
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Using inelastic neutron scattering spectroscopy to probe CO2 binding in grafted aminosilanes†
Matthew E.
Potter
 *ab,
Hamish
Cavaye
c,
Joshua J. M.
Le Brocq
d,
Luke L.
Daemen
e and
Yongqiang
Cheng
e
*ab,
Hamish
Cavaye
c,
Joshua J. M.
Le Brocq
d,
Luke L.
Daemen
e and
Yongqiang
Cheng
e
aInstitute for Sustainability, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: mep61@bath.ac.uk
bDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK
cISIS Neutron and Muon Source, STFC, Rutherford Appleton Laboratory, Chilton, OX11 0QX, UK
dDepartment of Chemistry, University of Southampton, Highfield Campus, Southampton, SO17 1BJ, UK
eNeutron Scattering Division, Oak Ridge National Laboratory, Oak Ridge Tennessee 37831, USA
First published on 1st October 2024
Abstract
While a range of in situ characterisation techniques are available to probe CO2 adsorption processes, inelastic neutron scattering is scarcely used, primarily due to the reliance on hydrogeneous modes. Materials capable of adsorbing CO2, such as solid supported-amines contain a range of C–H and N–H species, which can be probed to explore the adsorption of CO2. Here we show the benefits of using inelastic neutron spectroscopy to probe CO2 adsorption with solid supported-amines, and the complementarity that can be achieved using different world-leading spectrometers.
Introduction
To meet global and national net-zero targets it is believed that, globally, 10 gigatons of CO2 must be captured annually up to 2050.1,2 Despite this, global CO2 emissions increase year-on-year. Many reports suggest that the only way to reach to reach these targets is to embrace carbon capture technology, not just for industrial emissions (post-combustion), but also direct air capture (DAC).1–3 The former is a well-established technology, where aqueous liquid amines (e.g. monoethanolamine) are regularly used to capture CO2 from flue-gas, typically with 8–15 vol% of CO2.4 However liquid amines are highly volatile, and as such are prone to evaporate, causing them to corrode equipment along the industrial process.2 In comparison many technologies are being considered for DAC, including carbon looping with CaO, and capture with metal organic frameworks and supported-amine species.5By incorporating amine-species onto a solid matrix, forming supported-amines, many of the benefits of liquid amine sorbents are retained, particularly the strong amine–CO2 interactions that selectively adsorb CO2 from ultra-dilute streams in DAC. Further, supported-amines are easier to separate than liquid amines, are less susceptible to evaporation/sorbent loss, with improved heat-transfer properties.2,5 A variety of supported-amine systems exist, typically divided into four classes.6 Class 1 species contain aminopolymers within a porous support, leading to high amine densities, and strong CO2–amine interactions, though often suffer from polymer leaching due to the weak interactions with the support. Class 2 species have chemical bonds that link the amine to the porous support. An example of this is the grafting of aminosilanes onto surface hydroxyl groups, leaving an amine chain anchored to the support.6 Previous work by Jones et al. shows that amine spacing is a key metric in determining CO2 binding strength.7 3-aminopropyl groups were introduced onto a mesoporous silica support; SBA-15, and more dispersed amines (<1.2 mmol N g−1) resulted in weaker CO2 interactions of 30–45 kJ mol−1. However higher amine densities (>1.3 mmol N g−1) caused binding energies (at low uptake) to drastically increased to 90 kJ mol−1.7 Class 3 species contain a tethered aminopolymer, which is polymerised in situ, though this is difficult to control, making sorbent design challenging. Finally, class 4 species are a combination of class 1 and 2, containing both tethered aminosilanes and polyamine species in the same sorbent.2,6 While other classes of sorbents may have higher CO2 uptakes, class 2 sorbents are often used as model systems to understand the behaviour of supported-amines, as the identity and chemical environment of each amine is known.
Ex situ characterisation of supported amines can quantify the number of amines and investigate the nature of the amine, using techniques such as solid state nuclear magnetic resonance, elemental analysis and thermogravimetric analysis.8 However, the mild conditions used in carbon capture (temperatures < 150 °C, binary CO2-gas mixtures, atmospheric pressure), mean a wide range of spectroscopic techniques can operate during carbon capture. Due to fast acquisition times and intense C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O signals, infra-red9 and Raman10 are often used to monitor in situ carbon capture of aminosilanes. The 1200–1800 cm−1 region is particularly of interest, as features assigned to alkylammonium carbamates, carbamic acids and silyl carbamates can be observed, with species such as paired carbamic acids also hypothesised.11 Being able to observe the appearance and disappearance of these features, as a function of CO2 pressure, uptake and desorption temperature has provided significant insight into the binding strength of different species.
O signals, infra-red9 and Raman10 are often used to monitor in situ carbon capture of aminosilanes. The 1200–1800 cm−1 region is particularly of interest, as features assigned to alkylammonium carbamates, carbamic acids and silyl carbamates can be observed, with species such as paired carbamic acids also hypothesised.11 Being able to observe the appearance and disappearance of these features, as a function of CO2 pressure, uptake and desorption temperature has provided significant insight into the binding strength of different species.
Whilst Raman and infrared spectroscopies are readily accessible, the spectra are often dominated by the support matrix (e.g. silica), making it challenging to see contributions from organic components. Inelastic neutron scattering (INS) is a complementary vibrational technique to Raman and infrared, where instead of photons exciting quantised vibrational modes, neutrons transfer energy to the sample.12,13 X-ray cross sections increase with the number of electrons, thus heavier elements provide a greater contribution to the signal. However, for INS hydrogen (1H) has by far the largest neutron cross-section, meaning INS is particularly sensitive to hydrogen.14
A variety of neutron techniques have probed supported-amines. Quasi-elastic neutron scattering has been utilised to investigate the motion of confined aminopolymers under CO2 capture conditions.15 These studies concluded that polymer motion is a key descriptor for CO2 uptake, as rigid/immobile polymers create inaccessible amine sites, lowering CO2 uptake. Small angle neutron scattering has provided significant insights into the location of aminopolymers (polyethyleneimine; PEI) within a mesoporous SBA-15 silica support. It was shown that PEI initially forms a thin coat on the internal walls of the mesopores, with the primary amines interacting with the hydroxylated pore walls. However, once the walls are coated, PEI forms a plug inside the pore, with a greater proportion of available primary amines, thus improving amine efficiency (mmol CO2 adsorbed per mmol amines).16 Neutron diffraction has also probed the interconnectivity and porosity of PEI, supported on dendritic fibrous nanosilica,17 whilst neutron reflectivity has probed the morphology of thin amine-containing films for CO2 capture.18a CO2 is challenging to observe directly using INS, due to the lack of hydrogen, thus it is not widely used for observing CO2 capture.18b The few existing examples focus on metal–organic framework (MOF) sorbents, particularly examining differences to hydrogen-containing groups before and after CO2 adsorption. Previous groups have compared the difference spectra of MOFs to obtain difference spectra, to distinguish between computational models of different binding sites.19a,19b Whilst supported aminosilanes have a lower hydrogen content than MOFs, they still possess hydrogen environments that can be probed, e.g., the CH2 groups on the aminopropyl chain, and hydrogens in the amine group. The latter have shown quite drastic shifts when interacting with CO2. As such in this work we observe the behaviour of aminosilane species grafted onto a mesoporous silica support, during CO2 capture using in situ INS techniques, from three leading INS spectrometers, namely VISION at ORNL, and MAPS and TOSCA, both at ISIS. Such systems can investigate vibrational spectra up to 4000 cm−1, with emphasis on wavenumbers < 1000 cm−1, which are rarely probed by infrared spectroscopy. The Q–E coverage (Fig. S1, ESI†) and energy resolution (Table S1, ESI†) are compared in the ESI.†
Both TOSCA20,21 and VISION22 are indirect geometry spectrometers, meaning they utilise a “white” incident neutron beam and the final energy of the neutrons is fixed. This system allows for better resolution at lower energy transfers, but a fixed trajectory through Q,ω-space and therefore poor signal at higher energy transfers. The difference between the instruments is due to the neutron flux, with VISION having a much greater flux than TOSCA, allowing for a better signal-to-noise ratio. MAPS is a direct geometry spectrometer,23 meaning the incident neutron beam is monochromated but scattered neutrons are detected at a range of scattering angles, leading to a wider range of Q,ω-space being interrogated, allowing data to be collected at higher energy transfers, typically above 2000 cm−1, at the cost of overall energy resolution. All three spectrometers will be utilised in this study to explore the benefits and potential of INS on exploring CO2 uptake with supported aminosilane CO2 sorbents.
Experimental methods
Synthesis
The bare silica gel (herein known as “bare silica”) support material (Davisil Grade 643, pore size 150 Å, 200–425 mesh, purchased from Sigma-Aldrich) was dried at 120 °C under 20 mbar of vacuum overnight to dry the species before grafting.10 g of dried bare silica was suspended in 200 mL of anhydrous toluene (Sigma Aldrich), with 7.2 g of 3-aminopropyl trimethoxysilane (APS; Sigma-Aldrich) added dropwise, giving a theoretical amine value of 4 mmol g−1 of silica gel, to create “APS-silica”. 4 mmol g−1 is beyond what can be accommodated on the silica gel, however, is used to maximise the amine-grafting on the surface. A separate species was also synthesised using the secondary amine precursor; N-methylaminopropyl trimethoxysilane (MeAPS; Sigma-Aldrich) using an identical procedure, except 7.7 g (4 mmol g−1 intended also) of MeAPS was used instead of 7.2 g of APS, creating “MeAPS-silica”. The system was then left to reflux for 24 hours at 110 °C. After this the system was cooled, filtered, and washed with 1 litre each of toluene, hexane, and ethanol, before being left to dry overnight at 70 °C. After this the system was dried further at 110 °C overnight, under reduced pressure (50 mbar).
Nitrogen physisorption
Prior to BET analysis, the sample was dried at reduced pressure of 30 mBar, at 120 °C overnight. Analysis was performed on a Micromeritics Gemini 2375 surface area analyser. Surface area was calculated using the BET model,24 while the pore width distribution was calculated with the BJH model.25Elemental analysis
CHN analysis was performed through a commercial service from the London Metropolitan University: “https://www.londonmet.ac.uk/services-and-facilities/elemental-analysis-service/”.Inelastic neutron scattering
Results and discussions
Physicochemical characterisation
Elemental (CHN) analysis confirmed the concentration of carbon and nitrogen for APS-silica and MeAPS-silica (Table S2, ESI†). APS-silica contained 6.51 wt% of carbon, and 2.25 wt% of nitrogen, giving an amine density of 1.61 mmol g−1, and a C/N molar ratio of 3.4. As expected, this is slightly higher than the ideal value of three (from –C3H6NH2), though this is typical of class 2 species, due to incomplete anchoring to the surface, where only two Si–O–Si bonds form, leaving one alkoxy group.30Similarly, MeAPS-silica contained 7.03 wt% of carbon and 1.88 wt% of nitrogen respectively, translating to an amine density of 1.34 mmol g−1 (Table S2, ESI†). This is lower than APS-silica, likely due to bulkier secondary amine forcing greater amine separation. The C/N molar ratio is 4.4, again above the ideal value of four for MeAPS (–C3H6NHCH3).
Nitrogen porosity explored the influence of grafting aminosilanes to the support. All three systems show a type V hysteresis (Fig. S2A, ESI†), associated with mesoporous systems. Grafting APS and MeAPS onto the silica gel, shows a notable decrease in porosity from the isotherms from bare silica (295 m2 g−1), though APS-silica and MeAPS-silica, are near identical (203 and 198 m2 g−1 respectively, Table S3, ESI†). The mesopore volume also decreased from 1.16 for bare silica, to 0.78 and 0.79 cm3 g−1 for APS-silica and MeAPS-silica respectively. None of the systems showed any significant micropore volume or microporosity. Comparing the BJH pore distribution (Fig. S2B, ESI†) shows that all three systems have a strong feature from 100 and 350 Å, with only subtle variations in the centre, showing grafting has not significantly influenced the pore size.
Combining the BET surface area with the amine density suggests each amine in APS-silica covers an average circular area of 21 Å2, translating to an amine–amine distance (diameter) of 5.2 Å (Table S4, ESI†). In contrast each amine in the MeAPS-silica covers an average circular area of 25 Å2, correlating to an amine–amine distance of 5.6 Å (Table S4, ESI†). This suggests both in both systems the amines should be sufficiently close to form alkylammonium carbamate species.
INS grafting data
The ISIS indirect inelastic neutron scattering spectrometer TOSCA has excellent resolution at low energy transfers (<2000 cm−1), and focusses on the spectral changes in this region on grafting APS and MeAPS to the silica gel. Four distinct features of bare silica at low wavenumbers; 102, 189, 254 and 453 cm−1, are attributed to the empty inconcel cell (“empty can”, Fig. S3, ESI†). The differences between the two spectra are subtle, with the main difference being the appearance of peaks at 849 cm−1 and 985 cm−1 in bare silica. 849 cm−1 has previously been attributed to symmetric Si–O vibrations, from tetrahedral Si–O–Si units, whilst 985 cm−1 is attributed to silanol species.31 Comparing empty can and bare silica shows no notable change in the ice region; 645 cm−1, validating our drying approach.32 Overall, bare silica contains hydroxyl species, as expected.Grafting APS onto the system (Fig. 1 and Fig. S4A, ESI†) results in many more signals, particularly in the 20 to 300 cm−1 (Fig. S4B, ESI†) and the 600 to 1800 cm−1 regions (Fig. 1 and Fig. S4C, S4D, ESI†) that TOSCA is well suited to. Here “pure APS” (C3H9O3SiC3H6NH2) is compared with APS grafted on silica “APS-silica” and the bare support (bare silica) to identify new features. Pure APS naturally has a much more intense signal due to the greater hydrocarbon contribution (Table S2, ESI†); 40.2 wt% C, 9.6 wt% H, and 9.3 wt% N, compared to the APS-silica species (6.51 wt% C and 2.25 wt% N), and as such will be compared on scaled axis. Similarly, the APS-silica species is more intense than bare silica, again due to higher quantities of hydrocarbon, lower surface areas and pore volumes, resulting in more matter for the neutrons to scatter from.
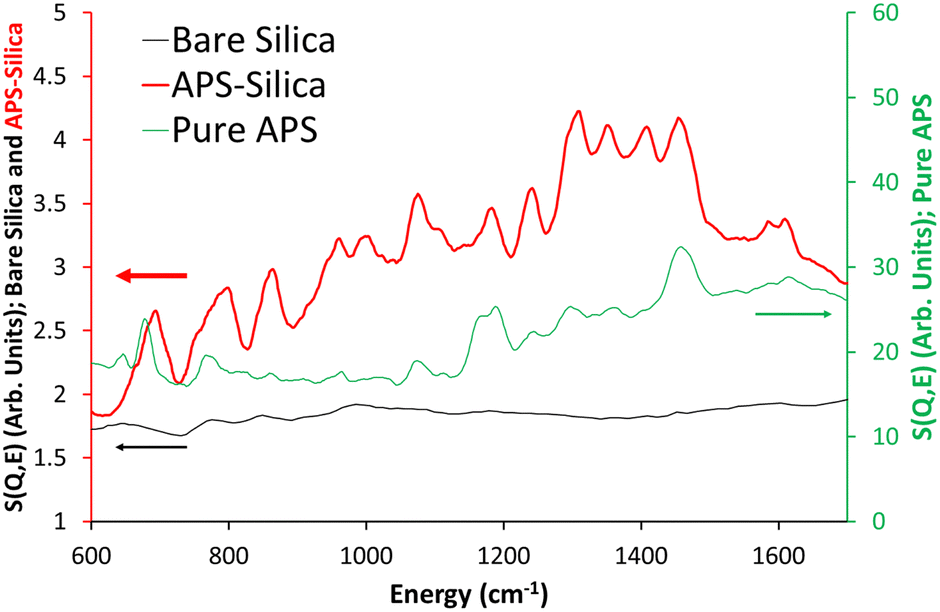 | ||
| Fig. 1 Comparing the TOSCA INS spectra of the bare silica gel (bare silica), APS grafted on silica (APS-silica) and the ungrafted APS molecule (pure APS) focussing on the 600 to 1700 cm−1 region. | ||
In the 20 to 600 cm−1 region, introducing APS to the system leads to new signals at 145 and 285 cm−1. Literature values, and our own DFT findings (Fig. S5 and Table S5, ESI†), assigned these as deformation of the C–C–N skeleton as previously seen in polyamine species.33 Specifically, they are attributed to in-plane (145 cm1) and out-of-plane (285 cm−1) C–C–N deformation. As detailed in the ESI,† the computational model is a simplified system, but is sufficient to identify the different hydrogeneous bands in the aminosilane species. The intensity of these species differs from APS-silica (Fig. S4, ESI†) to MeAPS-silica (Fig. S6, ESI†), particularly at 182 and 252 cm−1 where the MeAPS-silica is more intense (Fig. S6B, ESI†), as these features include contributions from the methyl group. Curiously in this region there is little agreement between the pure APS and APS-silica spectra (Fig. S4B, ESI†). Likely this is due to the pure APS being more concentrated, leading to significant hydrogen bonding and different environments, whereas the APS on silica will be more isolated.
A variety of sharp signals appear in the 600 to 1700 cm−1 region (Fig. 1 and Fig. S4C, S4D, ESI†), attributed to the grafted amine species (Table S5, ESI†). Features at 692 and 795 cm−1 are assigned to CH2 rocking34 for the C not bonded to N, and bonded to N, respectively, within the propyl chain, based on our DFT findings. There are similar, more muted features in pure APS, likely these are supressed due to the larger contributions from the three methoxy groups. 862 and 963 cm−1 are assigned as NH2 wagging,35 whereas the features at 1000 and 1075 cm−1 are due to CH2 twisting, as seen from our DFT findings. This is much weaker (compared to other features) in the pure APS. 1181 and 1241 cm−1 were found to be both a mixture of CH2 twisting and NH2 twisting.35 Similarly, the features at 1308 and 1352 cm−1 are a combination of CH2 wagging, CH2 twisting and NH2 twisting modes.36
The features at 1411 and 1457 cm−1 were found to be CH2 scissoring, in good agreement with the literature,37 whilst the less intense pair of features at 1584 and 1610 cm−1 are attributed to NH2 scissoring modes.35,38 MeAPS-silica shares many features with APS-silica, particularly below 600 cm−1 (Fig. S6A and B, ESI†), but there are notable differences at higher wavenumbers. The 862 cm−1 signal loses intensity (Fig. 2 and Fig. S6C, ESI†), as the methyl group hinders NH2 wagging, supporting the assignment. Similarly, in MeAPS-silica the signals at 963 and 1000 cm−1 combine, leading to a single signal centred at 980 cm−1, while the APS-silica signal at 1075 cm−1 also shifts to a higher wavenumber at 1107 cm−1 (Fig. 2 and Fig. S6C, ESI†). The APS-silica features at 1181 and 1241 cm−1 also begin to merge in MeAPS-silica and settle at 1200 and 1221 cm−1 (Fig. 2 and Fig. S6D, ESI†), likely due to a reduction in the contribution from the amine twisting component. The APS-silica 1308 cm−1 signal drops slightly in intensity, suggesting this signal contains only a small NH2 twisting contribution, whereas the 1352 cm−1 band drops markedly in intensity, suggesting a larger NH2 twisting component (Fig. 2 and Fig. S6D, ESI†). The feature at 1411 cm−1 shows a subtle shift, however the intensity of the 1457 cm−1 feature grows, suggesting it now includes modes from the methyl-amine group, which contribute to the C–H deformation (Fig. 2 and Fig. S6D, ESI†). Finally, bands at 1584 and 1610 cm−1, associated with amine scissoring also are not as prominent in the MeAPS-silica spectra, again due to the lack of primary amine (Fig. 2 and Fig. S6D, ESI†).
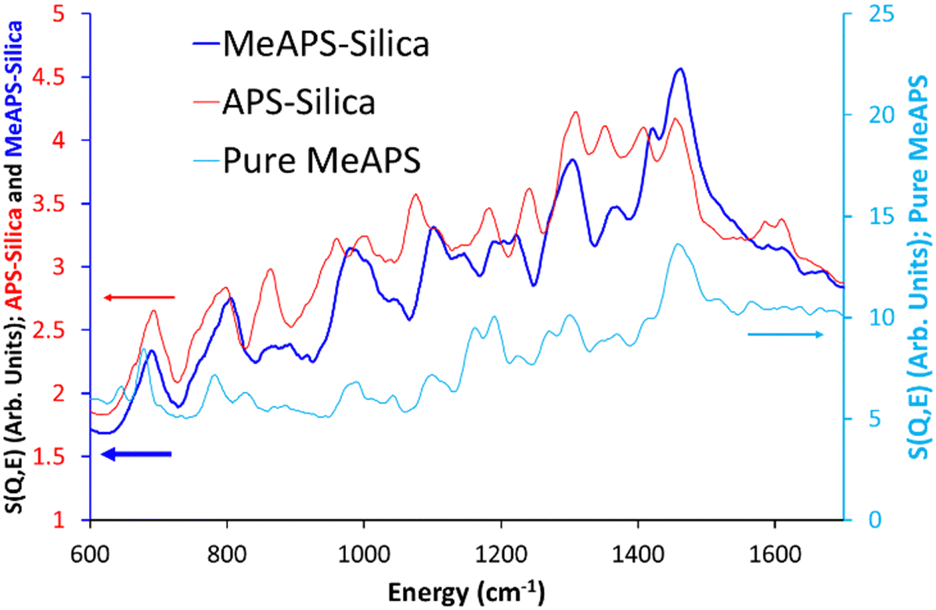 | ||
| Fig. 2 Comparing the TOSCA INS spectra of APS grafted on silica (APS-silica), MeAPS grafted on silica (MeAPS-silica) and the ungrafted MeAPS molecule (pure MeAPS) focussing on the 600 to 1700 cm−1 region. | ||
The ISIS spectrometer MAPS was used to observe the investigate higher wavenumbers (>2000 cm−1) for the empty can, bare silica, APS-silica and MeAPS-silica (Fig. 3). The empty can is featureless in this area, whereas bare silica shows a slight feature at ∼3710 cm−1, due to the presence of silanol species.31 This feature is less pronounced in APS-silica and MeAPS-silica, as the silanol species are consumed by the grafting process, and also obscured by the higher INS background, due to a greater quantity of scattering species (hydrogen) in the grafted samples. MAPS data shows signals due to C–H vibration, from both APS-silica and MeAPS-silica, at 2899 and 2907 cm−1 respectively (Fig. 3).39
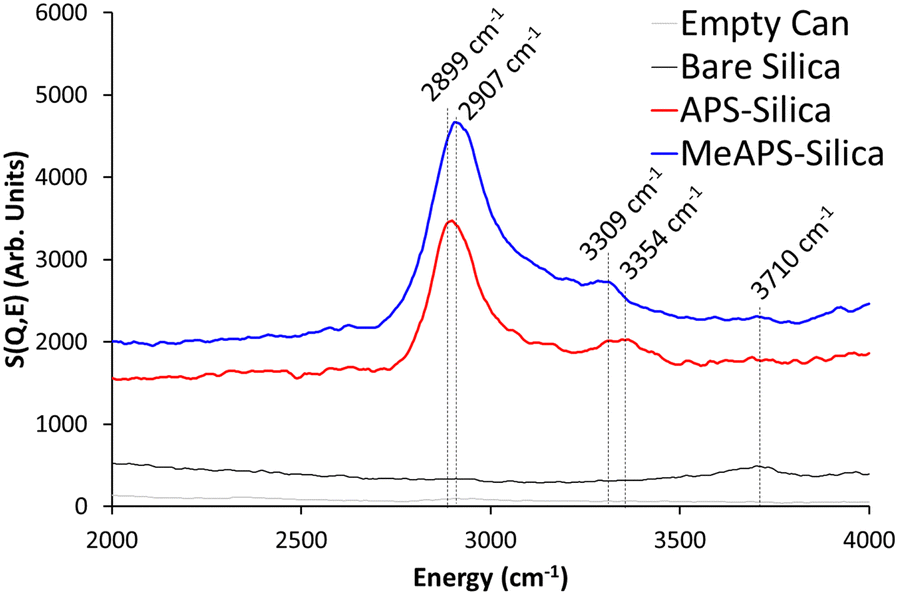 | ||
| Fig. 3 Comparing the MAPS INS spectra of APS grafted on silica (APS-silica), MeAPS grafted on silica (MeAPS-silica) to the bare silica support (bare silica) and the empty can focussing on the 2000 to 4000 cm−1 region. | ||
Broad N–H vibrations can also be seen, here APS-silica is centred at 3354 cm−1, likely due to a combination of symmetric and asymmetric modes39 whereas the secondary MeAPS-silica is notably lower at 3309 cm−1, given the breadth of these feature it is challenging to explain this. Further, while INS is clearly able to show signals in this area, it must be noted that, in this case, infrared measurements are often better suited to observing the C–H and N–H stretches in this region, due to the greater accessibility, faster collection time and better resolution, assuming the inorganic matrix does not dominate the spectra. Despite this, the data above shows the benefits of using INS to observe signals below 2000 cm−1.
Introduction of CO2
Introducing 1 bar of CO2 to bare silica showed little change in the spectrum (Fig. S7, ESI†), as the system adsorbed just 0.23 mmol of CO2 per g of silica gel (Tables S6 & S7, ESI†). This is slightly higher than reported values for mesoporous silicas such as SBA-15, as here 1 bar of pure CO2 is used, whereas literature uptake values are measured using dilute streams. Further the greater number of silanol groups on silica gel, compared to mesoporous silicas, will lead to greater interactions and uptake with the CO2. The inclusion of CO2 into the system slightly supresses some signals, particularly 849 and 909 cm−1 (symmetric Si–O vibrational stretch; Fig. S7C, ESI†), and also 1736 and 1806 cm−1 (SiO–H vibrations; Fig. S7D, ESI†), suggesting there is a weak interaction between the CO2 and the silanol species (summarised in Table S8, ESI†).Adding 1 bar of CO2 to APS-silica saw the system adsorb 0.84 mmol of CO2 per g of sorbent (Tables S6 and S7, ESI†), a significant improvement over bare silica due to the amines present. The calculated amine efficiency here was 0.52 (Table S7, ESI†), which is above the theoretical maximum of 0.5 under dry conditions. However, the bare silica support can adsorb CO2, thus it is unlikely the CO2 uptake is purely due to the amine. As per our previous work, a modified amine efficiency was calculated as 0.38, which assumes the amine is only responsible for the difference in CO2 uptake between the bare support, and amine-supported system, normalised by the amine density.6
The traditional amine efficiency and modified amine efficiency can be treated as upper and lower limits, with the true value being between the two. In the TOSCA data below 2000 cm−1; (Fig. 4A and Fig. S8, ESI†) of APS-silica gel the region below 600 cm−1 is relatively unchanged on addition of CO2, despite reduced intensity at 384 cm−1 (skeletal deformation), and an associated increase at 575 cm−1, which has previously been attributed to the formation of ammonium species (summarised in Table S8, ESI†). However more pronounced changes occur in the 600 to 1800 cm−1 region (Fig. 4A and Fig. S8C, S8D, ESI†), as features decrease in intensity, or shift position. It is expected that the largest changes will occur in modes involving the nitrogen atom, as this is where the most significant chemical changes are on adsorbing CO2. The feature at 862 cm−1 decreases in intensity, likely due to the R–NH2 groups adsorbing CO2 (Table S5, ESI†), preventing the NH2 wagging from occurring. Further the signal at 1075 cm−1 has shifted down to 1060 cm−1. Both the 1181 and 1241 cm−1 signals decrease on adsorbing CO2 (Fig. 4A and Fig. S8C, S8D, ESI†). Likely this is due to the adsorbed CO2 causing greater interactions either between amines (alkylammonium carbamate species) or the support (silylpropyl carbamate),6 restricting methylene and NH2 twisting. The signal at 1308 cm−1 (Fig. 4A and Fig. S8D, ESI†) is similarly associated with CH2 twisting motions, however unlike the previous features, it retains its intensity, which may be due to a concomitant rise in NCOO−, in a similar area. Further there are slight decreases in intensity in the 1352 and 1411 cm−1 features (Fig. 4A and Fig. S8D, ESI†), again suggesting the methylene motions are being hampered. Between 1500 and 1700 cm−1 (Fig. 4A and Fig. S8D, ESI†) the APS-silica gel had two features (1584 and 1610 cm−1), however small features appear in this region, at 1521, 1565, and 1639 cm−1, which are assigned to R–NH3+ symmetric and asymmetric modes, as well as deformation, highlighting the formation of an ammonium species, due to the formation of an alkyl carbamate species (Fig. S8E, ESI†).39
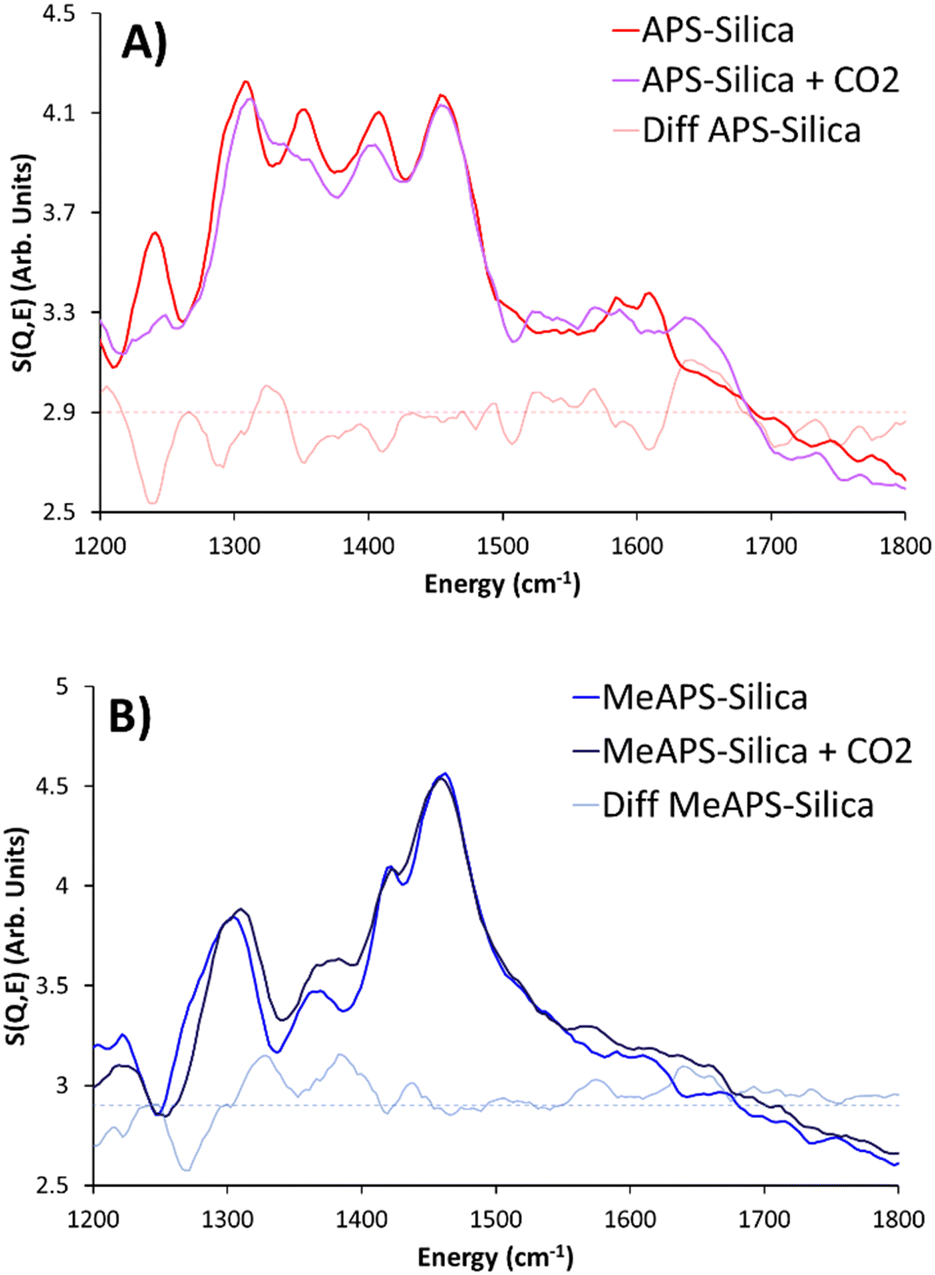 | ||
| Fig. 4 Comparing the TOSCA INS spectra of (A) APS-silica and (B) MeAPS-silica, before and after being exposed to 1 bar of CO2, focussing on the 1200 to 1800 cm−1 region, including artificially increased difference spectra. | ||
The secondary amine; MeAPS-silica species has a lower CO2 uptake than APS-silica, as expected based on previous CO2 uptake data, where primary amines typically outperform secondary amines. Here MeAPS-silica adsorbed 0.65 mmol of CO2 per g of sorbent (Tables S6 and S7, ESI†), corresponding to a lower amine efficiency of 0.49, and a lower modified amine efficiency of 0.32 (Table S7, ESI†). As a result, the TOSCA INS spectra of MeAPS-silica before and after CO2 adsorption showed fewer differences than the APS-silica species (Fig. 4B and Fig. S9, summarised in Table S8, ESI†). Below 600 cm−1 the two main differences for MeAPS-silica are a reduction in the 249 cm−1 signal and a new signal emerging at 411 cm−1 (Fig. S9B, ESI†) There is significantly less literature on vibrational spectroscopy of secondary amines, particularly at these low wavenumbers, as such the exact nature of this feature is unknown. Prior work on protonated primary amines has attributed this to a deformation involving a N–C–C species, however, to confidently assign this further spectroscopic and computational studies are required. In the 600 to 1200 cm−1 region (Fig. S9C, ESI†) there is very little change in intensity, however there is a notable shift in peak position as the 1107 cm−1 C–C vibration shifts to 1121 cm−1 on adsorbing CO2. Similar behaviour to the APS-silica species is seen in the 1500 to 1700 cm−1 region (Fig. 4B and Fig. S9D, ESI†), where the two original N–H bands, split into four species on adsorbing CO2, at 1571, 1613, 1639 and 1662 cm−1.39 Again, this is likely due to the protonated MeNH2+ species forming, though further analysis is required to probe this in greater detail (Fig. S9E, ESI†). Using MAPS to consider the higher wavenumbers, shows some differences on capturing CO2 (Fig. 5 and Fig. S10, ESI†). On binding to CO2, the C–H signal in APS-silica has shifted from 2899 cm−1 to 2910 cm−1, suggesting the C–H bonds have been strengthened as CO2 binds to the system.
 | ||
| Fig. 5 Comparing the MAPS INS spectra of APS-silica and MeAPS-silica, before and after being exposed to 1 bar of CO2, focussing on the 2000 to 4000 cm−1 region. The spectra have been vertically aligned for ease of comparison. | ||
As expected, the N–H signal at 3354 cm−1, associated with the primary amine, has diminished, as the amine either interacts with CO2, or becomes protonated by neighbouring amines. The MeAPS-silica system shows subtle differences in this region, in keeping with the rest of the spectra. There is a small shift in the C–H peak at 2907 cm−1, and a slight decrease in the 3309 cm−1 signal indicating the loss of N–H species.
To mimic infrared and Raman studies, VISION at ORNL was used to explore the change in INS spectra, on addition of small amounts of CO2. Whilst TOSCA has superb resolution for identifying distinct bands, the higher flux of VISION, and associated improved signal-to-noise ratio, is well suited to identifying small differences in the spectra (Fig. S11, ESI†). Here 0.053, 0.15, 0.5 and 1.5 mmol of CO2 per gram of was introduced into the system, corresponding to 6, 18, 60 and 180%, respectively, of the total CO2 uptake used in the MAPS and TOSCA experiments (Fig. 6 and Fig. S12, S13, ESI†).
 | ||
| Fig. 6 Comparing VISION INS difference spectra of APS-silica, on adsorbing different quantities of CO2. Each series is labelled with the quantity of CO2 (in mmol g−1) introduced to the sorbent, with the TOSCA INS spectra of APS-silica with no CO2. | ||
A range of differences can be seen in APS-silica on adsorbing CO2, which typically progress as the CO2 quantities increase, however as expected, the APS-silica 0.50 and APS-silica 1.50 spectra are similar, suggesting that the species was saturated at this point. A key difference between the spectra is the decrease in intensity below 200 cm−1, which was also seen in the TOSCA data (Fig. 6 and Fig. S12, S13, ESI†). This intensity change is very sensitive to the presence of CO2, as it can be clearly observed at low (0.053 mmol g−1) CO2 volumes. Similarly, there are notable decreases in intensity at 278, 383 and 494 cm−1 (Fig. 6 and Fig. S13B, ESI†). In the 600 to 1200 cm−1 region (Fig. S13C, ESI†) there are few changes seen, with a subtle increase at 673 cm−1 being observed, whilst the CH2 twisting at 1098 cm−1 decreases. A final, very subtle feature is the reduction of signal at 1253 cm−1 (Fig. S13D, ESI†), which has previously been attributed to CH2 twisting. At this stage it is challenging to accurately identify these stretches. However, noticeable changes in the INS spectra are occurring, even at low CO2 uptakes, suggesting there is potential for INS to be used to assign features, and verify chemical environments, alongside Raman and infrared spectroscopy.
Conclusions
CO2 adsorption is a vital technology that must be optimised in the coming years, requiring an in-depth understanding of CO2 sorbents. Given the lack of hydrogen in CO2, inelastic neutron spectroscopy has not traditionally been used to perform in situ adsorption measurements. However, in this work we have shown the benefits of this technique, namely the ability to identify subtle changes in hydrogenous modes of aminosilane sorbents, on adsorbing CO2, and not being constrained by selection rules, to identify a wide range of vibrational features. Further we utilise three of the leading inelastic neutron scattering spectrometers; TOSCA and MAPS both from ISIS, and VISION from ORNL, to provide greater detail of this process. Each spectrometer has unique advantages, be it the energy range studied, or measurement speed, which allowed complimentary information to be collected.Author contributions
MEP wrote the manuscript, designed the study, and collected physicochemical characterisation data, and INS data on TOSCA and MAPS. HC helped collect and analyse the INS data on TOSCA and MAPS. JJMLB helped collect the INS data on TOSCA and MAPS. LLD collected and analyse the INS data on VISION. YC performed the complementary DFT study and analysed the DFT data. All authors have given their approval for the submitted version.Data availability
Data for this article, including N2 physisorption, INS spectra, and calculated DFT spectra are available at Open Science Framework at https://doi.org/10.17605/OSF.IO/TDVJC.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
MEP acknowledges the University of Bath Prize Fellow scheme for funding. JJMLB acknowledges TotalEnergies “Consortium on metal nanocatalysts” for funding. The authors would like to thank the Science & Technology Facilities Council for the provision of neutron beamtime (experiments RB1820003; DOI: https://doi.org/10.5286/ISIS.E.RB1820003 and RB1920061; DOI: https://doi.org/10.5286/ISIS.E.RB1920061). A portion of this research used resources at Spallation Neutron Source, a DOE Office of Science User Facility operated by the Oak Ridge National Laboratory. The beam time was allocated to VISION BL-16B on proposal number IPTS-24094.1. Prof R. Raja is acknowledged as JJMLB's PhD supervisor.Notes and references
- https://www.iea.org/reports/world-energy-outlook-2020/achieving-net-zero-emissions-by-2050 .
- S. A. Didas, S. Choi, W. Chaikittisilp and C. W. Jones, Acc. Chem. Res., 2015, 48, 2680–2687 CrossRef CAS PubMed.
- K. Akimoto, F. Sano, J. Oda, H. Kanaboshi and Y. Nakano, Energy Clim. Change, 2021, 2, 100057 CrossRef CAS.
- A. K. Morken, S. Pedersen, S. O. Nesse, N. E. Flø, K. Johnsen, J. K. Feste, T. de Cazenove, L. Faramarzi and K. Vernstad, Int. J. Greenhouse Gas Control, 2019, 82, 175–183 CrossRef CAS.
- E. S. Sanz-Perez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef CAS PubMed.
- M. E. Potter, K. M. Cho, J. J. Lee and C. W. Jones, ChemSusChem, 2017, 10, 2192–2201 CrossRef CAS PubMed.
- M. A. Alkhabbaz, P. Bollini, G. S. Foo, C. Sievers and C. W. Jones, J. Am. Chem. Soc., 2014, 136, 13170–13173 CrossRef CAS PubMed.
- C.-T. Hung, C.-F. Yang, J.-S. Lin, S.-J. Huang, Y.-C. Chang and S.-B. Liu, Microporous Mesoporous Mater., 2017, 238, 2–13 CrossRef CAS.
- R. A. Khatri, S. S. C. Chuang, Y. Soong and M. Gray, Energy Fuels, 2006, 20, 1514–1520 CrossRef CAS.
- C. J. Keturakis, F. Ni, M. Spicer, M. G. Beaver, H. S. Caram and I. E. Wachs, ChemSusChem, 2014, 7, 3459–3466 CrossRef CAS PubMed.
- J. Yu and S. S. C. Chuang, Energy Fuels, 2016, 30, 7579–7587 CrossRef CAS.
- L. Lin, A. M. Sheveleva, I. da Silva, C. M. A. Parlett, Z. Tang, Y. Liu, M. Fan, X. Han, J. H. Carter, F. Tuna, E. J. L. McInnes, Y. Cheng, L. L. Daemen, S. Rudic, A. J. Ramirez-Cuesta, C. C. Tang and S. Yang, Nat. Mater., 2020, 19, 86–93 CrossRef CAS PubMed.
- A. P. Hawkins, A. J. O’Malley, A. Zachariou, P. Collier, R. A. Ewings, I. P. Silverwood, R. F. Howe, S. F. Parker and D. Lennon, J. Phys. Chem. C, 2018, 123, 417–425 CrossRef.
- S. F. Parker and D. Lennon, Physchem, 2021, 1, 95–120 CrossRef.
- A. Holewinski, M. A. Sakwa-Novak, J. Y. Carrillo, M. E. Potter, N. Ellebracht, G. Rother, B. G. Sumpter and C. W. Jones, J. Phys. Chem. B, 2017, 121, 6721–6731 CrossRef CAS PubMed.
- A. Holewinski, M. A. Sakwa-Novak and C. W. Jones, J. Am. Chem. Soc., 2015, 137, 11749–11759 CrossRef CAS PubMed.
- J. Bahadur, S. Mehta, S. Singh, A. Das, A. Maity, T. Youngs, D. Sen and V. Polshettiwar, Mater. Adv., 2022, 3, 6506–6517 RSC.
- (a) R. A. Campbell and K. J. Edler, Soft Matter, 2011, 7, 11125–11132 RSC; (b) K. W. B. Hunvik, P. Loch, L. P. Cavalcanti, K. K. Seljelid, P. M. Røren, S. Rudić, D. Wallacher, A. Kirch, K. D. Knudsen, C. R. Miranda, J. Breu, H. N. Bordallo and J. O. Fossum, J. Phys. Chem. C, 2020, 124, 26222–26231 CrossRef CAS.
- (a) S. Yang, J. Sun, A. J. Ramirez-Cuesta, S. K. Callear, W. I. David, D. P. Anderson, R. Newby, A. J. Blake, J. E. Parker, C. C. Tang and M. Schroder, Nat. Chem., 2012, 4, 887–894 CrossRef CAS PubMed; (b) P. Zhao, H. Fang, S. Mukhopadhyay, A. Li, S. Rudić, I. J. McPherson, C. C. Tang, D. Fairen-Jimenez, S. C. E. Tsang and S. A. T. Redfern, Nat. Commun., 2019, 10, 999 CrossRef CAS PubMed.
- Z. A. Bowden, M. Celli, F. Cilloco, D. Colognesi, R. J. Newport, S. F. Parker, F. P. Ricci, V. Rossi-Albertini, F. Sacchetti, J. Tomkinson and M. Zoppi, Physica B, 2000, 276–278, 98–99 CrossRef CAS.
- R. S. Pinna, S. Rudić, S. F. Parker, J. Armstrong, M. Zanetti, G. Škoro, S. P. Waller, D. Zacek, C. A. Smith, M. J. Capstick, D. J. McPhail, D. E. Pooley, G. D. Howells, G. Gorini and F. Fernandez-Alonso, Nucl. Instrum. Methods Phys. Res., Sect. A, 2018, 896, 68–74 CrossRef CAS.
- P. A. Seeger, L. L. Daemen and J. Z. Larese, Nucl. Instrum. Methods Phys. Res., 2009, 604, 719–728 CrossRef CAS.
- R. A. Ewings, J. R. Stewart, T. G. Perring, R. I. Bewley, M. D. Le, D. Raspino, D. E. Pooley, G. Skoro, S. P. Waller, D. Zacek, C. A. Smith and R. C. Riehl-Shaw, Rev. Sci. Instrum., 2019, 90, 035110 CrossRef CAS PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- M. E. Potter, Probing the binding of CO2 on supported aminosilane sorbents with INS, STFC ISIS Neutron and Muon Source, 2020 DOI:10.5286/ISIS.E.RB1820003.
- M. E. Potter, Probing the binding of CO2 on supported aminosilane sorbents with INS, STFC ISIS Neutron and Muon Source, 2020 DOI:10.5286/ISIS.E.RB1920061.
- https://www.mantidproject.org/ DOI:10.5286/SOFTWARE/MANTID .
- O. Arnold, J. C. Bilheux, J. M. Borreguero, A. Buts, S. I. Campbell, L. Chapon, M. Doucet, N. Draper, R. Ferraz Leal, M. A. Gigg, V. E. Lynch, A. Markvardsen, D. J. Mikkelson, R. L. Mikkelson, R. Miller, K. Palmen, P. Parker, G. Passos, T. G. Perring, P. F. Peterson, S. Ren, M. A. Reuter, A. T. Savici, J. W. Taylor, R. J. Taylor, R. Tolchenov, W. Zhou and J. Zikovsky, Nucl. Instrum. Methods Phys. Res., Sect. A, 2014, 764, 156–166 CrossRef CAS.
- S. Bali, J. Leisen, G. S. Foo, C. Sievers and C. W. Jones, ChemSusChem, 2014, 7, 3145–3156 CrossRef CAS PubMed.
- A. Burneau and C. d Carteret, Phys. Chem. Chem. Phys., 2000, 2, 3217–3226 RSC.
- C. Corsaro, V. Crupi, D. Majolino, S. F. Parker, V. Venuti and U. Wanderlingh, J. Phys. Chem. A, 2006, 110, 1190–1195 CrossRef CAS PubMed.
- M. P. M. Marques, L. A. E. Batista de Carvalho and J. Tomkinson, J. Phys. Chem. A, 2002, 106, 2473–2482 CrossRef CAS.
- I. Ospino, A. Luquin, M. Jiménez-Ruiz, J. I. Pérez-Landazábal, V. Recarte, J. C. Echeverría, M. Laguna, A. A. Urtasun and J. J. Garrido, J. Phys. Chem. C, 2017, 121, 22836–22845 CrossRef CAS.
- S. R. O. Mendes, A. M. Amado, J. Tomkinson, M. P. M. Marques and L. A. E. Batista de Carvalho, New J. Chem., 2017, 41, 10132–10147 RSC.
- M. K. Wong, M. A. Bustam and A. M. Shariff, Int. J. Greenhouse Gas Control, 2015, 39, 139–147 CrossRef CAS.
- I. P. Silverwood, N. G. Hamilton, A. R. McFarlane, J. Kapitan, L. Hecht, E. L. Norris, R. M. Ormerod, C. D. Frost, S. F. Parker and D. Lennon, Phys. Chem. Chem. Phys., 2012, 14, 15214–15225 RSC.
- C. T. Johnston, D. L. Bish, J. Eckert and L. A. Brown, J. Phys. Chem. B, 2000, 104, 8080–8088 CrossRef CAS.
- A. Danon, P. C. Stair and E. Weitz, J. Phys. Chem. C, 2011, 115, 11540–11549 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: including details of physicochemical characterisation, and further inelastic neutron scattering data, including computational details. See DOI: https://doi.org/10.1039/d4cp02316a |
| This journal is © the Owner Societies 2024 |