
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWater oxidation using earth-abundant transition metal catalysts: opportunities and challenges
Markus D.
Kärkäs
* and
Björn
Åkermark
*
Department of Organic Chemistry, Arrhenius Laboratory, Stockholm University, SE-106 91 Stockholm, Sweden. E-mail: markus.karkas@su.se; bjorn.akermark@su.se
First published on 1st April 2016
Abstract
Catalysts for the oxidation of H2O are an integral component of solar energy to fuel conversion technologies. Although catalysts based on scarce and precious metals have been recognized as efficient catalysts for H2O oxidation, catalysts composed of inexpensive and earth-abundant element(s) are essential for realizing economically viable energy conversion technologies. This Perspective summarizes recent advances in the field of designing homogeneous water oxidation catalysts (WOCs) based on Mn, Fe, Co and Cu. It reviews the state of the art catalysts, provides insight into their catalytic mechanisms and discusses future challenges in designing bioinspired catalysts based on earth-abundant metals for the oxidation of H2O.
 Markus D. Kärkäs | Markus D. Kärkäs received his M.Sc. degree in Chemistry from Stockholm University in 2008, where he conducted undergraduate research in the laboratory of Prof. Åkermark. In the same year, he began his Ph.D. studies under the guidance of Professor Björn Åkermark and Professor Jan-Erling Bäckvall. In 2013, he obtained his Ph.D. degree after conducting research on the development of artificial water oxidation catalysts. He is currently a Swedish Research Council Postdoctoral Fellow at the University of Michigan in the group of Professor Corey Stephenson where he is developing methods for the valorization of lignin. |
 Björn Åkermark | Björn Åkermark is Professor Emeritus in Organic Chemistry at the Royal Institute of Technology, Stockholm, and has, since his retirement, been employed at Stockholm University as Guest Professor and research leader. He received his Ph.D. from the Royal Institute of Technology in 1967 under the direction of Professor Holger Erdtman and Professor Carl-Axel Wachtmeister and became Assistant Professor at the Royal Institute of Technology the same year. During 1967–1968 he was a visiting scholar with Professor Eugen van Tamelen at Stanford University. He then returned to the Royal Institute of Technology where he was promoted to Associate Professor in 1972 and subsequently to Professor in 1980. He has received several prestigious awards, such as the Zorn Fellowship from the Sweden–America Foundation in 1977, the Arrhenius Medal in 1978 and the Bror Holmberg Medal in 2009, both from the Swedish Chemical Society, and Ulla and Stig Holmquist's Prize in 2009 from Uppsala University. His research interest is in the field of homogeneous catalysis and has, since 1989, focused on artificial photosynthesis. |
1. Introduction
Our society's growing energy demands and the impact of rising CO2 emissions have resulted in the awareness of the necessity of developing sustainable and carbon-neutral alternatives to fossil fuels.1–3 Of the possible sustainable energy sources, only solar energy has the potential to meet the global energy demands. In this context, an attractive way of harnessing solar energy is artificial photosynthesis, where solar energy is employed to split H2O into O2 and a fuel, H2 (eqn (1)), thus storing the energy provided by the sun in chemical bonds. Therefore, methods for converting and storing solar energy are being intensively pursued:4–6| 2H2O → O2 + 2H2 | (1) |
Owing to its natural abundance, H2O would be the ideal source of reducing equivalents, i.e. protons and electrons. However, the development of efficient catalysts for the oxidation of H2O remains a bottleneck in the development of technologies for the conversion of solar energy into fuels.7 The natural photosynthesis provides a brilliant strategy for harnessing sunlight and using it to oxidize H2O to O2 in order to generate the necessary reducing equivalents for producing biomass by reducing CO2 to carbohydrates. This process, which takes place in the photosynthetic machinery of green plants and cyanobacteria, is the basis for a major part of our global energy supplies and is thus a crucial reaction in order to sustain life on earth.8,9
1.1. Photosynthesis—light-harvesting for the oxidation of H2O
The initial step in the photosynthetic machinery involves light absorption aided by photosynthetic pigments. The energy is subsequently funneled to the central P680 reaction center, resulting in charge separation to give the oxidized P680 (P680+). Reduction of P680+ occurs by abstraction of an electron from the Mn4Ca cluster located in the oxygen-evolving complex (OEC). After four consecutive electron transfers, the Mn4Ca cluster is regenerated by formally oxidizing two molecules of H2O to O2 (eqn (2)):10,11| 2H2O → O2 + 4H+ + 4e− | (2) |
X-ray crystallography has revealed that the OEC is composed of a cuboidal Mn3Ca core with a dangling Mn center (Fig. 1).12–15 The mechanism by which this Mn cluster catalyzes the oxidation of H2O is currently fairly well understood. However, some questions still remain unsolved and it is therefore still actively studied with the view that further insight into the mechanism is important for the design of artificial systems for solar energy to fuel conversion.16–22
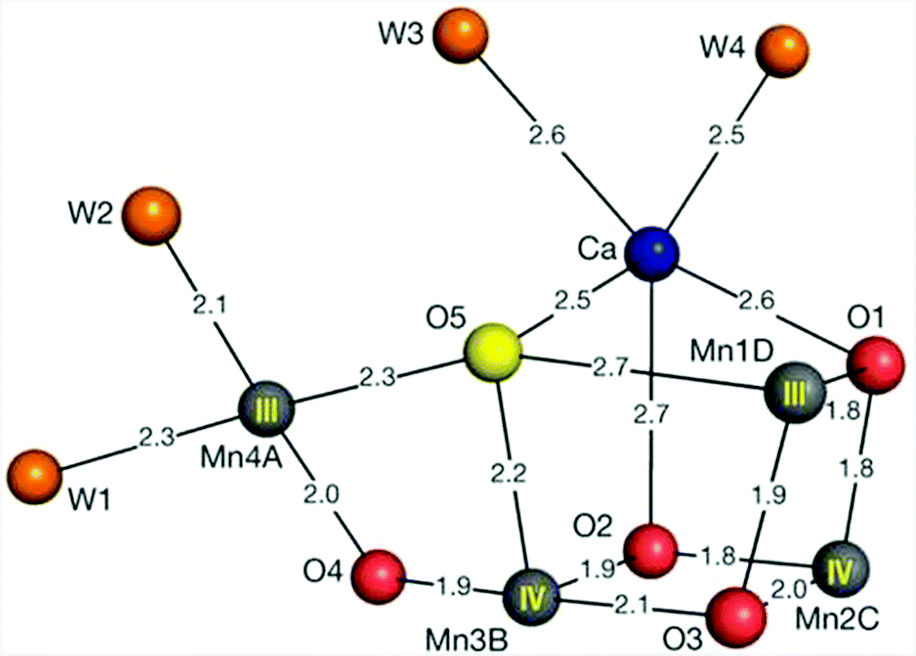 | ||
| Fig. 1 Structure of the Mn4Ca cluster in the oxygen-evolving complex (OEC). Distances for Mn–O, Ca–O, Mn–water and Ca–water are given in Ångstrom. Reprinted with permission from ref. 12. Copyright 2015 Macmillan Publishers Ltd. | ||
1.2. Artificial photosynthesis
In contrast to natural photosynthesis where the protons and electrons produced from H2O oxidation are used to convert CO2 to biomass, in artificial systems they are converted into simpler chemical fuels, such as H2. The light-driven H2O splitting requires the orchestration of several intricate processes, such as light absorption, charge separation, oxidation of H2O and reduction of the generated protons.4 In artificial photosynthetic systems, catalysts for the oxidation of H2O are currently the bottleneck. To catalyze such an energy demanding reaction as H2O oxidation, the catalysts should be able to accumulate several oxidizing equivalents, forming the essential O–O bond, and operate close to the thermodynamic potential.7 The design of artificial water oxidation catalysts (WOCs) thus represents a fundamental challenge in schemes for artificial photosynthesis and has therefore been the major focus of research during the past few decades.23The majority of homogeneous WOCs are based on the second- and third-row transition metals Ru and Ir.24 Complexes based on these metals have been shown to produce robust and highly active WOCs, and are, in general, thought to generate the vital O–O bond either via the water nucleophilic attack (WNA) mechanism or the interaction of two metal–oxo units (I2M) (see Fig. 2).25–28
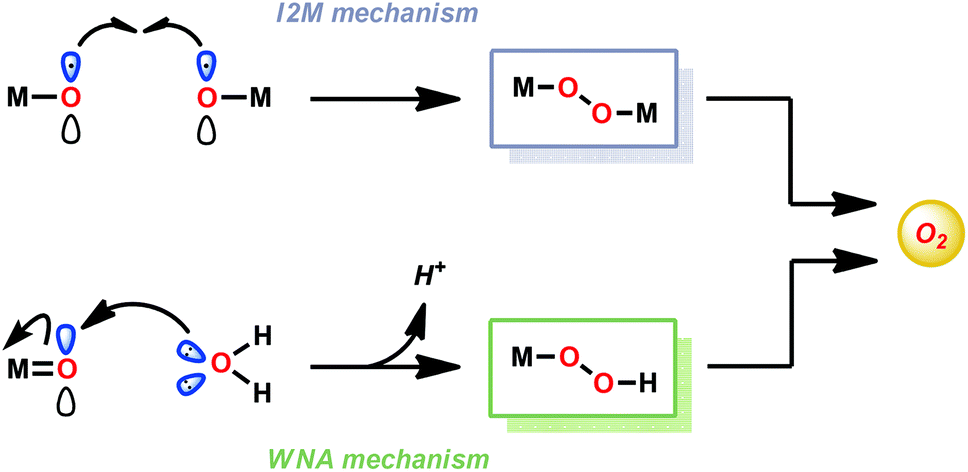 | ||
| Fig. 2 O–O bond formation pathways for Ru- and Ir-based water oxidation catalysts (WOCs). | ||
If water splitting is to become economically viable on a global scale, the high cost and low abundance associated with transition metals, such as Ru and Ir, may become a problem. The construction of WOCs based on earth-abundant first-row transition metals is therefore highly desirable. Despite considerable efforts, the development of robust WOCs incorporating first-row elements that operate under mild conditions has proven particularly challenging.29–32 However, if successful, this could have a widespread impact on the construction of low-cost devices for solar energy to fuel conversion. This Perspective summarizes recent advances in the design of WOCs based on earth-abundant first-row transition metals with an emphasis on homogeneous systems based on Mn, Fe, Co and Cu. An examination of the advantages and current limitations of the highlighted catalytic systems is also presented, along with potential future directions of this expanding research area.
2. Methods for evaluating water oxidation catalysts
There exist essentially three different methods for evaluating whether a complex is capable of oxidizing H2O. The first method is chemical oxidation and involves the use of sacrificial oxidants, allowing for rapid screening of potential WOCs. The most widely used one-electron oxidant is ceric ammonium nitrate (CAN, Ce(NH4)2(NO3)6, CeIV), which has a redox potential of ∼1.70 V vs. NHE (normal hydrogen electrode).33 However, the O2 studies with CeIV (eqn (3)) need to be performed in acidic aqueous media, due to the instability of CeIV at higher pH, and this limits the evaluation to homogeneous catalysts that are resistant to decomposition under acidic conditions:34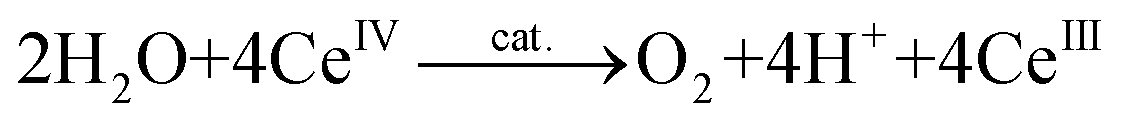 | (3) |
A number of two-electron oxidants have also been used for the evaluation of WOCs, including potassium peroxymonosulfate (Oxone®)35 and sodium periodate (NaIO4),36 which have redox potentials of 1.82 V and 1.60 V vs. NHE, respectively.37,38 The main advantage with these oxidants is that they can be used under neutral conditions, thus allowing the examination of acid labile catalysts.39 The two-electron nature of these oxidants limits their relevance to one-electron conditions, which might occur in photoelectrochemical cells. Oxone® and NaIO4 are also known oxo-transfer reagents, which precludes that both oxygen atoms in the generated O2 originate from H2O; however, this can be easily determined by mass spectrometry using 18O isotopically labeled H2O (H218O). It should be noted that recent studies on CeIV suggest that this oxidant might also function as an oxo-transfer reagent.40,41
An alternative to CeIV is the one-electron oxidant [Ru(bpy)3]3+ (1+, Fig. 3; bpy = 2,2′-bipyridine), which has a redox potential of 1.26 V vs. NHE. Although this and related complexes have not been widely used as oxidants (eqn (4)), perhaps due to their high cost, the advantages of these oxidants are their activity under neutral conditions and the fact that they can be photochemically generated from the corresponding [Ru(bpy)3]2+-type complexes.42 The photochemistry of these Ru polypyridyl complexes has been extensively studied and their photophysical properties are well documented:43–45
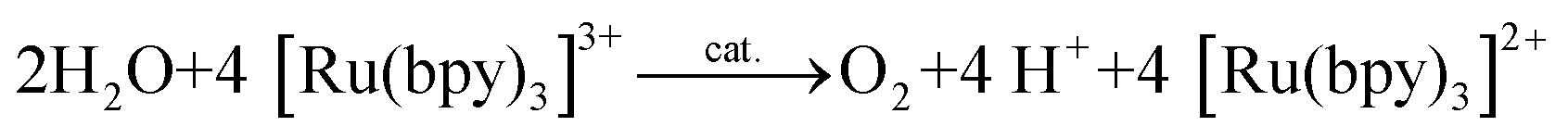 | (4) |
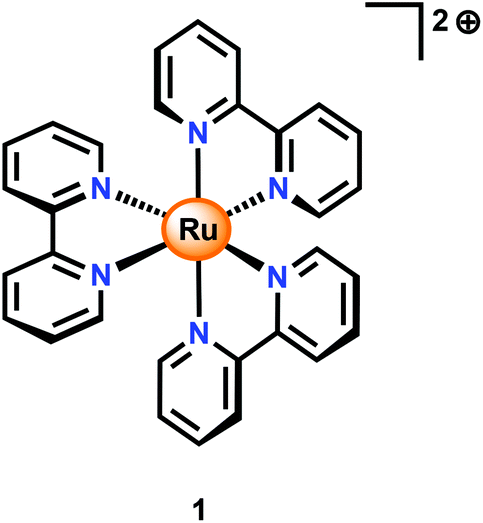 | ||
| Fig. 3 Structure of [Ru(bpy)3]2+ (1). bpy = 2,2′-bipyridine. | ||
In combination with a sacrificial electron acceptor, [Ru(bpy)3]2+ can be used for light-driven H2O oxidation. Visible-light absorption of the [Ru(bpy)3]2+ photosensitizer results in the formation of the singlet excited state (1[Ru(bpy)3]2+*). This undergoes rapid intersystem crossing (ISC) and is converted into an excited triplet state (3[Ru(bpy)3]2+*). In the presence of a sacrificial electron acceptor, such as sodium persulfate (Na2S2O8), 3[Ru(bpy)3]2+* is oxidatively quenched, which generates [Ru(bpy)3]3+, a sulfate ion (SO42−) and a sulfate radical (SO4−˙). The sulfate radical is a strong oxidant (E = ∼2.4 V vs. NHE46) and has the ability to directly oxidize another equivalent of [Ru(bpy)3]2+ to [Ru(bpy)3]3+. The overall light-induced process is summarized in eqn (5) and Fig. 4.43–45,47
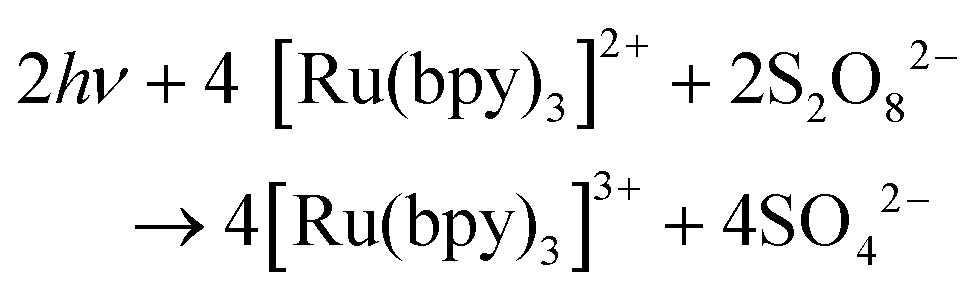 | (5) |
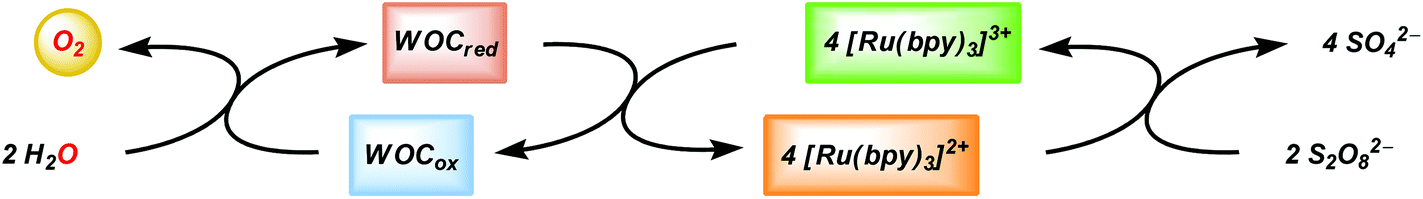 | ||
| Fig. 4 Three-component system for light-driven H2O oxidation. | ||
In addition to the aforementioned (photo)chemical techniques for studying WOCs, (photo)electrochemical methods are also available. Here, an important parameter is the so-called overpotential, which is the potential that has to be applied in an electrolysis cell in addition to the thermodynamical potential for a given half-reaction.48 These electrochemical studies have a major advantage, namely that the studied system resembles the conditions that would be employed in the envisioned formation of solar fuels.49
3. Distinguishing between heterogeneous and homogeneous species
Earth-abundant WOCs are especially prone to undergo irreversible structural modifications, leading to transformation of the initially homogeneous complex into heterogeneous metal oxide species. The designed metal complexes merely act as precursors for the in situ formation of metal nanoparticles that might be the true active catalytic species. An essential element for researchers working in the field is thus to determine whether the observed activity is derived from the pristine metal complex or whether nanoparticles, produced during the oxidation process, are responsible for converting H2O to O2. Several distinct physical techniques exist for establishing whether the starting well-defined molecular complex is the true catalytic species or merely a precursor and include X-ray photoelectron spectroscopy (XPS), energy-dispersive X-ray spectroscopy (EDX), transmission electron microscopy (TEM), and dynamic light scattering (DLS). In combination, these techniques provide a powerful toolbox for assessing the real catalytic entity. One also needs to consider that a slight change in the reaction conditions may have a great effect on both the stability of WOCs and the operating mechanism.50,514. Artificial Mn complexes as water oxidation catalysts
4.1. Dinuclear Mn complexes containing multidentate ligand scaffolds
Inspired by the Mn4Ca cluster located in the OEC, initial attempts to develop earth-abundant WOCs focused on Mn.52 One of the seminal examples of dinuclear Mn complexes capable of oxidizing H2O is the Mn porphyrin dimers 2–4 (Fig. 5) developed by Naruta and co-workers.53,54 In aqueous MeCN solutions containing nBu4NOH, all three Mn dimers showed a similar irreversible increase in current at potentials >1.4 V vs. NHE. Of the three studied Mn porphyrin dimers, complex 4 showed the highest activity, affording a TON of 9.2. The O2 evolution rate was also found to be proportional to the concentration of the Mn dimers. The analogous monomeric Mn porphyrin complexes did not promote O2 evolution and in the absence of the Mn dimers the onset potential was observed at >2.5 V, highlighting the necessity for the dinuclear Mn scaffold.53 Due to the proximity of the Mn centers, the authors proposed that O–O bond formation occurred through nucleophilic attack of OH− on a dimeric MnV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species, thus generating a MnIV–OO–MnIV species from which O2 could subsequently be liberated.54 A sulfonated Mn porphyrin complex has also been incorporated into a thin layer of poly(terthiophene) (PTTh) for attachment to indium tin oxide (ITO) electrodes. The resulting electrodes were capable of catalyzing photoelectrochemical H2O oxidation with low overpotentials.55
O species, thus generating a MnIV–OO–MnIV species from which O2 could subsequently be liberated.54 A sulfonated Mn porphyrin complex has also been incorporated into a thin layer of poly(terthiophene) (PTTh) for attachment to indium tin oxide (ITO) electrodes. The resulting electrodes were capable of catalyzing photoelectrochemical H2O oxidation with low overpotentials.55
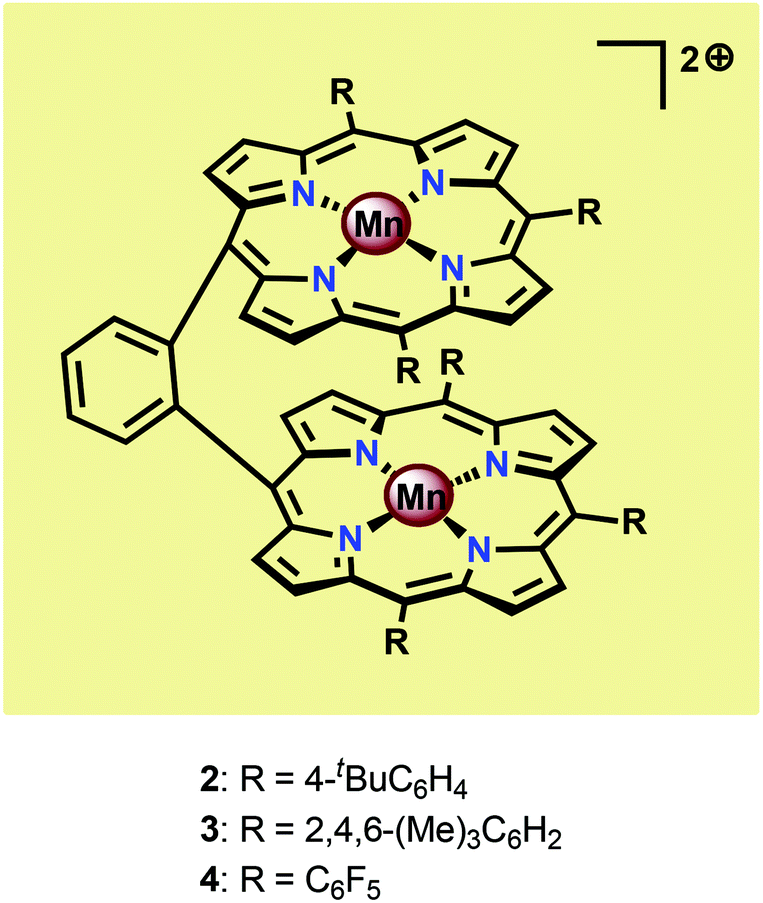 | ||
| Fig. 5 Mn porphyrin dimers 2–4. | ||
Perhaps the most well-studied Mn complex capable of mediating H2O oxidation is the dimeric [(tpy)(OH2)Mn(μ-O)2Mn(OH2)(tpy)]3+ complex (5, Fig. 6; tpy = 2,2′:6′,2′′-terpyridine) reported by Crabtree's and Brudvig's groups.35,56,57 The crystal structure of the dinuclear Mn complex 5 revealed that it consisted of a mixed valence dimer with one of the Mn centers being in the +III state and the other in the +IV state. The two exchangeable aqua ligands are essential for the catalytic activity of the Mn complex. The initial evaluation of the oxidation capability of the complex was carried out with NaOCl as a chemical oxidant. Using this oxidant at pH 8.6, the dimeric Mn complex 5 afforded a TON of 4 and a TOF of 0.0033 s−1. The low efficiency of the Mn dimer was ascribed to the ligand decomposition, resulting in the formation of catalytically active MnO4−, as monitored by UV-vis spectroscopy.35 Studies have also suggested that H2O oxidation with Mn complex 5 can be driven using Oxone®58,59 or CeIV (ref. 60 and 61) as a chemical oxidant. Deposition of the dimeric Mn complex 5 onto different materials, such as kaolin,62 mica,63–65 montmorillonite66 or TiO2,67,68 has also been carried out. Additionally, attempts to extend the lifetime of Mn complex 5 by its incorporation into metal–organic frameworks (MOFs) have been reported.69,70 A variety of analogs of Mn complex 5 have been synthesized and evaluated as WOCs.71–75
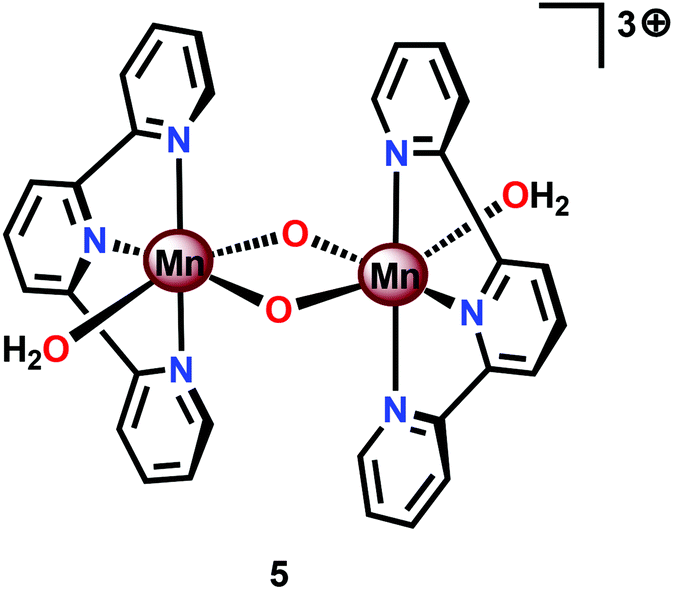 | ||
| Fig. 6 Structure of the dimeric MnIII,IV2 complex [(terpy)(OH2)Mn(μ-O)2Mn(OH2)(terpy)]3+ (5). | ||
Based on UV-vis spectroscopy and electron paramagnetic resonance (EPR) experiments, the authors proposed a simplified mechanism, depicted in Scheme 1, for O2 formation from the reaction of Mn complex 5 with NaClO. The proposed mechanism involves an initial oxidation of dimeric Mn complex 5 to produce a MnIV,IV2 species (6), which can react with the oxidant to generate MnVO (7). This species is the key intermediate and is responsible for promoting O–O bond formation via nucleophilic attack of OH−. The formed Mn–peroxo intermediate is subsequently oxidized, which results in the release of O2 and ultimately regenerates the dimeric MnIV,IV2 complex 6.35,76 It can be concluded that the mechanism by which Mn complex 5 mediates the oxidation of H2O is intricate and is highly dependent on the reaction conditions.77,78 This is further supported by findings concerning the possible involvement of a tetrameric Mn species79–81 while another report suggests that heterogeneous Mn–oxides are generated.82
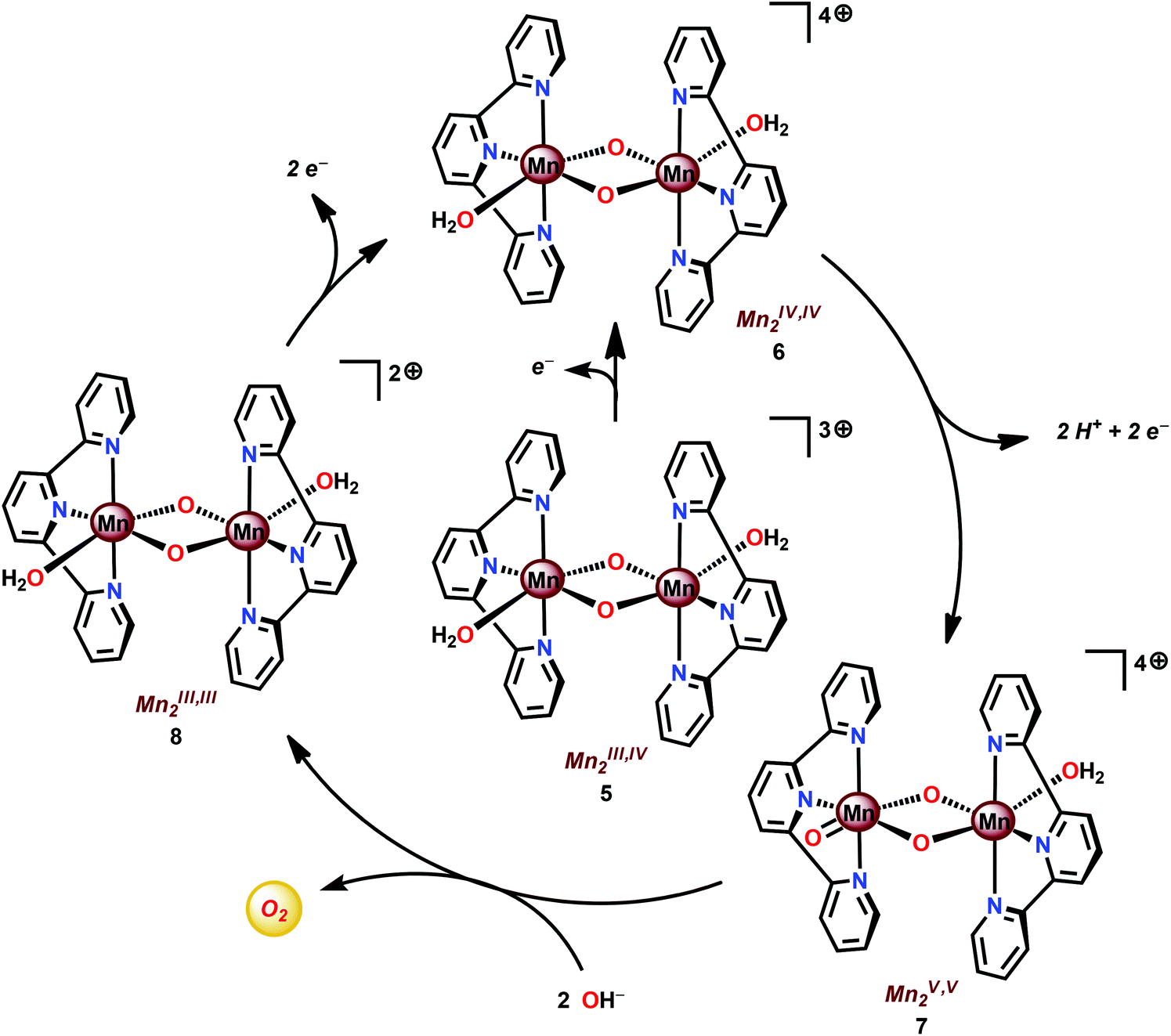 | ||
| Scheme 1 Simplified proposed mechanism for H2O oxidation by MnIII,IV2 complex 5. | ||
Based on previous findings,83 McKenzie and co-workers evaluated the dimeric Mn complex [MnII,II2(mcbpen)2(OH2)2]2+ (9; mcbpen = N-methyl-N′-carboxymethyl-N,N′-bis(2-pyridylmethyl)ethane-1,2-diamine) as a WOC (Fig. 7). The dimeric Mn complex 9 was able to evolve O2 when driven using tBuOOH or CeIV as a chemical oxidant.84 Using tBuOOH as an oxidant afforded high turnovers of up to 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mol of O2 per mol of Mn.85 However, when using isotopically labeled H2O (H218O), the evolved O2 was shown to be exclusively 34O2. This highlights that one of the oxygen atoms in the generated O2 originates from the tBuOOH oxidant, demonstrating the ability of this two-electron oxidant to act as an oxygen atom transfer reagent. In the case when CeIV is used as an oxidant, the only available source of oxygen is the nitrate counterion of the oxidant, thus suggesting the active involvement of the nitrate counterion in the catalytic cycle.84
000 mol of O2 per mol of Mn.85 However, when using isotopically labeled H2O (H218O), the evolved O2 was shown to be exclusively 34O2. This highlights that one of the oxygen atoms in the generated O2 originates from the tBuOOH oxidant, demonstrating the ability of this two-electron oxidant to act as an oxygen atom transfer reagent. In the case when CeIV is used as an oxidant, the only available source of oxygen is the nitrate counterion of the oxidant, thus suggesting the active involvement of the nitrate counterion in the catalytic cycle.84
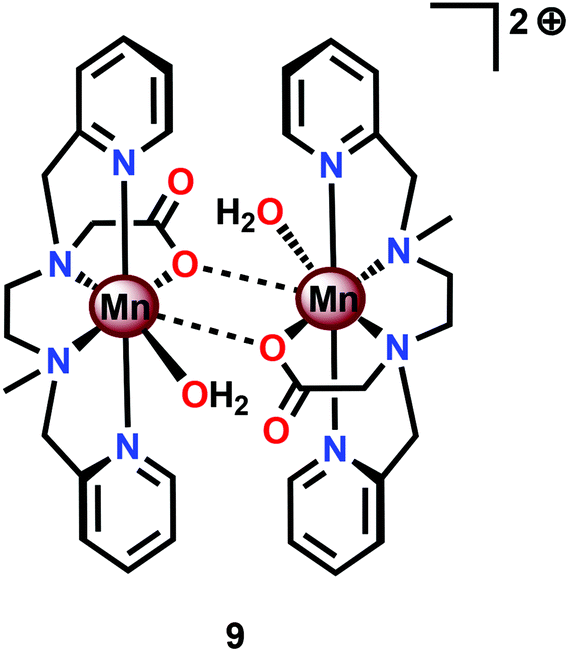 | ||
| Fig. 7 Structure of the dimeric MnII,II2 complex [MnII,II2(mcbpen)2(OH2)2]2+ (9). | ||
The mcbpen ligand scaffold with its carboxylate and pyridine motifs attached to the ethylenediamine backbone was suggested to provide an ideal environment for coordination to Mn in various redox states. It was initially proposed that the catalytic cycle for O2 generation involved the coupling of two μ-oxo units.84 However, a subsequent computational study suggested that the key species consisted of a MnIII(μ-O)MnIV–O˙ species (12), a mixed valent oxyl radical species, which is in equilibrium with the isomeric diamond-core MnIV(μ-O)2MnIV (13). This mixed valent MnIII(μ-O)MnIV–O˙ species (12) is responsible for mediating O–O bond formation through nucleophilic attack of H2O, resulting in a MnIII–OOH species (14). The other Mn center, which has thus far acted as a spectator, functions as the additional oxidizing equivalent, liberating O2 to regenerate the monomeric MnII complex 10 without the need for an additional oxidation event (Scheme 2).86
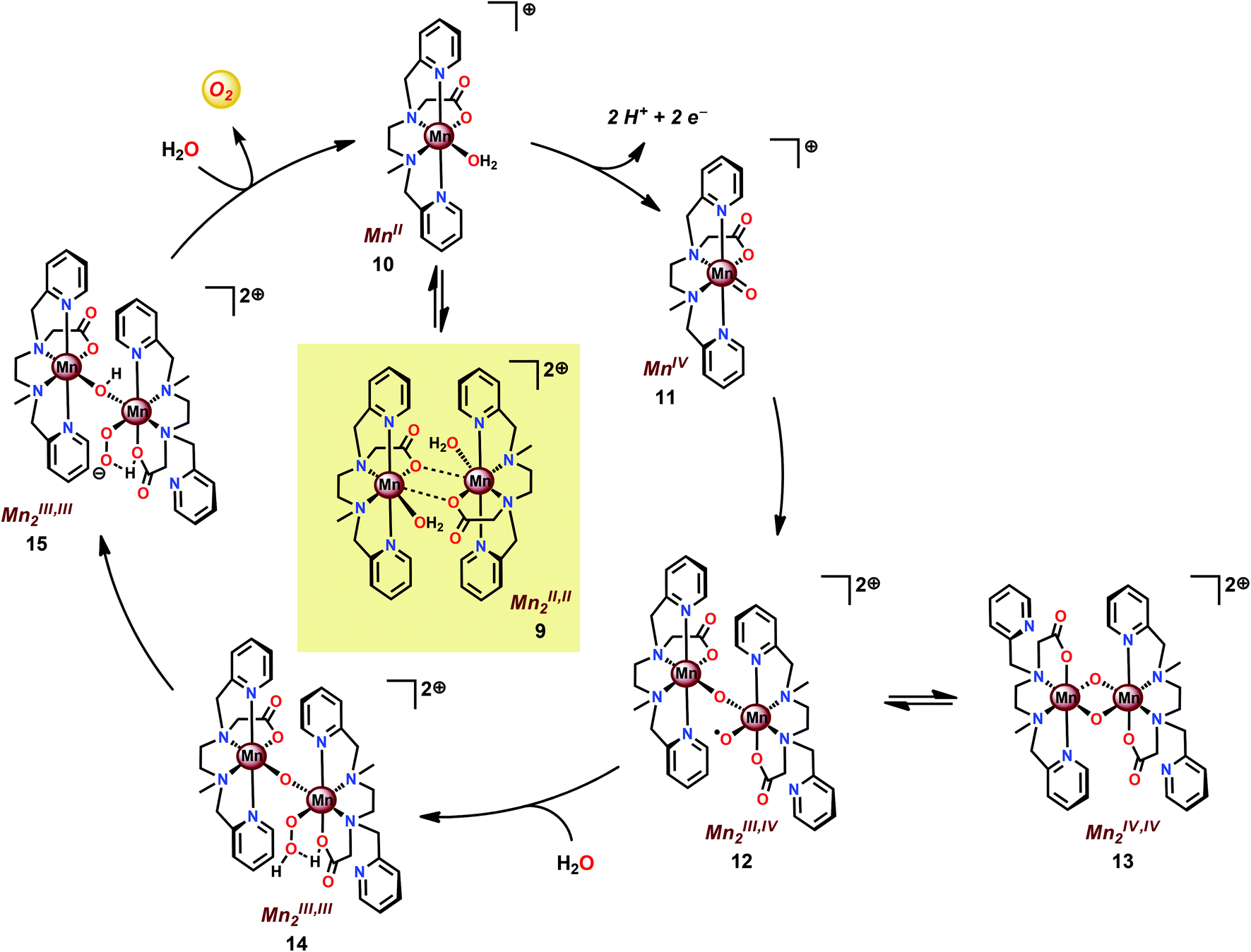 | ||
| Scheme 2 Proposed mechanism for O2 evolution catalyzed by the [MnII,II2(mcbpen)2(OH2)2]2+ complex 9. | ||
Several Mn complexes have been covalently linked to [Ru(bpy)3]2+-type chromophores for mimicking the electron transfer events occurring in Photosystem II. Light absorption of the RuII photosensitizer core triggers electron transfer from the photoexcited state (Ru2+*) to a sacrificial electron transfer. The oxidized photosensitizer subsequently abstracts an electron from the attached Mn complex.87–92 Of the examined Ru–Mn assemblies, assembly 16 (Fig. 8) displayed promising properties as it was able to undergo three consecutive electron transfers, which converted the initial MnII,II2 core to MnIII,IV2. Although Ru–Mn2 assembly could accumulate three oxidizing equivalents, it was not able to evolve O2 under the studied reaction conditions.93
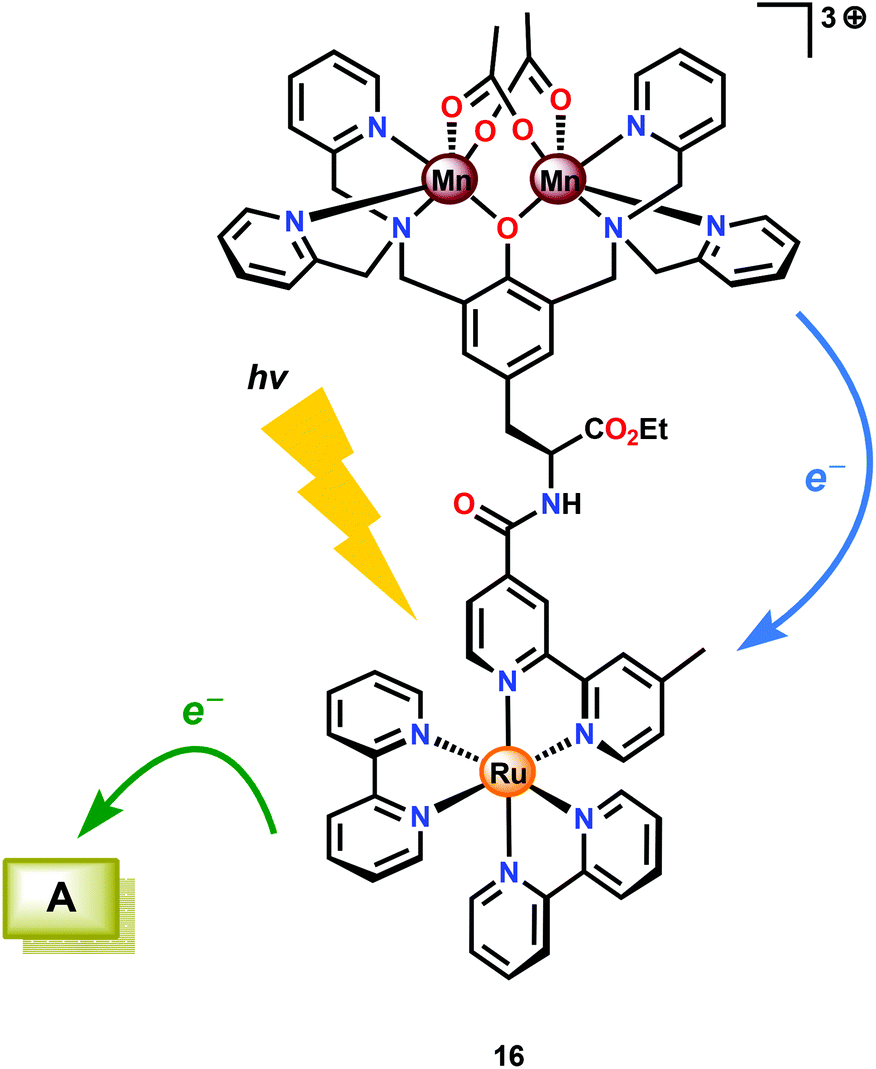 | ||
| Fig. 8 Ru–Mn2 assembly 16 capable of accumulating three oxidizing equivalents. A = acceptor. | ||
In light of the attractive features of the dinuclear Mn core in assembly 16, the properties and O2 evolving activities of several different dinuclear Mn complexes (17–20) housing ligand scaffolds based on benzylic amines have been studied (Fig. 9).61c,94–98 It could be concluded that the one-electron oxidants CeIV and [Ru(bpy)3]3+ were inefficient oxidants in H2O oxidation. However, with two-electron oxidants, such as H2O2, tBuOOH, Oxone® or NaClO, i.e. oxygen-transfer oxidants, a more efficient O2 evolution was observed.61c,96
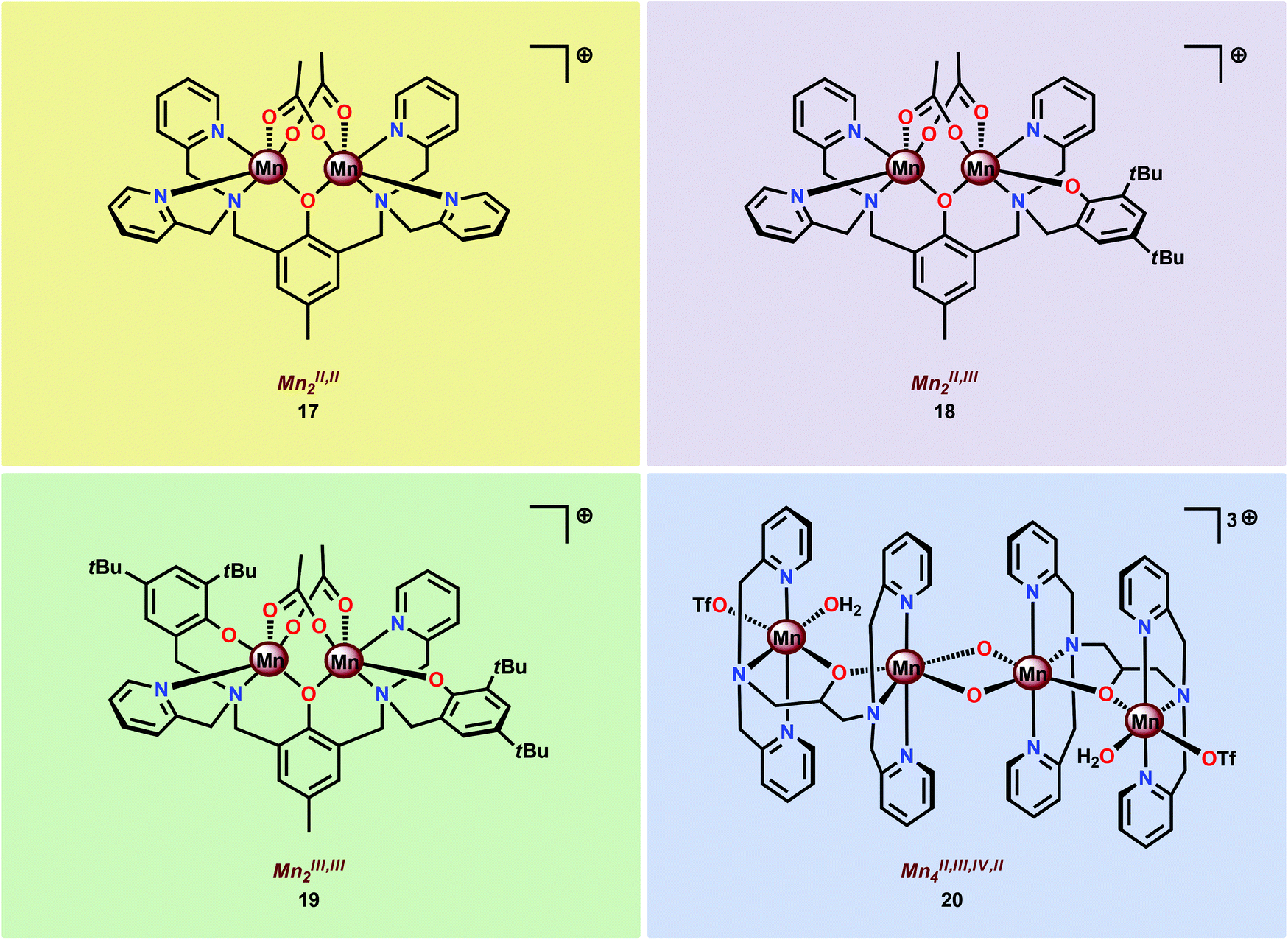 | ||
| Fig. 9 Structures of Mn complexes 17–20. | ||
The fact that imidazole, carboxylate and phenol moieties are essential features of the OEC inspired the group of Åkermark to design the dinuclear Mn complex 21 (Fig. 10, left) housing a bioinspired ligand scaffold with imidazole, carboxylate and phenolic groups.99 In the solid state, the crystal structure revealed that Mn complex 21 adopts a tetrameric structure (22, Fig. 10, right), resembling the tetranuclear Mn4Ca core in the OEC. An additional advantage of the incorporation of the negatively charged groups was the lowering of the redox potentials, an effect previously observed for other metal complexes,100,101 allowing for H2O oxidation to be driven by [Ru(bpy)3]3+. Experiments were initially carried out with a 480-fold excess of the chemical oxidant [Ru(bpy)3]3+ under neutral conditions. Upon addition of an aqueous solution containing Mn complex 21, O2 evolution was immediately observed, affording a TON of ∼25 and a TOF of ∼0.027 s−1. Light-driven H2O oxidation was also carried out using [Ru(bpy)3]2+-type photosensitizers and Na2S2O8 as a sacrificial electron acceptor. When employing [Ru(bpy)2(deeb)]2+ as a photosensitizer (deeb = diethyl 2,2′-bipyridine-4,4′-dicarboxylate), a TON of 4 was reached. Experiments with isotopically labeled H2O (H218O) confirmed that both of the oxygen atoms in the produced O2 originated from the solvent H2O.99
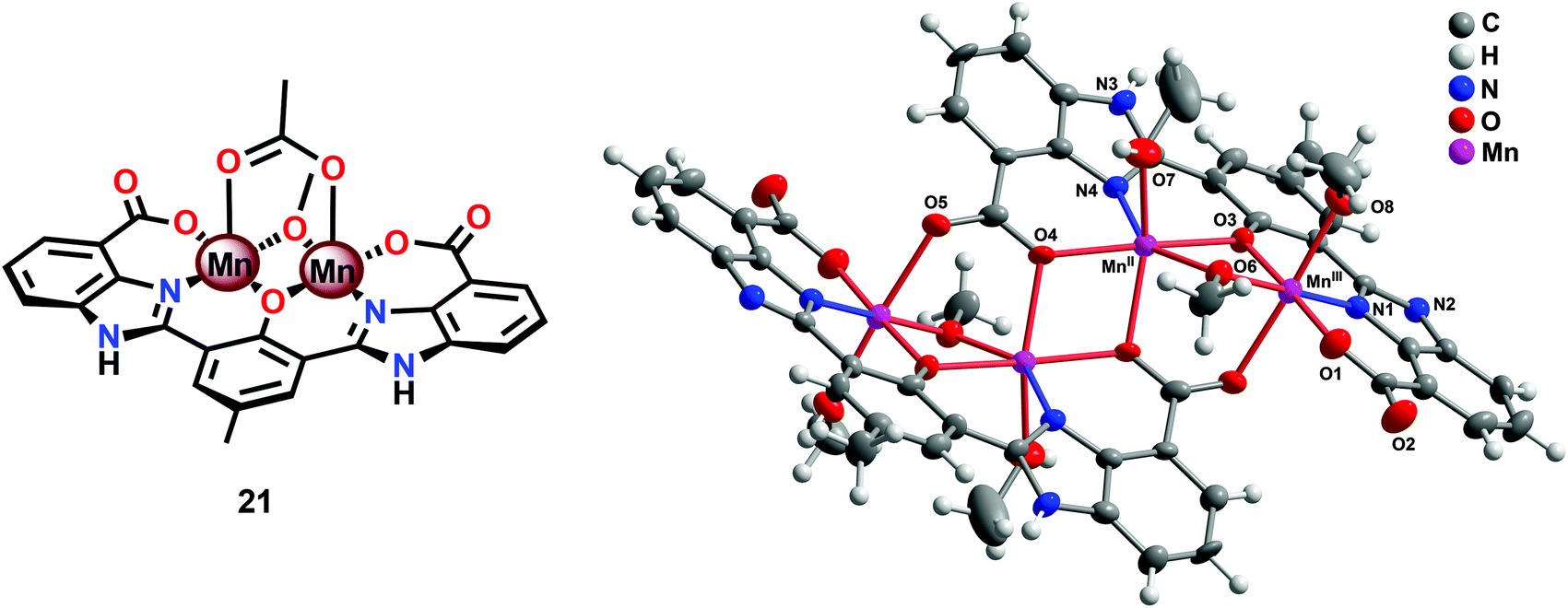 | ||
| Fig. 10 (Left) Molecular structure of the dinuclear MnII,III2 complex 21 and (right) crystal structure of the tetranuclear Mn species 22 generated from complex 21 (reprinted with permission from ref. 104. Copyright 2015 American Chemical Society). | ||
Subsequent work involved the preparation of a Ru–Mn2 assembly (23, Fig. 11) where the dinuclear MnII,III2 complex 21 was covalently linked to a [Ru(bpy)3]2+-type photosensitizer.102 Although Ru–Mn2 assembly 23 was able to mediate chemical oxidation of H2O, the photophysical properties of the assembly did not allow for photochemical H2O oxidation in the presence of a sacrificial electron acceptor (Na2S2O8). It was revealed that the excited state of the dyad 23 was too short-lived and could not be modelled with a mono-exponential decay function. Computational studies suggested the presence of an intricate excited-state manifold, along with possible large effects of different protonation states, which may have contributed to the observed intricate decay pattern.
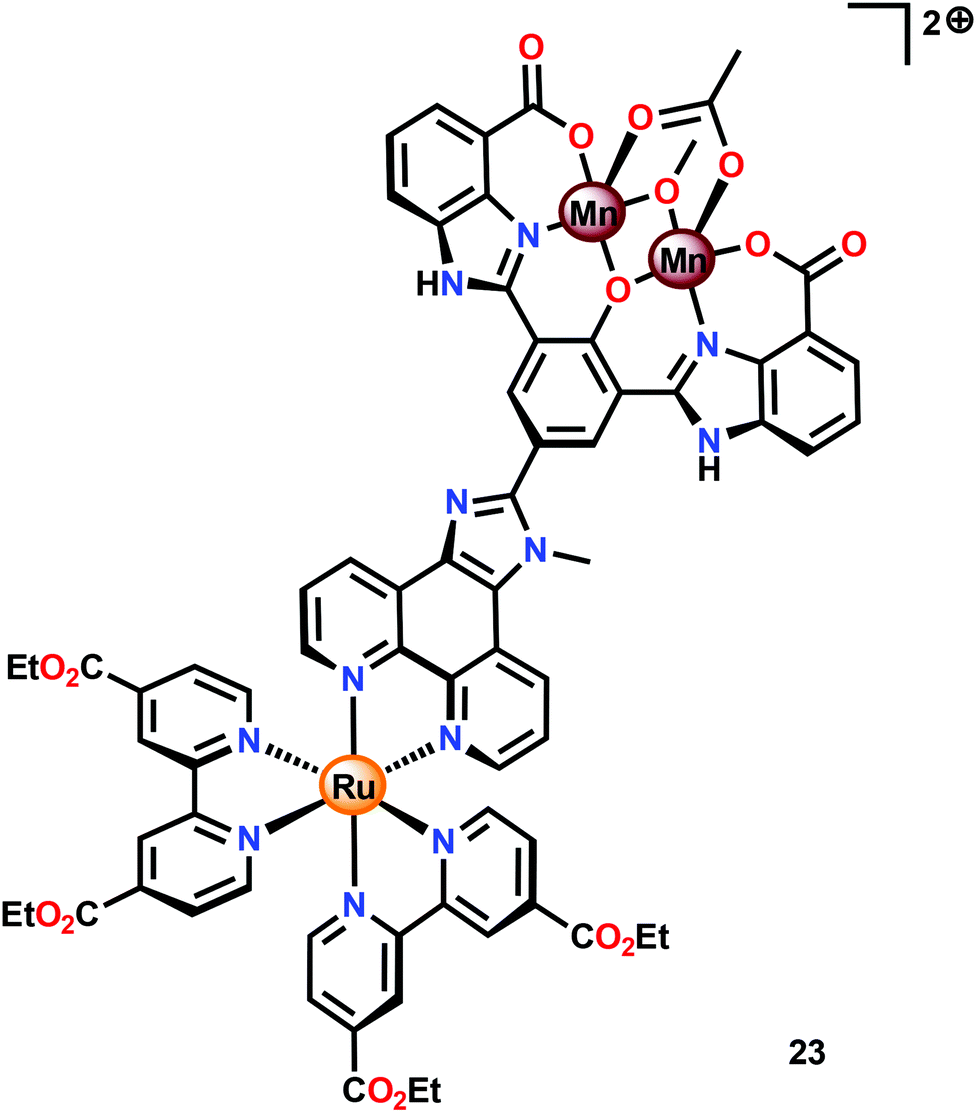 | ||
| Fig. 11 Structure of Ru–Mn2 assembly 23. | ||
A series of dinuclear derivatives of Mn complex 21 have also been synthesized.103 The prepared ligand scaffolds contained a variety of different substituents and the electronic and catalytic features of the corresponding Mn complexes were systematically examined. Of the synthesized Mn complexes, complex 24 possessing the distal carboxylate group (Fig. 12, left) was shown to produce O2 more efficiently than the other Mn complexes. This effect was proposed to originate from pre-orientation of the incoming H2O molecule where the introduced carboxylate moiety directs the incoming H2O through hydrogen bonding (Fig. 12, right). This probably facilitates proton-coupled electron transfer (PCET) and assists the nucleophilic attack on a high-valent Mn–oxyl/Mn–oxo species. Incorporation of non-innocent distal groups into ligand backbones could be a general method for enhancing the catalytic activity of WOCs.
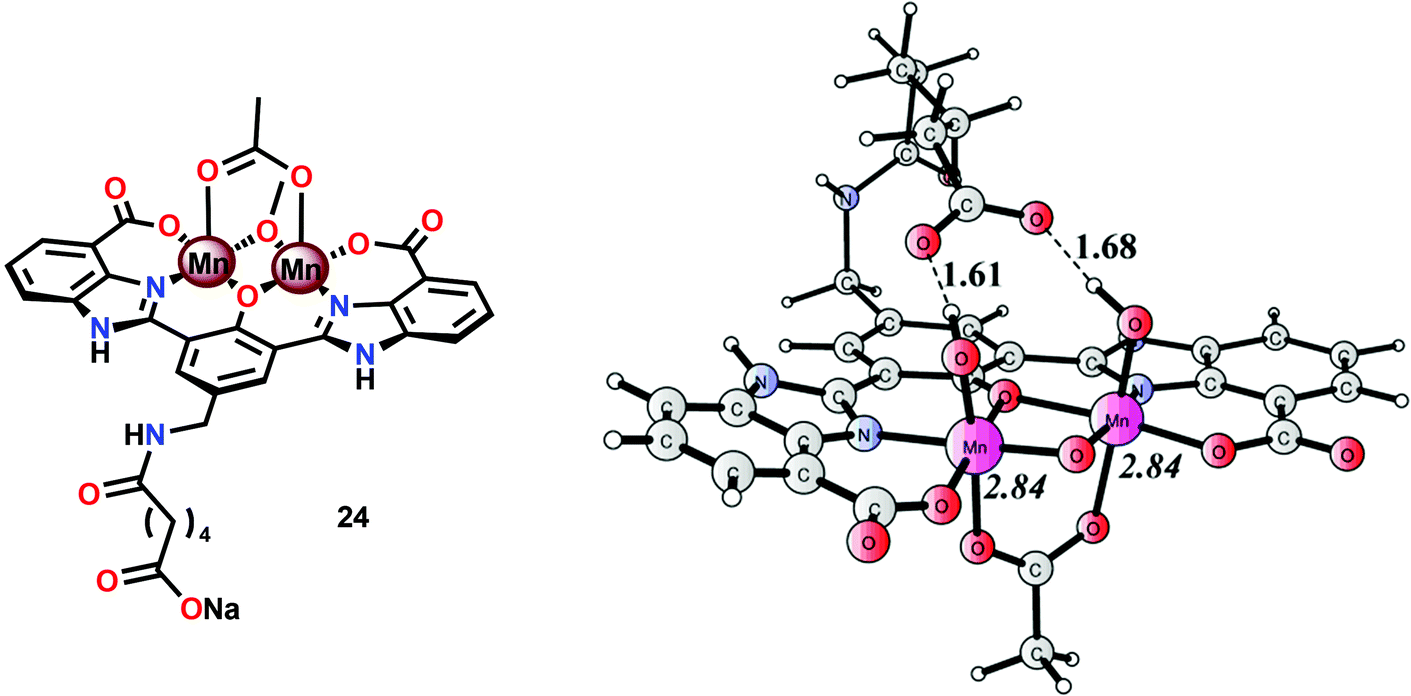 | ||
| Fig. 12 (Left) Molecular structure of dinuclear MnII,III2 complex 24 and (right) calculated structure of the complex in its formal MnIV,V2 state, showing the hydrogen-bonding interaction between the distal carboxylate moiety and the Mn bound hydroxide(s) (adapted with permission from ref. 103. Published by the PCCP Owner Societies). | ||
A computational study has been performed on the mechanism by which the dinuclear MnII,III2 complex 21 mediates O–O bond formation.104 The study suggested an OEC-like mechanism where tetrameric Mn species are involved throughout the catalytic cycle (Fig. 13). The catalytic cycle was suggested to begin with the formal MnIII,III,III,IV4 species, which is better described as having all four Mn centers in the +III state with a ligand radical. Four PCET events transform the initial formal MnIII,III,III,IV4 species into a MnIII,IV,IV,IV4–L˙–L˙ species. Intramolecular proton transfer subsequently generates a MnIV-bound oxyl radical that is responsible for creating the O–O bond with the Mn1–Mn2 bridging oxo group (Fig. 13). The proposed mechanistic pathway contains several features that are important clues to understand how the OEC catalyzes H2O oxidation.19 Examples of design principles that were obtained from the study are: (1) a Mn-bound terminal aqua molecule is essential for the formation of the crucial MnIV–O˙ species, (2) nucleophilic attack on the MnIV–O˙ species is less favorable compared to a route featuring the coupling between this species and a bridging oxo moiety, and (3) the presence of a redox-active ligand backbone decreases the redox potentials for the individual redox events and alleviates the metal centers from being too heavily oxidized.104
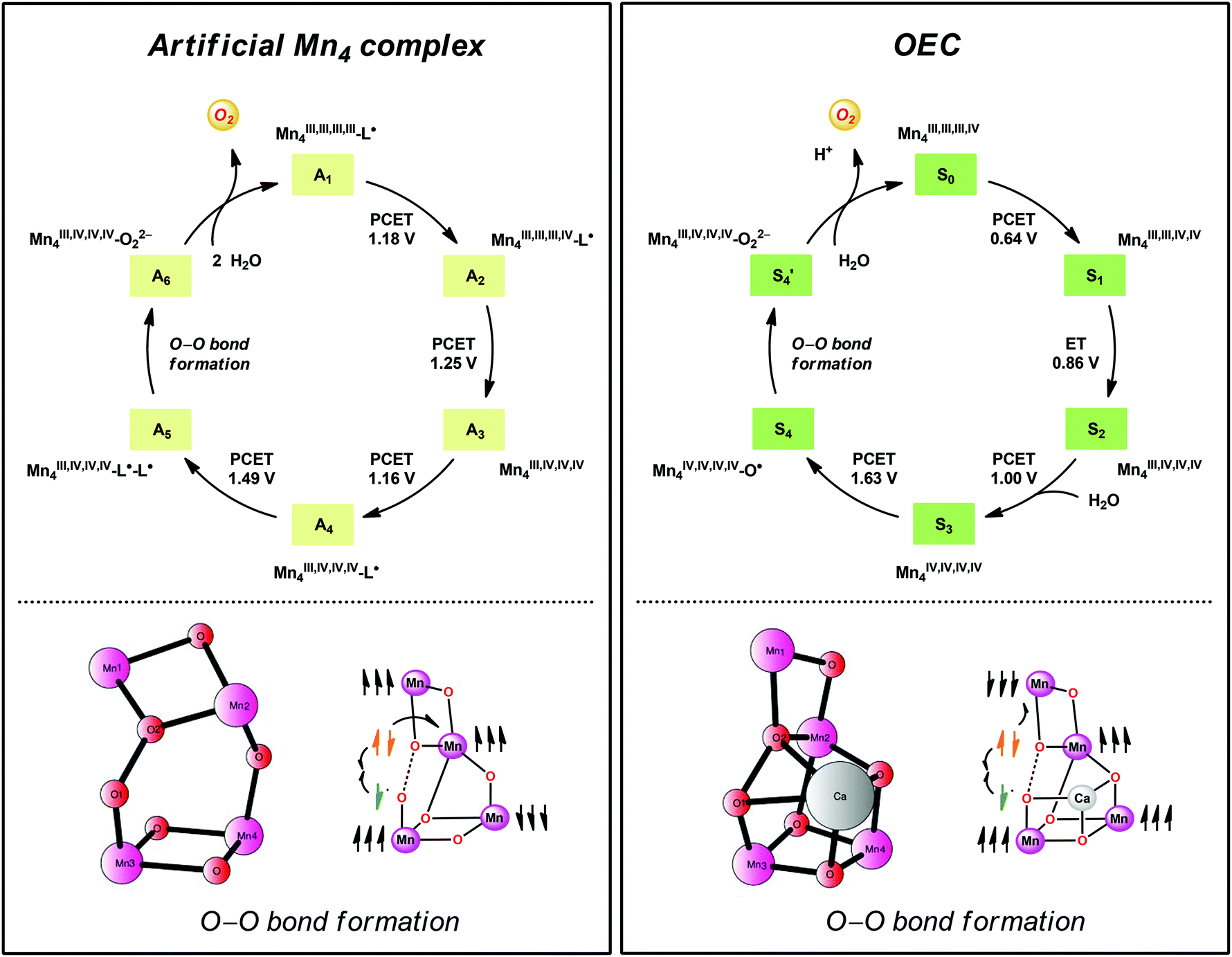 | ||
| Fig. 13 Comparison of the catalytic cycles and transition-states for O–O bond formation for the tetranuclear Mn complex derived from Mn complex 21 and the tetranuclear Mn4Ca cluster in the OEC. Adapted with permission from ref. 104. Copyright 2015 American Chemical Society. | ||
4.2. Mononuclear Mn complexes: O–O bond formation at single metal centers
A vital feature for metal complexes to mediate H2O oxidation is the necessity for these catalysts to generate high-valent metal–oxyl/oxo intermediates, which subsequently trigger O–O bond formation. Several formal high-valent Mn–oxo complexes have been prepared using various chemical oxidants, such as hydrogen peroxide (H2O2),105 iodosylbenzene (PhIO),106 or m-chloroperbenzoic acid (m-CPBA),107 or photochemically generated [Ru(bpy)3]3+.108Although a variety of high-valent Mn–oxo complexes have been prepared,109 only a few mononuclear Mn complexes have been reported to mediate H2O oxidation. An early example of a mononuclear Mn complex capable of evolving O2 is the Mn Schiff base complex [Mn(salpd)(OH2)]+ (25, Fig. 14; H2salpd = N,N′-bis(salicylidene)propane-1,3-diamine). This MnIII Schiff base complex was reported to liberate O2 and reduce p-benzoquinone to hydroquinone when irradiated with visible-light. The catalytic activity was shown to be dependent on the quinone concentration, the wavelength of light, temperature and pH, affording TONs of 0.02–0.06 depending on the reaction conditions. Although the exact mechanism has not been elucidated, the rate of O2 evolution followed first-order kinetics with respect to Mn complex 25 and [{Mn(salpd)}2O] was identified as a deposit during irradiation.110 Related Mn Schiff base complexes have also been studied for O2 evolution by several research groups.111–117
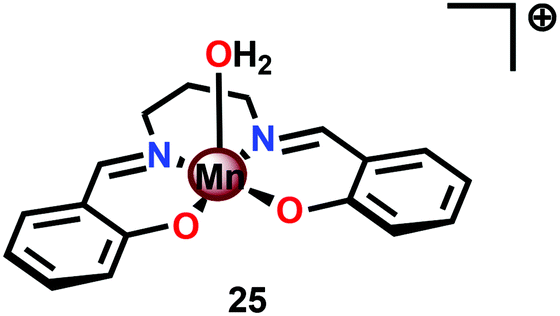 | ||
| Fig. 14 Structure of Mn Schiff base complex [Mn(salpd)(OH2)]+25. | ||
An interesting study was conducted by the group of Åkermark and Sun where the mononuclear MnIV corrole xanthene complex 26 (Fig. 15) was shown to promote electrochemical H2O oxidation.118 In aqueous solutions in the presence of nBu4NOH, O2 was detected at quite low oxidation potentials. Electrochemical measurements of Mn corrole complex 26 further revealed that high-valent redox states were accessed at relatively low potentials. A computational study suggested that O–O bond formation occurred via nucleophilic attack of OH− or H2O on a formal MnVO intermediate.119
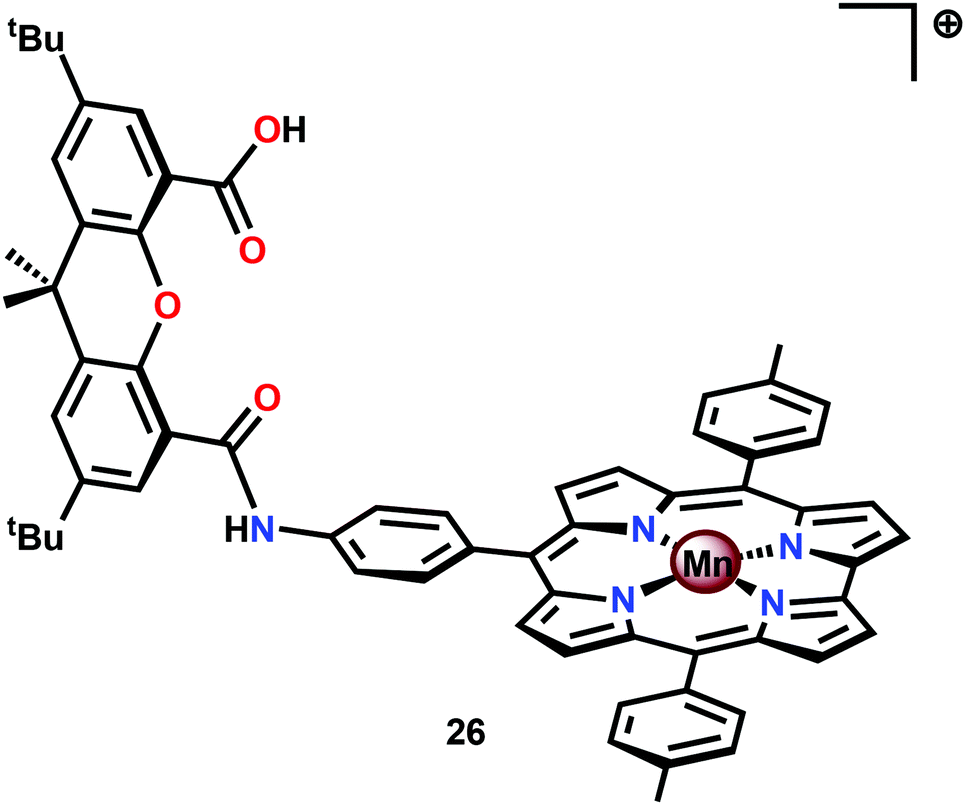 | ||
| Fig. 15 Structure of mononuclear MnIII corrole xanthene complex 26. | ||
In attempts to obtain experimental support for a mechanistic pathway involving nucleophilic addition of OH− or H2O, a subsequent study focused on MnIII corrole 27 (Fig. 16).120 The Mn corrole complex 27 was chosen as it was believed to produce a sufficiently stable MnV species (28). Treating Mn complex 27 with tBuOOH furnished the corresponding MnV species, as shown by UV-vis spectroscopy. Additional support for the generation of the MnVO species 28 was obtained from high-resolution mass spectrometry (HRMS). Upon addition of 2 equivalents of nBu4OH, rapid O2 evolution ensued and a peak ascribed to a MnIV species (30) appeared, which is most likely generated from oxidation of the MnIII–hydroperoxo species 29. This peak gradually decreased with the emergence of a peak corresponding to the MnIII–OH complex 31. The use of isotopically labeled H2O (H218O) supported that the evolved O2 is produced by addition of 18O to the unlabeled MnVO species. This study demonstrates that nucleophilic attack of hydroxide on a high-valent Mn species is a viable mechanistic scenario for triggering O–O bond formation in the conversion of H2O to O2 (Scheme 3).
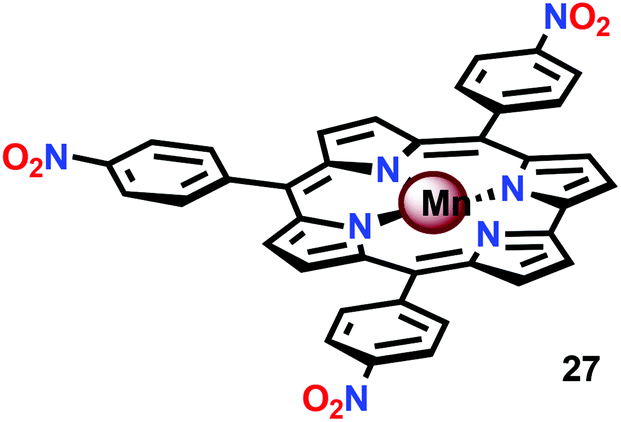 | ||
| Fig. 16 Structure of mononuclear Mn corrole complex 27. | ||
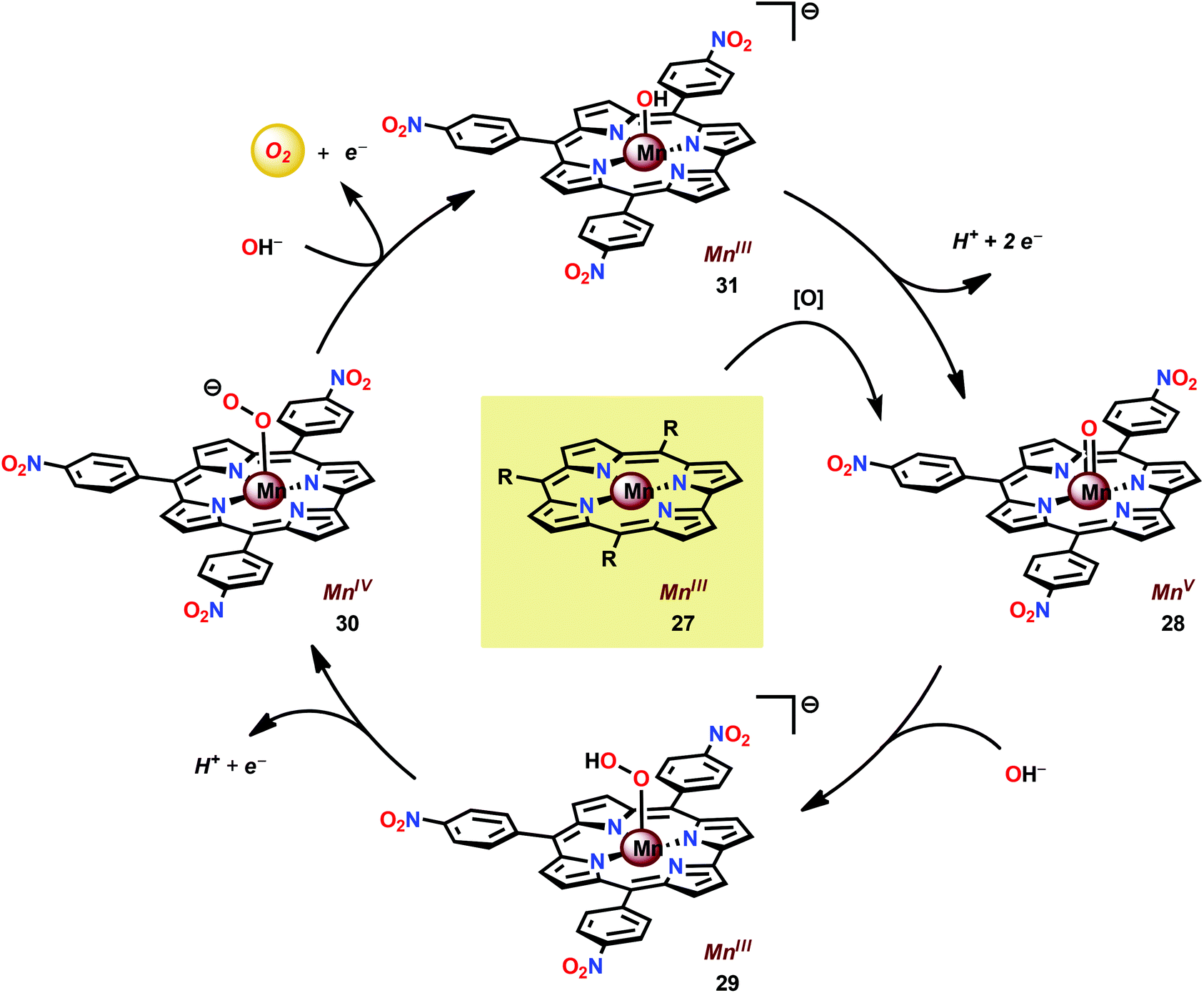 | ||
| Scheme 3 Proposed mechanism for O2 evolution catalyzed by mononuclear Mn corrole complex 27. | ||
The group of Smith found that small changes in the ligand backbone of MnII pyridinophane complexes (Fig. 17) had a dramatic influence on the catalytic properties of these complexes.121 Mn complexes 32 and 33, possessing small substituents, were shown to disproportionate H2O2 in aqueous solutions.122 However, for Mn complex 34 with the sterically encumbered tBu group, catalase activity is turned off and the complex instead mediates electrocatalytic H2O oxidation. When carrying out the electrocatalytic experiments at pH 12.2 with a potential of 1.23 V vs. NHE, TONs of 16–24 were obtained with Faradaic efficiencies of 74–81%. A number of experiments were also undertaken to exclude the involvement of heterogeneous nanoparticles: (1) no spectral changes were observed for the electrolysis solutions, (2) DLS measurements provided no evidence for nanoparticles, (3) the catalytic current does not increase over successive scans, which would be expected if a catalytic deposit was produced, and (4) EDX measurements showed no evidence of the formation of Mn-containing deposits on the electrode. Collectively, these results point to a homogeneous pathway for the oxidation of H2O. Mechanistic studies revealed that the catalytic current varies linearly with the concentration of Mn complex 34, indicating that O–O bond formation occurs at a single metal center.121 The results highlight that a ligand-controlled switch in catalytic reactivity has implications for the design of novel Mn-based WOCs.
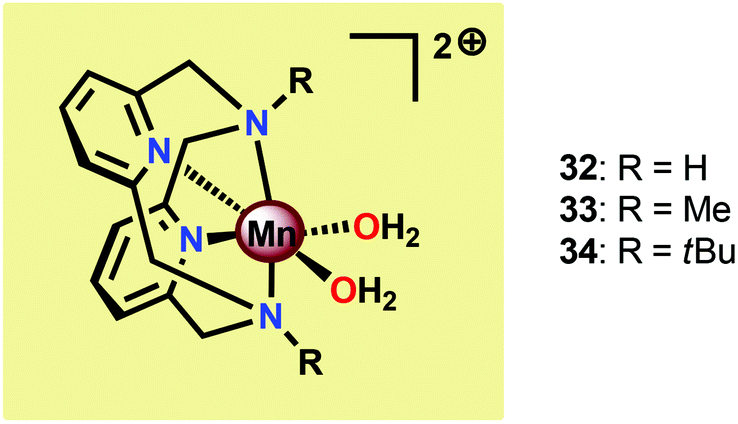 | ||
| Fig. 17 Structure of MnII pyridinophane complexes 32–34. | ||
Brudvig and co-workers recently investigated the H2O oxidation ability of the three mononuclear Mn complexes depicted in Fig. 18; [Mn(PaPy3)(NO3)]+ (35; HPaPy3123 = N,N-bis(2-pyridylmethyl)-amine-N-ethyl-2-pyridine-2-carboxamide), [Mn(N4Py)(OTf)]+ (36; N4Py124,125 = N,N-bis(2-pyridyl-methyl)-N-bis(2-pyridyl)methylamine) and Mn(PY5)(OH2)]2+ (37; PY5126,127 = 2,6-bis(methoxydi(pyridin-2-yl)methyl)pyridine).128 To facilitate access to the key high-valent Mn–oxo species required to initiate O–O bond formation, an electron-rich ligand scaffold is essential. The PaPy3− ligand was chosen as it has been shown that deprotonated carboxamido units are strong electron donors and have previously been employed for isolation of high-valent MnVO compounds.129–131 It was envisioned that the inclusion of the carboxamido unit at the trans position to the oxo site could favor a reactive species by weakening the Mn–oxo bond. Catalytic experiments revealed that Mn complex [Mn(PaPy3)(NO3)]+ (35) was able to catalyze O2 formation using either Oxone® or H2O2 as a two-electron oxidant and kinetic studies showed that the reaction was first-order in the catalyst. However, Mn complex [Mn(N4Py)(OTf)]+ (36) was shown to evolve O2 only in the presence of Oxone® while Mn complex Mn(PY5)(OH2)]2+ (37) was found to be inactive.128
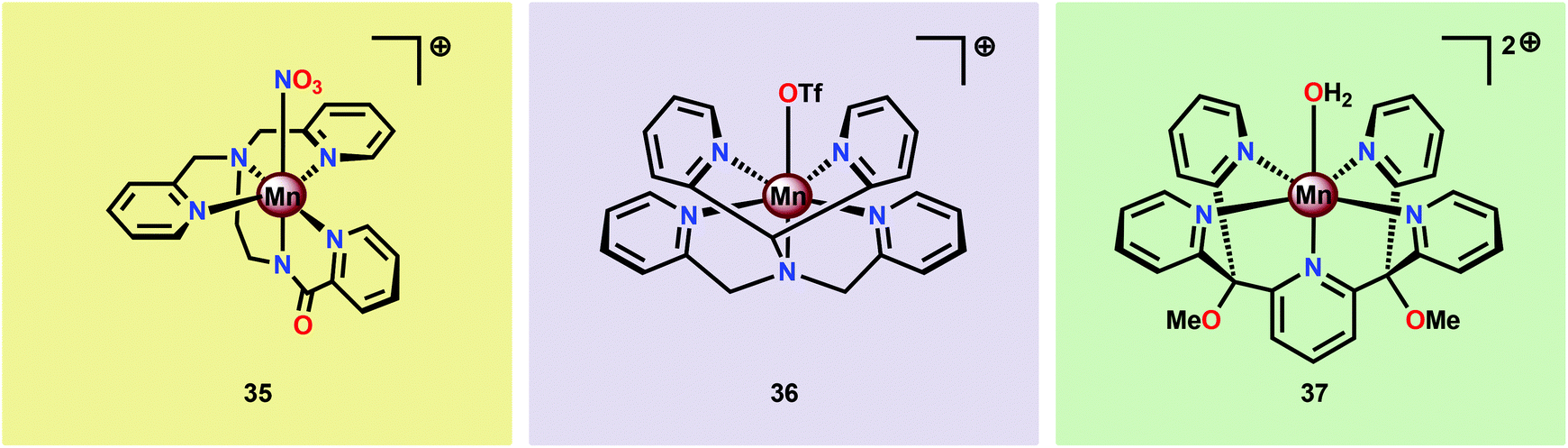 | ||
| Fig. 18 Structures of mononuclear Mn complexes [Mn(PaPy3)(NO3)]+ (35), [Mn(N4Py)(OTf)]+ (36) and Mn(PY5)(OH2)]2+ (37). | ||
The use of the anionic carboxamido ligand in Mn complex 35 is most apparent in the comparison of the O2 evolution rate, where complex 35 had a significantly higher rate than that of Mn complex 36. The different features of the anionic PaPy3− ligand compared to the neutral ligand backbones are further highlighted by the fact that complexation with Mn produces the MnIII complex 35 in air. However, the corresponding MnIII complex Mn(PY5)(OH2)]3+ (37+) requires oxidation with e.g. PhIO.128 The MnIII complex 37+ is stable in air but decomposes rapidly in aqueous solutions to heterogeneous Mn oxides.132 The stability difference between the two MnIII complexes 35 and 37+ demonstrates the power of using anionic ligand backbones for stabilizing high-valent Mn species, a critical feature for the activation of H2O.
4.3. Mn cubanes—bioinspired mimics of the Mn4Ca core in Photosystem II
Molecular mimics of the Mn4Ca cluster core in the OEC have attracted significant attention from researchers. Such artificial Mn clusters are highly desired and could facilitate the understanding of the chemical features of the OEC, and could ultimately be used for designing artificial molecular Mn-based WOCs.133 Numerous multinuclear Mn clusters have been developed as artificial OEC mimics,134–140 for example, the [Mn13Ca2O10(OH)2(OMe)2(O2CPh)18(OH2)4] cluster (38, Fig. 19, top), which contains [Mn4CaO4] sub-units (Fig. 19, bottom),141 and the [MnIV3Ca2O4(O2CtBu)8(tBuCO2H)4] cubane.142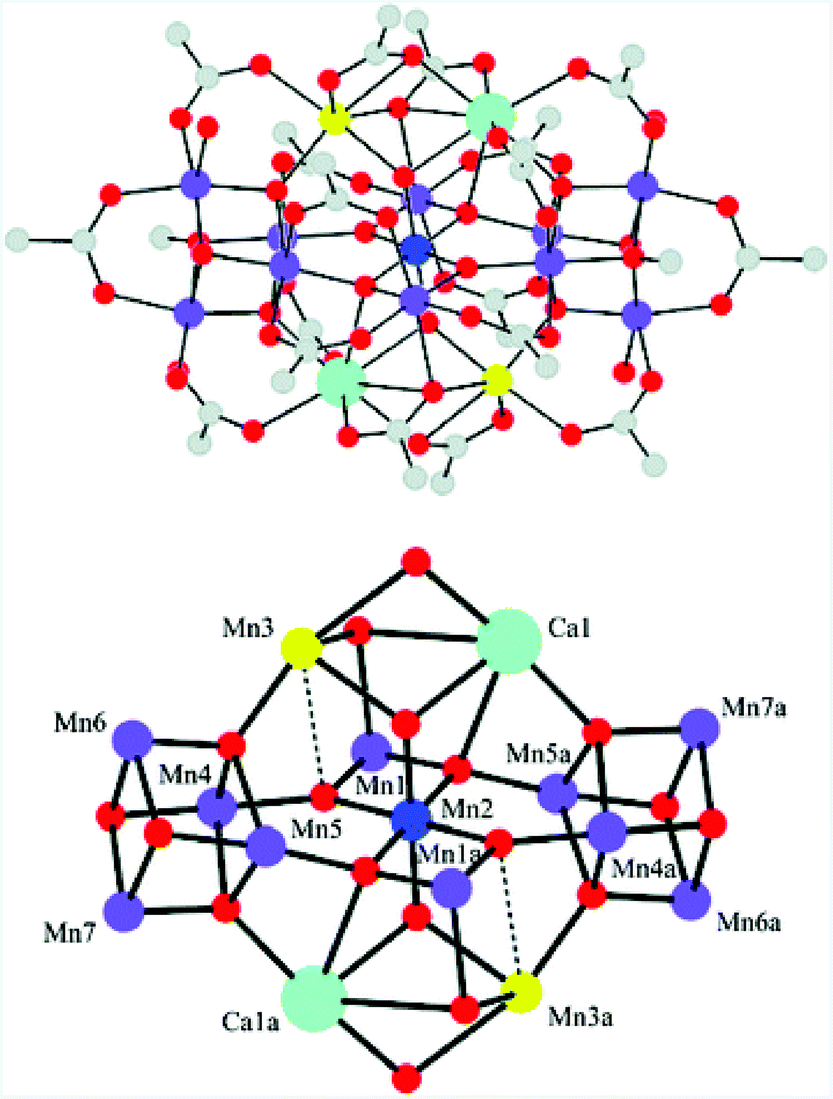 | ||
| Fig. 19 Structure of the [Mn13Ca2O10(OH)2(OMe)2(O2CPh)18(OH2)4] cluster (38) containing [Mn4CaO4] sub-units. Color code: cyan (Ca), blue (MnIV), violet (MnIII), yellow (MnII), red (O), and grey (C). Reprinted from ref. 141 with permission from the Royal Society of Chemistry. | ||
The heterometallic [MnIII3CaNa] cluster 39 based on a Schiff base ligand (Fig. 20) was recently reported by Reedijk and co-workers.143 This Mn3Ca cluster was found to evolve O2 using NaOCl, Oxone® or tBuOOH as a chemical oxidant. However, the molecular nature of the developed cluster was not thoroughly investigated in order to rule out the formation of heterogeneous metal oxides as the active catalytic entity.
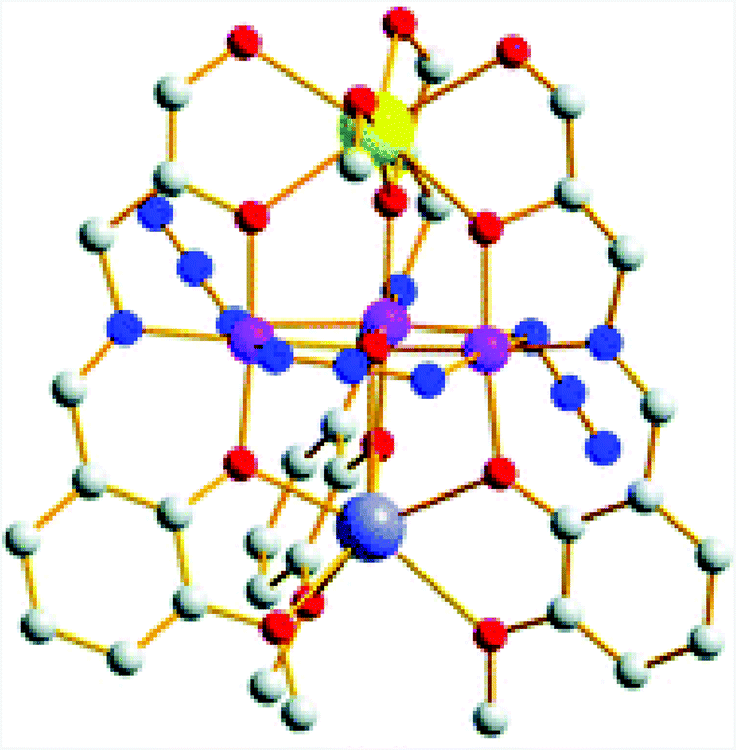 | ||
| Fig. 20 Structure of the heterometallic [MnIII3CaNa] cluster 39. Hydrogen atoms are omitted for clarity. Color code: blue (N), large, dark-gray (Na), gray (C), red (O), violet (Mn), and yellow (Ca). Adapted from ref. 143 with permission from the Royal Society of Chemistry. | ||
Perhaps the most well-studied tetranuclear Mn mimic is the [Mn4O4(dpp)6] cubane 40 (dpp = diphenylphosphinate), which contains a cubical [Mn4O4]6+ core surrounded by six facially bridging bidentate diphenylphosphinate groups coordinated to the Mn centers (Fig. 21).144–146 Mn cubanes, such as the [Mn4O4(dpp)6] cubane 40, self-assemble from mononuclear precursors145 or from dimeric complexes housing a [Mn2O2]3+ core in the presence of phosphinate ligands.144 The three oxo units and the three anionic phosphinate ligands provide an electron-rich environment for the Mn centers. The electrochemical properties of the [Mn4O4(dpp)6] cubane 40 have also been investigated.147 Electrochemical oxidation of the [Mn4O4]6+ core in cubane 40 occurs at a potential of ∼0.94 V vs. NHE and generates the one-electron oxidized MnIIIMnIV3 core. The redox potentials are ligand-dependent, indicating that they can be tuned using supporting phosphinate ligands with stronger electron donation.148,149
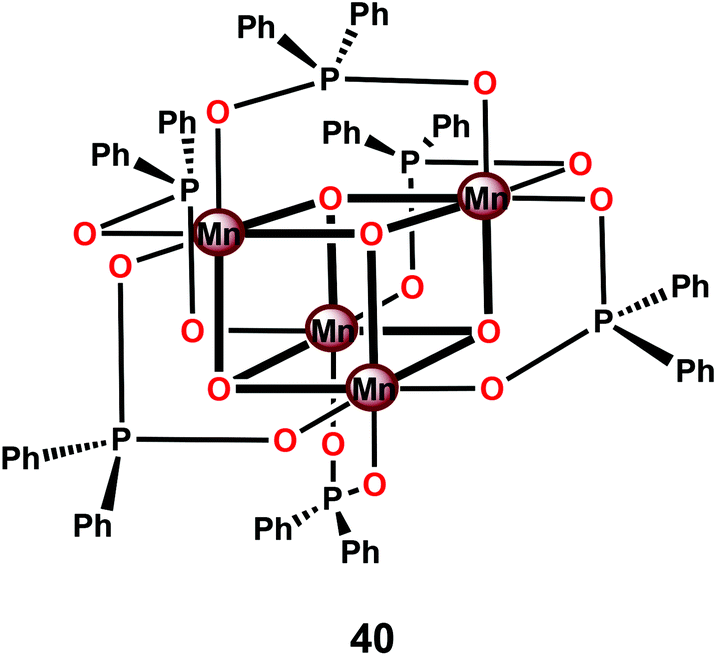 | ||
| Fig. 21 Structure of the [Mn4O4(dpp)6] cubane 40. dpp = diphenylphosphinate. | ||
The [Mn4O4(dpp)6] cubane 40 has also been reported to liberate stoichiometric amounts of O2 upon UV-light irradiation. The necessity for one of the phosphinate ligands to dissociate before O2 evolution can occur was demonstrated by isotopic labeling experiments in combination with mass spectrometry, with O–O bond formation occurring via direct coupling of two bridging oxo groups.150–153 Furthermore, Mn cubane 40 has been suspended in Nafion, a proton-conducting polymer matrix, for sustained O2 evolution. The generated Nafion154 polymer films were deposited on a variety of electrodes and reached TONs of >1000 and rates of 0.075 O2 molecules per s per Mn4 cluster at an overpotential of 0.38 V at pH 6 when illuminated with light of wavelength 275–750 nm.155–159 A photoelectrochemical cell has also been designed where the Nafion film embedded with cubane 40 was coupled to a [Ru(bpy)3]2+-type sensitized TiO2 layer (Fig. 22). The produced photoelectrochemical cell was able to catalyze the photooxidation of H2O using visible-light as the sole energy source, reaching TONs of 13 O2 molecules per Mn cluster and TOFs of ∼0.013 O2 molecules per s per Mn cluster.160 However, a recent report suggests that bulk electrolysis of the Nafion embedded cubane causes it to dissociate into MnII compounds. These compounds are subsequently reoxidized to produce nanoparticles composed of MnIII/IV oxides; thus the original Mn cubane cluster merely serves as a precursor to the catalytically active nanoparticle material.161
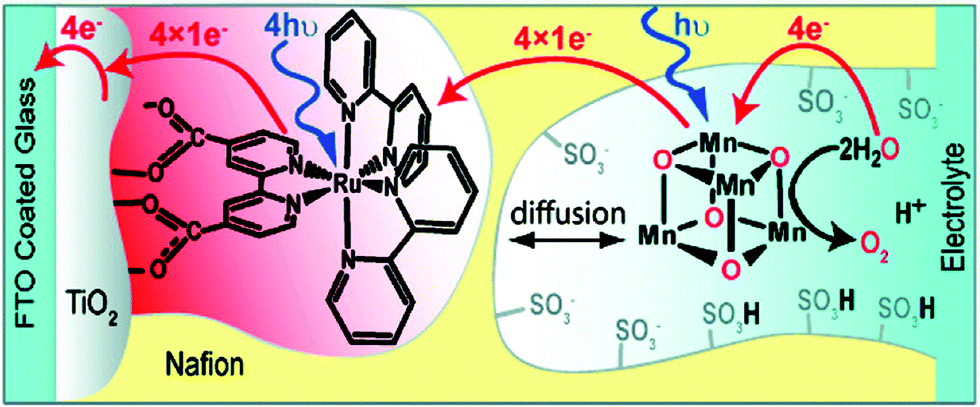 | ||
| Fig. 22 Schematic depiction of the photoelectrochemical cell containing Mn cubane 40. Reprinted with permission from ref. 160. Copyright 2010 American Chemical Society. | ||
Ca2+ is essential in the natural OEC for triggering O–O bond formation and O2 release and tuning the electrochemical properties of the Mn4Ca cluster.162–164 Agapie and co-workers therefore recently synthesized a [Mn3CaO4]6+ cubane (41) that models the Mn4Ca core of the OEC. Structural and electrochemical comparison between the [Mn3CaO4]6+ cubane 41 (Fig. 23, left) and a related Mn4O4 cubane (42, Fig. 23, right) demonstrated that the redox-inactive metal facilitates access to higher redox states and the assembly of the cluster.165 Subsequent work focused on the synthesis of a series of [Mn3M(μ4-O)(μ2-O)] clusters166 (where M = Na+, Ca2+, Sr2+, Zn2+ or Y3+) and [Mn3M′O4] clusters167 (where M′ = Sr2+, Zn2+, Sc3+, Y3+; see Fig. 24 for the structure of the Mn3ScO4 cubane 43). Here, the redox-inactive metal was shown to modulate the redox potentials of the heterobimetallic Mn clusters. These studies have provided a methodology with unprecedented structural control for the rational synthesis of Mn-based mimics of the Mn4Ca core of the OEC.168–171 The [Mn3M′O4] cubanes have recently been dropcast on ITO or glassy carbon disk electrodes and used as precursors to heterogeneous electrocatalysts for H2O oxidation.172 Multinuclear clusters of the type [Mn12O12] have also been demonstrated to be viable H2O oxidation electrocatalysts.173
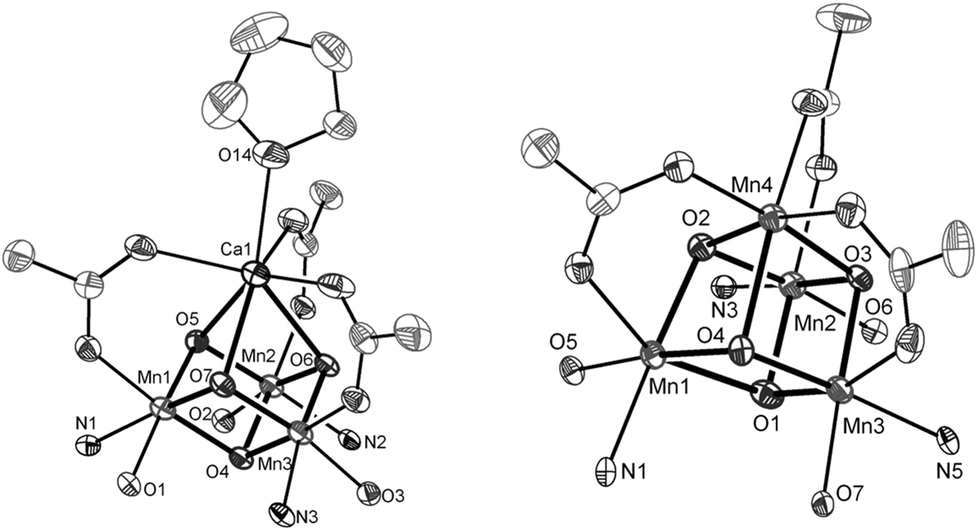 | ||
| Fig. 23 Structures of (left) the [Mn3CaO4]6+ cubane core in 41 and (right) the Mn4O4 cubane core (42). Adapted with permission from ref. 165. Copyright 2011 American Association for the Advancement of Science (AAAS). | ||
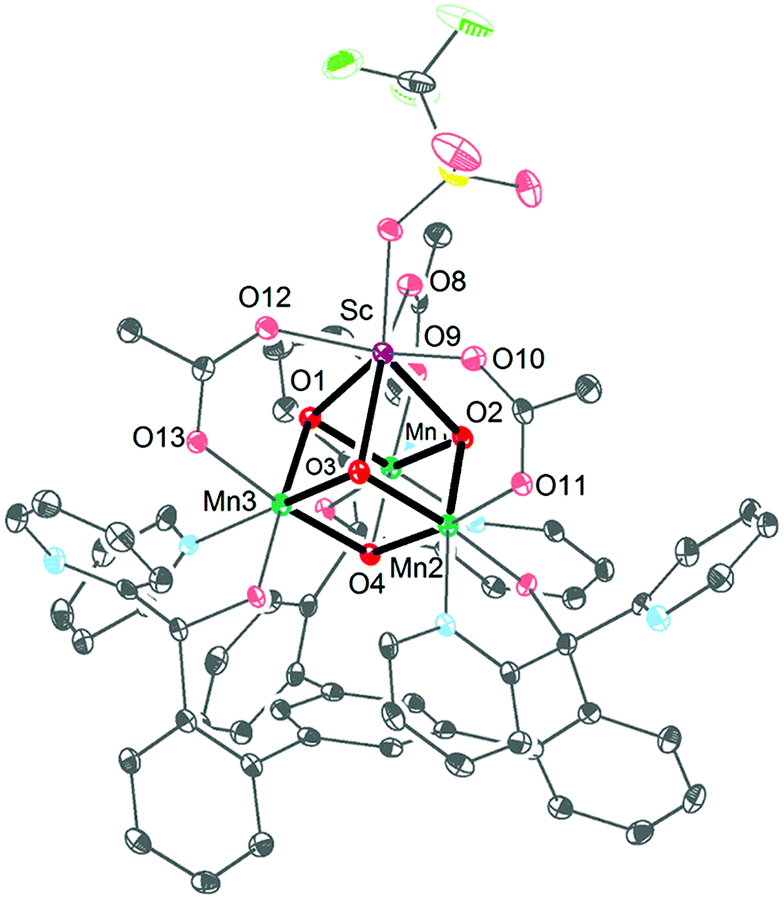 | ||
| Fig. 24 Structure of the Mn3ScO4 cubane 43. Reprinted with permission from ref. 169. Copyright 2013 American Chemical Society. | ||
Mechanistic understanding of the biological Mn4Ca cluster in the OEC at a molecular level is highly desirable for the rational design of viable WOCs. The collective work on the Mn-based model clusters has provided valuable insight into the properties and function of the natural OEC. Given the interesting properties associated with the Mn cluster mimics, it is expected that this research area will continue to expand and thus offers a promising future for producing artificial model systems for activation of small molecules, such as H2O.
5. Homogeneous Fe-based complexes for water oxidation
Fe is an essential element in a variety of enzymes and perhaps the most important transition metal from a biological point of view. Fe catalysis has emerged as a prominent research field and in recent years there has been a tremendous development in this area.174 Fe is known to have a rich redox chemistry and to adopt a variety of redox states. In addition to having access to this broad spectrum of redox states, Fe-complexes have the ability to engage in both one- and two-electron processes—essential features for H2O oxidation catalysis. The large number of spin states accessible to Fe should also make it amenable to tuning, by varying the ligand environment. Although Fe is associated with these attractive properties, relatively few examples exist of Fe-catalyzed H2O oxidation.5.1. Mononuclear Fe-based catalysts
Seminal work on Fe-catalyzed H2O oxidation has already been carried out in the 1980s.175 However, the first examples of homogeneous Fe-based WOCs were reported by Collins and co-workers in 2010.176 In order to stabilize the Fe center, the authors employed tetraamido macrocyclic ligands (TAMLs). These ligands have previously been shown to efficiently activate O2 and peroxides to produce FeIV2–μ-oxo species.177–179 In aqueous solutions, the FeIV2–μ-oxo species have been shown to participate in a pH-dependent equilibrium with a monomeric FeIV–oxo complex.180 Furthermore, the initial FeIII–TAML complexes react with peracids to produce the corresponding FeV–oxo complexes.181 The diverse and high reactivity associated with the Fe–TAML complexes thus rendered these catalysts attractive targets for use in H2O oxidation.Fe–TAML complexes 44–48 (Fig. 25) were therefore investigated toward H2O oxidation. In the presence of CeIV as a chemical oxidant, fast O2 evolution was observed for complexes 45–48. However, no O2 production was detected for the least acid stable Fe–TAML complex 44. The rate of O2 evolution was shown to be highly dependent on the electron-withdrawing ability of the macrocyclic ligand. Fe–TAML complex 48 gave the highest TOF, 1.3 s−1, and was shown to display a first-order dependence on the catalyst concentration, implying that O–O bond formation occurs at a single metal center. H2O oxidation could also be driven using NaIO4 as a chemical oxidant. Mechanistic studies using NaIO4 as an oxidant also concluded that an FeIV–μ-oxo–FeIV species is involved in the oxidation process.176 Although this seminal work highlighted that Fe-based WOCs could be designed, the catalysts were prone to undergo fast deactivation/decomposition, highlighting that more robust WOCs need to be targeted. In an attempt to improve the longevity of the Fe–TAML complexes, they were subsequently immobilized on electrodes for electrocatalytic H2O oxidation.182
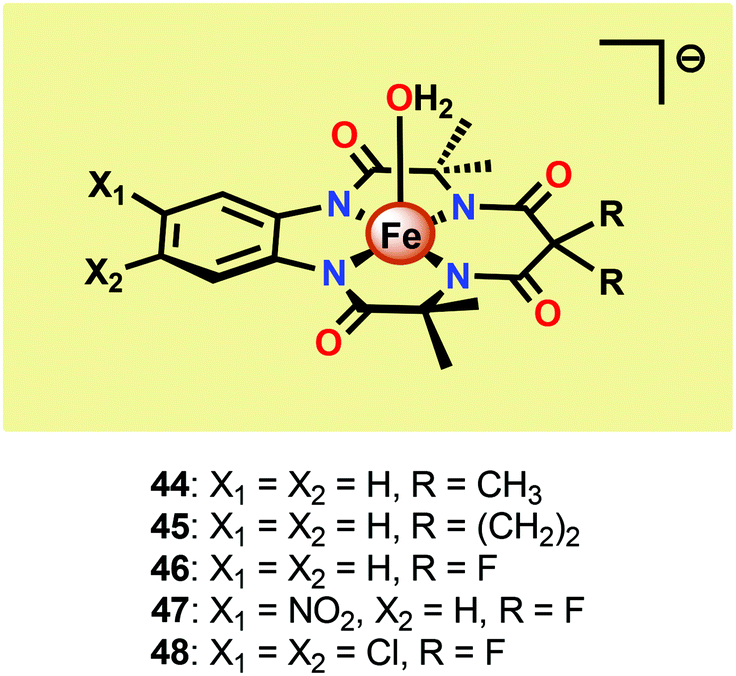 | ||
| Fig. 25 Structure of FeIII–TAML complexes 44–48. | ||
Dhar and co-workers subsequently employed the modified Fe–TAML complex 49 (Fig. 26) to carry out light-driven H2O oxidation using [Ru(bpy)3]2+ as a photosensitizer and Na2S2O8 as a sacrificial electron acceptor.183 The photocatalytic system was shown to produce a TON of 220 and a TOF of 0.67 s−1. An FeV–oxo intermediate was proposed to be generated in this system as supported by EPR, UV-vis and HRMS studies. Based on these results and related studies,184–188 the authors proposed a mechanism, outlined in Scheme 4, where the generated FeV–oxo species 52 undergoes nucleophilic attack by a H2O molecule to form a FeIII–hydroperoxo (FeIII–OOH) species (53). Subsequent oxidation of this FeIII–OOH species leads to O2 liberation and regenerates the starting aqua complex 49, FeIII–OH2.183
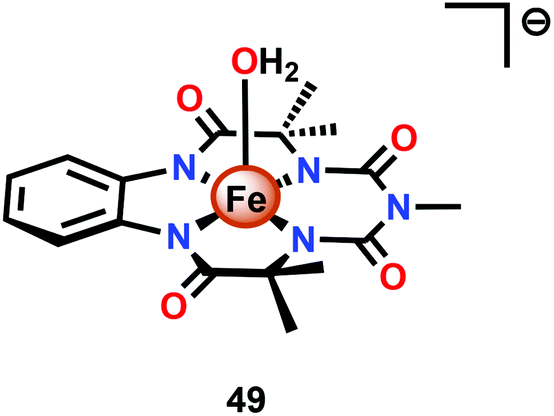 | ||
| Fig. 26 Structure of modified Fe–TAML complex 49. | ||
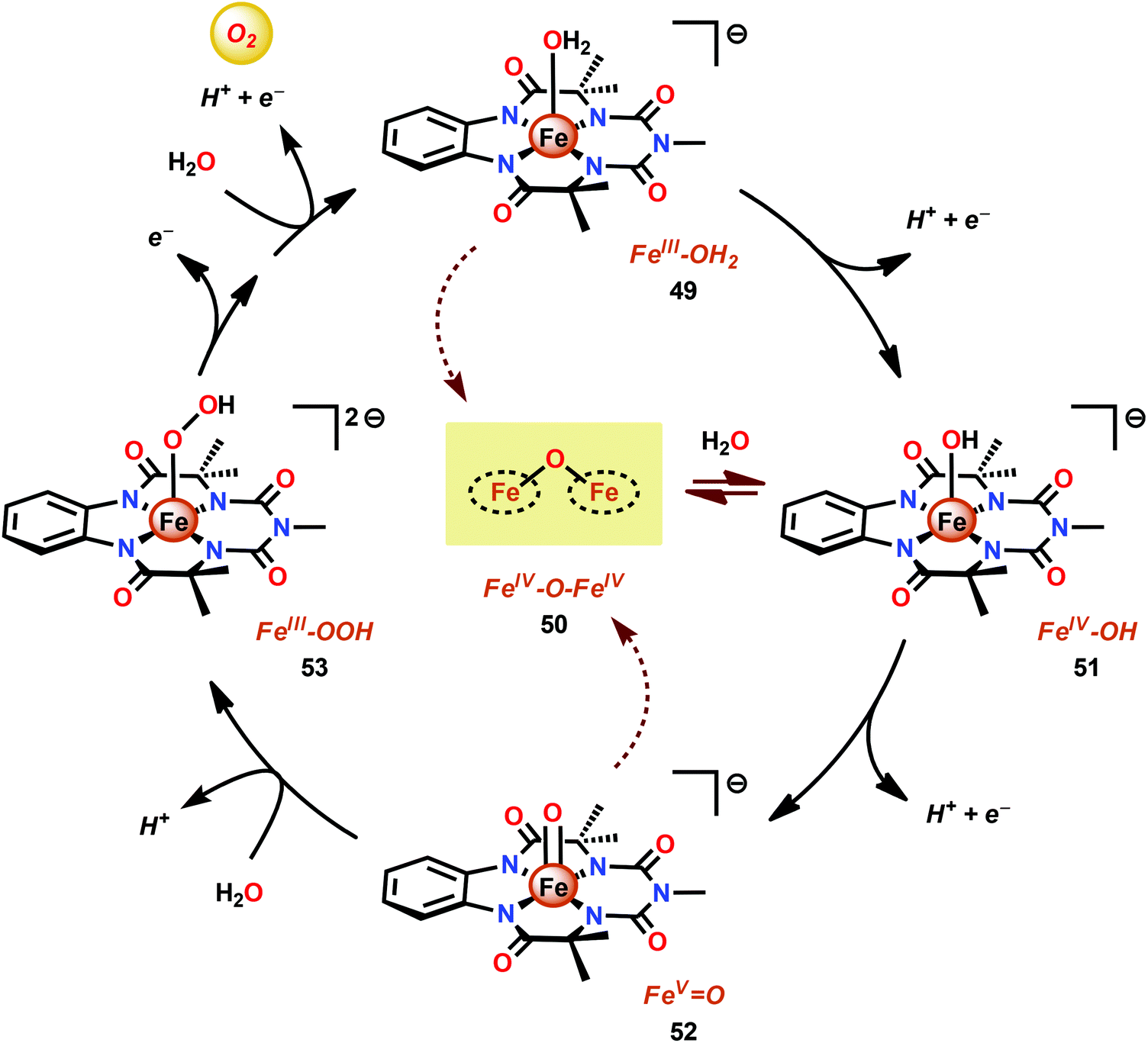 | ||
| Scheme 4 Proposed mechanism for H2O oxidation catalyzed by modified Fe–TAML complex 49. | ||
An alternative mechanism for the oxidation of H2O by the Fe–TAML complexes was presented by Liao and Siegbahn.189 This mechanism was proposed to involve the formation of a formal FeVI–oxo species, which upon further inspection was better described as having an FeV center with a ligand cation radical. Three distinct ligand modification pathways were also realized: (1) H2O or (2) nitrate (the anion originating from the CeIV oxidant) attack on the ligand framework, and (3) amide oxidation. The pathway involving H2O attack on the ligand was associated with a low barrier and results in the opening of the benzene ring. The observed reactivity pattern for the Fe–TAML complexes 44–48 could also be rationalized as it was found that the barrier for O–O bond formation decreased with electron-withdrawing substituents. However, the introduction of electron-withdrawing units increases the redox potentials and adds an additional energetic penalty. These effects need to be carefully balanced in future designs of ligands.
Another class of Fe-based WOCs was described by Lloret-Fillol and co-workers.190 The authors evaluated the catalytic activity of a series of Fe complexes based on tetra- and pentadentate ligand motifs (Fig. 27). From the study, it could be shown that Fe complexes 54–58 having two open coordination sites in a cis configuration were competent WOCs. The structural requirement was further confirmed by complexes 59 and 60, with a trans configuration or possessing only a single coordination site, which were found to be inactive. Of the evaluated catalysts, Fe complex 55α afforded a TON of >1000 and a TOF of 0.062 s−1 using NaIO4 as a chemical oxidant. Mechanistic investigations employing the dimeric FeIII,III2(μ-OH)(μ-O) complex of catalyst 55α showed that the dimeric complex was associated with a lower reaction rate and a different kinetic behavior, discarding such dimeric structures as essential active intermediates in the studied Fe-based system. Addition of 6 equivalents of CeIV oxidant resulted in the appearance of new bands in the UV-vis, a species characteristic of an FeIV–oxo intermediate. The persistence of this FeIV–oxo indicated that this species is not responsible for mediating O–O bond formation, and that species of higher valency are needed for O–O bond forming events.
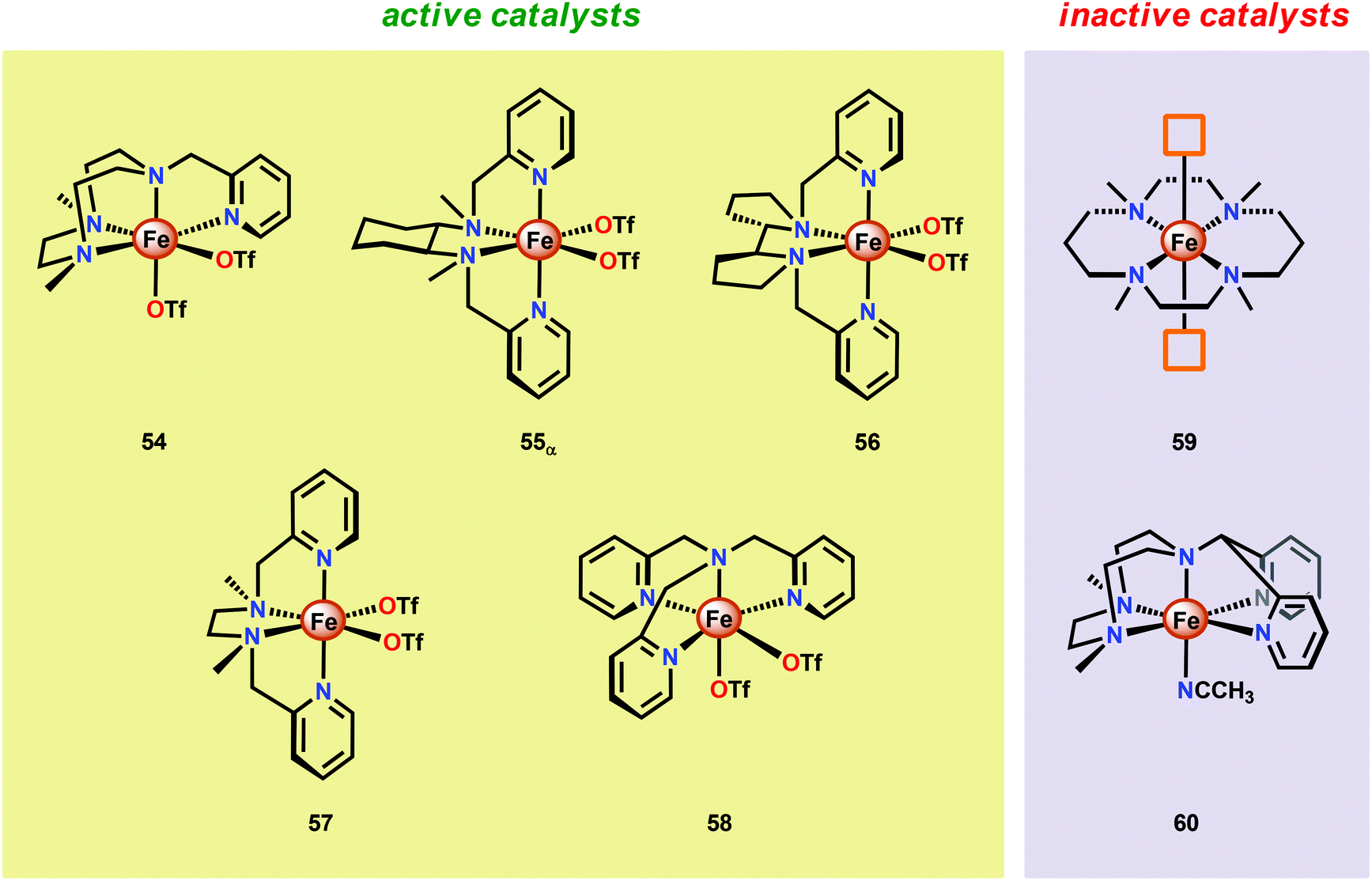 | ||
| Fig. 27 Structures of Fe complexes 54–60 containing tetra- and pentadentate nitrogen-based ligands. | ||
Several mechanistic studies on Fe complexes of the type 54–58 have also been carried out.191–195 A follow-up study by Lloret-Fillol and co-workers involved the electronic tuning of the Fe-based catalyst 54 in which a family of substituted Fe complexes (61–65, Fig. 28) were synthesized. Using Hammett parameters, the rate of O2 evolution was found to correlate with the electronic nature of the introduced substituents, with electron-withdrawing substituents favoring O2 production. The non-innocent effect of the CeIV cation was also observed in which a FeIV(OH)(O–CeIV) adduct was proposed to be involved prior to the O–O bond forming step.191
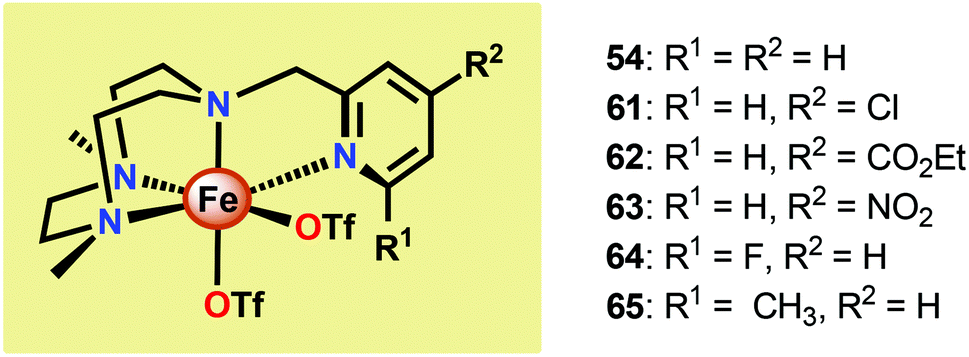 | ||
| Fig. 28 Fe complexes 54 and 61–65. | ||
A recent study identified FeIV(O)(O–CeIV) as the key reaction intermediate in H2O oxidation using Fe complex 55.193 Here, CeIV was found not to act merely as an inner-sphere oxidant but was revealed to generate an FeIV(μ-O)CeIV species, crucial for the catalytic reaction to proceed. The oxidation of H2O with Fe-based complexes such as 55 not only requires the presence of two open coordination sites in a cis configuration but also a framework that allows the generation of FeIV(μ-O)CeIV. The isomeric Fe complexes 55α and 55β also displayed different reactivity, arising either from steric encumbrance or electronic effects. While the complex 55β did not evolve O2, the α-isomer was found to produce O2 according to the pathways highlighted in Scheme 5. These findings could thus provide valuable information for future evaluations of earth-abundant WOCs.
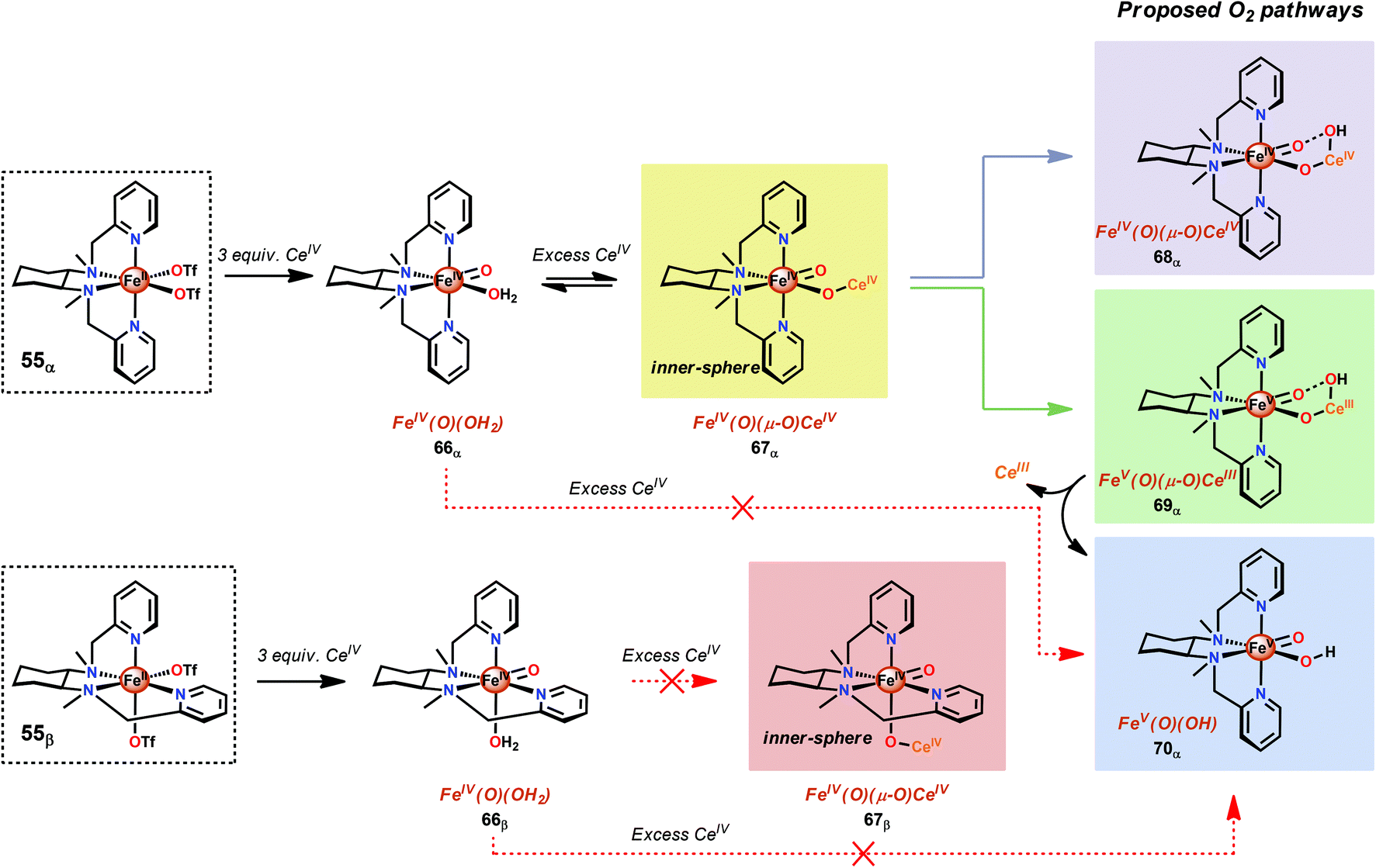 | ||
| Scheme 5 Reactivity differences between the isomeric Fe complexes 55α and 55β. | ||
The relative ease by which Fe complexes housing tetra- and pentadentate nitrogen-based ligands can be accessed has attracted attention from several research groups.196–198 The phosphonate modified Fe complex 71 (Fig. 29) has for example been covalently anchored to WO3 electrodes for photoelectrochemical H2O oxidation. The modified electrodes exhibited an increase in photocurrent of ∼60% whereas electrodes modified with FeCl2 or with the pristine tetradentate nitrogen ligand did not display any increase in photocurrent. Although the authors could not preclude that the phosphonate containing Fe complex 71 tethered to WO3 could potentially be a mere precatalyst for H2O oxidation, the observed rate enhancement is noteworthy for the modified WO3 electrodes.199
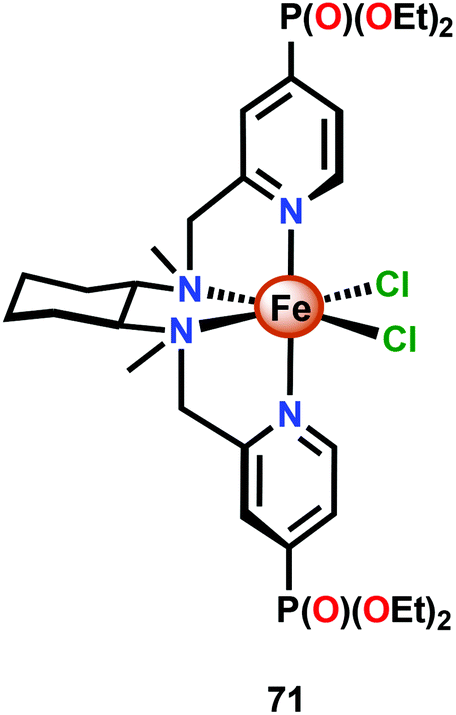 | ||
| Fig. 29 Structure of the phosphonate-functionalized Fe complex 71. | ||
The group of Meyer developed the mononuclear [Fe(dpaq)(OH2)]2+ complex (72, Fig. 30; dpaq = 2-[bis(pyridine-2-ylmethyl)]amino-N-quinolin-8-yl-acetamido),200 which has previously been reported to mediate alkane hydroxylation with H2O2 as an oxidant.201 Meyer and co-workers found that the [Fe(dpaq)(OH2)]2+ complex 72 was able to catalyze electrochemical H2O oxidation in propylene carbonate–water mixtures. Electrochemical examination revealed that the Fe catalyst exhibited a quasi-reversible one-electron wave at 0.38 V vs. NHE corresponding to the [FeIII–OH2]2+/[FeII–OH2]+ redox couple. At higher potentials (1.58 V), an irreversible two-electron wave appeared, which was assigned to the oxidation of [FeIII(OH2)]2+ to [FeV(O)]2+. Following this wave, an increase in current was observed. The peak current was shown to display a first-order dependence on the catalyst concentration, suggesting a rate-limiting reaction between [FeV(O)]2+ and H2O. This is consistent with a single-site mechanism for H2O oxidation as depicted in Scheme 6.200
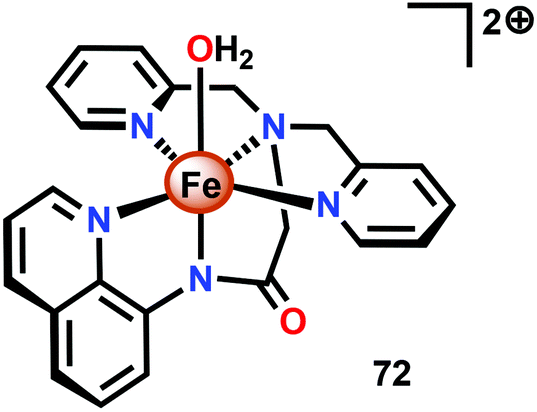 | ||
| Fig. 30 Structure of the [Fe(dpaq)(OH2)]2+ complex 72. | ||
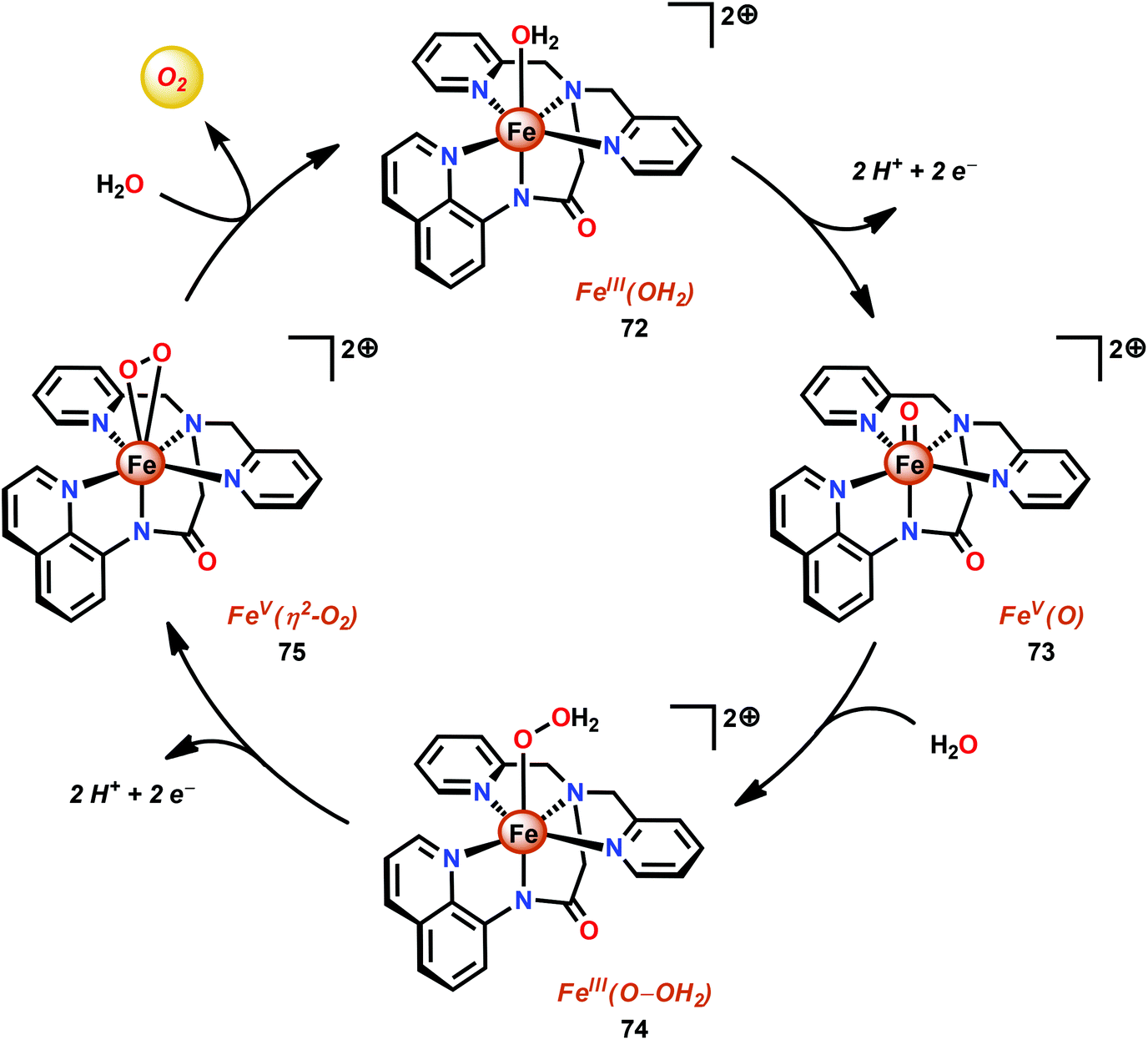 | ||
| Scheme 6 Proposed mechanism for electrocatalytic H2O oxidation by the [Fe(dpaq)(OH2)]2+ complex 72. | ||
A series of Fe-complexes containing pendant bases have also been synthesized by Yang and co-workers (76–82, Fig. 31).202 The authors envisioned that incorporation of bases into the second coordination sphere would facilitate PCET events and lead to enhanced reaction kinetics. However, Fe complexes 78–80 were shown to be inactive catalysts as they were not able to reach the critical FeIVO state. For Fe complexes 81 and 82, an undesirable change in coordination took place, in contrast to what was observed for the analogous complexes lacking the introduced heteroatoms. Of the developed Fe complexes, only the pyridazine containing complexes 76 and 77 displayed H2O oxidation activity similar to the analogous complexes lacking the ancillary proton relays. Although the introduction of proton relays did not improve the catalytic activity of the Fe complexes, the concept of designing ligand motifs containing strategically functionalized moieties able to participate in PCET is an attractive feature for accessing improved Fe-based WOCs.
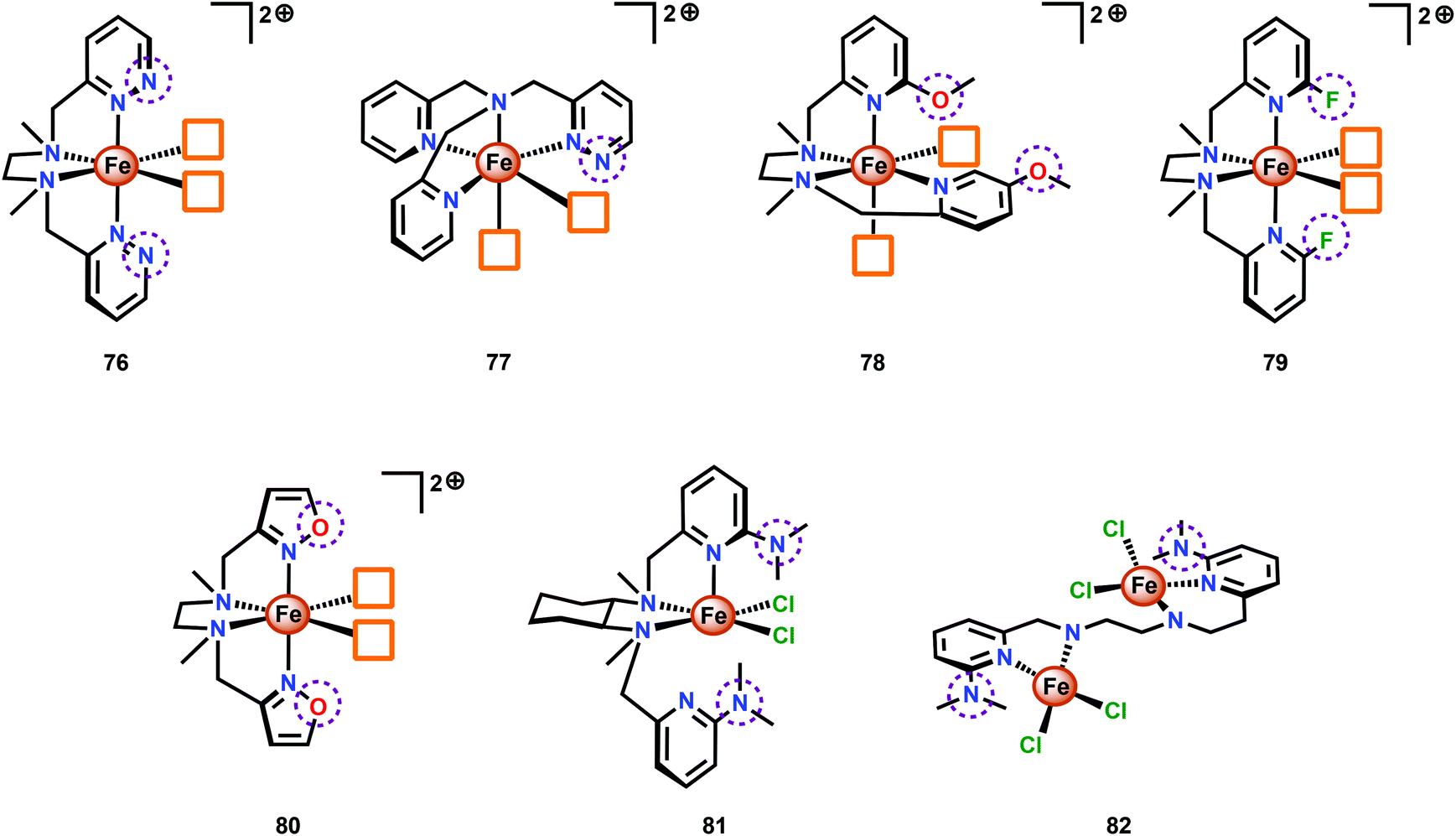 | ||
| Fig. 31 Structures of Fe-based complexes 76–82 containing proton relays. | ||
It needs to be stressed that the molecular nature of Fe-based complexes is highly dependent on the reaction conditions.203 Additionally, the catalytic activity of Fe-based complexes and the formation of metal nanoparticles are extremely susceptible to electronic and structural features. This intricate behavior is elegantly illustrated by recent examinations conducted by the groups of Lau204 and Fukuzumi.205 In these studies, the Fe-based complexes were shown to act as homogeneous catalysts under more acidic conditions while more basic conditions facilitated the formation of Fe–oxide nanoparticles.
The rational construction of ligand scaffolds, which allow high-valent Fe species to be generated and stabilized, still remains an essential challenge that needs to be addressed if more robust Fe-based catalysts are to be developed. As further insight into the mechanistic details of Fe-catalyzed H2O oxidation emerges, new ways to design more efficient Fe-based systems will most certainly be devised.
5.2. Dinuclear Fe-based catalysts
Although a variety of mononuclear Fe-based complexes have been shown to catalyze H2O oxidation, examples of dinuclear Fe complexes capable of mediating this transformation are currently limited. Initial observations on dinuclear Fe species being involved in the catalytic process were reported by Najafpour and co-workers.206 The authors selected the tris(2-pyridylmethyl)amine (tpa) ligand and synthesized the dinuclear FeIII,III2 complex [(tpa)(OH2)Fe(μ-O)Fe(OH2)(tpa)]4+ (83, Fig. 32) which contains a μ-oxo bridge. When the dinuclear Fe complex 83 was examined as a WOC using CeIV as a chemical oxidant, the rate for O2 evolution was found to be significantly higher206 than for its mononuclear analog 58 (see Fig. 27).190 The rate was found to be first order in both CeIV and catalyst 83, suggesting that O–O bond formation occurs within the dinuclear framework.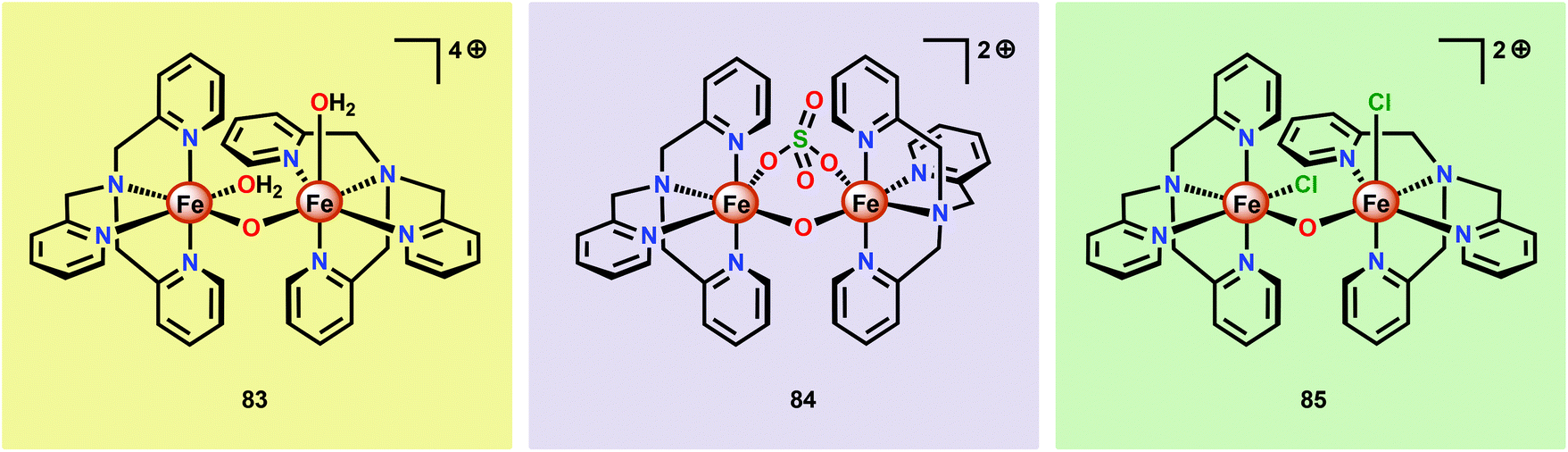 | ||
| Fig. 32 Structures of the dinuclear Fe complexes [(tpa)(OH2)Fe(μ-O)Fe(OH2)(tpa)]4+ (83), [(tpa)Fe(μ-O)(μ-SO4)Fe(tpa)]2+ (84) and [(tpa)(Cl)Fe(μ-O)Fe(Cl)(tpa)]2+ (85). | ||
Subsequent studies on dinuclear Fe-based WOCs by the groups of Sakai207 and Ma197 also centered on using the tpa ligand. The group of Sakai synthesized the dinuclear Fe complex [(tpa)Fe(μ-O)(μ-SO4)Fe(tpa)]2+ (84) and established that it mediated H2O oxidation using NaIO4 as a chemical oxidant. DLS analysis suggested that the dinuclear Fe complex served as a homogeneous WOC and that Fe–oxide nanoparticles were not formed under the investigated reaction conditions. Kinetic experiments showed a first-order dependence on the catalyst concentration. Unexpectedly, the rate of O2 evolution exhibited a dramatic increase when the pH was decreased. Such an effect is uncommon since the rate-determining step is usually nucleophilic attack of H2O or coupling of two oxyl units. It was speculated that the observed rate enhancement could be due to reduced anation at low pH.207 Ma and co-workers subsequently investigated the catalytic activity of the Fe complex [(tpa)(Cl)Fe(μ-O)Fe(Cl)(tpa)]2+ (85). In aqueous acetate buffer solutions, using Oxone® as a chemical oxidant, Fe complex 85 exhibited high O2 evolution activity with TONs reaching 2380 and TOFs of 2.2 s−1. HRMS suggested that upon dissolution in acetate buffer solutions, the acetate bridged dinuclear Fe species [(tpa)Fe(μ-O)(μ-OAc)Fe(tpa)]3+ was formed. This was proposed to be essential because the two free coordination sites are constrained in a cis fashion, allowing for efficient H2O oxidation.197
Thummel and co-workers recently revealed that the dinuclear FeIII,III2 complex [(ppq)(OH2)Fe(μ-O)Fe(Cl)(ppq)]3+ (86, Fig. 33; ppq = 2-(pyrid-2′-yl)-8-(1′′,10′′-phenanthrolin-2′′-yl)-quinoline) was able to mediate H2O oxidation.208 The tetradentate ppq ligand was previously employed in the synthesis of a Co-based complex for H2 evolution.209 Treating FeCl3 with the ppq ligand afforded the μ-oxo bridged dinuclear FeIII,III2 complex 86 in 28% yield. Electrochemical analysis of Fe complex 86 revealed a reversible wave at ∼0.21 V vs. NHE and another quasi-reversible wave at ∼0.69 V. The first event could be assigned to a two-electron process and was tentatively attributed to the simultaneous oxidation of the two FeIII centers, FeIII,III2 → FeIV,IV2. This produced [FeIVFeIV–OH2] could undergo disproportionation with loss of two protons to afford a [FeIIIFeVO] species. H2O oxidation was evaluated using CeIV as a chemical oxidant. Upon addition of the oxidant to a solution of complex 86, rapid O2 evolution was observed. As a comparison, the mononuclear Fe complex 87 (Fig. 33) was synthesized and examined as a WOC. Although the mononuclear complex 87 was able to oxidize H2O, the dinuclear Fe complex 86 was found to be more active, affording a TON of ∼1000 and a TOF of 2.2 s−1. An essential question was whether the dinuclear Fe complex 86 retains its dimeric structure or whether it dissociates to yield a monomeric species in solution. UV-vis analysis and the apparent first-order dependence on the catalyst concentration suggested that the Fe complex remains intact during the catalytic oxidation. The involvement of nanoparticles in the studied catalytic system could successfully be discarded based upon DLS measurements.208 The collective work on dinuclear Fe-based catalysts highlights that dinuclear species have the potential of generating more active catalytic systems compared to their mononuclear counterparts. The continued endeavors in this field will likely result in a rapid expansion of viable, and more robust, dinuclear Fe-based catalysts. This is further highlighted by the fact that a recently developed homogeneous pentanuclear Fe complex is able to facilitate O–O bond formation with an impressive efficiency.210
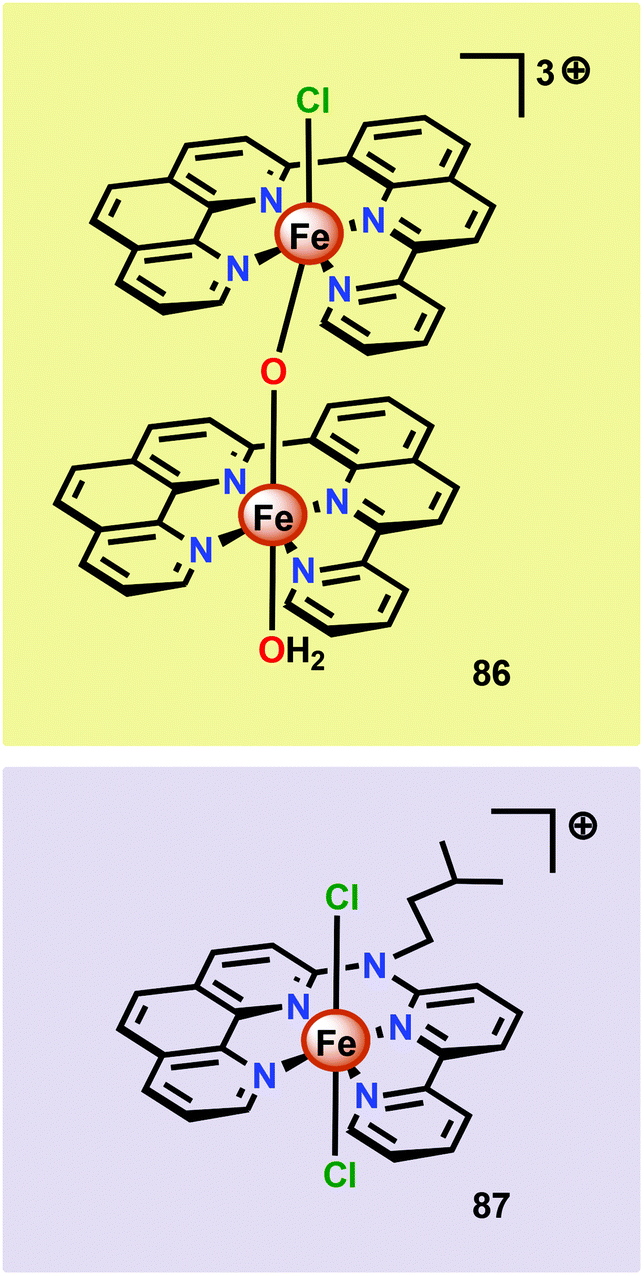 | ||
| Fig. 33 Structure of the dinuclear [(ppq)(OH2)Fe(μ-O)Fe(Cl)(ppq)]3+ complex 86 and the mononuclear Fe complex 87. | ||
6. Co-based systems for water oxidation
Inorganic Co salts have been studied as potential WOCs since the late 1960s.175,211–213 Although these early attempts were rather unsuccessful due to precipitation of heterogeneous species, they suggested the involvement of CoIV species during the catalytic process.214 Spurred by the findings of Nocera and co-workers that CoOx species (CoPi catalyst, Fig. 34),215–229 generated in situ under neutral conditions in phosphate buffer, exhibited O2 evolution activity, several research groups have attempted to prepare homogeneous molecularly defined Co-based catalysts for the oxidation of H2O. | ||
| Fig. 34 (Left) Depiction of the Mn4Ca cluster in the OEC. (Middle) Structure of the CoPi catalyst as determined by extended X-ray absorption fine structure (EXAFS). (Right) The CoPi structure rotated by 45° to highlight the edge sharing octahedra. Adapted with permission from ref. 225. Copyright 2012 American Chemical Society. | ||
6.1. Co polyoxometalates (POMs)—structurally defined all-inorganic catalysts
In 2010, Hill and co-workers reported that Co-based polyoxometalates (POMs) were able to evolve O2 when driven by pregenerated or photochemically generated [Ru(bpy)3]3+.230–234 The all-inorganic Co-POM catalysts can be considered as the homogeneous alternative to metal oxides with the advantage of being well-defined and consisting of only inorganic elements. Prior to the report dealing with the Co-POMs, the groups of Hill and Bonchio had simultaneously shown that their Ru-analogs were efficient catalysts for the oxidation of H2O.235,236 With the goal of developing an earth-abundant POM, Hill and co-workers synthesized the tetranuclear Co-POM [Co4(OH2)2(α-PW9O34)2]10− (88, Fig. 35) and showed that it was capable of driving H2O oxidation with [Ru(bpy)3]3+.230 Subsequent studies revealed that Co-POM 88 also catalyzed photoinduced H2O oxidation using [Ru(bpy)3]2+ as a photosensitizer and persulfate (S2O82−) as a sacrificial electron acceptor, generating a TON of 224 and a quantum yield of 30%.231 Co-POM 88's H2O oxidation activity and other essential features have also been studied by other research groups.237–240 A recent report by the Hill group has shown that the structurally analogous Co-POM [Co4(H2O)2(VW9O34)2]10− is an extremely efficient WOC, reaching TOFs >1000 s−1 with quantum yields approaching 68%.241 Additional Co-POM derivatives have also been synthesized by the Wang group and shown to mediate photocatalytic H2O oxidation.242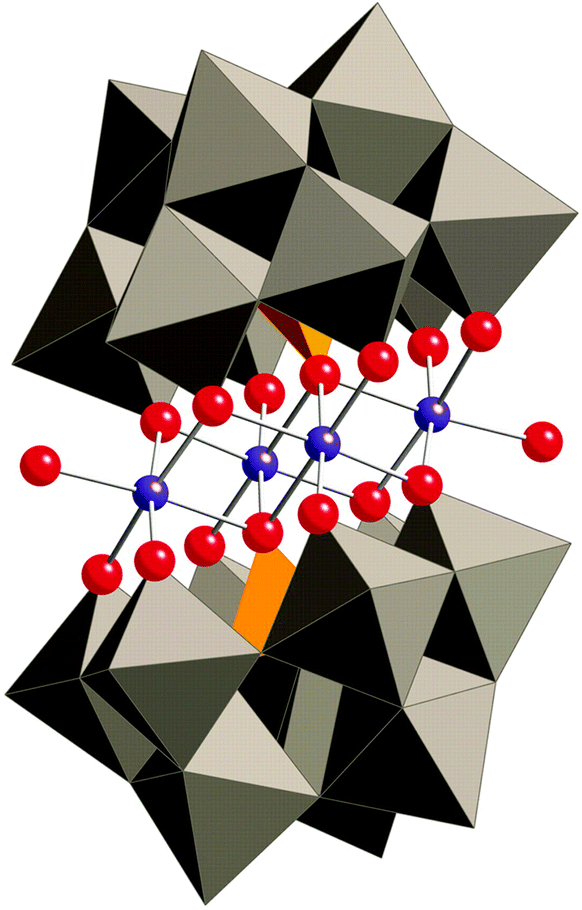 | ||
| Fig. 35 Structure of the tetranuclear Co-POM 88 ([Co4(OH2)2(α-PW9O34)2]10−). Reprinted with permission from ref. 230. Copyright 2010 American Association for the Advancement of Science. | ||
The original report on Co-POM 88 contained a rigorous evaluation of whether the observed catalytic activity originated from the homogeneous Co-POM 88 or whether decomposition resulted in Co oxides which were the real catalytic entities. From these extensive experiments it was suggested that Co-POM 88 did not decompose and operated via a homogeneous mechanism.230 Although Hill and co-workers concluded that Co-POM 88 was stable under the studied reaction conditions, subsequent reports challenged these conclusions, showing that POM 88 decomposed to Co oxides as the dominant catalyst.239,243 Additional studies revealed that the slight modifications of the reaction conditions influenced the operating mechanism and indicated that the conclusions from the original publication on Co-POM 88 were indeed correct.244,245 They further highlight that the homogeneity of WOCs is highly dependent on the reaction conditions, such as the buffer and pH, which needs to be considered when determining the involvement of heterogeneous metal oxide species during catalysis.246
The group of Galán-Mascarós subsequently reported on a high-nuclearity Co-POM, [Co9(OH)3(OH2)6(HPO4)2(PW9O34)3]16− (89, Fig. 36).247 The nonanuclear Co core is stabilized by three hydroxo and two hydrogen phosphate bridges. The nonanuclear Co-POM 89 was found to catalyze H2O oxidation using NaClO as a chemical oxidant. Experiments supported a homogeneous operating mechanism under chemical oxidation whereas slow release of Co was observed when electrochemical H2O oxidation was performed. However, formation of a Co oxide film on the electrode could be avoided by addition of excess 2,2′-bipyridine, which functions as a chelating agent for the liberated CoII or CoIII. A TON of ∼400 and a TOF of ∼0.1 were measured, with maintained O2 evolution activity for several days without any sign of fatigue or decomposition.247 The same group also synthesized an insoluble salt (Cs salt) of Co-POM 89 for integration into amorphous carbon paste electrodes. The catalytic activity of the modified electrodes was maintained in the solid state with constant rates for several hours.248
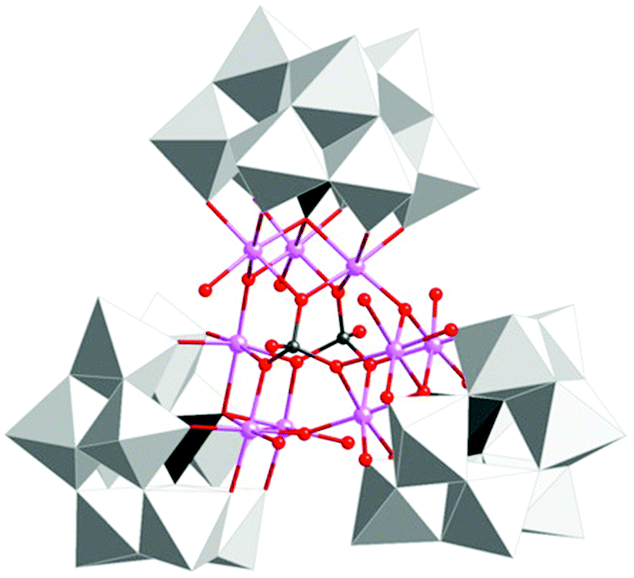 | ||
| Fig. 36 Structure of the nonanuclear Co-POM [Co9(OH)3(OH2)6(HPO4)2(PW9O34)3]16− (89). Adapted with permission from ref. 247. Copyright 2012 American Chemical Society. | ||
Additional examples of Co-based POMs that have been reported to mediate visible-light driven H2O oxidation include the mixed dinuclear Co-POM [CoIICoIII(OH2)W11O39]7− (90),249 the octanuclear Co-POM [(A-α-SiW9O34)2Co8(OH)6(OH2)2(CO3)3]16− (91),250 [CoII(bpy)3]6(H2bpy)[(CoIIbpy)2-(PMoVI8MoV4O40)]3[(CoIIbpy)(PMoVI8MoV4O40)] (92),251 [Co2Bi2(α-B-CoW9O34)2]14− (93),252 [Co2Mo10O38H4]6− (94)253 and [{Co(OH2)3}2{CoBi2W19O66(OH)4}]10− (95).254 From the plethora of studies conducted on Co-based POMs, it is clear that these catalysts can operate as robust and efficient molecular catalytic entities for the oxidation of H2O.
6.2. Co cubanes as mimics of the catalytic core in the oxygen-evolving complex
Inspired by the structural rearrangement of the Mn4Ca core in the OEC, researchers have devoted considerable efforts to synthesize complexes with similar cubane core structures. Early examples of such complexes include the Co cubanes Co4O4(pyr)4(OAc)4 (96)255,256 and [Co4O4(bpy)4(OAc)2]2+ (97) (Fig. 37).257,258 The Co4O4(pyr)4(OAc)4 (96) cubane can be easily prepared from Co(NO3)2, NaOAc and pyridine and contains a Co4O4 core, which makes this cubane and related systems attractive as potential WOCs. A seminal report by Dismukes and co-workers showed that cubane 96 indeed was able to promote light-driven H2O oxidation using a three-component system consisting of [Ru(bpy)3]2+ as a photosensitizer and Na2S2O8 as a sacrificial electron acceptor. This photocatalytic system generated a TON of 40 and a TOF of 0.02 s−1.259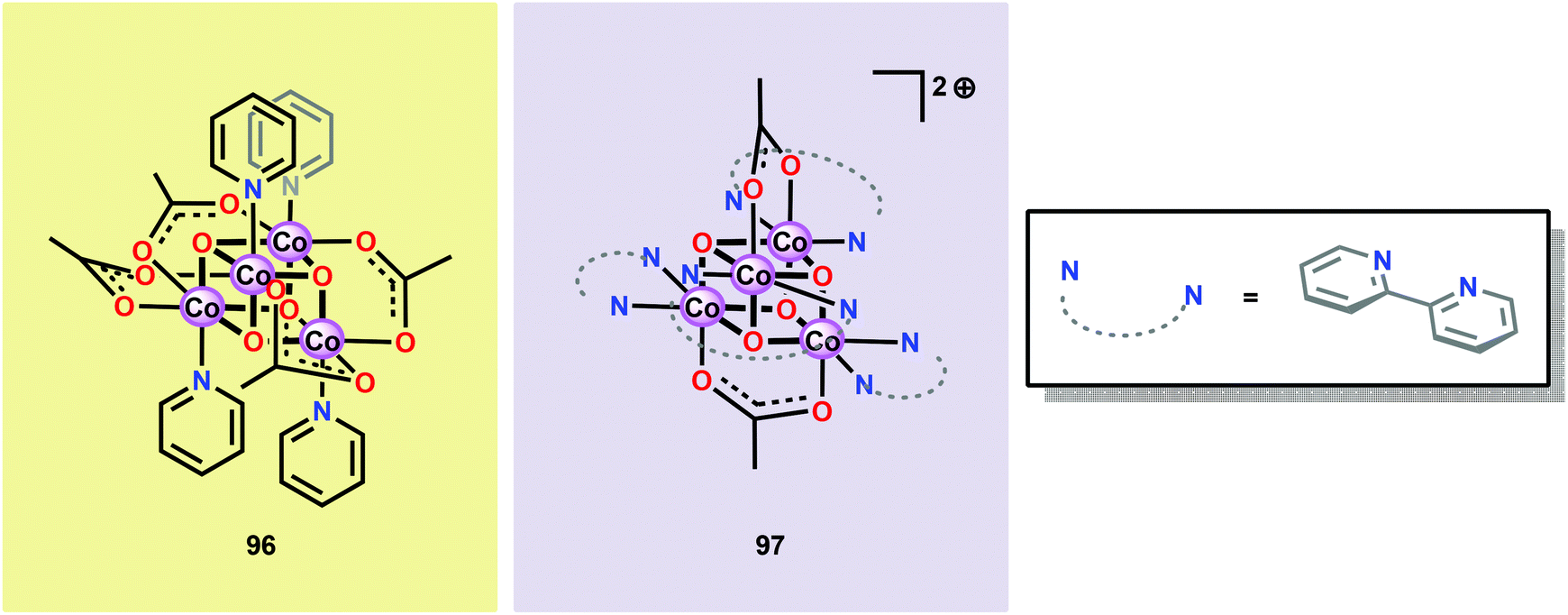 | ||
| Fig. 37 Structure of Co cubanes Co4O4(pyr)4(OAc)4 (96) and [Co4O4(bpy)4(OAc)2]2+ (97). | ||
Subsequent studies by Bonchio and co-workers involved the examination of isostructural analogs of Co cubane 96.260,261 The authors targeted Co cubanes 98–103 (Fig. 38) in order to study catalyst tuning for boosting the photocatalytic efficiency in the three-component system previously used by Dismukes and co-workers. Hammett linear free energy plots were employed and revealed a correlation between the photoinduced electron transfer constants and the electron-donating ability of the ligand. However, under the explored conditions all of the investigated cubanes reached similar TONs (∼140), with the difference being the rate by which O2 was produced.
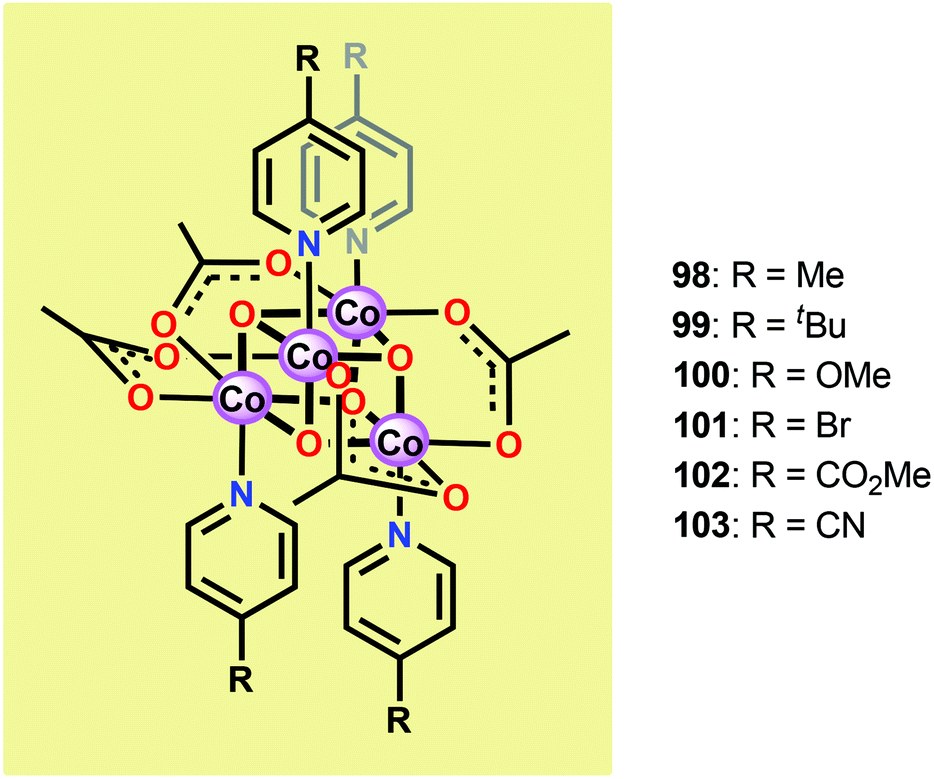 | ||
| Fig. 38 Structures of Co cubanes 98–103. | ||
Ofoli and co-workers recently immobilized Co cubane 96 on ITO and showed that the cubane catalyst retained its catalytic activity similarly to the homogeneous catalytic entity.262 Sun and co-workers have also coupled Co cubane 96 to [Ru(bpy)3]2+-type photosensitizers to generate two supramolecular assemblies, assembly 104 depicted in Fig. 39 and the cyclic assembly 105 consisting of two Co cubane and two [Ru(bpy)3]2+ units (not shown).263 The carboxylate motif was chosen since it has been shown that Co cubane 96 can be immobilized onto carboxylate functionalized silica via carboxylate exchange.264
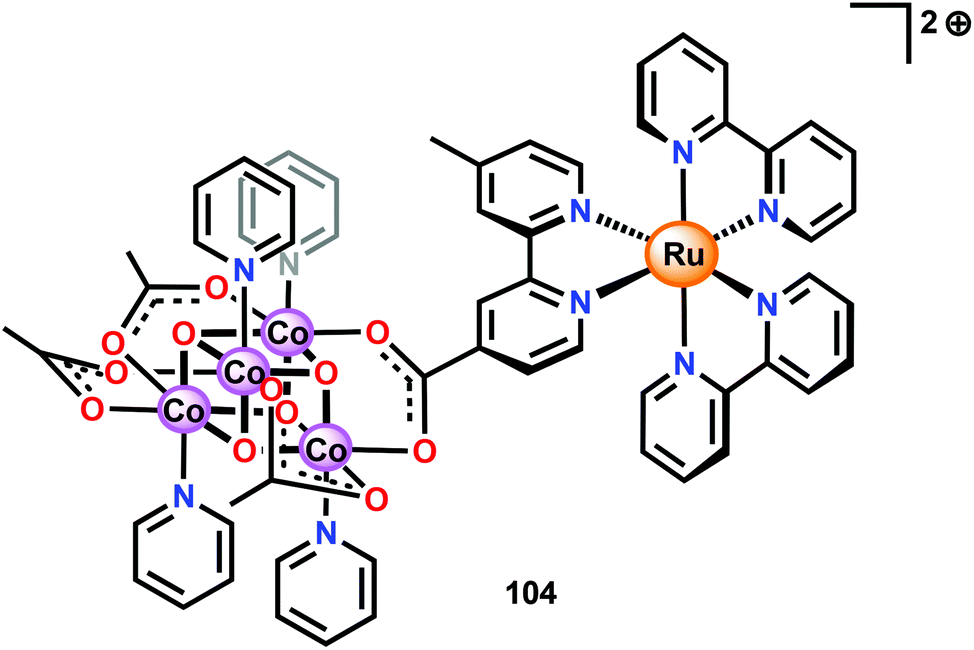 | ||
| Fig. 39 Depiction of Ru–Co assembly 104. | ||
Electrochemical measurements revealed that assembly 104 displayed two redox peaks at 1.01 V and 1.28 V vs. NHE. The cyclic assembly 105 also exhibited two redox peaks; however, these were positively shifted and appeared at 1.10 V and 1.34 V vs. NHE. In comparison, a solution containing a mixture of the separate components, Co cubane 96 and [Ru(bpy)3]2+ (1), displayed two waves at 0.91 V and 1.26 V vs. NHE. The two assemblies were subsequently evaluated toward light-driven H2O oxidation using Na2S2O8 as a sacrificial electron acceptor. When exposed to light, the two assemblies 104 and 105 rapidly evolved O2. Here, the cyclic assembly 105 proved to be a more efficient catalyst than assembly 104, producing a TOF of 0.023 s−1 compared to 0.0067 s−1 for the linear assembly 104. Although the reason for the striking difference in activity for the cyclic assembly 105 is still not clear, the authors speculated that assembly 105 was more robust than the linear counterpart. This hypothesis originated from the fact that cyclic assembly 105 exhibited fewer changes in the UV-vis spectrum during the photocatalytic experiments compared to assembly 104.263
A mechanistic investigation conducted by Tilley and co-workers on the Co4O4(pyr)4(OAc)4 cubane (96) suggested that the stoichiometric reaction of the one-electron oxidized cubane (96+, [CoIII3CoIV]) with OH− produces O2 with quantitative regeneration of cubane 96. The experimental results also suggested that the CoIII3CoIV species (96+) undergoes disproportionation to generate an intermediate with an even higher oxidation state, a formal CoIII3CoV or CoIII2CoIV2 species. The proposed mechanism for the reduction of 96+ by OH− is depicted in Scheme 7.265 The mechanism by which Co cubanes mediate H2O oxidation has also been investigated by density functional theory (DFT) calculations.266,267 Several reaction pathways were considered; however, the lowest energy pathway for Co cubane 96 was found to proceed through a formal CoV species, which is perhaps better described as a CoIV center coupled to an oxygen radical. The mechanism with the lowest energy involved water nucleophilic attack on the formed oxygen radical.266 In a related study, the pathway was observed to involve germinal coupling of a formal CoV–oxo unit with bridging oxo sites. In this study the examined models were revealed to be sensitive to positions of the ligands and the hydrogen bonding environment, resulting in distinct isomers with different energies.267,268 The studies described above highlight the necessity of designing Co cubanes, and other Co-based WOCs, that are capable of accessing high-valent redox states. A multimetallic system is well-suited to stabilize such high redox states but is not a prerequisite for designing active Co-based WOCs since the ancillary ligands can also be an essential feature for tuning the redox potentials of the metal complexes.
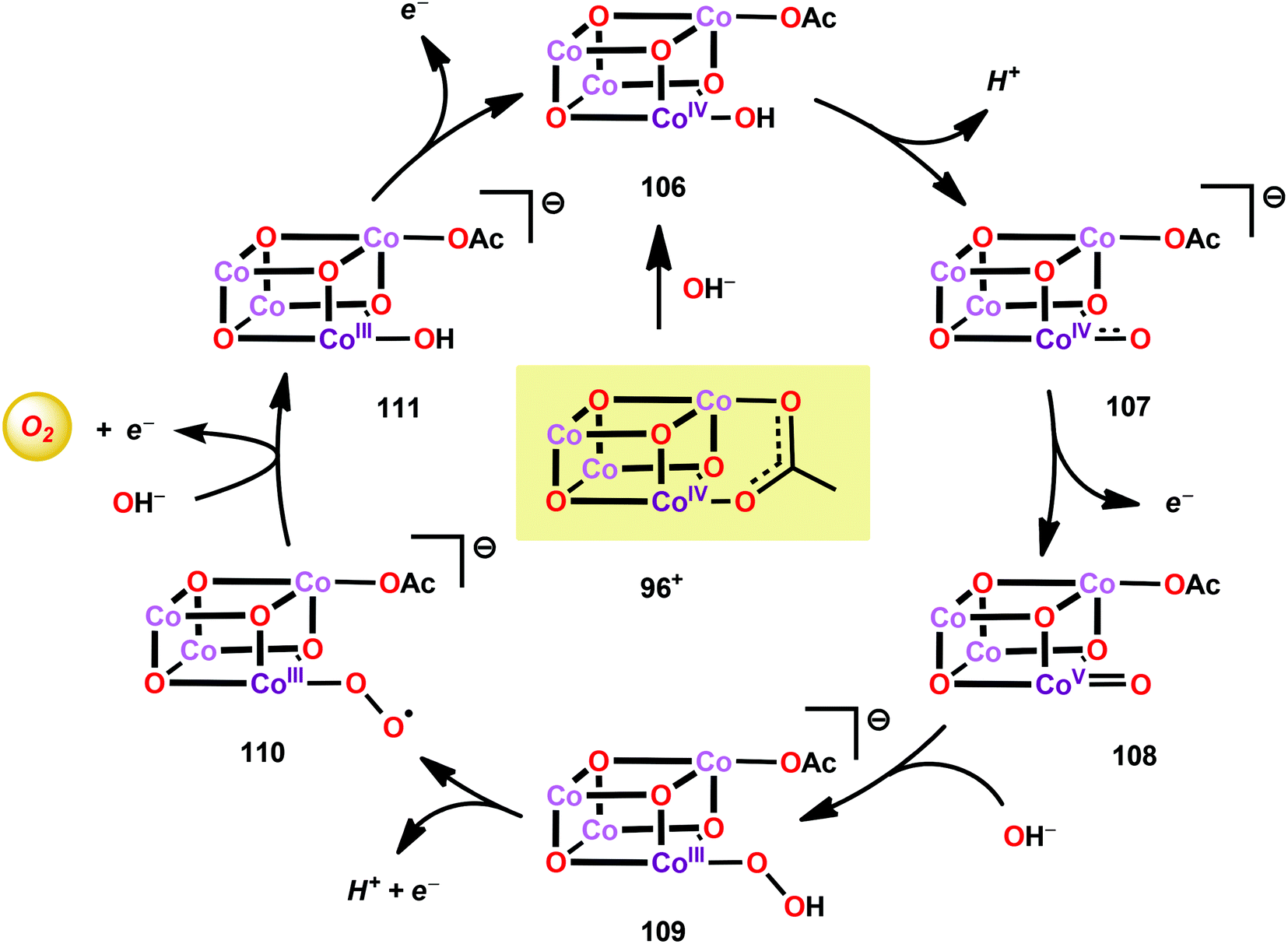 | ||
| Scheme 7 Proposed mechanism for the reaction of cubane 96+ with OH−. | ||
Co cubane [CoII4(hmp)4(μ-OAc)2(μ2-OAc)2(H2O)2] (112, Fig. 40; hmp = 2-(hydroxymethyl)pyridine) was synthesized by Patzke and co-workers as the first CoII-based cubane WOC.269 The catalytic activity of Co cubane 112 was evaluated in light-driven H2O oxidation using [Ru(bpy)3]2+ as a photosensitizer and Na2S2O8 as a sacrificial electron acceptor. From these experiments it was established that the O2 evolution performance was strongly dependent on the pH. The TOF was shown to change notably over the studied pH range, from 1.8 s−1 at pH 7 to 7 s−1 at pH 9. Several different techniques were used to determine the extent of nanoparticle formation and suggested that the Co cubane core of 112 remains intact. The high catalytic activity of Co cubane 112 was attributed to its flexible architecture consisting of monodentate acetate and aqua ligands in combination with a robust core. A recent computational study suggests that O–O bond formation occurs either through a single-site pathway involving H2O attack on a Co–oxo species or through an oxo–oxo coupling pathway.270
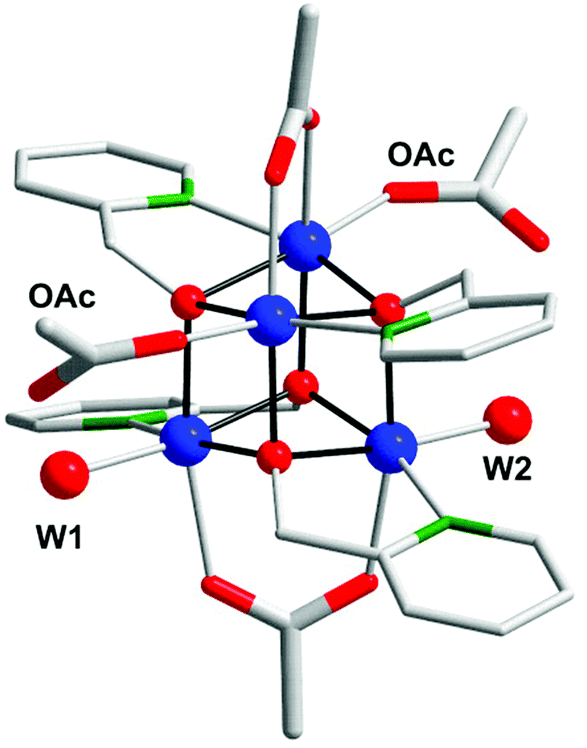 | ||
| Fig. 40 Crystal structure of Co cubane [CoII4(hmp)4(μ-OAc)2(μ2-OAc)2(H2O)2] (112). Co, blue; O, red; N, green; C, gray; H atoms are omitted for clarity. Adapted with permission from ref. 269. Copyright 2013 American Chemical Society. | ||
Subsequent work on biomimetic Co cubanes consisted of the synthesis and examination of a series of isostructural Co cubanes (113–116) with the general formula [CoII3Ln(hmp)4(OAc)5H2O] (Fig. 41, where Ln = Ho, Er, Tm, Yb).271 These bioinspired Co cubanes show several design features, such as ligand flexibility and redox-inert Ln3+ metal ions for electrochemical tuning. The catalytic performance of the lanthanide cubane series (113–116) was studied using [Ru(bpy)3]2+ as a photosensitizer and Na2S2O8 as a sacrificial electron acceptor in order to enable a direct comparison with the previously developed Co cubane 112. O2 evolution peaked at pH 8–9, which was also observed for the [CoII4(hmp)4(μ-OAc)2(μ2-OAc)2(H2O)2] cubane (112). Table 1 summarizes the catalytic activity of the biomimetic lanthanide cubanes [CoII3Ln(hmp)4(OAc)5H2O]. The stability of the lanthanide containing Co cubanes was subsequently assessed employing a variety of different techniques, including UV-vis aging tests, DLS, and extended X-ray absorption fine structure (EXAFS) and X-ray absorption near-edge structure (XANES) solution phase tests, which supported the structural integrity of the [CoII3Ln(hmp)4(OAc)5H2O] cubane core under the catalytic conditions.
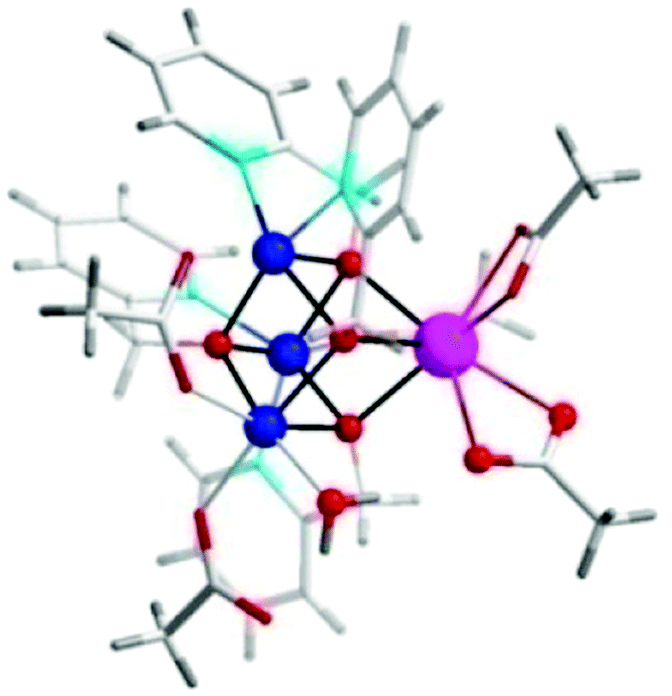 | ||
| Fig. 41 Representative crystal structure of Co cubanes [CoII3Ln(hmp)4(OAc)5H2O] (where Ln = Ho, Er, Tm, Yb). Co, blue; Ln, pink; O, red; C, white; H, gray. Adapted with permission from ref. 271. Copyright 2015 American Chemical Society. | ||
| pH | Catalyst concentration (μM) | TON | TOF (s−1) |
|---|---|---|---|
| a Photochemical experiments were carried out with a catalyst (12 μM), [Ru(bpy)3]Cl2 (1 mM) and Na2S2O8 (5 mM) in aqueous phosphate buffer solutions (pH 7, 40 mM Na2HPO4/NaH2PO4 buffer). b Experiments were performed with a catalyst (10 μM), [Ru(bpy)3]Cl2 (1 mM) and Na2S2O8 (5 mM) in aqueous borate buffer solutions (pH 8, adjusted with HCl addition to 50 mM borate buffer). c Experiments were performed with a catalyst (12 μM), [Ru(bpy)3]Cl2 (1 mM) and Na2S2O8 (5 mM) in aqueous borate buffer solutions (pH 9, adjusted with HCl addition to 50 mM borate buffer). | |||
| [CoII3Ho(hmp) 4 (OAc) 5 H 2 O] ( 113 ) | |||
| 7a | 12 | 7 | 0.87 |
| 8b | 10 | 163 | 5.84 |
| 9c | 12 | 135 | 9.55 |
| [CoII3Er(hmp) 4 (OAc) 5 H 2 O] ( 114 ) | |||
| 7a | 12 | 2 | 2.06 |
| 8b | 10 | 211 | 5.65 |
| 9c | 12 | 108 | 5.02 |
| [CoII3Tm(hmp) 4 (OAc) 5 H 2 O] ( 115 ) | |||
| 7a | 12 | 24 | 1.55 |
| 8b | 10 | 92 | 5.34 |
| 9c | 12 | 64 | 3.48 |
| [CoII3Yb(hmp) 4 (OAc) 5 H 2 O] ( 116 ) | |||
| 7a | 12 | 1.1 | 1.08 |
| 8b | 10 | 160 | 6.54 |
| 9c | 12 | 120 | 5.65 |
Although the stability of the various Co cubane WOCs was assessed and pointed to a homogeneous H2O oxidation pathway operating with the cubane core being intact, a report from the Nocera group suggested that the catalytic activity emanates from a CoII impurity. The CoII impurity was proposed to act as a source for the generation of heterogeneous Co species that are the real catalysts. These heterogeneous species are believed to be formed if the potential is sufficiently high to oxidize the CoII to CoIII in the presence of a proton accepting electrolyte, such as phosphate and carbonate.272 A recent report also questions the homogeneity of the Co-based cubane complexes, and highlights the dependence on the initial structure and the catalytic methodology being used.273
From the conflicting results regarding the homogeneity of Co cubanes it is clear that care must be exercised when examining whether a specific WOC operates through a homogeneous mechanism or whether heterogeneous species are produced in situ, and the studied molecular complex merely acts as a precursor. However, deriving a definite conclusion for the examined catalytic system is not always straightforward and can be highly sensitive to the methodologies and experimental conditions being employed.
6.3. Mononuclear Co catalysts
The early observation that CoIV species could be involved in the catalytic oxidation of H2O using simple inorganic salts214 suggested that one key to realizing molecular Co WOCs might be to design ligands that permit stabilization of high-valent Co intermediates. Two seminal reports by the groups of Berlinguette274 and Nocera275 showed that this principle was indeed viable. Berlinguette and co-workers utilized the pyridine-based pentacoordinating ligand Py5,126,127 giving Co-based complex [Co(Py5)(OH2)]2+ (117, Fig. 42) with a single open coordination site. The Py5 ligand was chosen as the authors envisioned it to be a good motif for withstanding the harsh oxidative conditions needed to carry out oxidation of H2O. Furthermore, the Py5 ligand lacks β-hydrogens, which prevents that the corresponding metal complex undergoes undesirable elimination processes. Electrochemical measurements showed that Co complex 117 displayed two redox events. The second event results in a significant rise in the current, suggesting that a catalytic reaction takes place. The catalytic current was demonstrated to increase with decreasing scan rate (ν), attesting that a chemical process, such as O–O bond formation, is the rate-determining step. The authors attributed this event to nucleophilic attack of H2O on a high-valent formal CoIV–oxo/hydroxo species, generated after the initial two redox steps.274 A subsequent study conducted on the [Co(Py5)(OH2)]2+ complex 117 supported that the catalytic current was molecular in origin and that the H2O oxidation did not emanate exclusively from in situ formed heterogeneous Co species.276 A detailed computational mechanistic study on Co complex 117 suggested that the resting state of the catalyst contained a CoIV–oxyl species. The O–O bond formation was suggested to proceed via nucleophilic attack of OH− on this formal CoIV–oxyl species.277 The structurally related CoII complex [Co(Py5OH)(Cl)]+ (118, Fig. 42) has also recently been reported to mediate H2O oxidation.278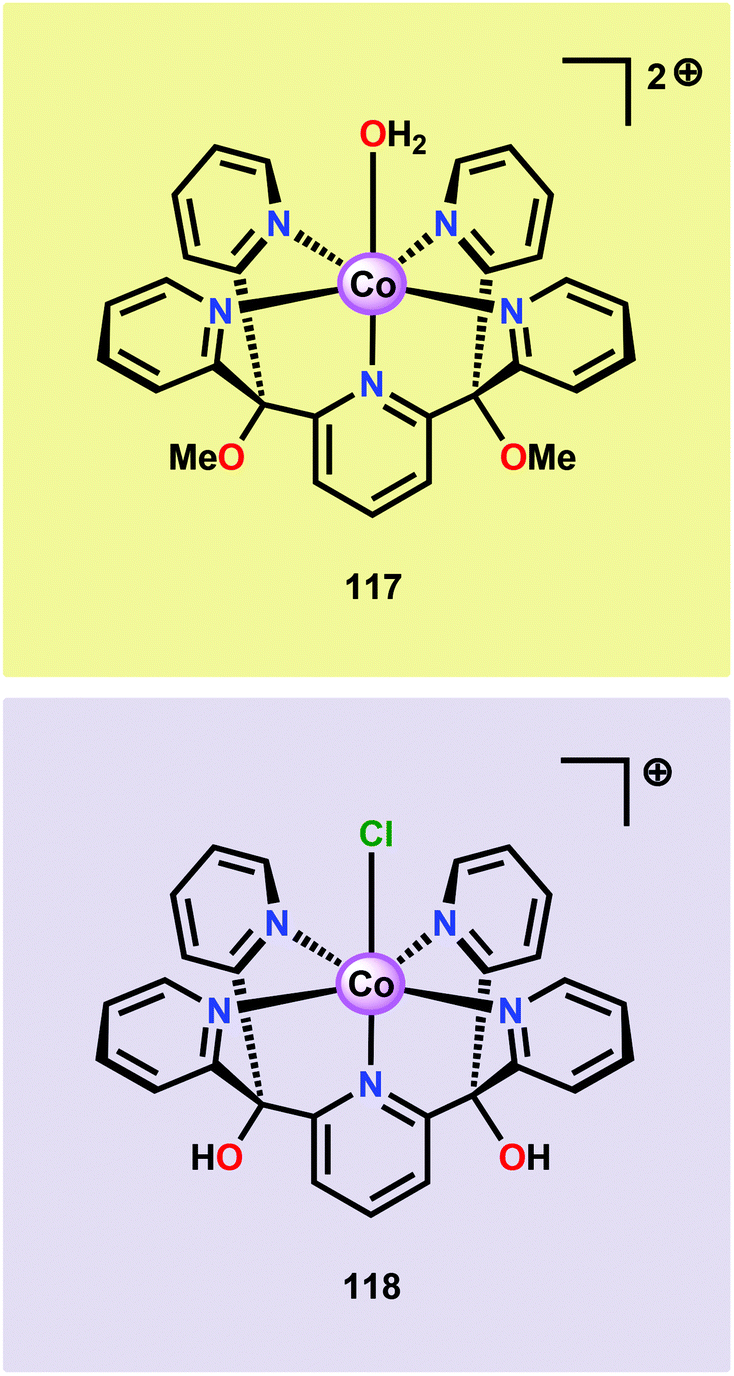 | ||
| Fig. 42 Structures of Co-based complexes [Co(Py5)(OH2)]2+ (117) and [Co(Py5OH)(Cl)]+ (118). | ||
Based on previous observations that CoII hangman porphyrin complexes were able to activate O2,279 Nocera and co-workers decided to investigate whether the related CoIII corrole complexes 119 and 120 (Fig. 43) could promote the opposite reaction—oxidation of H2O.275 These Co xanthene hangman corrole complexes have two vacant coordination sites and contain a proximal base. Measurements revealed that they could indeed drive H2O oxidation electrochemically, with the β-octafluoro Co complex 120 being the more active of the two complexes. The importance of the hangman cleft is believed to be its ability to arrange one H2O molecule in the primary coordination sphere of the Co center and another molecule in the secondary coordination sphere via hydrogen bonding to the xanthene hangman motif (see Fig. 43).275 Subsequent quantum chemical modeling supported the feature of the carboxylate unit in functioning as a general base to activate the attack of the incoming H2O molecule on the metal–oxo species. Additional key features in the catalytic cycle that were uncovered included the non-innocent role of the corrole backbone and that fluorination of the ligand backbone modulates the electrophilicity of the metal–oxo moiety and alleviates the decomposition of the produced corrole radical cations.280,281
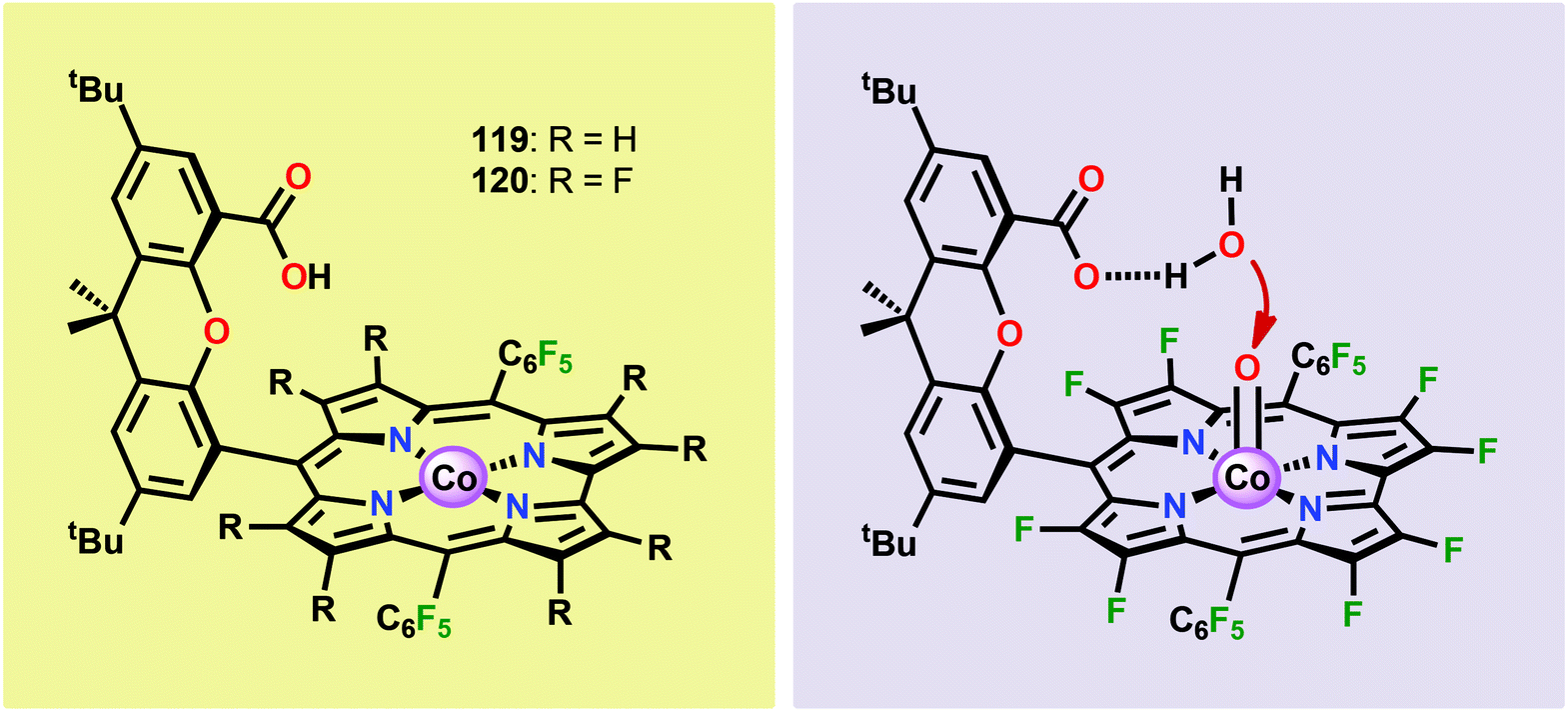 | ||
| Fig. 43 (Left) Co corrole complexes 119 and 120, and (right) organization of H2O within the Co hangman cleft. | ||
Another Co corrole complex that has been reported to catalyze electrocatalytic H2O oxidation is [Co(tpfc)(pyr)2] (121, Fig. 44; where tpfc = 5,10,15-tris(pentafluorophenyl)corrole and pyr = pyridine).282 Stability tests suggested that Co corrole 121 did operate through a homogeneous mechanism. The catalytic rate of H2O oxidation for Co complex 121 was enhanced by increasing the phosphate concentration, indicative of the importance of proton transfer for O–O bond formation, which was also calculated to constitute the rate-determining step.
 | ||
| Fig. 44 Structure of Co corrole complex [Co(tpfc)(pyr)2] 121. tpfc = 5,10,15-tris(pentafluorophenyl)corrole, pyr = pyridine. | ||
Co porphyrins have also been employed as WOCs and are depicted in Fig. 45.283,284 Sakai and co-workers recently reported that Co porphyrins 122–124 could function as active WOCs in photochemical oxidation of H2O. Of the studied catalysts, Co porphyrin 124 was found to be the most efficient one (see Table 2). DLS measurements were also carried out to provide information whether nanoparticles are generated during the course of the catalysis. After the reactions, the solutions did not show any dispersion due to nanoparticle formation upon irradiation, which is in contrast to [Co(bpy)3]2+ that was shown to efficiently produce nanoparticles when irradiated. The second order catalyst dependence suggests a bimolecular radical coupling event as the rate-determining step. Two pathways for O–O bond formation were therefore proposed (Scheme 8), which featured either radical coupling of two formal CoIV–oxyl or two CoIII–oxyl species, generated from the disproportionation of two CoIV species.283
 | ||
| Fig. 45 Co porphyrins 122–127 employed in H2O oxidation. | ||
 | ||
| Scheme 8 Proposed pathways for O–O bond formation for Co porphyrins 122–124. | ||
| Catalyst | TON | TOF |
|---|---|---|
| a Photochemical experiments were carried out with a catalyst (10 μM), [Ru(bpy)3](NO3)2 (1 mM) and Na2S2O8 (5 mM) in aqueous phosphate buffer solutions (0.1 M, pH 11). | ||
| Co porphyrin 122 | 88.7 | 0.118 |
| Co porphyrin 123 | 103.4 | 0.138 |
| Co porphyrin 124 | 121.8 | 0.170 |
Although nanoparticle formation was not observed for Co porphyrins 122–124, the catalytic activity of these catalysts diminished significantly in subsequent runs after addition of fresh Na2S2O8. The authors attributed this effect to decomposition of the Co porphyrins into catalytically less active or inactive entities. However, these entities must still contain the Co center; otherwise nanoparticles should have been observed. Mass spectrometry analysis of the reaction products revealed that oxidative cleavage of the porphyrin rings occurs at the meso positions, which produces species such as 134 (Fig. 46).283
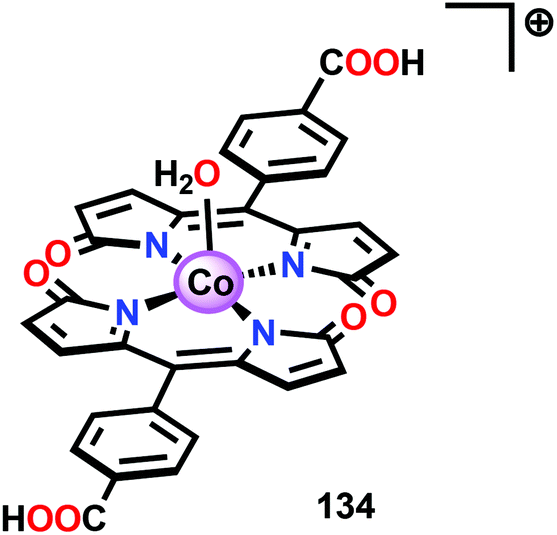 | ||
| Fig. 46 Example of the observed decomposition product (134) of Co porphyrin 123. | ||
In a previous report, meso-substituted Co porphyrin complexes have been shown to react with singlet oxygen (1O2), which is a powerful oxidant produced during H2O oxidation.285 This observation suggests that the studied photosystem can undergo undesired side reactions with 1O2, generated by the triplet state of either the Co-porphyrin or [Ru(bpy)3]2+. With this in mind, Sakai and co-workers examined the resistance of Co porphyrins towards 1O2.286 By introducing fluorine substituents at the 2- and 6-positions of the porphyrin aryl units, to afford the fluorinated Co porphyrin 135 (Fig. 47), the attack of 1O2 was effectively blocked. Comparing the fluorinated Co porphyrin complex 135 with the previously developed Co porphyrin 124 in light-sensitized H2O oxidation confirmed that the fluorinated catalyst was more resistant towards decomposition, and operates without loss of catalytic activity. In contrast to the earlier Co porphyrin WOCs 122–124, porphyrin 135 exhibited a first order dependence on the catalyst concentration. This disparity implies that the rate-determining step for catalyst 135 is H2O nucleophilic attack on a formal high-valent CoV species rather than oxyl coupling between two Co–oxyl units (Scheme 9). This study established the importance of resistance towards 1O2 during light-driven H2O oxidation and that the rational design of ligands can deliver dramatically improved WOCs.
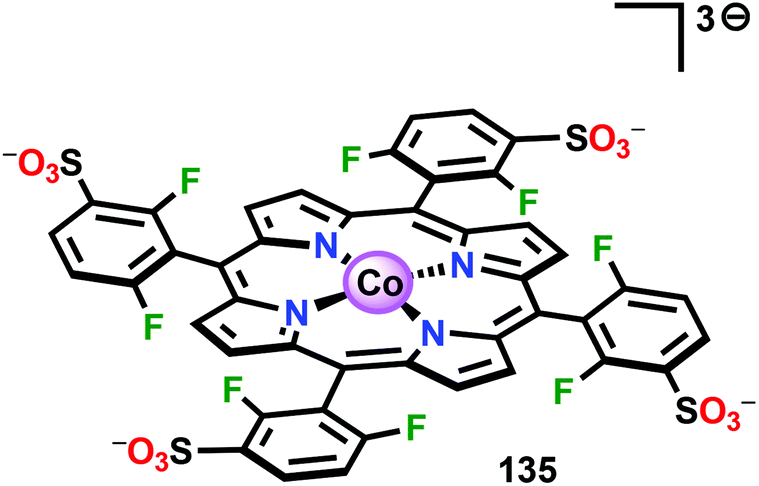 | ||
| Fig. 47 Structure of fluorinated Co porphyrin 135. | ||
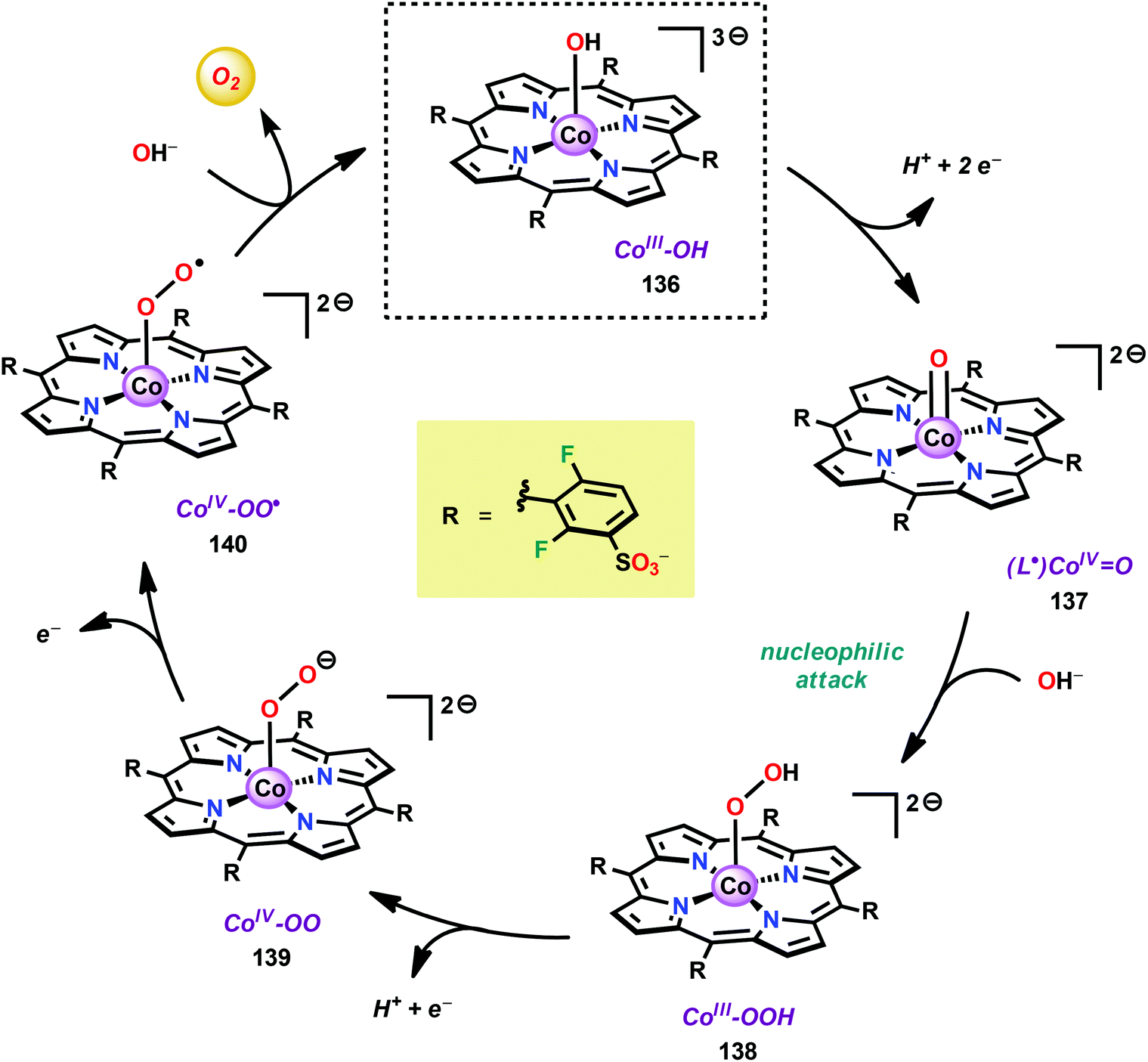 | ||
| Scheme 9 Proposed mechanism for H2O oxidation catalyzed by the fluorinated Co porphyrin 135. | ||
Groves and Wang have also synthesized a series of cationic Co-based porphyrin complexes (125–127) for H2O oxidation (Fig. 45).284 These porphyrin complexes were shown to mediate electrochemical H2O oxidation, with Co porphyrin 125, housing the electron-deficient ligand, being the most efficient catalyst. The key species for these WOCs was proposed to be a CoIV–oxo species containing an oxidized radical porphyrin ligand ([(L˙)CoIV–oxo]), which can be considered as a formal CoV–oxo species. Support of homogeneous O–O bond formation at a single metal center was also given, thus resembling the mechanistic pathway for the fluorinated Co porphyrin 135 (Scheme 9).
Additional mononuclear Co-based WOCs that have been proposed to promote light-sensitized H2O oxidation through a homogeneous pathway are depicted in Fig. 48 and include the [CoII(qpy)(OH2)2]2+ complex 141 (qpy = 2,2′:6′,2′′:6′′,2′′′-quaterpyridine),287 the CoII salophen complex 142288 and related salophen complexes,289 and the CoII complex [Co(tpa)Cl]+143 (tpa = tris(2-pyridylmethyl)amine).290
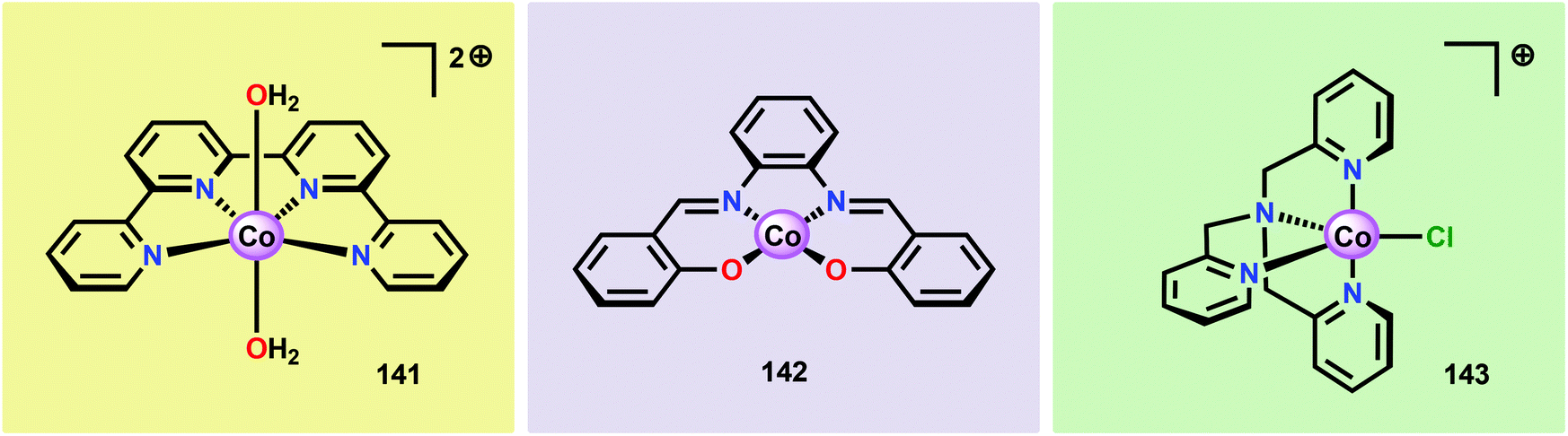 | ||
| Fig. 48 Structures of Co complexes 141–143. | ||
Several Co-based complexes with organic ligand frameworks have been shown to act as precatalysts to heterogeneous Co nanoparticles (Fig. 49).291–293 Although the initial well-defined Co complexes are transformed into heterogeneous materials, the carbonaceous residues originating from the ligand frameworks act as modifiers or capping agents of the generated nanoparticles. This indicates that the ligands might affect the structure and efficiency of the nanoparticular catalysts and suggests a ligand dependent route to efficient and robust catalytic materials, opening an appealing avenue for future research.
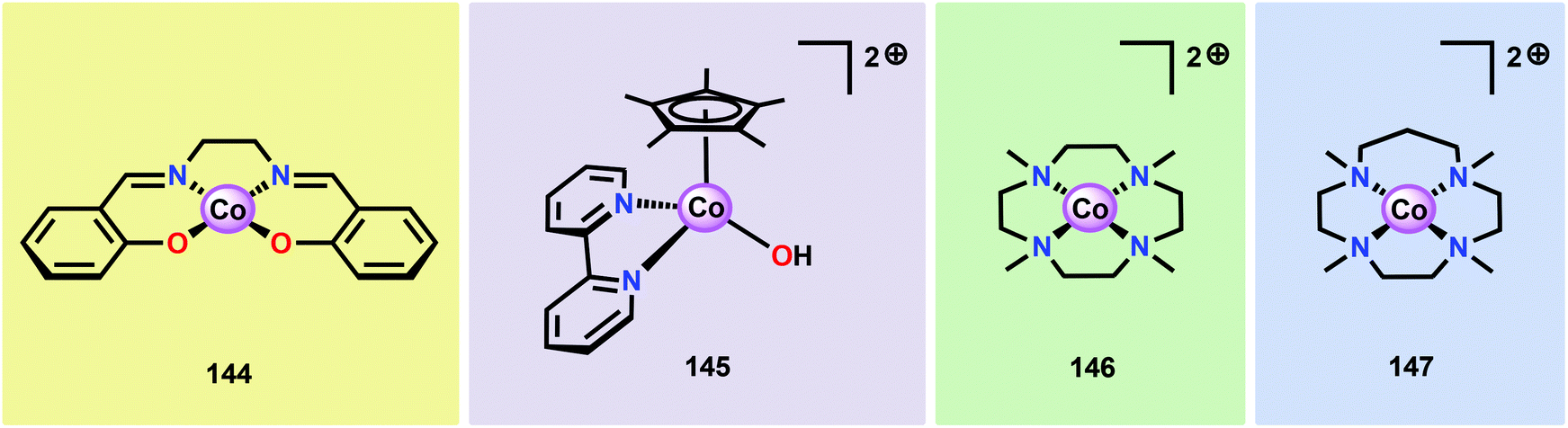 | ||
| Fig. 49 Co-based complexes employed as precatalysts. | ||
6.4. Dinuclear Co complexes as water oxidation catalysts
A relatively unexplored area is the development of dinuclear Co-based WOCs. Although efforts have been devoted to preparing dinuclear Co-based WOCs,100,294 only a few examples of active dinuclear Co catalysts exist. Two of these Co catalysts (148 and 149, Fig. 50) are based on the bridging bispyridylpyrazolate (bpp) ligand. The bpp ligand was chosen since it was considered to provide an environment for the two Co centers which is stable towards hydrolysis and ligand decomposition. Indeed, as confirmed by electrochemical analysis, the two Co–bpp complexes were able to evolve O2 under acidic conditions.295 Another example of an active dinuclear Co WOC is [(tpa)Co(μ-OH)(μ-O2)Co(tpa)]3+ (150), which catalyzes light-induced H2O oxidation using [Ru(bpy)3]2+ as a photosensitizer and Na2S2O8 as a sacrificial electron acceptor.296,297 The dinuclear [(tpa)Co(μ-OH)(μ-O2)Co(tpa)]3+ complex 150 was obtained by treating the previously developed mononuclear [Co(tpa)Cl]+ complex 143 with LiClO4 and O2, resulting in oxidation and formation of the CoIII,III2 complex 150. A pH of 8 was found to be optimal for the light-driven O2 evolution experiments, giving a TON of 58 with a TOF of ∼1.4 s−1. The molecular integrity of the dinuclear Co complex 150 during catalysis was assessed by DLS and suggested that no Co oxide colloids were produced. The proposed catalytic cycle for the oxidation of H2O by [(tpa)Co(μ-OH)(μ-O2)Co(tpa)]3+ complex 150 is depicted in Scheme 10 and revolves around the generation of a (O)CoIVCoIV(O) intermediate as the key species for intramolecular O–O bond formation.296 Further mechanistic investigations will likely result in a fundamental understanding of the catalytic process and enable the design of more efficient dinuclear Co-based WOCs.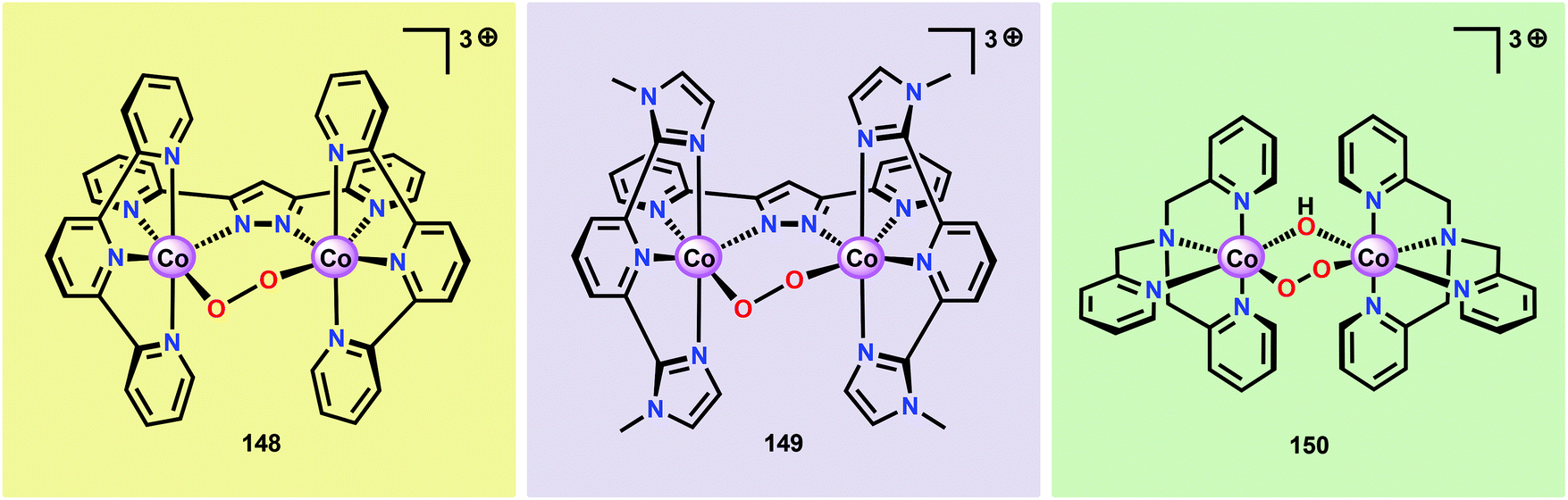 | ||
| Fig. 50 Structures of dinuclear Co-based WOCs 148–150. | ||
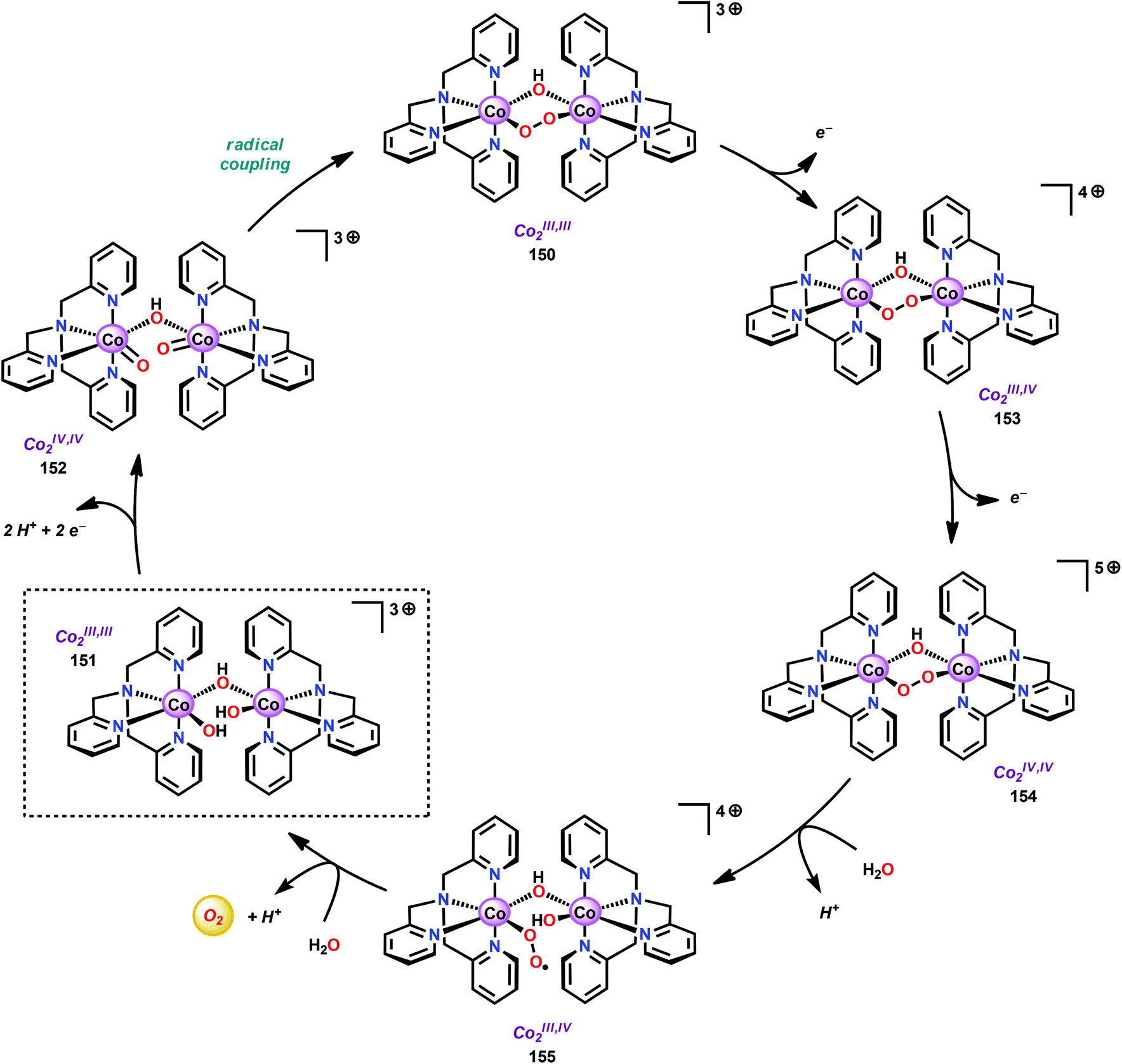 | ||
| Scheme 10 Proposed mechanism for H2O oxidation catalyzed by the dinuclear [(tpa)Co(μ-OH)(μ-O2)Co(tpa)]3+ complex 150. tpa = tris(2-pyridylmethyl)amine. | ||
7. Homogeneous Cu-based systems for catalytic water oxidation
Copper is a common catalyst for mediating aerobic oxidations using molecular oxygen as the terminal oxidant.298 However, only a handful of Cu-based catalysts are known to be capable of promoting the opposite reaction—H2O oxidation. This can perhaps be explained by the metal's preference to carry out one-electron redox events rather than two-electron processes, which are at the core of H2O oxidation.Early investigations on Cu-catalyzed H2O oxidation was conducted by Elizarova and co-workers.175 However, the first well-defined homogeneous Cu-based complex was reported by Mayer and co-workers in 2012.299 The authors examined the ability of the [Cu(bpy)(OH)2] complex 156 (Fig. 51), a self-assembling Cu complex, to mediate electrochemical H2O oxidation. EPR measurements and control studies with Cu oxide implied that the [Cu(bpy)(OH)2] complex was homogeneous in nature. However, the stability of this Cu complex was severely hampered as it merely afforded a TON of ∼30, highlighting that more robust catalysts need to be designed in order to access more efficient Cu-based WOCs.
 | ||
| Fig. 51 Structure of the [Cu(bpy)(OH)2] complex 156. | ||
Lin and co-workers recently studied the catalytic activity of 4,4′- and 6,6′-substituted bipyridine-based Cu complexes. The authors found that Cu complex 157, [Cu(bpyOH)(OH)2], housing the 6,6′-dihydroxy-2,2′-bipyridine ligand provided an efficient WOC (Fig. 52).300 This ligand has previously been employed for other transformations, such as CO2 reduction301 and carbonylation reactions,302 where it was proposed to have a non-innocent role during the catalytic transformations. Lin and co-workers therefore utilized the 6,6′-dihydroxy-2,2′-bipyridine ligand, envisioning that the ligand could be employed as a redox-active entity to modify the reactivity of the metal center to enhance the catalytic activity.300
 | ||
| Fig. 52 Structure of the [Cu(bpyOH)(OH)2] complex 157. | ||
For Cu complex 157, O2 evolution was shown to occur in controlled potential electrolysis (CPE) with an overpotential of ∼640 mV, giving a TON of ∼400. The 4,4′-dihydroxy-2,2′-bipyridine analogous Cu complex showed a significantly higher overpotential for H2O oxidation than Cu complex 156. Both experimental and computational studies suggested the involvement of the ligand framework in electron transfer and proton transfer events, thus enhancing the catalytic activity of the Cu-based WOC.300 The chemistry of 6,6′-substituted 2,2′-bipyridine-based Cu complexes in H2O oxidation catalysis has also been investigated by the group of Papish.303
The group of Meyer has recently developed a self-assembling Cu tetrapeptide-based complex (158, Fig. 53).304 Cu complex 158 contains a triglycylglycine ligand (H4tgg), which creates a suitable environment for the coordination of Cu.305,306 At pH 11, CPE showed that the catalytic current was maintained for ∼5 h, resulting in a TON of ∼13 based on the initial amount of Cu complex 158 in solution. Several lines of evidence supported a homogeneous mechanism: (1) no spectroscopic change of the electrolysis solution was observed, during CPE only minor changes in the peak current or wave shape were observed, (2) an electrode subjected to CPE at 1.32 V vs. NHE with Cu catalyst 158 resulted in no catalytic response when subjected to a fresh catalyst-free electrolyte solution, and (3) no precipitation or film generation was observed by SEM and XPS.304
 | ||
| Fig. 53 Structures of peptide-based Cu complexes 158–160. | ||
The catalytic peak current for H2O oxidation was found to vary linearly with the concentration of Cu complex 158, suggesting a mechanism involving O–O bond formation at a single-site Cu center. The proposed mechanism for oxidation of H2O by Cu complex 158 is shown in Scheme 11 and involves oxidation to produce a CuIII species. A subsequent oxidation event furnishes a CuIV–oxo (or a CuIII–oxyl) species, which is believed to be responsible for mediating O–O bond formation. The produced hydroperoxide species, CuII–OOH, is subsequently oxidized to CuIII–OO˙ from which O2 is liberated, regenerating the CuII catalyst 158 and closing the catalytic cycle.304,307 The two related Cu tetrapeptide complexes 159 and 160 (Fig. 53) have also recently been synthesized and studied in H2O oxidation.308
 | ||
| Scheme 11 Proposed mechanism for H2O oxidation catalyzed by Cu complex 158. | ||
Llobet and co-workers recently prepared a family of Cu complexes (161–164) based on tetra-anionic tetradentate amidate ligands.309 The prepared CuII complexes (Fig. 54) were found to be four-coordinate with a square planar geometry. Electrochemical measurements for the CuII complexes revealed a significant electrocatalytic current corresponding to H2O oxidation together with two redox waves. The first quasi-reversible wave was assigned to a one-electron process, generating a CuIII species (eqn (6)). The subsequent pH-dependent wave was proposed to be associated with a ligand-based oxidation, furnishing a formal aryl radical cation (eqn (7)):
| [(L)CuII]2− → [(L)CuIII]− + e− | (6) |
| [(L)CuIII]− + OH− → [(L˙)CuIII(OH)]− | (7) |
 | ||
| Fig. 54 CuII complexes 161–164 based on tetradentate amidate ligands. | ||
The proposed mechanism for the oxidation of H2O by Cu complexes 161–164 involves initial formation of the [(L˙)CuIII(OH)]− species, which subsequently reacts with OH− to produce a Cu–peroxo species, [(L)CuII(HOOH)]2−. This species undergoes a metal-based oxidation to form [(L)CuIII(HOOH)]−. A final proton-coupled oxidation gives a [(L)CuIII(HOO˙)]− species from which O2 is liberated, thus regenerating the starting complex, [(L)CuII]2−.309 The use of tailored ligands which are able to donate electrons—being non-innocent—during the catalytic oxidation process can be essential for designing novel molecular WOCs where this cooperative effect can alleviate the metal center from being too highly oxidized during the catalytic process.310
The Cu-based WOCs described thus far require alkaline conditions in order to mediate electrochemical oxidation of H2O. However, the dinuclear CuII,II2 complex 165 (Fig. 55) based on the 2,7-[bis(2-pyridylmethyl)aminomethyl]-1,8-naphthyridine ligand (bpman) ligand has been reported to operate under neutral conditions.311 Reacting the dinucleating bpman ligand312,313 with 2 equivalents of Cu(CF3SO3)2 produced the CuII,II2 complex 165 in 71% yield. Electrochemical measurements of Cu complex 165 showed an onset potential for catalytic H2O oxidation starting at ∼1.6 V vs. NHE. The catalytic activity of the dinuclear Cu complex 165 was assessed by CPE at 1.87 V vs. NHE to give a TOF of ∼0.6 s−1 with a Faradaic efficiency of ∼98%. Computational studies suggested that O–O bond formation proceeded through a cooperative interaction between the two CuIII centers rather than by the formation of a CuIVO unit in which a CuIII–O(H) moiety couples with a μ-oxo unit.311 The proposed O–O bond forming step is different from the previously reported Cu-based WOCs and could provide new routes for the activation of H2O.
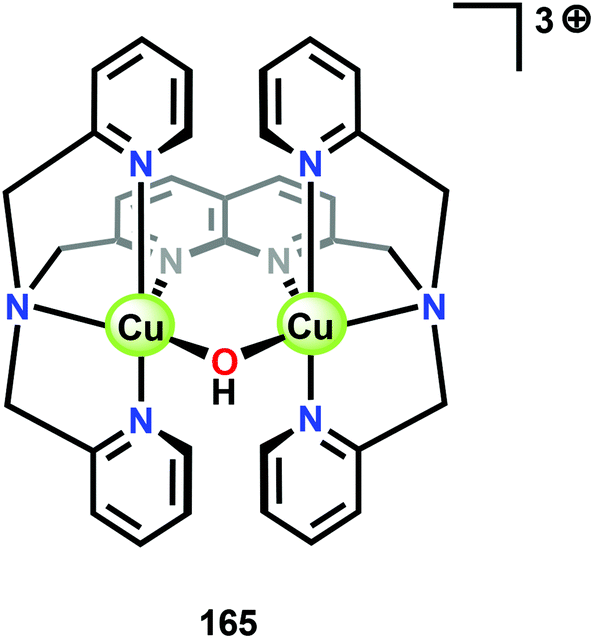 | ||
| Fig. 55 Structure of the dinuclear CuII,II2 complex [CuII,II2(bpman)(μ-OH)]3+ (165). bpman = 2,7-[bis(2-pyridylmethyl)aminomethyl]-1,8-naphthyridine. | ||
Ding and co-workers recently reported that the Cu-containing POM [Cu5(OH)4(OH2)2(A-α-SiW9O33)2]10− (166) functions as a catalyst for visible light-driven water oxidation.314 The Cu-substituted POM 166 (Fig. 56) was originally developed by Kortz and co-workers315 and consists of two A-α-[SiW9O34]10− Keggin moieties that are linked together through two W–O–W bonds and stabilized by a central [Cu5(OH)4(OH2)2]6+ unit. Of the evaluated Cu POMs, POM 166 was the only complex that exhibited water oxidation activity. Using a system consisting of Cu POM 166, [Ru(bpy)3]2+ as the photosensitizer and S2O82− as the sacrificial electron acceptor afforded a TON of 91 under optimized conditions.314 Several molecular Cu-based complexes have been reported to act as precursors to catalytically active heterogeneous materials,316–318 highlighting the importance of determining the true nature of the catalytic entity when studying Cu-based WOCs. For Cu POM 166, multiple experiments, including DLS, UV/vis spectroscopy and nanosecond laser flash photolysis experiments, suggest that Cu POM 166 is the dominant species under the studied catalytic conditions and that it operates through a homogeneous mechanism.314
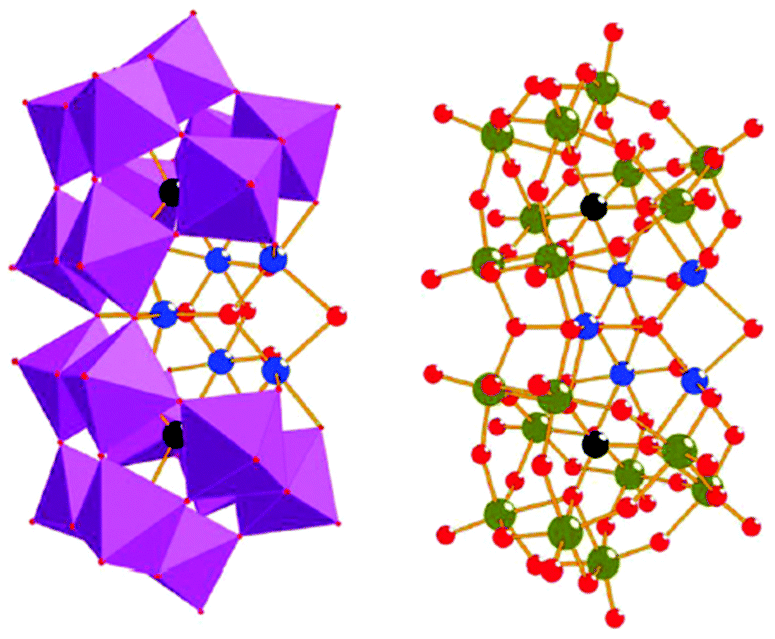 | ||
| Fig. 56 Structure of the Cu-containing POM [Cu5(OH)4(OH2)2(A-α-SiW9O33)2]10− (166). Color code: copper, turquoise; tungsten, green; silicon, black; oxygen, red. Reproduced from ref. 314 with permission from the Royal Society of Chemistry. | ||
8. Conclusions & outlook
Artificial photosynthesis is a competitive and rapidly expanding research field which offers routes to carbon-neutral and renewable fuels. Here, the development of robust catalysts for the oxidation of H2O is currently the bottleneck. Due to their low cost, catalysts based on earth-abundant first-row transition metals are highly attractive and have received considerable attention. This Perspective summarizes advances in the development of molecular WOCs composed of earth-abundant metals.Although relatively few water oxidation catalysts based on first-row transition metals had been reported before the early 2000s, recent years have seen a dramatic increase in the number of such catalysts. However, a majority of these catalysts require two-electron oxo-transfer oxidants to drive H2O oxidation. Despite considerable progress in recent years, a limiting feature encountered with these first-row transition metal WOCs is their relative lability compared to catalysts based on second- and third-row transition metals, such as Ru and Ir. Due to the highly oxidizing conditions required to oxidize H2O, the rational design of robust and efficient WOCs based on first-row transition metals still remains a crucial challenge.
A common topic in the discussion of molecular earth-abundant metal WOCs is the nature of the real catalytic entity—their homogeneity. Their propensity to form metal oxide nanoparticles under the catalytic conditions is a feature that requires particular attention. The generation of heterogeneous materials from the initially well-defined metal complexes is facile and highly dependent on the reaction conditions where small changes can affect both the mechanistic pathway and the stability of the examined metal complex.
The structural knowledge gained from studying the Mn4Ca cluster in the OEC has inspired researchers to design small metal-based model clusters of various shapes and nuclearities. These seminal studies have afforded considerable insight for the rational synthesis of closely related artificial metal-based cubane mimics of the natural photosynthetic system. For a long time, Nature has been a great source of inspiration in the development of molecular photosynthetic mimics for solar energy to fuel conversion and will certainly continue to stimulate the design of molecular mimics based on earth-abundant metals.
Acknowledgements
Financial support from the Swedish Research Council (621-2013-4872), the Knut and Alice Wallenberg Foundation and the Carl Trygger Foundation is gratefully acknowledged.References
- N. S. Lewis, ChemSusChem, 2009, 2, 383–386 CrossRef CAS PubMed.
- A. Cho, Science, 2010, 329, 786–787 CrossRef CAS PubMed.
- R. Schlögl, ChemSusChem, 2010, 3, 209–222 CrossRef PubMed.
- L. Sun, L. Hammarström, B. Åkermark and S. Styring, Chem. Soc. Rev., 2001, 30, 36–49 RSC.
- S. K. Ravi and S. C. Tan, Energy Environ. Sci., 2015, 8, 2551–2573 CAS.
- J. Marshall, Nature, 2014, 510, 22–24 CrossRef CAS PubMed.
- H. Inoue, T. Shimada, Y. Kou, Y. Nabetani, D. Masui, S. Takagi and H. Tachibana, ChemSusChem, 2011, 4, 173–179 CAS.
- D. W. Lawlor, Photosynthesis, Springer-Verlag, New York, 2001 Search PubMed.
- R. Croce and H. van Amerongen, Nat. Chem. Biol., 2014, 10, 492–501 CrossRef CAS PubMed.
- J. Yano and V. Yachandra, Chem. Rev., 2014, 114, 4175–4205 CrossRef CAS PubMed.
- M. Mahdi Najafpour, A. N. Moghaddam, S. I. Allakhverdiev and Govindjee, Biochim. Biophys. Acta, 2012, 1817, 1110–1121 CrossRef PubMed.
- M. Suga, F. Akita, K. Hirata, G. Ueno, H. Murakami, Y. Nakajima, T. Shimizu, K. Yamashita, M. Yamamoto, H. Ago and J.-R. Shen, Nature, 2015, 517, 99–103 CrossRef CAS PubMed.
- Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS PubMed.
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831–1838 CrossRef CAS PubMed.
- A. Zouni, H.-T. Witt, J. Kern, P. Fromme, N. Krauss, W. Saenger and P. Orth, Nature, 2001, 409, 739–743 CrossRef CAS PubMed.
- J.-R. Shen, Annu. Rev. Plant Biol., 2015, 66, 23–48 CrossRef CAS PubMed.
- N. Cox, M. Retegan, F. Neese, D. A. Pantazis, A. Boussac and W. Lubitz, Science, 2014, 345, 804–808 CrossRef CAS PubMed.
- H. Nilsson, F. Rappaport, A. Boussac and J. Messinger, Nat. Commun., 2014, 5, 4305 CAS.
- P. E. M. Siegbahn, Biochim. Biophys. Acta, 2013, 1827, 1003–1019 CrossRef CAS PubMed.
- N. Cox, D. A. Pantazis, F. Neese and W. Lubitz, Acc. Chem. Res., 2013, 46, 1588–1596 CrossRef CAS PubMed.
- H. Bao and R. L. Burnap, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E6139–E6147 CrossRef CAS PubMed.
- P. E. M. Siegbahn, Acc. Chem. Res., 2009, 42, 1871–1880 CrossRef CAS PubMed.
- For recent reviews on water splitting, see for example: (a) H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS; (b) M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed; (c) K. S. Joya, Y. F. Joya, K. Ocakoglu and R. van de Krol, Angew. Chem., Int. Ed., 2013, 52, 10426–10437 CrossRef CAS PubMed; (d) D. J. Wasylenko, R. D. Palmer and C. P. Berlinguette, Chem. Commun., 2013, 49, 218–227 RSC; (e) J. R. McKone, N. S. Lewis and H. B. Gray, Chem. Mater., 2014, 26, 407–414 CrossRef CAS; (f) T. Takata, C. Pan and K. Domen, ChemElectroChem, 2016, 3, 31–37 CrossRef CAS; (g) M. G. Mali, H. Yoon, H. Kim, B. Joshi, S. S. Al-Deyab and S. S. Yoon, ChemPhysChem, 2015, 16, 3450–3457 CrossRef CAS PubMed.
- For recent reviews on Ru- and Ir-based water oxidation catalysts, see: (a) M. D. Kärkäs, O. Verho, E. V. Johnston and B. Åkermark, Chem. Rev., 2014, 114, 11863–12001 CrossRef PubMed; (b) J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974–13005 CrossRef CAS PubMed; (c) M. Okamura and S. Masaoka, Chem. – Asian J., 2015, 10, 306–315 CrossRef CAS PubMed; (d) L. Duan, L. Wang, F. Li, F. Li and L. Sun, Acc. Chem. Res., 2015, 48, 2084–2096 CrossRef CAS PubMed; (e) Q. Zeng, F. W. Lewis, L. M. Harwood and F. Hartl, Coord. Chem. Rev., 2015, 304–305, 88–101 CrossRef CAS; (f) M. D. Kärkäs and B. Åkermark, Chem. Rec., 2016, 16, 940–963 CrossRef PubMed.
- T. Privalov, B. Åkermark and L. Sun, Chem. – Eur. J., 2011, 17, 8313–8317 CrossRef CAS PubMed.
- X. Sala, S. Maji, R. Bofill, J. García-Antón, L. Escriche and A. Llobet, Acc. Chem. Res., 2014, 47, 504–516 CrossRef CAS PubMed.
- M. G. Mavros, T. Tsuchimochi, T. Kowalczyk, A. McIsaac, L.-P. Wang and T. Van Voorhis, Inorg. Chem., 2014, 53, 6386–6397 CrossRef CAS PubMed.
- T. Kikuchi and K. Tanaka, Eur. J. Inorg. Chem., 2014, 607–618 CrossRef CAS.
- P. Du and R. Eisenberg, Energy Environ. Sci., 2012, 5, 6012–6021 CAS.
- A. Singh and L. Spiccia, Coord. Chem. Rev., 2013, 257, 2607–2622 CrossRef CAS.
- S. Fukuzumi, D. Hong and Y. Yamada, J. Phys. Chem. Lett., 2013, 4, 3458–3467 CrossRef CAS.
- A. R. Parent and K. Sakai, ChemSusChem, 2014, 7, 2070–2080 CrossRef CAS PubMed.
- E. Wadsworth, F. R. Duke and C. A. Goetz, Anal. Chem., 1957, 29, 1824–1825 CrossRef CAS.
- For selected examples of water oxidation catalysts evaluated with CeIV, see: (a) D. J. Wasylenko, C. Ganesamoorthy, B. D. Koivisto, M. A. Henderson and C. P. Berlinguette, Inorg. Chem., 2010, 49, 2202–2209 CrossRef CAS PubMed; (b) A. Savini, G. Bellachioma, G. Ciancaleoni, C. Zuccaccia, D. Zuccaccia and A. Macchioni, Chem. Commun., 2010, 46, 9218–9219 RSC; (c) J. D. Blakemore, N. D. Schley, D. Balcells, J. F. Hull, G. W. Olack, C. D. Incarvito, O. Eisenstein, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2010, 132, 16017–16029 CrossRef CAS PubMed; (d) M. D. Kärkäs, E. V. Johnston, E. A. Karlsson, B.-L. Lee, T. Åkermark, M. Shariatgorji, L. Ilag, Ö. Hansson, J.-E. Bäckvall and B. Åkermark, Chem. – Eur. J., 2011, 17, 7953–7959 CrossRef PubMed; (e) L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418–423 CrossRef CAS PubMed; (f) J. J. Concepcion, D. K. Zhong, D. J. Szalda, J. T. Muckerman and E. Fujita, Chem. Commun., 2015, 51, 4105–4108 RSC; (g) I. López, S. Maji, J. Benet-Buchholz and A. Llobet, Inorg. Chem., 2015, 54, 658–666 CrossRef PubMed; (h) L. Tong, R. Zong, R. Zhou, N. Kaveevivitchai, G. Zhang and R. P. Thummel, Faraday Discuss., 2015, 185, 87–104 RSC.
- For an example of using Oxone® to evaluate water oxidation activity, see: J. Limburg, J. S. Vrettos, L. M. Liable-Sands, A. L. Rheingold, R. H. Crabtree and G. W. Brudvig, Science, 1999, 283, 1524–1527 CrossRef CAS PubMed.
- For an example of using NaIO4 to evaluate water oxidation activity, see: A. R. Parent, T. P. Brewster, W. De Wolf, R. H. Crabtree and G. W. Brudvig, Inorg. Chem., 2012, 51, 6147–6152 CrossRef CAS PubMed.
- A. Robert and B. Meunier, New J. Chem., 1988, 12, 885–896 CAS.
- L. K. Weavers, I. Hua and M. R. Hoffmann, Water Environ. Res., 1997, 69, 1112–1119 CrossRef CAS.
- For a recent review discussing the advantages of different chemical oxidants used in the evaluation of water oxidation catalysts, see: A. R. Parent, R. H. Crabtree and G. W. Brudvig, Chem. Soc. Rev., 2013, 42, 2247–2252 RSC.
- A. Ikeda-Ohno, S. Tsushima, C. Hennig, T. Yaita and G. Bernhard, Dalton Trans., 2012, 41, 7190–7192 RSC.
- T. J. Demars, M. K. Bera, S. Seifert, M. R. Antonio and R. J. Ellis, Angew. Chem., Int. Ed., 2015, 54, 7534–7538 CrossRef CAS PubMed.
- For examples of water oxidation catalysts evaluated with [Ru(bpy)3]3+, see: (a) L. Duan, Y. Xu, P. Zhang, M. Wang and L. Sun, Inorg. Chem., 2010, 49, 209–215 CrossRef CAS PubMed; (b) M. D. Kärkäs, T. Åkermark, E. V. Johnston, S. R. Karim, T. M. Laine, B.-L. Lee, T. Åkermark, T. Privalov and B. Åkermark, Angew. Chem., Int. Ed., 2012, 51, 11589–11593 CrossRef PubMed; (c) M. D. Kärkäs, T. Åkermark, H. Chen, J. Sun and B. Åkermark, Angew. Chem., Int. Ed., 2013, 52, 4189–4193 CrossRef PubMed; (d) T. M. Laine, M. D. Kärkäs, R.-Z. Liao, T. Åkermark, B.-L. Lee, E. A. Karlsson, P. E. M. Siegbahn and B. Åkermark, Chem. Commun., 2015, 51, 1862–1865 RSC; (e) T. M. Laine, M. D. Kärkäs, R.-Z. Liao, P. E. M. Siegbahn and B. Åkermark, Chem. – Eur. J., 2015, 21, 10039–10048 CrossRef CAS PubMed; (f) W. Rabten, M. D. Kärkäs, T. Åkermark, H. Chen, R.-Z. Liao, F. Tinnis, J. Sun, P. E. M. Siegbahn, P. G. Andersson and B. Åkermark, Inorg. Chem., 2015, 54, 4611–4620 CrossRef CAS PubMed; (g) W. Rabten, T. Åkermark, M. D. Kärkäs, H. Chen, J. Sun, P. G. Andersson and B. Åkermark, Dalton Trans., 2016, 45, 3272–3276 RSC; (h) M. D. Kärkäs, R.-Z. Liao, T. M. Laine, T. Åkermark, S. Ghanem, P. E. M. Siegbahn and B. Åkermark, Catal. Sci. Technol., 2016, 6, 1306–1319 RSC.
- S. Campagna, F. Puntoriero, F. Nastasi, G. Bergamini and V. Balzani, Top. Curr. Chem., 2007, 280, 117–214 CrossRef CAS.
- D. M. Roundhill, in Photochemistry and Photophysics of Metal Complexes, ed. J. P. Fackler, Jr., Springer-Verlag, New York, 1st edn, 1994, vol. 5, pp. 165–215 Search PubMed.
- F. Teplý, Collect. Czech. Chem. Commun., 2011, 76, 859–917 CrossRef.
- B. C. Gau, H. Chen, Y. Zhang and M. L. Gross, Anal. Chem., 2010, 82, 7821–7827 CrossRef CAS PubMed.
- For a recent review on molecular chromophore–catalyst assemblies, see: D. L. Ashford, M. K. Gish, A. K. Vannucci, M. K. Brennaman, J. L. Templeton, J. M. Papanikolas and T. J. Meyer, Chem. Rev., 2015, 115, 13006–13049 CrossRef CAS PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons Inc., New York, 2nd edn, 2001 Search PubMed.
- For a recent review on electrochemical water oxidation with earth-abundant transition-metal catalysts, see: J. R. Galán-Mascarós, ChemElectroChem, 2015, 2, 37–50 CrossRef.
- For discussions on distinguishing homogeneous from heterogeneous catalysis, see: (a) J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317–341 CrossRef CAS; (b) R. H. Crabtree, Chem. Rev., 2012, 112, 1536–1554 CrossRef CAS PubMed; (c) V. Artero and M. Fontecave, Chem. Soc. Rev., 2013, 42, 2338–2356 RSC; (d) S. Fukuzumi and D. Hong, Eur. J. Inorg. Chem., 2014, 645–659 CrossRef CAS; (e) X. Wu, F. Li, B. Zhang and L. Sun, J. Photochem. Photobiol., C, 2015, 25, 71–89 CrossRef CAS; (f) S. Fukuzumi, J. Jung, Y. Yamada, T. Kojima and W. Nam, Chem. – Asian J., 2016, 11, 1138–1150 CrossRef CAS PubMed.
- (a) A. Asraf, H. A. Younus, M. Yusubov and F. Verpoort, Catal. Sci. Technol., 2015, 5, 4901–4925 RSC; (b) C. Lu, J. Du, X.-J. Su, M.-T. Zhang, X. Xu, T. J. Meyer and Z. Chen, ACS Catal., 2016, 6, 77–83 CrossRef CAS.
- For a review on Mn-based complexes with relevance to Photosystem II, see: S. Mukhopadhyay, S. K. Mandal, S. Bhaduri and W. H. Armstrong, Chem. Rev., 2004, 104, 3981–4026 CrossRef CAS PubMed.
- Y. Naruta, M.-a. Sasayama and T. Sasaki, Angew. Chem., Int. Ed., 1994, 33, 1839–1841 CrossRef.
- Y. Shimazaki, T. Nagano, H. Takesue, B.-H. Ye, F. Tani and Y. Naruta, Angew. Chem., Int. Ed., 2004, 43, 98–100 CrossRef PubMed.
- J. Chen, P. Wagner, L. Tong, G. G. Wallace, D. L. Officer and G. F. Swiegers, Angew. Chem., Int. Ed., 2012, 51, 1907–1910 CrossRef CAS PubMed.
- J. Limburg, J. S. Vrettos, H. Chen, J. C. de Paula, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2001, 123, 423–430 CrossRef CAS PubMed.
- C. W. Cady, K. E. Shinopoulos, R. H. Crabtree and G. W. Brudvig, Dalton Trans., 2010, 39, 3985–3989 RSC.
- H. Chen, R. Tagore, G. Olack, J. S. Vrettos, T.-C. Weng, J. Penner-Hahn, R. H. Crabtree and G. W. Brudvig, Inorg. Chem., 2007, 46, 34–43 CrossRef CAS PubMed.
- R. Tagore, R. H. Crabtree and G. W. Brudvig, Inorg. Chem., 2008, 47, 1815–1823 CrossRef CAS PubMed.
- R. Tagore, H. Chen, H. Zhang, R. H. Crabtree and G. W. Brudvig, Inorg. Chim. Acta, 2007, 360, 2983–2989 CrossRef CAS PubMed.
- Several reports suggest that the dimeric Mn complex 5 is not able to produce O2 without the help of an oxygen-donating oxidant, see: (a) M. Yagi and K. Narita, J. Am. Chem. Soc., 2004, 126, 8084–8085 CrossRef CAS PubMed; (b) C. Baffert, S. Romain, A. Richardot, J.-C. Leprêtre, B. Lefebvre, A. Deronzier and M.-N. Collomb, J. Am. Chem. Soc., 2005, 127, 13694–13704 CrossRef CAS PubMed; (c) P. Kurz, G. Berggren, M. F. Anderlund and S. Styring, Dalton Trans., 2007, 4258–4261 RSC.
- K. Narita, T. Kuwabara, K. Sone, K.-i. Shimizu and M. Yagi, J. Phys. Chem. B, 2006, 110, 23107–23114 CrossRef CAS PubMed.
- H. Yamazaki, T. Nagata and M. Yagi, Photochem. Photobiol. Sci., 2009, 8, 204–209 CAS.
- M. Yagi, M. Toda, S. Yamada and H. Yamazaki, Chem. Commun., 2010, 46, 8594–8596 RSC.
- M. Hirahara, H. Yamazaki, S. Yamada, K. Matsubara, K. Saito, T. Yui and M. Yagi, Catal. Sci. Technol., 2013, 3, 1776–1781 CAS.
- M. Yagi, K. Narita, S. Maruyama, K. Sone, T. Kuwabara and K.-i. Shimizu, Biochim. Biophys. Acta, Bioenerg., 2007, 1767, 660–665 CrossRef CAS PubMed.
- G. Li, E. M. Sproviero, R. C. Snoeberger, III., N. Iguchi, J. D. Blakemore, R. H. Crabtree, G. W. Brudvig and V. S. Batista, Energy Environ. Sci., 2009, 2, 230–238 CAS.
- G. Li, E. M. Sproviero, W. R. McNamara, R. C. Snoeberger, III., R. H. Crabtree, G. W. Brudvig and V. S. Batista, J. Phys. Chem. B, 2010, 114, 14214–14222 CrossRef CAS PubMed.
- B. Nepal and S. Das, Angew. Chem., Int. Ed., 2013, 52, 7224–7227 CrossRef CAS PubMed.
- R. E. Hansen and S. Das, Energy Environ. Sci., 2014, 7, 317–322 CAS.
- J. Limburg, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 1997, 119, 2761–2762 CrossRef CAS.
- H. Chen, R. Tagore, S. Das, C. Incarvito, J. W. Faller, R. H. Crabtree and G. W. Brudvig, Inorg. Chem., 2005, 44, 7661–7670 CrossRef CAS PubMed.
- M. M. Najafpour and V. McKee, Catal. Commun., 2010, 11, 1032–1035 CrossRef CAS.
- H. Yamazaki, S. Igarashi, T. Nagata and M. Yagi, Inorg. Chem., 2012, 51, 1530–1539 CrossRef CAS PubMed.
- H. Yamazaki, T. Ueno, K. Aiso, M. Hirahara, T. Aoki, T. Nagata, S. Igarashi and M. Yagi, Polyhedron, 2013, 52, 455–460 CrossRef CAS.
- M. Hatakeyama, H. Nakata, M. Wakabayashi, S. Yokojima and S. Nakamura, J. Phys. Chem. A, 2012, 116, 7089–7097 CrossRef CAS PubMed.
- I. Rivalta, K. R. Yang, G. W. Brudvig and V. S. Batista, ACS Catal., 2015, 5, 2384–2390 CrossRef CAS.
- S. Khan, K. R. Yang, M. Z. Ertem, V. S. Batista and G. W. Brudvig, ACS Catal., 2015, 5, 7104–7113 CrossRef CAS.
- H. Chen, J. W. Faller, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2004, 126, 7345–7349 CrossRef CAS PubMed.
- H. Chen, M.-N. Collomb, C. Duboc, G. Blondin, E. Rivière, J. W. Faller, R. H. Crabtree and G. W. Brudvig, Inorg. Chem., 2005, 44, 9567–9573 CrossRef CAS PubMed.
- Y. Gao, R. H. Crabtree and G. W. Brudvig, Inorg. Chem., 2012, 51, 4043–4050 CrossRef CAS PubMed.
- M. M. Najafpour, A. N. Moghaddam, H. Dau and I. Zaharieva, J. Am. Chem. Soc., 2014, 136, 7245–7248 CrossRef CAS PubMed.
- C. Baffert, M.-N. Collomb, A. Deronzier, S. Kjærgaard-Knudsen, J.-M. Latour, K. H. Lund, C. J. McKenzie, M. Mortensen, L. P. Nielsen and N. Thorup, Dalton Trans., 2003, 1765–1772 RSC.
- A. K. Poulsen, A. Rompel and C. J. McKenzie, Angew. Chem., Int. Ed., 2005, 44, 6916–6920 CrossRef CAS PubMed.
- R. K. Seidler-Egdal, A. Nielsen, A. D. Bond, M. J. Bjerrum and C. J. McKenzie, Dalton Trans., 2011, 40, 3849–3858 RSC.
- W. M. C. Sameera, C. J. McKenzie and J. E. McGrady, Dalton Trans., 2011, 40, 3859–3870 RSC.
- L. Sun, H. Berglund, R. Davydov, T. Norrby, L. Hammarström, P. Korall, A. Börje, C. Philouze, K. Berg, A. Tran, M. Andersson, G. Stenhagen, J. Mårtensson, M. Almgren, S. Styring and B. Åkermark, J. Am. Chem. Soc., 1997, 119, 6996–7004 CrossRef CAS.
- L. Hammarström, L. Sun, B. Åkermark and S. Styring, Spectrochim. Acta, Part A, 2001, 57, 2145–2160 CrossRef.
- A. Johansson, M. Abrahamsson, A. Magnuson, P. Huang, J. Mårtensson, S. Styring, L. Hammarström, L. Sun and B. Åkermark, Inorg. Chem., 2003, 42, 7502–7511 CrossRef CAS PubMed.
- A. Magnuson, M. Anderlund, O. Johansson, P. Lindblad, R. Lomoth, T. Polivka, S. Ott, K. Stensjö, S. Styring, V. Sundström and L. Hammarström, Acc. Chem. Res., 2009, 42, 1899–1909 CrossRef CAS PubMed.
- C. Herrero, A. Quaranta, S. Protti, W. Leibl, A. W. Rutherford, R. Fallahpour, M.-F. Charlot and A. Aukauloo, Chem. – Asian J., 2011, 6, 1335–1339 CrossRef CAS PubMed.
- C. E. Castillo, S. Romain, M. Retegan, J.-C. Leprêtre, J. Chauvin, C. Duboc, J. Fortage, A. Deronzier and M.-N. Collomb, Eur. J. Inorg. Chem., 2012, 5485–5499 CrossRef CAS.
- P. Huang, A. Magnuson, R. Lomoth, M. Abrahamsson, M. Tamm, L. Sun, B. van Rotterdam, J. Park, L. Hammarström, B. Åkermark and S. Styring, J. Inorg. Biochem., 2002, 91, 159–172 CrossRef CAS PubMed.
- G. Berggren, A. Thapper, P. Huang, P. Kurz, L. Eriksson, S. Styring and M. F. Anderlund, Dalton Trans., 2009, 10044–10054 RSC.
- G. Berggren, A. Thapper, P. Huang, L. Eriksson, S. Styring and M. F. Anderlund, Inorg. Chem., 2011, 50, 3425–3430 CrossRef CAS PubMed.
- K. Beckmann, H. Uchtenhagen, G. Berggren, M. F. Anderlund, A. Thapper, J. Messinger, S. Styring and P. Kurz, Energy Environ. Sci., 2008, 1, 668–676 CAS.
- P. Kurz, Dalton Trans., 2009, 6103–6108 RSC.
- H.-M. Berends, A.-M. Manke, C. Näther, F. Tuczek and P. Kurz, Dalton Trans., 2012, 41, 6215–6224 RSC.
- E. A. Karlsson, B.-L. Lee, T. Åkermark, E. V. Johnston, M. D. Kärkäs, J. Sun, Ö. Hansson, J.-E. Bäckvall and B. Åkermark, Angew. Chem., Int. Ed., 2011, 50, 11715–11718 CrossRef CAS PubMed.
- B.-L. Lee, M. D. Kärkäs, E. V. Johnston, A. K. Inge, L.-H. Tran, Y. Xu, Ö. Hansson, X. Zou and B. Åkermark, Eur. J. Inorg. Chem., 2010, 5462–5470 CrossRef CAS.
- T. Norrby, A. Börje, B. Åkermark, L. Hammarström, J. Alsins, K. Lashgari, R. Norrestam, J. Mårtensson and G. Stenhagen, Inorg. Chem., 1997, 36, 5850–5858 CrossRef CAS PubMed.
- E. A. Karlsson, B.-L. Lee, R.-Z. Liao, T. Åkermark, M. D. Kärkäs, V. Saavedra Becerril, P. E. M. Siegbahn, X. Zou, M. Abrahamsson and B. Åkermark, ChemPlusChem, 2014, 79, 936–950 CrossRef CAS.
- W. A. A. Arafa, M. D. Kärkäs, B.-L. Lee, T. Åkermark, R.-Z. Liao, H.-M. Berends, J. Messinger, P. E. M. Siegbahn and B. Åkermark, Phys. Chem. Chem. Phys., 2014, 16, 11950–11964 RSC.
- R.-Z. Liao, M. D. Kärkäs, B.-L. Lee, B. Åkermark and P. E. M. Siegbahn, Inorg. Chem., 2015, 54, 342–351 CrossRef CAS PubMed.
- I. Garcia-Bosch, A. Company, C. W. Cady, S. Styring, W. R. Browne, X. Ribas and M. Costas, Angew. Chem., Int. Ed., 2011, 50, 5648–5653 CrossRef CAS PubMed.
- D. E. Lansky, B. Mandimutsira, B. Ramdhanie, M. Clausén, J. Penner-Hahn, S. A. Zvyagin, J. Telser, J. Krzystek, R. Zhan, Z. Ou, K. M. Kadish, L. Zakharov, A. L. Rheingold and D. P. Goldberg, Inorg. Chem., 2005, 44, 4485–4498 CrossRef CAS PubMed.
- W. J. Song, M. S. Seo, S. DeBeer George, T. Ohta, R. Song, M.-J. Kang, T. Tosha, T. Kitagawa, E. I. Solomon and W. Nam, J. Am. Chem. Soc., 2007, 129, 1268–1277 CrossRef CAS PubMed.
- X. Wu, X. Yang, Y.-M. Lee, W. Nam and L. Sun, Chem. Commun., 2015, 51, 4013–4016 RSC.
- G. Yin, Acc. Chem. Res., 2013, 46, 483–492 CrossRef CAS PubMed.
- F. M. Ashmawy, C. A. McAuliffe, R. V. Parish and J. Tames, J. Chem. Soc., Dalton Trans., 1985, 1391–1397 RSC.
- M. K. Awad and A. B. Anderson, J. Am. Chem. Soc., 1989, 111, 802–806 CrossRef CAS.
- A. Garcia-Deibe, A. Sousa, M. R. Bermejo, P. P. Mac Rory, C. A. McAuliffe, R. G. Pritchard and M. Helliwell, J. Chem. Soc., Chem. Commun., 1991, 728–729 RSC.
- N. Aurangzeb, C. E. Hulme, C. A. McAuliffe, R. G. Pritchard, M. Watkinson, M. R. Bermejo, A. Garcia-Deibe, M. Rey, J. Sanmartin and A. Sousa, J. Chem. Soc., Chem. Commun., 1994, 1153–1155 RSC.
- M. Watkinson, A. Whiting and C. A. McAuliffe, J. Chem. Soc., Chem. Commun., 1994, 2141–2142 RSC.
- M. M. Najafpour and D. M. Boghaei, Transition Met. Chem., 2009, 34, 367–372 CrossRef CAS.
- G. González-Riopedre, M. I. Fernández-García, A. M. González-Noya, M. Á. Vázquez-Fernández, M. R. Bermejo and M. Maneiro, Phys. Chem. Chem. Phys., 2011, 13, 18069–18077 RSC.
- S. Kal, L. Ayensu-Mensah and P. H. Dinolfo, Inorg. Chim. Acta, 2014, 423, 201–206 CrossRef CAS.
- Y. Gao, J. Liu, M. Wang, Y. Na, B. Åkermark and L. Sun, Tetrahedron, 2007, 63, 1987–1994 CrossRef CAS.
- T. Privalov, L. Sun, B. Åkermark, J. Liu, Y. Gao and M. Wang, Inorg. Chem., 2007, 46, 7075–7086 CrossRef CAS PubMed.
- Y. Gao, T. Åkermark, J. Liu, L. Sun and B. Åkermark, J. Am. Chem. Soc., 2009, 131, 8726–8727 CrossRef CAS PubMed.
- W.-T. Lee, S. B. Muñoz, III., D. A. Dickie and J. M. Smith, Angew. Chem., Int. Ed., 2014, 53, 9856–9859 CrossRef CAS PubMed.
- W.-T. Lee, S. Xu, D. A. Dickie and J. M. Smith, Eur. J. Inorg. Chem., 2013, 3867–3873 CrossRef CAS.
- J. M. Rowland, M. Olmstead and P. K. Mascharak, Inorg. Chem., 2001, 40, 2810–2817 CrossRef CAS PubMed.
- M. Lubben, A. Meetsma, E. C. Wilkinson, B. Feringa and L. Que, Jr., Angew. Chem., Int. Ed. Engl., 1995, 34, 1512–1514 CrossRef CAS.
- R. A. Geiger, D. F. Leto, S. Chattopadhyay, P. Dorlet, E. Anxolabéhère-Mallart and T. A. Jackson, Inorg. Chem., 2011, 50, 10190–10203 CrossRef CAS PubMed.
- R. T. Jonas and T. D. P. Stack, J. Am. Chem. Soc., 1997, 119, 8566–8567 CrossRef CAS.
- M. E. de Vries, R. M. La Crois, G. Roelfes, H. Kooijman, A. L. Spek, R. Hage and B. L. Feringa, Chem. Commun., 1997, 1549–1550 RSC.
- K. J. Young, M. K. Takase and G. W. Brudvig, Inorg. Chem., 2013, 52, 7615–7622 CrossRef CAS PubMed.
- T. J. Collins and S. W. Gordon-Wylie, J. Am. Chem. Soc., 1989, 111, 4511–4513 CrossRef CAS.
- G. Berggren, F. B. Kaynak, M. F. Anderlund, L. Eriksson and B. Åkermark, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m2672–m2673 CAS.
- T. Taguchi, R. Gupta, B. Lassalle-Kaiser, D. W. Boyce, V. K. Yachandra, W. B. Tolman, J. Yano, M. P. Hendrich and A. S. Borovik, J. Am. Chem. Soc., 2012, 134, 1996–1999 CrossRef CAS PubMed.
- C. R. Goldsmith, A. P. Cole and T. D. P. Stack, J. Am. Chem. Soc., 2005, 127, 9904–9912 CrossRef CAS PubMed.
- For a review on molecular Mn mimics, see: C. S. Mullins and V. L. Pecoraro, Coord. Chem. Rev., 2008, 252, 416–443 CrossRef CAS PubMed.
- S. Wang, K. Folting, W. E. Streib, E. A. Schmitt, J. K. McCusker, D. N. Hendrickson and G. Christou, Angew. Chem., Int. Ed. Engl., 1991, 30, 305–306 CrossRef.
- C. Gedye, C. Harding, V. McKee, J. Nelson and J. Patterson, J. Chem. Soc., Chem. Commun., 1992, 392–394 RSC.
- S. Wang, H.-L. Tsai, K. S. Hagen, D. N. Hendrickson and G. Christou, J. Am. Chem. Soc., 1994, 116, 8376–8377 CrossRef CAS.
- C. R. Hamilton, R. A. Baglia, A. D. Gordon and M. J. Zdilla, J. Am. Chem. Soc., 2011, 133, 4208–4211 CrossRef CAS PubMed.
- S. Vaddypally, S. K. Kondaveeti and M. J. Zdilla, Inorg. Chem., 2012, 51, 3950–3952 CrossRef CAS PubMed.
- C. R. Hamilton, M. R. Gau, R. A. Baglia, S. F. McWilliams and M. J. Zdilla, J. Am. Chem. Soc., 2014, 136, 17974–17986 CrossRef CAS PubMed.
- C. Zhang, C. Chen, H. Dong, J.-R. Shen, H. Dau and J. Zhao, Science, 2015, 348, 690–693 CrossRef CAS PubMed.
- A. Mishra, W. Wernsdorfer, K. A. Abboud and G. Christou, Chem. Commun., 2005, 54–56 RSC.
- S. Mukherjee, J. A. Stull, J. Yano, T. C. Stamatatos, K. Pringouri, T. A. Stich, K. A. Abboud, R. D. Britt, V. K. Yachandra and G. Christou, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2257–2262 CrossRef CAS PubMed.
- S. Nayak, H. P. Nayek, S. Dehnen, A. K. Powell and J. Reedijk, Dalton Trans., 2011, 40, 2699–2702 RSC.
- W. F. Ruettinger, C. Campana and G. C. Dismukes, J. Am. Chem. Soc., 1997, 119, 6670–6671 CrossRef CAS.
- T. G. Carrell, S. Cohen and G. C. Dismukes, J. Mol. Catal. A: Chem., 2002, 187, 3–15 CrossRef CAS.
- W. F. Ruettinger, D. M. Ho and G. C. Dismukes, Inorg. Chem., 1999, 38, 1036–1037 CrossRef CAS PubMed.
- T. G. Carrell, E. Bourles, M. Lin and G. C. Dismukes, Inorg. Chem., 2003, 42, 2849–2858 CrossRef CAS PubMed.
- R. Brimblecombe, A. M. Bond, G. C. Dismukes, G. F. Swiegers and L. Spiccia, Phys. Chem. Chem. Phys., 2009, 11, 6441–6449 RSC.
- J.-Z. Wu, E. Sellitto, G. P. A. Yap, J. Sheats and G. C. Dismukes, Inorg. Chem., 2004, 43, 5795–5797 CrossRef CAS PubMed.
- W. Ruettinger, M. Yagi, K. Wolf, S. Bernasek and G. C. Dismukes, J. Am. Chem. Soc., 2000, 122, 10353–10357 CrossRef CAS.
- M. Yagi, K. V. Wolf, P. J. Baesjou, S. L. Bernasek and G. Charles Dismukes, Angew. Chem., Int. Ed., 2001, 40, 2925–2928 CrossRef CAS.
- J.-Z. Wu, F. De Angelis, T. G. Carrell, G. P. A. Yap, J. Sheats, R. Car and G. C. Dismukes, Inorg. Chem., 2006, 45, 189–195 CrossRef CAS PubMed.
- R.-Z. Liao and P. E. M. Siegbahn, J. Photochem. Photobiol., B, 2015, 152, 162–172 CrossRef CAS PubMed.
- A. J. Seen, J. Mol. Catal. A: Chem., 2001, 177, 105–112 CrossRef CAS.
- R. Brimblecombe, G. F. Swiegers, G. C. Dismukes and L. Spiccia, Angew. Chem., Int. Ed., 2008, 47, 7335–7338 CrossRef CAS PubMed.
- R. Brimblecombe, D. R. J. Kolling, A. M. Bond, G. C. Dismukes, G. F. Swiegers and L. Spiccia, Inorg. Chem., 2009, 48, 7269–7279 CrossRef CAS PubMed.
- R. Brimblecombe, A. M. Bond, G. C. Dismukes, G. F. Swiegers and L. Spiccia, Phys. Chem. Chem. Phys., 2009, 11, 6441–6449 RSC.
- R. Brimblecombe, A. Koo, G. C. Dismukes, G. F. Swiegers and L. Spiccia, ChemSusChem, 2010, 3, 1146–1150 CrossRef CAS PubMed.
- R. Brimblecombe, J. Chen, P. Wagner, T. Buchhorn, G. C. Dismukes, L. Spiccia and G. F. Swiegers, J. Mol. Catal. A: Chem., 2011, 338, 1–6 CAS.
- R. Brimblecombe, A. Koo, G. C. Dismukes, G. F. Swiegers and L. Spiccia, J. Am. Chem. Soc., 2010, 132, 2892–2894 CrossRef CAS PubMed.
- R. K. Hocking, R. Brimblecombe, L.-Y. Chang, A. Singh, M. H. Cheah, C. Glover, W. H. Casey and L. Spiccia, Nat. Chem., 2011, 3, 461–466 CAS.
- D. F. Ghanotakis, G. T. Babcock and C. F. Yocum, FEBS Lett., 1984, 167, 127–130 CrossRef CAS.
- G. W. Brudvig, Philos. Trans. R. Soc. London, Ser. B, 2008, 363, 1211–1219 CrossRef CAS PubMed.
- J. Dasgupta, R. T. van Willigen and G. C. Dismukes, Phys. Chem. Chem. Phys., 2004, 6, 4793–4802 RSC.
- J. S. Kanady, E. Y. Tsui, M. W. Day and T. Agapie, Science, 2011, 333, 733–736 CrossRef CAS PubMed.
- E. Y. Tsui, R. Tran, J. Yano and T. Agapie, Nat. Chem., 2013, 5, 293–299 CrossRef PubMed.
- E. Y. Tsui and T. Agapie, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10084–10088 CrossRef CAS PubMed.
- E. Y. Tsui, J. S. Kanady and T. Agapie, Inorg. Chem., 2013, 52, 13833–13848 CrossRef CAS PubMed.
- J. S. Kanady, J. L. Mendoza-Cortes, E. Y. Tsui, R. J. Nielsen, W. A. Goddard, III and T. Agapie, J. Am. Chem. Soc., 2013, 135, 1073–1082 CrossRef CAS PubMed.
- J. S. Kanady, P.-H. Lin, K. M. Carsch, R. J. Nielsen, M. K. Takase, W. A. Goddard, III and T. Agapie, J. Am. Chem. Soc., 2014, 136, 14373–14376 CrossRef CAS PubMed.
- P.-H. Lin, M. K. Takase and T. Agapie, Inorg. Chem., 2015, 54, 59–64 CrossRef CAS PubMed.
- S. Suseno, C. C. L. McCrory, R. Tran, S. Gul, J. Yano and T. Agapie, Chem. – Eur. J., 2015, 21, 13420–13430 CrossRef CAS PubMed.
- Y. Yan, J. S. Lee and D. A. Ruddy, Inorg. Chem., 2015, 54, 4550–4555 CrossRef CAS PubMed.
- For recent reviews on Fe catalysis, see: (a) M. Darwish and M. Wills, Catal. Sci. Technol., 2012, 2, 243–255 RSC; (b) D. Bézier, J.-B. Sortais and C. Darcel, Adv. Synth. Catal., 2013, 355, 19–33 CrossRef; (c) K. Riener, S. Haslinger, A. Raba, M. P. Högerl, M. Cokoja, W. A. Herrmann and F. E. Kühn, Chem. Rev., 2014, 114, 5215–5272 CrossRef CAS PubMed; (d) I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed.
- G. L. Elizarova, L. G. Matvienko, N. V. Lozhkina, V. N. Parmonand and K. I. Zamaraev, React. Kinet. Catal. Lett., 1981, 16, 191–194 CrossRef CAS.
- W. C. Ellis, N. D. McDaniel, S. Bernhard and T. J. Collins, J. Am. Chem. Soc., 2010, 132, 10990–10991 CrossRef CAS PubMed.
- A. D. Ryabov and T. J. Collins, Adv. Inorg. Chem., 2009, 61, 471–521 CrossRef CAS.
- A. Ghosh, F. T. de Oliveira, T. Yano, T. Nishioka, E. S. Beach, I. Kinoshita, E. Münck, A. D. Ryabov, C. P. Horwitz and T. J. Collins, J. Am. Chem. Soc., 2005, 127, 2505–2513 CrossRef CAS PubMed.
- L. L. Tang, W. A. Gunderson, A. C. Weitz, M. P. Hendrich, A. D. Ryabov and T. J. Collins, J. Am. Chem. Soc., 2015, 137, 9704–9715 CrossRef CAS PubMed.
- A. Chanda, X. Shan, M. Chakrabarti, W. C. Ellis, D. L. Popescu, F. T. de Oliveira, D. Wang, L. Que, Jr., T. J. Collins, E. Münck and E. L. Bominaar, Inorg. Chem., 2008, 47, 3669–3678 CrossRef CAS PubMed.
- F. T. de Oliveira, A. Chanda, D. Banerjee, X. Shan, S. Mondal, L. Que Jr., E. L. Bominaar, E. Münck and T. J. Collins, Science, 2007, 315, 835–838 CrossRef PubMed.
- E. L. Demeter, S. L. Hilburg, N. R. Washburn, T. J. Collins and J. R. Kitchin, J. Am. Chem. Soc., 2014, 136, 5603–5606 CrossRef CAS PubMed.
- C. Panda, J. Debgupta, D. Díaz Díaz, K. K. Singh, S. S. Gupta and B. B. Dhar, J. Am. Chem. Soc., 2014, 136, 12273–12282 CrossRef CAS PubMed.
- M. Ghosh, K. K. Singh, C. Panda, A. Weitz, M. P. Hendrich, T. J. Collins, B. B. Dhar and S. S. Gupta, J. Am. Chem. Soc., 2014, 136, 9524–9527 CrossRef CAS PubMed.
- M. Z. Ertem, L. Gagliardi and C. J. Cramer, Chem. Sci., 2012, 3, 1293–1299 RSC.
- K. K. Singh, M. k. Tiwari, M. Ghosh, C. Panda, A. Weitz, M. P. Hendrich, B. B. Dhar, K. Vanka and S. S. Gupta, Inorg. Chem., 2015, 54, 1535–1542 CrossRef CAS PubMed.
- K. K. Singh, M. k. Tiwari, B. B. Dhar, K. Vanka and S. S. Gupta, Inorg. Chem., 2015, 54, 6112–6121 CrossRef CAS PubMed.
- Q. Ren, Y. Guo, M. R. Mills, A. D. Ryabov and T. J. Collins, Eur. J. Inorg. Chem., 2015, 1445–1452 CrossRef CAS.
- R.-Z. Liao, X.-C. Li and P. E. M. Siegbahn, Eur. J. Inorg. Chem., 2014, 728–741 CrossRef CAS.
- J. Lloret-Fillol, Z. Codolà, I. Garcia-Bosch, L. Gómez, J. J. Pla and M. Costas, Nat. Chem., 2011, 3, 807–813 CrossRef PubMed.
- Z. Codolà, I. Garcia-Bosch, F. Acuña-Parés, I. Prat, J. M. Luis, M. Costas and J. Lloret-Fillol, Chem. – Eur. J., 2013, 19, 8042–8047 CrossRef PubMed.
- F. Acuña-Parés, Z. Codolà, M. Costas, J. M. Luis and J. Lloret-Fillol, Chem. – Eur. J., 2014, 20, 5696–5707 CrossRef PubMed.
- Z. Codolà, L. Gómez, S. T. Kleespies, L. Que, Jr., M. Costas and J. Lloret-Fillol, Nat. Commun., 2015, 6, 5865 CrossRef PubMed.
- E. E. Kasapbasi and M.-H. Whangbo, Inorg. Chem., 2012, 51, 10850–10855 CrossRef CAS PubMed.
- F. Acuña-Parés, M. Costas, J. M. Luis and J. Lloret-Fillol, Inorg. Chem., 2014, 53, 5474–5485 CrossRef PubMed.
- B. Zhang, F. Li, F. Yu, H. Cui, X. Zhou, H. Li, Y. Wang and L. Sun, Chem. – Asian J., 2014, 9, 1515–1518 CrossRef CAS PubMed.
- Y. Liu, R. Xiang, X. Du, Y. Ding and B. Ma, Chem. Commun., 2014, 50, 12779–12782 RSC.
- W.-P. To, T. W.-S. Chow, C.-W. Tse, X. Guan, J.-S. Huang and C.-M. Che, Chem. Sci., 2015, 6, 5891–5903 RSC.
- B. M. Klepser and B. M. Bartlett, J. Am. Chem. Soc., 2014, 136, 1694–1697 CrossRef CAS PubMed.
- M. K. Coggins, M.-T. Zhang, A. K. Vannucci, C. J. Dares and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 5531–5534 CrossRef CAS PubMed.
- Y. Hitomi, K. Arakawa, T. Funabiki and M. Kodera, Angew. Chem., Int. Ed., 2012, 51, 3448–3452 CrossRef CAS PubMed.
- W. A. Hoffert, M. T. Mock, A. M. Appel and J. Y. Yang, Eur. J. Inorg. Chem., 2013, 3846–3857 CrossRef CAS.
- K. G. Kottrup and D. G. H. Hetterscheid, Chem. Commun., 2016, 52, 2643–2646 RSC.
- G. Chen, L. Chen, S.-M. Ng, W.-L. Man and T.-C. Lau, Angew. Chem., Int. Ed., 2013, 52, 1789–1791 CrossRef CAS PubMed.
- D. Hong, S. Mandal, Y. Yamada, Y.-M. Lee, W. Nam, A. Llobet and S. Fukuzumi, Inorg. Chem., 2013, 52, 9522–9531 CrossRef CAS PubMed.
- M. M. Najafpour, A. N. Moghaddam, D. J. Sedigh and M. Hołyńska, Catal. Sci. Technol., 2014, 4, 30–33 CAS.
- A. R. Parent, T. Nakazono, S. Lin, S. Utsunomiya and K. Sakai, Dalton Trans., 2014, 43, 12501–12513 RSC.
- L. D. Wickramasinghe, R. Zhou, R. Zong, P. Vo, K. J. Gagnon and R. P. Thummel, J. Am. Chem. Soc., 2015, 137, 13260–13263 CrossRef CAS PubMed.
- L. Tong, R. Zong and R. P. Thummel, J. Am. Chem. Soc., 2014, 136, 4881–4884 CrossRef CAS PubMed.
- M. Okamura, M. Kondo, R. Kuga, Y. Kurashige, T. Yanai, S. Hayami, V. K. K. Praneeth, M. Yoshida, K. Yoneda, S. Kawata and S. Masaoka, Nature, 2016, 530, 465–468 CrossRef CAS PubMed.
- M. Anbar and I. Pecht, J. Am. Chem. Soc., 1967, 89, 2553–2556 CrossRef CAS.
- V. Y. Shafirovich and V. V. Strelets, Nouv. J. Chim., 1978, 2, 199–201 CAS.
- V. Y. Shafirovich, N. K. Khannanov and V. V. Strelets, Nouv. J. Chim., 1980, 4, 81–84 CAS.
- B. S. Brunschwig, M. H. Chou, C. Creutz, P. Ghosh and N. Sutin, J. Am. Chem. Soc., 1983, 105, 4832–4833 CrossRef CAS.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- Y. Surendranath, M. Dincă and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 2615–2620 CrossRef CAS PubMed.
- D. A. Lutterman, Y. Surendranath and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 3838–3839 CrossRef CAS PubMed.
- A. J. Esswein, Y. Surendranath, S. Y. Reece and D. G. Nocera, Energy Environ. Sci., 2011, 4, 499–504 CAS.
- M. Risch, V. Khare, I. Zaharieva, L. Gerencser, P. Chernev and H. Dau, J. Am. Chem. Soc., 2009, 131, 6936–6937 CrossRef CAS PubMed.
- M. W. Kanan, J. Yano, Y. Surendranath, M. Dincă, V. K. Yachandra and D. G. Nocera, J. Am. Chem. Soc., 2010, 132, 13692–13701 CrossRef CAS PubMed.
- J. G. McAlpin, Y. Surendranath, M. Dincă, T. A. Stitch, S. A. Stoian, W. H. Casey, D. G. Nocera and R. D. Britt, J. Am. Chem. Soc., 2010, 132, 6882–6883 CrossRef CAS PubMed.
- Y. Surendranath, M. W. Kanan and D. G. Nocera, J. Am. Chem. Soc., 2010, 132, 16501–16509 CrossRef CAS PubMed.
- J. B. Gerken, J. G. McAlpin, J. Y. C. Chen, M. L. Rigsby, W. H. Casey, R. D. Britt and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 14431–14442 CrossRef CAS PubMed.
- P. Du, O. Kokhan, K. W. Chapman, P. J. Chupas and D. M. Tiede, J. Am. Chem. Soc., 2012, 134, 11096–11099 CrossRef CAS PubMed.
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef CAS PubMed.
- X. L. Hu, S. Piccinin, A. Laio and S. Fabris, ACS Nano, 2012, 6, 10497–10504 CAS.
- C. L. Farrow, D. K. Bediako, Y. Surendranath, D. G. Nocera and S. J. L. Billinge, J. Am. Chem. Soc., 2013, 135, 6403–6406 CrossRef CAS PubMed.
- D. K. Bediako, C. Costentin, E. C. Jones, D. G. Nocera and J.-M. Savéant, J. Am. Chem. Soc., 2013, 135, 10492–10502 CrossRef CAS PubMed.
- A. M. Ullman and D. G. Nocera, J. Am. Chem. Soc., 2013, 135, 15053–15061 CrossRef CAS PubMed.
- Q. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342–345 CrossRef CAS PubMed.
- Z. Huang, Z. Luo, Y. V. Geletii, J. W. Vickers, Q. Yin, D. Wu, Y. Hou, Y. Ding, J. Song, D. G. Musaev, C. L. Hill and T. Lian, J. Am. Chem. Soc., 2011, 133, 2068–2071 CrossRef CAS PubMed.
- For reviews on polyoxometalates (POMs) as water oxidation catalysts, see: (a) H. Lv, Y. V. Geletii, C. Zhao, J. W. Vickers, G. Zhu, Z. Luo, J. Song, T. Lian, D. G. Musaev and C. L. Hill, Chem. Soc. Rev., 2012, 41, 7572–7589 RSC; (b) J. M. Sumliner, H. Lv, J. Fielden, Y. V. Geletii and C. L. Hill, Eur. J. Inorg. Chem., 2014, 635–644 CrossRef CAS.
- For a recent review on polyoxometalate (POM) catalyzed reactions, see: S.-S. Wang and G.-Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed.
- For examples of Ni-based POMs as water oxidation catalysts, see: (a) G. Zhu, E. N. Glass, C. Zhao, H. Lv, J. W. Vickers, Y. V. Geletii, D. G. Musaev, J. Song and C. L. Hill, Dalton Trans., 2012, 41, 13043–13049 RSC; (b) X.-B. Han, Y.-G. Li, Z.-M. Zhang, H.-Q. Tan, Y. Lu and E.-B. Wang, J. Am. Chem. Soc., 2015, 137, 5486–5493 CrossRef CAS PubMed.
- Y. V. Geletii, B. Botar, P. Kögerler, D. A. Hillesheim, D. G. Musaev and C. L. Hill, Angew. Chem., Int. Ed., 2008, 47, 3896–3899 CrossRef CAS PubMed.
- A. Sartorel, M. Carraro, G. Scorrano, R. D. Zorzi, S. Geremia, N. D. McDaniel, S. Bernhard and M. Bonchio, J. Am. Chem. Soc., 2008, 130, 5006–5007 CrossRef CAS PubMed.
- D. Lieb, A. Zahl, E. F. Wilson, C. Streb, L. C. Nye, K. Meyer and I. Ivanović-Burmazović, Inorg. Chem., 2011, 50, 9053–9058 CrossRef CAS PubMed.
- C. A. Ohlin, S. J. Harley, J. G. McAlpin, R. K. Hocking, B. Q. Mercado, R. L. Johnson, E. M. Villa, M. K. Fidler, M. M. Olmstead, L. Spiccia, R. D. Britt and W. H. Casey, Chem. – Eur. J., 2011, 17, 4408–4417 CrossRef CAS PubMed.
- M. Natali, S. Berardi, A. Sartorel, M. Bonchio, S. Campagna and F. Scandola, Chem. Commun., 2012, 48, 8808–8810 RSC.
- J. Wu, L. Liao, W. Yan, Y. Xue, Y. Sun, X. Yan, Y. Chen and Y. Xie, ChemSusChem, 2012, 5, 1207–1212 CrossRef CAS PubMed.
- H. Lv, J. Song, Y. V. Geletii, J. W. Vickers, J. M. Sumliner, D. G. Musaev, P. Kögerler, P. F. Zhuk, J. Bacsa, G. Zhu and C. L. Hill, J. Am. Chem. Soc., 2014, 136, 9268–9271 CrossRef CAS PubMed.
- X.-B. Han, Z.-M. Zhang, T. Zhang, Y.-G. Li, W. Lin, W. You, Z.-M. Su and E.-B. Wang, J. Am. Chem. Soc., 2014, 136, 5359–5366 CrossRef CAS PubMed.
- J. J. Stracke and R. G. Finke, J. Am. Chem. Soc., 2011, 133, 14872–14875 CrossRef CAS PubMed.
- J. J. Stracke and R. G. Finke, ACS Catal., 2013, 3, 1209–1219 CrossRef CAS.
- J. W. Vickers, H. Lv, J. M. Sumliner, G. Zhu, Z. Luo, D. G. Musaev, Y. V. Geletii and C. L. Hill, J. Am. Chem. Soc., 2013, 135, 14110–14118 CrossRef CAS PubMed.
- S. Goberna-Ferrón, J. Soriano-López, J. R. Galán-Mascarós and M. Nyman, Eur. J. Inorg. Chem., 2015, 2833–2840 CrossRef.
- S. Goberna-Ferrón, L. Vigara, J. Soriano-López and J. R. Galán-Mascarós, Inorg. Chem., 2012, 51, 11707–11715 CrossRef PubMed.
- J. Soriano-López, S. Goberna-Ferrón, L. Vigara, J. J. Carbó, J. M. Poblet and J. R. Galán-Mascarós, Inorg. Chem., 2013, 52, 4753–4755 CrossRef PubMed.
- F. Song, Y. Ding, B. Ma, C. Wang, Q. Wang, X. Du, S. Fu and J. Song, Energy Environ. Sci., 2013, 6, 1170–1184 CAS.
- J. Wei, Y. Feng, P. Zhou, Y. Liu, J. Xu, R. Xiang, Y. Ding, C. Zhao, L. Fan and C. Hu, ChemSusChem, 2015, 8, 2630–2634 CrossRef CAS PubMed.
- C. Zhang, X. Lin, Z. Zhang, L.-S. Long, C. Wang and W. Lin, Chem. Commun., 2014, 50, 11591–11594 RSC.
- D. Guo, S. Teng, Z. Liu, W. You and L. Zhang, J. Cluster Sci., 2013, 24, 549–558 CrossRef CAS.
- S. Tanaka, M. Annaka and K. Sakai, Chem. Commun., 2012, 48, 1653–1655 RSC.
- F. Evangelisti, P.-E. Car, O. Blacque and G. R. Patzke, Catal. Sci. Technol., 2013, 3, 3117–3129 CAS.
- J. K. Beattie, T. W. Hambley, J. A. Klepetko, A. F. Masters and P. Turner, Polyhedron, 1998, 17, 1343–1354 CrossRef CAS.
- R. Chakrabarty, S. J. Bora and B. K. Das, Inorg. Chem., 2007, 46, 9450–9462 CrossRef CAS PubMed.
- K. Dimitrou, K. Folting, W. E. Streib and G. Christou, J. Am. Chem. Soc., 1993, 115, 6432–6433 CrossRef CAS.
- K. Dimitrou, A. D. Brown, T. E. Concolino, A. L. Rheingold and G. Christou, Chem. Commun., 2001, 1284–1285 RSC.
- N. S. McCool, D. M. Robinson, J. E. Sheats and G. C. Dismukes, J. Am. Chem. Soc., 2011, 133, 11446–11449 CrossRef CAS PubMed.
- S. Berardi, G. La Ganga, M. Natali, I. Bazzan, F. Puntoriero, A. Sartorel, F. Scandola, S. Campagna and M. Bonchio, J. Am. Chem. Soc., 2012, 134, 11104–11107 CrossRef CAS PubMed.
- G. La Ganga, F. Puntoriero, S. Campagna, I. Bazzan, S. Berardi, M. Bonchio, A. Sartorel, M. Natali and F. Scandola, Faraday Discuss., 2012, 155, 177–190 RSC.
- H. Yuan, D. L. Newton, L. A. Seymour, A. Metz, D. Cropek, A. A. Holder and R. Y. Ofoli, Catal. Commun., 2014, 56, 76–80 CrossRef CAS.
- X. Zhou, F. Li, H. Li, B. Zhang, F. Yu and L. Sun, ChemSusChem, 2014, 7, 2453–2456 CrossRef CAS PubMed.
- P. Sarmah, R. Chakrabarty, P. Phukan and B. K. Das, J. Mol. Catal. A: Chem., 2007, 268, 36–44 CrossRef CAS.
- A. I. Nguyen, M. S. Ziegler, P. Oña-Burgos, M. Sturzbecher-Hohne, W. Kim, D. E. Bellone and T. D. Tilley, J. Am. Chem. Soc., 2015, 137, 12865–12872 CrossRef CAS PubMed.
- X. Li and P. E. M. Siegbahn, J. Am. Chem. Soc., 2013, 135, 13804–13813 CrossRef CAS PubMed.
- A. Fernando and C. M. Aikens, J. Phys. Chem. C, 2015, 119, 11072–11085 CAS.
- For a recent mechanistic study on the Co4O4(pyr)4(OAc)4 cubane, see: P. F. Smith, L. Hunt, A. B. Laursen, V. Sagar, S. Kaushik, K. U. D. Calvinho, G. Marotta, E. Mosconi, F. De Angelis and G. C. Dismukes, J. Am. Chem. Soc., 2015, 137, 15460–15468 CrossRef CAS PubMed.
- F. Evangelisti, R. Güttinger, R. Moré, S. Luber and G. R. Patzke, J. Am. Chem. Soc., 2013, 135, 18734–18737 CrossRef CAS PubMed.
- F. H. Hodel and S. Luber, ACS Catal., 2016, 6, 1505–1517 CrossRef CAS.
- F. Evangelisti, R. Moré, F. Hodel, S. Luber and G. R. Patzke, J. Am. Chem. Soc., 2015, 137, 11076–11084 CrossRef CAS PubMed.
- A. M. Ullman, Y. Liu, M. Huynh, D. K. Bediako, H. Wang, B. L. Anderson, D. C. Powers, J. J. Breen, H. D. Abruña and D. G. Nocera, J. Am. Chem. Soc., 2014, 136, 17681–17688 CrossRef CAS PubMed.
- X. Li, E. B. Clatworthy, A. F. Masters and T. Maschmeyer, Chem. – Eur. J., 2015, 21, 16578–16584 CrossRef CAS PubMed.
- D. J. Wasylenko, C. Ganesamoorthy, J. Borau-Garcia and C. P. Berlinguette, Chem. Commun., 2011, 47, 4249–4251 RSC.
- D. K. Dogutan, R. McGuire and D. G. Nocera, J. Am. Chem. Soc., 2011, 133, 9178–9180 CrossRef CAS PubMed.
- D. J. Wasylenko, R. D. Palmer, E. Schott and C. P. Berlinguette, Chem. Commun., 2012, 48, 2107–2109 RSC.
- D. W. Crandell, S. Ghosh, C. P. Berlinguette and M.-H. Baik, ChemSusChem, 2015, 8, 844–852 CrossRef CAS PubMed.
- B. Das, A. Orthaber, S. Ott and A. Thapper, Chem. Commun., 2015, 51, 13074–13077 RSC.
- R. McGuire, Jr., D. K. Dogutan, T. S. Teets, J. Suntivich, Y. Shao-Horn and D. G. Nocera, Chem. Sci., 2010, 1, 411–414 RSC.
- M. Z. Ertem and C. J. Cramer, Dalton Trans., 2012, 41, 12213–12219 RSC.
- W. Lai, R. Cao, G. Dong, S. Shaik, J. Yao and H. Chen, J. Phys. Chem. Lett., 2012, 3, 2315–2319 CrossRef CAS PubMed.
- H. Lei, A. Han, F. Li, M. Zhang, Y. Han, P. Du, W. Lai and R. Cao, Phys. Chem. Chem. Phys., 2014, 16, 1883–1893 RSC.
- T. Nakazono, A. R. Parent and K. Sakai, Chem. Commun., 2013, 49, 6325–6327 RSC.
- D. Wang and J. T. Groves, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15579–15584 CrossRef CAS PubMed.
- A. M. S. Silva, M. G. P. M. S. Neves, R. R. L. Martins, J. A. S. Cavaleiro, T. Boschi and P. Tagliatesta, J. Porphyrins Phthalocyanines, 1998, 2, 45–51 CrossRef CAS.
- T. Nakazono, A. R. Parent and K. Sakai, Chem. – Eur. J., 2015, 21, 6723–6726 CrossRef CAS PubMed.
- C.-F. Leung, S.-M. Ng, C.-C. Ko, W.-L. Man, J. Wu, L. Chen and T.-C. Lau, Energy Environ. Sci., 2012, 5, 7903–7907 CAS.
- E. Pizzolato, M. Natali, B. Posocco, A. Montellano López, I. Bazzan, M. Di Valentin, P. Galloni, V. Conte, M. Bonchio, F. Scandola and A. Sartorel, Chem. Commun., 2013, 49, 9941–9943 RSC.
- A. Asraf, H. A. Younus, C. I. Ezugwu, A. Mehta and F. Verpoort, Catal. Sci. Technol. 10.1039/c5cy02157j.
- H. Wang, Y. Lu, E. Mijangos and A. Thapper, Chin. J. Chem., 2014, 32, 467–473 CrossRef CAS.
- S. Fu, Y. Liu, Y. Ding, X. Du, F. Song, R. Xiang and B. Ma, Chem. Commun., 2014, 50, 2167–2169 RSC.
- H. Chen, Z. Sun, X. Liu, A. Han and P. Du, J. Phys. Chem. C, 2015, 119, 8998–9004 CAS.
- D. Hong, J. Jung, J. Park, Y. Yamada, T. Suenobu, Y.-M. Lee, W. Nam and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 7606–7616 CAS.
- J. Luo, N. P. Rath and L. M. Mirica, Inorg. Chem., 2011, 50, 6152–6157 CrossRef CAS PubMed.
- M. L. Rigsby, S. Mandal, W. Nam, L. C. Spencer, A. Llobet and S. S. Stahl, Chem. Sci., 2012, 3, 3058–3062 RSC.
- H.-Y. Wang, E. Mijangos, S. Ott and A. Thapper, Angew. Chem., Int. Ed., 2014, 53, 14499–14502 CrossRef CAS PubMed.
- For a study on the related [(tpa)Co(μ-OH)2Co(tpa)]4+ complex, see: T. Ishizuka, A. Watanabe, H. Kotani, D. Hong, K. Satonaka, T. Wada, Y. Shiota, K. Yoshizawa, K. Ohara, K. Yamaguchi, S. Kato, S. Fukuzumi and T. Kojima, Inorg. Chem., 2016, 55, 1154–1164 CrossRef CAS PubMed.
- For reviews on Cu-catalyzed aerobic oxidations, see: (a) J. Piera and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2008, 47, 3506–3523 CrossRef CAS PubMed; (b) Camilla Parmeggiani and Francesca Cardona, Green Chem., 2012, 14, 547–564 RSC; (c) S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234–6458 CrossRef CAS PubMed; (d) Q. Cao, L. M. Dornan, L. Rogan, N. L. Hughes and M. J. Muldoon, Chem. Commun., 2014, 50, 4524–4543 RSC; (e) B. L. Ryland and S. S. Stahl, Angew. Chem., Int. Ed., 2014, 53, 8824–8838 CrossRef CAS PubMed; (f) S. D. McCann and S. S. Stahl, Acc. Chem. Res., 2015, 48, 1756–1766 CrossRef CAS PubMed.
- S. M. Barnett, K. I. Goldberg and J. M. Mayer, Nat. Chem., 2012, 4, 498–502 CrossRef CAS PubMed.
- T. Zhang, C. Wang, S. Liu, J.-L. Wang and W. Lin, J. Am. Chem. Soc., 2014, 136, 273–281 CrossRef CAS PubMed.
- W.-H. Wang, J. F. Hull, J. T. Muckerman, E. Fujita and Y. Himeda, Energy Environ. Sci., 2012, 5, 7923–7926 CAS.
- C. M. Conifer, R. A. Taylor, D. J. Law, G. J. Sunley, A. J. P. White and G. J. P. Britovsek, Dalton Trans., 2011, 40, 1031–1033 RSC.
- D. L. Gerlach, S. Bhagan, A. A. Cruce, D. B. Burks, I. Nieto, H. T. Truong, S. P. Kelley, C. J. Herbst-Gervasoni, K. L. Jernigan, M. K. Bowman, S. Pan, M. Zeller and E. T. Papish, Inorg. Chem., 2014, 53, 12689–12698 CrossRef CAS PubMed.
- M.-T. Zhang, Z. Chen, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2013, 135, 2048–2051 CrossRef CAS PubMed.
- M. K. Kim and A. E. Martell, J. Am. Chem. Soc., 1966, 88, 914–918 CrossRef CAS PubMed.
- N. V. Nagy, T. Szabó-Plánka, A. Rockenbauer, G. Peintler, I. Nagypál and L. Korecz, J. Am. Chem. Soc., 2003, 125, 5227–5235 CrossRef CAS PubMed.
- S. G. Winikoff and C. J. Cramer, Catal. Sci. Technol., 2014, 4, 2484–2489 CAS.
- J. S. Pap, Ł. Szyrwiel, D. Srankó, Z. Kerner, B. Setner, Z. Szewczuk and W. Malinka, Chem. Commun., 2015, 51, 6322–6324 RSC.
- P. Garrido-Barros, I. Funes-Ardoiz, S. Drouet, J. Benet-Buchholz, F. Maseras and A. Llobet, J. Am. Chem. Soc., 2015, 137, 6758–6761 CrossRef CAS PubMed.
- For a recent study on Cu-based water oxidation catalysts that highlights the dramatic effect that ligand frameworks can have on the catalytic activity, see: R.-J. Xiang, H.-Y. Wang, Z.-J. Xin, C.-B. Li, Y.-X. Lu, X.-W. Gao, H.-M. Sun and R. Cao, Chem. – Eur. J., 2016, 22, 1602–1607 CrossRef CAS PubMed.
- X.-J. Su, M. Gao, L. Jiao, R.-Z. Liao, P. E. M. Siegbahn, J.-P. Cheng and M.-T. Zhang, Angew. Chem., Int. Ed., 2015, 54, 4909–4914 CrossRef CAS PubMed.
- C. He and S. J. Lippard, Tetrahedron, 2000, 56, 8245–8252 CrossRef CAS.
- C. He, J. L. DuBois, B. Hedman, K. O. Hodgson and S. J. Lippard, Angew. Chem., Int. Ed., 2001, 40, 1484–1487 CrossRef CAS.
- L. Yu, X. Du, Y. Ding, H. Chen and P. Zhou, Chem. Commun., 2015, 51, 17443–17446 RSC.
- S. Nellutla, J. van Tol, N. S. Dalal, L.-H. Bi, U. Kortz, B. Keita, L. Nadjo, G. A. Khitrov and A. G. Marshall, Inorg. Chem., 2005, 44, 9795–9806 CrossRef CAS PubMed.
- W.-B. Yu, Q.-Y. He, X.-F. Ma, H.-T. Shi and X. Wei, Dalton Trans., 2015, 44, 351–358 RSC.
- M. Mahdi Najafpour, F. Ebrahimi, R. Safdari, M. Z. Ghobadi, M. Tavahodi and P. Rafighi, Dalton Trans., 2015, 44, 15435–15440 RSC.
- T.-T. Li, S. Cao, C. Yang, Y. Chen, X.-J. Lv and W.-F. Fu, Inorg. Chem., 2015, 54, 3061–3067 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |