
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biomass-derived carbonaceous positive electrodes for sustainable lithium-ion storage†
Tianyuan
Liu
a,
Reza
Kavian
a,
Zhongming
Chen
b,
Samuel S.
Cruz
a,
Suguru
Noda
b and
Seung Woo
Lee
 *a
*a
aGeorge W. Woodruff School of Mechanical Engineering, Georgia Institute of Technology Atlanta, Georgia 30332, USA. E-mail: seung.lee@me.gatech.edu
bDepartment of Applied Chemistry, Waseda University, 3-4-1 Okubo, Shinjuku-ku, Tokyo 169-8555, Japan
First published on 5th January 2016
Abstract
Biomass derived carbon materials have been widely used as electrode materials; however, in most cases, only electrical double layer capacitance (EDLC) is utilized and therefore, only low energy density can be achieved. Herein, we report on redox-active carbon spheres that can be simply synthesized from earth-abundant glucose via a hydrothermal process. These carbon spheres exhibit a specific capacity of ∼210 mA h gCS−1, with high redox potentials in the voltage range of 2.2–3.7 V vs. Li, when used as positive electrode in lithium cells. Free-standing, flexible composite films consisting of the carbon spheres and few-walled carbon nanotubes deliver high specific capacities up to ∼155 mA h gelectrode−1 with no obvious capacity fading up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles, proposing to be promising positive electrodes for lithium-ion batteries or capacitors. Furthermore, considering that the carbon spheres were obtained in an aqueous glucose solution and no toxic or hazardous reagents were used, this process opens up a green and sustainable method for designing high performance, environmentally-friendly energy storage devices.
000 cycles, proposing to be promising positive electrodes for lithium-ion batteries or capacitors. Furthermore, considering that the carbon spheres were obtained in an aqueous glucose solution and no toxic or hazardous reagents were used, this process opens up a green and sustainable method for designing high performance, environmentally-friendly energy storage devices.
Introduction
Replacing fossil fuels with various types of clean and renewable energy sources is vital in order to effectuate the world's sustainable development.1 However, the inconstant and location dependent natures of renewable energy sources such as wind or solar require the integration with electrical energy storage systems for improving reliability.2,3 Electrochemical energy storage systems, including rechargeable lithium-ion batteries and electrochemical capacitors (ECs), have several advantages for effective renewable energy storage, such as compact size, long cycle life, high efficiency, short charge/discharge time and pollution free operation.2,4 Despite these merits, the high cost of electrode materials, such as transition metal oxides, limits their market penetration for large-scale energy storage applications.2,5 Thus, it is critical to develop low-cost, sustainable, and high-performance electrode materials to support electrochemical energy storage devices in line with the green and renewable energy sources.6–8Redox-active organic electrode materials derived from low-cost and sustainable sources have attracted extensive attention as replacements for transition metal based electrodes.6,9 In particular, organic carbonyl molecules have shown high capacities (up to 580 mA h g−1 for LixC6O6),10 by employing the reversible redox reaction between the carbonyl group and lithium ion.9 However, dissolution of these organic molecules into organic electrolytes during cycling limits their cycling stability within a few hundred cycles.11–14 In addition, the insulating nature of organic molecules requires mixing with a large amount of conductive carbons during the electrode fabrication process to support electrical conductivity.9,15–17 An alternative approach to resolving these challenges is to incorporate redox-active oxygen functional groups on a conductive carbon matrix such as carbon nanotubes or graphene, via a surface oxidation process.18–22 In addition, these functionalized carbon materials can be fabricated as free-standing electrodes. Free-standing carbon electrodes are free of metal current collectors, which can improve the electrochemical performance of practical devices.22 Free-standing oxidized carbon nanotubes or reduced graphene oxide electrodes show enhanced rate capability as well as cycling stability over 1000 cycles owing to the covalent-bonded oxygen functional groups on the conductive carbon matrix.18,20
Biomass is renewable, cheap, and is the most abundant carbon source for synthesizing sustainable functional carbon materials.23–25 Among various conversion methods, a hydrothermal carbonization (HTC) process has shown promising outcome in converting biomass to various functional carbon materials, utilizing its unique advantages including being an environmentally benign process, having relatively low reaction temperature and versatile morphology control of carbon products.23 In recent studies, carbon spheres have been synthesized from carbohydrates such as glucose, sucrose, or cellulose via the HTC process, and can be used as catalyst support26 or active electrode materials for electrochemical double layer capacitor (EDLC) applications.27,28 However, the redox-active properties of these carbon spheres have not been explored intensively. If comprehensively studied and rationally utilized, these properties can open up new possibilities for developing renewable electrode materials for lithium-ion batteries or lithium-ion capacitors.
In this study, we demonstrate for the first time that carbon sphere products from glucose, via a HTC process, have redox-active properties with lithium ions, which enable those carbon spheres to be active electrode components for lithium-ion batteries or capacitors. We assembled free-standing composite electrodes by mixing the carbon spheres with sub-millimeter long few-walled carbon nanotubes (FWNTs)29,30 using a vacuum-filtration process. In addition, we showed that a microwave treatment can improve connectivity between the carbon spheres and FWNTs. After the microwave process, the composite electrode exhibits a high capacity of ∼155 mA h gelectrode−1, with significantly enhanced rate capability compared to that before the microwave treatment. Moreover, the electrode shows an excellent cycling stability up to 10000 cycles. Considering that the redox-active carbon spheres are prepared from earth-abundant biomass via a scalable HTC process, this approach provides a promising green route to developing sustainable organic electrodes for large-scale energy storage devices.
Results and discussion
The carbon spheres were prepared from an aqueous solution of glucose (3 mg mL−1) via a HTC process at 200 °C for 18 h, during which the transparent glucose solution was transformed into a brownish colloidal dispersion (Fig. 1a). The HTC process consists of complex chemical transformation cascades, including dehydration, condensation, polymerization and aromatization reactions, which result in the formation of carbon-rich organic compounds (carbon spheres).23,31–33 The sizes of synthesized carbon spheres are mainly in the range of 60–100 nm as shown in Fig. S1c.† Free-standing composite films were fabricated via filtering the aqueous mixture of carbon spheres and FWNTs, where sub-millimeter long FWNTs were employed as a conductive agent (Fig. 1c inset). Pristine FWNT films were also prepared via a vacuum-filtration process for comparison. The FWNT films exhibited an intertwined network structure with a high electrical conductivity of ∼209 Scm2 g−1 (Fig. 1b and Table S1†). The density of a composite film with 40 wt% of carbon spheres (CS-0.4) increased to 0.37 g cm−3 from 0.24 g cm−3 of the pristine FWNT, whereas electrical conductivity of the composite films decreased to ∼93 Scm2 g−1 (Table S1†). Incorporating more carbon spheres into the composite electrode, up to 68 wt% (CS-0.68), further increased the density to 0.41 g cm−3 and decreased the electrical conductivity to ∼64.4 Scm2 g−1 (Table S1†). Most of the carbon spheres were attached on the surface FWNTs in the composite film with a lower loading (CS-0.4) (Fig. 1c), while considerable amount of the carbon spheres were separated from FWNTs in the film with a higher loading (CS-0.68) (Fig. 1d and S1b†). Irradiating nanocarbon materials with microwaves has been widely used as a rapid heat treatment method.34–36 After a microwave process for 30 s under Ar environment, the carbon spheres in the film (CS-0.68) were ‘glued’ together forming peanut-shaped nanorod structures, creating a dense network structure that was well threaded by FWNTs (Fig. 1e and f, CS-0.68-MW). During the microwave treatment, FWNTs in the composite films strongly absorbed the microwaves and acted as an internal heat source that can be heated up to 2000 °C.37–39 Therefore, we postulated that the heated FWNTs enable the reconstruction of the interface between CSs and FWNTs, giving rise to the fusion of CSs and FWNTs. The electrical conductivity of the film after the microwave process (CS-0.68-MW) was found to increase to ∼75.7 Scm2 g−1, owing to the improved connectivity between CSs and FWNTs during the process. | ||
| Fig. 1 (a) Digital images of the aqueous glucose solution (3 mg mL−1, left) and the colloidal dispersion of synthesized carbon spheres (CSs) after the hydrothermal carbonization (HTC) process (right). SEM images of (b) the pristine few-walled carbon nanotube (FWNT) film and composite films consisting of (c) 40 wt% of CSs (CS-0.4), (d) 68 wt% of CSs (CS-0.68). The digital image of the composite film (CS-0.4) fabricated by the vacuum-filtration process is shown in the inset of (c). (e) Low- and (f) high-magnification SEM images of the composite film (CS-0.68) after the microwave process at 1250 W for 30 s in Ar. | ||
The chemical structure of the carbon spheres was investigated by Raman spectroscopy and X-ray photoelectron spectroscopy (XPS). The Raman spectrum of the carbon spheres exhibited two broad overlapping bands at ∼1350 cm−1 (D mode) and ∼1580 cm−1 (G mode) (Fig. 2a), which are completely different from that of glucose (Fig. S2†). These two peaks resemble the characteristics of carbon materials40–42 that include sp2 hybridized carbon atoms in benzene or condensed benzene structures.42 The pristine FWNT film displayed much sharper peaks, and the spectrum of the composite films showed blended characteristics between the carbon sphere and FWNT (Fig. 2a and S3a†). Chemical element analysis of the carbon sphere and composite films was conducted using XPS (Fig. 2b and S3b†). As-synthesized carbon spheres showed significant oxygen content with an atomic ratio of oxygen to carbon (O/C ratio) of 0.32. According to the LaMer model, hydrothermally synthesized carbon spheres have a core–shell type structure consisting of a highly dehydrated hydrophobic core and an oxygen-rich hydrophilic shell.23,31 Thus, the high O/C ratio of the carbon spheres can be attributed to a large number of oxygen functional groups on the surface.24 On the other hand, pristine FWNT film exhibited negligible oxygen content with an O/C ratio of 0.03, which was probably due to the adsorbed oxygen or introduced defects during the dispersion process. The O/C ratios of the composite films were found to increase from 0.10 to 0.14 as the loading of the carbon sphere increased from 40 wt% (CS-0.4) to 68 wt% (CS-0.68) (Fig. 2b). Detailed chemical structures of the composite films were further analyzed by fitting the high resolution C 1s peaks (Fig. 2c).19 The main peak at 284.5 eV was attributed to sp2 hybridized graphitic carbon,19 and the binding energy for sp3 hybridized diamond-like carbon shifted to a higher binding energy at 285.2 ± 0.1 eV. C 1s spectra of the carbon sphere exhibited a dominant sp3 hybridized carbon peak with a minor sp2 hybridized graphitic carbon peak, while the FWNT film showed a major sp2 carbon peak with a minor sp3 carbon peak (Fig. S3c†). In addition, various oxygen functional groups, including hydroxyl/phenolic (C–O centered at 286.0 ± 0.1 eV), carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O centered at 286.9 ± 0.1 eV), and carboxylic/carboxylate (COOH/COOR, centered at 288.9 ± 0.1 eV),22 were found for the carbon sphere, which is consistent with the LaMer model. Therefore, the ratio of sp3 peak to sp2 peak, and the amount of oxygen functional groups, progressively increased with higher loading of the carbon spheres in the composite films (Fig. 2c). After the microwave process, the O/C ratios of the composite films were found to slightly decrease from 0.10 to 0.08 for CS-0.4, and from 0.14 to 0.12 for CS-0.68 (Fig. 2b and S3b†), presumably due to further carbonization of CSs. Fourier transform infrared (FTIR) spectroscopy comparison of the composite films before (CS-0.68) and after (CS-0.68-MW) the microwave treatment showed a significant decrease of peaks in the range of 1000–1300 cm−1, which correspond to the C–OH stretching and OH bending vibrations,31 further confirming the carbonization process of the carbon spheres (Fig. S4†).
O centered at 286.9 ± 0.1 eV), and carboxylic/carboxylate (COOH/COOR, centered at 288.9 ± 0.1 eV),22 were found for the carbon sphere, which is consistent with the LaMer model. Therefore, the ratio of sp3 peak to sp2 peak, and the amount of oxygen functional groups, progressively increased with higher loading of the carbon spheres in the composite films (Fig. 2c). After the microwave process, the O/C ratios of the composite films were found to slightly decrease from 0.10 to 0.08 for CS-0.4, and from 0.14 to 0.12 for CS-0.68 (Fig. 2b and S3b†), presumably due to further carbonization of CSs. Fourier transform infrared (FTIR) spectroscopy comparison of the composite films before (CS-0.68) and after (CS-0.68-MW) the microwave treatment showed a significant decrease of peaks in the range of 1000–1300 cm−1, which correspond to the C–OH stretching and OH bending vibrations,31 further confirming the carbonization process of the carbon spheres (Fig. S4†).
 | ||
| Fig. 2 (a) Raman spectra, (b) X-ray photoelectron spectroscopy (XPS) wide scan survey of as-synthesized carbon sphere (CS), pristine FWNT, and composite films at different loadings of CSs. (c) The high resolution C 1s spectra of the CS and composite films. | ||
Cyclic voltammetry (CV) measurements were conducted to investigate the charge storage characteristics of the carbon spheres in lithium cells. The first CV scan of the pristine FWNT electrode displayed a rectangular shape (Fig. 3a) and subsequent potential cycling showed negligible difference (Fig. S5a†), indicative of charge storage based on double-layer capacitance. In contrast, the first CV scan of the composite electrodes including the carbon spheres exhibited oxidation current with an onset potential of ∼2.5 V vs. Li and a strong oxidation wave at an onset potential of ∼3.7 V vs. Li (Fig. 3a and S5b–d†). The strong oxidation wave may be attributed to the anodic polymerization of aromatic hydrocarbons.4,43–46 Upon subsequent potential cycling, the anodic oxidation current gradually decreased and finally arrived at steady-state, which was accompanied by the formation of a broad redox peak in the voltage range of 2.2–3.7 V vs. Li (Fig. 3b). The remaining redox peak of the composite electrodes can be ascribed to the surface redox reactions between oxygen functional groups on the carbon spheres and lithium ions. Comparison of steady-state CV scans showed that the composite electrodes have higher gravimetric current densities compared to that of pristine FWNT, owing to the additional surface redox reactions on top of double layer capacitance (Fig. 3c). Interestingly, there was a little difference in CV scans between the CS-0.4 and CS-0.68 (Fig. 3c). We postulate that a significant amount of carbon spheres were not effectively utilized in the composite film with high loading (68 wt%), since many carbon spheres were separated from the conductive FWNT matrix (Fig. 1d). Comparison of steady-state CV scans after the microwave process displayed a significant increase in current for the CS-0.68-MW (Fig. S6a†), resulting in higher gravimetric current density compared to that of the CS-0.4-MW (Fig. 3d). Moreover, the CS-0.68-MW retained its redox behavior at a high scan rate of 100 mV s−1 (Fig. S6b†). Such enhanced charge storage performance after the microwave treatment can be attributed to the increased redox-active surface as well as electrical conductivity on the connected carbon nanorod structure that was well threaded by conductive FWNTs (Fig. 1e and f).
 | ||
| Fig. 3 Cyclic voltammetry (CV) scan comparisons of the FWNT and composite electrodes at 1 mV s−1 in lithium cells with 1 M LiPF6 in a mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) (3:7 volume ratio). (a) Comparison of initial CV scans of the pristine FWNT electrode and CS-0.4. (b) CV scans of the CS-0.4 as a function of cycle number. Comparisons of steady-state CV scans for different composite electrodes (c) before and (d) after the microwave process. CV scans of the pristine FWNT electrodes were utilized as guidelines. The weight of the total positive electrode (carbon spheres + FWNTs) was considered in the current density calculation. | ||
Charge storage performance of the electrodes was further evaluated using rate-dependent galvanostatic charge and discharge tests (Fig. 4a–c and S7†). The pristine FWNT electrode delivered a low specific capacity of ∼40 mA h gelectrode−1 at 0.05 A g−1, while the capacities of the composite electrodes were significantly increased to ∼103 mA h gelectrode−1 for CS-0.4 and ∼117 mA h gelectrode−1 for CS-0.68 (Fig. 4d). After the microwave treatment, the FWNT and CS-0.4 showed negligible changes in their capacities, whereas the CS-0.68 exhibited a considerable increase in capacity from ∼117 to ∼155 mA h gelectrode−1, owing to the improved utilization of the carbon spheres for charge storage (Fig. 4d). This capacity is higher than those of previously reported carbon based electrodes (38–135 mA h g−1)21,22,47–49 and comparable to those of recently reported redox-active graphene electrodes (∼160 mA h g−1)18 (Table S2†). Based on the rule of mixtures, the specific capacity of the carbon spheres in the CS-0.68-MW was estimated to be ∼210 mA h gcarbon sphere−1, which is higher than those of previously reported redox-active polyimides50–52 and carbonyl compounds.9,53 Microwave treatment also significantly improved the rate capability of the composite electrode (CS-0.68). The capacities of the CS-0.68 were compared before and after microwave treatment as a function of discharge rate (Fig. 4e). The rate dependent capacity curve of the CS-0.68-MW was almost parallel to that of the pristine FWNT electrode, while the curve of the CS-0.68 dropped rapidly when the current densities were higher than 1 A g−1 (Fig. 4e). The CS-0.68-MW retained ∼59% of the capacity when the current density increased from 0.05 to 1 A g−1, and ∼15% at an extremely high current density of 50 A g−1. The significant enhancement in both capacity and rate performance after the microwave process can be attributed to the transformation of isolated carbon spheres into connected peanut-shaped nanorods penetrated by FWNTs (Fig. 1e and f), which facilitates electronic transportation from the FWNT network to redox-active sites on the carbon spheres.
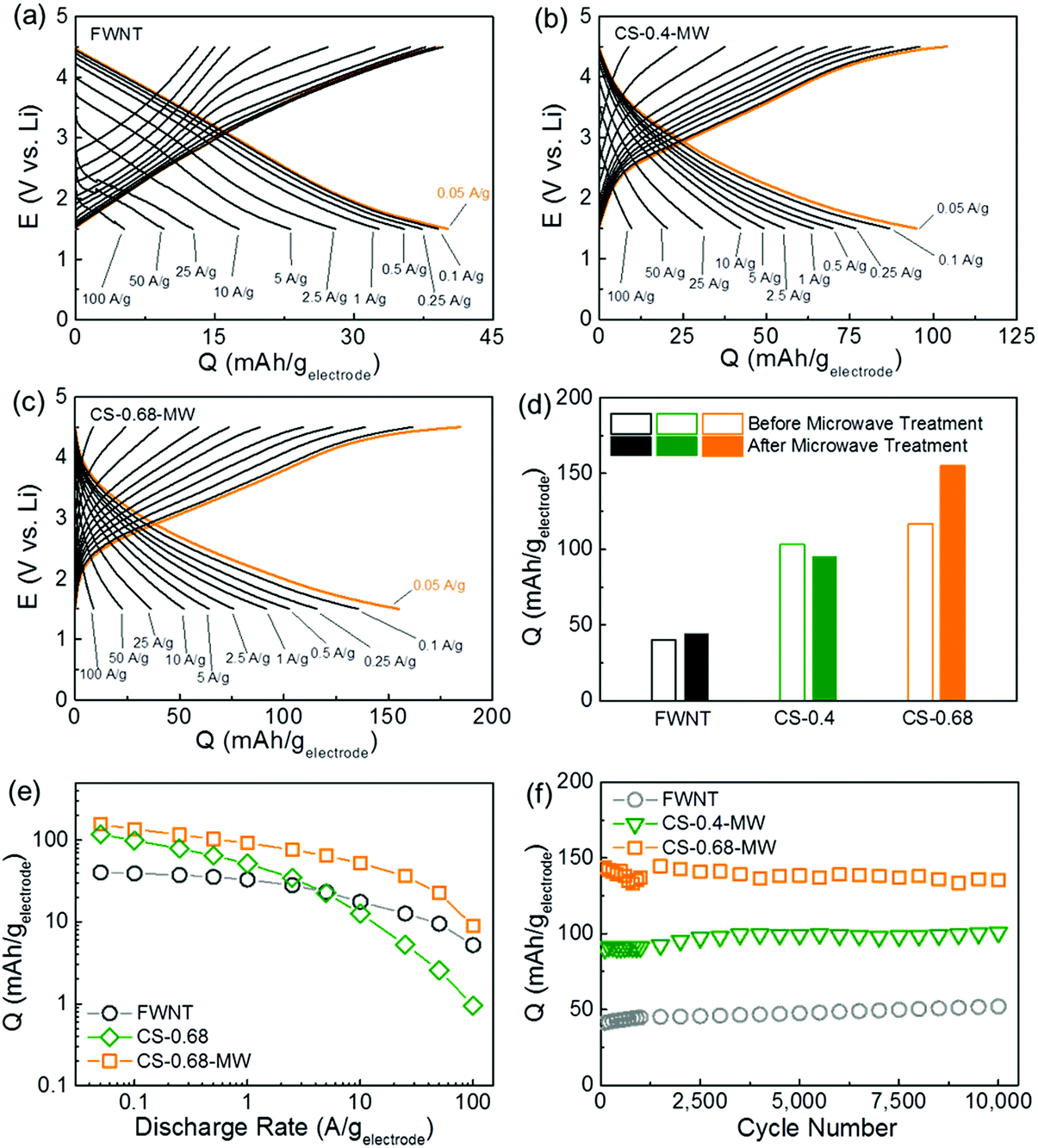 | ||
| Fig. 4 Rate-dependent galvanostatic charge and discharge curves of (a) the pristine FWNT electrode, (b) CS-0.4-MW, and (c) CS-0.68-MW in lithium cells. (d) Discharge capacity comparisons of the FWNT and composite electrodes at 0.05 A g−1 before and after microwave treatments. (e) Specific capacity comparison of the FWNT, CS-0.68 and CS-0.68-MW as a function of discharge current densities. (e) Specific capacities of the FWNT, CS-0.4-MW and CS-0.68-MW as a function of cycle number up to 10000 cycles using an accelerating cycling method.20 Data points indicate specific discharge capacities of the electrodes measured at a slow rate of 0.1 A g−1. Between each slow measurement, the cells were cycled at a high current density of 10 A g−1. | ||
It should be noted that the composite electrodes have sloped charge and discharge profiles. Our recent density functional theory (DFT) calculation study on similar redox reactions of the reduced graphene oxide showed that various oxygen functional groups on the graphene oxide have different redox potentials depending on their oxygen chemistry, as well as local chemical environments.18 Thus, the sloped profiles of the composite electrode can be attributed to overlap of double-layer capacitance and the redox reactions of various oxygen functional groups on the carbon spheres.
Cycling stability of the electrodes was evaluated at 0.1 A g−1 for up to 100 cycles (Fig. S8†), which showed no obvious capacity fading. In order to evaluate the long term stability of the electrodes, an accelerated cycling method was also conducted according to our previous works.18,20 Specific capacities displayed in Fig. 4f were measured at a slow current density of 0.1 A g−1, while the cells were accelerated at a high current density of 10 A g−1 between each measurement. The pristine FWNT and composite electrodes exhibited a negligible capacity decrease, up to 10000 cycles (Fig. 4f and S9†). The excellent cycling stability of the composite electrodes, which is superior to most of the organic electrode materials,9,11,13,15–17,53,54 can be attributed to the covalently bonded redox-active oxygen functional groups on the carbon spheres.
Conclusions
In summary, redox-active carbon spheres were synthesized from an aqueous glucose solution via the HTC process. The free-standing composite electrodes were fabricated by mixing the carbon spheres with sub-millimeter long FWNTs using a vacuum-filtration process. The composite electrodes exhibited broad redox peaks in the voltage range of 2.2–3.7 V vs. Li, and delivered high specific capacities up to ∼155 mA h gelectrode−1 in lithium cells. In addition, these electrodes maintained their high capacity up to 10000 cycles. Moreover, we demonstrated that microwave treatment can improve the utilization of carbon spheres for effective charge storage. The simple HTC conversion process from an earth-abundant biomass source to redox-active electrode material enables the development of high-performance lithium-ion storage systems, which can provide a sustainable solution towards clean and renewable energy storage.
Experimental
Carbon sphere synthesis
3 mg mL−1 of D-(+)-Glucose (Sigma Aldrich) were dissolved in DI-water by sonication for 30 min. The mixture was sealed in a Teflon-lined autoclave and maintained at 200 °C for 18 h for a hydrothermal carbonization (HTC) process, forming carbon spheres. The autoclave was naturally cooled down to room temperature as the HTC process was completed.Few-walled carbon nanotube (FWNT)
FWNTs were synthesized by a chemical vapor deposition method according to previous papers.29,30 The FWNTs were dispersed in a mixture of ethanol and DI-water (1:1 volume ratio) by short sonication. Pristine FWNT films were prepared by a vacuum-filtration process.
Composite film fabrication
The aqueous dispersion of carbon spheres was mixed with different amounts of FWNTs by short sonication for ∼10 min. Composite films were fabricated via vacuum-filtration of the mixture. The films were dried in a vacuum oven at 70 °C overnight. Microwave treatment was carried out for 30 s at a power of 1250 W under Ar protection using a commercial microwave (Panasonic).Characterization
The microstructures of the films were characterized using a scanning electron microscope (SEM) (Hitachi SU8010, operated at 5 kV). The Raman spectra were collected by a Thermo Nicolet Almega XR Dispersive Raman Spectrometer using a 488 nm wavelength laser. X-ray photoelectron spectroscopy (XPS, Thermal Scientific K-alpha XPS instrument) was employed to analyze the chemical composition of the films. High-resolution C 1s peaks were fitted using XPSPEAKS 4.1 software. Electrical conductivities of the films were measured by a standard four-point probe configuration (Signatone).Electrochemical measurements
Swagelok cells were assembled in an Argon filled glovebox (MBraun). The carbon (FWNT or composite) films and pieces of Li foil were used as the positive and negative electrodes, respectively. The negative and positive electrodes were separated by two pieces of Celgard 2500 separators. 1 M LiPF6 in a mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) (3:7 volume ratio, BASF) was used as an electrolyte. The electrochemical characteristics of the carbon films were evaluated using a Bio-Logic VMP3 potentiostat/galvanostat at room temperature. The voltage window was kept at 1.5–4.5 V vs. Li for cyclic voltammetry and galvanostatic charge/discharge tests. Current densities were controlled from 0.05 to 100 A g−1 during the galvanostatic tests. At the end of each charging or discharging process, the voltage was held for 30 min at either 4.5 V or 1.5 V vs. Li. The cycling stability of the carbon electrodes were tested via an accelerated cycling method up to 10000 cycles based on a previous report.20 The loading density of the positive electrodes was in the range of 0.9–3 mg cm−2.
Acknowledgements
S. W. L acknowledges the financial support of the startup fund from the Georgia Institute of Technology. This work is supported in part by the Samsung Advanced Institute of Technology (SAIT)'s Global Research Outreach (GRO) Program. This work was performed in part at the Georgia Tech Institute for Electronics and Nanotechnology, a member of the National Nanotechnology Infrastructure Network, which is supported by the National Science Foundation.Notes and references
- P. Poizot and F. Dolhem, Energy Environ. Sci., 2011, 4, 2003–2019 CAS.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- Z. Yang, J. Zhang, M. C. W. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- J. C. Bachman, R. Kavian, D. J. Graham, D. Y. Kim, S. Noda, D. G. Nocera, Y. Shao-Horn and S. W. Lee, Nat. Commun., 2015, 6, 7040 CrossRef PubMed.
- F. Cheng, J. Liang, Z. Tao and J. Chen, Adv. Mater., 2011, 23, 1695–1715 CrossRef CAS PubMed.
- Z. Song and H. Zhou, Energy Environ. Sci., 2013, 6, 2280–2301 CAS.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS PubMed.
- C. Liu, F. Li, L.-P. Ma and H.-M. Cheng, Adv. Mater., 2010, 22, E28–E62 CrossRef CAS PubMed.
- Y. Liang, P. Zhang, S. Yang, Z. Tao and J. Chen, Adv. Energy Mater., 2013, 3, 600–605 CrossRef CAS.
- H. Chen, M. Armand, G. Demailly, F. Dolhem, P. Poizot and J.-M. Tarascon, ChemSusChem, 2008, 1, 348–355 CrossRef CAS PubMed.
- M. Yao, H. Senoh, S.-I. Yamazaki, Z. Siroma, T. Sakai and K. Yasuda, J. Power Sources, 2010, 195, 8336–8340 CrossRef CAS.
- H. Senoh, M. Yao, H. Sakaebe, K. Yasuda and Z. Siroma, Electrochim. Acta, 2011, 56, 10145–10150 CrossRef CAS.
- Z. Song, H. Zhan and Y. Zhou, Chem. Commun., 2009, 448–450, 10.1039/B814515F.
- X. Han, C. Chang, L. Yuan, T. Sun and J. Sun, Adv. Mater., 2007, 19, 1616–1621 CrossRef CAS.
- Z. Song, Y. Qian, X. Liu, T. Zhang, Y. Zhu, H. Yu, M. Otani and H. Zhou, Energy Environ. Sci., 2014, 7, 4077–4086 CAS.
- W. Walker, S. Grugeon, O. Mentre, S. Laruelle, J.-M. Tarascon and F. Wudl, J. Am. Chem. Soc., 2010, 132, 6517–6523 CrossRef CAS PubMed.
- S. Wang, L. Wang, K. Zhang, Z. Zhu, Z. Tao and J. Chen, Nano Lett., 2013, 13, 4404–4409 CrossRef CAS PubMed.
- T. Liu, K. C. Kim, R. Kavian, S. S. Jang and S. W. Lee, Chem. Mater., 2015, 27, 3291–3298 CrossRef CAS.
- S. W. Lee, N. Yabuuchi, B. M. Gallant, S. Chen, B.-S. Kim, P. T. Hammond and Y. Shao-Horn, Nat. Nanotechnol., 2010, 5, 531–537 CrossRef CAS PubMed.
- T. Liu, R. Kavian, I. Kim and S. W. Lee, J. Phys. Chem. Lett., 2014, 5, 4324–4330 CrossRef CAS PubMed.
- H. R. Byon, B. M. Gallant, S. W. Lee and Y. Shao-Horn, Adv. Funct. Mater., 2013, 23, 1037–1045 CrossRef CAS.
- S. W. Lee, B. M. Gallant, Y. Lee, N. Yoshida, D. Y. Kim, Y. Yamada, S. Noda, A. Yamada and Y. Shao-Horn, Energy Environ. Sci., 2012, 5, 5437–5444 CAS.
- B. Hu, K. Wang, L. Wu, S.-H. Yu, M. Antonietti and M.-M. Titirici, Adv. Mater., 2010, 22, 813–828 CrossRef CAS PubMed.
- M. Sevilla and A. B. Fuertes, Chem. – Eur. J., 2009, 15, 4195–4203 CrossRef CAS PubMed.
- Z. Wen, Q. Wang and J. Li, Adv. Funct. Mater., 2008, 18, 959–964 CrossRef CAS.
- J. Matos, M. Rosales, R. Demir-Cakan and M. M. Titirici, Appl. Catal., A, 2010, 386, 140–146 CrossRef CAS.
- L. Zhao, L.-Z. Fan, M.-Q. Zhou, H. Guan, S. Qiao, M. Antonietti and M.-M. Titirici, Adv. Mater., 2010, 22, 5202–5206 CrossRef CAS PubMed.
- L. Wei, M. Sevilla, A. B. Fuertes, R. Mokaya and G. Yushin, Adv. Energy Mater., 2011, 1, 356–361 CrossRef CAS.
- Z. Chen, D. Y. Kim, K. Hasegawa, T. Osawa and S. Noda, Carbon, 2014, 80, 339–350 CrossRef CAS.
- D. Y. Kim, H. Sugime, K. Hasegawa, T. Osawa and S. Noda, Carbon, 2011, 49, 1972–1979 CrossRef CAS.
- X. Sun and Y. Li, Angew. Chem., Int. Ed., 2004, 43, 597–601 CrossRef PubMed.
- M.-M. Titirici, M. Antonietti and N. Baccile, Green Chem., 2008, 10, 1204–1212 RSC.
- P. Zhang, Z.-A. Qiao and S. Dai, Chem. Commun., 2015, 51, 9246–9256 RSC.
- D. Krishnan, K. Raidongia, J. Shao and J. Huang, ACS Nano, 2014, 8, 449–457 CrossRef CAS PubMed.
- M. D. Stoller, S. Murali, N. Quarles, Y. Zhu, J. R. Potts, X. Zhu, H.-W. Ha and R. S. Ruoff, Phys. Chem. Chem. Phys., 2012, 14, 3388–3391 RSC.
- Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537–1541 CrossRef CAS PubMed.
- A. L. Higginbotham, P. G. Moloney, M. C. Waid, J. G. Duque, C. Kittrell, H. K. Schmidt, J. J. Stephenson, S. Arepalli, L. L. Yowell and J. M. Tour, Compos. Sci. Technol., 2008, 68, 3087–3092 CrossRef CAS.
- T. J. Imholt, C. A. Dyke, B. Hasslacher, J. M. Perez, D. W. Price, J. A. Roberts, J. B. Scott, A. Wadhawan, Z. Ye and J. M. Tour, Chem. Mater., 2003, 15, 3969–3970 CrossRef CAS.
- C. Y. Wang, T. H. Chen, S. C. Chang, S. Y. Cheng and T. S. Chin, Adv. Funct. Mater., 2007, 17, 1979–1983 CrossRef CAS.
- A. Cuesta, P. Dhamelincourt, J. Laureyns, A. Martínez-Alonso and J. M. D. Tascón, Carbon, 1994, 32, 1523–1532 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- J. Schwan, S. Ulrich, V. Batori, H. Ehrhardt and S. R. P. Silva, J. Appl. Phys., 1996, 80, 440–447 CrossRef CAS.
- G. Lu and G. Shi, J. Electroanal. Chem., 2006, 586, 154–160 CrossRef CAS.
- B. Lu, J. Yan, J. Xu, S. Zhou and X. Hu, Macromolecules, 2010, 43, 4599–4608 CrossRef CAS.
- B. Lu, J. Xu, C. Fan, H. Miao and L. Shen, J. Phys. Chem. B, 2009, 113, 37–48 CrossRef CAS PubMed.
- B. Fan, L. Qu and G. Shi, J. Electroanal. Chem., 2005, 575, 287–292 CrossRef CAS.
- S. Dsoke, B. Fuchs, E. Gucciardi and M. Wohlfahrt-Mehrens, J. Power Sources, 2015, 282, 385–393 CrossRef CAS.
- A. Jain, V. Aravindan, S. Jayaraman, P. S. Kumar, R. Balasubramanian, S. Ramakrishna, S. Madhavi and M. P. Srinivasan, Sci. Rep., 2013, 3, 3002 Search PubMed.
- S. H. Ha, Y. S. Jeong and Y. J. Lee, ACS Appl. Mater. Interfaces, 2013, 5, 12295–12303 CAS.
- Z. Song, H. Zhan and Y. Zhou, Angew. Chem., Int. Ed., 2010, 49, 8444–8448 CrossRef CAS PubMed.
- Y. Meng, H. Wu, Y. Zhang and Z. Wei, J. Mater. Chem. A, 2014, 2, 10842–10846 CAS.
- Z. Song, T. Xu, M. L. Gordin, Y.-B. Jiang, I.-T. Bae, Q. Xiao, H. Zhan, J. Liu and D. Wang, Nano Lett., 2012, 12, 2205–2211 CrossRef CAS PubMed.
- M. Lee, J. Hong, H. Kim, H.-D. Lim, S. B. Cho, K. Kang and C. B. Park, Adv. Mater., 2014, 26, 2558–2565 CrossRef CAS PubMed.
- Y. Liang, P. Zhang and J. Chen, Chem. Sci., 2013, 4, 1330–1337 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr07064c |
| This journal is © The Royal Society of Chemistry 2016 |