
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantum chemical elucidation of the luminescence mechanism in a europium(III) co-doped UiO-66 chemosensor selective to mercury(II)†
Yoan
Hidalgo-Rosa
 ab,
Yoslainy
Echevarria-Valdés
c,
Mario
Saavedra-Torres
d,
Dayán
Páez-Hernández
ce,
Eduardo
Schott
*f and
Ximena
Zarate
*d
ab,
Yoslainy
Echevarria-Valdés
c,
Mario
Saavedra-Torres
d,
Dayán
Páez-Hernández
ce,
Eduardo
Schott
*f and
Ximena
Zarate
*d
aCentro de Nanotecnología Aplicada, Facultad de Ciencias, Ingeniería y Tecnología, Universidad Mayor, Camino La Pirámide 5750, Huechuraba, Santiago, Chile. E-mail: yoanhrj@gmail.com
bEscuela de Ingeniería del Medio Ambiente y Sustentabilidad, Facultad de Ciencias, Ingeniería y Tecnología, Universidad Mayor, Camino La Pirámide 5750, Huechuraba, 8580745 Santiago, Chile
cDoctorado en Fisicoquímica Molecular, Facultad de Ciencias Exactas, Universidad Andrés Bello, República 275, Santiago 8370146, Chile
dInstituto de Ciencias Aplicadas, Facultad de Ingeniería, Universidad Autónoma de Chile, Av. Pedro de Valdivia 425, Santiago, Chile. E-mail: ximena.zarate@uautonoma.cl
eCenter of Applied Nanosciences (CANS), Universidad Andres Bello, Ave. República #275, 8370146 Santiago de Chile, Chile
fDepartamento de Química Inorgánica, Facultad de Química y de Farmacia, Centro de Energía UC, Centro de Investigación en Nanotecnología y Materiales Avanzados CIEN-UC, Pontificia Universidad Católica de Chile, Vicuña Mackenna 4860, Macul, 7820436 Santiago, Chile. E-mail: maschotte@gmail.com; edschott@uc.cl
First published on 19th March 2025
Abstract
Lanthanide(III) ions can be incorporated into metal–organic frameworks (MOFs) to form Ln@MOFs through post-synthetic procedures. This makes the MOFs efficient luminescent chemical sensors for detecting trace amounts of heavy metals. In this report, a quantum chemical theoretical protocol has been carried out to elucidate the detection principle of the turn-off luminescence mechanism in a Eu@UiO-66(DPA)-type MOF selective to Hg2+ ions. UiO-66(DPA) is an iso-reticular MOF of UiO-66 constructed from the Zr6-cluster [Zr6(μ3-O)4(μ3-OH)4]12+ and the ligands 1,4-benzenedicarboxylate (BDC) and 2,6-pyridinedicarboxylate (DPA) as linkers. The sensitization and energy transfer (ET) in UiO-66(DPA) doped with Eu3+ were analyzed using multireference ab initio CASSCF/NEVPT2 methods and time-dependent density functional theory (TD-DFT). The cluster model used in the calculations comprises the Z6-cluster/BDC/DPA fragments with the DPA ligand coordinating to Eu3+ or Hg2+ ions. The proposed sensitization pathway involves intersystem crossing from S1(DPA) to T1(DPA), a plausible subsequent energy transfer from T1(DPA) to the 5D1 state of Eu3+, and then vibrational relaxation to the emissive 5D0 state. These results also suggest that the electronic states of the BDC ligand can be strengthened by the population of the T1 electronic states of the DPA antenna via ET. Periodic DFT calculations confirm the electronic state mixture of BDC and DPA linkers in the conduction bands, just above the electronic state of Eu3+ ions, which is in concordance with the proposed Eu3+ sensitization pathways. The assessed optical properties (absorption and emission) of Hg2+@UIO-66(DPA) explain the experimental behavior of this chemosensor when the Hg2+ ion replaces the Eu3+ ion and the luminescence diminishes.
Introduction
In recent years, the use of chemical methods for detecting metal ions has gained considerable importance.1 This shift is driven by growing concerns over environmental pollution and the health risks posed by metal ion contamination.2,3 Chemical detection methods play a crucial role in monitoring and controlling the metal ion levels in various settings, including water sources, food products, and industrial processes.3 Chemical detection relies on chemical transformation or a change in the physical property induced by specific interactions between target analytes (such as metal ions, small molecules, biomarkers, and others) and the chemosensor.4 Optical chemosensors have gained significant attention in the development of selective and efficient tools for metal ion detection, due to their ability to provide instantaneous data regarding a particular analyte. Many ions (like Mg2+, Ca2+, K+, F−, etc.) are essential in biological systems and play a crucial role in different biochemical processes.3 For example, fluoride is one type of valuable trace element of the human body; nevertheless, excessive fluoride in the organism causes serious health issues such as dental and skeletal fluorosis. Metal ions constitute biological electrolytes that must be controlled for people on Earth and also for astronauts in space. At certain concentrations, other metals such as copper (Cu), chromium and zinc are necessary for the suitable function of the human body. However, when the homeostatic concentration of these metal ions is exceeded, they turn into a poison. In contrast, lead, cadmium and mercury (Hg), even at trace concentrations, cause irreversible alterations in the health of living beings. So, for the protection of human health, not only the knowledge of their effects but also their detection and monitoring is necessary.5,6Optical sensors can be based on various principles, including absorbance, transmittance, polarization of light or refractive index.7 Advances in chemical detection technologies have allowed for more sensitive and selective detection of metal ions, even at trace levels.8 This has enabled researchers and environmental agencies to better understand metal ion distribution, sources, and potential health risks. Furthermore, developing portable chemical detection devices has significantly enhanced on-site monitoring and rapid screening of metal ion contamination.9 In 2020, Yuxiu Xiao et al. reported an analytical device based on a one-to-two logic gate utilizing a Eu-MOF. They designed a Eu-MOF-loaded fiber paper microsensor for the rapid detection of water in solid pharmaceuticals using ratiometric sensing and a portable visual device.10 Building on this advancement, the integration of metal–organic frameworks (MOFs) with smartphone technology has further unlocked new possibilities for real-time, on-site chemical detection.11–13 Lanthanide-based MOFs (Ln-MOFs) have shown exceptional promise for visual detection applications through smartphone-assisted systems,13–15 leveraging their distinctive luminescence properties to enhance sensitivity and enable accurate quantification via straightforward and interpretable colorimetric or luminescence changes.11,14
In this regard, luminescent metal–organic frameworks (L-MOFs) are promising alternatives in the development of chemosensors.16 These materials are composed of metal ions or clusters (nodes) connected by organic ligands (linkers), generating two-dimensional or three-dimensional structures.17 The combination of these metallic and organic building components (nodes and linkers) in L-MOFs leads to the appearance of unique luminescence properties. The hybrid nature of their components enables a wide range of photophysical processes that govern their luminescence properties.18 Metal-to-ligand charge transfer (MLCT), ligand-to-metal charge transfer (LMCT), ligand-to-ligand charge transfer (LLCT), metal localized emission and ligand-centered emission are some of the involved mechanisms.19 These materials exhibit luminescence changes in the presence of specific analytes, allowing the detection and recognition of target substances.20 Other features of MOFs that make them stand out as optical chemosensors are related to their tunable structures, high surface areas, and easy functionalization post-synthesis.21,22 These properties make L-MOFs highly useful in fields such as environmental monitoring,23 biomedical research,24 and industrial applications.25 Therefore, by understanding and tuning the luminescence changes in L-MOFs, researchers would be able to design highly sensitive and selective chemosensors for a wide range of analytes.26
In this sense, linkers can be designed or functionalized by post-synthetic modifications (PSM) to exhibit specific properties, such as desirable optical properties, and/or induce certain chemosensor–analyte interactions.27 A powerful PSM strategy is to include lanthanide ions (Ln3+) into MOF hosts to activate the emission properties of the material and generate new emission signals that are lanthanide ion-centered.28 The sharp line emissions, high color purity, high luminescence quantum yield, and large Stokes shifts, attributed to the 4f–4f transitions and relativistic effects of Ln ions, might undergo improvements in co-doped MOFs.29 Their luminescence arises from a sensitization process carried out via energy transfer (ET) from a suitable organic ligand to the Ln3+ ions, which is called the “antenna effect”. Thus, a careful selection and design of the linkers and nodes might tune the emission properties of L-MOFs. This makes L-MOFs promising materials for the development of efficient and sensitive chemosensors.30–32 Due to their structural and optical properties, L-MOFs functionalized with Ln3+ ions have been used to detect metal ions.33,34 Mechanisms such as energy transfer from the ligand to the Ln3+ center and metal–ligand charge transfer are well accepted in a detection process to explain the change in the optical properties of functionalized L-MOFs.34,35 Several studies indicate that the quenching effect is not completely clear in all cases. In this context, a recent systematic literature review by Shuangyan Wu (2024)36 concluded that the principles underlying the current sensing mechanisms are unclear and limited to qualitative analysis. However, much research has been descriptive and based only on experimental data. Some works have successfully applied DFT-based methods to elucidate potential photophysical processes that induce luminescence alteration in Ln3+ co-doped L-MOFs.37–39 However, in Ln3+-based systems, a more sophisticated level of theory is required to accurately address the multireference character arising from the various low-lying states associated with the 4fn configurations.40 Predicting the correlation between the L-MOF structure and analyte-induced luminescence changes is challenging due to the large size of MOFs. However, advancements in computational chemistry have enabled accurate descriptions of their molecular and electronic properties.26,41–46 This report presents a computational protocol designed to understand the luminescence properties and sensing mechanisms of Ln3+ co-doped L-MOFs (Ln3+@L-MOFs). A comprehensive study was conducted, focusing on molecular and electronic properties, including the relative energies of the ground and excited states (S1 or T1), as well as the electronic band structures of the L-MOFs. Our theoretical protocol integrates periodic DFT, molecular DFT, and multireference calculations to provide a detailed assessment of the luminescence properties and sensing mechanism.26,43–45
Theoretical studies of Ln3+@L-MOF chemosensors, due to their computational complexity, numerous electrons and electron correlation effects, need high-performance computation resources. These studies are crucial in the investigation of Ln3+@L-MOFs, offering in-depth knowledge of the principles underlying detection events toward a target analyte, thereby influencing experimental work and aiding in creating new chemosensors of the Ln3+@L-MOF type.
Hence, to enhance the understanding and contribute to the knowledge, in the work reported herein, a theoretical procedure is proposed. In this theoretical procedure, multiconfigurational ab initio methods, along with molecular density functional theory (DFT), and periodic DFT calculations were combined. This approach aims to accurately determine the sensitization and emission channels for the previously experimentally reported UiO-66 MOFs47 doped with a Eu3+ selective chemical sensor to detect Hg2+in situ as well as, to the best of our knowledge, the sensing mechanism for the first time.
This MOF holds the Eu3+ atom in a free –COOH group (Eu@UiO-66(DPA)) and it is used to detect very low Hg2+ levels (lower than 10 nM, which is the maximum level of Hg2+ in drinking water according to the U.S. Environmental Protection Agency48). It is proposed that the detection mechanism is via the replacement of the Eu3+ atom by the Hg2+ atom due to the higher affinity of DPA toward Hg2+. This substitution induces the blocking of the Eu3+ antenna effect inducing then the chelation enhancement quenching (CHEQ) effect (Fig. 1).
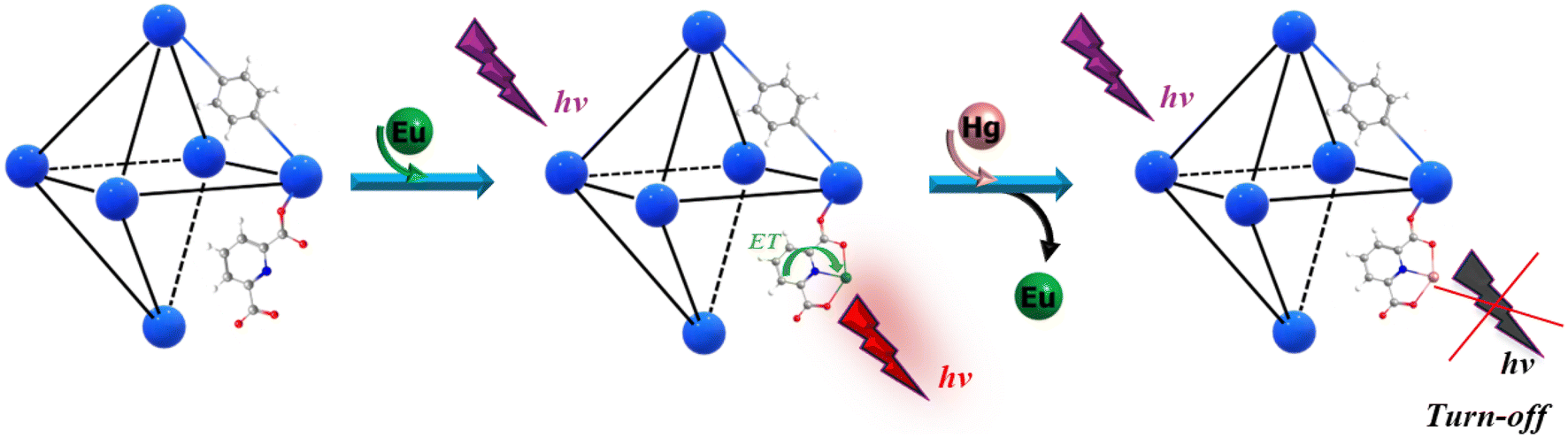 | ||
| Fig. 1 A simplified scheme showing the detection mechanism via the replacement of Eu3+ by Hg2+, inducing the blocking of the antenna effect of Eu3+ and then producing the CHEQ effect. | ||
Computational methods
Quantum chemistry offers reliable tools for gaining a deep understanding of the sensing mechanisms in co-doped MOFs. A careful examination of the detection principles is essential for designing new luminescent MOF-based chemosensors and facilitating their transition from experimental research to developing MOF-based portable devices.49 Consequently, rigorous investigations are still required to achieve a more rational design of Ln3+@L-MOF chemosensors.One of the most intriguing features of MOFs is their ability to fine-tune optical properties through slight structural modifications. These modifications can include the incorporation of functional groups50 or metal ions51 or be induced by the analyte,52 which involved a significant change in the optical properties of MOFs. The photophysical processes that dictate the optical behaviors of a luminescent sensor are intrinsically associated with electronic interactions between the sensor and the analyte.53 The density of states (DOS) method has been widely utilized to investigate the electronic structure and optical properties of MOF-based chemical sensors.52–57 DOS plots provide crucial insights into the nature of charge transport, helping to determine whether it occurs through ligand-to-ligand, metal-to-ligand, ligand-to-metal, or metal-to-metal processes involving the photophysical properties of materials.41,58 This analysis offers a straightforward approach for representing complex electronic structures while also providing valuable insights into the optical properties of materials.59 This method has been previously employed to gain insights into the photophysical processes associated with optical properties related to charge transfer and energy transfer mechanisms,60 such as ligand–ligand charge transfer (LLCT),61 metal-to-ligand charge transfer (MLCT),57 ligand-to-metal charge transfer (LMCT),62 and photoinduced electron transfer (PET).57 Additionally, it provides a detailed view of the atomic contributions to the occupied and unoccupied electronic states.60,61,63,64 Thus, DOS analysis was employed to further investigate the changes in the luminescence properties of the UiO-66(DPA) chemosensor induced by Eu3+ and Hg2+ ions, focusing on the structures Eu@UiO-66(DPA) and Hg@UiO-66(DPA).
On the other hand, to study the efficiency of Ln sensitization and emission in a MOF, the main processes involved must be considered. This process starts from the excitation of the linker with the final population of its first excited singlet state (S1) after no radiative processes followed by an intersystem crossing (ISC) between S1 and the first excited triplet state (T1) of the linker [linker (S1 → T1)]. Finally, there occurs an energy transfer process that populates the emissive state of the lanthanide ion [linker (T1) → Ln3+] from which emission occurs.26
Thus, to elucidate the origin of the sensitization and emission pathways and the sensing mechanism in Ln3+@L-MOF chemosensors, a detailed understanding of the electronic structure of the system and the effect of the presence of the analyte of interest must be obtained. This implies predicting the correct localization of the electronic state of the lanthanide ions and antenna both in the ground state and excited states.40 Advanced computational methods have been developed that enable a more in-depth examination of the electronic structure and excited states related to photophysical processes that govern luminescence properties.
Due to the intrinsic properties of heavy elements, such as lanthanides, their theoretical treatment requires meticulous evaluation.26,65 Three primary factors must be considered: (i) relativistic effects, including scalar relativistic contributions and spin–orbit coupling, (ii) electronic correlation, and (iii) the influence of the ligand field.66 In the case of lanthanides, spin–orbit interactions and electron correlation effects play a dominant role and must be explicitly accounted for in theoretical calculations. Notably, the quasi-degenerate nature of electronic configurations arising from the 4fn shell introduces significant static correlation, which is essential for accurately determining the energetic positions of both ground and excited states.40,67
One of the most important methods is the multi-configuration self-consistent field (MCSCF), which is employed to study the electronic structure of lanthanide ions.68 Therefore, our attention has been directed towards accomplishing a theoretical protocol to clarify the emission channels and understand the sensing mechanism in luminescent UiO-66(DPA) sensors using a cluster model. In the ESI,† we present a detailed description of structural models used for modeling these systems, both as extended solids and cluster models. Moreover, the computational methods are described in detail, such as software packages, theory levels, and theoretical approaches used for each of the calculations.
Results and discussion
Periodic modelling of Eu3+@UiO-66(DPA) and Hg2+@UiO-66(DPA)
Cell parameters were optimized, compared with the experimental crystal data,47 and modified in agreement with the herein studied models, i.e. to obtain periodic models of Eu3+@UiO-66(DPA) and Hg2+@UiO-66(DPA) shown in Fig. S1.† The obtained structures were finally reoptimized.For UiO-66(Zr) MOFs, it has been documented that their optical properties are governed by electronic transitions involving both non-functionalized BDC linkers and substituted BDC linkers. Matsuoka et al.69 demonstrated that the organic linker in NH2-UiO-66(Zr) absorbs light, as shown by in situ electron paramagnetic resonance (EPR) measurements conducted before and after exposure to visible light. The EPR measurements revealed no characteristic signals attributable to Zr3+ species after visible-light exposure, indicating that LMCT does not occur in this system. Jorge Gascon et al. reached a similar conclusion while investigating the electronic properties of NH2-UiO-66(Zr) and NH2-UiO-66(Hf) MOFs using DFT and EPR techniques. They emphasized that there is no evidence for the formation of M3+ species, such as Zr3+ or Hf3+ in the excited state.70 On the other hand, previous theoretical studies reported the electronic structure of the UiO-66(Zr), UiO-66(Hf) and UiO-(Th) MOFs via density of states (DOS) and projected DOS (PDOS) plots. In these previous studies, the contributions of ligand states near the conduction band minimum (CBM) and valence band maximum (VBM) were also shown.62,71 Accordingly, to gain deeper insights into the emission pathway of the sensing mechanism, the PDOSs of both the Eu3+@UiO-66(DPA) and Hg2+@UiO-66(DPA) systems were analyzed.
PDOS analysis by fragments is shown in Fig. 2. BDC fragments are predominantly present in the occupied range, particularly in the valence zone with higher contribution states between −7 and −1.8 eV (purple), while DPA (blue) and Zr6O4(OH)4 (red) are also present but with a small number of states. However, the Eu state emerged with a lone but highly populated band closer to the Fermi level at −0.1 eV. The lowest energy conduction band is populated almost entirely by Eu states (green curve) at 1.1 eV, and only with small contributions by the DPA linker at this energy level. Thus, doping UiO-66(DPA) with Eu3+ introduces 4f states that are sufficiently low in energy to fall below the electronic states of the BDC and DPA linkers. The rest of the fragments contribute above ∼1.1 eV, mainly DPA (between 1.1 and 2.2 eV) and BDC (mostly between 1.5 and 2 eV). The overlap between the electronic states of the DPA and BDC linkers next to Eu3+ (1.5 to 2.2 eV) suggests that both ligands are involved in the sensitization and emission pathways of the Eu3+@UiO-66(DPA) MOF.
 | ||
| Fig. 2 (a) Structure of Eu3+@UiO-66(DPA) identifying fragments: Zr6O4(OH)4 (red), BDC (violet), DPA (blue), and Eu (green). (b) PDOS by fragments. | ||
PDOS results obtained by elemental and orbital analyses presented in Fig. 3 are in agreement with the by-fragment results. It is observed that the valence band located closer to the Fermi level is populated mainly by Eu(f), followed by O(p) and C(p) between −2 and −7 eV, assigned previously to the BDC and Zr6O4(OH)4 fragments. However, O(p) mostly contributes at lower energy levels under −2 eV, attributed to the O atoms present on the Zr6O4(OH)4 and BDC/DPA terminal groups. The conduction band is also populated by Eu(f) at 1 eV, followed by C(p) from BDC and DPA, particularly between 1 and 2.2 eV. Over this range, Zr6O4(OH)4 shows an increase in the number of unoccupied states, through Zr(d), with Eu(d) and the rest of the elements showing values over 4 eV.
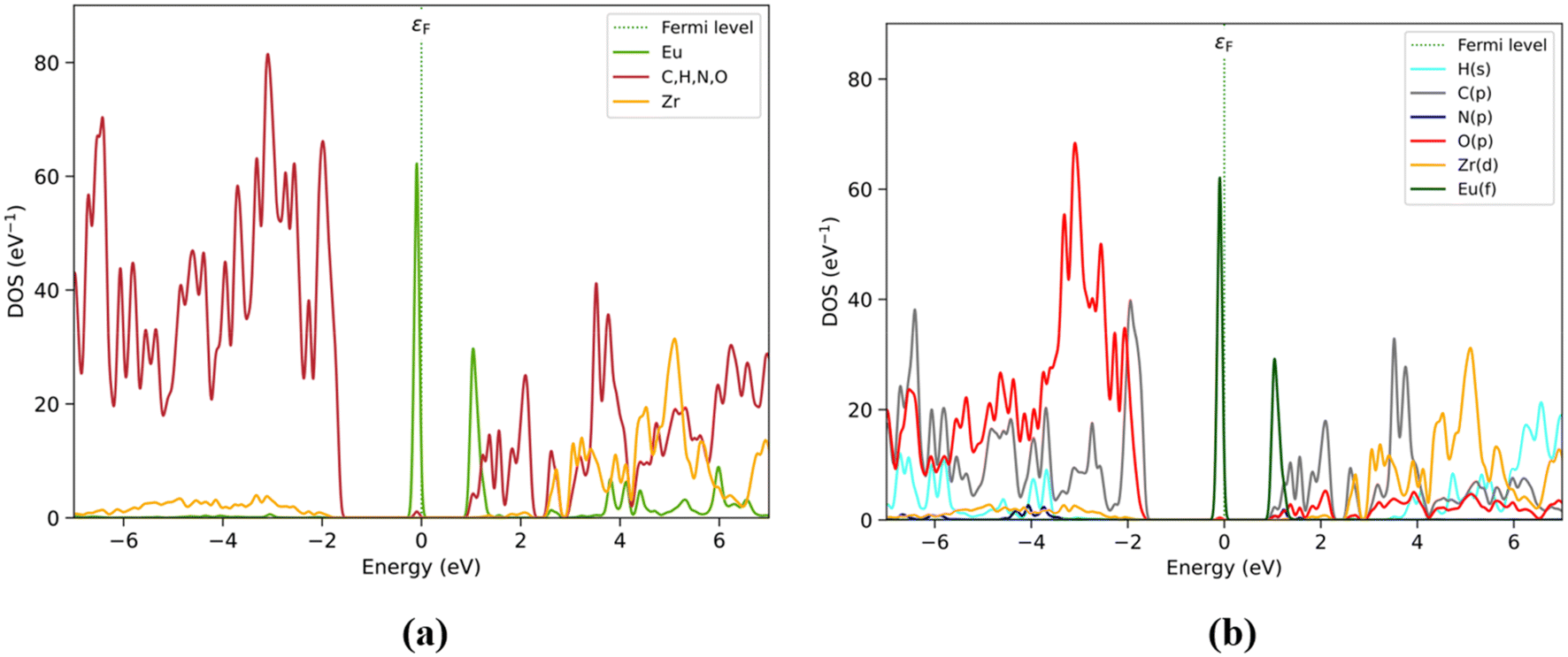 | ||
| Fig. 3 PDOS by elements (with organic elements C, H, N, and O as a single group) (a) and shells (b) considering those most relevant to Eu3+@UiO-66(DPA). Color labels for (a): Eu (green), organic group (red) and Zr (orange). For (b): H(s) (cyan), C(p) (grey), N(p) (blue), O(p) (red), Zr(d) (orange), and Eu(f) (dark green). | ||
Eu3+@UiO-66(DPA) exhibits alterations in the luminescence intensity after the introduction of various ions, with a particularly pronounced effect observed in the presence of Hg2+ ions. Experimental data suggest that the quenching mechanism may involve the substitution of Eu3+ by Hg2+ within the Eu3+@UiO-66(DPA) structure. This substitution leads to a reduction in the material luminescence.47 To get a deeper understanding of the sensing mechanism, PDOS analysis of Hg2+@UiO-66(DPA) was also performed.
PDOS by-fragment results (Fig. 4) show that at the Fermi level, the BDC fragments would have a relevant role with their occupied states in the valence zone around −2.0 eV (purple). Also, DPA (blue) is the fragment that maintains direct interactions with the Hg atom and this atom does not show a relevant contribution in this region. The BDC fragment also shows some contributions at around −1.8 and −2.5 eV, sharing the same energy range with the Zr6O4(OH)4 and DPA fragments. However, the Zr6O4(OH)4 and DPA fragments show much smaller contributions than BDC. On the other hand, at low energy levels, the conduction band is composed of a Hg atom (1.5 eV) and followed by the DPA fragment. Around this range, the DPA and BDC fragments maintain a relevant contribution up to 2.5 eV, especially the BDC fragment shows an important contribution between 1.8 and 2.5 eV. Over 3 eV, the Zr6O4(OH)4 and BDC fragments share their contributions to the PDOS, followed by the contribution of the DPA fragment with an overlap at larger energy values.
 | ||
| Fig. 4 (a) Structure of Hg2+@UiO-66(DPA) identifying fragments: Zr6O4(OH)4 (red), BDC (violet), DPA (blue), and Hg (brown). (b) PDOS by fragments. | ||
The PDOS obtained by elemental and shell analyses matches these results (Fig. 5), where the valence band is composed mainly of C(p) followed by O(p) at −2 eV. The O(p) contribution increases at lower energy levels assigned to the Zr6O4(OH)4 and BDC/DPA fragments. On the other hand, the conduction band is composed of C(p) and Hg(s) orbitals at ∼1.8 eV. Moreover, C(p) and O(p) unoccupied state contributions are present between the 1.8 and 2.5 eV energy range. Finally, over the 3 eV energy range, Zr(d) and H(s) contribute to those states.
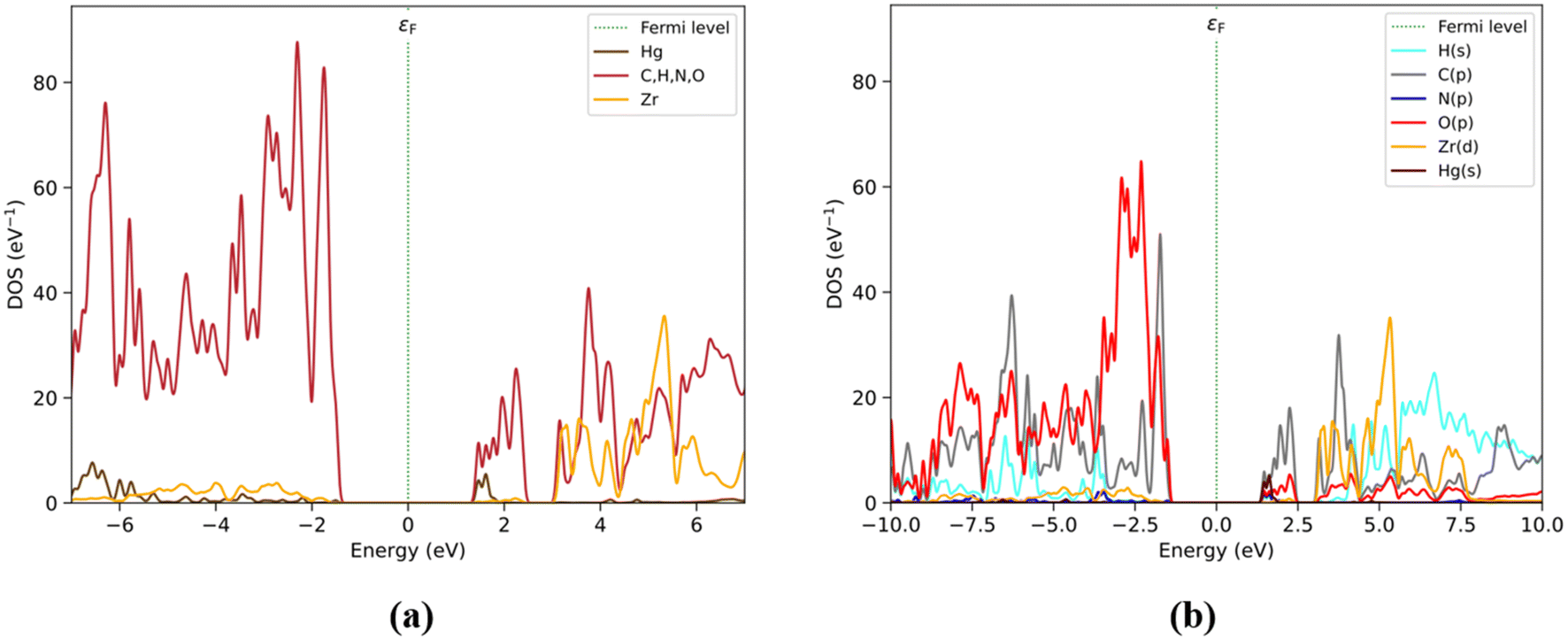 | ||
| Fig. 5 PDOS by elements (with organic elements C, H, N, and O as a single group) (a) and shells (b) considering those most relevant to Hg2+@UiO-66(DPA). Color labels for (a): Hg (brown), organic group (red) and Zr (orange). For (b): H(s) (cyan), C(p) (grey), N(p) (blue), O(p) (red), Zr(d) (orange), and Hg(s) (brown). | ||
The electronic states introduced by the Hg atoms and the DPA linkers generate unoccupied states at the conduction band edge, leading to the formation of a non-emissive excited state. According to our analysis, the Hg2+ and DPA composition of the conduction band edge in the Hg2+@UiO-66(DPA) system contributes to generate a ‘dark’ excited state after excitation and subsequent non-radiative deactivation. This ‘dark’ excited state leads to a turn-off in the luminescence process. This result is consistent with the experimental turn-off luminescence sensing mechanism of Eu3+@UiO-66(DPA) in the presence of Hg2+ ions.
Finally, for Eu3+@UiO-66(DPA) and Hg2+@UiO-66(DPA), the highest occupied levels are constituted by Eu3+ and DPA/BDC linkers, respectively. In particular, for Hg2+@UiO-66(DPA), the C(p) orbitals have an important contribution, i.e. the phenyl groups. On the other hand, both metals contribute primarily at low energy unoccupied levels of the conduction band. Specifically, the most notable orbital contribution of Eu has an energy of 1.1 eV, while for Hg2+@UiO-66(DPA), the same region of the conduction band is composed of Hg and DPA states (Fig. 4b). This difference in composition would lead to expected energy transfer channels from the BDC fragments to the metals (Eu3+ or Hg2+) and the DPA fragment. This charge transfer is particularly more favorable in the case of Eu2+@UiO-66(DPA) than in the case of Hg2+@UiO-66(DPA), which could be explained by the lower band gap in the electronic structure of the material with the lanthanide.
Periodic modelling of Eu3+Hg2+@UiO-66(DPA)
In a supercell model containing both Hg2+ and Eu3+ ions, the DPA linker was considered to possess vacant coordination sites, with the free –COOH groups and pyridine nitrogen atoms acting as coordination sites for metal ions. This theoretical investigation aligns with the experimentally proposed mechanism, which attributes the luminescence quenching to the substitution of Eu3+ ions by Hg2+ ions. According to Fig. S2,† and for this specific model where both ligands occupy the same site on each node, the metal–metal (Eu–Hg) distance is 14.68 Å. This value is close to the distance between replicas of two equivalent BDC linkers, which is 14.65 Å. This large separation between the metal centers suggests that any form of energy transfer (ET) between them is negligible, as there is no intervening electronic spacer to facilitate such a process. It is important to note that the energy transfer (ET) rates depend significantly on the donor–acceptor (D–A) separation distance (R) and the mechanism involved. For Förster resonance energy transfer (FRET), the ET rate decreases with increasing R, following an inverse sixth power relationship (1/R6). In contrast, the Dexter energy transfer mechanism exhibits an ET rate that decays exponentially with increasing D–A distance due to its dependence on wavefunction overlap. We concur with the mechanism proposed in the experimental work,47 which suggests that the substitution of Eu3+ ions by Hg2+ ions within the structure reduces the number of emission centers, thereby diminishing the material's luminescence as the Hg2+ ion concentration increases. Given that the mechanism is well supported by experimental evidence, our study focuses on characterizing and quantifying the luminescence response rather than investigating the substitution process in detail with a model involving both ions. This approach allows us to align our efforts with the primary goal of elucidating the emission mechanisms of Eu3+@UiO(DPA) and non-emissive states induced by the inclusion of Hg2+. Inspired by these results, the sensitization and emission of the Eu3+-doped UiO-66(DPA) MOF and the role of Hg2+ in the turn-off luminescence mechanism were investigated. To achieve this, accurate multiconfigurational ab initio methods were used along with DFT calculations using cluster models; see Fig. S3(a) and (b).† The next section provides a more insightful understanding of the luminescence properties of Eu2+@UiO-66(DPA) and its selective quenching response to Hg2+ ions.Optical properties of Eu3+@UiO-66(DPA)
The simulated UV–Vis absorption spectrum of Eu3+@UiO-66(DPA) displays two main vertical transitions based on the strength of the oscillator (f), which are very close in energy. The first one (labelled as A) appears centered at around 272 nm (f = 0.970), which corresponds to π → π* electronic transition. Table S1† shows that the electron density of the molecular orbitals involved in this electronic transition is located on the BDC linker. The second absorption band located at 278 nm (f = 0.175) is also assigned to linker-centered π → π* electronic transition and involves the active molecular orbitals of the DPA linkers. This result shows a difference of about 50 nm compared to the experimental value (305 nm),22 which falls within the usual error margin accepted for TD-DFT calculations. Based on this result, it is possible to apply the fragmentation scheme developed by Beltrán-Leiva et al.40 to evaluate the most likely sensitization and emission pathways in lanthanide complexes. In this approach, the ligand and the lanthanide fragments are calculated separately (at the same level of theory), using multiconfigurational methods. The author points out that it is crucial to consider that absorption is ligand-localized. This guarantees that excitation bands involved in the sensitization channel are not affected by the fragmentation procedure. Based on this approach, a definitive understanding of the possible sensitization and emission pathways of Eu3+@UiO-66(DPA) systems is provided in the next section.Sensitization and emission pathways of Eu3+@UiO-66(DPA)
The sensitization pathway and emission mechanisms of the Eu3+@UiO-66(DPA) system were studied through a multireference CASSCF/NEVPT2 approach. The ground states and excited electronic states of the Eu3+ fragment were explored via CAS(6,7)SCF/NEVPT2 calculations. At the theoretical level the calculations show that the emitting level (5D0) state and 5D1 of Eu3+ are located at 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 cm−1 and 20174 cm−1, respectively. Our results align with previous reports, which showed 5D0 and 5D1 values of 17830 and 19450 cm−1 respectively, based on CASSCF/XMCQDPT2/SO-CASSCF calculations.72 This is also consistent with the previously reported experimental values of 17300 and 19000 cm−1 for 5D0 and 5D1 of the Eu3+ion.73 The electronic structures of the BDC and DPA linkers, including the ground state (S0) and excited states (Sn and Tn), were investigated using the CAS(10,10)SCF/NEVPT2 method with an active space of ten electrons in ten molecular orbitals
600 cm−1 and 20174 cm−1, respectively. Our results align with previous reports, which showed 5D0 and 5D1 values of 17830 and 19450 cm−1 respectively, based on CASSCF/XMCQDPT2/SO-CASSCF calculations.72 This is also consistent with the previously reported experimental values of 17300 and 19000 cm−1 for 5D0 and 5D1 of the Eu3+ion.73 The electronic structures of the BDC and DPA linkers, including the ground state (S0) and excited states (Sn and Tn), were investigated using the CAS(10,10)SCF/NEVPT2 method with an active space of ten electrons in ten molecular orbitals
According to the CAS(10,10)SCF/NEVPT2 calculations for the DPA linker, the S1 electronic state is located at 31194 cm−1, whereas the T1 electronic state appears at 23828 cm−1. Therefore, the energy gap (S1–T1) is 7366 cm−1 which is not within the optimal range according to the Reinhoudt rule. Following this empirical rule (Reinhoudt rule), the energy gap between the S1 and T1 electronic states must be 5000 cm−1 to ensure an efficient inter-system crossing (ISC) S1 → T1.74 The CAS(10,10)SCF/NEVPT2 calculations showed that the S1 and T1 electronic states of the BDC linker are located at 27794 and 26558 cm−1, respectively. Thus, the energy gap (S1–T1) is 1236 cm−1.
In this regard, for both ligands (BDC and DPA), the S1–T1 energy difference calculated at the CAS(10,10)SCF/NEVPT2 level of theory shows that the ISC process is totally efficient, according to the empirical rule. However, for UiO-66(DPA), it has been reported that after the PSM with Eu3+ (generating Eu3+@UiO-66(DPA)), a characteristic emission peak assigned to the Eu3+ ions appears. This suggests that the population of the T1 electronic state of the antenna and its subsequent energy transfer pathway may take another route from the commonly observed mechanism. We support this hypothesis with two arguments. First, the periodic DFT calculations indicate that the conduction band (CB) consists of a mixture of electronic states from BDC and DPA linkers just above the electronic state associated with Eu3+ ions. Second, the strong spin–orbit coupling (SOC) effect of Eu3+ ions in this material induces a mixture of electronic states with different multiplicities.
According to the Latva rule, an energy gap between 2500 and 4000 cm−1 can facilitate efficient energy transfer from the T1 electronic state of the antenna to a resonance excited level of Ln3+ ions. This energy gap is adequate for an efficient ET channel when ΔE (T → Ln*), according to the Latva rule. A posterior analysis of CASSCF/NEVPT2 calculations showed that the T1 electronic state of the DPA antenna is located 6224 cm−1 higher in energy than the Eu3+ ion 5D0 state (17600 cm−1). The T1(DPA) electronic state is located 3554 cm−1 higher than the 5D1 electronic state (20174 cm−1) of the Eu3+ ion. The T1 electronic state of the BDC linker is located at 8988 and 6384 cm−1 higher than the 5D0 and 5D1 electronic states of the Eu3+ ion. Based on these results, it can be considered that an energy transfer from the T1 electronic state of the BDC or DPA linker to the emissive electronic state of 5D0 Eu ions has a low probability of occurring. Thus, the most important argument is that the most probable sensitization pathway involves the ISC process of DPA (S1 → T1), followed by an energy transfer channel that is activated from the T1 electronic state of the DPA linker to the 5D1 electronic state of Eu3+ ions, T1 (DPA) → 5D1 (Eu3+). Then, a vibrational relaxation (VR) from 5D1 can result in the emissive 5D0 state, 5D1 → 5D0 (Eu3+). Additionally, the excited electronic states of the BDC antenna could enhance the population of the DPA antenna T1 electronic states through the ET process, as shown in Fig. 6. Finally, the radiative deactivation from 5D0 electronic states to the Eu3+ 7FJ electronic states is produced (see Fig. 6).
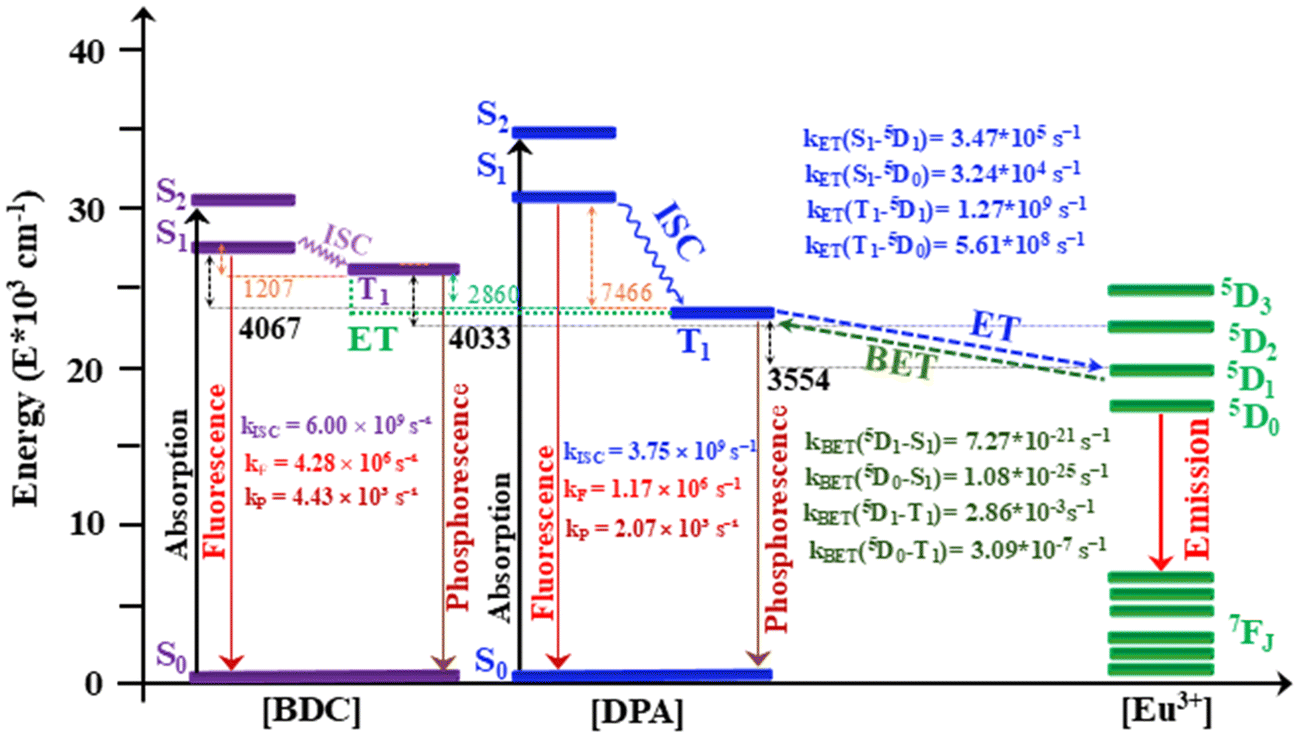 | ||
| Fig. 6 The most likely sensitization and emission channels are depicted in this energy level diagram. Intersystem crossing, phosphorescence, fluorescence, energy transfer and back energy transfer rates are represented by the letters kISC, kP, kF, kET and kBET, respectively. | ||
A more detailed analysis of the excited electronic states was carried out. Table S2† lists the active orbitals, occupation numbers and low-lying excited electronic state configurations computed for the BDC antenna. As it is possible to appreciate, the S1 electronic state has 50% contribution of a configuration with 1(π → π*) character and 38% contribution of a configuration with 1(n → π*), whereas the T1 electronic state of this antenna has 78% contribution of a configuration with 3(π → π*) character. Thus, multireference characteristics of these excited electronic states, which show a large change in the orbital type, could favor the ISC process (S1(BDC) → T1(BDC)), according to the El-Sayed rule.75,76 However, in the case of the DPA antenna, as shown in Table S3,† the electronic configurations of the S1 and T1 electronic states do not indicate any change in the orbital type within the active space. The S1 electronic state has 50% contribution of a configuration with 1(π → π*) character and 20% contribution of a configuration with 1(π → π*), whereas the T1 electronic state of the DPA antenna has 78% contribution of a configuration also with 3(π → π*) character. Thus, according to El-Sayed's rule, the electronic configurations of S1 and T1 are more likely to result in a slow intersystem crossing (ISC) due to the absence of a change in the orbital type.
Finally, the radiative and non-radiative processes involved in sensitization and ET channels were analyzed based on their rate constants. In the case of the BDC linker, the ISC rate kISC (S1 → T1) = 6.00 × 109 s−1 is larger than the fluorescence rate (three orders of magnitude) of kF (S1 → S0) = 4.28 × 106 s−1. As can be seen in Fig. 6, the ISC pathway from the DPA linker has an ISC rate of kISC (S1 → T1) = 3.75 × 109 s−1, which is also three orders of magnitude greater than the fluorescence rate kF (S1 → S0) = 1.17 × 106 s−1. Thus, based on the rate constant of these photophysical processes for both linkers, it is likely that the population of the T1 electronic state occurs through an intersystem crossing (ISC) process before radiative decay. On the other hand, these calculations also reveal a slow phosphorescence (kP) rate for both linkers. As shown in Fig. 6, the DPA linker has a kP (T1 → S0) value of 2.07 × 103 s−1, while the BDC linker has a kP (T1 → S0) value of 4.43 × 103 s−1. Both linkers exhibit long-lived populations in their T1 electronic states, with nonradiative pathways such as ET to sensitize the Eu3+ ion. The LUMPAC software77 was used to compute the energy transfer rates (kET) and the back energy transfer rates (kBET). As illustrated in Fig. 6, ET rates kET that are within the order of 104–109 s−1 lead to a probable ET channel, both from the S1 and T1 electronic states. Although the T1 electronic state of the DPA antenna falls outside the energy range stipulated by Latva's rule, it still exhibits adequate energy transfer rates for T1 (DPA) → 5DJ(Eu3+). The kET(S1–5D1) = 3.47 × 105 s−1 and kET(S1–5D0) = 3.24 × 104 s−1 values are lower than the corresponding values for probable ET from the T1 electronic state, with kET(T1–5D1) = 1.27 × 109 s−1 and kET(T1–5D0) = 5.61 × 108 s−1. Furthermore, the analysis of kBET values indicates that the BET mechanism is not competitive in this system. The observed results support the proposed sensitization and emission mechanism between the DPA linker and the Eu3+ ion. The ET pathway from the ligand to the lanthanide dominates, as evidenced by the significantly lower and non-competitive BET rates: kBET(5D1–T1) = 2.86 × 10−3 s−1 and kBET(5D0–T1) = 3.09 × 10−7 s−1, both of which are too low to represent a predominant process. Likewise, the BET rates kBET(5D1–S1) = 7.27 × 10−21 s−1 and kBET(5D0–S1) = 1.08 × 10−25 s−1 confirm the negligible role of back transfer in this system.
Luminescence quenching pathway in Hg2+@UiO-66(DPA)
The proposed sensing mechanism of Eu3+@UiO-66(DPA)22 is based on the high binding affinity of the DPA ligand for Hg2+ ions, which facilitates the replacement of Eu3+ by Hg2+, resulting in luminescence quenching. The Eu3+@UiO-66(DPA) chemosensor exhibits a decrease in luminescence intensity (turn-off response) in the presence of Hg2+ ions, achieving a detection limit of 8.26 nM. According to the authors, in the presence of Hg2+, the Eu3+ ions in Eu3+@UiO-66(DPA) are replaced by Hg2+ ions, leading to luminescence quenching as the concentration of Hg2+ increases. This proposed sensing mechanism aligns with previous reports. For instance, Hongliang Tan et al.78 developed a lanthanide-based luminescent probe for Hg2+ detection using Eu-DPA chelates immobilized on SiO2 nanoparticles. This system demonstrated excellent selectivity towards Hg2+, with a detection limit as low as 7.07 nM. In their study, the authors also suggested that the luminescence quenching is due to the replacement of Eu3+ by Hg2+, primarily attributed to the significantly higher binding constant of Hg2+ (K = 1026.4) compared to Eu3+ (K = 1022.39).78 Similar strategies leveraging the selectivity of these ligand (i.e., DPA) interactions have been reported in other Hg2+ sensing systems. For instance, a gold nanoparticle-based sensor functionalized with DPA demonstrated exceptional selectivity for Hg2+ ions over other metal ions in aqueous solutions. In this system, the high affinity of DPA for Hg2+ supports stable complex formation, leading to reliable detection. Such findings highlight the critical role of ligand design and binding affinity in achieving selectivity and sensitivity for Hg2+, reinforcing the probability of the proposed mechanism in Eu3+@UiO-66(DPA).To investigate the luminescence quenching pathway in the Hg2+@UiO-66(DPA) system, the S0 and S1 electronic states were studied in terms of energy and structure. According to the Franck–Condon principle and selection rules, we support the hypothesis that, upon photoexcitation, the electron relaxes until reaching the first excited electronic state, presumably involved with the Hg2+ ions. Thus, in the first step, the vertical excitation of the Hg2+@UiO-66(DPA) (based on cluster models) was computed. This absorption spectrum displays three main transitions regarding the f values, one at 250 nm, a more intense one at 258 nm, and the last one at 306 nm.
The bands centered at 306 nm (f = 0.02) correspond to π → π* electronic transitions from the HOMO to the LUMO+1, which are labeled as H and L+1 in Fig. 7. The MOs involved in this absorption band are localized at the bridging oxygen atoms of the [Zr6(OH)4O4] node and DPA linker; see the left panel in Fig. 7. The more intense bands are located at 258 nm (f = 1.02) and are composed of π → π* electronic transitions (HOMO−3 → LUMO+2). These electronic transitions exhibit an inter-ligand charge-transfer (ILCT) character. The MOs involved in this absorption band are mainly localized on the BDC linker. The band at 250 nm is associated with a π → π* electronic transition (HOMO−3 → LUMO+1), which also presents an ILCT character; see Fig. 7.
 | ||
| Fig. 7 Molecular orbital diagram based on the S0 (absorption) and S1 (emission) states of the Hg2+@UiO-66(DPA) system, where λ is the theoretical wavelength of absorption (left panel) and of emission (right panel), respectively; krad is the emission radiative rate; τrad is the emission times; and f is the oscillator strength. | ||
In the second step, the probable emissive S1 state of Hg2+@UiO-66(DPA) was explored. This step begins with the optimization of the S1 electronic state, which was subsequently used as input data to calculate the electronic transitions associated with the emission spectrum by means of TD-DFT methods. All electronic transitions that correspond to the absorption process involve the molecular orbital just above the LUMO (orbital located on the Hg atom). Thus, considering the Franck–Condon principle and selection rules, it is more likely that, after excitation, the electron relaxes until reaching the S1 electronic state located on the Hg atom. Therefore, radiative deactivation could arise from the S1 electronic state, following Kasha's rule. The radiative rate (krad) and radiative lifetime (τrad) computed for this electronic transition are in the range of fluorescence. As displayed in the right panel in Fig. 7, τrad and krad present values which are in fluorescence for all calculations (less than 10−6 seconds and 106 seconds−1, respectively).79 However, if the intensity of these electronic transitions based on the f magnitude (f = 0.002) is analyzed, the presence of Hg ions induces a “dark” excited state.
In summary, the most probable fluorescence quenching pathway of the Hg2+@UiO-66(DPA) system is supported by the hypothesis that after direct photoexcitation from the S0 electronic state, the electron relaxes to reach the first excited electronic state S1 located in the Hg2+ ion. For this reason, it is possible to state that the S1 electronic state decays to the S0 electronic state through a non-radiative mechanism.
Conclusions
In summary, this report outlines a theoretical protocol that successfully provided an accurate explanation for the selective detection of Hg2+ ions using a Eu3+-post functionalized UiO-66(DPA) chemosensor. The elucidation of the sensing mechanism was addressed considering two points: the host–guest interaction (in this case, Hg is the host) and the photophysical properties of the material. First, the study of the Eu3+@UiO-66(DPA) system using rigorous CASSCF/NEVPT2 calculations was carried out to analyze the most probable sensitization and energy transfer pathway. This involves multireference ab initio methods using a truncated structural model (cluster model). In the Eu3+@UiO-66(DPA) system, CASSCF/NEVPT2 calculations predict that the most probable sensitization pathway is the DPA linker (S1 → T1) and DPA (T1) → Eu(5D1). Next, vibrational relaxation (VR) from the 5D1 state can lead to the population of the emissive 5D0 state, which subsequently emits from 7FJ. Furthermore, our investigation of the excited-state dynamics of the antenna supports the proposed sensitization and emission pathways. The intersystem crossing (ISC) process is significantly faster than the deactivation of the S1 electronic state via fluorescence (kisc is three orders of magnitude greater than kF). This result confirms the feasibility of populating the T1 electronic state through ISC. The calculated rate constants for the phosphorescence process (kP) indicate that the DPA antenna exhibits a prolonged population in its T1 electronic state, which favors the energy transfer (ET) process to the Eu3+ ion. Lastly, the evaluation of radiative and non-radiative processes using the LUMPAC software demonstrates that ET is the predominant mechanism over back energy transfer (BET), further reinforcing our conclusions regarding the proposed emission mechanism for the Eu3+@UiO-66(DPA) system. From these results, an important finding is that the T1 electronic states of the BDC linker can reinforce the population of the T1 electronic states of the DPA antenna. The electronic density of states (DOS) analysis of Eu3+@UiO-66(DPA), obtained via periodic DFT calculations, evidences the mixture of states of the linkers (BDC and DPA) and Eu3+ ions. This agrees with the sensitization and energy transfer mechanism proposed by the CASSCF/NEVPT2 results. Second, both the electronic and photophysical properties were assessed to determine the correlation between the chemosensor structure and the luminescence change prompted by the analyte. For the Hg2+@UiO-66(DPA) system, employing the optimized geometry of the S0 and S1 electronic states, the absorption and emission spectra were modelled based on the vertical transitions of the Franck–Condon and Kasha rules. The results indicate that following direct photoexcitation from the S0 state, the electron relaxes to the S1 state within the Hg2+ ion, subsequently decaying non-radiatively to the S0 state. For this system, the calculated emission time (τrad = 2.51 × 10−7) and radiative rate (krad = 3.99 × 106) are evidence of a probable deactivation of the S1 electronic state via the fluorescence process. However, the strength of the oscillator supports the hypothesis of a dark emissive state due to a low emission intensity (f magnitude of 0.002) and deactivation from the Hg molecular orbital. The proposed mechanism is also in good agreement with the electronic DOS analysis of Hg2+@UiO-66(DPA) obtained via periodic DFT. DOS analysis evidences an overlap of states of both linkers (BDC and DPA) and Hg2+ ions at the edge of the conduction band, which agrees with the turn-off induced by Hg2+ ions. These findings are particularly relevant for advancing the design of MOF-based sensors, enhancing their efficacy in selective Hg2+ detection. Finally, we accomplished a computational procedure combining a multireference ab initio method with periodic DFT calculations which are not frequently taken into account by previous reports. Thus, this methodology is accurate to be applied to different lanthanide-doped MOF chemosensor systems to study the electronic properties that govern the sensing mechanisms, or to design new sensors based on MOFs.Data availability
Data are available upon request from the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful for FONDECYT projects 1231194, 1241917, and 1220442 and ANID Postdoctoral 3230141 (ANID/FONDAP/1523A0006). This material is based on the work supported by the Air Force Office of Scientific Research under award number FA8655-25-1-8759.References
- Z. Ali, R. Ullah, M. Tuzen, S. Ullah, A. Rahim and T. A. Saleh, Colorimetric Sensing of Heavy Metals on Metal Doped Metal Oxide Nanocomposites: A Review, Trends Environ. Anal. Chem., 2023, 37, e00187, DOI:10.1016/J.TEAC.2022.E00187.
- S. Mishra, R. N. Bharagava, N. More, A. Yadav, S. Zainith, S. Mani and P. Chowdhary, Heavy Metal Contamination: An Alarming Threat to Environment and Human Health, in Environmental Biotechnology: For Sustainable Future, Springer, Singapore, 2019, pp. 103–125. DOI:10.1007/978-981-10-7284-0_5.
- L. A. Malik, A. Bashir, A. Qureashi and A. H. Pandith, Detection and Removal of Heavy Metal Ions: A Review, Environ. Chem. Lett., 2019, 17(4), 1495–1521, DOI:10.1007/s10311-019-00891-z.
- J. F. Olorunyomi, S. T. Geh, R. A. Caruso and C. M. Doherty, Metal-Organic Frameworks for Chemical Sensing Devices, Mater. Horiz., 2021, 8(9), 2387–2419, 10.1039/d1mh00609f.
- T. Elias Abi-Ramia Silva, F. Burisch and A. T. Güntner, Gas Sensing for Space: Health and Environmental Monitoring, TrAC, Trends Anal. Chem., 2024, 177, 117790, DOI:10.1016/j.trac.2024.117790.
- X.-Y. Xu and B. Yan, Fabrication and Application of a Ratiometric and Colorimetric Fluorescent Probe for Hg 2+ Based on Dual-Emissive Metal–Organic Framework Hybrids with Carbon Dots and Eu 3+, J. Mater. Chem. C, 2016, 4(7), 1543–1549, 10.1039/C5TC04002G.
- J. R. Askim, M. Mahmoudi and K. S. Suslick, Optical Sensor Arrays for Chemical Sensing: The Optoelectronic Nose, Chem. Soc. Rev., 2013, 42(22), 8649–8682, 10.1039/c3cs60179j.
- L. Guan, Z. Jiang, Y. Cui, Y. Yang, D. Yang and G. Qian, An MOF-Based Luminescent Sensor Array for Pattern Recognition and Quantification of Metal Ions, Adv. Opt. Mater., 2021, 9(9), 1–7, DOI:10.1002/adom.202002180.
- C. W. Huang, C. Lin, M. K. Nguyen, A. Hussain, X. T. Bui and H. H. Ngo, A Review of Biosensor for Environmental Monitoring: Principle, Application, and Corresponding Achievement of Sustainable Development Goals, Bioengineered, 2023, 14(1), 58–80, DOI:10.1080/21655979.2022.2095089.
- L. Yu, Q. Zheng, H. Wang, C. Liu, X. Huang and Y. Xiao, Double-Color Lanthanide Metal–Organic Framework Based Logic Device and Visual Ratiometric Fluorescence Water Microsensor for Solid Pharmaceuticals, Anal. Chem., 2020, 92(1), 1402–1408, DOI:10.1021/acs.analchem.9b04575.
- Y. Zhao, M. Liu, S. Zhou, Z. Yan, J. Tian, Q. Zhang and Z. Yao, Smartphone-Assisted Ratiometric Sensing Platform for on-Site Tetracycline Determination Based on Europium Functionalized Luminescent Zr-MOF, Food Chem., 2023, 425, 136449, DOI:10.1016/j.foodchem.2023.136449.
- T. Wang, J. Zhang, Y. Wu, S. Wang, X. Jiang, Z. Zhang and S. Li, Smartphone - Integrated Ratiometric Fluorescence Sensing Platform Based on Bimetallic Metal – Organic Framework Nanowires for Anthrax Biomarker Detection, Microchim. Acta, 2023, 190(12), 1–9, DOI:10.1007/s00604-023-06065-7.
- K. F. Kayani, N. N. Mohammad, D. A. Kader and S. J. Mohammed, Ratiometric Lanthanide Metal-Organic Frameworks (MOFs) for Smartphone-Assisted Visual Detection of Food Contaminants and Water: A Review, ChemistrySelect, 2023, 8, 202303472, DOI:10.1002/slct.202303472.
- X. Zeng, J. Hu, M. Zhang, F. Wang, L. Wu and X. Hou, Visual Detection of Fluoride Anions Using Mixed Lanthanide Metal − Organic Frameworks with a Smartphone, Anal. Chem., 2020, 92, 2097–2102, DOI:10.1021/acs.analchem.9b04598.
- X. Zhang, Z. Li, Y. Zhang, C. Jiao, H. Zheng, Y. Zhu and Z. Sun, Ultrastable Lanthanide Metal − Organic Frameworks for Smartphone-Assisted Ratiometric Fluorescent Sensing of Toluenediamines and Tunable Luminescence, Inorg. Chem., 2024, 63, 16418–16428, DOI:10.1021/acs.inorgchem.4c02461.
- W. Wu, Y. Li, P. Song, Q. Xu, N. Long, P. Li, L. Zhou, B. Fu, J. Wang and W. Kong, Metal-Organic Framework (MOF)-Based Sensors for Exogenous Contaminants in Food: Mechanisms, Advances, and Prospects, Trends Food Sci. Technol., 2023, 138, 238–271, DOI:10.1016/j.tifs.2023.06.016.
- H. Y. Li, S. N. Zhao, S. Q. Zang and J. Li, Functional Metal-Organic Frameworks as Effective Sensors of Gases and Volatile Compounds, Chem. Soc. Rev., 2020, 49(17), 6364–6401, 10.1039/c9cs00778d.
- M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Luminescent Metal-Organic Frameworks, Chem. Soc. Rev., 2009, 38(5), 1330–1352, 10.1039/b802352m.
- W. P. Lustig, S. Mukherjee, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Metal–Organic Frameworks: Functional Luminescent and Photonic Materials for Sensing Applications, Chem. Soc. Rev., 2017, 46(11), 3242–3285, 10.1039/C6CS00930A.
- L. Fan, D. Zhao, B. Li, F. Wang, Y. Deng, Y. Peng, X. Wang and X. Zhang, Luminescent Binuclear Zinc(II) Organic Framework as Bifunctional Water-Stable Chemosensor for Efficient Detection of Antibiotics and Cr(VI) Anions in Water, Spectrochim. Acta, Part A, 2022, 264, 120232, DOI:10.1016/j.saa.2021.120232.
- J. Jin, J. Xue, Y. Liu, G. Yang and Y. Y. Wang, Recent Progresses in Luminescent Metal-Organic Frameworks (LMOFs) as Sensors for the Detection of Anions and Cations in Aqueous Solution, Dalton Trans., 2021, 50(6), 1950–1972, 10.1039/d0dt03930f.
- Z. Xiaoxiong, Z. Wenjun, L. Cuiliu, Q. Xiaohong and Z. Chengyu, Eu 3+ -Postdoped UIO-66-Type Metal–Organic Framework as a Luminescent Sensor for Hg 2+ Detection in Aqueous Media, Inorg. Chem., 2019, 58(6), 3910–3915, DOI:10.1021/acs.inorgchem.8b03555.
- L. Fan, D. Zhao, B. Li, X. Chen, F. Wang, Y. Deng, Y. Niu and X. Zhang, An Exceptionally Stable Luminescent Cadmium(II) Metal-Organic Framework as a Dual-Functional Chemosensor for Detecting Cr(vi) Anions and Nitro-Containing Antibiotics in Aqueous Media, CrystEngComm, 2021, 23(5), 1218–1225, 10.1039/d0ce01721c.
- T. Zhang, H. Chen, H. Lv, Q. Li and X. Zhang, Nanochannel-Based Heterometallic {ZnIIHoIII}-Organic Framework with High Catalytic Activity for the Chemical Fixation of CO2, RSC Adv., 2021, 11(16), 9731–9739, 10.1039/d1ra00590a.
- G. Li and C. Tong, Dual-Functional Lanthanide Metal Organic Frameworks for Visual and Ultrasensitive Ratiometric Fluorescent Detection of Phosphate Based on Aggregation-Induced Energy Transfer, Anal. Chim. Acta, 2020, 1133, 11–19, DOI:10.1016/j.aca.2020.07.066.
- Y. Hidalgo-Rosa, M. A. Treto-Suárez, E. Schott, X. Zarate and D. Páez-Hernández, Sensing Mechanism Elucidation of a Europium(III) Metal–Organic Framework Selective to Aniline: A Theoretical Insight by Means of Multiconfigurational Calculations, J. Comput. Chem., 2020, 41(22), 1956–1964, DOI:10.1002/JCC.26365.
- J. Dong, D. Zhao, Y. Lu and W.-Y. Sun, Photoluminescent Metal-Organic Frameworks and Their Application for Sensing Biomolecules, J. Mater. Chem. A, 2019, 7, 22744–22767, 10.1039/C9TA07022B.
- B. Yan, Lanthanide-Functionalized Metal-Organic Framework Hybrid Systems to Create Multiple Luminescent Centers for Chemical Sensing, Acc. Chem. Res., 2017, 50(11), 2789–2798, DOI:10.1021/acs.accounts.7b00387.
- S. Zhao, G. Wang and D. Poelman, Luminescent Lanthanide MOFs : A Unique Platform for Chemical Sensing, Materials, 2018, 11(4), 572, DOI:10.3390/ma11040572.
- Y. Cui, Y. Yue, G. Qian and B. Chen, Luminescent Functional Metal–Organic Frameworks, Chem. Rev., 2012, 112(2), 1126–1162, DOI:10.1021/cr200101d.
- S. M. Cohen, Postsynthetic Methods for the Functionalization of Metal-Organic Frameworks, Chem. Rev., 2012, 112(2), 970–1000, DOI:10.1021/cr200179u.
- S. Mandal, S. Natarajan, P. Mani and A. Pankajakshan, Post-Synthetic Modification of Metal–Organic Frameworks Toward Applications, Adv. Funct. Mater., 2021, 31(4), 1–22, DOI:10.1002/adfm.202006291.
- S. Mandal, S. Natarajan, P. Mani and A. Pankajakshan, Post–Synthetic Modification of Metal–Organic Frameworks Toward Applications, Adv. Funct. Mater., 2021, 31(4), 1–22, DOI:10.1002/adfm.202006291.
- Y. N. Zeng, H. Q. Zheng, J. F. Gu, G. J. Cao, W. E. Zhuang, J. D. Lin, R. Cao and Z. J. Lin, Dual-Emissive Metal-Organic Framework as a Fluorescent “Switch” for Ratiometric Sensing of Hypochlorite and Ascorbic Acid, Inorg. Chem., 2019, 58(19), 13360–13369, DOI:10.1021/acs.inorgchem.9b02251.
- J. N. Hao and B. Yan, Ag+-Sensitized Lanthanide Luminescence in Ln3+ Post-Functionalized Metal-Organic Frameworks and Ag+ Sensing, J. Mater. Chem. A, 2015, 3(9), 4788–4792, 10.1039/c4ta06462c.
- S. Yang, Z. Zhou, Z. Zhao, J. Liu, Y. Sun, Y. Zhang, E. Gao, M. Zhu and S. Wu, Synthesis of Multi-Emission MOF Composites for Multi-Dimensional Sensing Application, Microchem. J., 2024, 196, 109660, DOI:10.1016/j.microc.2023.109660.
- C. Xia, Y. Xu, M. M. Cao, Y. P. Liu, J. F. Xia, D. Y. Jiang, G. H. Zhou, R. J. Xie, D. F. Zhang and H. L. Li, A Selective and Sensitive Fluorescent Probe for Bilirubin in Human Serum Based on Europium(III) Post-Functionalized Zr(IV)-Based MOFs, Talanta, 2020, 212, 120795, DOI:10.1016/j.talanta.2020.120795.
- Z. Yu, S. Kang, M. Tai, J. Wang, Q. Tian, D. Jin and L. Wang, Synthesis, Modulation, and Characterization of Ln3+ Ions Doped Metal−organic Frameworks for WLED Applications, Dyes Pigm., 2023, 209, 110897, DOI:10.1016/j.dyepig.2022.110897.
- H. Liu, Y. Liu, Y. Meng, X. Shi, J. Sun, L. Zhao, D. Chen, H. Hao, D. Li, J. Dou and J. Han, Di-Functional Luminescent Sensors Based on Y3 + doped Eu3 + and Tb3 + coordination Polymers: Fast Response and Visible Detection of Cr3+, Fe3 + ions in Aqueous Solutions and Acetone, RSC Adv., 2020, 10(53), 32232–32240, 10.1039/d0ra06407f.
- M. J. Beltrán-Leiva, P. Cantero-López, C. Zúñiga, A. Bulhões-Figueira, D. Páez-Hernández and R. Arratia-Pérez, Theoretical Method for an Accurate Elucidation of Energy Transfer Pathways in Europium(III) Complexes with Dipyridophenazine (Dppz) Ligand: One More Step in the Study of the Molecular Antenna Effect, Inorg. Chem., 2017, 56(15), 9200–9208, DOI:10.1021/acs.inorgchem.7b01221.
- J. L. Mancuso, A. M. Mroz, K. N. Le and C. H. Hendon, Electronic Structure Modeling of Metal-Organic Frameworks, Chem. Rev., 2020, 120(16), 8641–8715, DOI:10.1021/acs.chemrev.0c00148.
- Y. Hidalgo-Rosa, J. Santoyo-Flores, M. A. Treto-Suárez, E. Schott, D. Páez-Hernández and X. Zarate, Tuning the Sensitization Pathway T 1 → 5 D J in Eu-Based MOF through Modification of the Antenna Ligand. A Theoretical Approach via Multiconfigurational Quantum Calculations, J. Lumin., 2023, 260, 119896, DOI:10.1016/j.jlumin.2023.119896.
- Y. Hidalgo-Rosa, K. Mena-Ulecia, M. A. Treto-Suárez, E. Schott, D. Páez-Hernández and X. Zarate, Expanding the Knowledge of the Selective-Sensing Mechanism of Nitro Compounds by Luminescent Terbium Metal-Organic Frameworks through Multiconfigurational Ab Initio Calculations, J. Phys. Chem. A, 2022, 126(39), 7040–7050 CrossRef CAS PubMed.
- Y. Hidalgo-Rosa, K. Mena-Ulecia, M. A. Treto-Suárez, E. Schott, D. Páez-Hernández and X. Zarate, Insights into the Selective Sensing Mechanism of a Luminescent Cd(II)-Based MOF Chemosensor toward NACs: Roles of the Host–Guest Interactions and PET Processes, J. Mater. Sci., 2021, 56(24), 13684–13704, DOI:10.1007/s10853-021-06196-3.
- Y. Hidalgo-Rosa, M. Saavedra-Torres, B. D. Koivisto, M. A. Treto-Suárez, D. Páez-Hernández, X. Zarate and E. Schott, Rare-Earth-Based Metal–Organic Frameworks with Improved Visible-Light-Harvesting Properties: A Quantum Chemistry Study, J. Mater. Sci., 2023, 58(21), 8862–8877, DOI:10.1007/s10853-023-08581-6.
- Y. Hidalgo-Rosa, M. A. Treto-Suárez, E. Schott, X. Zarate and D. Páez-Hernández, Sensing Mechanism Elucidation of a Chemosensor Based on a Metal-Organic Framework Selective to Explosive Aromatic Compounds, Int. J. Quantum Chem., 2020, 120(23), e26404, DOI:10.1002/qua.26404.
- Z. Xiaoxiong, Z. Wenjun, L. Cuiliu, Q. Xiaohong and Z. Chengyu, Eu 3+ -Postdoped UIO-66-Type Metal–Organic Framework as a Luminescent Sensor for Hg 2+ Detection in Aqueous Media, Inorg. Chem., 2019, 58(6), 3910–3915, DOI:10.1021/acs.inorgchem.8b03555.
- J. Guo, C. Dong, X. Zhang, Y. Liu, Y. Leng, G. Wang and Z. Chen, Colorimetric Sensors Constructed with One Dimensional PtNi Nanowire and Pt Nanowire Nanozymes for Hg2+ Detection, Anal. Chim. Acta, 2024, 1321, 343039, DOI:10.1016/j.aca.2024.343039.
- Y. Zhao and D. Li, Lanthanide-Functionalized Metal-Organic Frameworks as Ratiometric Luminescent Sensors, J. Mater. Chem. C, 2020, 8(37), 12739–12754, 10.1039/d0tc03430d.
- M. Treger, A. Hannebauer, A. Schaate, J. L. Budde, P. Behrens and A. M. Schneider, Tuning the Optical Properties of the Metal-Organic Framework UiO-66 via Ligand Functionalization, Phys. Chem. Chem. Phys., 2023, 25(8), 6333–6341, 10.1039/d2cp03746g.
- E. A. Dolgopolova, A. J. Brandt, O. A. Ejegbavwo, A. S. Duke, T. D. Maddumapatabandi, R. P. Galhenage, B. W. Larson, O. G. Reid, S. C. Ammal, A. Heyden, M. Chandrashekhar, V. Stavila, D. A. Chen and N. B. Shustova, Electronic Properties of Bimetallic Metal-Organic Frameworks (MOFs): Tailoring the Density of Electronic States through MOF Modularity, J. Am. Chem. Soc., 2017, 139(14), 5201–5209, DOI:10.1021/jacs.7b01125.
- J.-C. Jin, J. Wu, Y.-X. He, B.-H. Li, J.-Q. Liu, R. Prasad, A. Kumar and S. R. Batten, A 3D Luminescent Zn(ii) MOF for the Detection of High Explosives and the Degradation of Organic Dyes: An Experimental and Computational Study, CrystEngComm, 2017, 19(43), 6464–6472, 10.1039/C7CE01341H.
- B. Sun, T. Tao, L. Liu, R. Ding and Y. Mao, Electron Transfer Facilitated by π–π Stacking during the Nitrobenzene Recognition Process of an MOF Sensor, J. Phys. Chem. C, 2021, 125(22), 12433–12440, DOI:10.1021/acs.jpcc.1c02942.
- L. Liu, R. Ding, Y. Mao and B. Sun, Theoretical Investigations on the Nitro-Explosive Sensing Process of a MOF Sensor: Roles of Hydrogen Bond and π-π Stacking, Chem. Phys. Lett., 2022, 793, 139393, DOI:10.1016/j.cplett.2022.139393.
- S. Ghosh, R. Lipin, A. Ngoipala, N. Ruser, D. M. Venturi, A. Rana, M. Vandichel and S. Biswas, Hf-Based MOF for Rapid and Selective Sensing of a Nerve Agent Simulant and an Aminophenol: Insights from Experiments and Theory, Inorg. Chem., 2023, 62(36), 14632–14646, DOI:10.1021/acs.inorgchem.3c01777.
- D. Yan, Y. Tang, H. Lin and D. Wang, Tunable Two-Color Luminescence and Host–Guest Energy Transfer of Fluorescent Chromophores Encapsulated in Metal-Organic Frameworks, Sci. Rep., 2014, 4(1), 4337, 10.1039/C3TA14328G.
- Y. Luo, D. Lei, M. Li, Y. Ge, J. Li, B. Zu, J. Yao and X. Dou, Fluorophore Branching Boosted Photo-Induced Energy Transfer in UiO-66 for Ultrasensitive and Instant Hydrazine Sensing, J. Mater. Chem. A, 2024, 12(20), 12088–12097, 10.1039/d4ta01549e.
- K. Leong, M. E. Foster, B. M. Wong, E. D. Spoerke, D. Van Gough, J. C. Deaton and M. D. Allendorf, Energy and Charge Transfer by Donor–Acceptor Pairs Confined in a Metal–Organic Framework: A Spectroscopic and Computational Investigation, J. Mater. Chem. A, 2014, 2(10), 3389–3398, 10.1039/C3TA14328G.
- M. Y. Toriyama, A. M. Ganose, M. Dylla, S. Anand, J. Park, M. K. Brod, J. M. Munro, K. A. Persson, A. Jain and G. J. Snyder, How to Analyse a Density of States, Mater. Today Electron., 2022, 1, 100002, DOI:10.1016/j.mtelec.2022.100002.
- K. Hendrickx, J. J. Joos, A. De Vos, D. Poelman, P. F. Smet, V. Van Speybroeck, P. Van Der Voort and K. Lejaeghere, Exploring Lanthanide Doping in UiO-66: A Combined Experimental and Computational Study of the Electronic Structure, Inorg. Chem., 2018, 57(9), 5463–5474, DOI:10.1021/acs.inorgchem.8b00425.
- F. Yuan, H. X. Ma, C. M. Yuan, C. S. Zhou, H. M. Hu, A. Kumar and M. Muddassir, Syntheses of a Series of Lanthanide Metal-Organic Frameworks for Efficient UV-Light-Driven Dye Degradation: Experiment and Simulation, CrystEngComm, 2021, 23(12), 2404–2413, 10.1039/d0ce01245a.
- X. P. Wu, L. Gagliardi and D. G. Truhlar, Cerium Metal-Organic Framework for Photocatalysis, J. Am. Chem. Soc., 2018, 140(25), 7904–7912, DOI:10.1021/jacs.8b03613.
- S. Yu, G. Jing, S. Li, Z. Li and X. Ju, Tuning the Hydrogen Storage Properties of MOF-650: A Combined DFT and GCMC Simulations Study, Int. J. Hydrogen Energy, 2020, 45(11), 6757–6764, DOI:10.1016/j.ijhydene.2019.12.114.
- L. Zhao, X. Ren, Y. Du, Z. Gao, H. Ma, H. Wang, Y. Li, Q. Wei, H. Ju and D. Wu, Europium-Based Metal–Organic Framework with N–H⋯π Interaction and Intramolecular Energy Transfer Mechanisms for Self-Electrochemiluminescence, Adv. Funct. Mater., 2024, 2410886, 1–7, DOI:10.1002/adfm.202410886.
- J. J. Santoyo-Flores and D. Páez-Hernández, Theoretical Study of 8-Hydroxyquinoline Derivatives as Potential Antennas in Lanthanide Complexes: Photophysical Properties and Elucidation of Energy Transfer Pathways, Int. J. Quantum Chem., 2022, 122(10), e26880, DOI:10.1002/qua.26880.
- C. Celis-Barros, D. Páez-Hernández, M. J. Beltrán-Leiva and R. Arratia-Perez, Ab Initio Calculations of Heavy-Actinide Hexahalide Compounds: Do These Heavy Actinides Behave like Their Isoelectronic Lanthanide Analogues?, Phys. Chem. Chem. Phys., 2018, 20(6), 4038–4049, 10.1039/c7cp06585j.
- Y. Hidalgo-Rosa, K. Mena-Ulecia, M. A. Treto-Suárez, E. Schott, D. Páez-Hernández and X. Zarate, Expanding the Knowledge of the Selective-Sensing Mechanism of Nitro Compounds by Luminescent Terbium Metal-Organic Frameworks through Multiconfigurational Ab Initio Calculations, J. Phys. Chem. A, 2022, 126(39), 7040–7050, DOI:10.1021/ACS.JPCA.2C05468.
- F. Kossoski and P. F. Loos, State-Specific Configuration Interaction for Excited States, J. Chem. Theory Comput., 2023, 19(8), 2258–2269, DOI:10.1021/acs.jctc.3c00057.
- Y. Horiuchi, T. Toyao, M. Saito, K. Mochizuki, M. Iwata, H. Higashimura, M. Anpo and M. Matsuoka, Visible-Light-Promoted Photocatalytic Hydrogen Production by Using an Amino-Functionalized Ti(IV) Metal-Organic Framework, J. Phys. Chem. C, 2012, 116(39), 20848–20853, DOI:10.1021/jp3046005.
- M. A. Nasalevich, C. H. Hendon, J. G. Santaclara, K. Svane, B. Van Der Linden, S. L. Veber, M. V. Fedin, A. J. Houtepen, M. A. Van Der Veen, F. Kapteijn, A. Walsh and J. Gascon, Electronic Origins of Photocatalytic Activity in D0 Metal Organic Frameworks, Sci. Rep., 2016, 6, 1–9, DOI:10.1038/srep23676.
- A. De Vos, K. Hendrickx, P. Van Der Voort, V. Van Speybroeck and K. Lejaeghere, Missing Linkers: An Alternative Pathway to UiO-66 Electronic Structure Engineering, Chem. Mater., 2017, 29(7), 3006–3019, DOI:10.1021/acs.chemmater.6b05444.
- Z. Abbas, S. Dasari, M. J. Beltrán-Leiva, P. Cantero-López, D. Páez-Hernández, R. Arratia-Pérez, R. J. Butcher and A. K. Patra, Luminescent Europium(III) and Terbium(III) Complexes of β-Diketonate and Substituted Terpyridine Ligands: Synthesis, Crystal Structures and Elucidation of Energy Transfer Pathways, New J. Chem., 2019, 43(38), 15139–15152, 10.1039/c9nj02838b.
- E. M. Gomes, J. P. De Oliveira Silva, M. V. Colaço, A. Cuin, D. F. Franco, S. L. Scarpari, D. O. De Souza, M. S. Ferreira, R. O. Freire and L. F. Marques, Two Highly Photoluminescent Eu3+ β-Diketonates Complexes with ε-Caprolactam as Ancillary Ligands: From Synthesis to the First Example as Gunshot Residue Markers, Opt. Mater., 2023, 137, 113527, DOI:10.1016/j.optmat.2023.113527.
- F. J. Steemers, W. Verboom, D. N. Reinhoudt, E. B. van der Tol and J. W. Verhoeven, New Sensitizer-Modified Calix[4]Arenes Enabling Near-UV Excitation of Complexed Luminescent Lanthanide Ions, J. Am. Chem. Soc., 1995, 117(37), 9408–9414, DOI:10.1021/ja00142a004.
- M. A. El-Sayed, The Triplet State: Its Radiative and Nonradiative Properties, Acc. Chem. Res., 1968, 1(1), 8–16, DOI:10.1021/ar50001a002.
- M. A. El-Sayed, Spin-Orbit Coupling and the Radiationless Processes in Nitrogen Heterocyclics, J. Chem. Phys., 1963, 38(12), 2834–2838, DOI:10.1063/1.1733610.
- J. D. L. Dutra, T. D. Bispo and R. O. Freire, LUMPAC Lanthanide Luminescence Software: Efficient and User Friendly, J. Comput. Chem., 2014, 35(10), 772–775, DOI:10.1002/jcc.23542.
- H. Tan, Q. Li, C. Ma, Y. Song, F. Xu, S. Chen and L. Wang, Lanthanide Based Dual-Emission Fluorescent Probe for Detection of Mercury(II) in Milk, Biosens. Bioelectron., 2015, 63, 566–571, DOI:10.1016/j.bios.2014.08.015.
- M. A. Treto-Suárez, Y. Hidalgo-Rosa, E. Schott, X. Zarate and D. Páez-Hernández, Radiative Decay Channel Assessment to Understand the Sensing Mechanism of a Fluorescent Turn-on Al3+ Chemosensor, Int. J. Quantum Chem., 2020, 120(3), 26083, DOI:10.1002/qua.26083.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt03285c |
| This journal is © The Royal Society of Chemistry 2025 |