
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bifunctional dye-based organocatalysts with enhanced activity in the conversion of CO2 into cyclic carbonates†
Jing
Chen
 and
Paolo P.
Pescarmona
*
and
Paolo P.
Pescarmona
*
Chemical Engineering Group, Engineering and Technology Institute Groningen (ENTEG), University of Groningen, Nijenborgh 3, 9747 AG Groningen, The Netherlands. E-mail: p.p.pescarmona@rug.nl
First published on 14th March 2025
Abstract
A series of novel dye-based organocatalysts was designed, synthesised and tested in the cycloaddition of CO2 to styrene oxide yielding styrene carbonate under mild reaction conditions (45 °C, 10 bar CO2). Tuning the acid strength and the geometry of the –OH functional group in the modified dyes allowed the generation of a tailored bifunctional catalyst (RhB-Ethyl-PhOH-I) with enhanced catalytic activity.
Cyclic carbonates are a versatile class of compounds finding application as green solvents for organic synthesis and for electrolytes in Li-batteries,1,2 and as reactants for the synthesis of a variety of products ranging from engineering plastics to speciality chemicals.1 These growing applications have led to a sizeable market, estimated to be around 100–200 ktonne per year.2 Their synthesis through the cycloaddition of CO2 to epoxides is particularly appealing as it is a 100% atom-efficient reaction and utilises a renewable and non-hazardous feedstock as carbon dioxide.1 In this context, the development of low-cost and efficient catalysts for the cycloaddition of CO2 to epoxides is of utmost importance towards the upscaling of the production of cyclic carbonates.3 Ideally, a catalyst should be not only active and selective but also inexpensive, readily available, and based on earth-abundant elements. In this sense, metal-free organocatalysts are preferable over metal-based catalysts that typically require costly multiple synthesis steps for their preparation and may rely on scarce elements.1 Organic halides, such as quaternary ammonium, phosphonium, and imidazolium salts, contain anions (e.g. Cl−, Br−, and I−) that can act as nucleophilic species to catalyse the conversion of CO2 and epoxides into cyclic carbonates (see Scheme S1 for the catalytic mechanism, ESI†). Organic halides with a bulky cation with delocalised positive charge are preferable, as this can reduce the ionic interaction energy, thus making the anion more readily available for acting as a nucleophile.4,5 However, organocatalysts are generally less active than metal-based catalysts due to the lack of metal Lewis acid sites that promote the reaction by activating the epoxides towards nucleophilic attack (Scheme S1, ESI†).5,6 One way to overcome this limitation is to introduce in the catalytic system a hydrogen bond donor (HBD) group that can act as Lewis acid (e.g. –OH and –COOH), either in the form of a separate compound or as a functional group within the same compound.7 The latter option is more attractive as it enables tuning the relative position of nucleophile and HBD, which in turn allows improving the cooperation between the two catalytically active species. So far, numerous HBD-functionalised quaternary onium salts have been studied as bifunctional organocatalysts (Fig. S1, ESI†), achieving the cycloaddition of CO2 to epoxides under mild reaction conditions (T ≤ 70 °C, pCO2 ≤ 40 bar).7 Recently, we reported the synthesis of a novel dye-based bifunctional organocatalyst (RhB-EtOH-I, 1a in Fig. 1A), which achieved the cycloaddition of CO2 to styrene oxide at 60 °C, 10 bar CO2, with nearly double carbonate yield compared to the unfunctionalised RhB-I catalyst.8 Additionally, the RhB-based catalysts could be separated from the cyclic carbonate product either by precipitation with diethyl ether or by nanofiltration, and retained their activity upon reuse.8 Inspired by these promising results, we designed new dye-based bifunctional organocatalysts with enhanced activity by varying the geometry and type of functional group (Fig. 1A). These metal-free homogeneous catalysts were prepared through a one-step, straightforward and affordable protocol involving the reaction of RhB base with an organic halide (Fig. 1A, see the ESI† for experimental details). The obtained organocatalysts were categorised based on the nature of the introduced functional groups (Fig. 1A). Type 1 comprises dyes that were modified with HBD groups containing linkers of varying lengths, with the purpose of tuning the geometry of the HBD group (–OH) relative to the nucleophile (I−). Minimising the distance between the HBD group and the nucleophile is expected to be beneficial for the cooperative action of the two active species (see Fig. 1B).8,9 However, since the positive charge in RhB is delocalised over the xanthene-based core, the iodide is not expected to be localised in a single, specific position, making it difficult to predict a priori the length of the linker leading to the optimum activity. Additionally, the functional groups in the type 1 dyes differ in terms of acid strength. This implies different strengths of the H-bond interaction, which in turn influences the activation of the epoxide (Fig. 1).10 Type 2 consists of organocatalysts without HBD groups. The functional groups in 2a and 2b are analogous to those in 1b and 1c, but without the –OH group acting as HBD. Therefore, these catalysts serve as references for evaluating the efficiency of the HBD-functionalised catalysts. Additionally, the difference in the polarity between the functional groups of types 1 and 2 is anticipated to affect their solubility in the epoxide, thereby impacting their catalytic activity. Among the type 2 catalysts, 2c and 2d were synthesised with the additional purpose of providing grafting sites for immobilisation on solid supports (e.g. by thiol–ene reaction with thiol-functionalised silica11). All the dyes in Fig. 1A were prepared with either chloride (Cl−) or bromide (Br−) as the halide. However, it has been shown that employing iodide (I−) as the halide leads to more active dye-based catalysts (due to the better leaving ability of I−).7,8,12 Therefore, the prepared dyes were converted to their iodide-form via a straightforward ion-exchange step with KI (Fig. 1A). The successful synthesis of the modified dyes was demonstrated by characterisation using NMR spectroscopy (1H NMR, 13C NMR, 1H–13C HSQC, and 1H–13C HMBC) and elemental analysis (C, H, O); see the ESI† for the spectra (Fig. S2–S49) and data. This set of organocatalysts in iodide form was tested for the conversion of CO2 and styrene oxide into styrene carbonate under mild conditions (45 °C, 10 bar CO2, 18 h). Styrene oxide was selected as the test compound because it is a rather challenging substrate for this reaction.13 The reaction conditions were chosen to achieve intermediate epoxide conversion, as this allows highlighting better the differences in catalytic activity. Both the type 1 and type 2 catalysts were able to convert CO2 and styrene oxide into styrene carbonate with complete selectivity towards the desired cyclic carbonate product (Table 1, entries 1–7). Among the type 1 catalysts, RhB-Ethyl-PhOH-I exhibited significantly higher activity than RhB-EtOH-I and RhB-PrOH-I, reaching a styrene carbonate yield of 41%, which corresponds to a 12% and 20% higher yield, respectively, compared to the other two HBD-containing catalysts (Table 1, entries 1–3). The superior activity of the RhB-Ethyl-PhOH-I catalyst is likely due to the higher acid strength of the phenol moiety (pKa,phenol = 9.9) than that of alcohol functional groups (pKa,ethanol = 15.9, pKa,1-propanol = 16.1), enabling the formation of stronger hydrogen bonds with epoxides.10 This result is consistent with the reported effectiveness of phenolic compounds as HBD additives in binary catalytic systems for the cycloaddition of CO2 to epoxides.4,10 On the other hand, the higher catalytic activity of RhB-EtOH-I compared to RhB-PrOH-I, despite the similar acidity of the alcohol groups, may be attributed to the different alkyl linker length,9 suggesting that a shorter linker is beneficial for the simultaneous interaction between the –OH group, the epoxide and the iodide, thereby facilitating the nucleophilic attack of the iodide on the epoxide (see Fig. 1B).9 Compared to the type 1 catalysts, the type 2 catalysts exhibited in general lower activity due to the absence of the HBD group enabling the activation of the epoxide, with styrene carbonate yields around 21–23% (entries 4–7 vs. entries 1 and 3). However, all type 2 catalysts displayed significantly higher activity than the parent RhB-I (Table 1, entry 8). This trend might be related to the incomplete solubility of RhB-I in the reaction mixture at the beginning of the catalytic test (see Table S1, ESI†), whereas the type 2 catalysts display full solubility under the same conditions (with the exception of 2a).4,8
 | ||
Fig. 1 (A) Design strategy and synthesis of modified dye organocatalysts. [a]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) The RhB-EtOH-I catalyst was previously reported by our group.8 (B) Schematic representation of the cooperative action of the nucleophilic and HBD species of the bifunctional dye-based organocatalysts. The RhB-EtOH-I catalyst was previously reported by our group.8 (B) Schematic representation of the cooperative action of the nucleophilic and HBD species of the bifunctional dye-based organocatalysts. | ||
| Entry | Organocatalyst | Cyclic carbonate yielda [%] | Cyclic carbonate selectivitya [%] |
|---|---|---|---|
| Reaction conditions: styrene oxide (20 mmol), organocatalyst (1 mol% relative to the epoxide), o-xylene (1.5 mmol) as internal standard, 45 °C, 10 bar CO2, 18 h.a The yield and selectivity values were determined by 1H NMR using o-xylene as internal standard (see the ESI for a representative spectrum).b 1 mol% of phenol relative to the epoxide was used as HBD.c 2 mol% of phenol relative to the epoxide was used as HBD. | |||
| 18 | RhB-EtOH-I (1a) | 29 | ≥99 |
| 2 | RhB-PrOH-I (1b) | 21 | ≥99 |
| 3 | RhB-Ethyl-PhOH-I (1c) | 41 | ≥99 |
| 4 | RhB-Pr-I (2a) | 21 | ≥99 |
| 5 | RhB-Ethyl-Ph-I (2b) | 23 | ≥99 |
| 6 | RhB-Allyl-I (2c) | 23 | ≥99 |
| 7 | RhB-Butenyl-I (2d) | 21 | ≥99 |
| 88 | RhB-I | 5 | ≥99 |
| 98 | RhB-I/H2O | 7 | 67 |
| 10b | RhB-I/phenol (1:1) |
9 | 84 |
| 11c | RhB-I/phenol (1:2) |
16 | 90 |
To gain more insight into the origin of the enhanced activity exhibited by RhB-Ethyl-PhOH-I, we investigated the effect of combining RhB-I with an HBD, such as H2O or phenol, to generate a binary catalytic system, which was tested under the same reaction conditions (Table 1, entries 9–11). All these RhB-I/HBD binary catalytic systems showed higher activity than RhB-I alone (compare entry 8 with entries 9–11). Increasing the phenol amount to 2 mol% (relative to the epoxide) boosted the styrene carbonate yield to 16% (entry 11). Nonetheless, even the optimum RhB-I/phenol binary catalytic system exhibited markedly lower catalytic activity than RhB-Ethyl-PhOH-I. This indicates that our strategy was successful in designing a bifunctional catalyst in which the proximity of the nucleophile and of the HBD within the same compound (RhB-Ethyl-PhOH-I) leads to their enhanced cooperation compared to a binary system (RhB-I/phenol), in which the same two catalytic species are present on two separate compounds.
Although several of our modified dyes already contain an HBD group, their performance might be further enhanced by combining them with an HBD additive.4,14 We chose water as an inexpensive, abundant, non-hazardous and thus green HBD.4,7,8 When a small amount of H2O (50 mg) was added as a co-catalyst, the activity of the modified dye catalysts (type 1 and type 2) improved, with an increase of up to 13% in styrene carbonate yield (Fig. 2). In line with logical expectations, the benefit of using H2O as HBD was in general more prominent in combination with the non-HBD-functionalised organocatalysts. However, when discussing these results it is necessary to consider that the presence of H2O can also lead to a decrease in the solubility of the dye-based catalysts in the reaction mixture at the beginning of the reaction (Table S1, ESI†), resulting in fewer active sites available for catalysing the reaction in solution, which partially offsets the beneficial effect of H2O as HBD. Among the dye-based catalysts assisted by H2O as HBD, RhB-Ethyl-PhOH-I again achieved the highest activity, yielding 49% styrene carbonate, i.e. 24% and 11% higher yield than the benchmark organocatalysts Bu4NI and PPNI,4 respectively (Fig. 2). Compared to the state-of-art bifunctional organocatalysts (Fig. S1, ESI†),15–28 RhB-Ethyl-PhOH-I exhibited competitive activity (Table S3, ESI†), particularly when considering that these results were obtained under mild reaction conditions (45 °C, 10 bar CO2) and with relatively low catalyst loading. The use of water as HBD can cause a slight decrease in selectivity towards the desired cyclic carbonate product, due to hydrolysis of the epoxide.4 However, the selectivity towards styrene carbonate remained high with the RhB-Ethyl-PhOH catalyst (96%), with only a minor amount of styrene diol (2%) formed as a by-product.4 For the rest of the dye-based catalysts, the styrene carbonate selectivity was slightly lower, but still ≥92% (Fig. 2). Increasing the amount of H2O to 100 mg led to a less notable improvement in the activity of the dye-based catalysts and resulted in a further decrease in the selectivity towards styrene carbonate, due to higher styrene diol yields (5 and 6%).16 The higher fraction of H2O also negatively affected the solubility of some of the dyes in the reaction mixture (Table S1, ESI†). Combining these observations, it was concluded that using 50 mg of H2O as HBD additive is preferable over a larger amount.
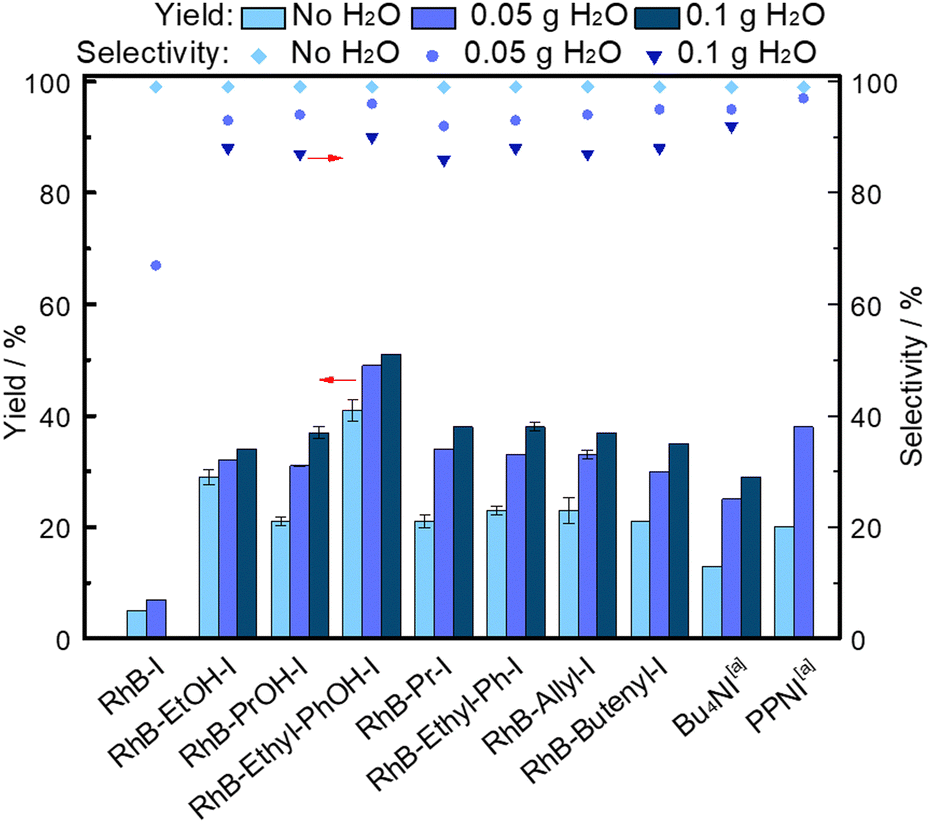 | ||
| Fig. 2 Effect of using H2O as additional HBD on the activity of modified dyes as organocatalysts for the conversion of CO2 and styrene oxide into styrene carbonate. Reaction conditions: styrene oxide (20 mmol), organocatalyst (1 mol% relative to the epoxide), o-xylene (1.5 mmol) as internal standard, 45 °C, 10 bar CO2, 18 h. aData taken from ref. 4. | ||
The versatility of RhB-Ethyl-PhOH-I was investigated with a range of substrates, comprising external and internal epoxides bearing groups with different electronic and steric effects (Table 2). For comparison, another catalyst of type 1 (RhB-EtOH-I) and one of type 2 (RhB-Ethyl-Ph-I) were tested, to determine whether the observed catalytic trends are correlated with the nature of the epoxide. Due to differences in their functional groups and the nature of the epoxides, these catalysts displayed different solubility in the reaction mixtures. In general, RhB-Ethyl-Ph-I and RhB-Ethyl-PhOH-I exhibited better solubility than RhB-EtOH-I (Table S2, ESI†). Following a previously reported approach, a small amount of propylene carbonate was added to solubilise the dyes that showed incomplete solubility.4,8 Propylene carbonate was chosen as it is a non-hazardous, polar aprotic solvent.4,8 Additionally, it does not need to be separated from the reaction mixture in the case of CO2 cycloaddition to propylene oxide, being the cyclic carbonate product.4 Most CO2 cycloaddition reactions were conducted under mild conditions (45 °C, 10 bar CO2, 18 h, entries 1–12), except for the more challenging cyclohexene oxide and limonene oxide substrates, for which harsher conditions were employed (120 °C, 30 bar CO2, 18 h, entries 13–18). Among the three dye-based catalysts, RhB-Ethyl-PhOH-I consistently displayed the highest activity, with good to excellent cyclic carbonate yields (46 to 84%, Table 2). However, it showed poor activity towards the CO2 cycloaddition to the refractory limonene oxide (entry 18).29 The highest product yield was obtained for propylene carbonate (entry 3). This is attributed to the small size of this epoxide,13 which allows easier access to the active sites of the catalyst. Electron-withdrawing groups, such as Cl in epichlorohydrin, promote the nucleophilic attack on the epoxide, resulting in high cyclic carbonate yields, which in this case were very similar for the three catalysts (entries 4–6). Notably, the catalysts were also effective in the conversion of an internal, challenging substrate such as cyclohexene oxide,13,29 though this required harsher reaction conditions (entries 13–15). Apart from the above-mentioned case of styrene oxide, the undesired hydrolysis of the epoxide into the corresponding diol was negligible, leading to nearly complete cyclic carbonate selectivity.
| Entry | Organocatalyst | Epoxide | Cyclic carbonate product | Solvent [mL] | Yielda [%] | Selectivitya [%] |
|---|---|---|---|---|---|---|
| Reaction conditions: epoxide (20 mmol), organocatalyst (1 mol% relative to the epoxide), o-xylene (1.5 mmol) as the internal standard, 50 mg H2O, 45 °C, 10 bar CO2, 18 h.a The yield and selectivity values were determined by 1H NMR using o-xylene as internal standard (see Fig. S50–S56, ESI for representative spectra).b Taken from ref. 8.c 120 °C, 30 bar CO2, 18 h; in entries 13–15, cis-cyclohexene oxide was used as the substrate and only the cis-form of cyclohexene carbonate was obtained (see Fig. S54, S55 and S57–S61, ESI for the NMR spectra). n.a. = not applicable. | ||||||
| 1 | RhB-Ethyl-Ph-I |
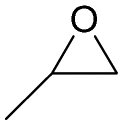
|

|
0 | 67 | ≥99 |
| 2 | RhB-EtOH-I | 0 | 65 | ≥99 | ||
| 3 | RhB-Ethyl-PhOH-I | 0 | 84 | ≥99 | ||
| 4 | RhB-Ethyl-Ph-I |
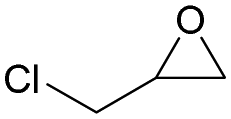
|
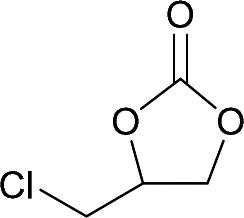
|
0 | 54 | ≥99 |
| 5 | RhB-EtOH-I | 0 | 51 | ≥99 | ||
| 6 | RhB-Ethyl-PhOH-I | 0 | 53 | ≥99 | ||
| 7 | RhB-Ethyl-Ph-I |

|

|
0 | 33 | 93 |
| 8 | RhB-EtOH-Ib | 0 | 32 | 93 | ||
| 9 | RhB-Ethyl-PhOH-I | 0 | 49 | 96 | ||
| 10 | RhB-Ethyl-Ph-I |

|
|
0.5 | 20 | ≥99 |
| 11 | RhB-EtOH-I | 0.5 | 12 | ≥99 | ||
| 12 | RhB-Ethyl-PhOH-I | 0.5 | 46 | ≥99 | ||
| 13 | RhB-Ethyl-Ph-Ic |

|

|
0 | 54 | ≥99 |
| 14 | RhB-EtOH-Ib,c | 0.5 | 50 | ≥99 | ||
| 15 | RhB-Ethyl-PhOH-Ic | 0 | 62 | ≥99 | ||
| 16 | RhB-Ethyl-Ph-Ic |
|

|
0.5 | <1 | n.a. |
| 17 | RhB-EtOH-Ib,c | 0.5 | <1 | n.a | ||
| 18 | RhB-Ethyl-PhOH-Ic | 0.5 | <1 | n.a. | ||
The RhB-Ethyl-PhOH-I catalyst can be recovered by precipitation using diethyl ether as an antisolvent. The 1H and 13C NMR spectra of the recovered RhB-Ethyl-PhOH-I (Fig. S62 and S63, ESI†) are analogous to those of the fresh catalyst and the activity was fully retained upon reuse (Fig. S64, ESI†). Finally, we demonstrated that nearly full conversion of propylene oxide with complete selectivity to propylene carbonate (98% yield, >99% selectivity, Fig. S65, ESI†), can be achieved with the RhB-Ethyl-PhOH-I catalyst by increasing the reaction temperature to 60 °C (with the other conditions as in Entry 3, Table 2). The obtained propylene carbonate can be isolated to achieve the product with high purity (see the ESI† for the procedure and Fig. S66–S68 for the NMR and GC-MS data).
In conclusion, we demonstrated that careful tuning of the nature and position of the HBD group in modified RhB dyes allows designing a bifunctional metal-free homogeneous catalyst with enhanced activity and high selectivity in the reaction of CO2 with a broad scope of epoxides to yield cyclic carbonates under mild reaction conditions.
We acknowledge the China Scholarship Council for the PhD grant of Jing Chen. We are grateful to Marcel de Vries, Rick van der Reijd, Henk van de Bovenkamp, Léon Rohrbach, Gert-Jan Boer and Hans van der Velde for technical and analytical support.
Data availability
Data for this article are available either in the ESI† or, in the case of the raw data, at Figshare (https://doi.org/10.6084/m9.figshare.28533206).Conflicts of interest
There are no conflicts to declare.Notes and references
- A. J. Kamphuis, F. Picchioni and P. P. Pescarmona, Green Chem., 2019, 21, 406–448 RSC.
- P. P. Pescarmona, Curr. Opin. Green Sustainable Chem., 2021, 29, 100457 CrossRef CAS.
- M. Aresta, A. Dibenedetto and E. Quaranta, J. Catal., 2016, 343, 2–45 CAS.
- Y. A. Alassmy and P. P. Pescarmona, ChemSusChem, 2019, 12, 3856–3863 CrossRef CAS PubMed.
- R. R. Shaikh, S. Pornpraprom and V. D’Elia, ACS Catal., 2018, 8, 419–450 CrossRef CAS.
- Q.-W. Song, Z.-H. Zhou and L.-N. He, Green Chem., 2017, 19, 3707–3728 RSC.
- M. Liu, X. Wang, Y. Jiang, J. Sun and M. Arai, Catal. Rev., 2019, 61, 214–269 CrossRef CAS.
- J. Chen, G. Chiarioni, G.-J. W. Euverink and P. P. Pescarmona, Green Chem., 2023, 25, 9744–9759 RSC.
- H. Büttner, K. Lau, A. Spannenberg and T. Werner, ChemCatChem, 2015, 7, 459–467 CrossRef.
- P. Yingcharoen, C. Kongtes, S. Arayachukiat, K. Suvarnapunya, S. V. C. Vummaleti, S. Wannakao, L. Cavallo, A. Poater and V. D’Elia, Adv. Synth. Catal., 2019, 361, 366–373 CAS.
- P. Agrigento, S. M. Al-Amsyar, B. Sorée, M. Taherimehr, M. Gruttadauria, C. Aprile and P. P. Pescarmona, Catal. Sci. Technol., 2014, 4, 1598–1607 RSC.
- M. Liu, X. Li, L. Liang and J. Sun, J. CO2 Util., 2016, 16, 384–390 CrossRef CAS.
- P. P. Pescarmona and M. Taherimehr, Catal. Sci. Technol., 2012, 2, 2169–2187 RSC.
- J. Sun, W. Cheng, Z. Yang, J. Wang, T. Xu, J. Xin and S. Zhang, Green Chem., 2014, 16, 3071–3078 RSC.
- R. Yan, K. Chen, Z. Li, Y. Qu, L. Gao, H. Tong, Y. Li, J. Li, Y. Hu and K. Guo, ChemSusChem, 2021, 14, 738–744 CrossRef CAS PubMed.
- C. Li, F. Liu, T. Zhao, J. Gu, P. Chen and T. Chen, Mol. Catal., 2021, 511, 111756 CrossRef CAS.
- D. Wei-Li, J. Bi, L. Sheng-Lian, L. Xu-Biao, T. Xin-Man and A. Chak-Tong, J. Mol. Catal. A: Chem., 2013, 378, 326–332 CrossRef.
- W. Natongchai, D. Crespy and V. D'Elia, Chem. Commun., 2025, 61, 419–440 CAS.
- S. Denizaltı, RSC Adv., 2015, 5, 45454–45458 Search PubMed.
- T. Wang, X. Zhu, L. Mao, Y. Liu, T. Ren, L. Wang and J. Zhang, J. Mol. Liq., 2019, 296, 111936 CAS.
- Y. Li, B. Dominelli, R. M. Reich, B. Liu and F. E. Kühn, Catal. Commun., 2019, 124, 118–122 CrossRef CAS.
- W. D. A. Bezerra, J. L. S. Milani, C. H. D. J. Franco, F. T. Martins, Â. de Fátima, Á. F. A. da Mata and R. P. das Chagas, Mol. Catal., 2022, 530, 112632 CAS.
- A. Zhang, C. Chen, C. Zuo, X. Xu, T. Cai, X. Li, Y. Yuan, H. Yang and G. Meng, Green Chem., 2022, 24, 7194–7207 CAS.
- J. Peng, S. Wang, H.-J. Yang, B. Ban, Z. Wei, L. Wang and B. Lei, Fuel, 2018, 224, 481–488 CrossRef CAS.
- W. Cheng, B. Xiao, J. Sun, K. Dong, P. Zhang, S. Zhang and F. T. T. Ng, Tetrahedron Lett., 2015, 56, 1416–1419 CrossRef CAS.
- L. Gao, Y. Zhou, Z. Li, J. He, Y. Qu, X. Zou, B. Liu, C. Ma, J. Sun and K. Guo, J. CO2 Util., 2022, 65, 102196 CrossRef CAS.
- A. Rostami, M. Mahmoodabadi, A. Hossein Ebrahimi, H. Khosravi and A. Al-Harrasi, ChemSusChem, 2018, 11, 4262–4268 CrossRef CAS PubMed.
- Y. Hu, Z. Wei, A. Frey, C. Kubis, C. Y. Ren, A. Spannenberg, H. Jiao and T. Werner, ChemSusChem, 2021, 14, 363–372 CrossRef CAS PubMed.
- A. J. Kamphuis, M. Tran, F. Picchioni and P. P. Pescarmona, Green Chem. Eng., 2022, 3, 171–179 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Scheme with the reaction mechanism, solubility tests, and NMR spectra. See DOI: https://doi.org/10.1039/d4cc06799a |
| This journal is © The Royal Society of Chemistry 2025 |