DOI:
10.1039/C5SC02018B
(Edge Article)
Chem. Sci., 2015,
6, 7213-7221
Highly efficient, selective, and durable photocatalytic system for CO2 reduction to formic acid†
Received
5th June 2015
, Accepted 28th August 2015
First published on 29th September 2015
Abstract
We discovered an extremely suitable sacrificial electron donor, 1,3-dimethyl-2-(o-hydroxyphenyl)-2,3-dihydro-1H-benzo[d]imidazole, for the selective photocatalytic reduction of CO2 to formic acid using a Ru(II)–Ru(II) supramolecular photocatalyst. The efficiency, durability, and rate of photocatalysis are significantly increased (ΦHCOOH = 0.46, TONHCOOH = 2766, TOFHCOOH = 44.9 min−1) in comparison with those using 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole or 1-benzyl-1,4-dihydronicotinamide.
Introduction
A tremendous amount of carbon dioxide, the most oxidized form of carbon, has been produced from the combustion of carbon-containing materials such as fossil fuels, plastics, and foods. Because carbon dioxide is a primary greenhouse gas that contributes significantly toward global warming, the production of CO2 must be reduced. Therefore, a technology that produces useful energy-rich chemicals from CO2 with solar light as an energy source should be developed in the future.
Although hydrogen gas has garnered attention as a carbon-free fuel, it is constrained by its physical and chemical properties such as difficulties in transportation because of its low density and risk of explosion. Formic acid, one of the two-electron reduced compounds of CO2, is a promising hydrogen storage medium because it can easily be split by catalysts into hydrogen and CO2 under mild conditions and it exists in the liquid form with a higher energy density at ambient temperature.1,2 The hydrogenation reaction of CO2 has been actively studied owing to its ability to produce formic acid.3–5 For example, Himeda, Fujita, and co-workers recently reported iridium(III) catalysts for efficient hydrogenation of CO2 under mild conditions.5 Another method for the production of formic acid without H2 is the photocatalytic reduction of CO2.
To the best of our knowledge, we were the first to report supramolecular photocatalysts for the selective reduction of CO2 to formic acid, which consist of [Ru(dmb)m(BL)3−m]2+ (dmb = 4,4′-dimethyl-2,2′-bipyridine, BL = 1,2-bis(4′-methyl-[2,2′-bipyridin]-4-yl)ethane, m = 0, 1, 2) as a photosensitizer unit and [Ru(dmb)n(BL)2−n(CO)2]2+ (n = 0, 1) as a catalyst unit.6 In the photocatalytic reactions, a model compound of coenzyme NAD(P)H called 1-benzyl-1,4-dihydronicotinamide (BNAH, Chart 1) was employed as a sacrificial electron donor. In particular, a trinuclear complex with two photosensitizer units and one catalyst unit (Ru2–Ru(CO), Chart 1) exhibited the highest photocatalytic activity. A high turnover number (TONHCOOH = 671) and turnover frequency (TOFHCOOH = 11.6 min−1) for the formation of formic acid were observed. However, the quantum yield for formic acid formation was relatively low (ΦHCOOH = 0.061), even compared with that for supramolecular photocatalytic systems employing the same photosensitizer units and cis,trans-[Re(BL)(CO)2(L)2]2+-type complexes as catalyst units. These systems selectively produced CO, with ΦCO values ranging from 0.10 to 0.15.7
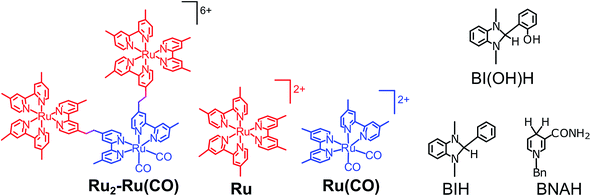 |
| | Chart 1 Structures and abbreviations of Ru2–Ru(CO), model mononuclear complexes, BI(OH)H, BIH, and BNAH. The counter anions of the complexes were PF6−. | |
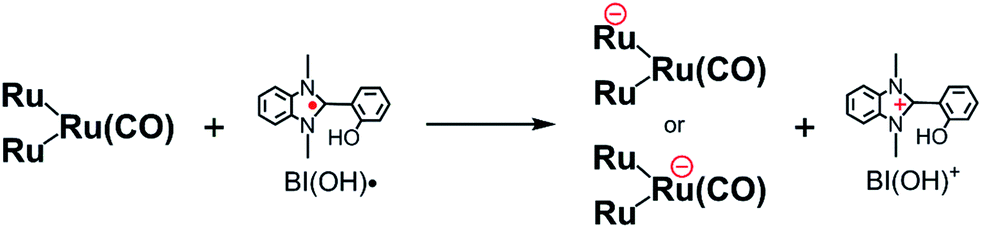
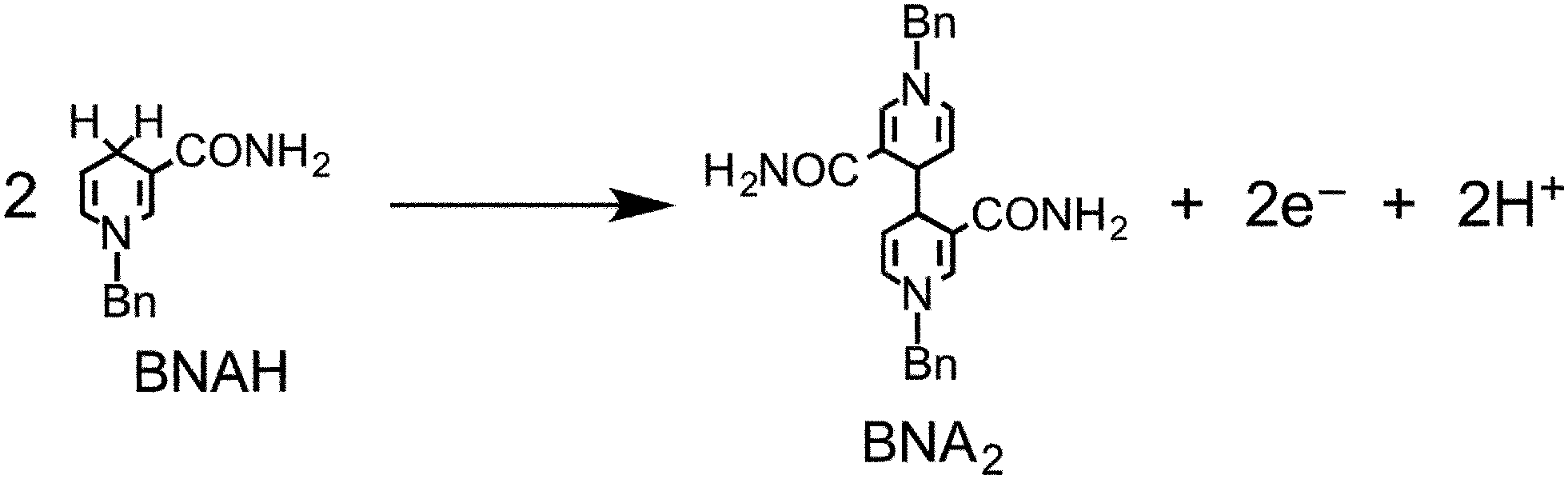
We also reported that BNAH has several disadvantages as a sacrificial electron donor for the improvement of photocatalysis in supramolecular systems.6–8 First, the efficiency of quenching the excited state of the Ru(II) photosensitizer unit with BNAH is relatively low. For example, even a 0.1 M solution of BNAH quenched only 59% of the excited photosensitizer unit in Ru2–Ru(CO). Second, a relatively fast back electron transfer occurs from the reduced form of the photosensitizer unit to the oxidized form of BNAH, i.e., BNAH˙+. Third, accumulation of the BNA dimers (BNA2), which were produced from the deprotonation of BNAH˙+ and following coupling processes, prevents further progression of the photocatalytic reaction. This is because BNA2 is more effective than BNAH at quenching the excited photosensitizer unit of the supramolecular photocatalysts. Owing to these limitations, we can conclude that the reported values of the photocatalytic abilities of Ru2–Ru(CO) do not reflect their actual potential.
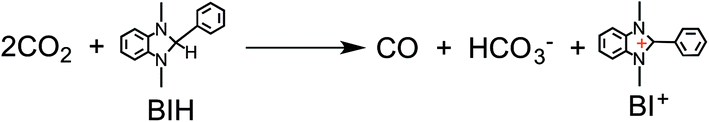
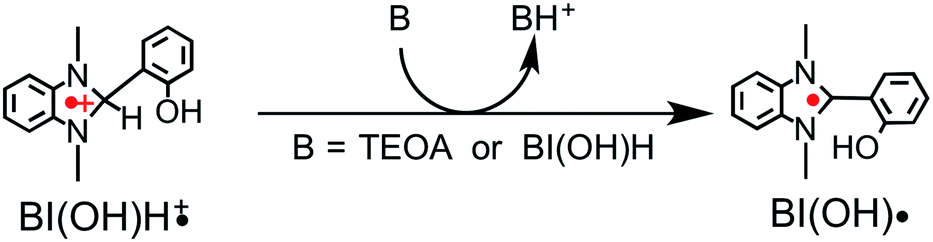
We recently discovered that compared with BNAH, a benzimidazoline derivative (BIH, Chart 1) can function as a much better sacrificial electron donor for the excited state of [Ru(N^N)3]2+-type (N^N = diimine ligand) complexes.8 The efficiency, durability, and formation rate of CO generated from the photocatalytic reduction of CO2 with the Ru(II)–Re(I) supramolecular photocatalyst (Ru–Re) were substantially improved with BIH (ΦCO = 0.45, TONCO = 3029, TOFCO = 35.7 min−1) compared to those with BNAH (ΦCO = 0.15, TONCO = 207, TOFCO = 4.7 min−1). BIH could quantitatively quench the excited state of the Ru(II)–Re(I) photocatalyst. BIH is more effective than BNAH in decreasing the efficiency of the back electron transfer from its oxidized form to the reduced form of the photocatalyst. BIH serves as a two-electron donor, whereas BNAH can only donate one electron to the photocatalyst because of the coupling reaction of BNA˙. The two-electron oxidized product of BIH (BI+) did not prevent the progress of the photocatalytic reaction.
We applied similar strategies in this study and implemented a new reductant to clarify the “real” photocatalytic abilities of Ru2–Ru(CO) for the reduction of CO2. We found that the photocatalysis of Ru2–Ru(CO) is significantly better than those employing BNAH or BIH if another benzimidazoline derivative with an –OH group at the o-position of the phenyl group, i.e., 1,3-dimethyl-2-(o-hydroxyphenyl)-2,3-dihydro-1H-benzo[d]imidazole9–12 (BI(OH)H, Chart 1), is used. In particular, this method gave the highest rate for the photocatalytic reduction of CO2 of all previously reported visible-light-driven photocatalytic systems (TOFHCOOH = 44.9 min−1).
Results
We determined the quantum yield (Φ), turnover number (TON), and turnover frequency (TOF) of the reduction product(s) to evaluate the photocatalysts. The following reaction conditions were selected. The details and justifications for these conditions are described in ESI.†6
Light irradiation condition 1 (LIC1) was used to determine the ΦHCOOH: a 480 nm monochromatic light with 5.0 × 10−9 einstein s−1 intensity was used for excitation. Quantum yields were determined using two different concentrations of the photocatalyst, i.e., 0.15 mM and 0.025 mM. When [Ru2–Ru(CO)] was 0.15 mM, the absorbance of the solution was greater than 3 with a 1 cm path length. The solution was mixed vigorously during the irradiation process. Using 0.025 mM Ru2–Ru(CO), the number of absorbed photons could be calculated from the change in absorbance at 480 nm during irradiation.
LIC2 was used for determining the TONHCOOH: a wavelength of light >500 nm was used for excitation. The photocatalyst concentration was 0.025 mM.
LIC3 was used for determining the TOFHCOOH: a wavelength of light >420 nm was used for excitation. The photocatalyst concentration was 0.025 mM.
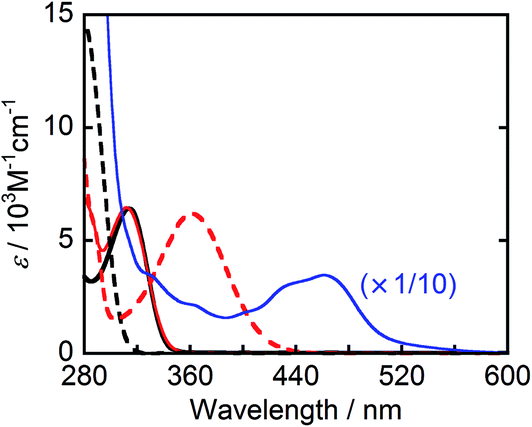
Fig. 1 shows the UV-vis absorption spectra of the reductants, BIH and BI(OH)H, and their respective oxidized compounds, BI+ and BI(O−)+ along with the photocatalyst, Ru2–Ru(CO). Because none of these compounds absorbed wavelength of light longer than 440 nm, the excitation light was selectively absorbed by Ru2–Ru(CO) in the LIC1 and LIC2 conditions in a mixed solvent system of N,N-dimethylformamide (DMF) and triethanolamine (TEOA) (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v), which was also used as the solvent in the photocatalytic reactions. Because the irradiated light was partially absorbed by BI(O−)+ in LIC3, the TOFHCOOH values were obtained in the first stage of the photocatalytic reaction when only a small amount of BI(O−)+ had accumulated in the reaction solution (10 min irradiation).
:1 v/v), which was also used as the solvent in the photocatalytic reactions. Because the irradiated light was partially absorbed by BI(O−)+ in LIC3, the TOFHCOOH values were obtained in the first stage of the photocatalytic reaction when only a small amount of BI(O−)+ had accumulated in the reaction solution (10 min irradiation).
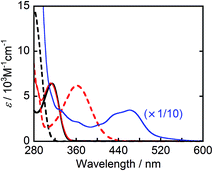 |
| | Fig. 1 UV-vis absorption spectra of BIH (black solid line), BI+ (black broken line), BI(OH)H (red solid line), BI(O−)+ (red broken line), and Ru2–Ru(CO) (blue line, 1/10 intensity) in DMF–TEOA (4:1 v/v). | |
We first attempted to use BIH as a reductant. A mixed solution of DMF and TEOA (4:1 v/v) containing Ru2–Ru(CO) (0.025 mM) and BIH (0.1 M) was irradiated (>500 nm, LIC2) under a CO2 atmosphere. The resulting photocatalytic products included both formic acid (TONHCOOH = 641) and CO (TONCO = 237). Note that the substitution of BIH with BNAH under this same reaction condition gives formic acid as a main product and CO as an extremely minor product (TONHCOOH = 562, TONCO = 29). Therefore, the yield for the selective formation of formic acid decreased from 91% to 72% when BIH was used as the reductant (Table 1). Markedly, BIH could function as a fascinating sacrificial electron donor for CO2 reduction with Ru–Re to give CO as the main product (ΦCO = 0.45, TONCO = 3029, TOFCO = 35.7 min−1). These values are substantially greater than those obtained with BNAH.8
Table 1 Photocatalytic properties using Ru2–Ru(CO)a
| Reductant |
TONb (selectivityc/%) |
Φ
HCOOH
|
TOFHCOOHe/min−1 |
k
q
/108 M−1 s−1 |
η
q
|
| HCOOH |
CO |
H2 |
0.15 mM |
0.025 mM |
|
A CO2 saturated DMF–TEOA (4:1 v/v) solution consisting of a sacrificial electron donor (0.1 M) and Ru2–Ru(CO) was irradiated. The concentration of Ru2–Ru(CO) was 0.15 mM or 0.025mM for the measurement of Φ (LIC1) and 0.025 mM for the measurement of TON and TOF (LIC2 and LIC3, respectively).
A 2 mL solution was irradiated in LIC2 for 20 h.
Selectivity of the products.
A 4 mL solution was irradiated using LIC1 (light intensity: 5.0 × 10−9 einstein s−1).
A 4 mL solution was irradiated using LIC3.
Quenching rate constant of emission from the photosensitizer unit by a sacrificial electron donor.
Quenching fractions of emission, which is calculated as ηq = kqτem[reductant]/(1 + kqτem[reductant]), from the photosensitizer unit using 0.1 M of a sacrificial electron donor, where the emission lifetime of Ru2–Ru(CO) (τem) was 726 ns (ref. 6).
|
| BI(OH)H |
2766 (87) |
215 (7) |
212 |
0.46 |
0.42 |
44.9 |
9.9 |
0.99 |
| BIH |
641 (72) |
237 (27) |
13 |
0.18 |
0.17 |
10.2 |
11.0 |
0.99 |
| BNAH |
562 (91) |
29 (5) |
29 |
0.06 |
0.04 |
7.8 |
0.20 |
0.59 |
The total reaction scheme for the photocatalytic reduction of CO2 to CO using Ru–Re and BIH was reported as follows.
The formation of formic acid from CO2 might require two protons along with the two required electrons. This may induce a shortage of protons in the solution during the photocatalytic reaction process.
Therefore, we substituted the reductant BIH with another benzimidazoline derivative with an OH group, BI(OH)H,13 which can supply two electrons and two protons from one molecule.9–11 This substitution of BIH with BI(OH)H using the same photochemical reaction condition (LIC2) produced formic acid with a much higher TON (TONHCOOH = 2766) and selectivity (87%) (eqn (1), Fig. 2a). When Ru2–Ru(CO) or irradiation was excluded from the experimental condition, no formic acid was produced. The amount of formic acid produced during the photocatalytic reaction without TEOA (TONHCOOH = 1096 after 8 h irradiation) was lower than that with TEOA (TONHCOOH = 1504) (Fig. S2†). Using a mixed system of model complexes (Chart 1), i.e., [Ru(dmb)3]2+ (Ru, dmb = 4,4′-dimethyl-2,2′-bipyridine) and cis-[Ru(dmb)2(CO)2]2+ (Ru(CO)), both the TON (TONHCOOH = 1969) and the selectivity (78%) of formic acid formation were also lower than those using Ru2–Ru(CO) (Fig. 2b). The quantum yields for formic acid formation exhibited very high values; ΦHCOOH = 0.46 with [Ru2–Ru(CO)] = 0.15 mM and ΦHCOOH = 0.42 with [Ru2–Ru(CO)] = 0.025 mM. The reaction rate was also very high (TOFHCOOH = 44.9 min−1). To the best of our knowledge, the turnover frequency value in particular is the highest of those reported for visible-light-driven photocatalysts for CO2 reduction. As shown in Fig. 2b and Table 1, the previously described photocatalysis of Ru2–Ru(CO) significantly increased when BI(OH)H was used instead of BIH or BNAH.
| |  | (1) |
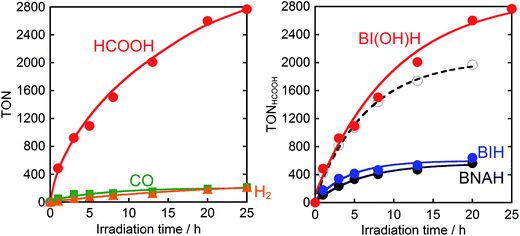 |
| | Fig. 2 (a) Photocatalytic formation of formic acid ( ), CO ( ), CO ( ), and H2 ( ), and H2 ( ) as a function of irradiation time. (b) Photocatalytic formation of formic acid using BI(OH)H ( ) as a function of irradiation time. (b) Photocatalytic formation of formic acid using BI(OH)H ( ), BIH ( ), BIH ( ) or BNAH ( ) or BNAH ( ) as a sacrificial electron donor: a CO2 saturated DMF–TEOA (4:1 v/v, 2 mL) solution containing Ru2–Ru(CO) (0.025 mM) and a sacrificial electron donor (0.1 M) was irradiated using λex > 500 nm light (LIC2). Open circles ( ) as a sacrificial electron donor: a CO2 saturated DMF–TEOA (4:1 v/v, 2 mL) solution containing Ru2–Ru(CO) (0.025 mM) and a sacrificial electron donor (0.1 M) was irradiated using λex > 500 nm light (LIC2). Open circles ( ) show the results obtained from the photocatalysis using BI(OH)H (0.1 M) and a mixed system of [Ru(dmb)3]2+ (0.05 mM) and cis-[Ru(dmb)2(CO)2]2+ (0.025 mM) instead of Ru2–Ru(CO). ) show the results obtained from the photocatalysis using BI(OH)H (0.1 M) and a mixed system of [Ru(dmb)3]2+ (0.05 mM) and cis-[Ru(dmb)2(CO)2]2+ (0.025 mM) instead of Ru2–Ru(CO). | |
In the photoinduced electron transfer process, the quenching rate constant of emission from the photosensitizer unit of Ru2–Ru(CO) by BI(OH)H (kq = 9.9 × 108 M−1 s−1) was significantly larger than that by BNAH (kq = 2.0 × 107 M−1 s−1). This result is reasonable because BI(OH)H (E1/2(BI(OH)H/BI(OH)H˙+) = −0.06 V)12 is a better reducing agent than BNAH (E°(BNAH/BNAH˙+) = 0.20 V).14 Under photocatalytic reaction conditions where the [reductant] = 0.1 M, the reductive quenching by BI(OH)H proceeded almost quantitatively (ηq = 99%) and was 1.7 times more efficient than that by BNAH (ηq = 59%). This could be one of the reasons BI(OH)H provides a higher photocatalytic efficiency than BNAH.15
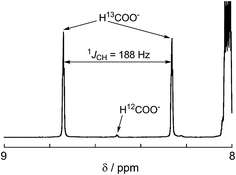
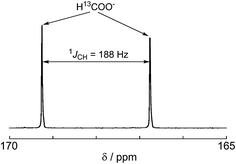
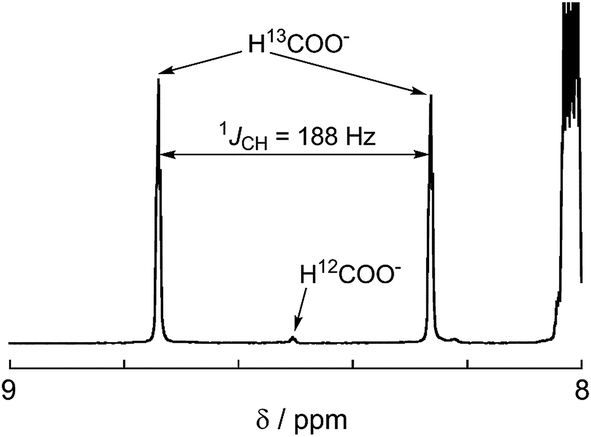
A labeling experiment using 13CO2 was conducted to determine the source of the carbon atoms in the produced formic acid. Prior to irradiation, a strong signal attributed to 13CO2 was observed at 125.6 ppm in a 13C{1H} NMR spectrum of a DMF-d7-TEOA (4:1 v/v) solution consisting of Ru2–Ru(CO) (0.5 mM) and BI(OH)H (0.1 M) under a 13CO2 (569 mmHg) atmosphere (Fig. 3a). Irradiation at λex > 420 nm for 13.5 h caused both a decrease in the 13CO2 signal and appearance of a strong signal at 168.2 ppm, which was attributed to an equilibrium mixture of H13COOH and H13COO− (Fig. 3b). In the 1H NMR spectrum of the same solution, a doublet (1JCH = 188 Hz) at 8.52 ppm was attributed to the proton of an equilibrium mixture of H13COOH and H13COO−, which should be coupled with the 13C atom. The peak area of a singlet at 8.52 ppm was approximately 1/100 of the peak area of the doublet and was attributed to the equilibrium mixture of H12COOH and H12COO− (Fig. 4). This peak area ratio was consistent with the contamination of 12CO2 in the used 13CO2 (99%). The same J value from the 13C–H coupling (188 Hz) was also observed in the 13C NMR spectrum that was measured without 1H decoupling (Fig. 5). These results clearly show that formic acid was produced through the reduction of CO2.
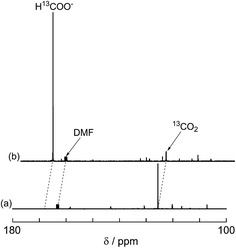 |
| | Fig. 3
13C{1H} NMR spectra of the reaction solution before (a) and after (b) 13.5 h of irradiation: DMF-d7-TEOA (4:1 v/v) solution containing BI(OH)H (0.1 M), Ru2–Ru(CO) (0.5 mM), and 13CO2 (569 mmHg) was irradiated at >420 nm using a xenon lamp. | |
 |
| | Fig. 4
1H NMR spectrum after 13.5 h irradiation for the same sample shown in Fig. 3. | |
 |
| | Fig. 5
13C NMR spectrum without 1H decoupling after 13.5 h irradiation for the same sample shown in Fig. 3. | |
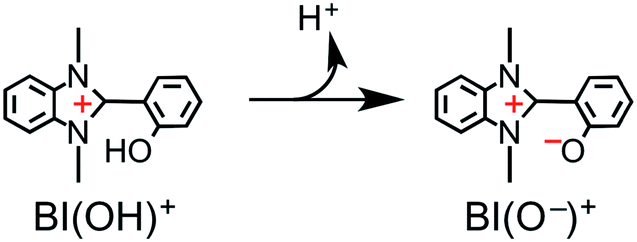
The simultaneous consumption of BI(OH)H and the production of its oxidized compound during the photocatalytic reaction were quantitatively analyzed by high-performance liquid chromatography (HPLC). The oxidized compound of BI(OH)H, a two-electron oxidized and two-proton releasing compound (BI(O−)+), was quantitatively produced (eqn (2)).
| | 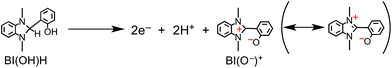 | (2) |
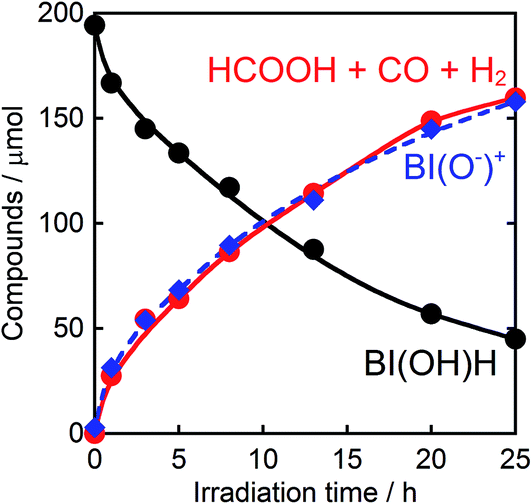
Fig. 6 shows that the amounts of consumed BI(OH)H and the generated BI(O−)+, and the sum of the generated two-electron reduced compounds (formic acid + CO + H2) were almost equivalent. For example, after a 3 h irradiation period, 55 μmol of BI(OH)H was consumed and 54 μmol of BI(O−)+ and 54 μmol of the two-electron reduced compounds were produced. Therefore, it is clear that BI(OH)H acts as both a two-electron donor and a two-proton donor, and the electrons and material balances in the photocatalytic reaction can be represented in eqn (3). Moreover, it should be noted that although BIH functions as a two-electron donor, only one proton was released in the two-electron oxidation process (eqn (4)).8
| |  | (3) |
| | 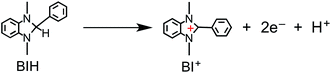 | (4) |
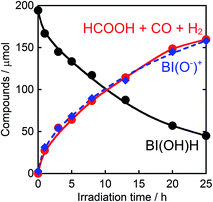 |
| | Fig. 6 Photocatalytic production of the reduction products (HCOOH + CO + H2,  ) and BI(O−)+ ( ) and BI(O−)+ ( ), and the consumption of BI(OH)H ( ), and the consumption of BI(OH)H ( ): a CO2 saturated DMF–TEOA (4:1 v/v, 2 mL) solution containing BI(OH)H (0.1 M) and Ru2–Ru(CO) (0.025 mM) was irradiated using λex > 500 nm light (LIC2). ): a CO2 saturated DMF–TEOA (4:1 v/v, 2 mL) solution containing BI(OH)H (0.1 M) and Ru2–Ru(CO) (0.025 mM) was irradiated using λex > 500 nm light (LIC2). | |
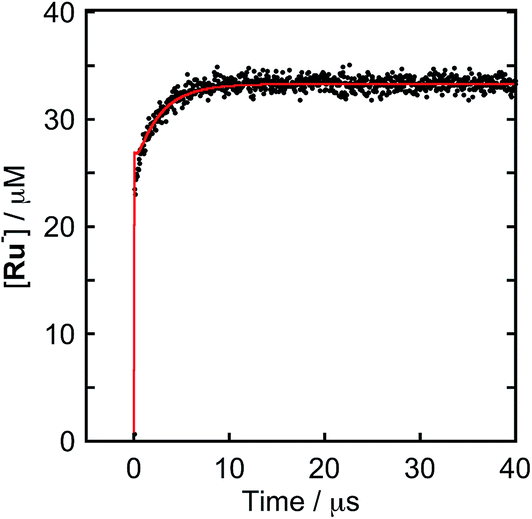
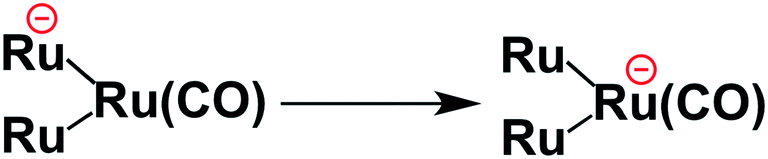
To clarify the two-electron donation processes of BI(OH)H, a laser flash photolysis method was employed. A DMF–TEOA mixed solution containing BI(OH)H (0.1 M) and Ru (0.3 mM), which was used as a model of the photosensitizer unit, was irradiated at λex = 532 nm using a Nd:YAG pulse laser. The time-resolved UV-vis absorption spectrum is shown in Fig. 7. At 100 ns after the laser excitation, new absorption was observed at 500–550 nm. This is attributed to the one-electron reduced species of Ru, i.e., Ru−,8 which should be produced via the reductive quenching of excited Ru by BI(OH)H. Fig. 8 shows the time course of [Ru−] measured at λabs = 550 nm, which increased in two steps. The initial rapid increase was completed within the laser pulse, and the concentration of Ru− increased up to approximately 10 μs. These results clearly indicate that BI(OH)H acts as a two-electron donor with one-photon excitation of Ru. Since BI(OH)˙, which can be produced via the deprotonation of BI(OH)H˙+, has strong reducing power (Eoxp = −1.96 V vs. Fc+/Fc),12 it should be able to donate an electron to another Ru molecule even in the ground state (Ered1/2 = −1.81 V).
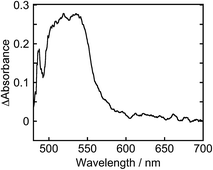 |
| | Fig. 7 Time-resolved UV-vis absorption spectrum at 100 ns after the laser excitation: a DMF–TEOA (5:1 v/v) solution containing Ru (0.3 mM) and BI(OH)H (0.1 M) was irradiated at λex = 532 nm with a Nd:YAG pulse laser under degassed conditions at room temperature. | |
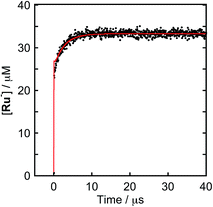 |
| | Fig. 8 Formation of Ru−: (a) DMF–TEOA (5:1 v/v) solution containing Ru (0.3 mM) and BI(OH)H (0.1 M) was irradiated at λex = 532 nm with a Nd:YAG pulse laser under degassed conditions at room temperature. The red line shows the fitting results (ESI†). | |
This time course of the Ru− production was fitted using numerical analysis based on kinetics, as shown in Scheme 1,8 where I0 is the light intensity absorbed by Ru, 1(*Ru) and 3(*Ru) are singlet and triplet metal-to-ligand charge-transfer (MLCT) excited states of Ru, respectively, (Ru−⋯BI(OH)H˙+) is an ion pair produced via the reductive quenching of 3(*Ru) by BI(OH)H, and B is a base, i.e., TEOA or BI(OH)H. The quenching rate constant of 3(*Ru) was obtained with a Stern–Volmer analysis of emission from 3(*Ru) (kq = 1.17 × 109 M−1 s−1). The increase in Ru− was fitted using eqn (5), for which the derivation is shown in the ESI.†
| | | d[Ru−]/dt = kesc[(Ru−⋯BI(OH)H˙+)] + ket[Ru][BI(OH)˙] − krec2[Ru−][BI(OH)H˙+] | (5) |
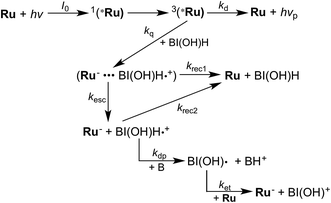 |
| | Scheme 1 Kinetics of Ru− production using BI(OH)H. | |
Based on these analyses, the rate constants of the electron transfer from BI(OH)˙ to the ground state of Ru (ket), of the back electron transfer from Ru− to BI(OH)H˙+ (krec2), and of the deprotonation of BI(OH)H˙+ (kdp) were 9 × 109 M−1 s−1, 5 × 109 M−1 s−1, and 2 × 105 s−1, respectively.
The formation quantum yields for the one-electron-reduced species (OERS) (ΦOERS) of the model of the photosensitizer unit, Ru, using BI(OH)H, BIH, and BNAH were also determined to compare their abilities as sacrificial electron donors (Table 2). A DMF–TEOA (5:1 v/v) solution containing Ru (0.1 mM) and one of the reductants (0.1 M) was irradiated with three different light intensities. The production of Ru− was monitored with a UV-vis absorption spectrometer. A greater light intensity induced a lower quantum yield in all cases (Table 2). Irradiation with a greater light intensity should result in higher concentrations of both Ru− and the one-electron oxidized species of the reductant (QH+). This should, in turn, induce more rapid back electron transfer from Ru− to QH+. When BI(OH)H was used as the reductant, the values of ΦOER were 5–7 times greater than those by using BNAH. This ratio is consistent with the quantum yields of production for formic acid, i.e., ΦHCOOH(BI(OH)H)/ΦHCOOH(BNAH) = 7.7 with a light intensity of 5.0 × 10−9 einstein s−1 (LIC1). Therefore, the main reasons for the significant increase in the ΦHCOOH value when BI(OH)H is substituted for BNAH are ascribed to an increase in the electron-donating ability and suppression of the back electron transfer (Table 2 and Fig. 7).8
Table 2 The accumulation quantum yields of OERS of Ru using BI(OH)H, BIH, or BNAH as a reductanta
| Light intensity/ 10−9 einstein s−1 |
Φ
OER
|
| BI(OH)H |
BIH |
BNAH |
|
A 4 mL Ar-saturated DMF–TEOA (5:1 v/v) solution consisting of the reductant (0.1 M) and Ru (0.1 mM) was irradiated at 480 nm.
|
| 11 |
0.45 |
0.59 |
0.066 |
| 4.3 |
0.54 |
0.74 |
0.087 |
| 1.1 |
0.64 |
0.95 |
0.14 |
Discussion
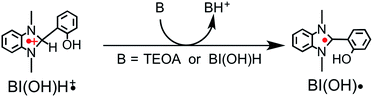
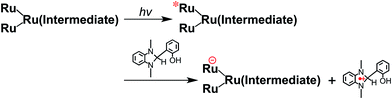
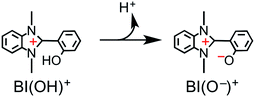
From the steady-state measurements of BI(OH)H decay and BI(O−)+ formation and the transient measurement of Ru− by laser flash photolysis, we can conclude that BI(OH)H acts as a two-electron donor with one-photon excitation of Ru2–Ru(CO) during photocatalysis. As the initial process, excited Ru2–Ru(CO) was reductively quenched by BI(OH)H giving the OERS of the photosensitizer unit and BI(OH)H˙+ at a rate constant of kq = 9.9 × 108 M−1 s−1, which was obtained from linear Stern–Volmer plots (Fig. 9, eqn (6)). The second step is most likely the deprotonation of BI(OH)H˙+ by a base (B), i.e., TEOA or BI(OH)H to produce BI(OH)˙ (eqn (7)). The intramolecular electron transfer should occur from the OERS of the photosensitizer unit to the catalyst unit (eqn (8)). Because BI(OH)˙ is a strong reducing agent, it can donate an additional electron to Ru2–Ru(CO) (Ered1/2 = −1.87 V) and/or possibly to a reaction intermediate derived from the OERS of Ru2–Ru(CO) (Ru2–Ru(Intermediate), eqn (10)) in the ground state (eqn (9) and (12)). Excitation of the photosensitizer unit of Ru2–Ru(Intermediate) might allow the acceptance of an additional electron from BI(OH)H (eqn (11)). In the last step, BI(OH)+ should release an additional proton to produce a zwitterion BI(O−)+ as a dead-end oxidized compound of BI(OH)H (eqn (13)). Therefore, a single-photon excitation of Ru2–Ru(CO) should allow BI(OH)H to donate two electrons and two protons via a sequence of electron transfer, deprotonation, electron transfer, and deprotonation processes.| | 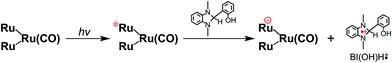 | (6) |
| |  | (7) |
| |  | (8) |
| | 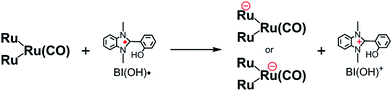 | (9) |
| | 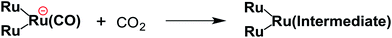 | (10) |
| |  | (11) |
| | 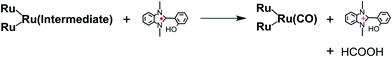 | (12) |
| |  | (13) |
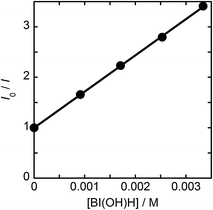 |
| | Fig. 9 Stern–Volmer plots of emission quenching of Ru2–Ru(CO) by BI(OH)H in DMF–TEOA (4:1 v/v); the excitation wavelength was 530 nm. | |
As described above, the photocatalytic CO2 reduction to formic acid proceeded using BI(OH)H without TEOA; however the TONHCOOH was lower than that using BI(OH)H with TEOA (Fig. S2†). As previously reported, TEOA does not quench the 3MLCT excited state of the photosensitizer unit of Ru2–Ru(CO), which means that it should function only as a base in the photocatalytic reaction. In the absence of TEOA, a proton from BI(OH)H˙+ should be captured by another molecule of BI(OH)H. Since the rates of the deprotonation process of BI(OH)H˙+ and the back electron transfer process from the OERS of Ru2–Ru(CO) to BI(OH)H˙+ compete with each other (Scheme 1), slower deprotonation should lower the electron-donating ability of BI(OH)H.
Although BIH has a slightly stronger electron-donating ability than BI(OH)H (Table 2), ΦHCOOH and the selectivity toward the formation of formic acid using BIH were both significantly lower than those in the case of BI(OH)H (Table 1). As previously reported, a system that used both Ru–Re as the photocatalyst and BIH as the reductant showed an extremely high efficiency, durability, and rate for the photocatalytic reduction of CO2 (ΦCO = 0.45, TONCO = 3029).8 A system that utilized Ru2–Ru(CO) as the photocatalyst and BIH as the electron donor showed an eight-fold increase in the production of CO (TONCO = 237), whereas the amount of formic acid (TONHCOOH = 641) was almost equivalent to the results obtained with BNAH (TONCO = 29, TONHCOOH = 562). As described above, the difference between BI(OH)H and BIH is most likely attributed to the number of released protons per donated electrons. A one-photon excitation of Ru2–Ru(CO) allows BI(OH)H to donate two electrons and two protons (eqn (2)) in a step-by-step process. Conversely, BIH can only donate two electrons and one proton (eqn (4)).8 In the case of BNAH, only one electron and one proton can be donated (eqn (14)), where the ratio between the donated electron and proton is similar to BI(OH)H.6,7 In other words, BIH can only donate half the amount of protons for every electron; however, both BI(OH)H and BNAH can donate an equal amount of protons for every electron.
| | 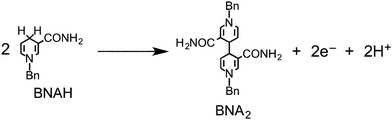 | (14) |
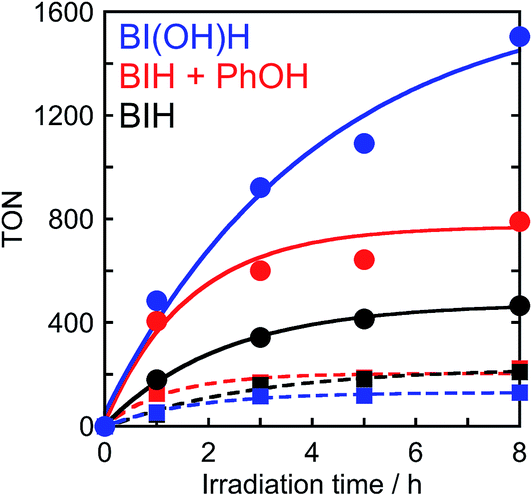
To determine the effects of the proton concentration in the solution on the photocatalysis of Ru2–Ru(CO), a photocatalytic reaction that uses BIH in the presence of a phenol (0.1 M) as a proton source was investigated (Fig. 10). In the initial stage of the photocatalytic reaction (1 h irradiation), the TONs for formic acid and CO were 406 and 122, respectively. This is similar to a system containing BI(OH)H (TONHCOOH = 484, TONCO = 53), where the TONHCOOH is 2.3 times larger than a system with BIH (TONHCOOH = 180, TONCO = 46). This result clearly indicates that BIH decreases the formation of formic acid, which is caused by a shortage of protons as BIH can only supply half the amount of protons for every donated electron but conversion of CO2 to HCOOH requires both two electrons and two protons.16,17
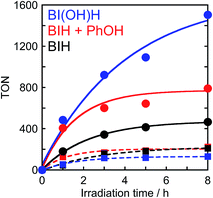 |
| | Fig. 10 Photocatalytic formation of formic acid ( ) and CO ( ) and CO ( ) using BI(OH)H (0.1 M, blue), BIH and phenol (0.1 M each, red) or BIH (0.1 M, black): a CO2 saturated DMF–TEOA (4:1 v/v, 2 mL) solution containing Ru2–Ru(CO) (0.025 mM), a sacrificial electron donor (0.1 M) and a proton source (0.1 M) was irradiated by λex > 500 nm light (LIC2). ) using BI(OH)H (0.1 M, blue), BIH and phenol (0.1 M each, red) or BIH (0.1 M, black): a CO2 saturated DMF–TEOA (4:1 v/v, 2 mL) solution containing Ru2–Ru(CO) (0.025 mM), a sacrificial electron donor (0.1 M) and a proton source (0.1 M) was irradiated by λex > 500 nm light (LIC2). | |
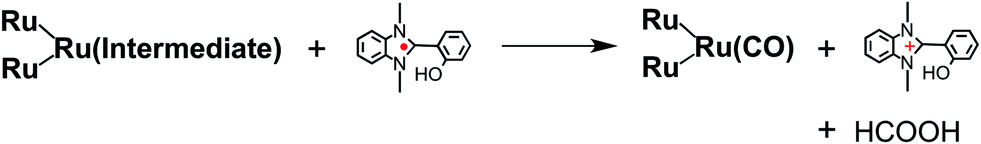
Candidates for the Ru2–Ru(Intermediate) might be formato and carboxylato complexes as the catalyst unit, i.e., [Ru(BL)2(CO)(OC(O)H)]18 and [Ru(BL)2(CO)(COOH)]19 (eqn (10)). Sullivan, Meyer, and co-workers reported an electrocatalytic reduction of CO2 to formic acid using the hydrido complex cis-[Ru(bpy)2(CO)H]+ (bpy = 2,2′-bipyridine).18 They suggested that the one-electron reduction of cis-[Ru(bpy)2(CO)H]+ and the subsequent insertion of CO2 gives cis-[Ru(bpy)2(CO)(OC(O)H)]0. A subsequent additional electron reduction leads to the loss of the formate anion and cis-[Ru(bpy)2(CO)H]+ is reformed. If a similar mechanism occurs in the photocatalytic reaction of Ru2–Ru(CO), the formation of the hydrido complex might become more favorable if BI(OH)H is used instead of BIH. This is because BI(OH)H can supply more protons during the photocatalytic reaction.
Experiments
General procedures
The UV-vis absorption spectra were measured with a JASCO V-565 spectrophotometer. 1H NMR spectra were obtained using a JEOL AL400 (400 MHz) system to identify the synthesized compounds in solutions of either CDCl3 or acetone-d6. The tetramethylsilane protons contained in CDCl3 and the residual protons of acetone-d6 were used as an internal standard for these measurements. The emission spectra were measured using a JASCO FP-6500 spectrofluorometer. The emission quenching experiments were performed on Ar-saturated solutions containing Ru2–Ru(CO) and five different concentrations of a sacrificial electron donor. Quenching rate constants kq were calculated from linear Stern–Volmer plots for the luminescence of the 3MLCT excited state of the photosensitizer units together with knowledge of their lifetimes.
Photocatalytic reactions
The experimental details of photo-irradiation conditions of the photocatalytic reactions are shown in the ESI.†6 All of the experiments used 0.1 M concentrations of BI(OH)H, BIH, or BNAH in a solvent mixture of DMF and TEOA (4:1 v/v). The solutions were purged with CO2 for 20 min before irradiation.
The concentrations of CO and H2 were analyzed by a GC-TCD (GL science GC323). Formic acid was analyzed using a capillary electrophoresis system (Otsuka Electronics Co. CAPI-3300I). To quantify the amount of formic acid, the photocatalytic reaction solution was pretreated by diluting the solution 10 times with H2O. As DMF is hydrolyzed to formic acid in the presence of a base,20 a nonirradiated photocatalytic reaction solution of saturated CO2, which suppresses the hydrolysis of DMF by acting as an acid, was employed as a reference. The reference solution was also measured before and after the quantification of formic acid, and its value was subtracted from the quantified formic acid. It should be emphasized that substantially smaller amounts of formic acid were produced by the hydrolysis of DMF in comparison with the amounts of formic acid produced by the photocatalytic reduction of CO2. This is probably due to the fact that CO2 works as an acid. For example, in the photocatalytic reaction that utilizes BI(OH)H under LIC2 (Fig. 2, Table 1), 138 μmol of formic acid was produced by the photocatalytic reduction of CO2 after a 20 h irradiation period. In contrast, only 2.0 μmol of formic acid was produced in the reference solution.
The HPLC analyses of BI(OH)H and BI(O−)+ were accomplished using a JASCO 880-PU pump with a Develosil ODS-UG-5 column (250 × 4.6 mm), a JASCO 880-51 degasser, and a JASCO UV-2070 detector. The column temperature was maintained at 30 °C with a JASCO 860-CO oven. The mobile phase was a 6:4 (v/v) mixture of acetonitrile and a NaOH–KH2PO4 buffer solution (pH 7) with a flow rate of 0.5 mL min−1. The retention times were 23.8 min (BI(OH)H) and 6.3 min (BI(O−)+).
Labeling experiments using 13CO2
The 13CO2 experiment was performed in a DMF-d7-TEOA solution containing Ru2–Ru(CO) (0.5 mM) and BI(OH)H (0.1 M). The tube was deaerated using the freeze–pump–thaw method before 13CO2 (569 mmHg) was introduced into the solution. 13C{1H}, 13C, and 1H NMR spectra were measured with a JEOL AL300 (75 MHz for 13C NMR and 300 MHz for 1H NMR) before and after 13.5 h of irradiation using light with a wavelength of more than 420 nm obtained using a 500 W xenon lamp with a cut-off filter. The residual carbon and proton of DMF-d7 was used as an internal standard for these measurements.
Time-resolved UV-vis absorption spectroscopy
Second-harmonic-generation light at 532 nm produced with a Spectra-Physics Quanta-Ray LAB-150-10 pulsed Nd:YAG laser was used for excitation (10 ns FWHM). An Ushio 300 W Xe arc lamp was operated in a pulse-enhanced mode (500 μs duration) using an XC-300 power supply and a YXP-300 light pulsar (Eagle Shoji) as a monitoring light source. The monitoring light beam was passed through the quartz cuvette (10 × 10 × 40 mm) that contained a sample and was directed into an R926 photomultiplier tube (Hamamatsu Photonics) on a Jobin-Yvon HR-320 monochromator. Time profiles of the monitoring light intensity were stored using a LeCroy WaveRunner 640zi oscilloscope (4 GHz bandwidth). Transient spectra were obtained with an Andor Technology iStar H320T-18F-03 (690 channels; minimum gate width: 5 ns) ICCD detector head mounted on the HR-320 monochromator. A DMF–TEOA (5:1 v/v) solution that contained Ru (0.3 mM), and BI(OH)H or BIH (0.1 M) was degassed by freeze–pump–thaw method prior to the laser flash photolysis.
Quantum yield measurements of the one-electron reduction of Ru
The quantum yield measurements were performed in a quartz cubic cell (light pass length: 1 cm) consisting of Ru (0.1 mM) and a sacrificial electron donor (0.1 M) in a DMF–TEOA (5:1 v/v, 4 mL) solution. After purging with Ar for 20 min, the solution was irradiated using a 500 W xenon lamp with a 480 nm (FWHM = 10 nm) band pass filter (Asahi Spectra Co.) and a 5 cm long CuSO4 solution (250 g L−1) filter. The UV-vis absorption spectral changes during irradiation were measured with a Photal MCPD-2000 spectrophotometer. During irradiation, the temperatures of the solutions were maintained at 25 °C using an IWAKI constant temperature system CTS-134A. The incident light intensity was determined using a K3Fe(C2O4)3 actinometer and the number of absorbed photons were calculated on the basis of the absorbance changes at an irradiation wavelength of 480 nm. The amounts of produced Ru− were calculated using the molar absorptivity of Ru−, which was obtained by the electrochemical reduction of Ru in an acetonitrile solution containing Et4NBF4 as an electrolyte.7
Materials
DMF was dried over molecular sieves 4A and distilled under reduced pressure (10–20 mm Hg). TEOA was also distilled under reduced pressure (<1 mm Hg). Both solvents were kept under Ar before use. All other reagents were of reagent-grade quality and used without further purification. Ru2–Ru(CO),6 BI(OH)H,9,12 BI(O−)+,21 and BIH9,12 were prepared according to the reported methods.
Conclusion
We found that BI(OH)H is a suitable sacrificial electron donor for the photochemical reduction of CO2 to formic acid by the utilization of Ru2–Ru(CO) as the photocatalyst. The efficiency, durability, and rate of the photocatalytic reaction using Ru2–Ru(CO) were all significantly increased when BI(OH)H was employed as a sacrificial reductant (ΦHCOOH = 0.46, TONHCOOH = 2766, TOFHCOOH = 44.9 min−1) relative to BIH or BNAH. The electron balance and the material balance of the photocatalysis was also clarified, where one molecule of BI(OH)H donates two electrons and two protons to one molecule of CO2 to produce one molecule each of BI(O−)+ and formic acid.
References
- S. Enthaler, ChemSusChem, 2008, 1, 801–804 CrossRef CAS PubMed.
- F. Joó, ChemSusChem, 2008, 1, 805–808 CrossRef PubMed.
- R. Tanaka, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14168–14169 CrossRef CAS PubMed.
- R. Langer, Y. Diskin-Posner, G. Leitus, L. J. Shimon, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2011, 50, 9948–9952 CrossRef CAS PubMed.
- J. F. Hull, Y. Himeda, W.-H. Wang, B. Hashiguchi, R. Periana, D. J. Szalda, J. T. Muckerman and E. Fujita, Nat. Chem., 2012, 4, 383–388 CrossRef CAS PubMed.
- Y. Tamaki, T. Morimoto, K. Koike and O. Ishitani, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15673–15678 CrossRef CAS PubMed.
- Y. Tamaki, K. Watanabe, K. Koike, H. Inoue, T. Morimoto and O. Ishitani, Faraday Discuss., 2012, 155, 115–127 RSC.
- Y. Tamaki, K. Koike, T. Morimoto and O. Ishitani, J. Catal., 2013, 304, 22–28 CrossRef CAS.
- E. Hasegawa, T. Seida, N. Chiba, T. Takahashi and H. Ikeda, J. Org. Chem., 2005, 70, 9632–9635 CrossRef CAS PubMed.
- E. Hasegawa, S. Takizawa, T. Seida, A. Yamaguchi, N. Yamaguchi, N. Chiba, T. Takahashi, H. Ikeda and K. Akiyama, Tetrahedron, 2006, 62, 6581–6588 CrossRef CAS.
- E. Hasegawa, H. Hirose, K. Sasaki, S. Takizawa, T. Seida and N. Chiba, Heterocycles, 2009, 77, 1147–1161 CrossRef CAS.
- X.-Q. Zhu, M.-T. Zhang, A. Yu, C.-H. Wang and J.-P. Cheng, J. Am. Chem. Soc., 2008, 130, 2501–2516 CrossRef CAS PubMed.
- Deprotonation of the hydroxy group of BI(OH)H should not proceed in the DMF–TEOA (4:1 v/v) solution because the π–π* absorption band of BI(OH)H observed at λmax = 275 nm did not change at all between in the presence and absence of TEOA (Fig. S1†).
- M. Patz, Y. Kuwahara, T. Suenobu and S. Fukuzumi, Chem. Lett., 1997, 567–568 CrossRef CAS.
- M. J. Berr, P. Wagner, S. Fischbach, A. Vaneski, J. Schneider, A. S. Susha, A. L. Rogach, F. Jäckel and J. Feldmann, Appl. Phys. Lett., 2012, 100, 223903 CrossRef.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, Science, 2012, 338, 90–94 CrossRef CAS PubMed.
- J. M. Smieja, M. D. Sampson, K. A. Grice, E. E. Benson, J. D. Froehlich and C. P. Kubiak, Inorg. Chem., 2013, 52, 2484–2491 CrossRef CAS PubMed.
- J. R. Pugh, M. R. M. Bruce, B. P. Sullivan and T. J. Meyer, Inorg. Chem., 1991, 30, 86–91 CrossRef CAS.
- H. Ishida, K. Tanaka and T. Tanaka, Organometallics, 1987, 6, 181–186 CrossRef CAS.
- A. Paul, D. Connolly, M. Schulz, M. T. Pryce and J. G. Vos, Inorg. Chem., 2012, 51, 1977–1979 CrossRef CAS PubMed.
- F. Montgrain, S. M. Ramos and J. D. Wuest, J. Org. Chem., 1988, 53, 1489–1492 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc02018b |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence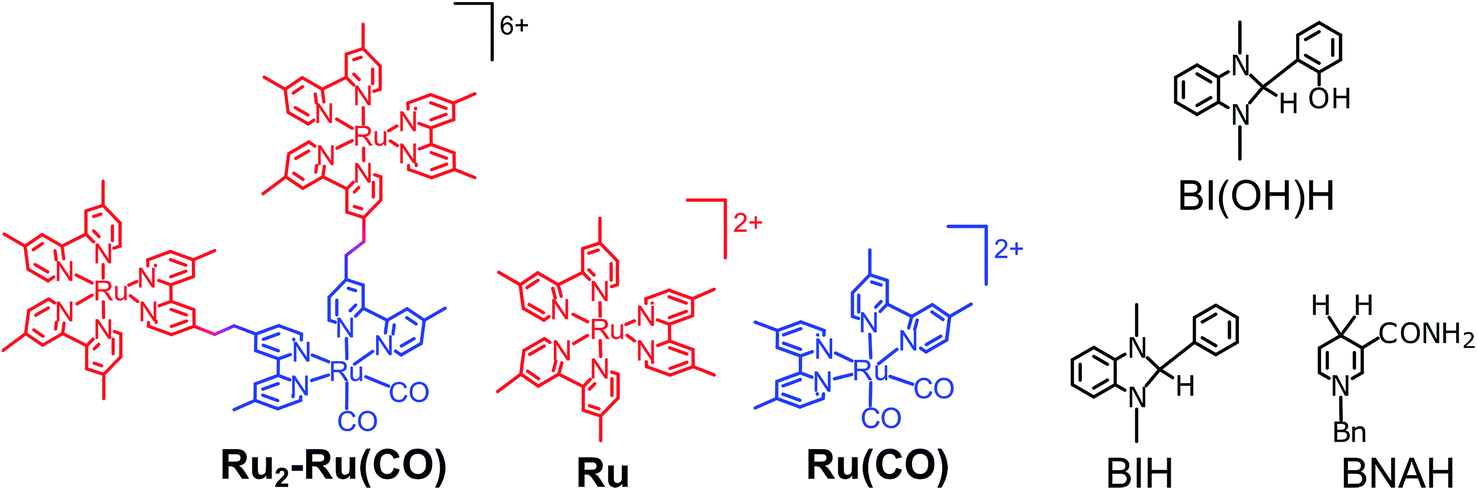


 ), CO (
), CO ( ), and H2 (
), and H2 ( ) as a function of irradiation time. (b) Photocatalytic formation of formic acid using BI(OH)H (
) as a function of irradiation time. (b) Photocatalytic formation of formic acid using BI(OH)H ( ), BIH (
), BIH ( ) or BNAH (
) or BNAH ( ) as a sacrificial electron donor: a CO2 saturated DMF–TEOA (4
) as a sacrificial electron donor: a CO2 saturated DMF–TEOA (4 ) show the results obtained from the photocatalysis using BI(OH)H (0.1 M) and a mixed system of [Ru(dmb)3]2+ (0.05 mM) and cis-[Ru(dmb)2(CO)2]2+ (0.025 mM) instead of Ru2–Ru(CO).
) show the results obtained from the photocatalysis using BI(OH)H (0.1 M) and a mixed system of [Ru(dmb)3]2+ (0.05 mM) and cis-[Ru(dmb)2(CO)2]2+ (0.025 mM) instead of Ru2–Ru(CO).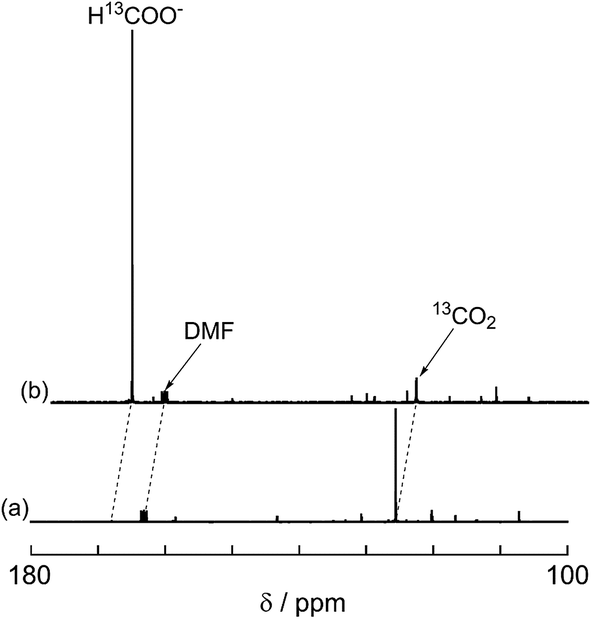

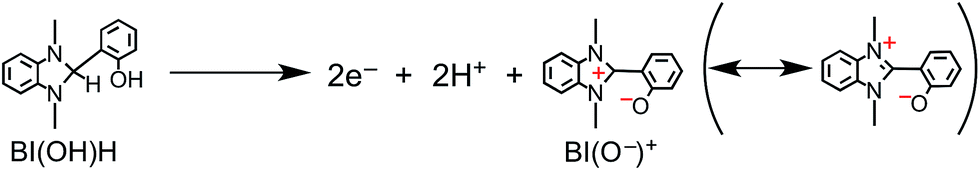
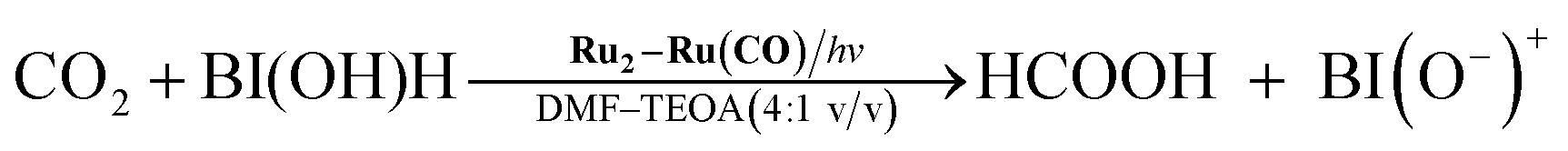
 ) and BI(O−)+ (
) and BI(O−)+ ( ), and the consumption of BI(OH)H (
), and the consumption of BI(OH)H ( ): a CO2 saturated DMF–TEOA (4
): a CO2 saturated DMF–TEOA (4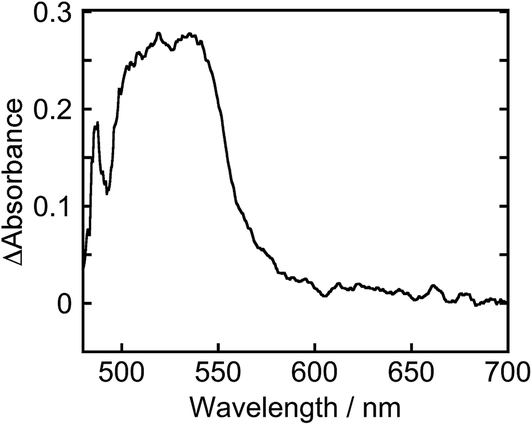





 ) and CO (
) and CO ( ) using BI(OH)H (0.1 M, blue), BIH and phenol (0.1 M each, red) or BIH (0.1 M, black): a CO2 saturated DMF–TEOA (4
) using BI(OH)H (0.1 M, blue), BIH and phenol (0.1 M each, red) or BIH (0.1 M, black): a CO2 saturated DMF–TEOA (4