
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluorine in fragrances: exploring the difluoromethylene (CF2) group as a conformational constraint in macrocyclic musk lactones†
Michael J.
Corr
a,
Rodrigo A.
Cormanich
b,
Cortney N.
von Hahmann
a,
Michael
Bühl
a,
David B.
Cordes
a,
Alexandra M. Z.
Slawin
a and
David
O'Hagan
*a
aSchool of Chemistry, University of St Andrews, North Haugh, St Andrews, KY16 9ST, UK. E-mail: do1@st-andrews.ac.uk
bChemistry Institute, University of Campinas, Campinas, SP 13083-970, Brazil
First published on 4th November 2015
Abstract
The CF2 group is incorporated into specific positions within the lactone ring of the natural musk lactone, (12R)-(+)-12-methyl-13-tridecanolide, a constituent of Angelica root oil, Angelica archangelica L. The approach is taken as it was anticipated that CF2 groups would dictate corner locations in the macrocycle and limit the conformational space available to the lactone. Three fluorine containing lactones are prepared by organic synthesis. One (8) has CF2 groups located at the C-6 and C-9 positions, another (9) with CF2 groups at the C-5 and C-9 positions, and a third (10) with a CF2 group at C-8. Two of the fluorine containing lactones (8 and 10) were sufficiently crystalline to obtain X-ray crystal structures which revealed that the CF2 groups do adopt corner locations. All three lactones were subject to computational analysis at the B3LYP-D3/6-311+G** level to assess the relative energies of different conformers. In all cases, the global minima and most of the lowest energy minima have squared/rectangular geometries and located the CF2 groups at the corners. The lowest energy structures for 8 and 10 closely approximated the observed X-ray structures, suggesting good convergence of theory and experiment in determining relevant low energy conformations. All three compounds retained a pleasant odour suggesting the rings retained sufficient conformational flexibility to access relevant olfactory conformations.
Introduction
The selective replacement of a hydrogen for a fluorine atom is a powerful method for altering the properties of organic compounds, and a strategy that is widely used in bio-organic and medicinal chemistry.1–3 Generally, this replacement has only a moderate steric effect, but the high electronegativity of the fluorine can have significant electronic influences.4 The difluoromethylene (CF2) group has received considerably less attention as a group for modifying the properties of organic molecules relative to the –F and –CF3 groups.5 However, we have recently reported6–8 that incorporation of a CF2 group can influence the conformation of organic molecules. In particular, cyclododecanes 1–3 were prepared with varied locations of two difluoromethylene groups (Fig. 1). X-ray crystal structure analysis showed that the CF2 groups only occupied corner positions. In the case of cyclododecanes 1 and 2, this stabilises the square like conformation. However, in the case of cyclododecane 3, incorporation of the difluoromethylene group 1,6 to each other results in considerable distortion of the ring conformation as the CF2 group avoids an unfavourable edge location.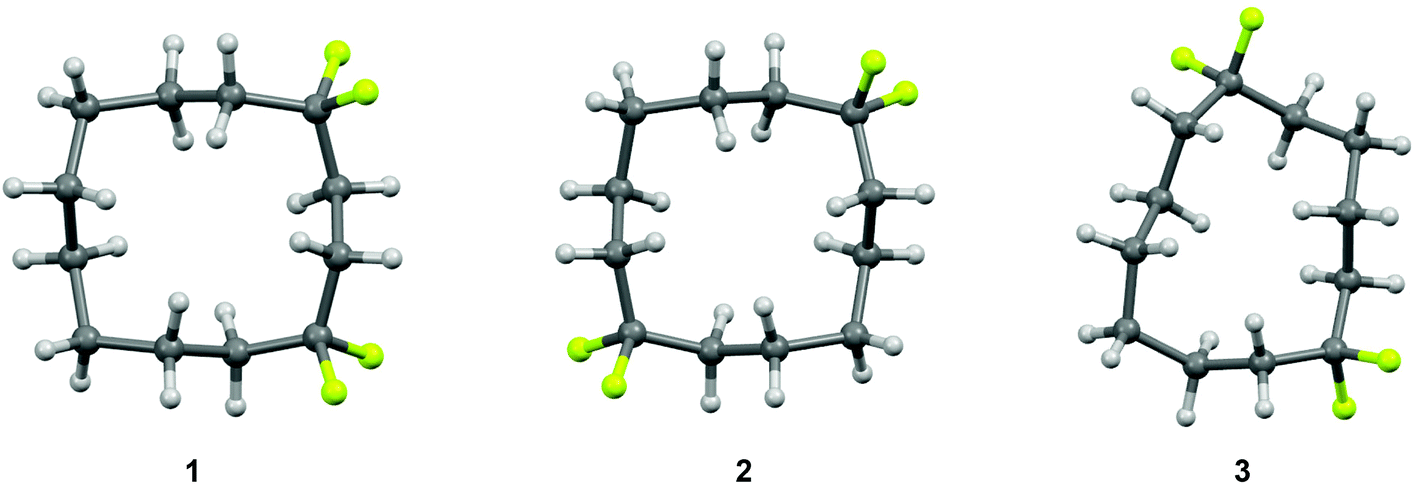 | ||
| Fig. 1 X-ray crystal structures of 1,1,4,4-(1), 1,1,7,7-(2) and 1,1,6,6-(3) tetrafluorocyclododecanes.6a The CF2 groups conspicuously adopt corner locations either reinforcing square like structures for 1 and 2 or a inducing a distorted ring for structure 3. | ||
Natural musks are large conformationally flexible macrocyclic lactones and we were interested to evaluate if the CF2 group might limit this conformational flexibility as a tool for exploring preferred conformations responsible for scent. (12R)-(+)-12-Methyl-13-tridecanolide 4 is a natural constituent of Angelica root oil, Angelica archangelica L.9 The (R)-enantiomer is described as having “a clean, crystal-clear musk top note with strong, elegant woody accents”.10,11 Macrocycle 4 possesses a high degree of conformational flexibility and could be expected to adopt a wide variety of conformations, not all of which are likely to be active musk odorants. Several syntheses of (R)-4 have been reported,10,12–14 but little work has been carried out on determining the active conformations of the musk lactone that contribute to its scent. One such study by Kraft and Cadalbert15 calculated the six lowest-energy conformations of (R)-4 (Fig. 3), all of which were within 0.9 kcal mol−1 of each other. In order to impart conformational restriction, they synthesised three musk lactone derivatives 5–7 (Fig. 2), featuring bridging methylene groups. Musk derivatives 5–7 were weaker odorants than the parent (R)-4, as the authors predicted, but did modify the olfactory properties resulting in slightly differing smell descriptions.
 | ||
| Fig. 2 (12R)-(+)-12-Methyl-13-tridecanolide 4 and bridged musk lactones 5–7. | ||
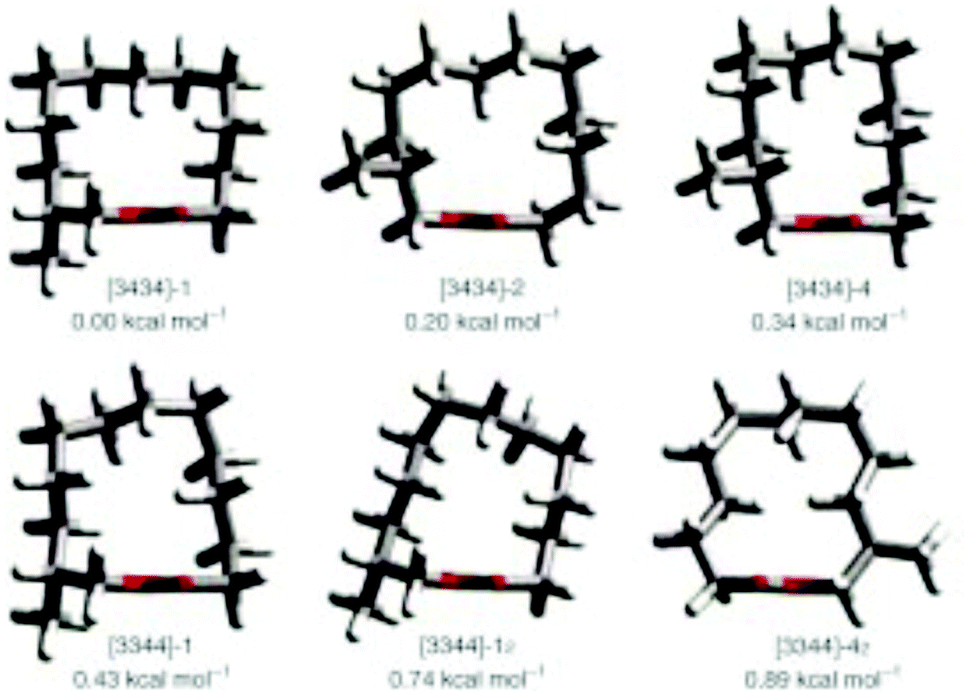 | ||
| Fig. 3 The six lowest-energy conformations of musk lactone (R)-4 as reported by Kraft and Cadalbert (MM3 force field).15 | ||
As a continuation of the observation that difluoromethylene groups can influence macrocyclic ring conformation, we decided to investigate the synthesis of CF2 analogues of (12R)-(+)-12-methyl-13-tridecanolide 4. The predicted lower energy conformers shown in Fig. 3 are largely rectangular structures with linear edges of four or five methylene groups. It was envisaged that strategic incorporation of CF2 groups into the macrolide would allow different ground state conformations to be more highly populated, and then different conformations could be assessed for their musk/odour properties.
In this respect lactones 8, 9 and 10 (Fig. 4) were selected for synthesis. In each case we anticipated that they would adopt different Kraft and Cadalbert conformations (Fig. 3). Conformation [3434]-4 has a four carbon edge running along the top of the ring which should be reinforced by the 1,4-CF2 groups in structure 8. Conformation [3434]-1, the lowest energy Kraft and Cadalbert structure should be reproduced by lactone 9 if the hypothesis holds. Finally the Kraft and Cadalbert structure [3344]-1, which is 0.74 kcal mol−1 above the ground state structure, is an asymmetric rectangular structure, with a methyl at a corner location, a conformation which should be preferred by lactone 10, which will locate a corner at C-8 of the lactone ring.
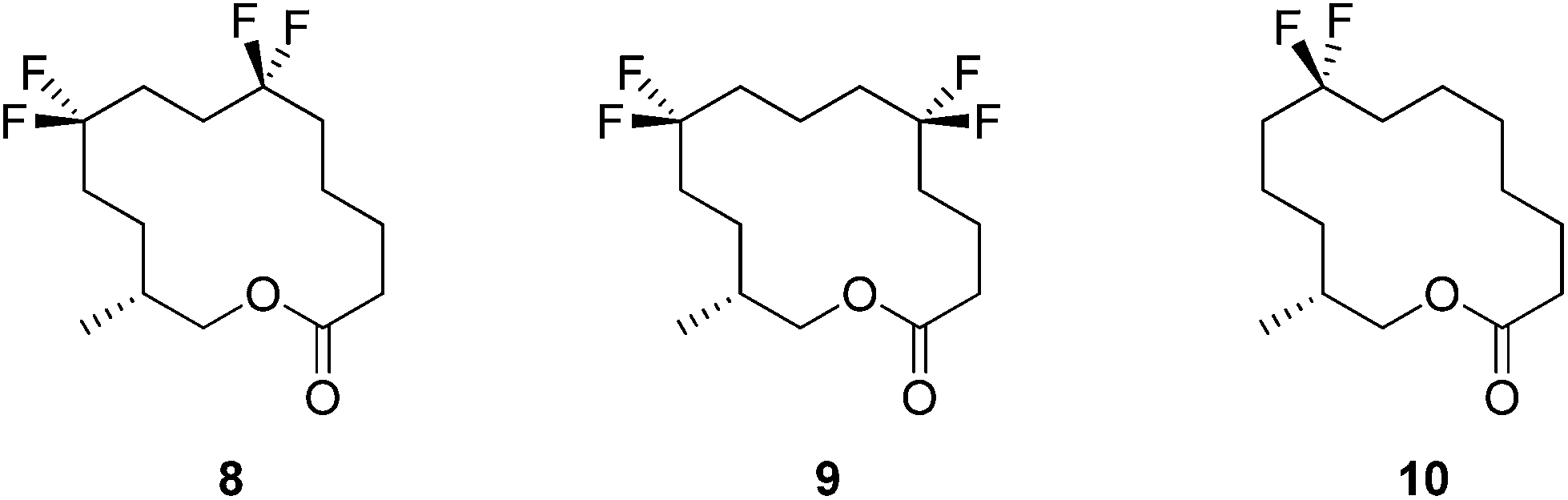 | ||
| Fig. 4 Target musk lactone analogues 8–10 incorporating CF2 moieties. | ||
Results and discussion
Synthesis
In order to prepare the CF2 containing musk lactones 8–10, a synthesis was devised that would allow their assembly from a common enantiomerically pure chiral fragment (Scheme 1). (R)-Olefin 11 was chosen as it could be progressed in three separate directions to aldehyde 12, epoxide 13 and aldehyde 14, respectively. It was envisaged that intermediates 12–14 could then be progressed to the individual musk lactones 8–10. A key strategy for incorporating the CF2 groups would exploit Grée's method of diethyaminosulfur trifluoride (DAST) treatment of propargylic ketones to generate propargylic difluoromethylene.16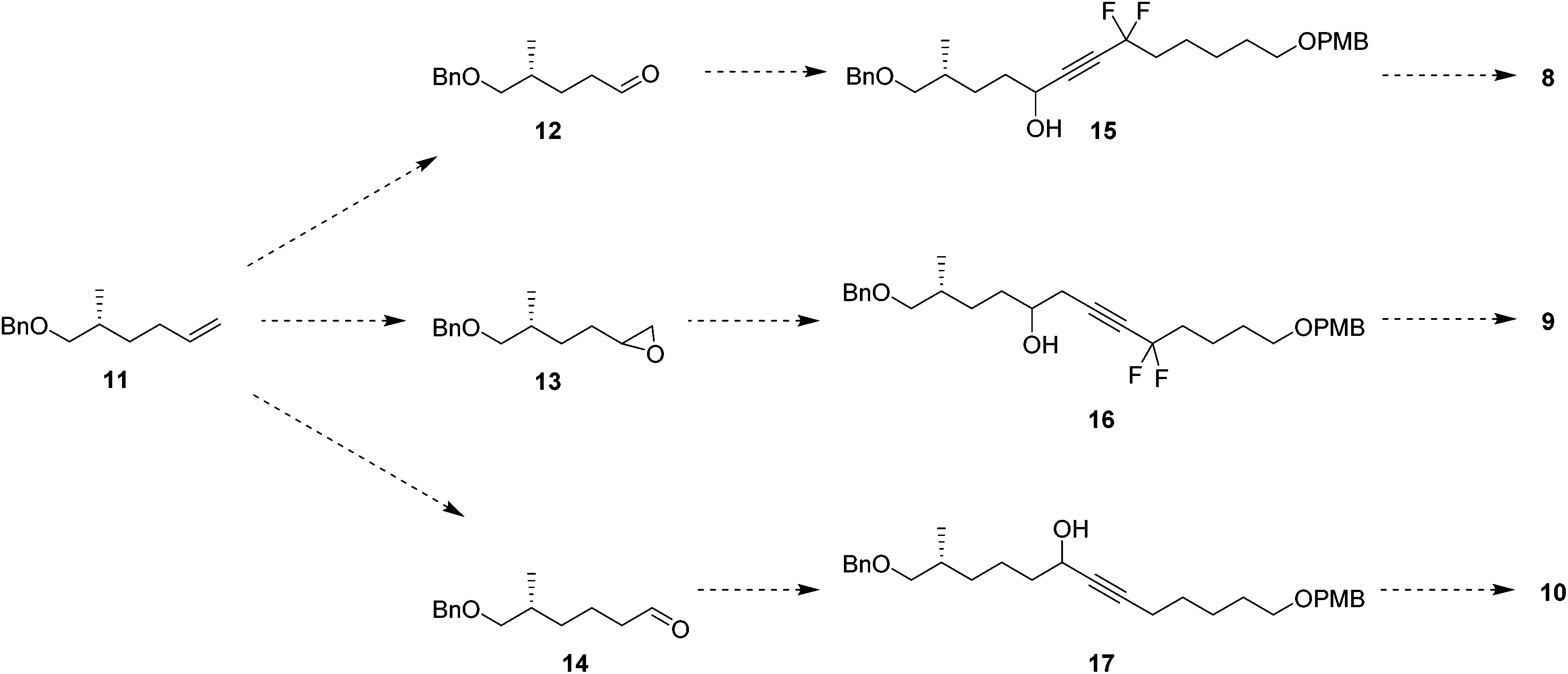 | ||
| Scheme 1 Planned routes to target lactones 8–10 from (R)-olefin 11. | ||
(R)-Olefin 11 was prepared as shown in Scheme 2. Firstly hexenoic acid 19 was obtained from cyclohexanone 18 by the method reported by Ogibin et al.17 Carboxylic acid 19 was then activated as a mixed anhydride and coupled18 to (R)-oxazolidinone 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19 to generate 21. Asymmetric methylation with iodomethane gave 22 as a single diastereoisomer and the stereochemistry was established by X-ray crystallographic analysis as shown in Fig. 5. Oxazolidinone 22 was then progressed to aldehyde 12 by a sequence of lithium aluminium hydride (LiAlH4) reduction to give alcohol 23, benzyl ether formation20 to give (R)-alkene 11 and then ozonolysis.
19 to generate 21. Asymmetric methylation with iodomethane gave 22 as a single diastereoisomer and the stereochemistry was established by X-ray crystallographic analysis as shown in Fig. 5. Oxazolidinone 22 was then progressed to aldehyde 12 by a sequence of lithium aluminium hydride (LiAlH4) reduction to give alcohol 23, benzyl ether formation20 to give (R)-alkene 11 and then ozonolysis.
 | ||
| Scheme 2 Synthesis of chiral fragments 11 and 12. Reagents and conditions: (a) (i) H2O2, MeOH, 30 min. (ii) FeSO4·7H2O, CuSO4·5H2O, H2O, 20%; (b) (i) Oxazolidinone 20, nBuLi, THF, −78 °C, 30 min. (ii) Pivaloyl chloride, NEt3, THF, 0 °C, 30 min, r.t., 1.5 h, 93%; (c) NaHMDS, MeI, THF, −78 °C, 3 h, 92%, dr >98:2; (d) LiAlH4, Et2O, 0 °C, 2 h, 66%; (e) NaH, BnBr, DME, r.t.-reflux, 20 h, 94%; (f) (i) O3, DCM, −78 °C. (ii) PPh3, −78 °C–r.t., 55%. | ||
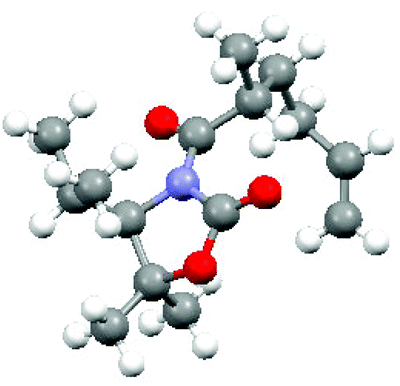 | ||
| Fig. 5 X-ray crystal structure of compound 22. | ||
Attention turned to the synthesis of the propargylic difluoromethylene fragments 28a and 28b in Scheme 3. Diols 24a and 24b were mono-protected21 as their 4-methoxybenzyl ethers to give alcohol 25a and 25b by modification of a previous procedure.5 Oxidation to the corresponding aldehydes,22 followed by reaction with ethynylmagnesium bromide gave propargylic alcohols 26a and 26b. Dess–Martin periodinane (DMP)23 oxidation gave propargyl ketones 27, which were treated with DAST in methodology developed by Grée16,24,25 to give the required difluoromethylene acetylenes 28a and 28b.
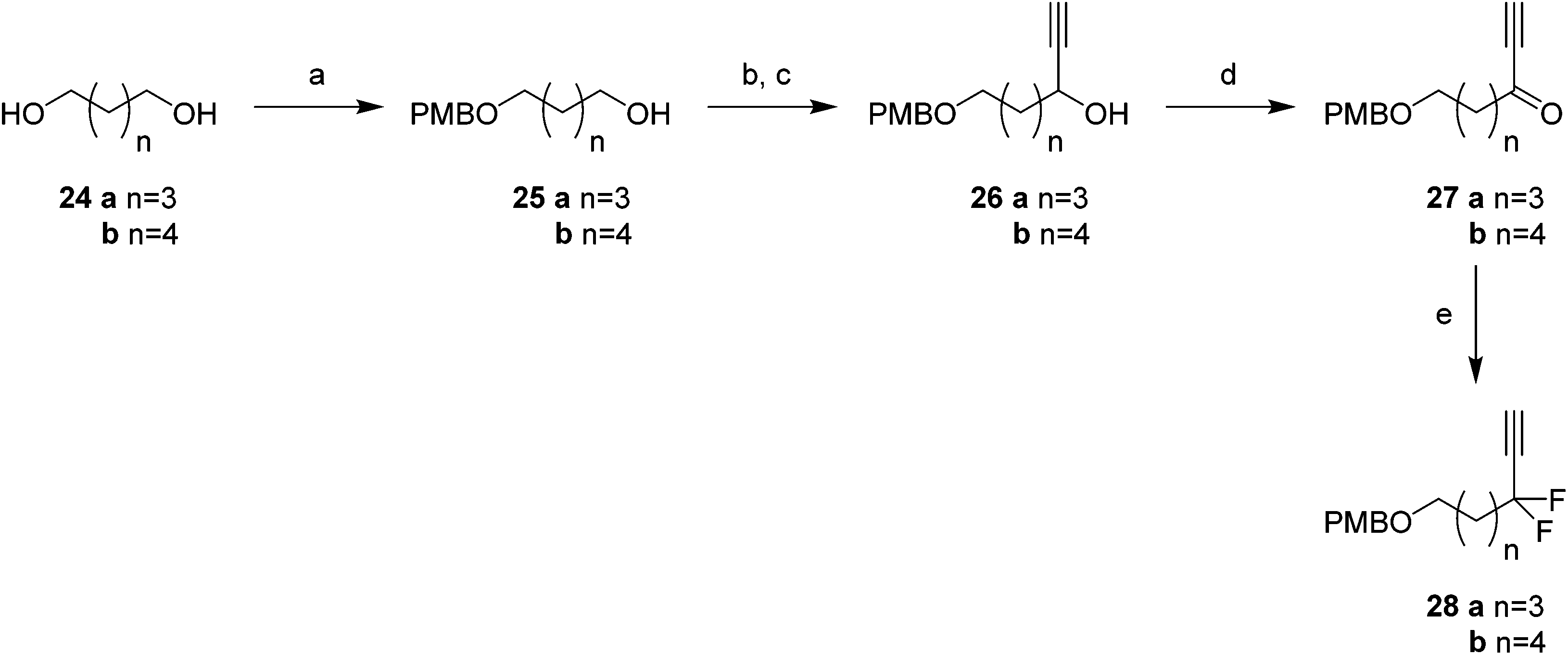 | ||
| Scheme 3 Synthesis of difluoro-alkyne fragments 28a and 28b. Reagents and conditions: (a) NaH, PMB–Cl, TBAI, THF, 0 °C-reflux, 19 h, 25a 85%, 25b 55%; (b) 25a: DMP, DCM, r.t., 18 h, 100%; 25b: oxalyl chloride, DMSO, NEt3, DCM, −78 °C–0 °C, 3 h, 100%; (c) ethynylmagnesium bromide, THF, −78 °C–r.t., 18 h, 26a 71%, 26b 77%; (d) DMP, DCM, r.t., 2 h, 27a 85%, 27b 71%; (e) DAST, 50 °C, 18 h, 28a 61%, 28b 51%. | ||
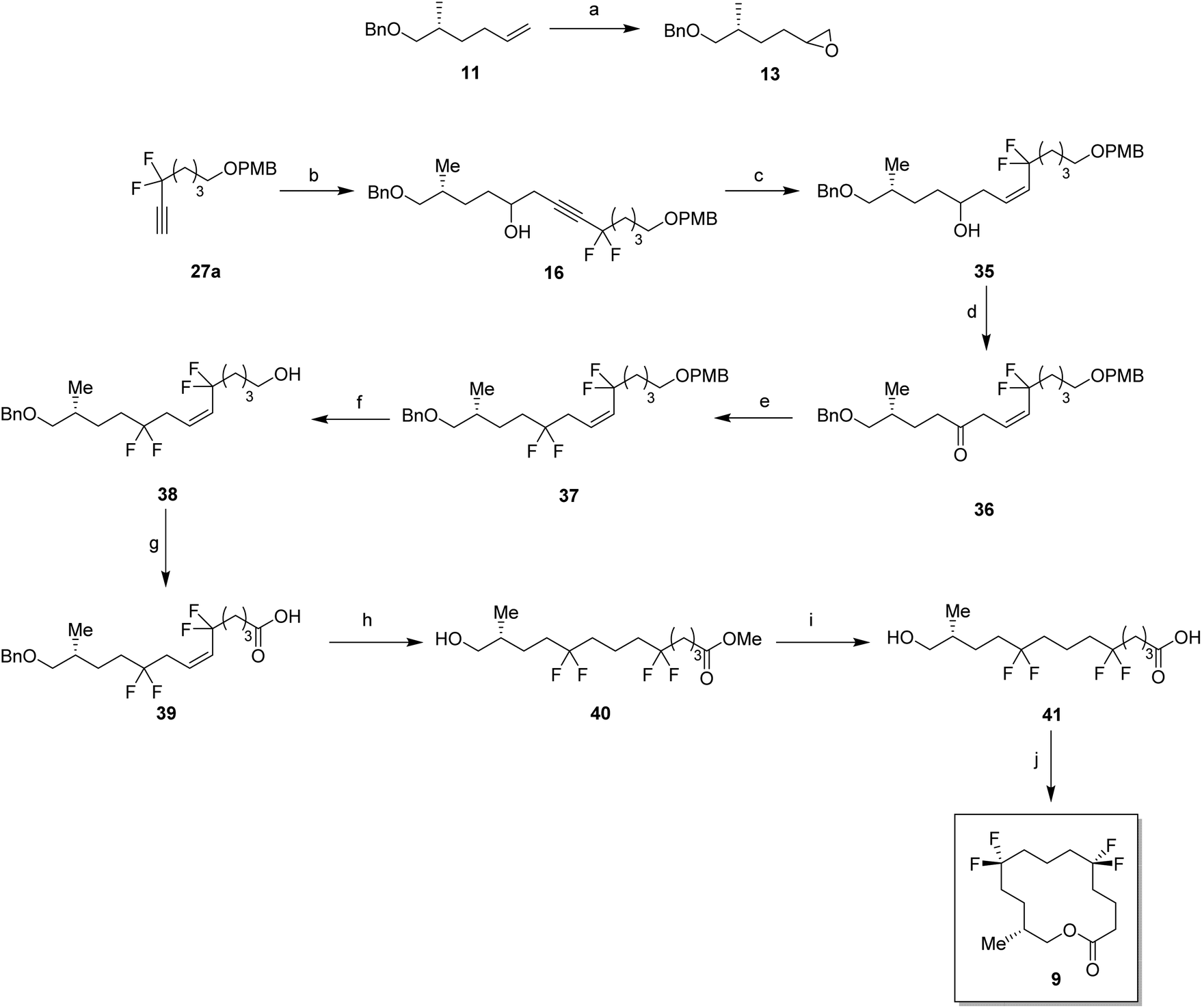
With the appropriate fragments in hand, attention turned to the synthesis of lactone 8 (Scheme 4). Lithiation of alkyne 27b with n-butyllithium, and then treatment with, aldehyde 12 gave alcohol 15 as a 1:1 mixture of diastereoisomers. After oxidation,23 ketone 29 was converted to alkyne 30 with DAST. 4-Methoxybenzyl cleavage26 and then oxidation with bis(acetoxy)iodobenzene (BAIB)/TEMPO27 generated carboxylic acid 32. Benzyl deprotection could not be achieved under standard conditions, proving resistant to hydrogenation in either ethyl acetate or THF. Hydrogenation in methanol was successful but ester formation occurred to give the saturated methyl ester 33. This proved satisfactory and then finally ester hydrolysis and macrolactonisation28 of seco-acid 34 gave lactone 8 as a white waxy semi crystalline solid. Lactone 8 proved amenable to X-ray crystal structure analysis and the structure is shown in Fig. 6.
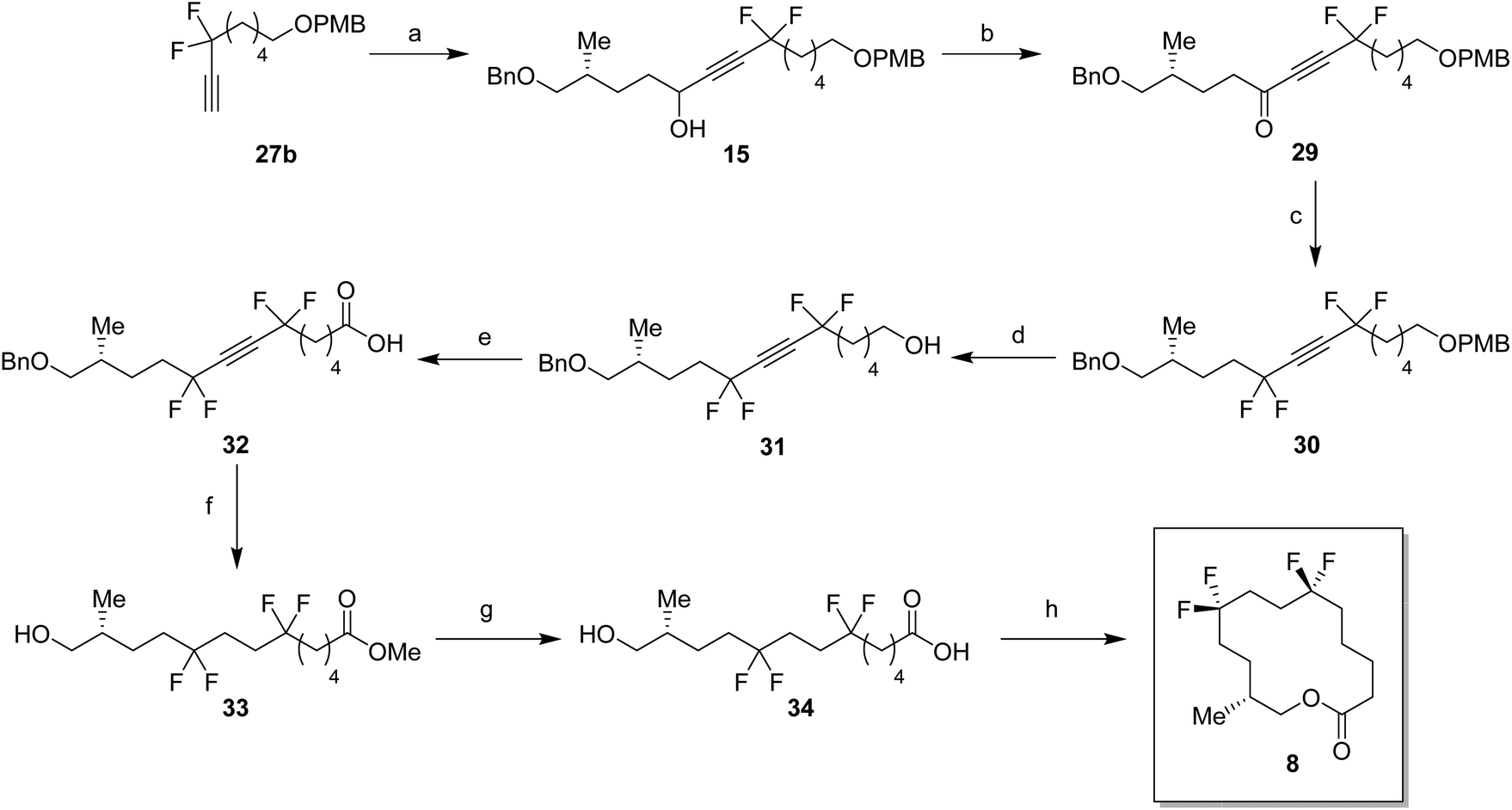 | ||
| Scheme 4 Synthesis of musk lactone 8. Reagents and conditions: (a) (i) nBuLi, THF, −78 °C, 30 min. (ii) Aldehyde 12, THF, −78 °C–r.t., 20 h, 61%; (b) DMP, DCM, r.t., 2 h, 89%; (c) DAST, 50 °C, 18 h, 71%; (d) DDQ, DCM, H2O, r.t., 2 h, 78%; (e) BAIB, TEMPO, CH3CN, H2O, r.t., 7 h, 58%; (f) palladium on carbon (10 wt%), H2, MeOH, r.t., 18 h, 65%; (g) LiOH, THF, H2O, r.t., 3 h, 94%; (h) 2,4,6-trichlorobenzoyl chloride, NEt3, THF, r.t., 2.5 h. (ii) 4-DMAP, toluene, r.t., 3.5 h, 54%. | ||
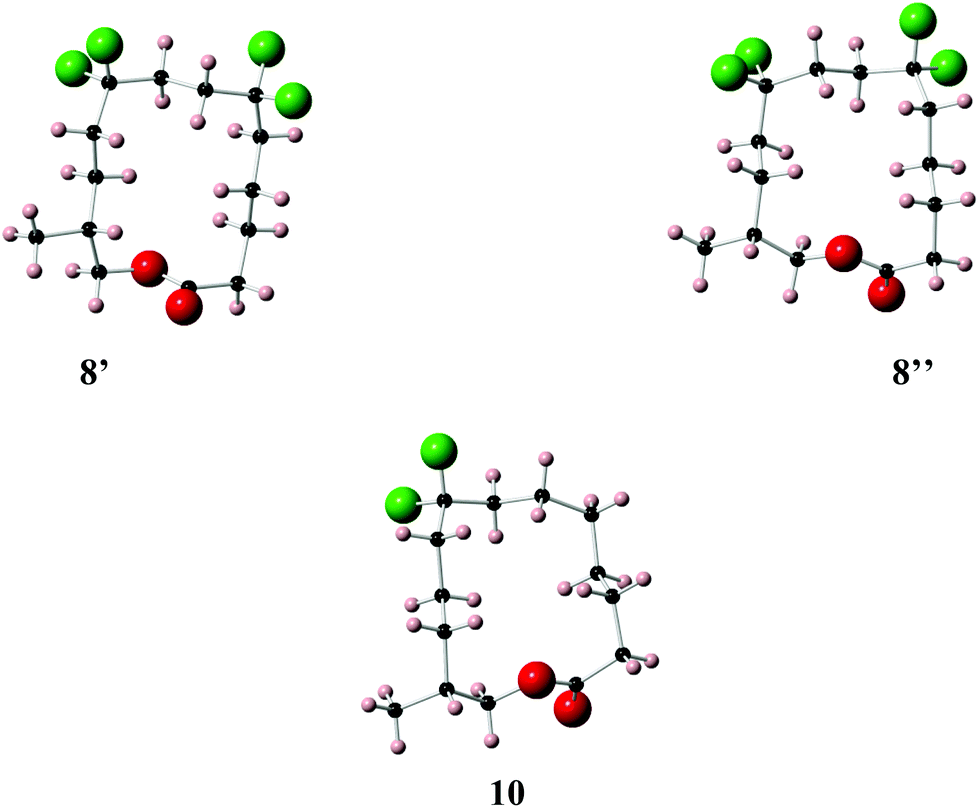 | ||
| Fig. 6 X-ray crystal structure of musk lactones 8 (two conformers 8′ and 8′′ are obvious in the unit cell) and 10. | ||
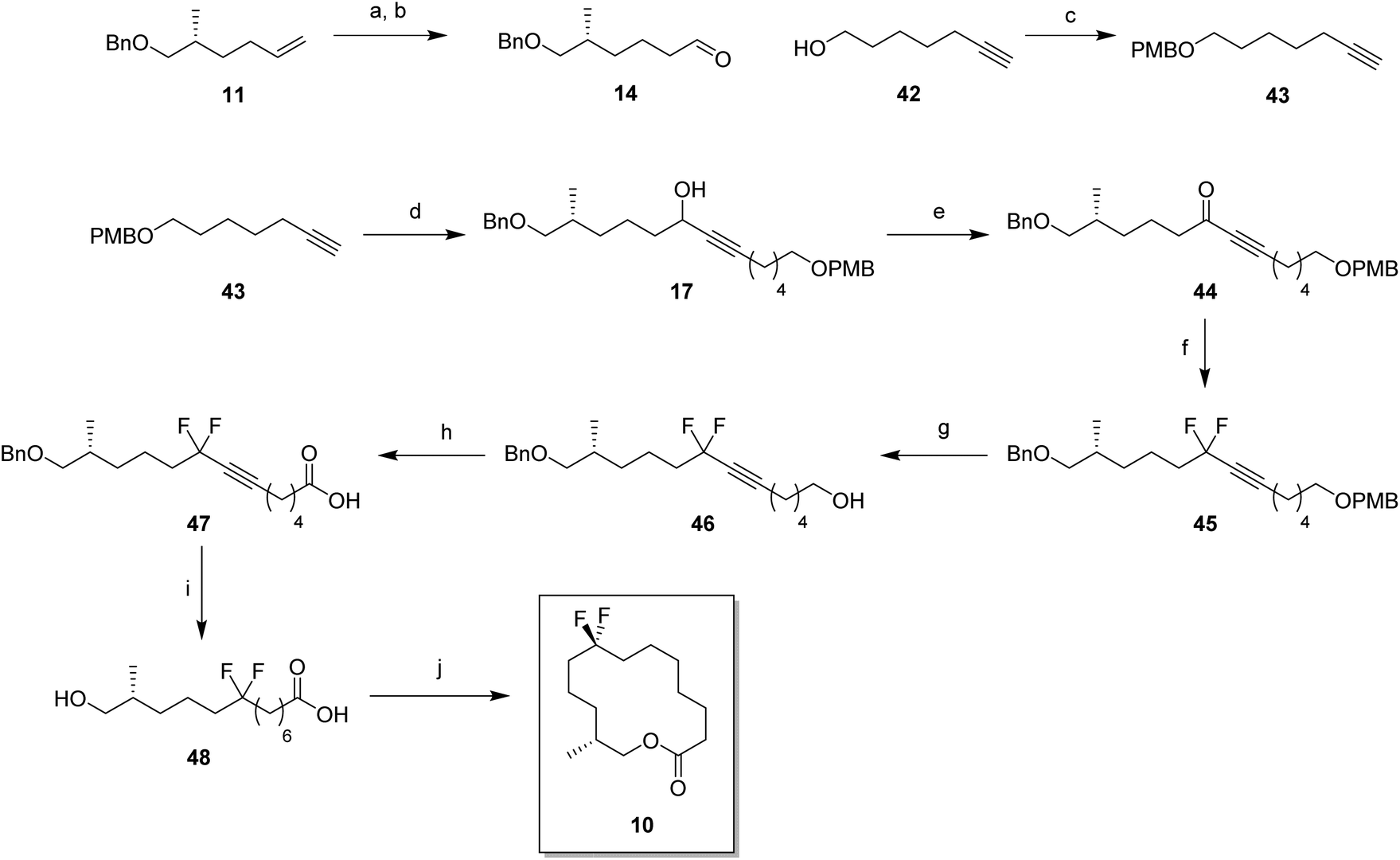
For the synthesis of lactone 9, alkene 11 was oxidised with meta-perbenzoic acid (mCPBA) to give epoxide 13 as a 1:1 mixture of diastereoisomers. Ring opening addition of lithiated alkyne 28a29 to epoxide 13 gave alcohol 16 in 60% yield. The acetylene could be selectively reduced to the (Z)-alkene with a poisoned palladium catayst,30 and this gave alkene 35 in 82% yield. This partial reduction was necessary as fluorinations of the ketone from the oxidised acetylene 16 lead to decomposition in reactions with DAST. From here, a similar sequence of reactions as describe for the conversion of 14 to 8 was used to convert alcohol 35 to lactone 9. This lactone was an oil at room temperature and it was not possible to obtain an X-ray structure.
For the synthesis of lactone 10, (R)-olefin 11 was again used as a starting material but this time converted to aldehyde 14. This was achieved by hydroboration,31 followed by oxidation with DMP (Scheme 6). The terminal acetylene 43 was prepared by protection of 6-heptyn-1-ol 42. A coupling of alkyne 43 and aldehyde 14 gave alcohol 16 in good yield. Following a similar sequence to the conversions in Scheme 4 and Scheme 5, alcohol 17 was converted to seco-acid 48, without having to progress through the corresponding methyl ester. Macrolactonisation28 of seco-acid 48 gave lactone 10 as a white solid. This lactone was also a wax but a suitable crystal was grown which proved amenable to X-ray structure analysis, and the resultant structure is shown in Fig. 6.
 | ||
| Scheme 5 Synthesis of musk lactone 9. Reagents and conditions: (a) mCPBA, DCM, r.t., 18 h, 55%; (b) (i) nBuLi, THF, −78 °C, 15 min. (ii) BF3·OEt2, 15 min. (iii) Epoxide 13, THF, −78 °C–r.t., 20 h, 60%; (c) palladium (5% on barium sulfate), quinoline, pyridine, H2 (1 atm), 22 h, 82% (d) DMP, DCM, r.t., 2 h, 57%; (e) DAST, 50 °C, 18 h, 27%; (f) DDQ, DCM, H2O, r.t., 2 h, 70%; (g) BAIB, TEMPO, CH3CN, H2O, r.t., 18 h, 49%; (h) palladium on carbon (10 wt%), H2, MeOH, r.t., 18 h, 59%; (i) LiOH, THF, H2O, r.t., 3 h, 70%; (j) 2,4,6-Trichlorobenzoyl chloride, NEt3, THF, r.t., 2.5 h. (ii) 4-DMAP, toluene, r.t., 3.5 h, 52%. | ||
 | ||
| Scheme 6 Synthesis of musk lactone 10. Reagents and conditions: (a) (i) 9-BBN dimer, THF, 0 °C–r.t., 23 h. (ii) EtOH, NaOH (2 N), H2O2, 0 °C–r.t., 3 h, 59%; (b) DMP, DCM, r.t., 1 h, 100%; (c) NaH, PMBCl, DMF, 0 °C–r.t., 18.5 h, 87%; (d) (i) nBuLi, THF, −78 °C, 30 min. (ii) Aldehyde 14, THF, −78 °C–r.t., 20 h, 80%; (e) DMP, DCM, r.t., 2 h, 61%; (f) DAST, 50 °C, 18 h, 62%; (g) DDQ, DCM, H2O, r.t., 2 h, 88%; (h) BAIB, TEMPO, CH3CN, H2O, r.t., 7 h, 48%; (i) palladium hydroxide on carbon (20 wt%), H2, THF, r.t., 18 h, 46%; (j) (i) 2,4,6-trichlorobenzoyl chloride, NEt3, THF, r.t., 2.5 h. (ii) 4-DMAP, toluene, r.t., 3.5 h, 67%. | ||
X-ray structures
Lactone 8 has two conformers 8′ and 8′′, which are present in a 1:1 ratio within the unit cell. It is immediately apparent that the CF2 groups locate at corner positions, thus the anticipated role of the CF2 group is as predicted. Also for X-ray structures 8′ and 10 adopt the anticipated Kraft and Cadalbert conformations (Fig. 3). Structure 8′ corresponds to the [3434]-4 structure which has the methyl group at an edge position, and structure 8′′ corresponds to the [3344]-1 structure with the methyl group at a corner. Presumably they are close in energy. For lactone 10 the predicted Kraft and Cadalbert [3344]-1 structure is also observed. Lactone 9 is a liquid at room temperature and we could not obtain any X-ray structural data, although we predict that the Kraft and Cadalbert structure [3434]-1, will be relevant (see theory study below).
Lactones 8–10 all smell rather similar and all retain a pleasant muskoid odour, with the additional descriptors, ‘weak hint of green, powdery’.32 A distinct smell differential between the compounds proved difficult to discern even for compound 8 despite its having four fluorine atoms. So we could not draw any significant conclusions from the smell analysis, except to conclude that they retained a pleasant odour.
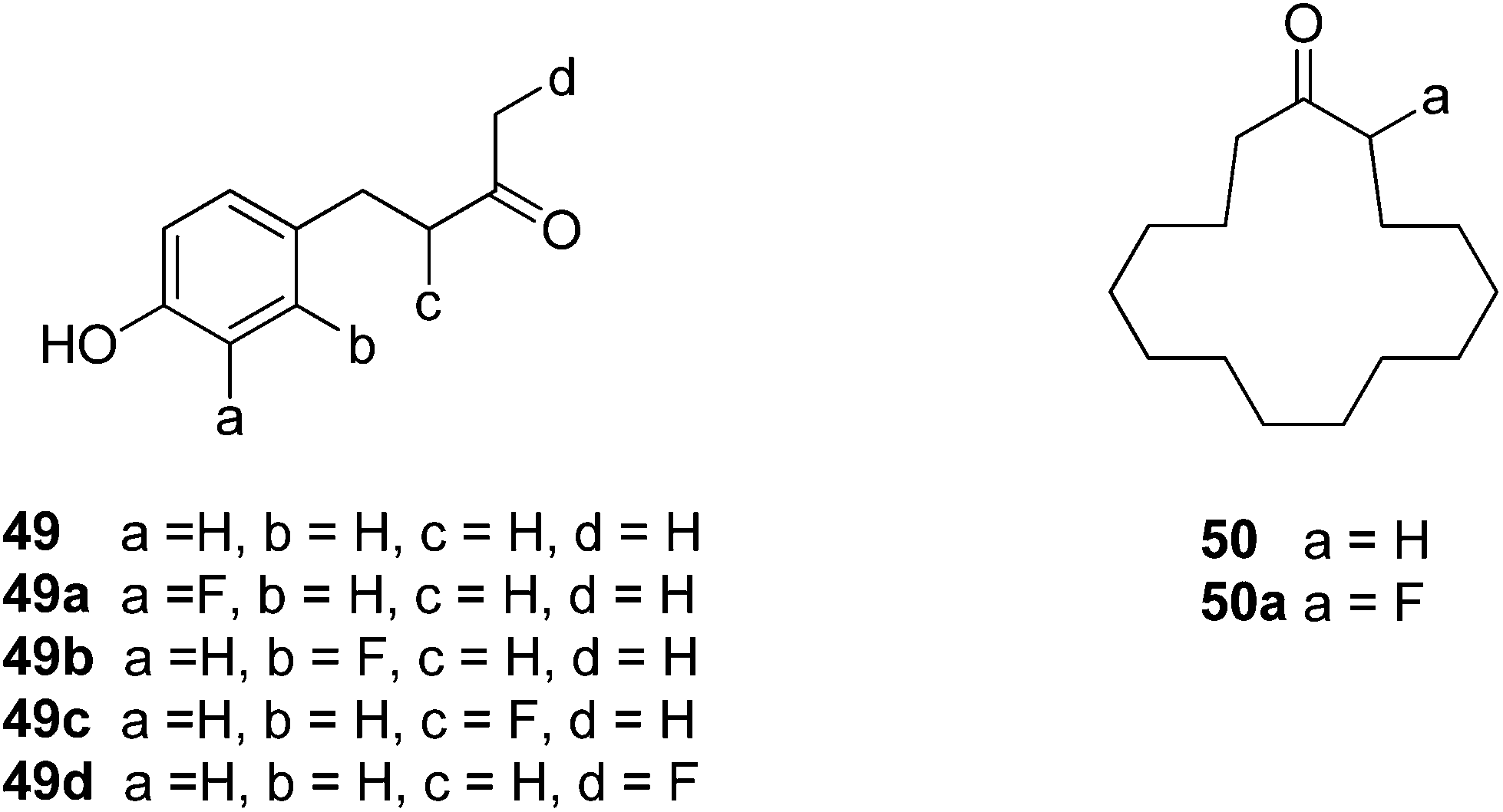
Michel and Schlosser report33,34 the only other examples that we are aware of where fluorine substitutions for hydrogen have been used as a probe to examine fragrance. They explored several flavour and fragrance compounds and in general found that there was no significant change in taste or smell when a hydrogen was replaced by a fluorine. For example, for the raspberry ketone 49 a systematic fluorine scan generated analogues 49a–d, however this resulted in little change to the smell, whereas the same systematic replacement by a methyl has a much more profound effect on the organoleptic properties.33 They concluded that the fluorine did not induce a significant conformational or steric/shape change in this case, whereas the methyl analogues do. In the case of musk lactone 50 and analogue 50a, the fluorine located alpha to the ketone made a significant difference in the smell. This was attributed to a conformational change induced by the fluorine, although no conformational analysis was carried out.34
Computational studies
Theory calculations have been carried out on the three lactones 8–10 in order to explore their conformations further. Minimum energy conformers of the lactones in the gas phase were located through a Monte Carlo conformational search at the MMFF level with the Spartan 14 program,35 and using an initial temperature of 5000 K in the simulated-annealing algorithm. This procedure found 9650 conformers for lactone 8, 9714 conformers for lactone 9 and 7948 conformers for lactone 10. The 30 lowest energy minima in the gas phase for each compound were optimised at the B3LYP-D3/6-311+G** level and frequency calculations were carried out at the same level by using the Gaussian 09 program, Revision D.01.36 No negative harmonic vibrational frequencies were observed and, hence the conformers are true energy minima. The same frequency calculations were used to obtain thermodynamic corrections affording enthalpies and Gibbs free energies at ambient, standard pressure and temperature for all conformers of 8–10 in the gas phase.The lowest energy conformers of lactones 8–10 in the gas phase, calculated at the B3LYP-D3/6-311+G** level corrected by Gibbs free energies, are shown in Fig. 7. The 30 lowest energy conformers for each lactone are detailed in the ESI (see Fig. S1–S3 and Tables S1–S3†). The lowest energy conformers for 8 all have square/rectangular ring shapes and all but conformer 8-1 have their CF2 groups located at corner positions. Notably, the calculated global minimum conformer in the gas phase matches the experimental X-ray structure 8′ (Fig. 6) and was the anticipated Kraft and Cadalbert structure at the outset. Conformer 8–27, which emerges as a higher energy conformer (1.35 kcal mol−1) in this study, matches the experimental X-ray structure 8′′. That only two calculated conformers, 8–14 and 8–27, were observed experimentally can be attributed to order and packing in the crystal, where a range of conformers is not expected. Similarly lactone 10 emerges from this study with many low energy conformers, all with square/rectangular ring geometries. Most of them have the CF2 groups locating at corner locations. Indeed, both the calculated global minimum 10-5 (0.00 kcal mol−1) and the second lowest energy conformer 10-7 have squared carbon ring geometries with CF2 groups at corners (Fig. 7). Conformer 10-7 has a geometry which matches the observed X-ray structure of 10. Taking structures 8 and 10 together, the outcomes suggest that the computational analysis was able to identify the experimental structures, as low energy stable conformers. These outcomes give some confidence that the calculated lowest energy geometries are relevant experimentally. For lactone 9 we have to rely on theory only. It emerges as a very flexible structure, with many calculated low energy conformers, however it is noteworthy that the lowest energy conformer, 9-6 is almost identical to the anticipated Kraft and Cadalbert [3434]-1 structure for musk lactone. Thus the predictive power of the CF2/corner hypothesis holds well.
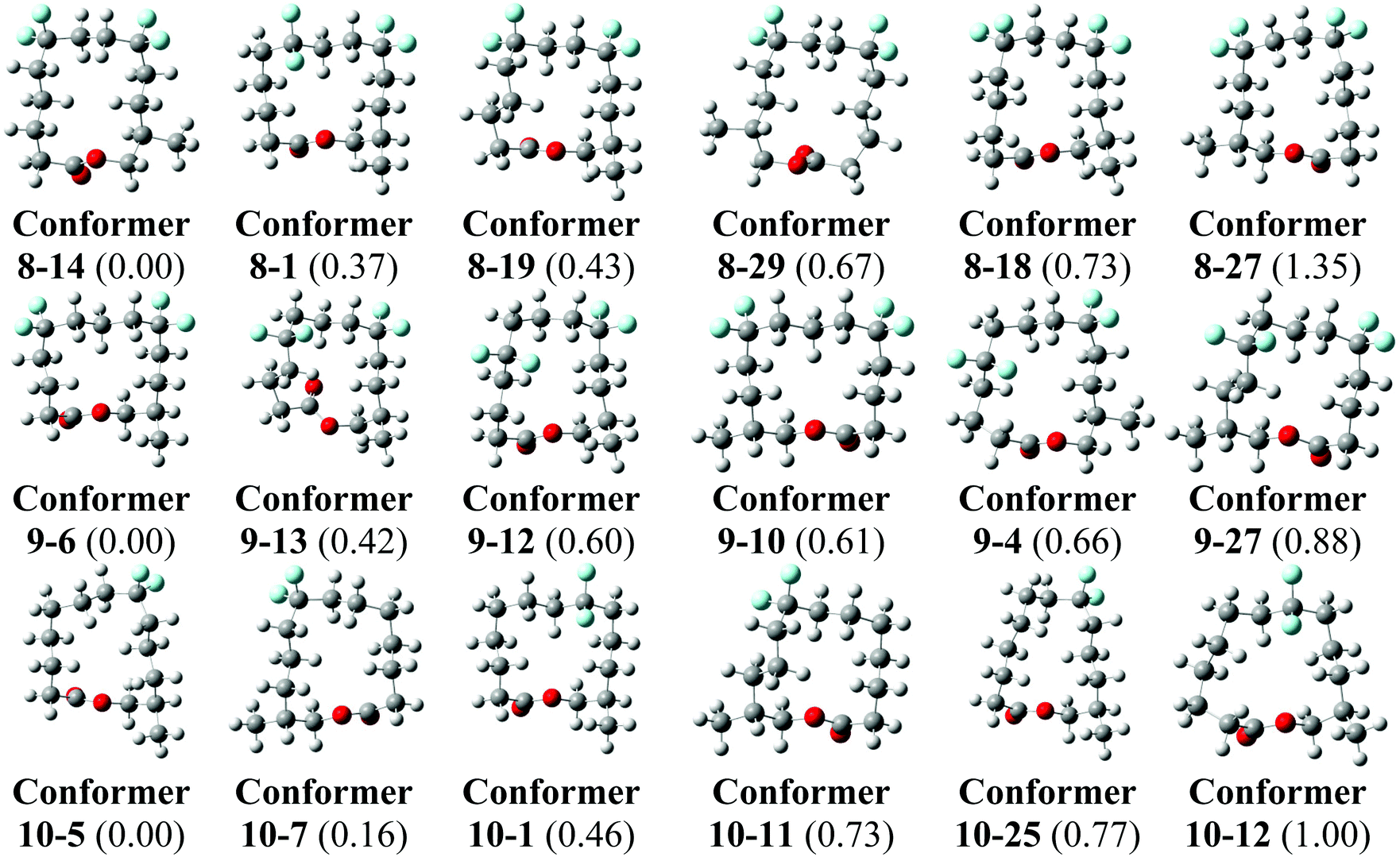 | ||
| Fig. 7 The calculated lowest energy geometries for 8, 9 and 10. Relative Gibbs free energies calculated at the B3LYP-D3/6-311+G** level in kcal mol−1 are given between parenthesis. For full details see ESI.† | ||
In summary we have carried out a synthesis and structural study on three CF2 containing analogues of musk lactone 4. The CF2 group was explored as a design feature to preferentially locate at corner positions of the ring. X-ray structures of lactones 8 and 10 showed this to be the case. In addition the conformations of the lactones 8–10 were explored by detailed computational analysis and this also revealed lowest energy conformers (from many) that had CF2 at the corner positions. The calculated lowest energy structures matched the X-ray structures for 8′ and 10. All three of the fluorinated lactones retained a pleasant odour, and it proved difficult to assign a particular analogue to a stronger fragrance. However, even with the CF2 constraints, it is clear from the conformational analysis that each lactone can access a range of low energy rectangular structures. These structures typically have the lactone moiety in the middle of a flat edge and with the methyl group at the corner of that edge, an arrangement which is presumably consistent with eliciting a fragrance response.
Acknowledgements
We thank EPSRC for a grant and we thank the EPSRC National Mass Spectrometry Service, Swansea and Mrs Caroline Horsburgh (University of St Andrews) for mass spectrometery analysis. CNvH also thanks the Fluorine Division of the American Chemical Society for a Moissan Summer Undergraduate Fellowship. RAC thank FAPESP for a fellowship (#2015/00975-7) and MB thanks EaStCHEM and the University of St Andrews for support access to a computing facility managed by Dr H. Fruchtl. We thank Prof. Paul Davey and Dominique Lelievre of Givaudan, Schweiz AG, Dübendorf, Switzerland, for faciliting the olfactive assessments.References
- K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- W. Liu, W. Huang, M.-J. Cheng, R. J. Neilson, W. A. Goddard III and J. T. Groves, Science, 2012, 337, 1322–1325 CrossRef CAS PubMed.
- S. Purser, P. R. Moore, S. Swallow and C. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- F. R. Leroux, B. Manteau, J.-P. Vors and S. Pazenok, Beilstein J. Org. Chem., 2008, 4, 13 Search PubMed.
- (a) M. Skibinski, Y. Wang, A. M. Z. Slawin, T. Lebl, P. Kirsch and D. O'Hagan, Angew. Chem., Int. Ed., 2011, 50, 10581–10584 CrossRef CAS PubMed; (b) M. Skibinski, C. A. Urbina-Blanco, A. M. Z. Slawin, S. P. Nolan and D. O'Hagan, Org. Biol. Chem., 2013, 11, 8209–8213 RSC.
- D. O'Hagan, Y. Wang, M. Skibinski and A. M. Z. Slawin, Pure Appl. Chem., 2012, 84, 1587–1595 CrossRef.
- C. A. Urbina-Blanco, M. Skibinski, D. O'Hagan and S. P. Nolan, Chem. Commun., 2013, 49, 7201–7203 RSC.
- K. Schultz and P. Kraft, J. Essent. Oil Res., 1997, 509–514 CrossRef CAS.
- P. Kraft and W. Tochtermann, Leibigs Ann. Chem., 1994, 1161–1164 CrossRef CAS.
- E. Brenna, C. Fuganti and S. Serra, Tetrahedron: Asymmetry, 2003, 14, 1–42 CrossRef CAS.
- A. Fürstner and K. Langemann, J. Org. Chem., 1996, 61, 3942–3943 CrossRef.
- A. Fürstner and K. Langemann, Synthesis, 1997, 792–803 CrossRef.
- P. Scafato, A. Larocca and C. Rosini, Tetrahedron: Asymmetry, 2006, 17, 2511–2515 CrossRef CAS.
- P. Kraft and R. Cadalbert, Chem. – Eur. J., 2001, 7, 3254–3262 CrossRef CAS.
- M. Prakesch, E. Kerouredan, D. Grée, R. Grée, J. DeChancie and K. N. Houk, J. Fluorine Chem., 2004, 125, 537–541 CrossRef CAS.
- Y. N. Ogibin, E. K. Starostin, A. V. Aleksandrov, K. K. Piunitsky and G. I. Nikishin, Synthesis, 1994, 901–903 CrossRef CAS.
- J. S. Yadav and N. Rami Reddy, Tetrahedron, 2010, 66, 3265–3274 CrossRef CAS.
- S. D. Bull, S. G. Davies, S. Jones and H. J. Sanganee, J. Chem. Soc., Perkin Trans. 1, 1999, 4, 387–398 RSC.
- D. F. Taber, R. P. Meagley and D. J. Doren, J. Org. Chem., 1996, 61, 5723–5728 CrossRef CAS.
- Using a modification of the procedure reported by: R. S. Narayan and B. Borhan, J. Org. Chem., 2006, 71, 1416–1429 CrossRef CAS.
- Swern oxidation in the case of 24a; K. Anyram and D. Swern, Tetrahedron, 1978, 34, 1651–1660 CrossRef ; oxidation with Dess–Martin Periodinane23 in the case of 24b.
- D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155–4156 CrossRef CAS.
- M. Prakesch, D. Grée, S. Chandrasekhar and R. Grée, Eur. J. Org. Chem., 2005, 1221–1232 CrossRef CAS.
- Y. Wang, R. Callejo, A. M. Z. Slawin and D. O'Hagan, Beilstein J. Org. Chem., 2014, 10, 18–25 CrossRef PubMed.
- K. Horita, T. Yoshioka, T. Tanaka, Y. Oikawa and O. Yonemitsu, Tetrahedron, 1986, 42, 3021–3028 CrossRef CAS.
- J. B. Epp and T. S. Widlanski, J. Org. Chem., 1999, 64, 293–295 CrossRef CAS.
- I. Paterson, E. A. Anderson, S. M. Dalby, J. Holim and P. Maltas, Org. Biomol. Chem., 2012, 10, 5873–5886 CAS.
- M. Yamaguchi and I. Hirao, Tetrahedron Lett., 1983, 24, 391–394 CrossRef CAS.
- W.-C. Sun, C.-S. Ng and G. D. Prestwich, J. Org. Chem., 1992, 57, 132–137 CrossRef CAS.
- S. Dickschat, E. Helmke and S. Schulz, Chem. Biodiversity, 2005, 2, 318–353 Search PubMed.
- Assessed at Givaudan Schweiz AG, Dübendorf, Switzerland.
- M. Schlosser and D. Michel, Tetrahedron, 1996, 52, 99–108 CrossRef CAS.
- D. Michel and M. Schlosser, Tetrahedron, 2000, 56, 4253–4260 CrossRef CAS.
- Y. Shao, L. F. Molnar, Y. Jung, J. Kussmann, C. Ochsenfeld, S. T. Brown, A. T. B. Gilbert, L. V. Slipchenko, S. V. Levchenko, D. P. O'Neill, R. A. DiStasio Jr., R. C. Lochan, T. Wang, G. J. O. Beran, N. A. Besley, J. M. Herbert, C. Y. Lin, T. Van Voorhis, S. H. Chien, A. Sodt, R. P. Steele, V. A. Rassolov, P. E. Maslen, P. P. Korambath, R. D. Adamson, B. Austin, J. Baker, E. F. C. Byrd, H. Dachsel, R. J. Doerksen, A. Dreuw, B. D. Dunietz, A. D. Dutoi, T. R. Furlani, S. R. Gwaltney, A. Heyden, S. Hirata, C.-P. Hsu, G. Kedziora, R. Z. Khalliulin, P. Klunzinger, A. M. Lee, M. S. Lee, W. Z. Liang, I. Lotan, N. Nair, B. Peters, E. I. Proynov, P. A. Pieniazek, Y. M. Rhee, J. Ritchie, E. Rosta, C. D. Sherrill, A. C. Simmonett, J. E. Subotnik, H. L. Woodcock III, W. Zhang, A. T. Bell, A. K. Chakraborty, D. M. Chipman, F. J. Keil, A. Warshel, W. J. Hehre, H. F. Schaefer, J. Kong, A. I. Krylov, P. M. W. Gill and M. Head-Gordon, Phys. Chem. Chem. Phys., 2006, 8, 3172–3191 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1427901–1427903. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob02023a. For access to research data and metadata see DOI: 10.17630/f6dc25c1-c693-4c59-a164-3ccce6e7ef15 |
| This journal is © The Royal Society of Chemistry 2016 |