
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThioether–NHC bidentate manganese complexes as efficient phosphine-free catalysts for hydrogenation at room temperature†
Mariia
Hruzd
ab,
Sabrina L.
Kleynemeyer
 c,
Christophe
Michon
a,
Stéphanie
Bastin
c,
Eric
Pollet
bd,
Vincent
Ritleng
*a and
Jean-Baptiste
Sortais
*bc
c,
Christophe
Michon
a,
Stéphanie
Bastin
c,
Eric
Pollet
bd,
Vincent
Ritleng
*a and
Jean-Baptiste
Sortais
*bc
aUniversité de Strasbourg, Ecole européenne de Chimie, Polymères et Matériaux, CNRS, LIMA, UMR 7042, 25 rue Becquerel, Strasbourg, France. E-mail: vritleng@unistra.fr
bUniversity of Strasbourg Institute for Advanced Study (USIAS), Strasbourg, France. E-mail: jean-baptiste.sortais@lcc-toulouse.fr
cLCC-CNRS, Université de Toulouse, CNRS, UPS, Toulouse, France
dUniversité de Strasbourg, Ecole européenne de Chimie, Polymères et Matériaux, CNRS, ICPEES, UMR 7515, 25 rue Becquerel, Strasbourg, France
First published on 14th January 2025
Abstract
A series of four original phosphine-free thioether–NHC manganese complexes have been synthesised and fully characterized. These complexes have been applied as efficient catalysts for the hydrogenation of alkenes and ketones at room temperature, with low catalyst loadings (TON up to 900).
Hydrogenation or hydrogen transfer reactions using manganese complexes as pre-catalysts have developed significantly in recent years.1 Among the advantages of these systems, the abundant and non-critical nature of manganese is an important criterion that explains the enthusiasm it has aroused in recent years for its use in sustainable catalytic systems, as is the ease with which pre-catalysts can be synthesised from the precursor Mn(CO)5Br and the stability of the resulting catalysts. These systems have mainly been investigated for the reduction of polar functional groups (ketones, imines, esters, aldehydes, nitriles, etc.), with tridentate ligands bearing either an acidic NH function2 or a central N-heterocycle being involved in the cooperative metal–ligand activation of the reductant (dihydrogen or iPrOH-type hydrogen donor),3,4 in line with the polar nature of the functional groups.
In contrast, apolar substrates, such as alkenes5–7 and alkynes,8,9 have been much less studied in homogeneous catalysis with manganese. In 2018, Jacobi von Wangelin used [Mn(N(SiMe3)2)2] in the presence of Dibal-H as a reductant to generate an Mn6 nanocluster that proved efficient for the hydrogenation of alkenes under mild conditions (Chart 1).10,11 A year later, the combination of the same complex with phenylacetylene also proved to be active for the hydrogenation of a few alkenes under relatively mild conditions, most probably through the generation of Mn nanoparticles.11 The same year, Kirchner described the first well-defined Mn complex for alkene hydrogenation, an Mn-alkyl complex bearing a bis-dialkylphosphine ligand.12 This complex, obtained following a reduction step with sodium, enables alkenes to be reduced in 24 h with a 2 mol% catalytic loading under 50 bar H2 at 25–60 °C. In 2020, Khusnutdinova reported that bidentate pyridine–phosphine ligands can also promote this reaction, but under harsher conditions.13 More recently, Beller's group illustrated the possibility of introducing bis-NHC as a phosphine-free ligand, which was a breakthrough, although the reaction temperature remained rather high (80–100 °C).14 Finally, this year, Kirchner showed that catalytic loadings could be reduced to 0.25 mol% with tridentate P–NHC–P ligands, provided that the reaction temperature was held at 100 °C.15
 | ||
| Chart 1 Manganese catalysts for hydrogenation of alkenes. | ||
In this article, we demonstrate that the use of NHC–thioether ligands enables the hydrogenation of alkenes at room temperature with catalytic loadings as low as 0.2 mol%. Having recently studied the catalytic activity of manganese complexes with highly coordinating bidentate NHC–phosphine16–18 and bis-NHC19 ligands for the hydrogenation of ketones and esters, as well as that of nickel complexes with hemilabile NHC–thioether ligands for the hydrosilylation of aldehydes,20 we turned our attention to thioether–NHC ligands for the hydrogenation of alkenes with manganese. Our working hypothesis was that, given the potentially hemilabile character of sulphur,20–27 it would be easier to generate a free coordination site, without loss of a CO ligand, on an active species of the [L2Mn(CO)3H] type14,18 and that it should therefore be possible to catalyse hydrogenation reactions under milder conditions than with P–NHC–P or bis-NHC Mn complexes, and target the reduction of alkenes.
The S-functionalized carbene precursors L1.HBr–L4.HBr were readily synthesised in two steps from N-substituted imidazoles (Fig. 1).20,28–32 Next, the manganese complexes 1–4 were prepared in a two-step, one-pot procedure via pre-coordination of the sulphur moiety followed by deprotonation of the imidazolium group with tBuOK to allow carbene formation and coordination. The four complexes 1–4 were obtained in good yields (66–88%) and fully characterised by solution and solid-state IR and NMR spectroscopy, HRMS and elemental analysis. In the 1H NMR spectra, the protons of the ethylene (1–3) and propylene (4) bridges are all diastereotopic, confirming the chelation of the ligand on the metal centre. In the IR spectra, three CO vibration bands are observed for all four complexes, which is characteristic of the facial coordination mode of the carbonyl ligands. The vibration frequencies (for example, 2014, 1932 and 1909 cm−1 for 1 in ATR) indicate a lower electron-donation of the thioether moieties, compared to phosphorus analogues such as [Mes-Im-(CH2)2-PPh2)Mn(CO)3Br] (2005, 1925, 1907 cm−1).18
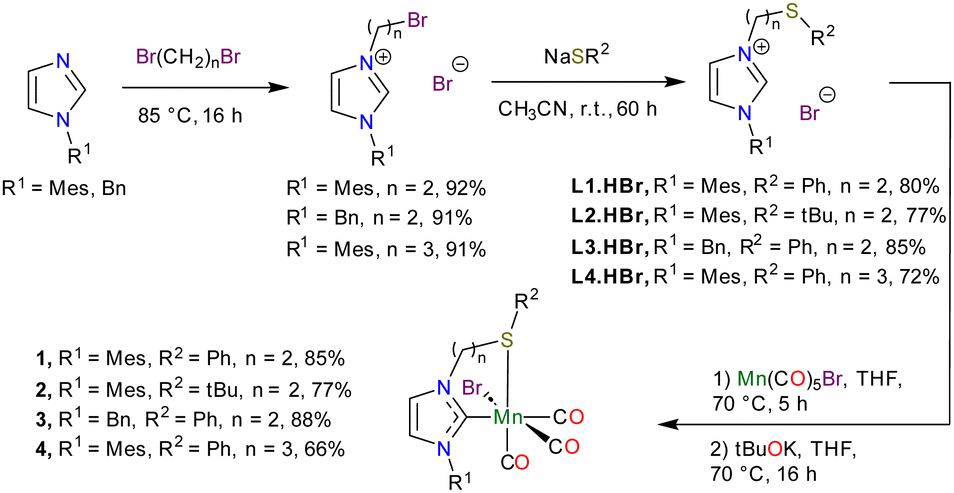 | ||
| Fig. 1 Synthesis of ligands and manganese complexes 1–4. | ||
The molecular structures of 1–4 with the facial arrangement of the carbonyl ligands and the formation of 6- (1–3) and 7-membered (4) CNHC,S-metallacycles were confirmed by single-crystal X-ray diffraction studies (Fig. 2). No significant variations were observed between all four complexes for the Mn–CNHC, Mn–S, and Mn–Br bond distances (Table S1, ESI†). In contrast, noticeable variations were observed for the CNHC–Mn–S (87.84(14) in 3vs. 91.22(13) in 2), CNHC–Mn–Br (86.16(12) in 2vs. 90.04(10) in 1), and Br–Mn–S (85.00(4) in 3vs. 90.44(11) in 4) bond angles (Table S1, ESI†), but no obvious correlation can be drawn between these variations and the catalytic activities of 1–4 (vide infra). The main difference between complexes 1 and 4, that only vary by the size of their metallacycle and give the highest catalytic activities (vide infra), is a greater distortion between the plane of the imidazolylidene ring and the Mn–S bond in 4, the N1CNHC–MnS twist angle being +29.4(3)° and −37.0(1)°, in 1 and 4, respectively, while the other structural parameters are similar (Table S1, ESI†).
 | ||
| Fig. 2 Perspective view of the single-crystal X-ray structure of complexes 1–4 with thermal ellipsoids drawn at the 30% probability level. | ||
The hydrogenation of styrene a1 was chosen as the model reaction to optimise the reaction conditions. Based on the conditions developed for ester hydrogenations,18 NaHBEt3 was first used to activate the pre-catalysts in 2-MeTHF. At 30 °C, in 18 h, under 50 bar of H2, with a catalyst loading of only 0.2 mol%, styrene a1 was reduced to ethylbenzene b1 with all complexes 1–4, without the formation of by-products (Table 1). The catalysts 1–3 incorporating an ethylene bridge led to yields of 71, 22 and 56%, respectively (entries 1–3), evidencing that the presence of the N-mesityl and S–Ph substituents led to a higher catalytic activity than that of a N-benzyl or S–tBu substituent. Under the same conditions, lengthening the carbon chain in complex 4 enabled full conversion of the alkene (entry 4). The latter pre-catalyst was therefore chosen for further optimisation. By lowering the catalytic loading to 0.1 mol%, good catalytic activity was maintained (90%, TON 900), but it fell to 38% when the loading was halved again (entries 5 and 6). The nature of the cation Na, K, and Li of the hydride donor was investigated, and found to have a moderate but noteworthy impact on catalysis, with yields of 90, 71 and 77%, respectively (entries 5, 7, and 8). On the other hand, the use of strong bases, such as KHMDS and tBuOK, did not promote the hydrogenation of styrene (entries 9 and 10). Of all the solvents tested, 2-MeTHF was by far the best for this transformation (entries 5, 11–13). Finally, control experiments confirmed that the presence of both the catalyst and the hydride, as well as a high pressure of H2, are all necessary to achieve good yields (entries 14–16).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst (mol%) | Additive | Solvent | Yield (%) in b1a |
| Typical reaction conditions: to a solution of 1–4 (0.05–0.2 mol%) in solvent (4 mL) were added the additive (1 mol%) and styrene (5 mmol), in this order. The reaction mixture was transferred into an autoclave and stirred for 18 h at 30 °C under H2 (50 bar).a Yield determined by GC using n-dodecane as an internal standard.b Styrene (10 mmol).c H2 (10 bar). | ||||
| 1 | 1 (0.2) | NaHBEt3 | 2-MeTHF | 71 |
| 2 | 2 (0.2) | NaHBEt3 | 2-MeTHF | 22 |
| 3 | 3 (0.2) | NaHBEt3 | 2-MeTHF | 36 |
| 4 | 4 (0.2) | NaHBEt3 | 2-MeTHF | 99 |
| 5 | 4 (0.1) | NaHBEt3 | 2-MeTHF | 90 |
| 6b | 4 (0.05) | NaHBEt3 | 2-MeTHF | 38 |
| 7 | 4 (0.1) | KHBEt3 | 2-MeTHF | 71 |
| 8 | 4 (0.1) | LiHBEt3 | 2-MeTHF | 77 |
| 9 | 4 (0.1) | KHMDS | 2-MeTHF | 2 |
| 10 | 4 (0.1) | tBuOK | 2-MeTHF | 2 |
| 11 | 4 (0.1) | NaHBEt3 | THF | 60 |
| 12 | 4 (0.1) | NaHBEt3 | Dioxane | 35 |
| 13 | 4 (0.1) | NaHBEt3 | Toluene | 11 |
| 14c | 4 (0.1) | NaHBEt3 | 2-MeTHF | 26 |
| 15 | — | NaHBEt3 | 2-MeTHF | 0 |
| 16 | 4 (0.1) | — | 2-MeTHF | 0 |

The scope of this transformation was then studied with a series of vinyl arenes, aliphatic alkenes, and cyclic alkenes (Fig. 3). Styrene (a1) and styrene derivatives with either electron-donor (p-methylstyrene, a2) or electron-acceptor groups (o-chlorostyrene, a5) or 1,1′-disubtituted (α-methyl styrene, a9) were fully converted within 18 h at 30 °C with optimised catalyst loadings ranging between 0.2 and 0.6 mol% and a NaHBEt3 loading of 1 mol%. The catalyst loading was then set by default at 1 mol% and that of NaHBEt3 at 2 mol% for the rest of the study. Under these conditions, the catalytic system was found tolerant to both electron-donating and electron-withdrawing groups as a1–a8 were all fully reduced in 18 h at 30 °C under 50 bar H2. More sterically crowded 1,1′- (a9 and a10) and 1,2-disubstituted (a11 and a12) styrenes were also reduced using this system. However, the limits of the system were reached with tri-substituted olefins such as a13 or a19, which gave little to no conversion, respectively. The hydrogenation of the aliphatic terminal olefins a14–a16 proceeded well without isomerisation of the terminal double bond33 and showed that the catalytic system is tolerant to halogens and esters, which remained intact. The reduction of conjugated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds in ethyl cinnamate (a17) and 3-phenylacrylonitrile (a18) was more difficult as the loading had to be increased to 5 mol%, but the reduction of the CC bond was selective, as no alcohol or amine was detected. Finally, diphenylacetylene (a20) was chosen as a model substrate for alkynes. Under the standard conditions (1 mol% 4, 2 mol% NaHBEt3, 50 bar H2, 30 °C, 18 h), the triple bond was reduced to 1,2-diphenylethane (b12) in 84% yield, presumably via the transient formation of trans-stilbene (a12), which was obtained in 16% yield at the end of the reaction.
C bonds in ethyl cinnamate (a17) and 3-phenylacrylonitrile (a18) was more difficult as the loading had to be increased to 5 mol%, but the reduction of the CC bond was selective, as no alcohol or amine was detected. Finally, diphenylacetylene (a20) was chosen as a model substrate for alkynes. Under the standard conditions (1 mol% 4, 2 mol% NaHBEt3, 50 bar H2, 30 °C, 18 h), the triple bond was reduced to 1,2-diphenylethane (b12) in 84% yield, presumably via the transient formation of trans-stilbene (a12), which was obtained in 16% yield at the end of the reaction.
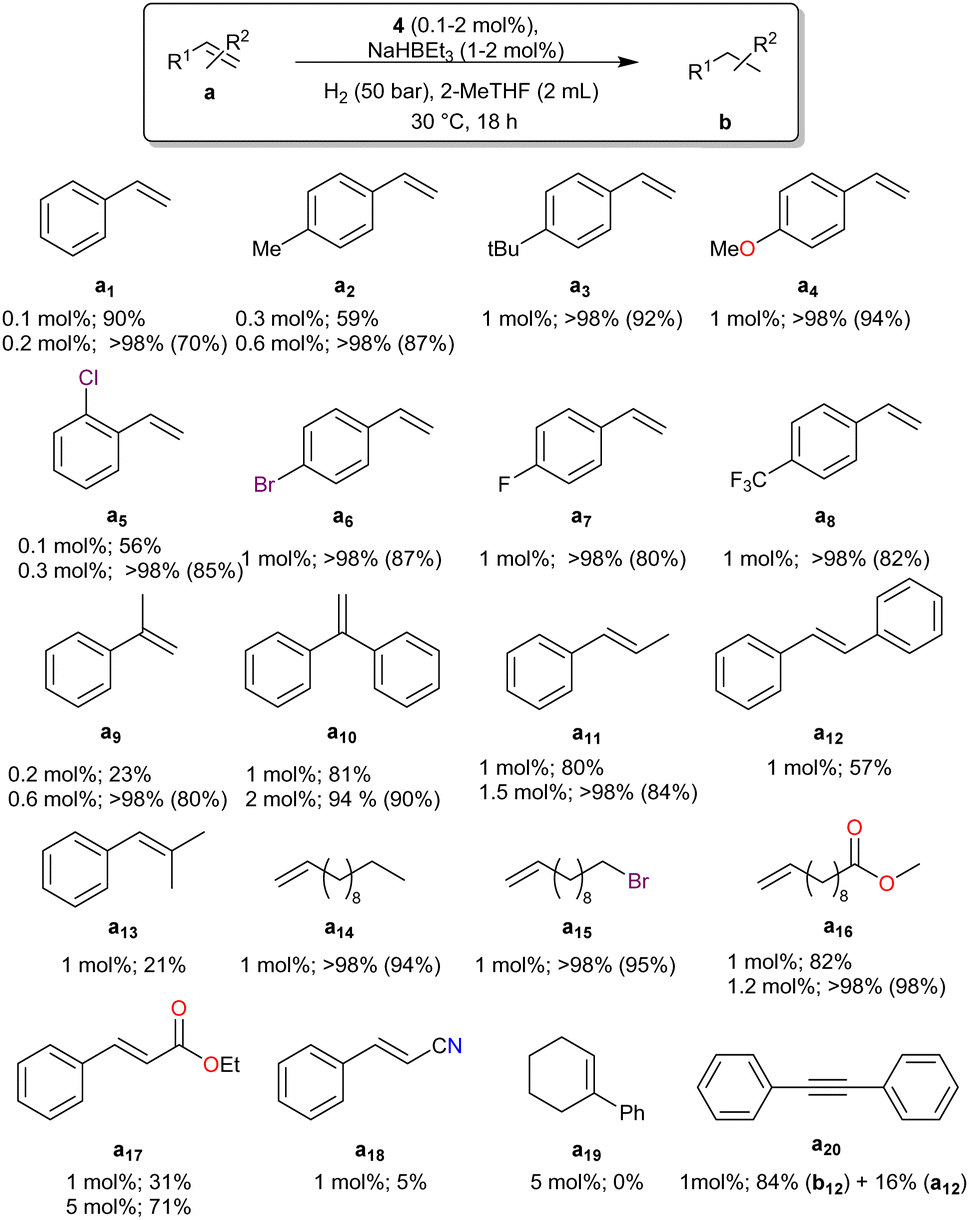 | ||
| Fig. 3 Scope of the hydrogenation of alkenes with the pre-catalyst 4. Conversion determined by GC and NMR (isolated yield). For exact conditions, see ESI.† | ||
Given the performance of this thioether–NHC based manganese catalyst for the hydrogenation of alkenes, we next studied its behaviour towards the reduction of ketones. Very few examples of hydrogenation of polar functional groups with Mn-based catalysts that are, a priori, non-cooperative have indeed been reported to date, the only examples being complexes bearing diphosphine,34–38 phosphine–NHC,16,17,19 and bis-NHC14,18 ligands. To that end, the reaction parameters were re-optimised for the reduction of acetophenone (see Table S2, ESI†). The catalyst loading had to be increased to 3 mol% and that of NaHBEt3 to 7 mol% to reach full conversion, but the hydrogenation still took place in 18 h at 30 °C under 50 bar H2.
With these conditions in hands, the limits of the system were then evaluated (Fig. 4). Increasing the steric hindrance on the alkyl side of acetophenone (c1 to c4) led to a moderate conversion of isobutyrophenone (c3), but the reductions of propiophenone (c2) and cyclopropylphenylketone (c4) both proceeded well, and no cyclopropyl ring opening was observed in the latter case. Similarly, 2′-methylacetophenone (c5) was fully reduced in 18 h while 2′,4′,6′-trimethylacetophenone (c6) was only partially converted to the corresponding alcohol (80%). As for alkenes, the catalytic system was found tolerant to electron-donating and withdrawing groups (c7–c11). It is worth noting that 4-(methylthio)acetophenone (c8) did not poison the catalyst and allowed the formation of the corresponding alcohol in 85% isolated yield. However, 4-cyano-acetophenone (c12) only gave d12 in 45% yield (at 60 °C). Finally, whereas 4-phenylpropan-2-one (c13), a representative example of aliphatic ketones, was fully converted to the corresponding alcohol d13, the reduction of the conjugated 4-phenylbutenone (c14) led to a mixture of d13 (33%), resulting from the full reduction of the enone, and 4-phenylprop-3-en-2-ol (d14, 67%), resulting from the sole reduction of the CO bond.
 | ||
| Fig. 4 Scope of the hydrogenation of ketones with the pre-catalyst 4. Conversion determined by GC and NMR (isolated yield).* = 4 (5 mol%), 60 °C; for exact conditions, see ESI.† | ||
In summary, we have developed a new series of phosphine-free manganese catalysts for the hydrogenation of alkenes and ketones, based on thioether–NHC ligands, able to work at room temperature with low catalyst loadings (TON up to 900 for styrene). The synthesis of ligands is straightforward in two steps and the complexes, which can be handled in air, were obtained in high yields. The scope of substrates showed a good tolerance toward several functional groups and moderate steric hindrance. The full rationalization of the operating mechanism is still under investigation and will be reported in due time.
This work was supported by the University of Strasbourg Institute for Advanced Study (USIAS) for Fellowships, within the French national program “Investment for the future” (IdEx-Unistra) (E.P., J.-B.S. as well as M. H. for postdoc).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- J.-B. Sortais, Manganese Catalysis in Organic Synthesis, Wiley-VCH GmbH, Weinheim, Germany, 2021 Search PubMed.
- S. Elangovan, C. Topf, S. Fischer, H. Jiao, A. Spannenberg, W. Baumann, R. Ludwig, K. Junge and M. Beller, J. Am. Chem. Soc., 2016, 138, 8809–8814 CrossRef CAS PubMed.
- F. Kallmeier, T. Irrgang, T. Dietel and R. Kempe, Angew. Chem., Int. Ed., 2016, 55, 11806–11809 CrossRef CAS PubMed.
- A. Bruneau-Voisine, D. Wang, T. Roisnel, C. Darcel and J.-B. Sortais, Catal. Commun., 2017, 92, 1–4 CrossRef CAS.
- P. L. Bogdan, P. J. Sullivan, T. A. Donovan and J. D. Atwood, J. Organomet. Chem., 1984, 269, c51–c54 CrossRef CAS.
- T. A. Weil, S. Metlin and I. Wender, J. Organomet. Chem., 1973, 49, 227–232 CrossRef CAS.
- R. H. Fish, A. D. Thormodsen and G. A. Cremer, J. Am. Chem. Soc., 1982, 104, 5234–5237 CrossRef CAS.
- R. A. Farrar-Tobar, S. Weber, Z. Csendes, A. Ammaturo, S. Fleissner, H. Hoffmann, L. F. Veiros and K. Kirchner, ACS Catal., 2022, 12, 2253–2260 CrossRef CAS PubMed.
- H. Schratzberger, B. Stöger, L. F. Veiros and K. Kirchner, ACS Catal., 2023, 13, 14012–14022 CrossRef CAS PubMed.
- U. Chakraborty, E. Reyes-Rodriguez, S. Demeshko, F. Meyer and A. Jacobi von Wangelin, Angew. Chem., Int. Ed., 2018, 57, 4970–4975 CrossRef CAS PubMed.
- U. Chakraborty, S. Demeshko, F. Meyer and A. Jacobi von Wangelin, Angew. Chem., Int. Ed., 2019, 58, 3466–3470 CrossRef CAS PubMed.
- S. Weber, B. Stöger, L. F. Veiros and K. Kirchner, ACS Catal., 2019, 9, 9715–9720 CrossRef CAS.
- S. M. W. Rahaman, D. K. Pandey, O. Rivada-Wheelaghan, A. Dubey, R. R. Fayzullin and J. R. Khusnutdinova, ChemCatChem, 2020, 12, 5912–5918 CrossRef CAS.
- N. F. Both, A. Spannenberg, H. Jiao, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2023, 62, e202307987 CrossRef CAS PubMed.
- D. P. Zobernig, M. Luxner, B. Stöger, L. F. Veiros and K. Kirchner, Chem. – Eur. J., 2024, 30, e202302455 CrossRef CAS PubMed.
- R. Buhaibeh, O. A. Filippov, A. Bruneau-Voisine, J. Willot, C. Duhayon, D. A. Valyaev, N. Lugan, Y. Canac and J.-B. Sortais, Angew. Chem., Int. Ed., 2019, 58, 6727–6731 CrossRef CAS PubMed.
- E. S. Gulyaeva, R. Buhaibeh, M. Boundor, K. Azouzi, J. Willot, S. Bastin, C. Duhayon, N. Lugan, O. A. Filippov, J.-B. Sortais, D. A. Valyaev and Y. Canac, Chem. – Eur. J., 2024, 30, e202304201 CrossRef CAS PubMed.
- K. Azouzi, R. Pointis, R. Buhaibeh, P. H. Fernández, L. Pedussaut, M. Boundor, A. Bonfiglio, A. Bruneau-Voisine, D. Wei, T. Roisnel, C. Duhayon, M. Á. Casado, D. A. Valyaev, Y. Canac, S. Bastin, C. Raynaud and J.-B. Sortais, J. Catal., 2024, 430, 115334 CrossRef CAS.
- K. Azouzi, L. Pedussaut, R. Pointis, A. Bonfiglio, R. Kumari Riddhi, C. Duhayon, S. Bastin and J.-B. Sortais, Organometallics, 2023, 42, 1832–1838 CrossRef CAS.
- F. Ulm, A. I. Poblador-Bahamonde, S. Choppin, S. Bellemin-Laponnaz, M. J. Chetcuti, T. Achard and V. Ritleng, Dalton Trans., 2018, 47, 17134–17145 RSC.
- A. Jana, K. Das, A. Kundu, P. R. Thorve, D. Adhikari and B. Maji, ACS Catal., 2020, 10, 2615–2626 CrossRef CAS.
- K. Das, K. Sarkar and B. Maji, ACS Catal., 2021, 11, 7060–7069 CrossRef CAS.
- K. Sarkar, K. Das, A. Kundu, D. Adhikari and B. Maji, ACS Catal., 2021, 11, 2786–2794 CrossRef CAS.
- A. Jana, C. B. Reddy and B. Maji, ACS Catal., 2018, 8, 9226–9231 CrossRef CAS.
- J. Grover, S. Maji, C. Teja, S. A. Al Thabaiti, M. M. M. Mostafa, G. K. Lahiri and D. Maiti, ACS Org. Inorg. Au, 2023, 3, 299–304 CrossRef CAS PubMed.
- N. F. Both, J. Fessler, A. Vicenzi, K. Andres, A. Spannenberg, K. Junge and M. Beller, ChemCatChem, 2024, 16, e202301562 CrossRef CAS.
- V. Zubar, J. Sklyaruk, A. Brzozowska and M. Rueping, Org. Lett., 2020, 22, 5423–5428 CrossRef CAS PubMed.
- W. Chen, J. Egly, A. I. Poblador-Bahamonde, A. Maisse-Francois, S. Bellemin-Laponnaz and T. Achard, Dalton Trans., 2020, 49, 3243–3252 RSC.
- V. Mechrouk, B. Leforestier, W. Chen, A. I. Poblador-Bahamonde, A. Maisse-Francois, S. Bellemin-Laponnaz and T. Achard, Chem. – Eur. J., 2024, 30, e202401390 CrossRef CAS PubMed.
- J. Egly, M. Bouché, W. Chen, A. Maisse-François, T. Achard and S. Bellemin-Laponnaz, Eur. J. Inorg. Chem., 2018, 159–166 CrossRef CAS.
- J. Wolf, A. Labande, J.-C. Daran and R. Poli, Eur. J. Inorg. Chem., 2007, 5069–5079 CrossRef CAS.
- C. Fliedel and P. Braunstein, Organometallics, 2010, 29, 5614–5626 CrossRef CAS.
- I. Blaha, S. Weber, R. Dülger, L. F. Veiros and K. Kirchner, ACS Catal., 2024, 14, 13174–13180 CrossRef CAS PubMed.
- S. Weber, B. Stöger and K. Kirchner, Org. Lett., 2018, 20, 7212–7215 CrossRef CAS PubMed.
- S. Weber, L. F. Veiros and K. Kirchner, Adv. Synth. Catal., 2019, 361, 5412–5420 CrossRef CAS PubMed.
- S. Weber, J. Brünig, L. F. Veiros and K. Kirchner, Organometallics, 2021, 40, 1388–1394 CrossRef CAS PubMed.
- J. A. Garduño, M. Flores-Alamo and J. J. García, ChemCatChem, 2019, 11, 5330–5338 CrossRef.
- J. A. Garduño and J. J. García, ACS Catal., 2019, 9, 392–401 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, characterization data, NMR spectra. CCDC 2406903–2406906. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc06627h |
| This journal is © The Royal Society of Chemistry 2025 |