
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDinitrogen reduction chemistry with scandium provides a complex with two side-on (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) N)2− ligands bound to one metal: (C5Me5)Sc[(µ-η2:η2-N2)Sc(C5Me5)2]2†
N)2− ligands bound to one metal: (C5Me5)Sc[(µ-η2:η2-N2)Sc(C5Me5)2]2†
Joshua D. Queen a,
Ahmadreza Rajabia,
Quinn E. Goudzwaarda,
Qiong Yuanb,
Dang Khoa Nguyena,
Joseph W. Zillera,
Filipp Furche*a,
Zhenfeng Xi*b and
William J. Evans*a
a,
Ahmadreza Rajabia,
Quinn E. Goudzwaarda,
Qiong Yuanb,
Dang Khoa Nguyena,
Joseph W. Zillera,
Filipp Furche*a,
Zhenfeng Xi*b and
William J. Evans*a
aDepartment of Chemistry, University of California, Irvine, 92697, California, USA. E-mail: jqueen@uci.edu; arajabis@uci.edu; qgoudzwa@uci.edu; khoan16@uci.edu; wevans@uci.edu; filipp.furche@uci.edu; jziller@uci.edu
bBeijing National Laboratory for Molecular Sciences (BNLMS), Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry, Peking University, Beijing 100871, China. E-mail: yuanqiongyin@pku.edu.cn; zfxi@pku.edu.cn
First published on 30th August 2024
Abstract
Although there are few reduced dinitrogen complexes of scandium, this metal has revealed a new structural type in reductive dinitrogen chemistry by reduction of bis(pentamethylcyclopentadienyl) scandium halides under N2. Reduction of 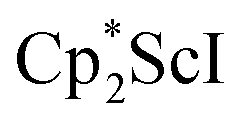 (Cp* = C5Me5) with potassium graphite (KC8) under dinitrogen generates the dark blue paramagnetic complex
(Cp* = C5Me5) with potassium graphite (KC8) under dinitrogen generates the dark blue paramagnetic complex 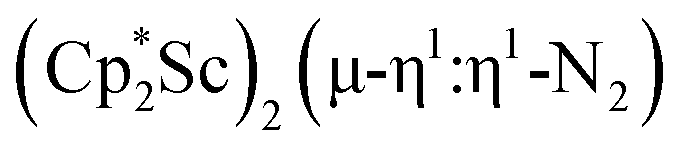 , 1. This end-on bridging (N
, 1. This end-on bridging (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N)2− complex is a diradical with a magnetic moment of 2.8µB. Upon further reduction of 1 with KC8, the orange diamagnetic trimetallic complex
N)2− complex is a diradical with a magnetic moment of 2.8µB. Upon further reduction of 1 with KC8, the orange diamagnetic trimetallic complex 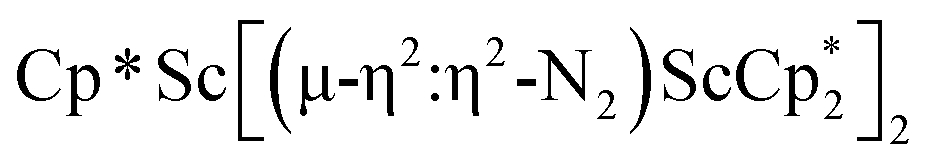 , 2, is obtained. This complex has an unprecedented structure in which two side-on bridging (NN)2− ligands are bound to the central (Cp*Sc)2+ moiety. Complex 2 can also be obtained directly from reduction of
, 2, is obtained. This complex has an unprecedented structure in which two side-on bridging (NN)2− ligands are bound to the central (Cp*Sc)2+ moiety. Complex 2 can also be obtained directly from reduction of  or a mixture of
or a mixture of  and
and  with KC8. The reaction of
with KC8. The reaction of 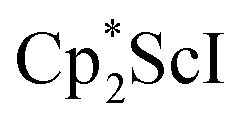 with KC8 in the presence of 18-crown-6 or 2.2.2-cryptand affords 2 along with small amounts of
with KC8 in the presence of 18-crown-6 or 2.2.2-cryptand affords 2 along with small amounts of 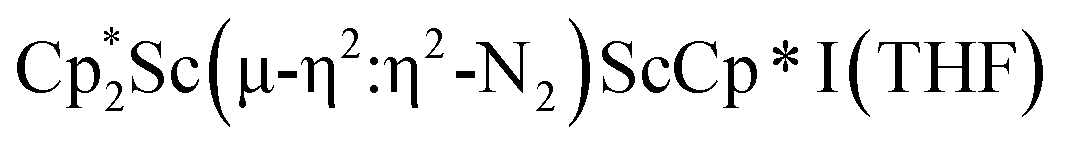 , 3, which is green at room temperature and purple at low temperature and displays a mixture of side-on and end-on bridging isomers in the crystal structure collected at −180 °C. Density functional theory (DFT) calculations are consistent with a triplet ground state for the end-on complex 1 and singlet ground states for the side-on complexes 2 and 3.
, 3, which is green at room temperature and purple at low temperature and displays a mixture of side-on and end-on bridging isomers in the crystal structure collected at −180 °C. Density functional theory (DFT) calculations are consistent with a triplet ground state for the end-on complex 1 and singlet ground states for the side-on complexes 2 and 3.
Introduction
Metal dinitrogen complexes are an important class of compounds that are of interest for chemical processes that require reduction of the inert N2 molecule.1–5 The first reported N2 complex, the monometallic [Ru(NH3)5(N2)]2+, was found to have a terminal end-on bound dinitrogen ligand.6 Subsequently, bimetallic complexes with bridging dinitrogen ligands were discovered which had both end-on and side-on coordination of the N2 unit.7–10 There are now hundreds of examples of crystallographically characterized metal dinitrogen compounds9 with approximately 600 terminal compounds, 500 bridging end-on examples, and almost 100 side-on complexes reported in the Cambridge Structural Database.11 With such an extensive effort on this topic, it is unusual to find new structural types of reduced dinitrogen complexes.Although metal dinitrogen chemistry has been heavily studied, for the first member of the transition metal series, scandium, only three structurally characterized examples of dinitrogen complexes have been reported to date, Fig. 1.12–14 The first of these, [(C5Me4H)2Sc]2(µ-η2:η2-N2),12 had a side-on bridging (NN)2− ligand with a planar Sc2N2 core that was the hallmark of rare-earth element (Sc, Y, and the lanthanides) dinitrogen complexes from 1988 until 2017.15–26
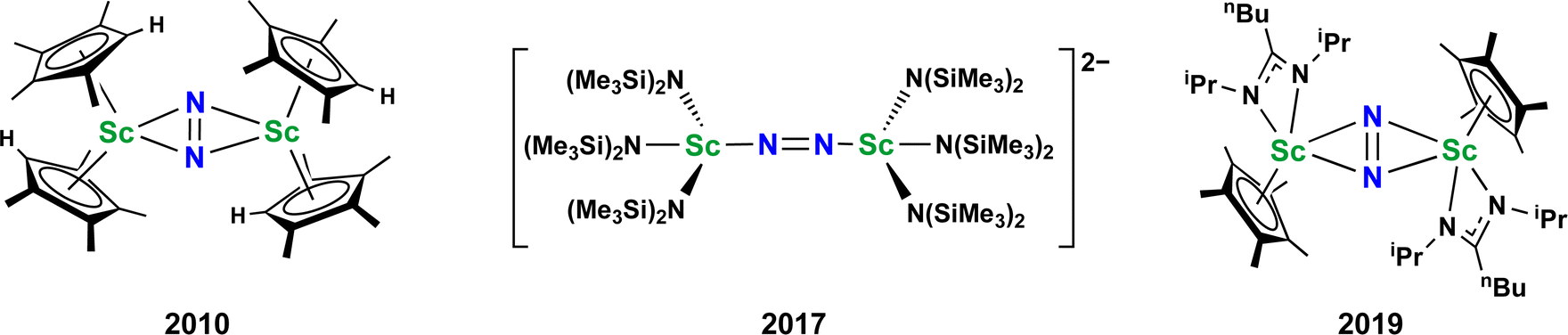 | ||
| Fig. 1 Previously reported dinitrogen compounds of scandium. In all cases the N2 is reduced to an (NN)2− ligand. | ||
However, the second example, [K(crypt)]2{[(R2N)3Sc]2(µ-η1:η1-N2)} (crypt = 2.2.2-cryptand, R = SiMe3), was ground-breaking in that it revealed the first end-on bridging coordination mode for a rare-earth (NN)2− complex13 and was found to readily revert to its Sc(II) amide precursor, [K(crypt)][Sc(NR2)3] with exposure to light. Subsequently, it was found with the larger rare-earth metal analogs of this amide-ligated compound that the dianonic {[(R2N)3Ln]2(µ-ηx:ηx-N2)}2− complexes could contain mixtures of end-on and side-on isomers in the solid state, i.e. x = 1,2 for Ln = Nd, Gd.27,28 It is relevant to results reported here that single crystals of the Nd compound were found to isomerize on the diffractometer at around −90 °C.
The third example of a reduced dinitrogen complex of scandium, a side-on complex with a mixed ligand cyclopentadienyl amidinate coordination environment, {[nBuC(NiPr)2]Cp*Sc}2(µ-η2:η2-N2) (Cp* = C5Me5),14 was also special. This complex could be reduced further to the (N2)3− species, [{[nBuC(NiPr)2]Cp*Sc}2(µ-η2:η2-N2)]1−,14 chemistry that had to that point only been observed for the bis(silyl)amido compounds {[(R2N)2(THF)Ln]2(µ-η2:η2-N2)}1−.29,30 Furthermore, the [{[nBuC(NiPr)2]Cp*Sc}2(µ-η2:η2-N2)]1− complex could be stepwise functionalized at the bridging nitrogen to release hydrazine derivatives.14
We now report a new structural type of reduced dinitrogen complex previously unobserved with any metal by studying the reduction of bis(pentamethylcyclopentadienyl) scandium halides. Recently we reported that treating  with KC8 generates a Sc(II) compound with spectroscopic features similar to those of the first stable scandium metallocene, Cpttt2Sc (Cpttt = C5H2tBu3).31 However, the product of the
with KC8 generates a Sc(II) compound with spectroscopic features similar to those of the first stable scandium metallocene, Cpttt2Sc (Cpttt = C5H2tBu3).31 However, the product of the  reaction, which forms red solutions at −78 °C under argon and is possibly the scandocene
reaction, which forms red solutions at −78 °C under argon and is possibly the scandocene 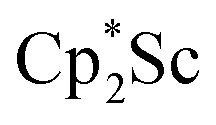 , is highly reactive with arene and ether solvents through C–H or C–O bond activation. To further understand the nature of the
, is highly reactive with arene and ether solvents through C–H or C–O bond activation. To further understand the nature of the  reduction product, we investigated its reactivity with N2. This has resulted in the isolation of the first neutral end-on bridging dinitrogen complex of scandium,
reduction product, we investigated its reactivity with N2. This has resulted in the isolation of the first neutral end-on bridging dinitrogen complex of scandium, 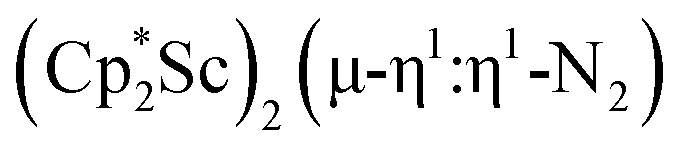 , 1.
, 1.
The subsequent treatment of 1 with KC8, rather than forming a stable compound containing an (N2)3− anion, gave the trimetallic complex  , 2. To our knowledge, this is the first example of a complex containing two side-on dinitrogen ligands attached to a single metal. Previously, a bimetallic mixed-valent titanium complex with two bridging (NN)2− ligands connecting the metals, {[(R2N)2Ti]2(µ-η2:η2-N2)2}1− (R = SiMe3) was reported,32 but reanalysis of this product revealed it to be the toluene inverse sandwich complex {[(R2N)2Ti]2(µ-C6H5Me)}1−.33 Complexes with two end-on dinitrogen ligands per metal are known 9 such as
, 2. To our knowledge, this is the first example of a complex containing two side-on dinitrogen ligands attached to a single metal. Previously, a bimetallic mixed-valent titanium complex with two bridging (NN)2− ligands connecting the metals, {[(R2N)2Ti]2(µ-η2:η2-N2)2}1− (R = SiMe3) was reported,32 but reanalysis of this product revealed it to be the toluene inverse sandwich complex {[(R2N)2Ti]2(µ-C6H5Me)}1−.33 Complexes with two end-on dinitrogen ligands per metal are known 9 such as 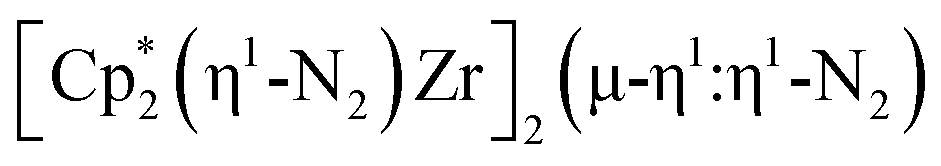 34 and [(Nacnac)Cr(µ-η1:η1-N2)]x (x = 3, 4; Nacnac = HC{MeCN(2,6-Me2C6H3)}2).35 However, multiple side-on coordination to a single metal center has so-far been elusive. In the course of these experiments we also identified the mixed ligand cyclopentadienyl halide complex,
34 and [(Nacnac)Cr(µ-η1:η1-N2)]x (x = 3, 4; Nacnac = HC{MeCN(2,6-Me2C6H3)}2).35 However, multiple side-on coordination to a single metal center has so-far been elusive. In the course of these experiments we also identified the mixed ligand cyclopentadienyl halide complex,  , 3 (x = 1, 2), which displays temperature dependent side-on/end-on isomerization and has primarily a side-on structure with some end-on disorder in the solid state.
, 3 (x = 1, 2), which displays temperature dependent side-on/end-on isomerization and has primarily a side-on structure with some end-on disorder in the solid state.
Results
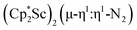 , 1
, 1
The end-on bridging dinitrogen complex, 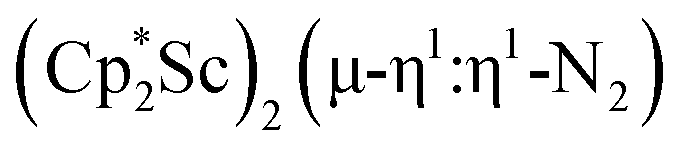
 , 1, was prepared by reduction of
, 1, was prepared by reduction of 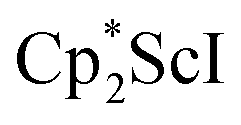 with potassium graphite (KC8) under a N2 atmosphere according to eqn (1). Upon addition of KC8 to a stirred yellow solution of
with potassium graphite (KC8) under a N2 atmosphere according to eqn (1). Upon addition of KC8 to a stirred yellow solution of 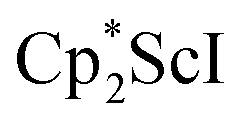 in Et2O, a dark blue solution immediately forms. Repeated extractions of the resulting mixture with Et2O followed by removal of the solvent under reduced pressure gives 1 as a dark blue powder in ca. 80% yield. Storage of a saturated Et2O solution of 1 at −35 °C afforded dark blue-purple crystals that were suitable for X-ray diffraction.
in Et2O, a dark blue solution immediately forms. Repeated extractions of the resulting mixture with Et2O followed by removal of the solvent under reduced pressure gives 1 as a dark blue powder in ca. 80% yield. Storage of a saturated Et2O solution of 1 at −35 °C afforded dark blue-purple crystals that were suitable for X-ray diffraction.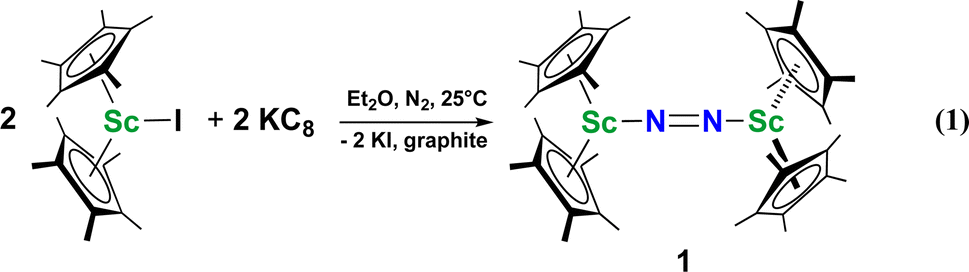
The structure of 1, Fig. 2, shows that the overall molecular structure has a tetrahedral arrangement of the four Cp* ligand ring centroids which are all equivalent through crystallographic symmetry. A small positional disorder of the Sc atom site in the structure of 1 suggests the presence of a co-crystallized ca. 5% impurity of 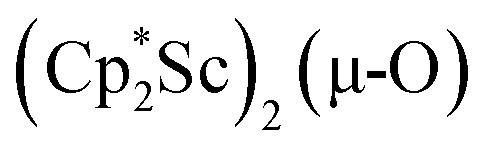 , which is discussed in a later section. Co-crystallization of (O)2− oxide side products with rare-earth element (NN)2− complexes has previously been observed28 and attempts to independently prepare this oxide are discussed later.
, which is discussed in a later section. Co-crystallization of (O)2− oxide side products with rare-earth element (NN)2− complexes has previously been observed28 and attempts to independently prepare this oxide are discussed later.
 | ||
Fig. 2 Thermal ellipsoid plot (30%) of 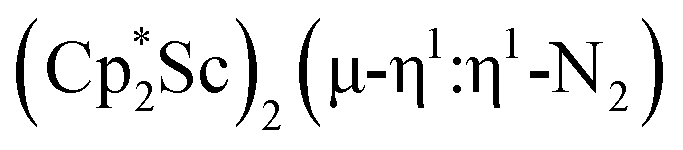 , 1, with hydrogen atoms and disordered Sc atoms not shown for clarity. , 1, with hydrogen atoms and disordered Sc atoms not shown for clarity. | ||
The Sc–Cent (Cent = Cp* ring centroid) distance in 1 is 2.17 Å with a Cent–Sc–Cent angle of 146.7°. The analogous metrical parameters in the side-on complex, [(C5Me4H)2Sc]2(µ-η2:η2-N2),12 are 2.20 Å and 131.0°. The Sc–N–N–Sc core in 1 is strictly linear, enforced by crystallographic symmetry, and has a 2.049(3) Å Sc–N distance. The 1.174(5) Å N–N distance is on the shorter end of the range of N–N distances reported for end-on bridging rare-earth element (N2)2− compounds (Table 1). In comparison, the end-on (NN)2− complex [K(crypt)]2{[(R2N)3Sc]2(µ-η1:η1-N2)} has Sc–N distances of 2.032(2) and 2.030(2) Å, an N–N bond length of 1.221(3) Å, and Sc–N–N angles of 178.2(2) and 179.7(2)°.
| Compound | N–N bond length (Å) | Raman shift (cm−1) | Ref. |
|---|---|---|---|
| a R = SiMe3. | |||
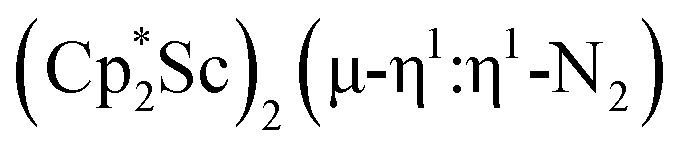 , 1 , 1 |
1.174(5) | 1595 | This work |
| [K(crypt)]2{[(R2N)3Sc]2(µ-η1:η1-N2)}a | 1.221(3) | 1644 | 13 |
| [K(crypt)]2{[(R2N)3Y]2(µ-η1:η1-N2)}a | 1.216(7), 1.229(7) | 1635 | 27 |
| [K(crypt)]2{[(R2N)3Tb]2(µ-η1:η1-N2)}a | 1.217(3) | 1623 | 28 |
| (Cpttt2Gd)2(µ-η1:η1-N2) | 1.130(8) | 1623 | 36 |
| (Cpttt2Tb)2(µ-η1:η1-N2) | 1.175(8) | 1621 | 36 |
| (Cpttt2Dy)2(µ-η1:η1-N2) | 1.215(13) | 1618 | 36 |
We note that the crystallographically determined N–N bond lengths in rare-earth metal dinitrogen compounds can be variable between crystals of the same compound, whereas the N–N stretching frequency is consistent between samples.37 The Raman spectrum of 1 shows the N–N stretching vibration at 1595 cm−1 which is less than that of {[(R2N)3Sc]2(µ-η1:η1-N2)}2− at 1644 cm−1 and closer to the ca. 1620 cm−1 stretch for the neutral end-on bridging complexes (Cpttt2Ln)2(µ-η1:η1-N2) (Ln = Gd, Tb, Dy),36 Table 1.
Compound 1 is a diradical and has a broad 1H NMR signal at 29 ppm in C6D6. The magnetic moment was determined by Evans' method NMR measurement38–40 to be 2.8µB, consistent with the 2.83µB spin-only magnetic moment expected for two unpaired electrons. Density functional theory calculations are in agreement with a predicted triplet ground state and are described further in the Computational Details section.
Compound 1 is soluble in hexane, benzene, toluene, and Et2O, but upon dissolving in THF the dark blue color immediately fades to pale yellow. The thermal stability of 1 in solution was investigated by heating a C6D6 solution of 1 inside a J-Young NMR tube sealed under N2. The filled portion of the NMR tube was immersed in a 100 °C oil bath and over 6 hours the intense blue solution faded to pale yellow. The 1H NMR spectrum of this solution revealed the presence of a new Cp* methyl resonance at 1.73 ppm consistent with the formation of  .41 Signals corresponding to
.41 Signals corresponding to 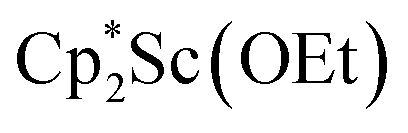 31 were also seen, which could result from reactions with trace amounts of Et2O present in the sample. We previously found that these phenyl and ethoxide products formed from C–H and C–O bond activation of benzene or Et2O by the
31 were also seen, which could result from reactions with trace amounts of Et2O present in the sample. We previously found that these phenyl and ethoxide products formed from C–H and C–O bond activation of benzene or Et2O by the 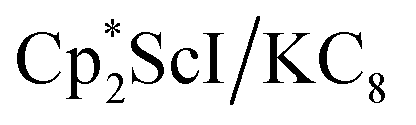 reduction system in these solvents.31 The other expected products from these reactions,
reduction system in these solvents.31 The other expected products from these reactions,  and
and  , were not observed as they are expected to react further with the C6D6 solvent.41 In the solid state, 1 was found to melt over the range of 183–187 °C during which the color changed to red. Gas evolution was observed and the red melt quickly decomposed to a white solid. These results are consistent with the liberation of dinitrogen upon thermolysis of 1 and concomitant formation of
, were not observed as they are expected to react further with the C6D6 solvent.41 In the solid state, 1 was found to melt over the range of 183–187 °C during which the color changed to red. Gas evolution was observed and the red melt quickly decomposed to a white solid. These results are consistent with the liberation of dinitrogen upon thermolysis of 1 and concomitant formation of  although this highly reactive species has not yet been isolated.
although this highly reactive species has not yet been isolated.
The stability of 1 towards photolysis was also examined, as [K(crypt)]2{[(R2N)3Sc]2(µ-η1:η1-N2)} was found to eliminate N2 under ambient light.13 Solutions of 1 in C6D6 sealed under N2 showed no decomposition products when the samples were left on the benchtop for a week. One of these samples in a quartz tube was irradiated with a mercury arc lamp with a 200–400 nm emission range for 4 hours and no formation of  or other products were observed in the 1H NMR spectrum.
or other products were observed in the 1H NMR spectrum.
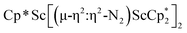 , 2
, 2
The reduction of complex 1 was attempted with the aim of forming a 
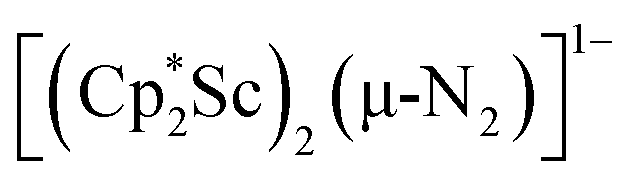 product containing an (N2)3− moiety.14,29,30 Treating an Et2O solution of 1 and 18-crown-6 with 1 equiv. of KC8 under a dinitrogen atmosphere resulted in a gradual color change to dark orange. Evaporation of the supernatant solution gave orange-brown crystals which were identified by X-ray diffraction as the trimetallic complex
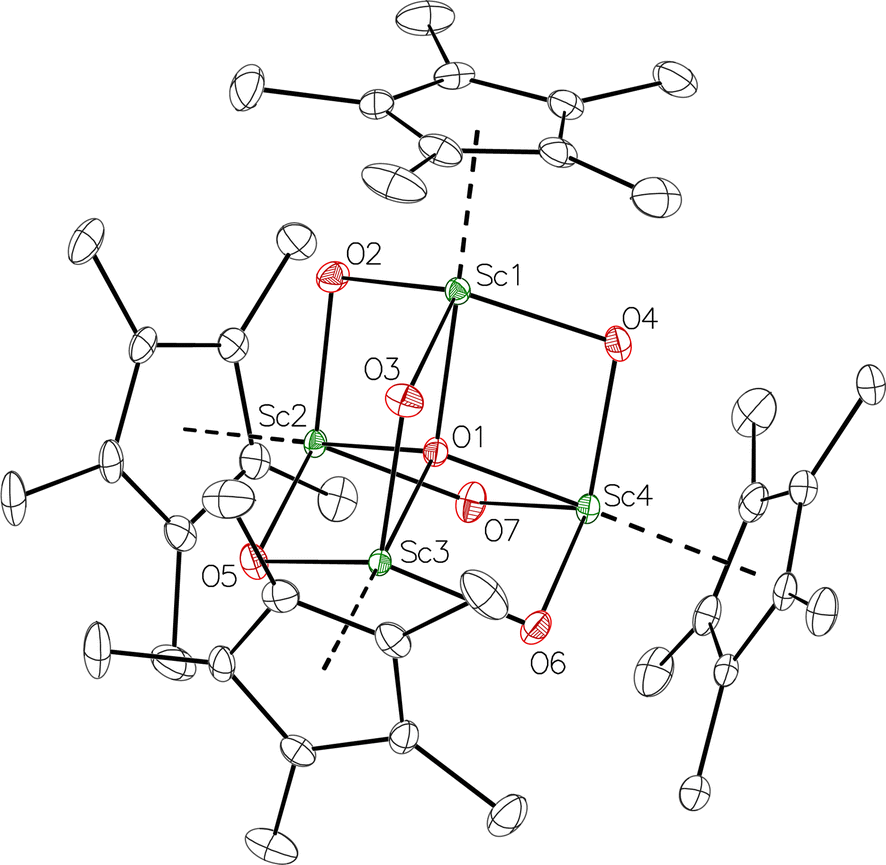
product containing an (N2)3− moiety.14,29,30 Treating an Et2O solution of 1 and 18-crown-6 with 1 equiv. of KC8 under a dinitrogen atmosphere resulted in a gradual color change to dark orange. Evaporation of the supernatant solution gave orange-brown crystals which were identified by X-ray diffraction as the trimetallic complex  , 2, Fig. 3. To our knowledge, complexes with two side-on dinitrogen ligands bound to a single metal have not been previously reported. The deliberate synthesis of 2 from 1 was found to proceed in high yield according to the stoichiometry in Scheme 1. Although the intermediates are not yet understood, this reaction can be balanced by the reduction of an additional equivalent of N2 to (NN)2− and elimination of KCp*.
, 2, Fig. 3. To our knowledge, complexes with two side-on dinitrogen ligands bound to a single metal have not been previously reported. The deliberate synthesis of 2 from 1 was found to proceed in high yield according to the stoichiometry in Scheme 1. Although the intermediates are not yet understood, this reaction can be balanced by the reduction of an additional equivalent of N2 to (NN)2− and elimination of KCp*.
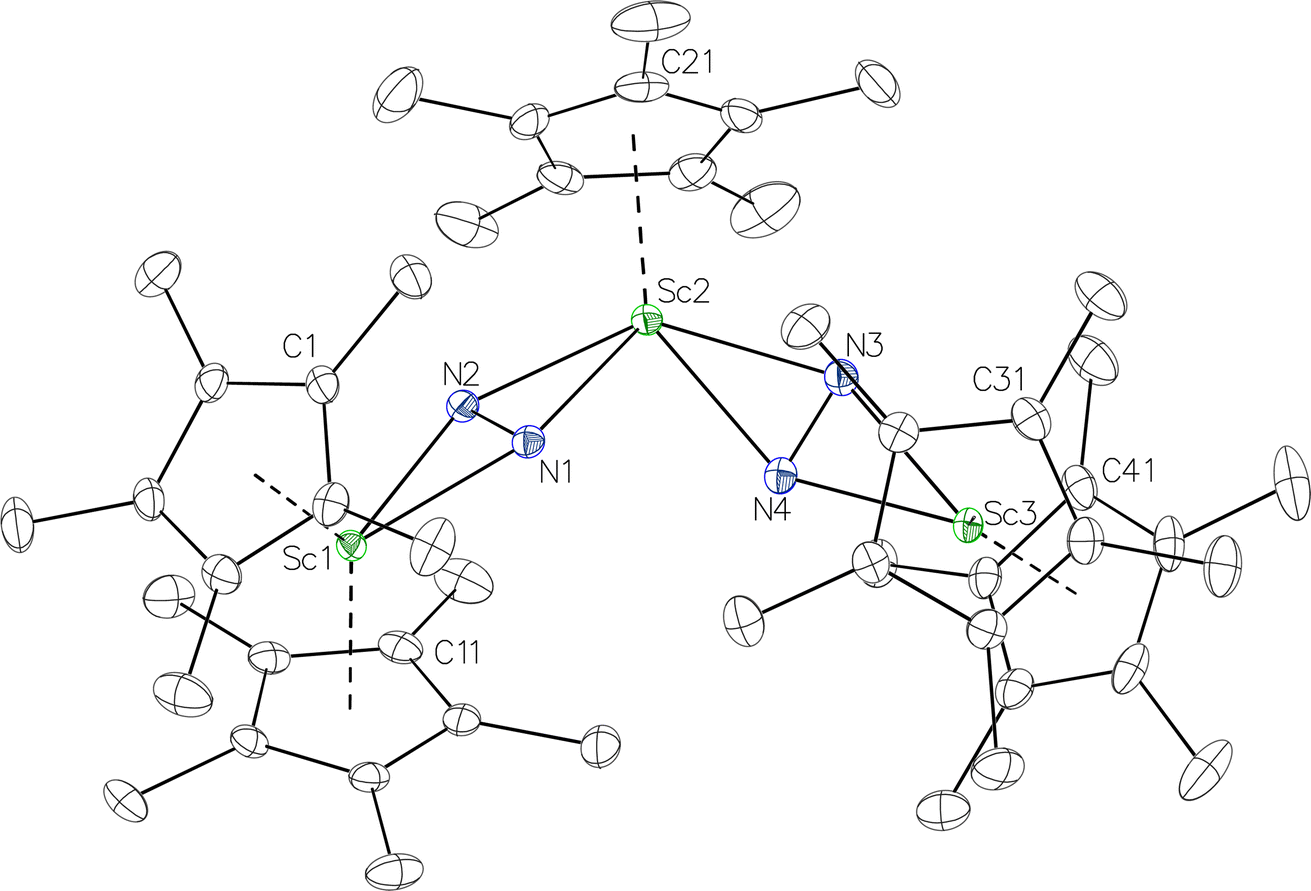 | ||
Fig. 3 Molecular structure of 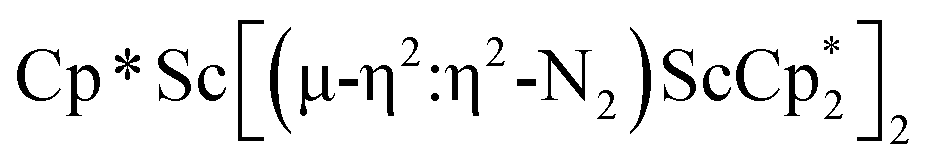 , 2 with thermal ellipsoids shown at 30% and hydrogen atoms omitted for clarity. , 2 with thermal ellipsoids shown at 30% and hydrogen atoms omitted for clarity. | ||
 | ||
Scheme 1 Synthetic routes to  , 2. , 2. | ||
Alternatively, 2 could be prepared in a “one-pot” fashion from halide precursors, Scheme 1. The reaction of 3 equiv. 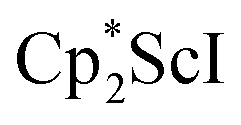 with 4 equiv. of KC8 in Et2O initially formed of a dark blue solution of 1 followed by a gradual change to orange. Complex 2 was also prepared independently by reduction of a mixture of the chloride precursors
with 4 equiv. of KC8 in Et2O initially formed of a dark blue solution of 1 followed by a gradual change to orange. Complex 2 was also prepared independently by reduction of a mixture of the chloride precursors  and
and  in toluene. Initially, ScCl3 was combined with 2 equiv. of KCp* in THF. Exchange of the reaction solvent to toluene and treatment of the resulting mixture with KC8 under dinitrogen at room temperature led to the isolation of complex 2. The reaction between ScCl3 and 2 equiv. of KCp* in THF yielded crystals of both the solvated and unsolvated complexes,
in toluene. Initially, ScCl3 was combined with 2 equiv. of KCp* in THF. Exchange of the reaction solvent to toluene and treatment of the resulting mixture with KC8 under dinitrogen at room temperature led to the isolation of complex 2. The reaction between ScCl3 and 2 equiv. of KCp* in THF yielded crystals of both the solvated and unsolvated complexes,  and
and 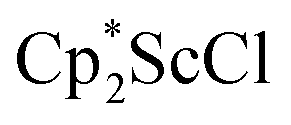 , which were characterized by X-ray diffraction (see ESI†) and by comparison of their 1H NMR spectra with literature values.41,42 Complex 2 was not obtained by reacting either of these two compounds separately with KC8 in toluene, but only reduction of the mixture of
, which were characterized by X-ray diffraction (see ESI†) and by comparison of their 1H NMR spectra with literature values.41,42 Complex 2 was not obtained by reacting either of these two compounds separately with KC8 in toluene, but only reduction of the mixture of 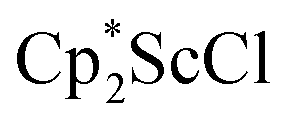 and
and  in toluene provided complex 2 in good yield as shown in Scheme 1. Evidently, the presence of some THF facilitates the reduction of the Sc(III) complexes without causing decomposition of intermediates such as 1.
in toluene provided complex 2 in good yield as shown in Scheme 1. Evidently, the presence of some THF facilitates the reduction of the Sc(III) complexes without causing decomposition of intermediates such as 1.
In the X-ray crystal structure of 2, the two 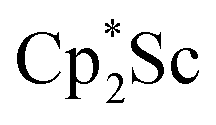 moieties are not related by symmetry and the two Sc2N2 cores do not have the rigorously planar side-on bridging structure that is usually seen in rare-earth element dinitrogen compounds.9,26 Rather the Sc2N2 cores have a small distortion toward a butterfly structure which has been seen in some Zr and Hf side-on bridging (NN)2− compounds.9,43 The Sc2N2 fold angles in 2 are 7.47° for the N1–N2 moiety and 15.13° for the N3–N4 moiety.
moieties are not related by symmetry and the two Sc2N2 cores do not have the rigorously planar side-on bridging structure that is usually seen in rare-earth element dinitrogen compounds.9,26 Rather the Sc2N2 cores have a small distortion toward a butterfly structure which has been seen in some Zr and Hf side-on bridging (NN)2− compounds.9,43 The Sc2N2 fold angles in 2 are 7.47° for the N1–N2 moiety and 15.13° for the N3–N4 moiety.
The structural details of 2 are summarized in Table 2. The Sc–N distances are all in the narrow range of 2.140(2)–2.189(2) Å. Interestingly, within this range each nitrogen has either two shorter distances (for N2 and N3) or two longer distances (for N1 and N4), i.e. there is no short/long combination for each nitrogen donor atom as is sometimes seen in bridging ligands. The 2.18–2.20 Å Sc–Cent distances for the outer two scandium atoms, Sc1 and Sc3, are similar to the 2.17 Å value for the central Sc2, although the coordination numbers of the two types of scandium atoms are different. The 140.1° and 141.5° Cent–Sc–Cent angles of Sc1 and Sc3 are similar and smaller than the 146.7° angle in end-on 1, but larger than the 131.0° angle in side-on [(C5Me4H)2Sc]2(µ-η2:η2-N2).12
 , 2, and the side-on isomer of
, 2, and the side-on isomer of 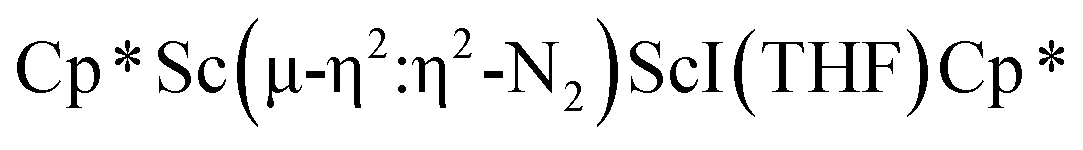 , 3
, 3
|
|
||||
|---|---|---|---|---|---|
| Sc1–N1 | 2.178(2) | N1–N2 | 1.230(2) | N1–N2 | 1.246(5) |
| Sc1–N2 | 2.160(2) | N3–N4 | 1.233(2) | Sc1–N1 | 2.125(3) |
| Sc2–N1 | 2.189(2) | Sc1–Cent | 2.20, 2.19 | Sc1–N2 | 2.168(4) |
| Sc2–N2 | 2.177(2) | Sc2–Cent | 2.17 | Sc2–N1 | 2.184(3) |
| Sc2–N3 | 2.158(2) | Sc3–Cent | 2.19, 2.18 | Sc2–N2 | 2.185(4) |
| Sc2–N4 | 2.178(2) | Cent–Sc1–Cent | 140.1 | Sc1–I1 | 2.881(1) |
| Sc3–N3 | 2.140(2) | Cent–Sc3–Cent | 141.5 | Sc1–O1 | 2.231(3) |
| Sc3–N4 | 2.176(2) | (Sc1N1N2)–(N1N2Sc2) | 7.47(5)° | Sc1–Cent | 2.21 |
| (Sc2N3N4)–(N3N4Sc3) | 15.13(6)° | Sc2–Cent | 2.20, 2.20 | ||
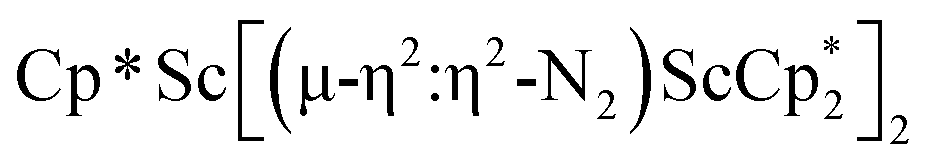
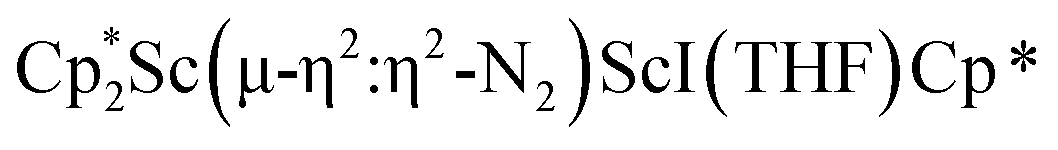
The 1H NMR spectrum of 2 shows two singlets corresponding to the Cp* methyl groups in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 ratio, indicating that the two
:4 ratio, indicating that the two  groups are equivalent in solution. The 15N NMR spectrum of the isotopically labeled compound
groups are equivalent in solution. The 15N NMR spectrum of the isotopically labeled compound 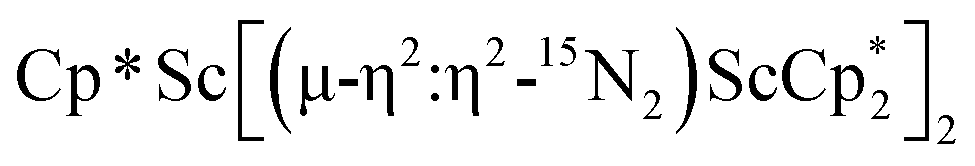 , 15N2, shows one signal at 771 ppm. This shift is further downfield than the 385 ppm shift of [(C5Me4H)2Sc]2(µ-η2:η2-N2)12 and the 495–569 range for [A2(THF)Ln]2(µ-η2:η2-N2) (A = Cp*, C5Me4H, N(SiMe3)2; Ln = Y, La, Lu),23 but these shifts obviously have a wide range.10 The Raman spectrum of 2 shows a single N–N stretch at 1460 cm−1 which is shifted to 1415 cm−1 in the 15N2 analogue. This stretching frequency is close in energy to those of other rare-earth metallocene side-on (NN)2− compounds which have N–N stretches in the ca. 1400–1470 cm−1 range.37
, 15N2, shows one signal at 771 ppm. This shift is further downfield than the 385 ppm shift of [(C5Me4H)2Sc]2(µ-η2:η2-N2)12 and the 495–569 range for [A2(THF)Ln]2(µ-η2:η2-N2) (A = Cp*, C5Me4H, N(SiMe3)2; Ln = Y, La, Lu),23 but these shifts obviously have a wide range.10 The Raman spectrum of 2 shows a single N–N stretch at 1460 cm−1 which is shifted to 1415 cm−1 in the 15N2 analogue. This stretching frequency is close in energy to those of other rare-earth metallocene side-on (NN)2− compounds which have N–N stretches in the ca. 1400–1470 cm−1 range.37
Isolation of 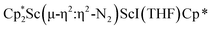 , 3
, 3
In previous attempts to isolate Cp* ligated Sc(II) species, 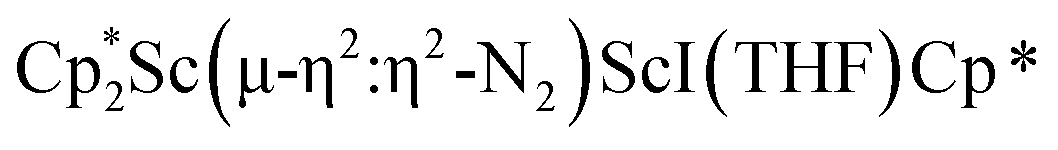
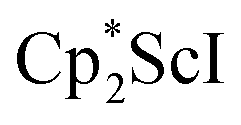 was treated with a combination of KC8 and 18-crown-6 under an argon atmosphere and a crystal of the ligand redistribution product
was treated with a combination of KC8 and 18-crown-6 under an argon atmosphere and a crystal of the ligand redistribution product  was isolated.31 The analogous reaction investigated under N2 atmosphere resulted again in the formation of 2. However, the black precipitates from this reaction, presumably graphite and KCp*, had a green hue and were extracted with THF to afford a green solution. Removal of the solvent and recrystallization of the residue from n-hexane at −35 °C gave green crystals, which turned purple under the cold stream of the X-ray diffractometer, and were identified to be
was isolated.31 The analogous reaction investigated under N2 atmosphere resulted again in the formation of 2. However, the black precipitates from this reaction, presumably graphite and KCp*, had a green hue and were extracted with THF to afford a green solution. Removal of the solvent and recrystallization of the residue from n-hexane at −35 °C gave green crystals, which turned purple under the cold stream of the X-ray diffractometer, and were identified to be 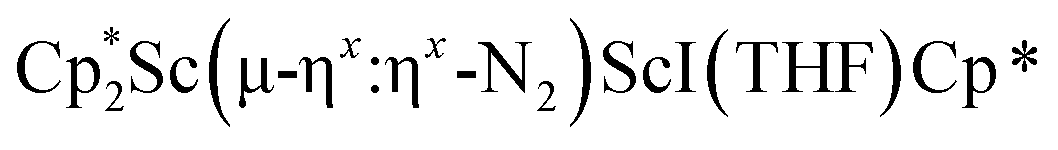 (x = 1, 2), 3, Fig. 4.
(x = 1, 2), 3, Fig. 4.
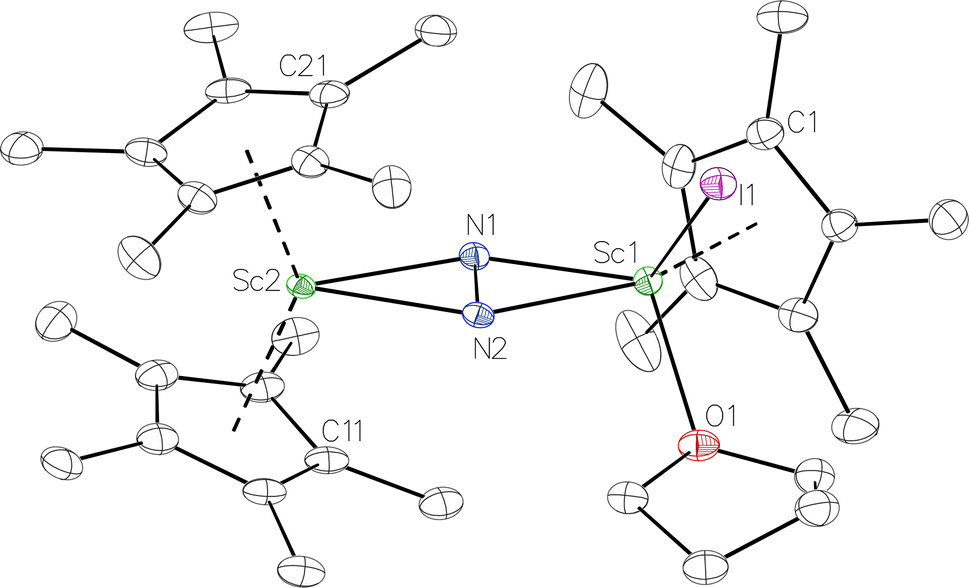 | ||
Fig. 4 Molecular structure of the major 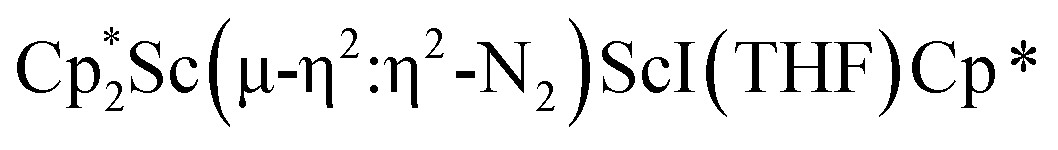 component of 3 with thermal ellipsoids shown at 30% and the Sc(µ-η1:η1-N2)Sc disordered core and hydrogen atoms omitted for clarity. component of 3 with thermal ellipsoids shown at 30% and the Sc(µ-η1:η1-N2)Sc disordered core and hydrogen atoms omitted for clarity. | ||
The structure of 3 shows a disordered Sc–N2–Sc core with 82% of the structure binding N2 in a side-on fashion and the remainder binding end-on. For the major side-on component of the structure of 3, 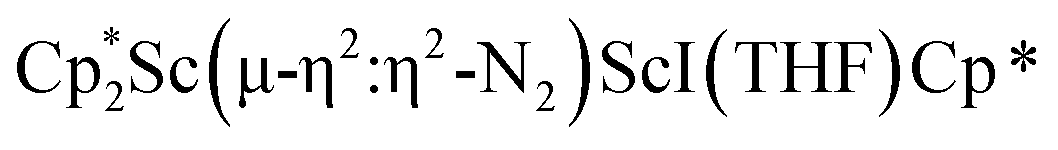 , the
, the 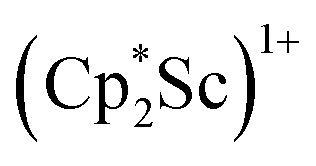 moiety involving Sc2 has metrical parameters that are very similar to those in complex 2, Table 2. The 1.246(5) Å N–N distance is also similar. The [Cp*ScI(THF)]1+ moiety has a similar Sc1–Cent distance and one similar Sc–N distance, but the 2.125(3) Å Sc1–N1 distance is shorter than the other Sc–N distances in 2 and 3. The N–N stretch of 3 appears at 1700 cm−1 in the Raman spectrum which is on the high end of the known rare-earth element (NN)2− compounds.37
moiety involving Sc2 has metrical parameters that are very similar to those in complex 2, Table 2. The 1.246(5) Å N–N distance is also similar. The [Cp*ScI(THF)]1+ moiety has a similar Sc1–Cent distance and one similar Sc–N distance, but the 2.125(3) Å Sc1–N1 distance is shorter than the other Sc–N distances in 2 and 3. The N–N stretch of 3 appears at 1700 cm−1 in the Raman spectrum which is on the high end of the known rare-earth element (NN)2− compounds.37
Small amounts of 3 could be reproducibly made by treating  with KC8 and 18-crown-6 in Et2O according to eqn (2) and it was also obtained when 2.2.2-cryptand was used instead. The 1H NMR spectrum of the material shows that 3 is isolated alongside varying amounts of an unidentified byproduct that appears at 1.93 ppm in C6D6. The use of a chelating agent appears necessary to generate 3, as only 2 was isolated from the reduction of
with KC8 and 18-crown-6 in Et2O according to eqn (2) and it was also obtained when 2.2.2-cryptand was used instead. The 1H NMR spectrum of the material shows that 3 is isolated alongside varying amounts of an unidentified byproduct that appears at 1.93 ppm in C6D6. The use of a chelating agent appears necessary to generate 3, as only 2 was isolated from the reduction of 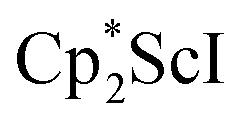 using 2 equiv. of KC8 without 2.2.2-cryptand or 18-crown-6.
using 2 equiv. of KC8 without 2.2.2-cryptand or 18-crown-6.

A green solution of 3 in toluene slowly changes color to red-purple when immersed in liquid nitrogen and reverts to green within several seconds of being removed from the liquid nitrogen. A movie of this transformation is in the ESI.† This temperature dependent change of color is reminiscent of the solid-state end-on to side-on yellow to green transformation observed for [(R2N)3Nd(µ-ηx:ηx-N2)]2− (x = 1,2).27 Although a high yield synthesis of this compound has not been pursued, it is described here to show the existence of such hetero-ligated reduced dinitrogen complexes and as another example of solid state isomerization system.
Cp*Sc oxides
The scandium atoms in the crystal structure of 1 are disordered with 5% lying closer to the center of the Sc2N2 core. This could be indicative of a small portion of side-on bridging N2 isomer in the crystal structure or a co-crystallized oxide product,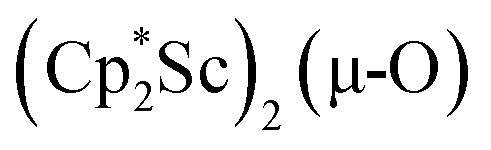 . Such
. Such 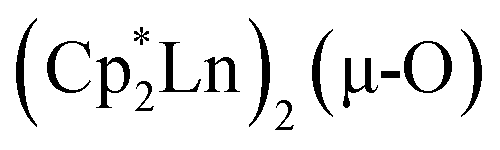 complexes are common side products in Cp* rare-earth metal chemistry23,44,45 and [(R2N)3Ln(µ-O)]2− species were previously found in crystal structures of [(R2N)3Ln(µ-ηx:ηx-N2)]2− complexes.27,28
complexes are common side products in Cp* rare-earth metal chemistry23,44,45 and [(R2N)3Ln(µ-O)]2− species were previously found in crystal structures of [(R2N)3Ln(µ-ηx:ηx-N2)]2− complexes.27,28
The 3.74(2) Å Sc⋯Sc distance in this minor component in the structure of 1 is much closer to the 3.80 Å Sc⋯Sc separation in the known oxide [(C5Me4H)2Sc]2(µ-O) than the 4.18 Å distance in the side-on (N2)2− complex [(C5Me4H)2Sc]2(µ-η2:η2-N2).12 Notably, the titanium oxide 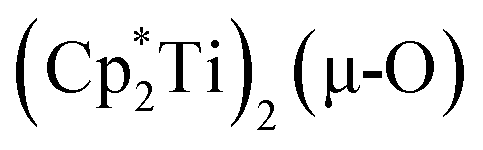 has a similar Ti⋯Ti separation (3.816 Å) and crystallizes in the same space group (I222) as 1 with similar unit cell parameters.46 A signal at 2.01 ppm is consistently present in the 1H NMR spectra of purified samples of 1 in C6D6 and is possibly due to this oxide,
has a similar Ti⋯Ti separation (3.816 Å) and crystallizes in the same space group (I222) as 1 with similar unit cell parameters.46 A signal at 2.01 ppm is consistently present in the 1H NMR spectra of purified samples of 1 in C6D6 and is possibly due to this oxide, 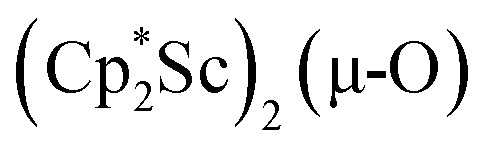 .
.
Since the reaction between 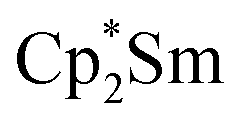 and N2O gave the bridging oxide product
and N2O gave the bridging oxide product 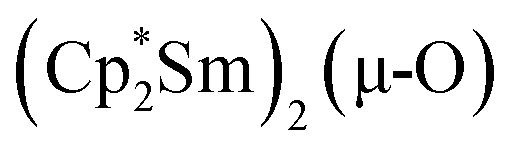 ,44 we attempted to generate the scandium oxide this way starting from 1. Treating a C6D6 solution of 1 with N2O led to a rapid color change from dark blue to pale yellow and formation of a single product with a 1H resonance at 2.01 ppm, lending further evidence that the oxide product is responsible for this signal. Similarly, the reaction of 2 with N2O in C6D6 resulted in two new 1H signals at 2.26 and 2.03 ppm in a 1:4 ratio indicating a different product is formed starting from 2.
,44 we attempted to generate the scandium oxide this way starting from 1. Treating a C6D6 solution of 1 with N2O led to a rapid color change from dark blue to pale yellow and formation of a single product with a 1H resonance at 2.01 ppm, lending further evidence that the oxide product is responsible for this signal. Similarly, the reaction of 2 with N2O in C6D6 resulted in two new 1H signals at 2.26 and 2.03 ppm in a 1:4 ratio indicating a different product is formed starting from 2.
We have not yet been successful in isolating single crystals of the complex 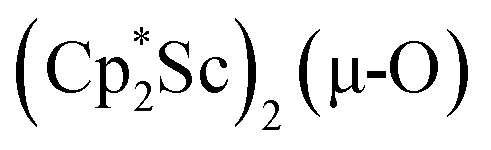 or of the side-on bridging N2 isomer of 1 for comparison with the crystal structure of 1 reported above. In one attempt to grow crystals by slow evaporation of a C6D6 solution containing presumably the oxide
or of the side-on bridging N2 isomer of 1 for comparison with the crystal structure of 1 reported above. In one attempt to grow crystals by slow evaporation of a C6D6 solution containing presumably the oxide 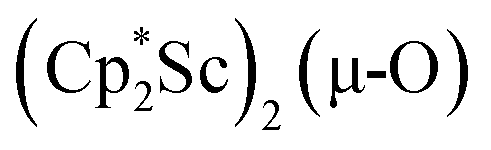 , several colorless crystals were obtained and identified by X-ray crystallography to be (Cp*Sc)4(µ4-O)(µ-OH)6, 4, Fig. 5. This polyhydroxy species has a diamondoid core with a central µ4-oxide and is further described in the ESI.† The formation of such polymetallic clusters containing [Cp*Ln]2+ vertices is common in rare-earth metal chemistry.47,48
, several colorless crystals were obtained and identified by X-ray crystallography to be (Cp*Sc)4(µ4-O)(µ-OH)6, 4, Fig. 5. This polyhydroxy species has a diamondoid core with a central µ4-oxide and is further described in the ESI.† The formation of such polymetallic clusters containing [Cp*Ln]2+ vertices is common in rare-earth metal chemistry.47,48
 | ||
| Fig. 5 Molecular structure of (Cp*Sc)4(µ4-O)(µ-OH)6, 4, with ellipsoids drawn at 30% probability and hydrogen atoms omitted for clarity. | ||
Computational studies
Electronic structure calculations were performed on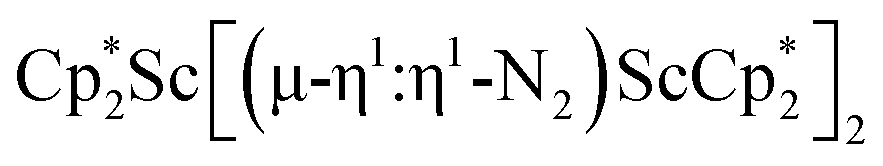 , 1,
, 1,  , 2, and
, 2, and 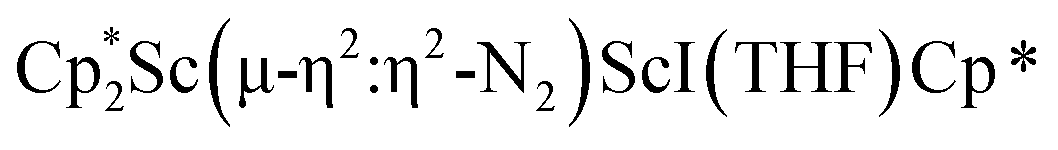 , 3 using density functional theory (DFT) with the TPSSh density functional49,50 with Grimme's D3 dispersion correction,51 including a Becke–Johnson damping function,52 in the gas phase. Additionally, the resolution of the identity (RI-J) approximation was utilized.53 Nonmetallic atoms were treated with def2-SV(P)54 basis sets, while Sc was treated with triple-zeta quality basis sets def2-TZVP.55,56 Additionally, each compound was optimized in the liquid phase using the TPSSh functional and the same basis sets as in the gas phase. The solvation effects of hexane were taken into account by applying the COSMO model57 with a dielectric constant of 1.887 and an index of refraction of 1.3727. The optimized structures with the COSMO model were verified as local minima through vibrational analysis. The electronic spectra of each optimized compound were simulated using time-dependent density functional theory (TDDFT) calculations employing the non-orthonormal Krylov subspace method.58 The validity of the computational protocol has been thoroughly tested and compared with X-ray structures and UV-visible spectra of rare-earth molecular complexes, as demonstrated in previous studies.59 All calculations in this work were performed using the TURBOMOLE quantum chemistry package (version 7.8).60 Complete details can be found in the ESI.†
, 3 using density functional theory (DFT) with the TPSSh density functional49,50 with Grimme's D3 dispersion correction,51 including a Becke–Johnson damping function,52 in the gas phase. Additionally, the resolution of the identity (RI-J) approximation was utilized.53 Nonmetallic atoms were treated with def2-SV(P)54 basis sets, while Sc was treated with triple-zeta quality basis sets def2-TZVP.55,56 Additionally, each compound was optimized in the liquid phase using the TPSSh functional and the same basis sets as in the gas phase. The solvation effects of hexane were taken into account by applying the COSMO model57 with a dielectric constant of 1.887 and an index of refraction of 1.3727. The optimized structures with the COSMO model were verified as local minima through vibrational analysis. The electronic spectra of each optimized compound were simulated using time-dependent density functional theory (TDDFT) calculations employing the non-orthonormal Krylov subspace method.58 The validity of the computational protocol has been thoroughly tested and compared with X-ray structures and UV-visible spectra of rare-earth molecular complexes, as demonstrated in previous studies.59 All calculations in this work were performed using the TURBOMOLE quantum chemistry package (version 7.8).60 Complete details can be found in the ESI.†
The initial structures of 1–3 for DFT calculations were obtained from single crystal X-ray diffraction. In each case, the structural parameters calculated for the ground state structures were within a few degrees for angles and within 0.02–0.03 Å in distances as is typical for this level of computation.59 The ground state assignment for each complex as described below is based on the relative energies of the different spin states, the agreement between calculated structural parameters and X-ray data, and a comparison between the simulated and experimental electronic spectra. Detailed information on the possible spin states for these complexes are given in the ESI.†
For the end-on complex, 1, DFT revealed that the triplet state is the ground state, while the singlet state is 0.26 eV (6.00 kcal mol−1) higher in energy (see Table S10†). The qualitative MO diagram in Fig. 6 shows two unpaired electrons in the highest occupied molecular orbitals that are two degenerate orbitals that are mainly N2 π* in character, but have Sc–N2–Sc π bonding components, Fig. 7a. This bonding picture matches that of the end-on complex [K(crypt)]2{[(R2N)3Sc]2(µ-η1:η1-N2)}.13 Analysis of the Kohn–Sham molecular orbitals suggests a strong interaction between the scandium 3d orbitals and the antibonding π* orbital of N2 in the ScN2Sc plane, leading to calculated and observed N–N and Sc–N bond lengths that are relatively shorter compared to other rare-earth dinitrogen complexes, Table 1. The analysis of spin density from the natural population indicates that the unpaired electrons are assigned to N p-orbitals and Sc d-orbitals with a value of 0.45 assigned to each orbital. This results in a triplet state and two-electron four-center Sc(dπ)–N2(π*) bonding interactions between two  fragments and N2.
fragments and N2.
 | ||
Fig. 6 Qualitative MO diagram for (a)  , 1, and (b) the side-on component of 3, , 1, and (b) the side-on component of 3,  . . | ||
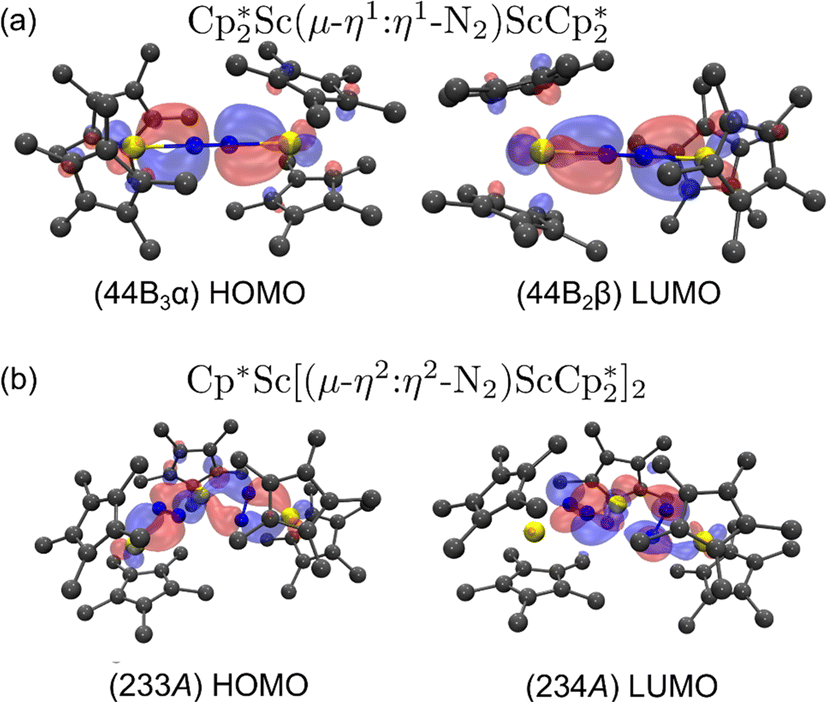 | ||
Fig. 7 (a) HOMO and LUMO of 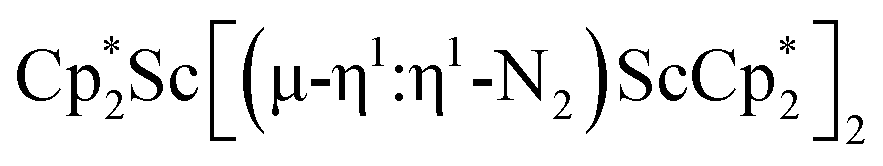 , 1; (b) HOMO and LUMO of , 1; (b) HOMO and LUMO of 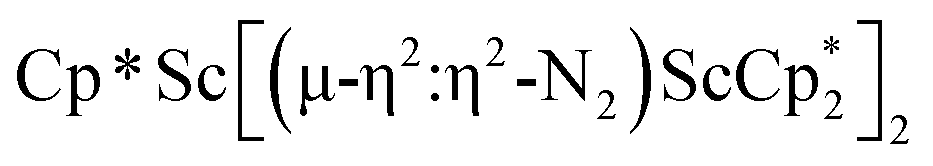 , 2, using a contour value of 0.04. Hydrogen atoms are omitted for clarity. , 2, using a contour value of 0.04. Hydrogen atoms are omitted for clarity. | ||
A calculation on the side-on variant of 1, i.e. 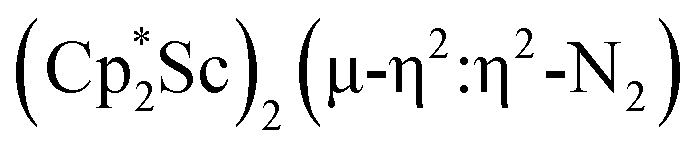 , indicated that it is 0.415 eV (9.57 kcal mol−1) higher in energy. In comparison, the side-on {[(R2N)3Sc]2(µ-η2:η2-N2)}2– was previously calculated to be 12 kcal mol−1 less stable than the observed end-on {[(R2N)3Sc]2(µ-η1:η1-N2)}2−.4
, indicated that it is 0.415 eV (9.57 kcal mol−1) higher in energy. In comparison, the side-on {[(R2N)3Sc]2(µ-η2:η2-N2)}2– was previously calculated to be 12 kcal mol−1 less stable than the observed end-on {[(R2N)3Sc]2(µ-η1:η1-N2)}2−.4
For the side-on component of 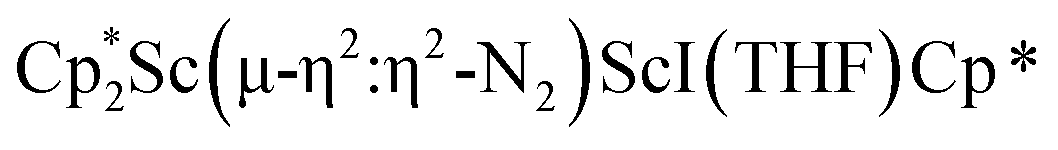 , 3, DFT calculations indicated that the singlet state is the ground state, while the triplet state is 1.16 eV (26.75 kcal mol−1) higher in energy (see Table S15†). The qualitative MO diagram in Fig. 6b is similar to that of [(C5Me4H)2Sc]2(µ-η2:η2-N2)12 and contains an electron pair in the HOMO formed from interaction of Sc 3d orbitals and an antibonding π* orbital of N2. However, the LUMO of side-on 3 exhibits mixing between a Sc d orbital and a π* orbital of N2 (see Fig. S17 and Table S9†), in contrast to the LUMO of [(C5Me4H)2Sc]2(µ-η2:η2-N2).12 Additionally, the end-on component of 3 was also evaluated by DFT calculations and is found to be 0.54 eV (12.45 kcal mol−1) higher in energy. In the case of the [(R2N)3Nd(µ-ηx:ηx-N2)]2− dianion (x = 1, 2), the end-on isomer is favored at low temperature and converts to the side-on at around −100 °C in the solid state.27
, 3, DFT calculations indicated that the singlet state is the ground state, while the triplet state is 1.16 eV (26.75 kcal mol−1) higher in energy (see Table S15†). The qualitative MO diagram in Fig. 6b is similar to that of [(C5Me4H)2Sc]2(µ-η2:η2-N2)12 and contains an electron pair in the HOMO formed from interaction of Sc 3d orbitals and an antibonding π* orbital of N2. However, the LUMO of side-on 3 exhibits mixing between a Sc d orbital and a π* orbital of N2 (see Fig. S17 and Table S9†), in contrast to the LUMO of [(C5Me4H)2Sc]2(µ-η2:η2-N2).12 Additionally, the end-on component of 3 was also evaluated by DFT calculations and is found to be 0.54 eV (12.45 kcal mol−1) higher in energy. In the case of the [(R2N)3Nd(µ-ηx:ηx-N2)]2− dianion (x = 1, 2), the end-on isomer is favored at low temperature and converts to the side-on at around −100 °C in the solid state.27
DFT calculations on the trimetallic complex, 2, indicated that the ground state is consistent with a singlet. Calculated structures with alternative triplet and quintet spin states are 0.97 (22.37 kcal mol−1) and 2.22 eV (51.19 kcal mol−1) higher in energy, respectively (see Table S13†). The HOMO of 2 displays characteristics consistent with two reduced N2 ligands (see Fig. 7b and Table S19†), which is consistent with the Sc–N bond distances (Tables 2 and S13†). Hence, this complex has an electron pair in each of the two reduced dinitrogen ligands. Since rare-earth metal complexes of (µ-η2:η2-N2)2− ligands have an extensive two-electron reduction chemistry,61–63 complex 2 could conceivably be a four-electron reductant.
The electronic spectra were simulated using TDDFT calculations and are compared with the experimental spectra in Fig. 8. Comparisons of the calculated spectra for different spin states are given in the ESI.† As shown in Fig. 8, the calculated spectra match the observed data well. For 1, the main absorption band is due to electronic transitions corresponding to degenerate (44B2 alpha) HOMO and (44B3 alpha) HOMO-1 with Sc(dπ)-N2(π*) character to (45B3 alpha) LUMO+6 and (45B2 alpha) LUMO+7 with predominantly d character, respectively. Similarly, for 2 and 3, the significant transitions are due to transitions with similar characters (see Tables S19–S22†).
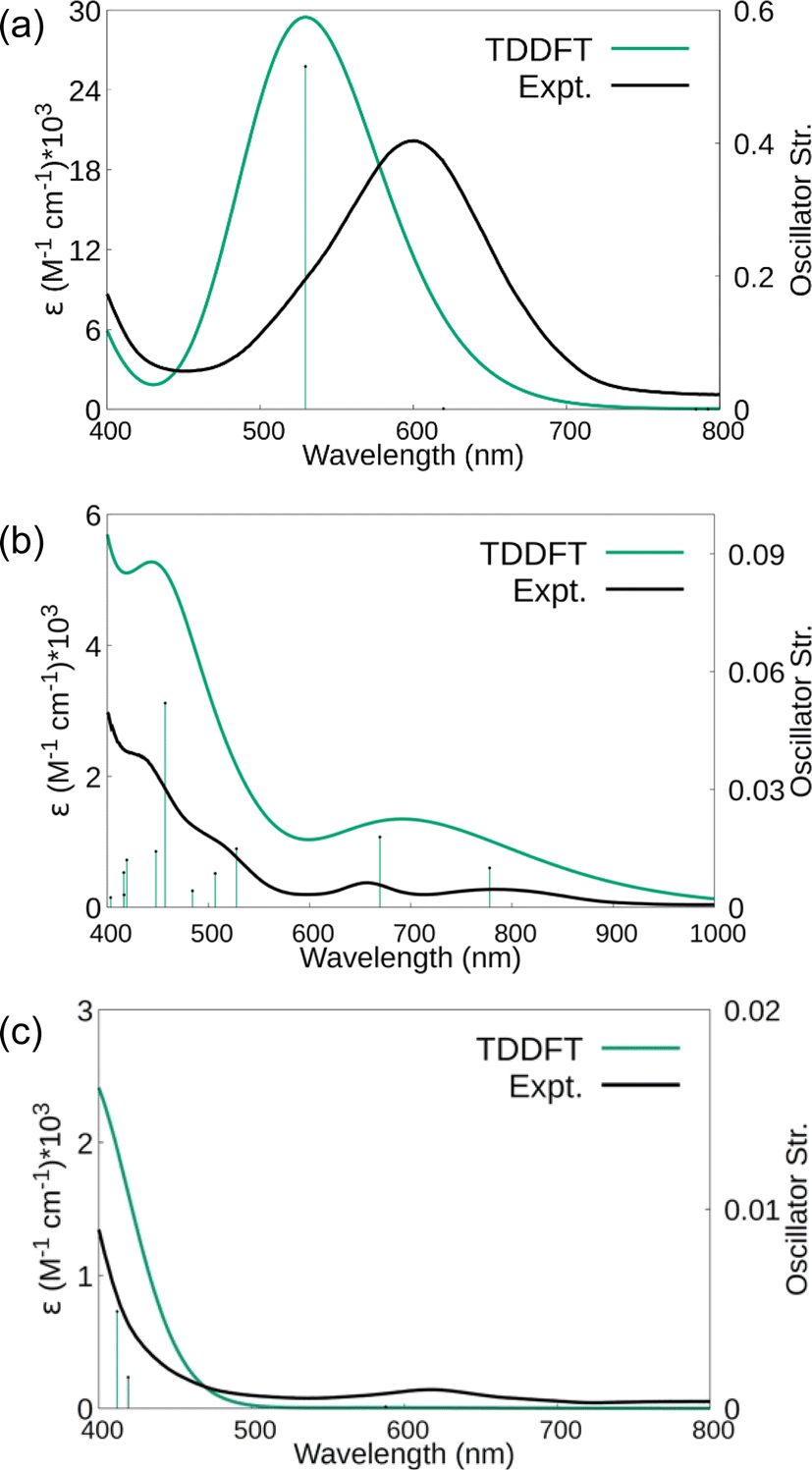 | ||
Fig. 8 Simulated and experimental (Et2O) UV-visible absorbance spectra of (a)  , 1, (b) , 1, (b)  , 2, and (c) the side-on isomer of 3, , 2, and (c) the side-on isomer of 3,  . A Gaussian spectral line shape with a width of 0.2 eV was employed in each case. . A Gaussian spectral line shape with a width of 0.2 eV was employed in each case. | ||
Raman shifts were also calculated for 1–3 and scaled by a factor of 0.95 to account for anharmonicities and basis set incompleteness.64 For end-on 1, the calculated value was 1661 cm−1 compared to the 1595 cm−1 experimental value. For side-on 2, the calculations indicated two stretching vibrations at lower energies, 1484 and 1544 cm−1. A lower energy absorbance is observed at 1460 cm−1, but only one band is observed under the experimental conditions. For 3, the 1700 cm−1 experimentally observed stretch is in between the 1515 cm−1 calculated estimate for the side-on isomer and the 1737 cm−1 calculated stretch for the end-on isomer. The observed band is closer to the end-on calculation and higher than that expected for the solid-state structure that was predominantly side-on. However, it is possible that laser heating of the sample under the Raman experimental conditions generates more end-on isomer in situ.
Discussion
The preliminary reactivity previously observed in reductions of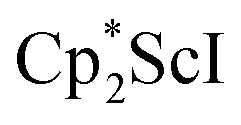 with KC8 under argon31 is further supported by the reactivity observed under dinitrogen reported here. Under argon, EPR and C–H bond reactivity data indicated the formation of a highly reactive red Sc(II) complex, possibly
with KC8 under argon31 is further supported by the reactivity observed under dinitrogen reported here. Under argon, EPR and C–H bond reactivity data indicated the formation of a highly reactive red Sc(II) complex, possibly  , but this species has not yet been isolated or crystallographically characterized.31 It would be expected that a species like
, but this species has not yet been isolated or crystallographically characterized.31 It would be expected that a species like 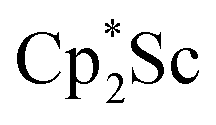 could reduce N2 and indeed the
could reduce N2 and indeed the  system forms the bimetallic (NN)2− complexes
system forms the bimetallic (NN)2− complexes  , 1, and
, 1, and 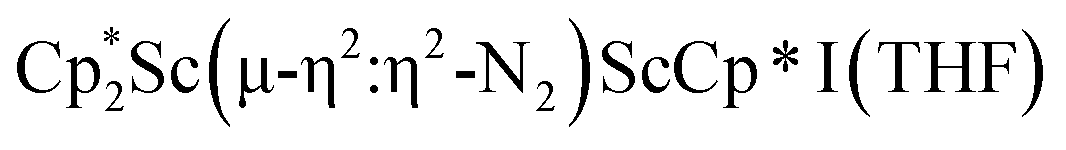 , 3, as well as the trimetallic species
, 3, as well as the trimetallic species 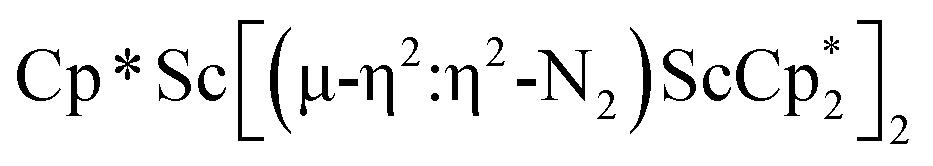 , 2. This doubles the number of previously characterized reduced dinitrogen complexes of scandium. In addition, to our knowledge complex 2 is the first dinitrogen complex of any metal in the periodic table that contains two side-on (NN)2− ligands bound to the same metal.32,33
, 2. This doubles the number of previously characterized reduced dinitrogen complexes of scandium. In addition, to our knowledge complex 2 is the first dinitrogen complex of any metal in the periodic table that contains two side-on (NN)2− ligands bound to the same metal.32,33
The end-on coordination mode found in  contrasts with the side-on binding found with the tetramethylcyclopentadienyl scandium analog, [(C5Me4H)2Sc]2(µ-η2:η2-N2), as well as the Cp* samarium analog,
contrasts with the side-on binding found with the tetramethylcyclopentadienyl scandium analog, [(C5Me4H)2Sc]2(µ-η2:η2-N2), as well as the Cp* samarium analog,  .15 Compared to either of these, there is more steric crowding in
.15 Compared to either of these, there is more steric crowding in 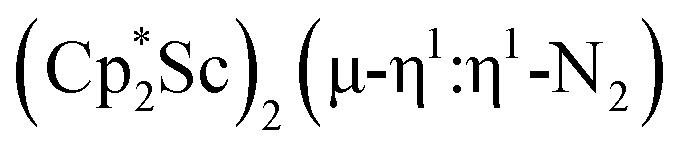 due to the larger ligand versus C5Me4H and the smaller metal versus Sm, which evidently leads to the end-on binding. The preference for side-on coordination in
due to the larger ligand versus C5Me4H and the smaller metal versus Sm, which evidently leads to the end-on binding. The preference for side-on coordination in  , 2, and
, 2, and  , 3, as well as in{[nBuC(NiPr)2](Cp*)Sc}2(µ-η2:η2-N2) is consistent with this steric rationale since all of these complexes have less bulky ligand sets. The end-on structure of [K(crypt)]2{[(R2N)3Sc]2(µ-η1:η1-N2)}, which has three bulky amide groups per scandium, also fits this picture.
, 3, as well as in{[nBuC(NiPr)2](Cp*)Sc}2(µ-η2:η2-N2) is consistent with this steric rationale since all of these complexes have less bulky ligand sets. The end-on structure of [K(crypt)]2{[(R2N)3Sc]2(µ-η1:η1-N2)}, which has three bulky amide groups per scandium, also fits this picture.
The Raman shift of the N–N stretch in 1 is on the low end of values observed for end-on rare-earth metal (NN)2− complexes, Table 1, but it is consistent with an NN double bond. All of these shifts for end-on coordination are higher in energy than shifts observed for side-on rare-earth metal species which are in the range of 1413–1473 cm−1,37 along with that of 2 (1460 cm−1).
The formation of the first example of a bis(side-on) (NN)2− trimetallic complex, 2, was unexpected. The fact that this can be made both sequentially from 1 and directly from 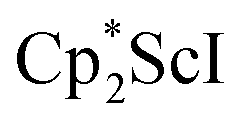 and
and  suggests it is a rather favorable reaction product in this system. The reaction of 1 to form 2 in Scheme 1 requires the loss of a Cp* ligand during the KC8 reduction. This may prove to be a general reaction of KC8 with rare-earth metal Cp* complexes, since several examples of mono-Cp* ligated complexes were recently reported from KC8 reductions of
suggests it is a rather favorable reaction product in this system. The reaction of 1 to form 2 in Scheme 1 requires the loss of a Cp* ligand during the KC8 reduction. This may prove to be a general reaction of KC8 with rare-earth metal Cp* complexes, since several examples of mono-Cp* ligated complexes were recently reported from KC8 reductions of 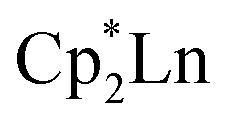 bipyridyl complexes.65 The mechanism for the conversion of 1 to 2 is not obvious since the net stoichiometry in Scheme 1 is complicated and end-on to side-on isomerization is involved. The DFT calculations show that although the LUMO of 1 is consistent with the targeted (N2)3− ligand, it is quite high in energy at 1.49 eV (34.36 kcal mol−1) above the HOMO. Hence, this species may be unstable with respect to further reactions to form 2. The LUMO of 1 also has a significant contribution from the scandium d-orbitals, while in compounds which have been successfully reduced, e.g. {[(Me3Si)2N]2(THF)Y}2(µ-η2:η2-N2)29 and {[nBuC(NiPr)2]Cp*Sc}2(µ-η2:η2-N2),14 the LUMOs are localized on the (NN)2− moiety.
bipyridyl complexes.65 The mechanism for the conversion of 1 to 2 is not obvious since the net stoichiometry in Scheme 1 is complicated and end-on to side-on isomerization is involved. The DFT calculations show that although the LUMO of 1 is consistent with the targeted (N2)3− ligand, it is quite high in energy at 1.49 eV (34.36 kcal mol−1) above the HOMO. Hence, this species may be unstable with respect to further reactions to form 2. The LUMO of 1 also has a significant contribution from the scandium d-orbitals, while in compounds which have been successfully reduced, e.g. {[(Me3Si)2N]2(THF)Y}2(µ-η2:η2-N2)29 and {[nBuC(NiPr)2]Cp*Sc}2(µ-η2:η2-N2),14 the LUMOs are localized on the (NN)2− moiety.
Conclusion
Although dinitrogen reduction chemistry with the first metal in the transition series, scandium, has not been extensively studied, this metal has generated unexpected results in this area. Reduction of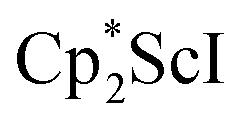 and
and 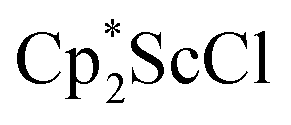 under dinitrogen leads not only to the bimetallic scandium (NN)2− complexes,
under dinitrogen leads not only to the bimetallic scandium (NN)2− complexes, 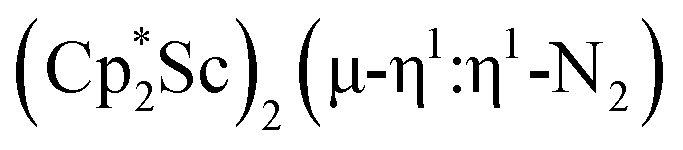 , 1, and
, 1, and 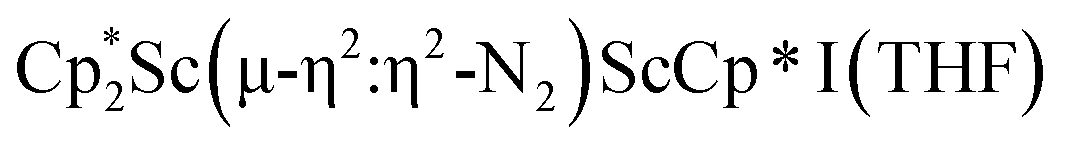 , 3, but also to the unprecedented trimetallic species
, 3, but also to the unprecedented trimetallic species 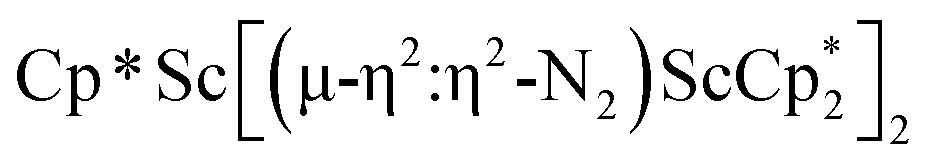 , 2. Clearly, scandium has a more extensive chemistry with dinitrogen than the three examples previously in the literature.12–14 Isolation of a Sc(II) precursor such as
, 2. Clearly, scandium has a more extensive chemistry with dinitrogen than the three examples previously in the literature.12–14 Isolation of a Sc(II) precursor such as 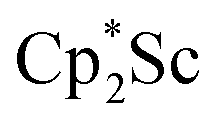 is not necessary to access the scandium dinitrogen complexes and it is anticipated that other Cp*-ligated scandium reduced dinitrogen complexes should be synthetically accessible. Complexes 1–3 are expected to have a rich reaction chemistry based on previous studies of (NN)2− reactivity.61–63
is not necessary to access the scandium dinitrogen complexes and it is anticipated that other Cp*-ligated scandium reduced dinitrogen complexes should be synthetically accessible. Complexes 1–3 are expected to have a rich reaction chemistry based on previous studies of (NN)2− reactivity.61–63
Data availability
Experimental procedures, X-ray crystallographic details, NMR and infrared spectroscopy data, and computational details are available in the ESI.† CCDC 2324189, 2344840, 2344841, and 2348488–2348490 contain the supplementary crystallographic data for this paper.Author contributions
JDQ, ZX, and WJE provided the original concept. JDQ and QEG synthesized and characterized the compounds at UCI under the supervision of WJE and collected X-ray data under the supervision of JWZ. QY synthesized and characterized compounds at PKU under the supervision of ZX. Computations were done by AR and DKN under the supervision of FF. JDQ, AR, QY, ZX, and WJE wrote the manuscript with contributions from all authors. Funding was obtained by ZX, FF, and WJE.Conflicts of interest
The authors declare no competing financial interest. Principal Investigator Filipp Furche has an equity interest in TURBOMOLE GmbH. The terms of this arrangement have been reviewed and approved by the University of California, Irvine, in accordance with its conflict of interest policies.Acknowledgements
We thank the U. S. National Science Foundation for support of this research under CHE-2154255 (to W. J. E for the experimental research) and CHE-2102568 (to F. F. for the theoretical studies) and National Natural Science Foundation of China (No. 21988101, to Z. X.). A. R. acknowledges support from an Eddleman Quantum Institute Fellowship. We also thank Dr D. Fishman and Marcus Marracci for collection of the Raman spectra.References
- J. Chatt, J. R. Dilworth and R. L. Richards, Chem. Rev., 1978, 78, 589–625 CrossRef CAS.
- Y. Nishibayashi, Inorg. Chem., 2015, 54, 9234–9247 CrossRef CAS PubMed.
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneikder and R. R. Schrock, Science, 2018, 360, eaar6611 CrossRef.
- S. Kim, F. Loose and P. J. Chirik, Chem. Rev., 2020, 120, 5637–5681 CrossRef CAS PubMed.
- M. J. Chalkley, M. W. Drover and J. C. Peters, Chem. Rev., 2020, 120, 5582–5636 CrossRef CAS.
- A. D. Allen and C. V. Senoff, Chem. Commun., 1965, 621–622 RSC.
- D. F. Harrison, E. Weissberger and H. Taube, Science, 1968, 159, 320–322 CrossRef CAS.
- K. Jonas, Angew. Chem., Int. Ed., 1973, 12, 997–998 CrossRef.
- D. Singh, W. R. Buratto, J. F. Torres and L. J. Murray, Chem. Rev., 2020, 120, 5517–5581 CrossRef CAS.
- E. A. MacLachlan and M. D. Fryzuk, Organometallics, 2006, 25, 1530–1543 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS.
- S. Demir, S. E. Lorenz, M. Fang, F. Furche, G. Meyer, J. W. Ziller and W. J. Evans, J. Am. Chem. Soc., 2010, 132, 11151–11158 CrossRef CAS PubMed.
- D. H. Woen, G. P. Chen, J. W. Ziller, T. J. Boyle, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2017, 139, 14861–14864 CrossRef CAS.
- Z.-J. Lv, Z. Huang, W.-X. Zhang and Z. Xi, J. Am. Chem. Soc., 2019, 141, 8773–8777 CrossRef CAS.
- W. J. Evans, T. A. Ulibarri and J. W. Ziller, J. Am. Chem. Soc., 1988, 110, 6877–6879 CrossRef CAS.
- W. J. Evans, N. T. Allen and J. W. Ziller, J. Am. Chem. Soc., 2001, 123, 7927–7928 CrossRef CAS PubMed.
- W. J. Evans, N. T. Allen and J. W. Ziller, Angew. Chem., Int. Ed., 2002, 41, 359–361 CrossRef CAS.
- W. J. Evans, G. Zucchi and J. W. Ziller, J. Am. Chem. Soc., 2003, 125, 10–11 CrossRef CAS.
- W. J. Evans, D. S. Lee and J. W. Ziller, J. Am. Chem. Soc., 2004, 126, 454–455 CrossRef CAS.
- W. J. Evans, D. S. Lee, D. B. Rego, J. M. Perotti, S. A. Kozimor, E. K. Moore and J. W. Ziller, J. Am. Chem. Soc., 2004, 126, 14574–14582 CrossRef CAS.
- W. J. Evans, D. S. Lee, C. Lie and J. W. Ziller, Angew. Chem., Int. Ed., 2004, 43, 5517–5519 CrossRef CAS PubMed.
- W. J. Evans, D. S. Lee, M. A. Johnston and J. W. Ziller, Organometallics, 2005, 24, 6393–6397 CrossRef CAS.
- W. J. Evans, D. B. Rego and J. W. Ziller, Inorg. Chem., 2006, 45, 10790–10798 CrossRef CAS PubMed.
- F. Jaroschik, A. Momin, F. Nief, X.-F. Le Goff, G. B. Deacon and P. C. Junk, Angew. Chem., Int. Ed., 2009, 48, 1117–1121 CrossRef CAS.
- B. M. Schmiege, J. W. Ziller and W. J. Evans, Inorg. Chem., 2010, 49, 10506–10511 CrossRef CAS PubMed.
- W. J. Evans and D. S. Lee, Can. J. Chem., 2005, 83, 375–384 CrossRef CAS.
- A. B. Chung, D. Rappoport, J. W. Ziller, R. E. Cramer, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2022, 144, 17064–17074 CrossRef CAS PubMed.
- A. J. Ryan, S. G. Balasubramani, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2020, 142, 9302–9313 CrossRef CAS.
- W. J. Evans, M. Fang, G. Zucchi, F. Furche, J. W. Ziller, R. M. Hoekstra and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 11195–11202 CrossRef CAS PubMed.
- M. Fang, J. E. Bates, S. E. Lorenz, D. S. Lee, D. B. Rego, J. W. Ziller, F. Furche and W. J. Evans, Inorg. Chem., 2011, 50, 1459–1469 CrossRef CAS PubMed.
- J. D. Queen, L. M. Anderson-Sanchez, C. R. Stennett, A. Rajabi, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2024, 146, 3279–3292 CrossRef CAS PubMed.
- R. Duchateau, S. Gambarotta, N. Beydoun and C. Bensimon, J. Am. Chem. Soc., 1991, 113, 8986–8988 CrossRef CAS.
- D. N. Huh, R. F. Koby, Z. E. Stuart, R. J. Dunscomb, N. D. Schley and I. A. Tonks, Chem. Sci., 2022, 13, 13330–13337 RSC.
- R. D. Sanner, J. M. Manriquez, R. E. Marsh and J. E. Bercaw, J. Am. Chem. Soc., 1976, 98, 8351–8357 CrossRef CAS.
- Y.-T. Hung, G. P. A. Yap and K. H. Theopold, Polyhedron, 2019, 157, 381–388 CrossRef CAS.
- A. Mondal, C. G. T. Price, J. Tang and R. A. Layfield, J. Am. Chem. Soc., 2023, 145, 20121–20131 CrossRef CAS PubMed.
- M. E. Fieser, D. H. Woen, J. F. Corbey, T. J. Mueller, J. W. Ziller and W. J. Evans, Dalton Trans., 2016, 45, 14634–14644 RSC.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- S. K. Sur, J. Magn. Reson., 1989, 82, 169–173 CAS.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532 CrossRef CAS.
- M. E. Thompson, S. M. Baxter, A. R. Bulls, B. J. Burger, M. C. Nolan, B. D. Santarsiero, W. P. Schaefer and J. E. Bercaw, J. Am. Chem. Soc., 1987, 109, 203–219 CrossRef CAS.
- W. Huynh, D. B. Culver, H. Tafazolian and M. P. Conley, Dalton Trans., 2018, 47, 13063–13071 RSC.
- M. Hirotsu, P. P. Fontaine, P. Y. Zavalij and L. R. Sita, J. Am. Chem. Soc., 2007, 129, 12690–12692 CrossRef CAS.
- W. J. Evans, J. W. Grate, I. Bloom, W. E. Hunter and J. L. Atwood, J. Am. Chem. Soc., 1985, 107, 405–409 CrossRef CAS.
- W. J. Evans, B. L. Davis, G. W. Nyce, J. M. Perotti and J. W. Ziller, J. Organomet. Chem., 2003, 677, 89–95 CrossRef CAS.
- J. Pinkas, I. Císařová, R. Gyepes, J. Kubišta, M. Horáček and K. Mach, Organometallics, 2013, 32, 6306–6314 CrossRef CAS.
- W. J. Evans, N. T. Allen, M. A. Greci and J. W. Ziller, Organometallics, 2001, 20, 2936–2937 CrossRef CAS.
- T. Shima and Z. Hou, J. Am. Chem. Soc., 2006, 128, 8124–8125 CrossRef CAS PubMed.
- V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, J. Chem. Phys., 2003, 119, 12129–12137 CrossRef CAS.
- M. K. Armbruster, F. Weigend, C. van Wüllen and W. Klopper, Phys. Chem. Chem. Phys., 2008, 10, 1748–1756 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 240, 283–290 CrossRef CAS.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- R. Gulde, P. Pollak and F. Weigend, J. Chem. Theory Comput., 2012, 8, 4062–4068 CrossRef CAS PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- F. Furche, B. T. Krull, B. D. Nguyen and J. Kwon, J. Chem. Phys., 2016, 144, 174105 CrossRef PubMed.
- A. Rajabi, R. Grotjahn, D. Rappoport and F. Furche, Dalton Trans., 2024, 53, 410–417 RSC.
- Y. J. Franzke, C. Holzer, J. H. Andersen, T. Begušić, F. Bruder, S. Coriani, F. Della Sala, E. Fabiano, D. A. Fedotov, S. Fürst, S. Gillhuber, R. Grotjahn, M. Kaupp, M. Kehry, M. Krstić, F. Mack, S. Majumdar, B. D. Nguyen, S. M. Parker, F. Pauly, A. Pausch, E. Perlt, G. S. Phun, A. Rajabi, D. Rappoport, B. Samal, T. Schrader, M. Sharma, E. Tapavicza, R. T. Treß, V. Voora, A. Wodyński, J. M. Yu, B. Zerulla, F. Furche, C. Hattig, M. Sierka, D. P. Tew and F. Weigend, J. Chem. Theory Comput., 2023, 19, 6859–6890 CrossRef CAS PubMed.
- W. J. Evans, D. S. Lee, J. W. Ziller and N. Kaltsoyannis, N. J. Am. Chem. Soc., 2006, 128, 14176–14184 CrossRef CAS PubMed.
- W. J. Evans, J. R. Walensky, T. M. Champagne, J. W. Ziller, J. W. A. G. DiPasquale and A. L. Rheingold, J. Organomet. Chem., 2009, 694, 1238–1243 CrossRef CAS.
- S. E. Lorenz, B. M. Schmiege, D. S. Lee, J. W. Ziller and W. J. Evans, Inorg. Chem., 2010, 49, 6655–6663 CrossRef CAS PubMed.
- K. P. Huber and G. Herzberg, Constants of Diatomic Molecules (data prepared by J. W. Gallagher, and R. D. Johnson III), in NIST Chemistry WebBook, NIST Standard Reference Database No. 69, ed. P. J. Linstrom and W. G. Mallard, National Institute of Standards and Technology, Gaithersburg MD, 2009, https://webbook.nist.gov/, (retrieved December 6, 2009) Search PubMed.
- C. R. Stennett, J. W. Ziller and W. J. Evans, Eur. J. Inorg. Chem., 2024, e202300732 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2324189, 2344840, 2344841 and 2348488–2348490. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc03977g |
| This journal is © The Royal Society of Chemistry 2024 |