
Enhanced electrocatalytic activity and stability of high performance symmetrical solid oxide fuel cells with praseodymium-doped SrCo0.2Fe0.8O3−δ electrodes†
Peng
Li‡
a,
Bing
Yang‡
a,
Jing
Chen
*a,
Bo
Li
a,
Lushan
Ma
a,
Mengjia
Wang
a,
Xuzhuo
Sun
*a,
Yunfeng
Tian
 *b and
Bo
Chi
c
*b and
Bo
Chi
c
aSchool of Chemistry and Chemical Engineering, Henan University of Technology, Zhengzhou 450001, China. E-mail: chenjing0504@haut.edu.cn; sunxuzhuo@haut.edu.cn
bSchool of Materials Science and Physics, China University of Mining and Technology, Xuzhou 221116, PR China. E-mail: yunfengup@cumt.edu.cn
cCenter for Fuel Cell Innovation, State Key Laboratory of Material Processing and Die & Mould Technology, School of Materials Science and Engineering, Huazhong University of Science and Technology, Wuhan, Hubei 430074, China
First published on 31st October 2024
Abstract
Symmetrical solid oxide fuel cells (SSOFCs) represent a promising path towards energy conversion and storage solutions characterized by reduced material costs, simplified manufacturing, and improved operational stability. The development of high-performance symmetrical electrodes with superior catalytic activity and durability remains a critical challenge. In this study, a series of praseodymium-doped SrCo0.2Fe0.8O3−δ perovskites, PrxSr1−xCo0.2Fe0.8O3−δ (x = 0, 0.1, 0.2, 0.4, 0.6, 0.8, 1) are systematically explored, to evaluate their potential as efficient symmetrical electrodes for SSOFCs. Among the compositions studied, Pr0.2Sr0.8Co0.2Fe0.8O3−δ (P0.2SCF) stands out, with an excellent crystal structure and microstructural stability demonstrated by XRD, XPS and SEM test results, and low polarization resistance values at 850 °C of 0.029 Ω cm2 for the cathode and 0.054 Ω cm2 for the anode. In particular, a single cell containing P0.2SCF achieved an impressive maximum power density of 719.08 mW cm−2 at 850 °C as well as good stability. The results of the DRT analysis show that the main rate-limiting steps of the cell are the gas diffusion and catalytic dissociation processes. These results underscore the potential of P0.2SCF as a highly effective perovskite electrode material for SSOFCs and highlight the key role of Pr doping in enhancing perovskite electrode performance and advancing sustainable energy technologies.
Introduction
Solid oxide fuel cells (SOFCs) can efficiently convert chemical energy into electrical energy. They have a wide range of fuel sources and offer advantages such as low pollution, all-solid-state design, and modular assembly.1–5 As a result, SOFCs are widely recognized as one of the most promising energy conversion technologies of the 21st century, with significant development potential. In conventional SOFCs, the materials used for the cathode and anode are different. In particular, nickel-based anodes face challenges such as carbon deposition and sulfur poisoning when using hydrocarbons or sulfides are used as fuels.6 Using the same material for both the cathode and anode, and constructing a symmetric sandwich structure can simplify the fabrication process, reduce costs, and minimize compatibility issues. This configuration also allows the catalytic activity of the anode to be restored by switching the air flow to remove carbon and sulfur-containing impurities from the anode surface.7 Therefore, symmetric SOFCs (SSOFCs) have garnered increasing research interest and attention.8–11As mentioned above, symmetrical cells use identical electrode materials, even though the anode and cathode play different roles in the electrochemical reactions. This places high demands on the electrode material. Currently, symmetrical electrodes for SSOFCs are mostly selected from SOFC cathode or anode materials,12 which are then modified by elemental doping or surface treatment to improve the catalytic activity and stability of the electrode materials in both oxidizing and reducing atmospheres.13 Among these materials, SrFeO3−δ-based perovskites have attracted the attention of researchers due to their excellent electrocatalytic activity and stability for the oxygen reduction reaction (ORR) and hydrogen oxidation reaction (HOR).14 They exhibit high oxygen vacancy concentration, surface oxygen exchange coefficient and excellent conductivity.15 The perovskite oxide SrFe0.9Mo0.1O3−δ has been evaluated as an electrode for SSOFCs with La0.9Sr0.1Ga0.8Mg0.2O3−δ (LSGM) electrolytes, with a maximum power density of 253 mW cm−2 at 700 °C.16 The SrFe0.8W0.2O3−δ (SFW) perovskite oxide has been developed as an electrode material for SSOFCs, with polarization resistance (Rp) values as low as 0.084 and 0.20 Ω cm2 in air and wet H2 at 800 °C, respectively.17 SrFe0.8Ti0.2O3−δ has been used in SSOFCs and shows the highest electrochemical performance with a maximum power density of 700 mW cm−2 and a lower electrode Rp value (0.573 Ω cm2) at 800 °C.18 In addition, SrFe0.9−xW0.1TixO3−δ has been prepared as a promising electrode material for SSOFCs, with the Rp of a symmetric single cell recorded at 0.28 Ω cm2.19 Other materials such as SrFe0.9Si0.1O3−δ,20 SrFe0.75Mo0.25O3−δ,21 and SrFe0.75Zr0.25O3−δ22 have also been investigated.
In addition to doping the B-site with high-valence metals to improve stability and electrocatalytic activity, introducing high-valence rare earth elements at the A-site is also an effective strategy due to their unique 4f orbital electron properties.23 This approach can significantly improve both electrocatalytic activity and stability. For example, the manipulation of the A-site deficiency in La0.3Sr0.7Ti0.3Fe0.7O3−δ-based electrodes for SSOFCs was investigated, and the Rp values of (La0.3Sr0.7)0.95Ti0.3Fe0.7O3−δ were found to be 0.032 and 0.110 Ω cm2 in air and wet H2 at 800 °C, respectively.24 In another study, SrFeO3−δ perovskite (Ce0.2Sr0.8Fe0.95Ni0.05O3) was modified with ceria and NiO doping on both the A- and B-sites to achieve a maximum power density of 580 mW cm−2 and an Rp of 0.21 Ω cm2 at 800 °C for SSOFCs.25 Pr0.6Sr0.4Fe0.8Cu0.1W0.1O3−δ has also been investigated as a symmetrical electrode material, yielding Rp values of 0.069 Ω cm2 under oxidizing conditions and 0.24 Ω cm2 under reducing conditions at 800 °C.26 Similarly, Pr0.4Sr0.6Co0.3Fe0.6Nb0.1O3−δ oxides have been evaluated as electrode materials for SSOFCs, with the lowest Rp values of 0.028 and 0.077 Ω cm2 at 900 °C in air and hydrogen, respectively.27
Although A-site doping with the rare earth element Pr can enhance the electrocatalytic activity, the specific mechanism of its effect remains unclear, and the optimal doping amount is also unknown. In this study, LSGM was chosen as the electrolyte support due to its higher ionic conductivity, better chemical compatibility with perovskite electrodes and chemical stability in both oxidizing and reducing environments. SrFeO3−δ-based perovskite oxides doped with a small amount of the element Co were used as the substrate. Pr-doped PrxSr1−xCo0.2Fe0.8O3−δ (PxSCF, x = 0, 0.1, 0.2, 0.4, 0.6, 0.8, 1) symmetric electrode materials were prepared using the sol–gel method. The impacts of different Pr doping level on the structure and electrochemical properties of the phases were investigated, and their AC impedance values were determined. The doping ratios with the best electrochemical performance were identified, and the stability of the crystal structure and microscopic morphology of these optimal ratios were studied in detail under oxidizing and reducing atmospheres.
Experimental
Powder synthesis
PrxSr1−xCo0.2Fe0.8O3−δ (PxSCF, x = 0, 0.1, 0.2, 0.4, 0.6, 0.8, 1) powders with different Pr doping level were synthesized using the sol–gel method. For example, to prepare the Pr doping level of x = 0.2, stoichiometric amounts of Pr(NO3)3·6H2O, Sr(NO3)2, Co(NO3)2·6H2O, and Fe(NO3)2·9H2O were weighed and dissolved in deionized water. Citric acid (CA) and ethylenediaminetetraacetic acid (EDTA) were added to the solution as complexing agents in a molar ratio of ions![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CA:EDTA = 1:2:1. The pH of the solution was adjusted to 7–8 using ammonia, then stirred at 80 °C until a gel formed, which was then heated to 180 °C to produce the precursor. The precursors were then calcined in air at 900 °C for 6 h to finalize the electrode powders with the appropriate Pr doping level. To simulate the actual conditions of the electrode powder in the anode working atmosphere, the electrode powder was calcined at 800 °C for 2 h in a 5% H2/95% Ar atmosphere to obtain the reduced powder, denoted as PxSCF@2. To further investigate the stability of its crystal structure and microstructure in a long-term high temperature reducing atmosphere, the electrode powder was calcined at 800 °C for 100 h in a 5% H2/95% Ar atmosphere, denoted as PxSCF@100. The La0.8Sr0.2Ga0.8Mg0.2O3−δ (LSGM) electrolyte was synthesized via a solid-state reaction. First, La2O3 was calcined at 1250 °C for 2 h, while MgO was calcined in air at 900 °C for 2 h. Then, stoichiometric amounts of La2O3, Ga2O3, MgO, and SrCO3 powders were ball-milled in ethanol for 12 h. The resulting powder mixture was dried and calcined in air at 1100 °C for 5 h. The calcined powder underwent a second 12-hour ball-milling. To produce dense LSGM electrolyte discs with a thickness of 400 μm, the pre-calcined powder was dry-pressed with 1 wt% polyvinyl butyral (PVB) and held in air at 1450 °C for 10 h.
:CA:EDTA = 1:2:1. The pH of the solution was adjusted to 7–8 using ammonia, then stirred at 80 °C until a gel formed, which was then heated to 180 °C to produce the precursor. The precursors were then calcined in air at 900 °C for 6 h to finalize the electrode powders with the appropriate Pr doping level. To simulate the actual conditions of the electrode powder in the anode working atmosphere, the electrode powder was calcined at 800 °C for 2 h in a 5% H2/95% Ar atmosphere to obtain the reduced powder, denoted as PxSCF@2. To further investigate the stability of its crystal structure and microstructure in a long-term high temperature reducing atmosphere, the electrode powder was calcined at 800 °C for 100 h in a 5% H2/95% Ar atmosphere, denoted as PxSCF@100. The La0.8Sr0.2Ga0.8Mg0.2O3−δ (LSGM) electrolyte was synthesized via a solid-state reaction. First, La2O3 was calcined at 1250 °C for 2 h, while MgO was calcined in air at 900 °C for 2 h. Then, stoichiometric amounts of La2O3, Ga2O3, MgO, and SrCO3 powders were ball-milled in ethanol for 12 h. The resulting powder mixture was dried and calcined in air at 1100 °C for 5 h. The calcined powder underwent a second 12-hour ball-milling. To produce dense LSGM electrolyte discs with a thickness of 400 μm, the pre-calcined powder was dry-pressed with 1 wt% polyvinyl butyral (PVB) and held in air at 1450 °C for 10 h.
Single-cell fabrication and fuel cell testing
The optimal Pr doping level was determined by fabricating PxSCF|LSGM|PxSCF symmetric cells and evaluating their electrochemical performance as both the cathode and anode. The PxSCF was combined with a binder in a 1:1 weight ratio and manually ground for 40 min to produce the electrode slurry. The paste was then screen-printed onto both sides of the LSGM electrolyte and calcined in air at 950 °C for 2 h, forming electrodes with an area of 0.28 cm2 on each side. A symmetric cell was assembled by applying DAD-87 silver paste and attaching silver wires to both sides. These PxSCF|LSGM|PxSCF symmetric cells were tested at open circuit voltage (OCV) with H2 supplied to the fuel electrode at a flow rate of 40 ml min−1 and the cathode exposed to ambient air. For the AC impedance test to evaluate anode performance, the cell was first reduced at 800 °C for 2 h in a 5% H2/95% Ar atmosphere to simulate the anode environment, followed by a polarization impedance measurement. Current–voltage curves (I–V) and electrochemical impedance spectroscopy (EIS) measurements were carried out in the temperature range of 700–850 °C using a Zahner electrochemical workstation (Zennium). The EIS measurements covered a frequency range of 0.1–106 Hz with an amplitude of 10 mV. The EIS data were analyzed using the Distribution of Relaxation Times (DRT) method.
Characterization
The crystal structure and structural stability of the powders were evaluated by X-ray diffractometry (XRD, Rigaku Ultima IV) using Cu Kα radiation with step widths of 0.02° and scan ranges from 20° to 90°. Detailed structural information was obtained by Rietveld refinement using the FullProf software. Scanning electron microscopy (SEM, ZEISS Sigma 300) was used to examine the microstructure and cross-sections of the samples before and after reduction. X-ray photoelectron spectroscopy (XPS, Thermo Scientific K-Alpha) was used to analyze the chemical valence states of the PxSCF samples. All XPS measurements were calibrated to a binding energy of 284.8 eV corresponding to the C 1s peak. Peak fitting was performed using XPS Avantage software to determine the valence states and chemical environments of the elemental ions. The conductivity of the electrode material in air was determined by the DC four-terminal method.Results and discussion
Materials screening
The PxSCF (x = 0, 0.1, 0.2, 0.4, 0.6, 0.8, 1) electrode powders with different Pr doping levels were first analyzed for their physical structures as depicted in Fig. 1(a), for x ≤ 0.4, the PxSCF samples exhibit a well-defined orthorhombic perovskite structure, with all diffraction peaks matching the standard reference (PDF#40-0906) and showing no impurity peaks. However, for x ≥ 0.6, a few impurity peaks were observed in P0.6SCF, P0.8SCF, and P1SCF samples, identified as PrFeO3 in comparison with the standard reference (PDF#19-1012). Consequently, only the electrode powders with Pr doping levels of 0, 0.1, 0.2, and 0.4 were selected for further electrochemical performance testing in half-cells. The Rp values for each doping ratio, derived from the tests are presented in Fig. 1(b)–(e). The Rp of PxSCF decreases with increasing temperature both in air and in a 5% H2/95% Ar atmosphere. This trend suggests that higher temperatures facilitate oxygen adsorption and desorption, as well as enhance the catalytic activity of the H2. From Fig. 1(f) and (g), the Rp values of P0.2SCF in air are lower than those of P0.1SCF, indicating its superior oxygen catalytic activity. However, in a reducing atmosphere, P0.1SCF has lower Rp values than P0.2SCF at lower temperatures. As the temperature increases, the difference in Rp between the two materials decreases. After careful consideration of these factors, the P0.2SCF electrode material with the highest catalytic activity was selected for further detailed study.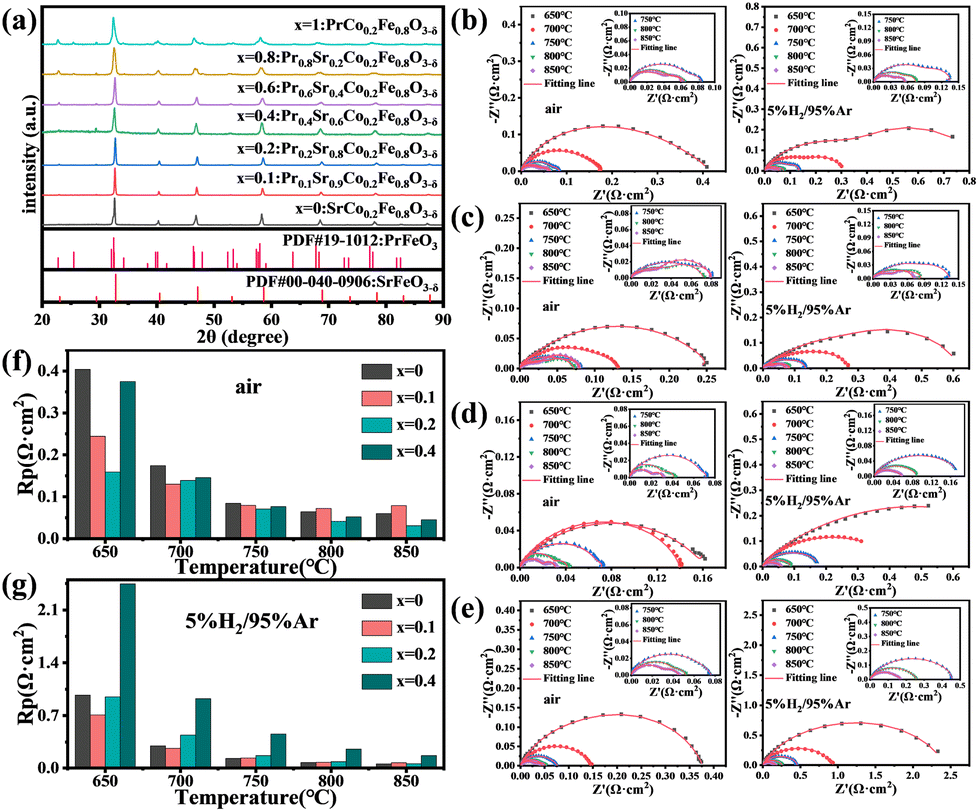 | ||
| Fig. 1 (a) XRD patterns of PxSCF (x = 0, 0.1, 0.2, 0.4, 0.6, 0.8, 1) calcined at 900 °C for 6 h in air; (b)–(e) impedance spectra of PxSCF (x = 0, 0.1, 0.2, 0.4) under air and 5% H2/95% Ar; (f) and (g) histograms of the Rp values of PxSCF (x = 0, 0.1, 0.2, 0.4) under air and 5% H2/95% Ar at different temperatures. | ||
Materials characterization
To further investigate the crystal structure stability of the P0.2SCF powder in reducing atmospheres, the powder was exposed to a 5% H2/95% Ar mixture at 800 °C for 2 h and 100 h, respectively. The orthorhombic perovskite structure of P0.2SCF maintained its structural stability after 2 h of reduction, as illustrated in Fig. 2(a). However, after 100 h of reduction, a localized magnification study of the diffraction angles revealed a slight shift to lower angles, indicating lattice expansion. This expansion is attributed to the reduction of Fe4+ to Fe3+ and the corresponding increase in lattice parameters in a reducing atmosphere, which is consistent with observations in similar electrode materials such as SrFe0.7Ti0.3O3−δ.28 Despite these changes, the orthorhombic perovskite structure remained intact, demonstrating that P0.2SCF has excellent crystal structure stability in oxidizing and prolonged reducing atmospheres. The Rietveld refinement of the P0.2SCF electrode powder in air and after reduction is shown in Fig. 2(b)–(d). The space group is P/mmm, and the refined cell parameters are provided in Table S1.† The results confirm that lattice expansion occurs after 100 h of reduction in a 5% H2/95% Ar atmosphere, with slight increases in the cell parameters a, b, and c. This expansion is attributed to the reduction-induced decrease in the valence states of the Co and Fe elements over a prolonged period. Fig. 2(e) shows the SEM images of the P0.2SCF electrode powder before reduction at different magnifications, revealing a loose, porous structure. Fig. 2(f) and (g) show the microscopic morphology of the P0.2SCF electrode powders reduced in a 5% H2/95% Ar atmosphere for 2 h and 100 h, respectively, at different magnifications. The images illustrate that the microscopic morphology remains unchanged regardless of the reduction time, maintaining a loose, porous structure. The micromorphology of P0.2SCF after 100 h of reduction, along with the elemental distribution, was obtained through X-ray energy dispersive spectroscopy (EDS) analysis of P0.2SCF@100. Fig. 2(h) shows that the elements Pr, Sr, Co, Fe, and O are uniformly distributed without any elemental enrichment. Combining the results of the XRD, SEM, and EDS tests, it can be concluded that P0.2SCF exhibits excellent crystal structure and microstructure stability under redox conditions. This stability meets the basic requirements for a symmetric electrode. | ||
| Fig. 2 (a) XRD patterns of P0.2SCF in oxidizing and reducing atmospheres; (b)–(d) XRD refinements of P0.2SCF with different reduction times; (e)–(g) microscopic morphology of P0.2SCF with different reduction times; (h) EDS patterns of P0.2SCF reduced for 100 h in a 5% H2/95% Ar atmosphere. | ||
The specific valence states of each element on the surface of P0.2SCF and P0.2SCF@100 electrode powders were analyzed by XPS as shown in Fig. 3(a). It indicates that the elemental composition of the powder remains unchanged, confirming the stability of its physical phase structure. Fig. 3(b) shows the XPS pattern of Pr 3d. The binding energy peaks of Pr3+ are located at 929.9 eV, 949.4 eV, 934.1 eV, and 955.2 eV,29 while the binding energy peaks of Pr4+ are at 932.6 eV and 952.6 eV. An increased content of Pr3+ in the P0.2SCF@100 powders, with binding energies at 934.1 eV and 955.2 eV, suggests a decrease in the average valence state of Pr from 3.52 to 3.42 after 100 h of reduction. In Sr containing materials, the Sr element migrates from the bulk to the surface, forming compounds such as Sr(OH), SrCO3, and SrO.30–33 The XPS pattern of Sr 3d is shown in Fig. 3(c), exhibiting a bimodal pattern corresponding to the 3d5/2 and 3d3/2 orbitals of Sr, with a peak separation of 1.75 eV.34 The binding energy peaks at 3d5/2 and 3d3/2 are attributed to the Sr2+ lattice (Srlattice) in both P0.2SCF and P0.2SCF@100 powders. The peaks at 133.1 eV and 134.9 eV are assigned to non-lattice Sr (Srnon-lattice) compounds formed by interaction with carbon dioxide and oxygen in air.31 Srnon-lattice forms an insulating layer on the surface, hindering electron transfer and degrading electrode performance. Semi-quantitative analysis of the Sr 3d spectra reveals that the Srlattice and Srnon-lattice contents are 45.55% and 54.45% in P0.2SCF, and 51.37% and 48.63% in P0.2SCF@100, respectively. This indicates no significant Sr segregation after prolonged high temperature reduction, suggesting the stability of the P0.2SCF electrode. The XPS pattern of Co 2p shown in Fig. 3(d) includes binding energy peaks at 780.6/795.6 eV, 779.9/794.6 eV, and 782.1/796.7 eV, corresponding to Co2+, Co3+, and Co4+, respectively.35 This indicates that Co exists in mixed valence states (Co2+/Co3+/Co4+) in both P0.2SCF and P0.2SCF@100 powders. After prolonged reduction, there is a slight increase in Co2+ and Co3+ contents, while Co4+ decreases, reducing the average valence state of Co from 2.88 to 2.82. The XPS pattern of Fe 2p shown in Fig. 3(e) includes binding energy peaks at 709.8/722.6 eV, 711.8/724.6 eV, and 715.1/727.8 eV, corresponding to Fe2+, Fe3+, and Fe4+, respectively.36 Fe exists in mixed valence states (Fe2+/Fe3+/Fe4+), predominantly as Fe2+ and Fe3+. The average valence state of Fe decreases from 2.74 to 2.59 after prolonged reduction. The XPS pattern of O 1s shown in Fig. 3(f) has two distinct peaks corresponding to lattice oxygen (Olat) at 528.5 eV and surface adsorbed oxygen (Oads) at 531.1 eV.37 Semi-quantitative analysis shows that the Oads and Olat contents in P0.2SCF are 59.72% and 40.28%, respectively, while in P0.2SCF@100 they are 68.84% and 31.16%, respectively. Prolonged high temperature reduction increases the surface adsorbed oxygen content, indicating the formation of additional oxygen vacancies, which enhances the electrocatalytic activity. Overall, the average valence states of Pr, Co, and Fe decrease in the P0.2SCF@100 powder, this reduction in valence states suggests improved catalytic performance due to increased oxygen vacancy concentration.
 | ||
| Fig. 3 XPS spectra of (b) Pr 3d, (c) Sr 3d, (d) Co 2p, (e) Fe 2p, and (f) O 1s in P0.2SCF and P0.2SCF@100 (a). | ||
Electrochemical performance
Fig. 4 presents the EIS and DRT analysis for P0.2SCF used as both the cathode and anode, with the ohmic impedance of the electrolyte subtracted for a clearer comparison of the Rp. When P0.2SCF is used as the cathode, the Rp values at 700 °C, 725 °C, 750 °C, 800 °C and 850 °C were 0.14 Ω cm2, 0.098 Ω cm2, 0.069 Ω cm2, 0.041 Ω cm2 and 0.029 Ω cm2 as shown in Fig. 4(a), respectively. The results of the DRT analysis showed that the resistive response frequency of P0.2SCF as a cathode is mainly in the mid-frequency range (102–104 Hz) as shown in Fig. 4(b), indicating that the process of absorption and desorption of oxygen is the rate-limiting step. As an anode, the Rp values at 700 °C, 725 °C, 750 °C, 800 °C and 850 °C were 0.34 Ω cm2, 0.24 Ω cm2, 0.17 Ω cm2, 0.086 Ω cm2 and 0.054 Ω cm2 as shown in Fig. 4(c), respectively. The resistive response frequency of P0.2SCF is mainly in the low to mid-frequency range (<102 Hz) as shown in Fig. 4(d), indicating that the rate-limiting step is mainly the diffusion and adsorption–desorption process of hydrogen on the electrode surface, with gas diffusion limitations being particularly prominent at low frequencies (1 Hz). The peak intensity decreases rapidly with increasing temperature, indicating that increasing temperature is favorable for increasing the electrocatalytic activity. | ||
| Fig. 4 (a) and (b) EIS and corresponding DRT plots of P0.2SCF in an air atmosphere; (c) and (d) EIS and corresponding DRT plots of P0.2SCF in 5% H2/95% Ar atmosphere. | ||
Both the ORR at the cathode and the HOR at the anode in SSOFCs involve complex reaction steps. To further clarify the kinetics of the ORR and the HOR catalyzed by the P0.2SCF electrode, the Rp of P0.2SCF as both the cathode and anode was examined under varying partial pressures of oxygen and hydrogen. This analysis aims to identify the rate-limiting steps of these reactions. Fig. 5(a) illustrates the Rp values at different partial pressure of oxygen (PO2) at 800 °C. In addition, EIS data at other temperatures and PO2 are shown in Fig. S1,† with the corresponding fitting results presented in Table S2.† The results show that a decrease in Rp with increasing PO2, highlighting the significant influence of PO2 on the ORR. In addition, temperature dependence tests show that Rp decreases with increasing temperature, suggesting that higher temperatures enhance electrode responses. The measured Rp values were fitted using Zsimpwin software and the equivalent circuit fitting model is shown in Fig. S2.† The contributions of the high frequency resistance (RH) and low frequency resistance (RL) to the overall Rp are critical. As shown in Fig. 5(c), for the P0.2SCF cathode at PO2 of 0.21 atm, the activation energies are 122.2 kJ mol−1 for RH and 134.68 kJ mol−1 for RL. This comparison reveals that the working temperature has a greater impact on the oxygen ion migration in the cathode, making RH more sensitive to temperature changes. In contrast, the adsorption/desorption process on the electrode surface is less temperature-dependent, resulting in a higher activation energy for RL. The relationships between PO2 and Rp can be described using the following equation Rp = R0PO2−n, where different values of n indicate different rate-determining steps in the electrochemical reaction: (1) n = 1, O2(g) → O2,ads, indicating oxygen adsorption on the electrode surface and diffusion in the organic phase. (2) n = 0.5, O2,ads → 2Oads, indicating the dissociation of adsorbed oxygen into atomic oxygen. (3) n = 0.25, 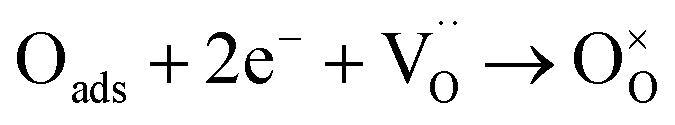 , indicating a charge transfer process. Fig. 5(e) shows the linear relationship between PO2 and Rp for the P0.2SCF cathode at different temperatures. In the 700–850 °C range, the n values are 0.2, 0.33, 0.34, and 0.35, which are close to 0.25. This suggests that charge transfer is the rate-limiting step for the ORR under these conditions.
, indicating a charge transfer process. Fig. 5(e) shows the linear relationship between PO2 and Rp for the P0.2SCF cathode at different temperatures. In the 700–850 °C range, the n values are 0.2, 0.33, 0.34, and 0.35, which are close to 0.25. This suggests that charge transfer is the rate-limiting step for the ORR under these conditions.
 | ||
| Fig. 5 (a) EIS for different PO2 at 800 °C; (b) EIS for different PH2 at 800 °C; (c) Arrhenius plots of RH and RL changes with temperature at PO2 = 0.21 atm; (d) Arrhenius plots of RH and RL changes with temperature at PH2 = 0.05 atm; (e) correlation between PO2 and Rp at different temperatures; (f) dependence of RH and RL on the PH2. | ||
As an anode, RH represents the charge transfer process at the electrode–electrolyte interface, while RL represents the hydrogen adsorption and dissociation process at the anode surface. Fig. 5(b) shows the Rp values at 800 °C for different hydrogen partial pressures (PH2). In addition, EIS data at other temperatures and PH2 are shown in Fig. S3,† with the corresponding fit results presented in Table S3.† The Rp decreases with increasing PH2, indicating the influence of the PH2 on the HOR. Temperature-dependent tests of the PH2 reveal that RH is consistently greater than RL, indicating that the P0.2SCF anode has a high dependence on the working temperature. According to the fitting results in Fig. 5(d), the activation energies of RH and RL are 80.21 kJ mol−1 and 88.1 kJ mol−1 at a PH2 of 0.05 atm, respectively. Fig. 5(f) shows the relationship between RH, RL, Rp, and PH2. The slope of the RL fitted line (−0.71) is smaller than the slope of the RH fitted line (−0.25), indicating that hydrogen adsorption and dissociation on the anode surface are highly sensitive to the PH2 during the reaction process at the P0.2SCF anode.
For an in-depth evaluation of the electrochemical stability of P0.2SCF as the anode, the long-term impedance stability test in a 5% H2/95% Ar atmosphere at 800 °C was performed, as shown in Fig. 6(a), the Rp of the P0.2SCF anode decreases from 0.088 to 0.077 Ω cm2 over 120 h and remains stable. This decrease in Rp suggests that P0.2SCF exhibits excellent catalytic activity for the HOR process when used as an anode, which is in good agreement with the XPS test results very well. Fig. 6(b) displays the SEM cross-sectional morphology of the P0.2SCF anode after the long-term stability test. The P0.2SCF anode retains a loosely packed structure and adheres tightly to the LSGM electrolyte without significant delamination or cracking, indicating good interfacial stability between the electrode and the electrolyte. These results demonstrate that P0.2SCF possesses excellent and stable electrocatalytic activity for the HOR in a reducing atmosphere, making it a promising candidate for use as an anode in SSOFCs.
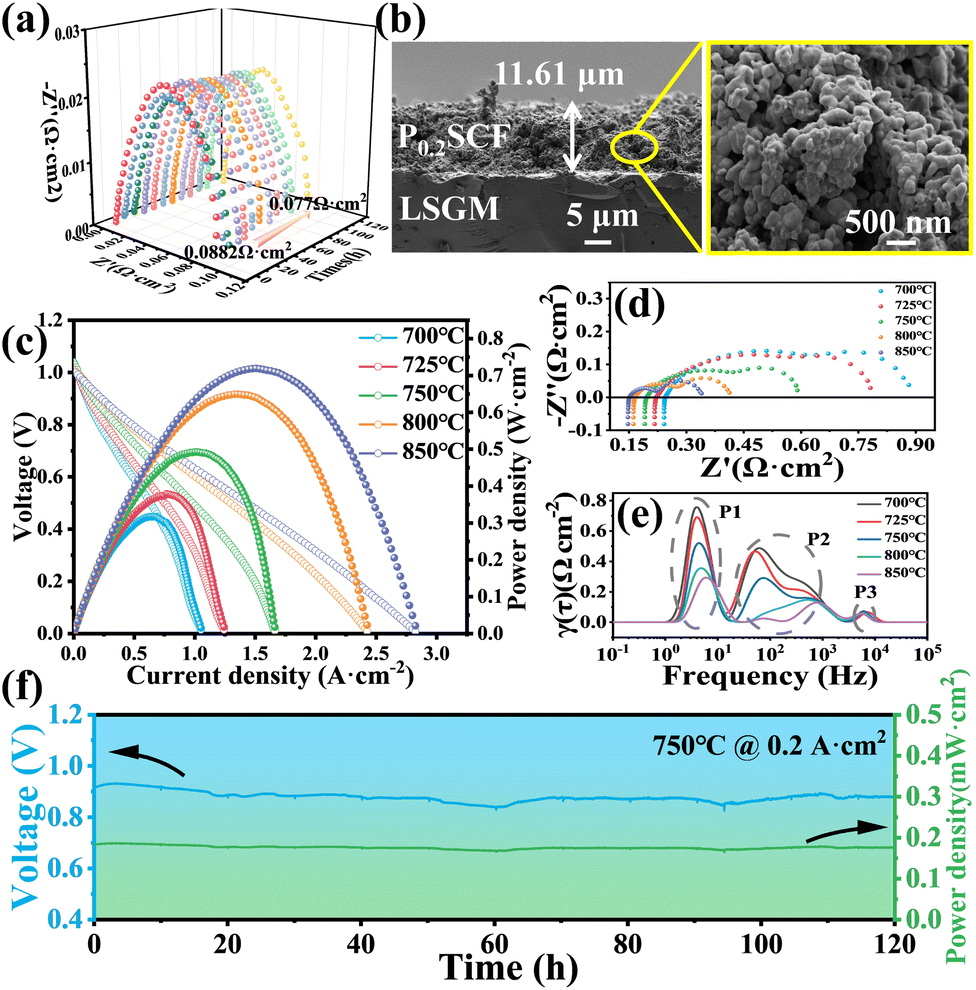 | ||
| Fig. 6 (a) 120 h stability test of the P0.2SCF anode in 5% H2 at 800 °C; (b) microscopic cross-section of a half-cell after the stability test; (c) I–V–P curves of the single cell with P0.2SCF in wet H2 at 700–850 °C; (d) polarization impedance values for a single cell; (e) DRT analysis of the P0.2SCF single point cell at 700–850 °C, (f) stability test for a single cell. | ||
The I–V–P curves of the symmetric cell P0.2SCF|LSGM|P0.2SCF are shown in Fig. 6(c). The maximum power density (MPD) of the cell increases with temperature, and the current and voltage exhibit a clear linear relationship, indicating minimal concentration and activation polarization. The MPD of the single cell reached 719.08 mW cm−2 at 850 °C. Fig. 6(d) presents the EIS data of the P0.2SCF single cell at different temperatures. The intercept of the high-frequency arc with the real axis represents the ohmic resistance (Ro), while the intercept of the low-frequency arc with the real axis indicates the total resistance (Rt). The Rp of the single cell is calculated as the difference between Rt and Ro. At 850 °C, the Ro and Rp of the single cell are 0.3 Ω cm2 and 0.42 Ω cm2, respectively. Due to the high conductivity of the electrode material (Fig. S4†), the primary source of the Ro of the cell is the electrolyte. These results highlight the potential of P0.2SCF as a high-performance electrode material in SSOFCs, demonstrating both excellent electrochemical stability and catalytic activity.
To elucidate the reaction kinetics of the P0.2SCF symmetric single cell, the EIS results of a symmetric single cell using the DRT method were analyzed. This analysis directly calculates the distribution function of the relaxation times and the relaxation amplitude of the impedance-dependent processes in Fig. 6(e). The DRT peaks at different response frequencies correspond to different electrochemical processes. The DRT results for the three groups of peaks were categorized, labeled P1 to P3, based on their response frequencies. The relative heights of these peaks indicate the extent to which each reaction process contributes to the overall reaction in the cell. Specifically, the high-frequency peak P3 (103 to 104 Hz) represents the ion transport processes within the P0.2SCF electrode, the LSGM electrolyte, and their interface. The mid-frequency peaks P2 (101 to 103 Hz) represent the HOR and ORR on the electrode surface, and the gas adsorption/desorption on the electrode material surface, respectively. The low-frequency peak P1 (<101 Hz) corresponds to that of hydrogen and oxygen on the porous electrode surface. Our observations indicate that the processes represented by P3 (ion transfer) are relatively insensitive to temperature. In contrast, P1 and P2 (gas diffusion and catalytic dissociation) decrease fast with increasing temperature in the range of 700–850 °C, indicating a strong temperature dependence. This suggests that the primary rate-limiting step for the single cell is the diffusion, adsorption and desorption of gas on the P0.2SCF electrode. These results highlight the critical role of gas diffusion, adsorption and desorption in determining the overall reaction rate, providing valuable insights for optimizing the performance of P0.2SCF-based SSOFCs. The cell showed no significant degradation during the 120 hours of operation, as shown in Fig. 6(f), providing an initial indication of its stability. However, slight voltage fluctuations were observed. Therefore, further testing over a longer period is necessary to confirm its stability.
Conclusions
This study investigated the effects of various Pr doping levels on the physical phase structure and electrochemical properties of Pr-doped SrCoFeO3−δ (PxSCF) materials. AC impedance measurements identified Pr0.2Sr0.8Co0.2Fe0.8O3−δ (P0.2SCF) as having the optimal electrochemical performance, maintaining a stable orthorhombic perovskite structure at doping ratios ≤0.4 and exhibiting the highest HOR and ORR activities. Reduction tests at 800 °C in a 5% H2/95% Ar atmosphere for 2 and 120 h demonstrated that P0.2SCF retains its crystal structure and micro-morphology, indicating excellent structural stability. The polarization impedance values at 850 °C were 0.029 Ω cm2 for the cathode and 0.054 Ω cm2 for the anode. Long-term stability tests showed a decrease in polarization impedance from 0.088 Ω cm2 to 0.077 Ω cm2 at 800 °C. The maximum power density of a single cell using LSGM as the electrolyte was 719.08 mW cm−2 at 850 °C. Overall, P0.2SCF exhibits outstanding high electrochemical catalytic activity, making it a promising candidate electrode for SSOFCs.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Peng Li: investigation, methodology, writing – original draft. Bing Yang: data curation, formal analysis, investigation, methodology. Jing Chen: methodology, data curation, supervision, writing – review & editing. Bo Li: methodology, funding acquisition. Lushan Ma: methodology, project administration, funding acquisition. Mengjia Wang: investigation. Xuzhuo Sun: methodology, project administration, funding acquisition, supervision. Yunfeng Tian: methodology, supervision, conceptualization writing – review & editing. Bo Chi: methodology, funding acquisition, project administration.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22279029 and 52302334), Key Scientific and Technological Research Project in Henan Province (No. 242102241022), Jiangsu Province (BZ2022027), the Innovative Funds Plan of Henan University of Technology (2022ZKCJ01) and Changzhou City (CZ20230010).References
- J. Zamudio-García, L. Caizán-Juanarena, J. M. Porras-Vázquez, E. R. Losilla and D. Marrero-López, J. Power Sources, 2022, 520, 230852 CrossRef.
- M. Zhang, Z. Du, Z. Sun and H. Zhao, J. Mater. Chem. A, 2023, 11, 21645 RSC.
- W. Zhang, J. Wei, F. Yin and C. Sun, Mater. Chem. Front., 2023, 7, 1943–1991 RSC.
- Q. Guo, J. Geng, J. Pan, L. Zou, Y. Tian, B. Chi and J. Pu, Energy Rev., 2023, 100037 CrossRef.
- M. Yuan, Z. Wang, J. Gao, H. Hao, Z. Lv, X. Lou, L. Xu, J. Li and B. Wei, J. Colloid Interface Sci., 2024, 787–796 CrossRef.
- T. Hays, A. M. Hussain, Y.-L. Huang, D. W. McOwen and E. D. Wachsman, ACS Appl. Energy Mater., 2018, 1, 1559–1566 CrossRef.
- T. Wei, Q. Zhang, Y.-H. Huang and J. B. Goodenough, J. Mater. Chem., 2012, 22, 225–231 RSC.
- X.-Y. Jiao, A.-Y. Geng, Y.-Y. Xue, X.-B. Wang, F.-J. Jin, Y.-H. Ling and Y.-F. Tian, Tungsten, 2023, 5, 598–606 CrossRef.
- Y. Tian, N. Abhishek, C. Yang, R. Yang, S. Choi, B. Chi, J. Pu, Y. Ling, J. T. Irvine and G. Kim, Matter, 2022, 5, 482–514 CrossRef.
- Y. Tian, Y. Liu, L. Jia, A. Naden, J. Chen, B. Chi, J. Pu, J. T. Irvine and J. Li, J. Power Sources, 2020, 475, 228620 CrossRef.
- C. Su, W. Wang, M. Liu, M. O. Tadé and Z. Shao, Adv. Energy Mater., 2015, 5, 1500188 CrossRef.
- W. N. A. W. Yusoff, N. A. Baharuddin, M. R. Somalu, A. Muchtar, N. P. Brandon and H. Fan, Int. J. Miner., Metall. Mater., 2023, 30, 1933–1956 CrossRef.
- M. Zhang, Z. Du, Y. Zhang and H. Zhao, ACS Appl. Energy Mater., 2022, 5, 13081–13095 CrossRef CAS.
- C. Yang, Y. Wang, Y. Tian, Z. Wang, J. Pu, F. Ciucci and B. Chi, Chem. Eng. J., 2024, 485, 149970 CrossRef CAS.
- P. Xiaokaiti, T. Yu, A. Yoshida, X. Du, X. Hao, Y. Kasai, A. Abudula and G. Guan, J. Alloys Compd., 2020, 831, 154738 CrossRef CAS.
- X. Han, P. Chen, M. Wu, Z. Song, C. Li, L. Zhang and L. Zhang, Ceram. Int., 2022, 48, 26440–26451 CrossRef CAS.
- Y. Cao, Z. Zhu, Y. Zhao, W. Zhao, Z. Wei and T. Liu, J. Power Sources, 2020, 455, 227951 CrossRef CAS.
- G. Hong, T. W. Kim, M. J. Kwak, J. Song, Y. Choi, S.-K. Woo, M. H. Han, C. H. Cho and S.-D. Kim, J. Alloys Compd., 2020, 846, 156154 CrossRef CAS.
- T. Su, T. Zhang, H. Xie, J. Zhong and C. Xia, Int. J. Hydrogen Energy, 2022, 47, 16272–16282 CrossRef CAS.
- J. M. Porras-Vazquez, T. Pike, C. A. Hancock, J. F. Marco, F. J. Berry and P. R. Slater, J. Mater. Chem. A, 2013, 1, 11834–11841 RSC.
- X. Meng, X. Liu, D. Han, H. Wu, J. Li and Z. Zhan, J. Power Sources, 2014, 252, 58–63 CrossRef CAS.
- L. dos Santos-Gómez, J. Compana, S. Bruque, E. Losilla and D. Marrero-López, J. Power Sources, 2015, 279, 419–427 CrossRef.
- X. Sun, Y. Xia, B. Wang, B. Li, L. Ma, J. Chen and B. Chi, Chem. Eng. J., 2024, 479, 147598 CrossRef CAS.
- L. Liu, M. Wu, X. Zhou, H. Chen, X. Qian, Z. Wang, F. He, Y. Sheng, J. Qian and L. Pan, ACS Appl. Energy Mater., 2022, 6, 841–855 CrossRef.
- B. Li, J. T. Irvine, J. Ni and C. Ni, Appl. Energy, 2022, 306, 118117 CrossRef CAS.
- S. Wang, Y. Wu, H. Chen, Y. Gao, J. Liu, M. Li, Z. Lü and B. Wei, J. Alloys Compd., 2023, 932, 167685 CrossRef CAS.
- P. Xiaokaiti, T. Yu, A. Yoshida, X. Du, X. Hao, Y. Kasai, A. Abudula and G. Guan, J. Alloys Compd., 2020, 831, 154738 CrossRef CAS.
- P. I. Cowin, R. Lan, C. T. G. Petit, D. Du, K. Xie, H. Wang and S. Tao, Solid State Ionics, 2017, 301, 99–105 CrossRef CAS.
- W. Zhang, M. Shiraiwa, N. Wang, T. Ma, K. Fujii, E. Niwa and M. Yashima, J. Ceram. Soc. Jpn., 2018, 126, 814–819 CrossRef CAS.
- H. Chen, Z. Guo, L. A. Zhang, Y. Li, F. Li, Y. Zhang, Y. Chen, X. Wang, B. Yu, J.-m. Shi, J. Liu, C. Yang, S. Cheng, Y. Chen and M. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 39785–39793 CrossRef CAS PubMed.
- Y. Chen, W. Jung, Z. Cai, J. J. Kim, H. L. Tuller and B. Yildiz, Energy Environ. Sci., 2012, 5, 7979 RSC.
- W. Jung and H. L. Tuller, Energy Environ. Sci., 2012, 5, 5370–5378 RSC.
- Z. Zhang, S. Chen, H. Zhang, C. Yao, W. Zhang, T. Qu, T. Wang, H. Wang, X. Lang and K. Cai, J. Colloid Interface Sci., 2024, 655, 157–166 CrossRef CAS PubMed.
- J. H. Kim, Appl. Surf. Sci., 2011, 258, 350–355 CrossRef.
- W. Wang, L. Shi, D. Lan and Q. Li, J. Power Sources, 2018, 377, 1–6 CrossRef.
- P. Zhang, G. Guan, D. S. Khaerudini, X. Hao, C. Xue, M. Han, Y. Kasai and A. Abudula, J. Power Sources, 2014, 266, 241–249 CrossRef.
- D. Zou, Y. Yi, Y. Song, D. Guan, M. Xu, R. Ran, W. Wang, W. Zhou and Z. Shao, J. Mater. Chem. A, 2022, 10, 5381 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta06084a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |