 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNucleobase-functionalized ABC triblock copolymers: self-assembly of supramolecular architectures†
Keren
Zhang
,
Gregory B.
Fahs
,
Motohiro
Aiba
,
Robert B.
Moore
and
Timothy E.
Long
*
Department of Chemistry, Macromolecules and Interfaces Institute, Virginia Tech, Blacksburg, VA 24061, USA. E-mail: telong@vt.edu
First published on 24th June 2014
Abstract
RAFT polymerization afforded acrylic ABC triblock copolymers with self-complementary nucleobase-functionalized external blocks and a low-Tg soft central block. ABC triblock copolymers self-assembled into well-defined lamellar microphase-separated morphologies for potential applications as thermoplastic elastomers. Complementary hydrogen bonding within the hard phase facilitated self-assembly and enhanced mechanical performance.
Nature broadly employs complementary hydrogen bonding of nucleobase pairs to encode our genetic profiles. Research interest in nucleobase-containing synthetic polymers and supramolecular polymers focuses on the unique molecular recognition between the purine and pyrimidine rings: adenine–thymine (uracil) and guanine–cytosine.1–3 Nucleobase incorporation significantly affects both solution and solid-state properties of polymers due to the formation of physically crosslinked networks.2,4,5 Nucleobase heterocycles also restrict the segmental motion of polymer chains, leading to higher glass transition temperatures (Tg). Most studies of nucleobase-containing polymers focus on the physical properties and self-assembly in solution, which enable template polymerizations, biosensors, and drug therapies.2,6–9 A fundamental understanding of the effect of noncovalent interactions on self-assembly in the absence of solvent is also critical to elucidate the solid-state properties of nucleobase-containing polymers. Nucleobase incorporation impacts polymer thermo-mechanical performance, solid-state self-assembly, and temperature-dependent melt viscosity.10–12
Many controlled polymerization methods provide nucleobase-containing polymers with well-defined architectures in the earlier literature. Common synthetic strategies to control the polymerization of nucleobase-functionalized monomers have included atom transfer radical polymerization (ATRP),9,13,14 nitroxide mediated polymerization (NMP),6,11,15 and ring-opening metathesis polymerization (ROMP).16–18 Reversible addition-fragmentation chain transfer (RAFT) polymerization represents a relatively new and promising method for controlling polymerization of nucleobase monomers due to superior solvent compatibility and functional group tolerance.19 Advances in controlled radical polymerization methods also allow sequence and structural control for synthetic polymers that contain complementary molecular recognition sites. For example, Bazzi et al. observed the effect of complementary hydrogen bonding on polymer self-assembly in solution.17 Tao et al. utilized a hydrogen-bonding mediator to control tacticity in radical polymerization.20 However, the solid-state properties of polymers bearing complementary molecular recognition sites remained unexplored.
To achieve biomimetic synthetic polymers with tailored sequence and specificity, we employed RAFT polymerization for the copolymerization of acrylic adenine and thymine monomers with commercially available acrylic comonomers. Physical characterization of the block copolymers focused on elucidating the effect of molecular recognition and copolymer sequence on the mechanical properties and bulk self-assembly. This manuscript reports unprecedented nucleobase-functionalized ABC triblock copolymers with well-defined structures and controlled sequences.
Long et al. previously synthesized poly(adenine acrylate) and poly(thymine acrylate) homopolymers with glass transition temperatures of 65 °C and 43 °C, respectively, which were significantly lower than styrenic and methacrylic analogs.10,11 The low Tg values aided in the elucidation of the effect of complementary hydrogen bonding on the bulk properties of nucleobase-containing copolymers. When the polymer Tg exceeds the dissociation temperature of the physical crosslinks, the polymer chains remain constrained despite disruption of the physical crosslinks. Thus, the influence of dissociation of physical crosslinks on the polymer thermo-mechanical properties is less clear. In addition, the flexible acrylic backbone and longer spacers from the backbone to the interactive nucleobase groups facilitate molecular recognition. The acrylic backbone also enables facile sequence control of nucleobase-functionalized sequences and common acrylic blocks through simply changing the order of monomer addition. More versatile monomer addition avoids common crossover inefficiencies of monomers with different propagating radical stabilities. In this study, sequential RAFT polymerization permitted the synthesis of nucleobase-functionalized ABC triblock copolymers with a fully acrylic backbone using adenine acrylate and thymine acrylate monomers (Scheme 1).
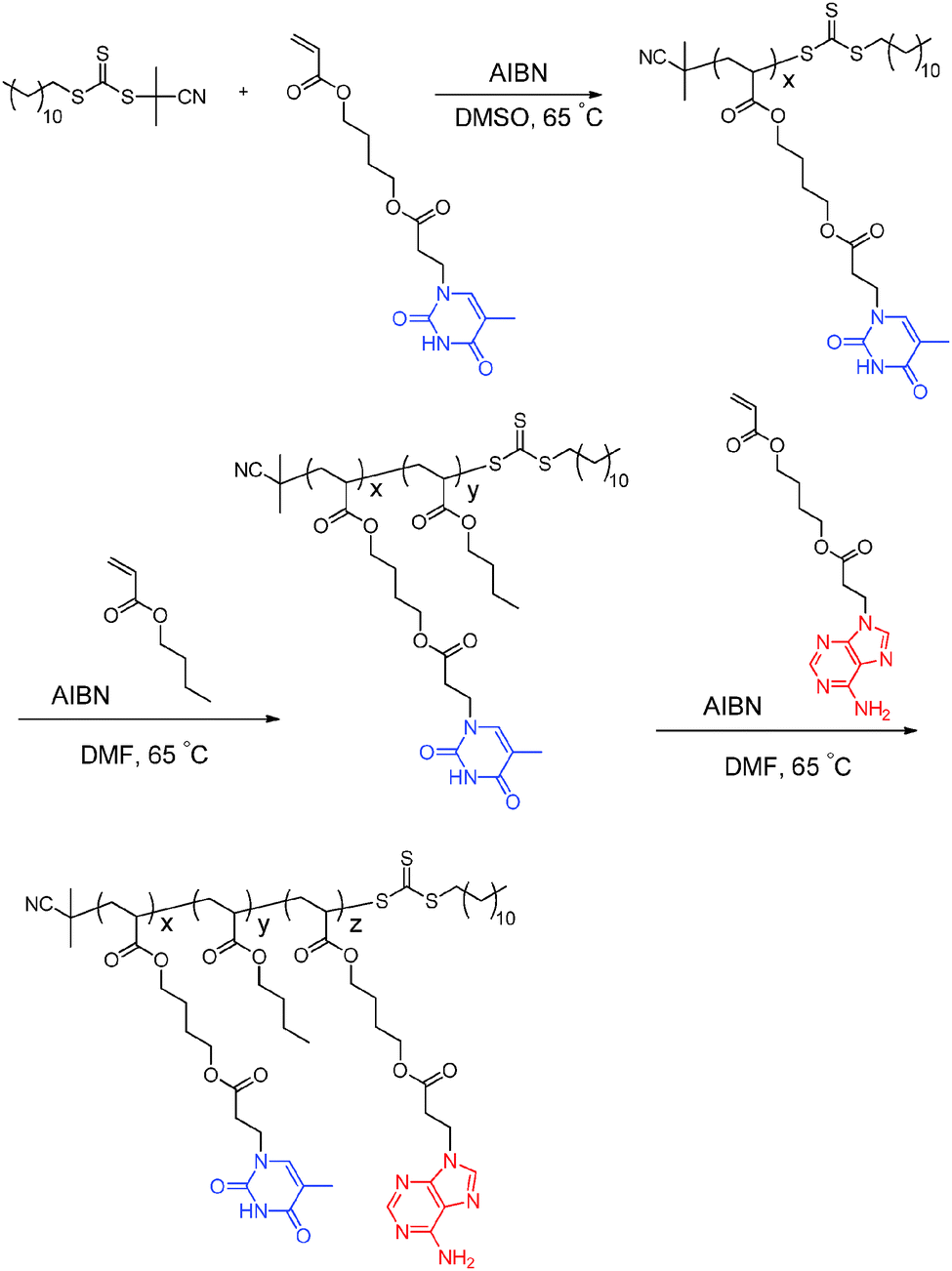 | ||
| Scheme 1 RAFT polymerization for the preparation of poly(ThA-b-nBA-b-AdA) ABC triblock copolymers. | ||
The poly(thymine acrylate-b-n-butyl acrylate-b-adenine acrylate), abbreviated as poly(ThA-b-nBA-b-AdA) (PTBA), represents the first example of an ABC triblock copolymer containing complementary nucleobase-functionalized external blocks. PTBA copolymers were also solution cast into free-standing films, enabling the physical characterization of self-assembly behavior in the absence of competing solvents. The self-complementary external blocks of PTBA copolymers resulted in unique self-assembled morphologies and thermo-mechanical performance compared to poly(ThA-b-nBA-b-ThA) (PTBT) and poly(AdA-b-nBA-b-AdA) (PABA) controls. RAFT polymerization afforded novel nucleobase-containing acrylic triblock copolymers for potential thermoplastic elastomer (TPE) applications.
Optimized RAFT polymerization conditions afforded ABC triblock copolymers using 2-cyano-2-propyl dodecyl trithiocarbonate as the chain transfer agent (CTA) (Scheme 1). Varying monomer conversions during the two chain extension steps provided three comparative compositions of PTBA triblock copolymers with differing block lengths. 1H NMR spectroscopy was the primary tool for monitoring monomer conversions and quantifying the degree of polymerization for each block. End-group analysis determined the number-average molecular weight of poly(ThA) macro-CTA. The challenging solubility of nucleobase copolymers limited the applicability of size exclusion chromatography. The PTBA triblock copolymer series consisted of three compositions: PTBA1 9.4–19.3–6.8 kDa, PTBA2 9.4–27.2–5.3 kDa, and PTBA3 9.4–31.5–4.6 kDa. Degree of polymerizations for ThA, nBA, and AdA blocks (x, y, and z in Scheme 1) were: PTBA 29–151–20, PTBA2 29–213–16, and PTBA3 29–246–14. The thymine![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :adenine molar ratio of PTBA 1, 2, and 3 was 1.5, 1.8, and 2.1, respectively. Corresponding weight percentages of nucleobase-functionalized monomers were 46 wt%, 35 wt%, and 31 wt%.
:adenine molar ratio of PTBA 1, 2, and 3 was 1.5, 1.8, and 2.1, respectively. Corresponding weight percentages of nucleobase-functionalized monomers were 46 wt%, 35 wt%, and 31 wt%.
Thermogravimetric analysis (TGA) determined 5% weight loss temperatures of PTBA copolymers near 290 °C, indicating potential tolerance for a high processing temperature. All three PTBA copolymers showed two Tg's from differential scanning calorimetry (DSC), as summarized in Table 1. The lower Tg at ca. −44 °C corresponded to the central poly(nBA) block. The central block lengths were sufficiently high to ensure that Tg remained constant with increasing molecular weight. The Tg's of poly(ThA) and poly(AdA) homopolymers were 43 °C and 65 °C, respectively.10 However, the external blocks only showed a single Tg at ca. 70 °C, rather than two distinct Tg's for adenine- and thymine-functionalized external blocks, presumably due to strong complementary hydrogen bonding between the adenine and thymine blocks, forming a single hard phase. The adenine block presumably restricted segmental motion of the thymine-functionalized block even above the thymine-functionalized block's Tg, resulting in a single observable Tg. The Tg of the hard phase also increased as the adenine-functionalized block length increased.
| Temperature (°C) |
T
1g (Tanδ) |
T 1g (DSC) | T 2g (DSC) | T flow |
|---|---|---|---|---|
| PTBA3 (9.4–31.5–4.6) kDa | −23 | −44 | 66 | 80 |
| PTBA2 (9.4–27.2–5.3) kDa | −21 | −45 | 67 | 84 |
| PTBA1 (9.4–19.3–6.8) kDa | −25 | −44 | 72 | 85 |
Fabrication of mechanically robust block copolymer TPEs demands a specific hard–soft–hard arrangement of hard (high Tg) and soft (low Tg) blocks.21 The triblock copolymer sequence of poly(ThA-b-nBA-b-AdA) was critical for preparing potential block copolymer TPEs. The nucleobase-functionalized external blocks provided mechanical reinforcement and served as physical crosslinks during the plateau window. The central poly(nBA) soft block provided flexibility above its Tg. The structural similarity of nucleobase-functionalized monomers and nBA led to similar propagating radical stability, enabling well-defined crossover at each chain extension step. The χ parameter is also important for microphase separation of block copolymers, where enthalpic interactions between the soft and hard blocks require minimization. The relatively hydrophobic nature, thermal stability, and low Tg of poly(nBA) all contributed to its suitability as the soft block for designing block copolymer TPEs.
Solution-cast films of PTBA copolymers showed microphase-separated morphologies and corresponding thermo-mechanical properties. Dynamic mechanical analysis (DMA) suggested microphase separation of PTBA copolymers as depicted in Fig. 1. The lowest thermal transitions in the storage modulus versus temperature curves were assigned to the glass transitions of the PnBA soft phase. The following regions in the storage modulus analysis revealed a plateau temperature window for each sample, demonstrating feasibility for TPE applications over a wide temperature range. The service temperature window of amorphous block copolymer TPEs usually ranges from the soft phase Tg to the hard phase Tg. Among the three compositions of PTBA copolymers, the plateau modulus increased with increasing nucleobase content, which was attributed to an increased physical crosslink density. The nucleobase-functionalized copolymers displayed tunable plateau moduli with varying nucleobase block or soft block lengths. The plateau moduli of PTBA 1, 2, and 3 were higher than rubbery plateau moduli of commercial TPEs, with typical values of 2 MPa. Future work will involve synthesis of triblock copolymers with shorter nucleobase blocks and longer central blocks for TPE applications. The soft block Tg's as determined from the tan delta curves remained constant with varying compositions, indicating that solution casting achieved microphase separation (Table 1). The microphase-separated morphologies and tunable plateau moduli both contribute to the potential of nucleobase-functionalized ABC triblock copolymers for TPE applications.
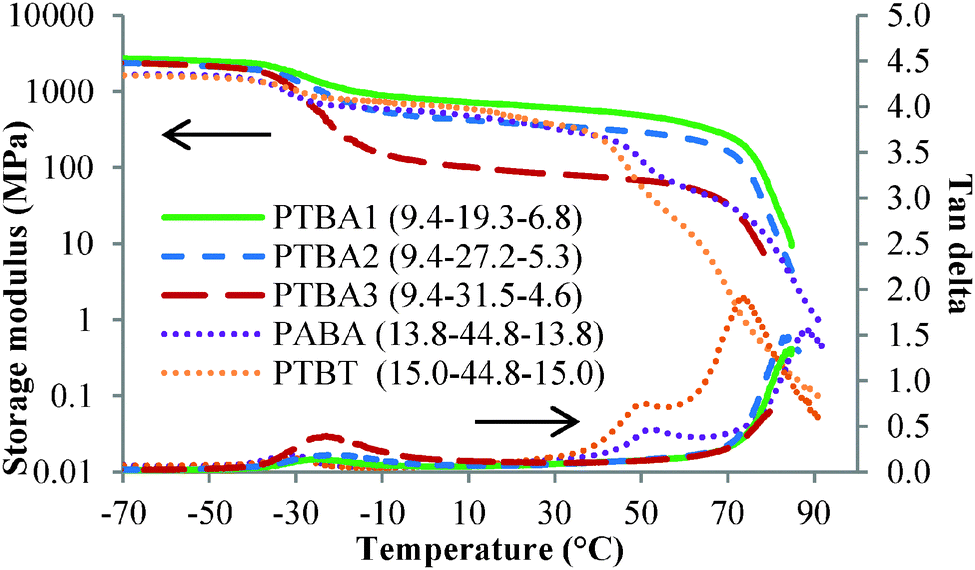 | ||
| Fig. 1 Dynamic mechanical temperature ramp of PTBA, copolymers with PABA and PTBT controls. | ||
Complementary hydrogen bonding between the adenine–thymine nucleobase pairs enhanced the thermo-mechanical performance of PTBA copolymer films compared to ABA triblock copolymer controls. PTBT and PABA with 40 wt% and 38 wt% nucleobase were synthesized in-parallel and adopted as controls. Identical film casting procedures and thermal histories were maintained to validate the comparisons. The plateau moduli of PTBT and PABA fell between PTBA1 and PTBA2 owing to their intermediate nucleobase contents. The plateau moduli largely depended on hard phase incorporation. The plateau region for the PTBT and PABA copolymers ended with a second glass transition belonging to the hard phase. However, PTBA did not show a second Tg in either the storage modulus or tan delta curves. This phenomenon presumably originated from the hypothesis that complementary hydrogen bonding maintained mechanical strength despite long range segmental motion of the hard blocks. The moduli of PTBA copolymer films eventually decreased upon reaching the dissociation temperature for hydrogen bonding near 80–90 °C. The complementary hydrogen bonding within the hard phase broadened the plateau window for the block copolymers and enhanced thermal response at high temperature. Thus, hydrogen bonding assisted self-assembly potentially solves the paradox of enhancing mechanical strength while improving processability of polymeric materials.
Complementary hydrogen bonding within the hard phase also contributed to a remarkably ordered microphase separated morphology of the ABC triblock copolymers. Small angle X-ray scattering (SAXS) was used to elucidate the microphase-separated bulk morphology of all three PTBA copolymers in Fig. 2. The phase-separated morphologies of asymmetric ABC copolymers are usually more complex than symmetric ABA triblock and AB diblock copolymers.22 However, all three PTBA triblock copolymers showed distinctive lamellar scattering profiles. SAXS profiles of PTBA2 and PTBA3 both exhibit primary scattering peaks and four additional secondary peaks, indicating well-organized lamellar morphologies with long-range order. The distinctive scattering peaks for ABC triblock copolymers resulted from the complementary molecular recognition, which combined two chemically different blocks into a single, precisely-ordered hard phase. Consequently, self-assembly of an ABC triblock copolymer with complementary external blocks resembled the self-assembly behavior of a symmetric ABA triblock copolymer. However, in sharp contrast, PABA with 38 wt% nucleobase content (similar to PTBA2 with 35 wt%) showed a less-ordered cylindrical microphase-separated morphology with broader SAXS scattering peaks. These analogous ABA triblock copolymers will be discussed in more detail in a forthcoming publication on nucleobase-functionalized symmetric ABA triblock copolymers. The degree of polymerization (N) difference provided one possible explanation for the morphological difference according to the phase diagram, since the morphology varies as a function of χN. However, the highly ordered phase separation of PTBA copolymers was still unique for an ABC or ABA triblock copolymer, considering the presence of the complementary hydrogen bonding between two dissimilar external blocks.
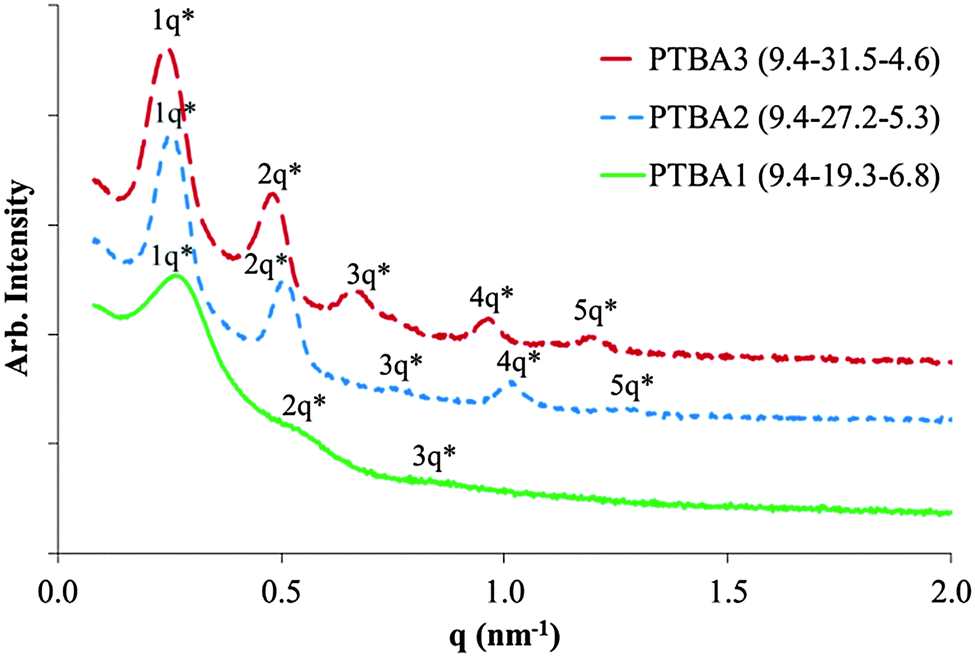 | ||
| Fig. 2 SAXS profiles of poly(ThA-b-nBA-b-AdA) triblock copolymers. | ||
The domain spacing of the lamellae were 25.9 nm, 25.0 nm, and 23.4 nm for PABT3, 2, and 1, respectively. Increasing central block lengths resulted in an increased spacing between lamellae. The SAXS profile of PTBA1 showed broader scattering peaks with fewer highly ordered peaks compared to the other two compositions, suggesting a broader distribution of lamellar thicknesses and/or interlamellar dimensions, and thus less ordered domains. Two hypotheses may account for the difference in self-assembled morphologies for PTBA triblock copolymers with various compositions. The less ordered phase separation of PTBA1 was possibly a consequence of a shorter central block. The central block length was insufficient to induce well-defined microphase separation. Secondly, the highly ordered lamellar packing possibly resulted from the fact that thymine:adenine molar ratios of PTBA2 and 3 were close to 2. In the second hypothesis, each thymine-functionalized block statistically paired with two adenine blocks, facilitating the aligning of alternating soft phase and hard phase lamellae. In the latter case, the phase separated morphology would not follow the general phase diagram of ABA triblock copolymers.23 Both inter-and intra-chain hydrogen bonding were presumably present according to the second hypothesis at a 2:1 thymine:adenine molar ratio. The inter-chain complexion also provided an explanation for enhanced mechanical performance compared to PABA and PTBT in Fig. 1, resulting from increased apparent chain lengths. However, elucidation of inter-and intra-chain hydrogen bonding in the solid state is challenging due to the complexity of polymer structure compared to low molar mass molecules. Preliminary atomic force microscopic (AFM) results did not reveal a well-defined surface morphology for copolymer films, differing from the observed lamellae bulk morphology in SAXS profiles. Tuning film casting conditions and applying transmission electron microscopy (TEM) will further elucidate the morphology of nucleobase-functionalized ABC triblock copolymers. Further investigations are underway to validate two hypotheses mentioned above and understand the details of self-assembly of ABC triblock copolymers with complementary external blocks.
RAFT polymerization afforded novel self-complementary nucleobase-functionalized ABC triblock copolymers with fully acrylic backbones. Nucleobase units converted low Tg acrylics into mechanically robust polymeric films through steric restriction of segmental motion coupled with physical crosslinking. Hard–soft–hard triblock copolymers self-assembled into lamellar microphase-separated morphologies. The molecular recognition between two dissimilar hard blocks contributed to a single hard phase, and the complementary hydrogen bonding within the hard phase reinforced and facilitated the microphase-separated morphology of ABC triblock copolymers. Physical crosslinks within the hard phase broadened the plateau temperature window and improved the processability of block copolymers for potential TPE applications.
The authors acknowledge Henkel Corporation for financial support. This material is also partially based upon work supported by the National Science Foundation under Grant No. DMR-0923107.
Notes and references
- S. T. Hemp and T. E. Long, Macromol. Biosci., 2012, 12, 29 CrossRef CAS PubMed.
- R. McHale and R. K. O'Reilly, Macromolecules, 2012, 45, 7665 CrossRef CAS.
- S. Sivakova and S. J. Rowan, Chem. Soc. Rev., 2005, 34, 9 RSC.
- A. S. Karikari, B. D. Mather and T. E. Long, Biomacromolecules, 2006, 8, 302 CrossRef PubMed.
- M. Tamami, S. T. Hemp, K. Zhang, M. Zhang, R. B. Moore and T. E. Long, Polymer, 2013, 54, 1588 CrossRef CAS PubMed.
- R. McHale, J. P. Patterson, P. B. Zetterlund and R. K. O'Reilly, Nat. Chem., 2012, 4, 491 CrossRef CAS PubMed.
- J.-F. Lutz, A. F. Thuenemann and K. Rurack, Macromolecules, 2005, 38, 8124 CrossRef CAS.
- P. K. Lo and H. F. Sleiman, Macromolecules, 2008, 41, 5590 CrossRef CAS.
- H. J. Spijker, A. J. Dirks and H. J. C. M. Van, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4242 CrossRef CAS.
- S. Cheng, M. Zhang, N. Dixit, R. B. Moore and T. E. Long, Macromolecules, 2012, 45, 805 CrossRef CAS.
- B. D. Mather, M. B. Baker, F. L. Beyer, M. A. G. Berg, M. D. Green and T. E. Long, Macromolecules, 2007, 40, 6834 CrossRef CAS.
- K. Yamauchi, J. R. Lizotte and T. E. Long, Macromolecules, 2002, 35, 8745 CrossRef CAS.
- H. J. Spijker, D. F. L. van and H. J. C. M. van, Macromolecules, 2007, 40, 12 CrossRef CAS.
- J.-F. Lutz, A. F. Thuenemann and R. Nehring, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4805 CrossRef CAS.
- B. D. Mather, M. B. Baker, F. L. Beyer, M. D. Green, M. A. G. Berg and T. E. Long, Macromolecules, 2007, 40, 4396 CrossRef CAS.
- H. S. Bazzi and H. F. Sleiman, Macromolecules, 2002, 35, 9617 CrossRef CAS.
- H. S. Bazzi, J. Bouffard and H. F. Sleiman, Macromolecules, 2003, 36, 7899 CrossRef CAS.
- P. K. Lo and H. F. Sleiman, J. Am. Chem. Soc., 2009, 131, 4182 CrossRef CAS PubMed.
- Y. Kang, A. Lu, A. Ellington, M. C. Jewett and R. K. O'Reilly, ACS Macro Lett., 2013, 581 CrossRef CAS.
- Y. Tao, K. Satoh and M. Kamigaito, Macromol. Rapid Commun., 2011, 32, 226 CrossRef CAS PubMed.
- R. J. Spontak and N. P. Patel, Curr. Opin. Colloid Interface Sci., 2000, 5, 334 CrossRef CAS.
- R. Stadler, C. Auschra, J. Beckmann, U. Krappe, I. Voight-Martin and L. Leibler, Macromolecules, 1995, 28, 3080 CrossRef CAS.
- A. M. Mayes and M. Olvera de la Cruz, J. Chem. Phys., 1989, 91, 7228 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cc03363a |
| This journal is © The Royal Society of Chemistry 2014 |