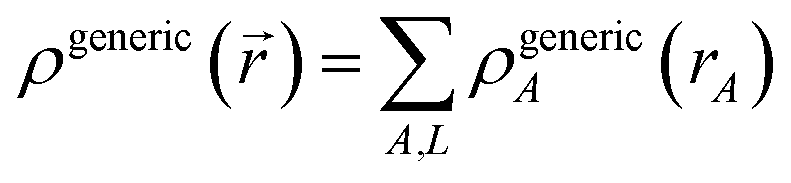
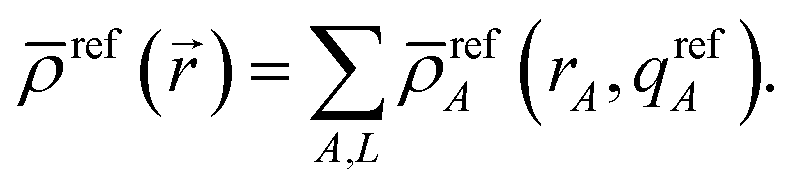
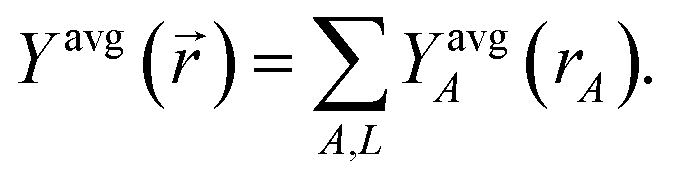
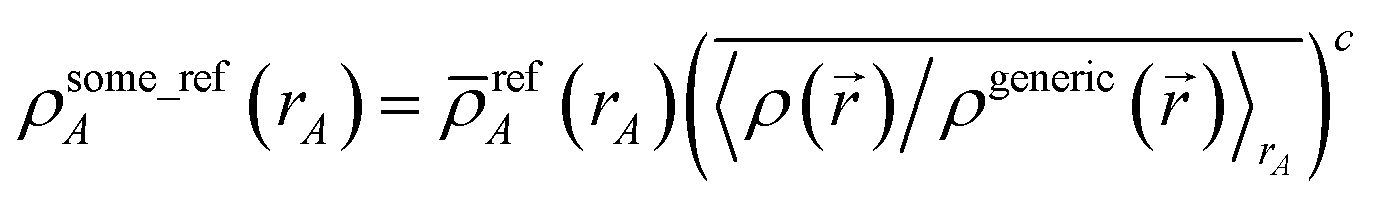
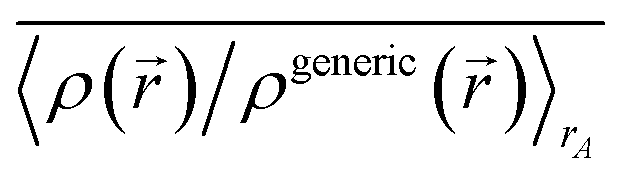 is a geometric average of all of the 〈ρ(
is a geometric average of all of the 〈ρ(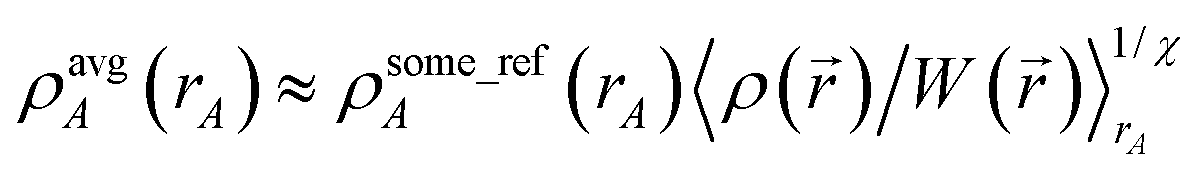
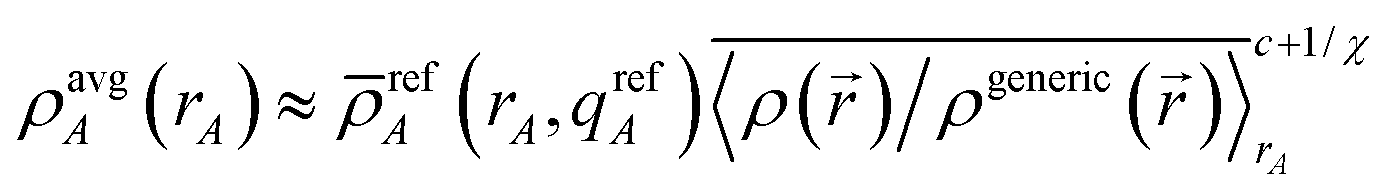
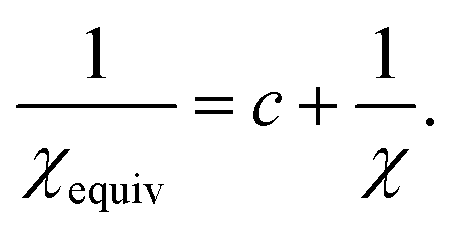
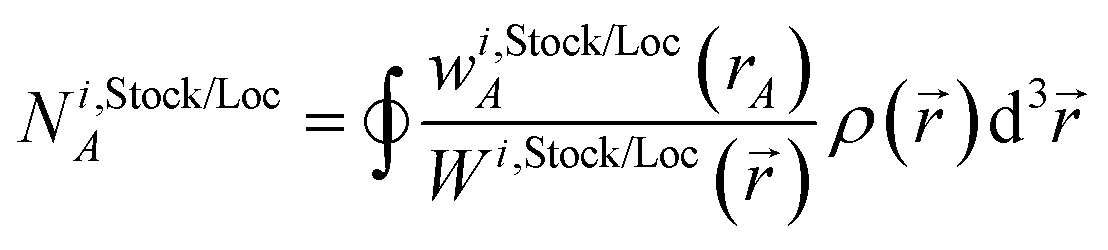
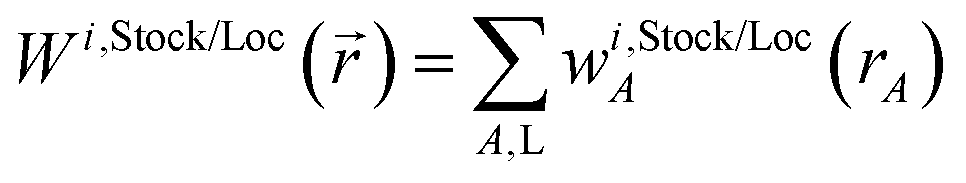
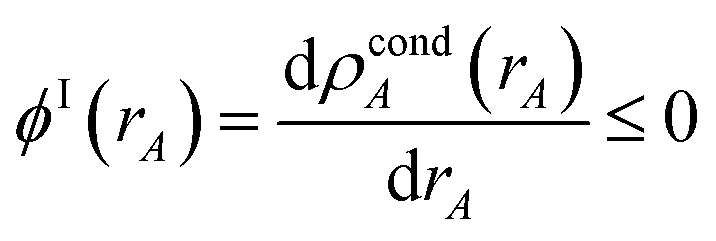
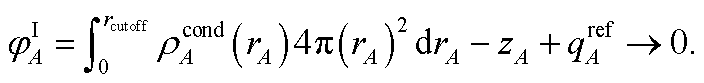

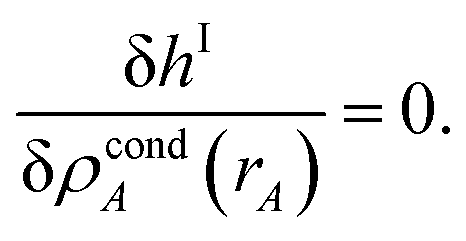
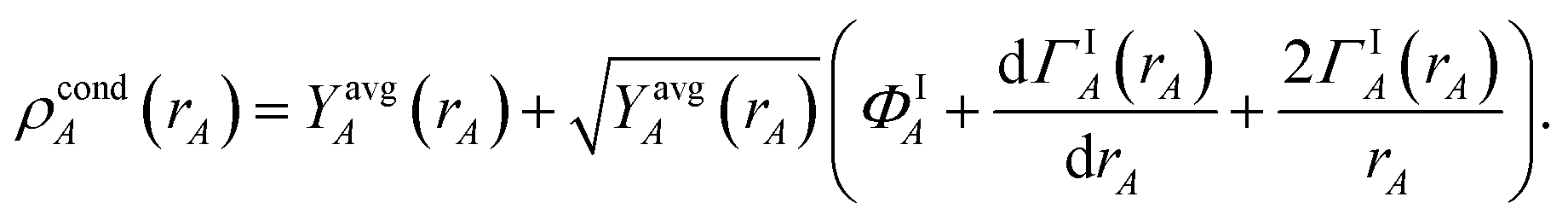
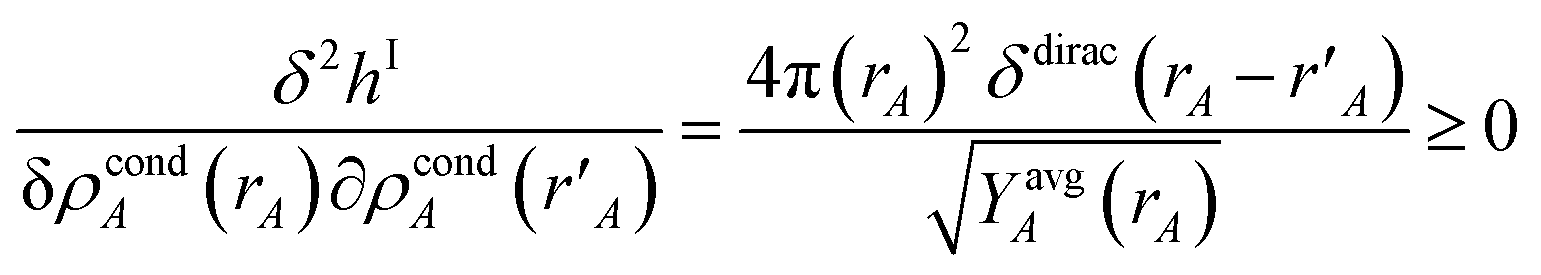
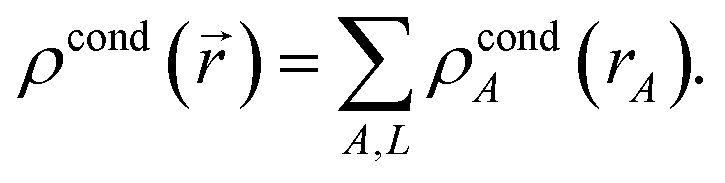
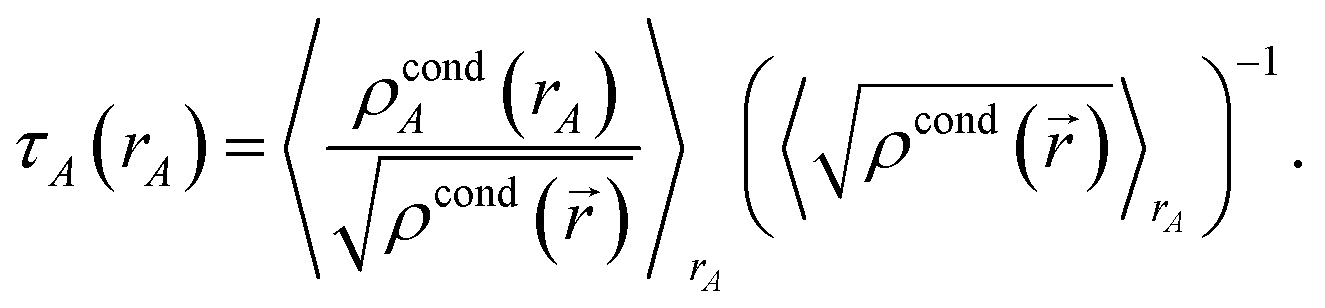
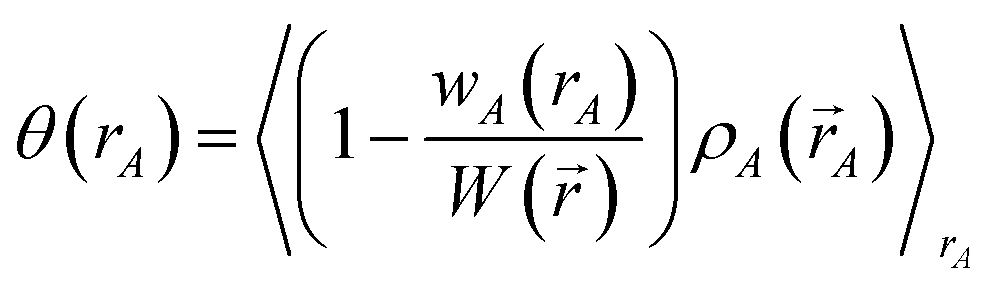
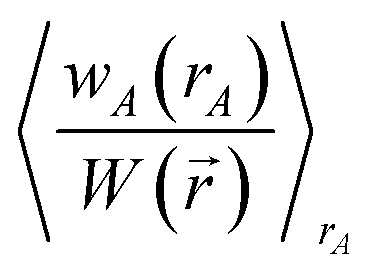
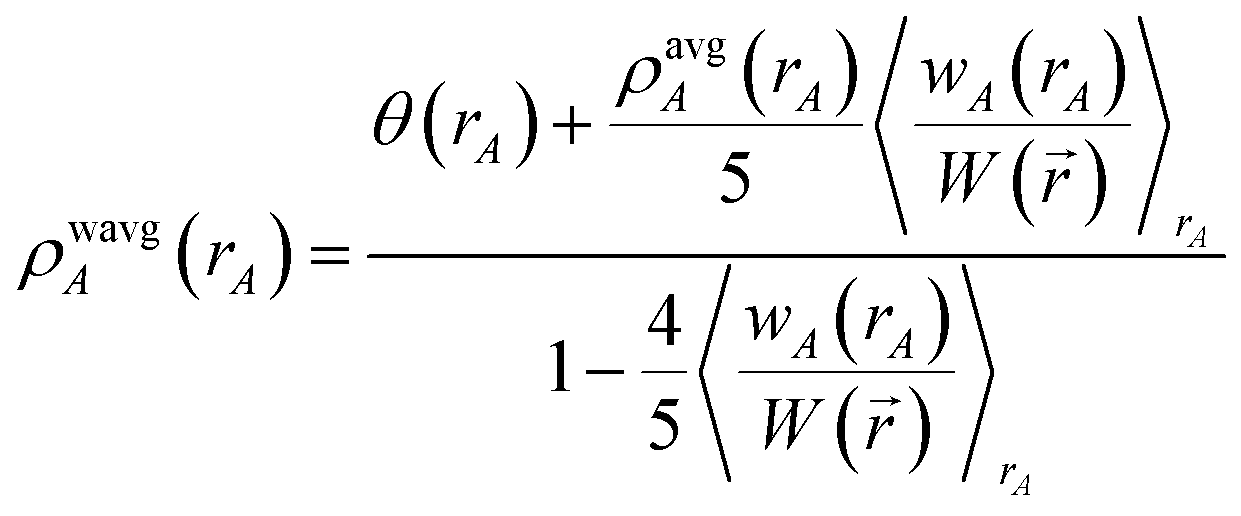
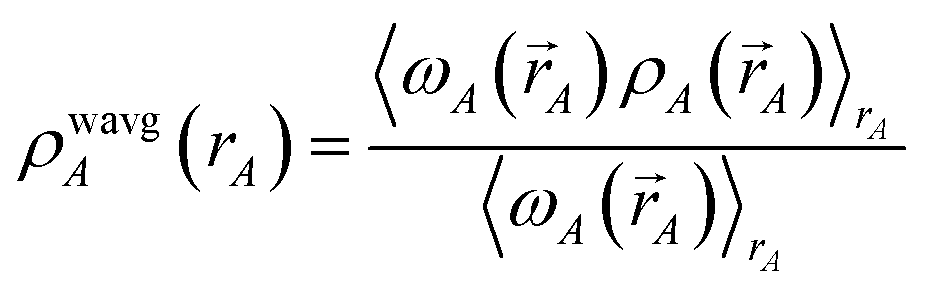
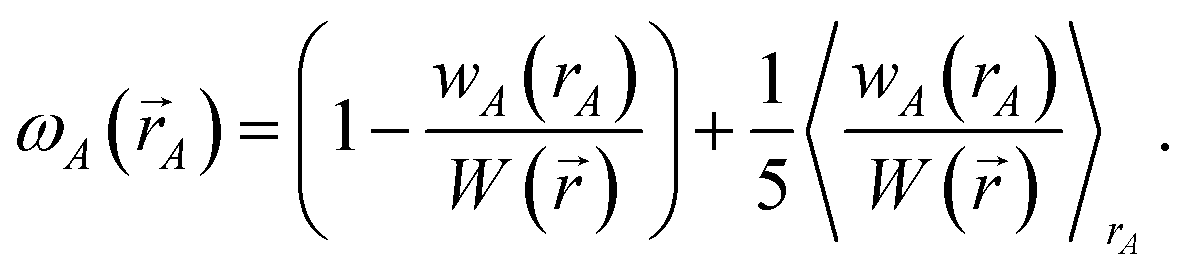
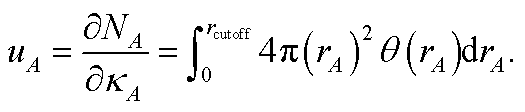
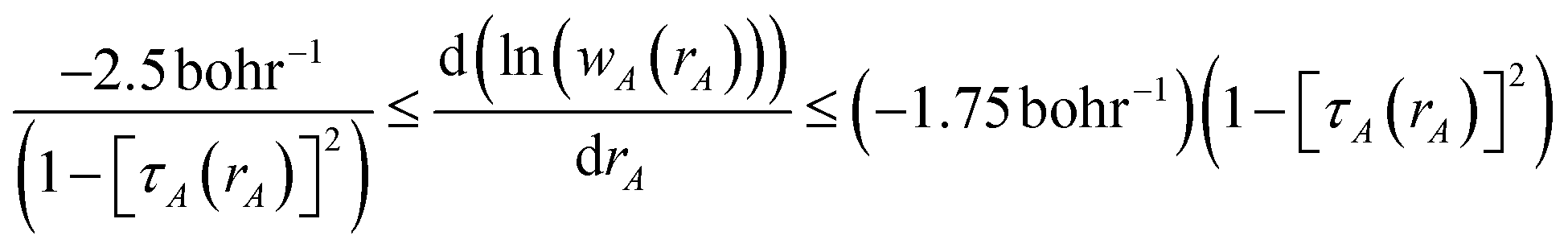
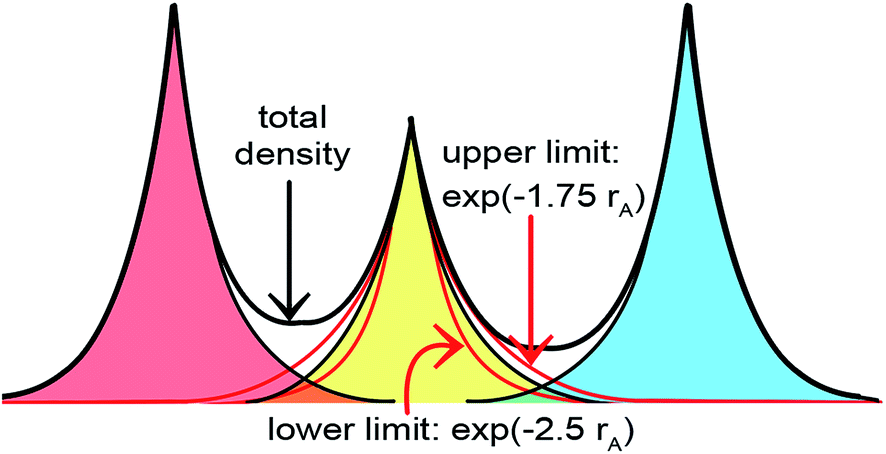
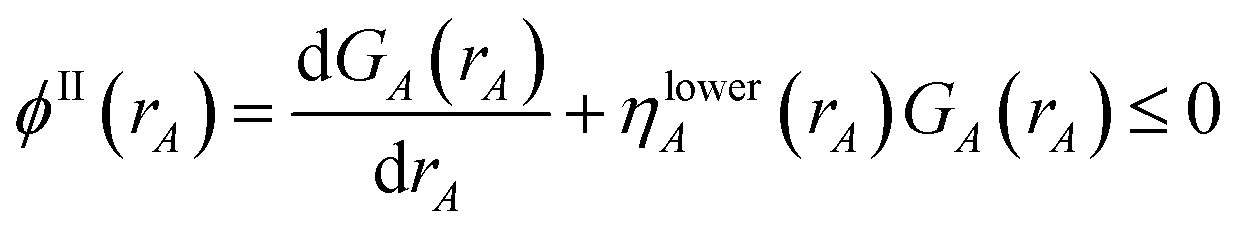
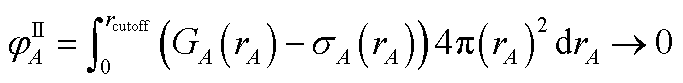
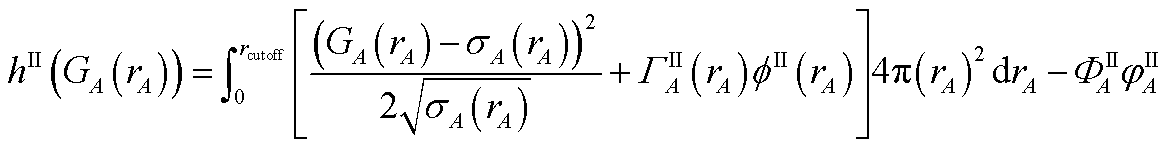
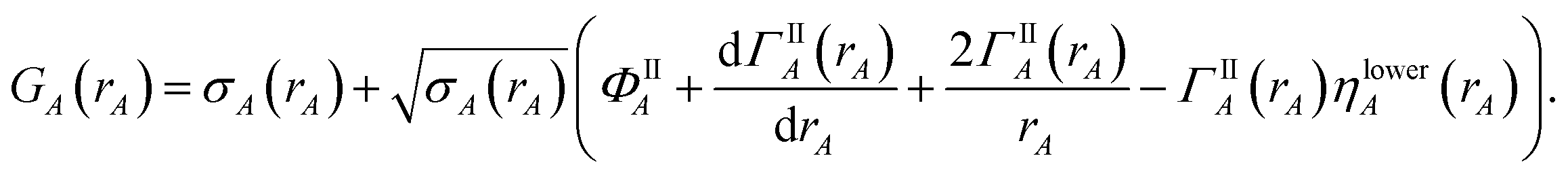
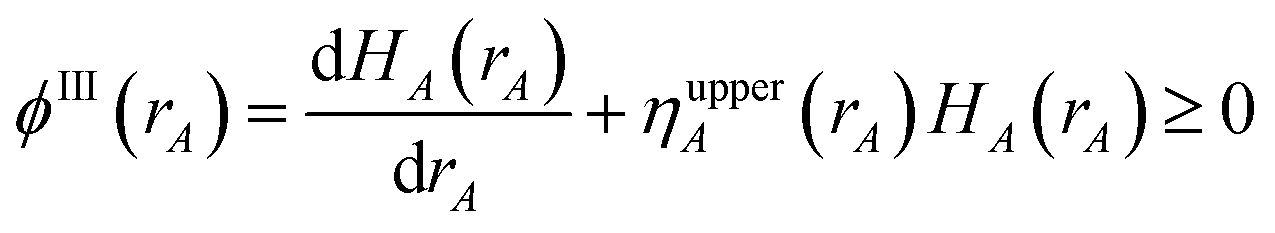
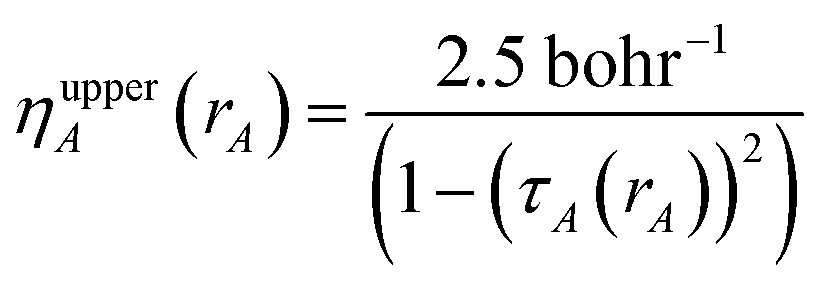
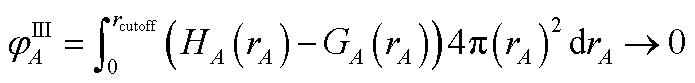
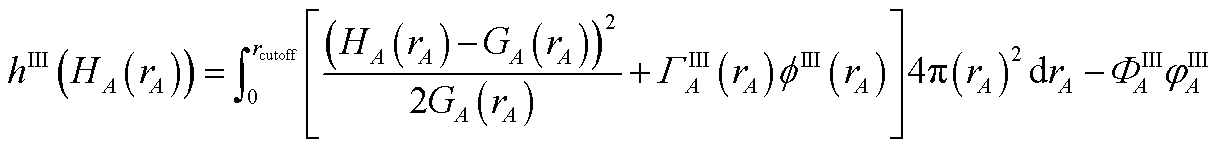
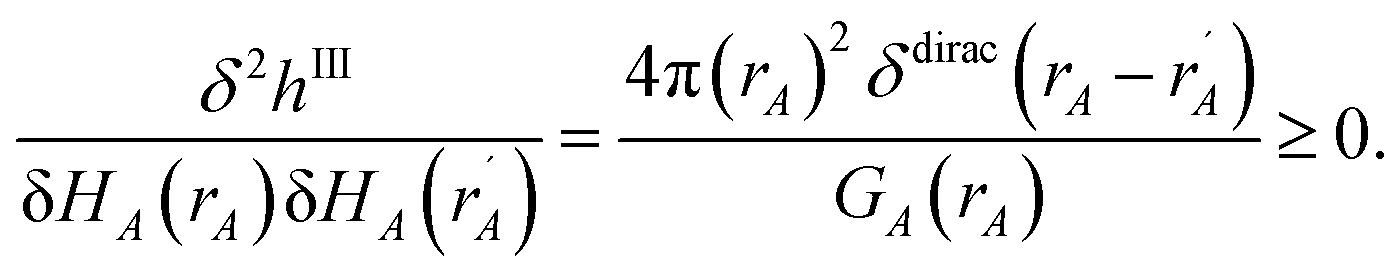
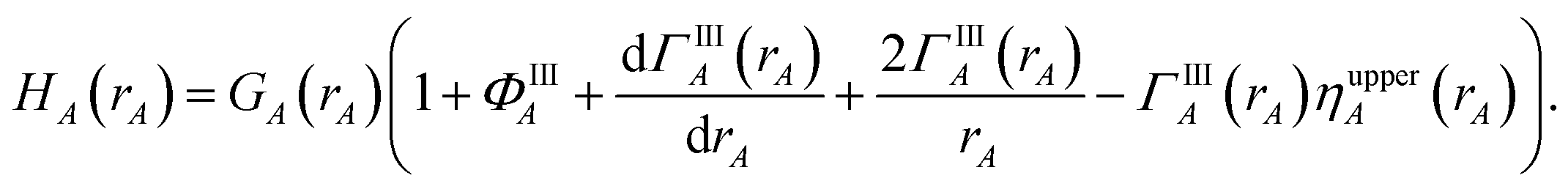
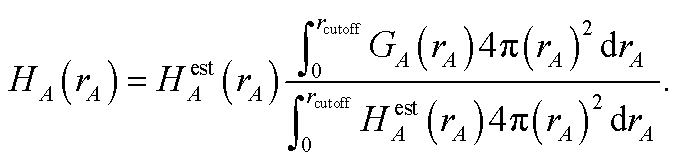
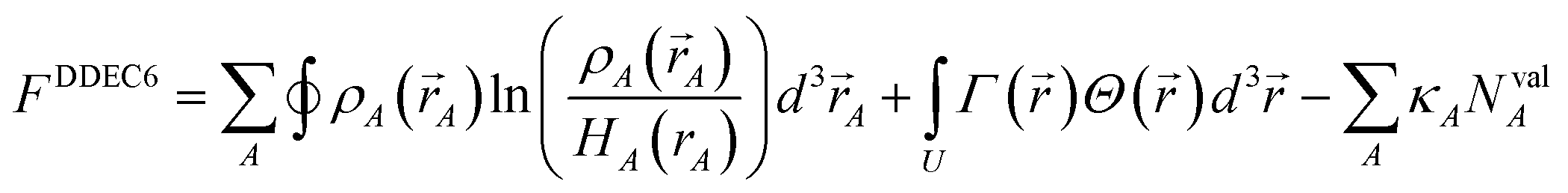
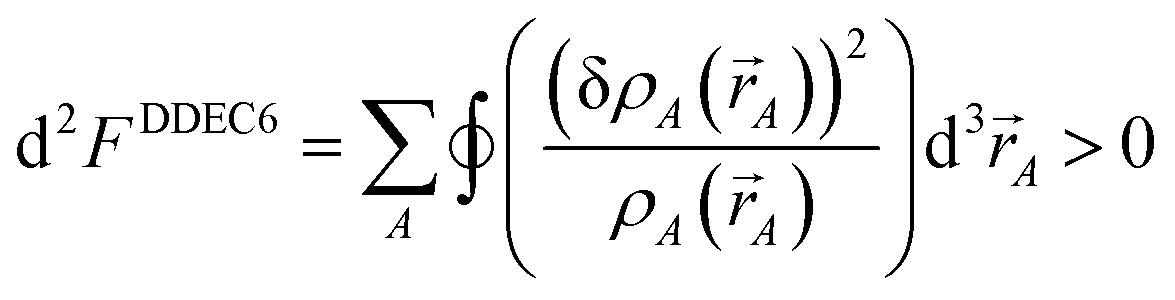
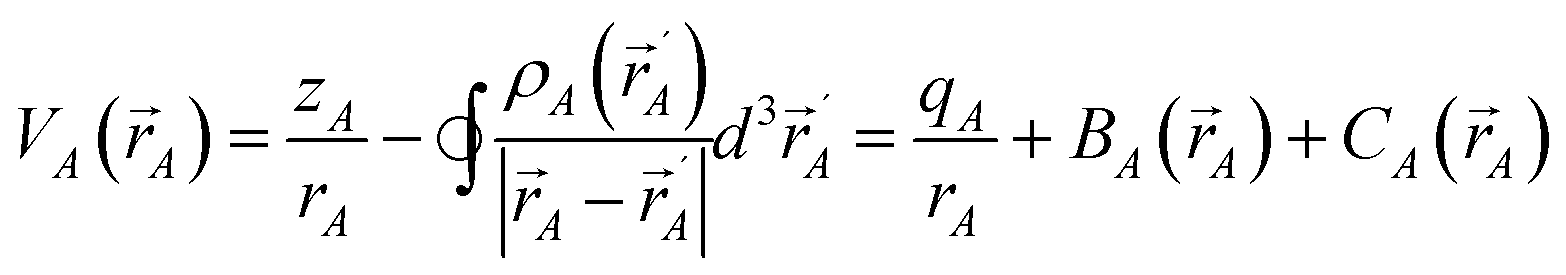
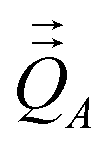 , is defined by
, is defined by 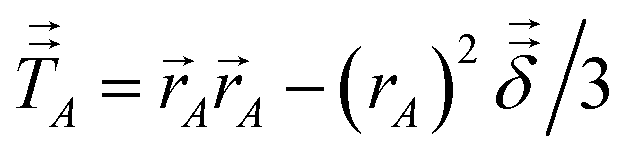 , where
, where 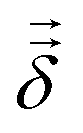 is the identity tensor. The three eigenvalues of
is the identity tensor. The three eigenvalues of 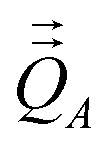 are independent of molecular orientation and coordinate system, and they sum to zero. For a spherical atom, all three quadrupole eigenvalues are zero. However, three zero quadrupole eigenvalues does not mean the atom is necessarily spherical, because such an atom could have non-zero dipole or higher order multipole (e.g., octapole) moments that indicate deviation from spherical symmetry. An oblate spheroidal density has one negative and two equal positive quadrupole eigenvalues. A prolate spheroidal density has one positive and two equal negative quadrupole eigenvalues. An ellipsoidal density can have three unequal quadrupole eigenvalues.
are independent of molecular orientation and coordinate system, and they sum to zero. For a spherical atom, all three quadrupole eigenvalues are zero. However, three zero quadrupole eigenvalues does not mean the atom is necessarily spherical, because such an atom could have non-zero dipole or higher order multipole (e.g., octapole) moments that indicate deviation from spherical symmetry. An oblate spheroidal density has one negative and two equal positive quadrupole eigenvalues. A prolate spheroidal density has one positive and two equal negative quadrupole eigenvalues. An ellipsoidal density can have three unequal quadrupole eigenvalues.
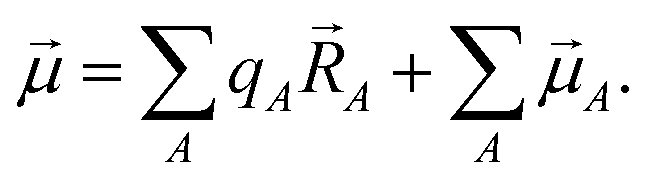
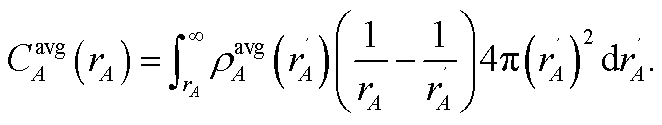
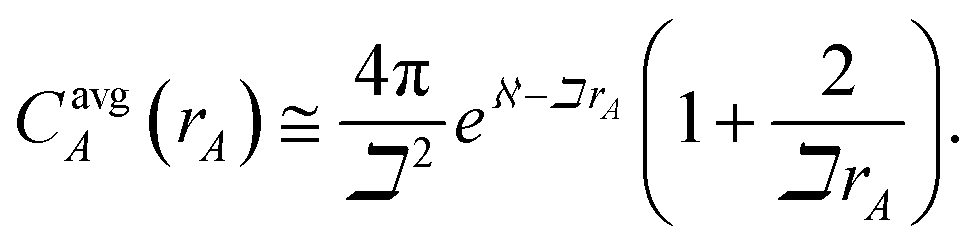
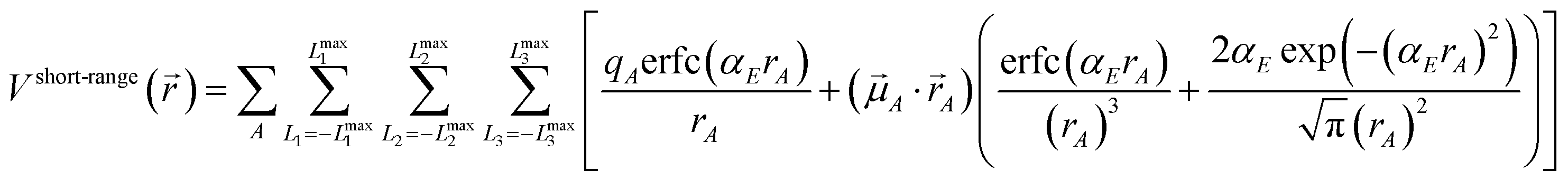
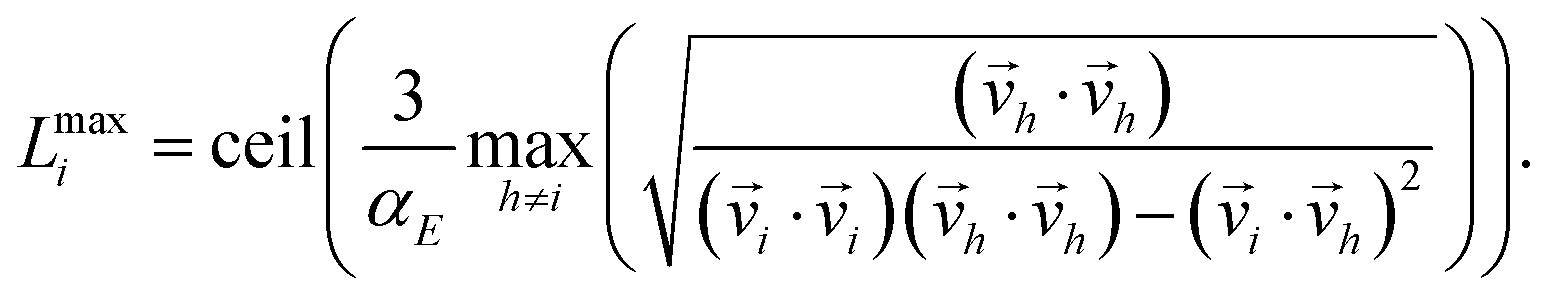
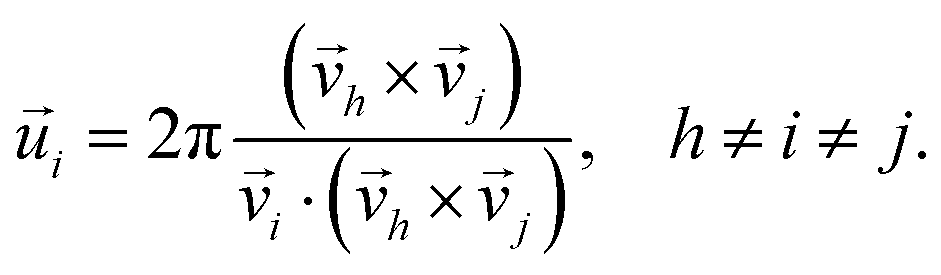
![[u with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0075_20d1.gif) 1 + b2
1 + b2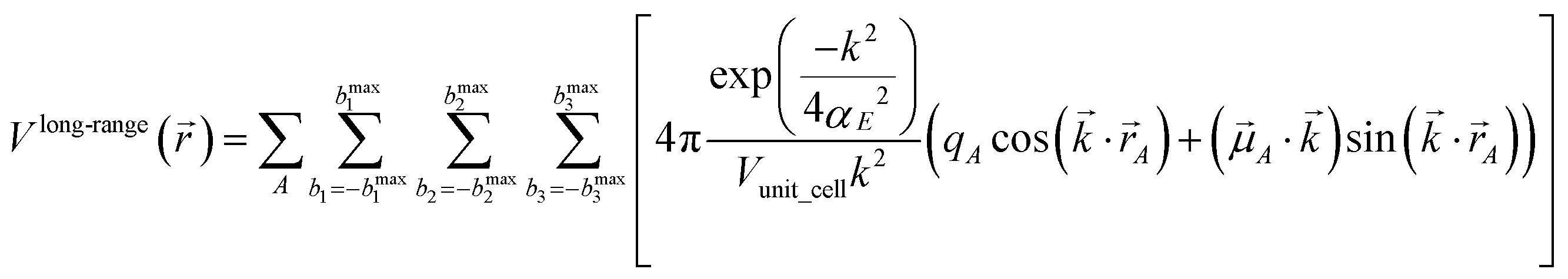
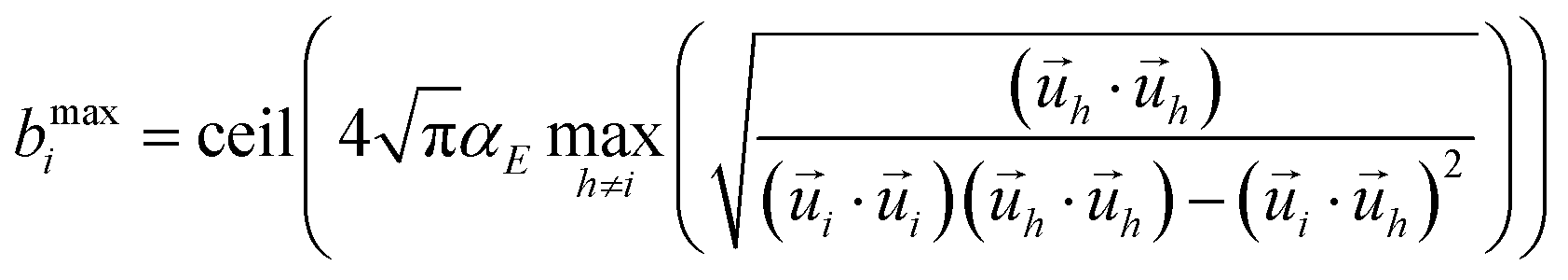
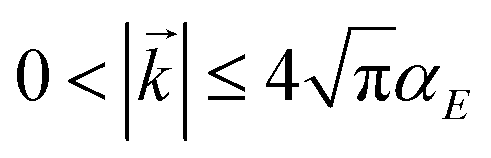 . Noting that each term in the reciprocal space term includes exp(−k2/(4αE2)) as a multiplier, our reciprocal space cutoff corresponds to exp(−k2/(4αE2)) ≈ exp(−4π) ≈ 3.5 × 10−6. Because it is a short-range effect, spherical charge penetration can be included entirely in the real space summation using the analytic potential of eqn (106). While the spherical cloud penetration effect is small over grid points used to compute RMSE, it becomes increasingly important for smaller rA values.
. Noting that each term in the reciprocal space term includes exp(−k2/(4αE2)) as a multiplier, our reciprocal space cutoff corresponds to exp(−k2/(4αE2)) ≈ exp(−4π) ≈ 3.5 × 10−6. Because it is a short-range effect, spherical charge penetration can be included entirely in the real space summation using the analytic potential of eqn (106). While the spherical cloud penetration effect is small over grid points used to compute RMSE, it becomes increasingly important for smaller rA values.