
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Precise control of nanoparticle surface by host–guest chemistry for delivery to tumor†
Hisato
Matsui
a,
Motoki
Ueda
b,
Isao
Hara
c and
Shunsaku
Kimura
*a
aDepartment of Material Chemistry, Graduate School of Engineering, Kyoto University, Kyoto-Daigaku-Katsura, Nishikyo-ku, Kyoto, 615-8510, Japan. E-mail: shun@scl.kyoto-u.ac.jp
bClinical Division of Diagnostic Radiology, Kyoto University Hospital, 54 Shogoin Kawara-cho, Sakyo-ku, Kyoto, 606-8507, Japan
cTechnology Research Laboratory, Shimadzu Corporation, Kyoto 619-0237, Japan
First published on 2nd April 2015
Abstract
Nanoparticles were prepared by host–guest chemistry using stereo-complex formation between right-handed and left-handed helical peptides. The host molecule is a 3rd generation polyamidoamine dendrimer having 16 terminated right-handed helices. Three types of guest molecules were examined: poly(sarcosine)-b-(D-Leu-Aib)6 (AB-LP), (poly(sarcosine))3-b-(D-Leu-Aib)6 (A3B-LP), and (D-Leu-Aib)6-b-(poly(sarcosine))3 (A3B-apLP). All the guest peptides associate stoichiometrically with the host dendrimer because of the stereo-complex formation. When A3B-apLP associates with the host dendrimer, the conjugate shows a hydrodynamic diameter of 27 nm, which is explainable by the fact that 16 guest peptides are incorporated in the host dendrimer with tight helix packing and an antiparallel helix dipole arrangement. The nanoparticles were labeled with indocyanine green fluorescence agent and were applied for tumor imaging. Among them, the conjugate with A3B-apLP shows a long life time in the blood stream and a good tumor/liver signal ratio. Furthermore, the conjugate does not trigger the accelerated blood clearance phenomenon. Although these nanoparticles that were modified by similar guest molecules should have similar surfaces, their in vivo disposition is significantly affected.
Introduction
Nanoparticles have been attracting considerable attention in the field of theranostics,1 which is currently considered to be essential for personalized medicine.2–5 They can load various types of imaging agents and therapeutic agents. Nanoparticles loading imaging probes can provide information on in vivo disposition, which can determine appropriate doses for individuals and can predict adverse effects of the nanoparticle loaded drugs. There is, however, a serious obstacle for nanoparticles to be used as a vehicle for imaging and therapeutic agents, which is the pharmacokinetic change of the nanoparticles between the first dose of the imaging nanoparticle and the following dose of the same nanoparticle or the therapeutic nanoparticle. Pharmacokinetic alterations can be caused by two reasons. One is the change in the physical properties (particle size, structural stability, surface density, thickness of hydrophilic shell, and half-life time in blood stream) of the nanoparticle upon labelling and loading different agents for the diagnosis and therapy on the nanoparticle. The other is the immune response to the nanoparticle, which is known as the accelerated blood clearance (ABC) phenomenon.6–9 In order to solve these difficulties, it is imperative to examine various types of the nanoparticles, and thus we prepared nanoparticles using host–guest chemistry.One typical example of nanoparticles is a polymeric micelle.10 It is easy to load hydrophobic agents at the hydrophobic core of core–shell type micelles and to insert functional polymers via the hydrophobic interaction. However, size control is a difficult task because of the swelling of the polymeric micelle when loading these agents10 or changing the hydrophilic–hydrophobic balance by binding functional molecules. Furthermore, the modification of nanoparticle surfaces with a diagnosis probe affects their life time in the blood stream.11 With the aim to solve these problems, we propose a novel molecular assembly using host–guest chemistry applied to a dendritic host scaffold, which makes it possible to construct one nanoparticle and keep a defined number of the constituent amphiphilic polymers and the core size unchanged upon the incorporation of functional groups. Accordingly, the dendritic core has eight amphiphilic polypeptides as a host molecule to incorporate eight amphiphilic polypeptides as a guest molecule.12 These polypeptides have a helical hydrophobic block with opposite helicity, and these two types of helices form a stereo-complex with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry.12–15 We successfully prepared a polymeric nanoconjugate containing sixteen amphiphilic polypeptides using host–guest chemistry. With the chemical modification of the guest amphiphilic polypeptide by a diagnostic or therapeutic agent, this type of nanoconjugate can therefore be functionalized by keeping the size unchanged due to the constant core size of the dendritic host molecule and the defined number of guest peptides. Here, we extended this scheme to a dendrimer with sixteen amphiphilic polypeptides in combination with three types of the guest amphiphilic polypeptides to achieve more precise molecular control of the nanoparticle (Fig. 1). In order to evaluate the nanoparticle properties precisely, we also examined the availability of the nanoparticle for tumor imaging.
:1 stoichiometry.12–15 We successfully prepared a polymeric nanoconjugate containing sixteen amphiphilic polypeptides using host–guest chemistry. With the chemical modification of the guest amphiphilic polypeptide by a diagnostic or therapeutic agent, this type of nanoconjugate can therefore be functionalized by keeping the size unchanged due to the constant core size of the dendritic host molecule and the defined number of guest peptides. Here, we extended this scheme to a dendrimer with sixteen amphiphilic polypeptides in combination with three types of the guest amphiphilic polypeptides to achieve more precise molecular control of the nanoparticle (Fig. 1). In order to evaluate the nanoparticle properties precisely, we also examined the availability of the nanoparticle for tumor imaging.
 | ||
| Fig. 1 Schematic illustration and chemical structure of host and guest molecules. Schematic illustration of the 3rd generation dendrimer core (a), amphiphilic dendrimer template, three amphiphilic guest molecules and host–guest nanoconjugate (b). Chemical structure of amphiphilic three guest molecules (AB-LP, A3B-LP, A3B-apLP) (c). | ||
Results and discussion
Self-assembly by host–guest chemistry
The association of the guest peptides (AB-LP, A3B-LP and A3B-apLP) with the host dendrimer (16RD) was analyzed by dynamic light scattering (DLS) measurements by varying the feed molar ratios of the guest peptides against the host. In these combinations, there were three types of molecular assemblies coexisting in the mixtures, the assemblies of the pure guest, the pure host, and a mixture of the guest and the host, because these guests and the host are amphiphilic by themselves. The former two types of molecular assemblies became relatively large having size over 200 nm. The guest molecules alone formed a curved sheet of ca. 200 nm square as previously reported.12,16–19 The host took on a small disk-like structure, which quickly grew into larger aggregates of ca. 200 nm size by itself, because the peripheral sixteen poly(sarcosine) chains of the host cannot shield the hydrophobic blocks inside.12 On the other hand, upon the association of the guest peptide with the host, the molecular assemblies became smaller (Fig. 2). | ||
| Fig. 2 Hydrodynamic diameters vs. feed molar ratios of AB-LP/16RD by DLS measurement (a) and TEM images (negative staining with uranyl acetate) of molecular assembly prepared from a mixture of AB-LP and 16RD at a molar ratio of 0:1 (b), 16:1 (c) and 32:1 (d). | ||
A mixture of AB-LP and 16RD at the feed molar ratio of 16:1 formed a molecular assembly having a minimum diameter of 49 nm (Fig. 2a), suggesting that they should associate in a stoichiometric manner of 1:1 between the right-handed helices of 16RD and the left-handed helices of AB-LP as expected. With a decrease in the ratios below 16:1, the hydrodynamic diameters became larger, up to 200 nm, and this size corresponds to the molecular assembly of pure 16RD. The hydrodynamic diameters also became larger with an increase in the ratios above 16:1 because the excess AB-LP generated curved sheets of 200 nm. Therefore, it is considered that 16RD cannot accommodate more than 16 mol equivalents of the guest peptides.
TEM images (Fig. 2b–d) supported the coexistence of these three types of molecular assemblies in the mixtures. Molecular assemblies of about 50 nm size were formed from a mixture of AB-LP and 16RD in a molar ratio of 16:1 (Fig. 2c). In addition to the size of the molecular assemblies, large curved sheets coexisted when AB-LP and 16RD were mixed in a molar ratio of 32:1. The mixed solutions were filtered through a cut-off membrane of 20 kDa to remove the molecular assemblies of 16RD or AB-LP with sizes over 200 nm, and the filtrates were subjected to CD measurements. The Cotton effects due to the remaining helicity of the guest (left-handed helix)–host (right-handed helix) conjugate decreased with an increase in the ratios of the guest up to 16. Upon further increasing the ratios to 24 and 32, the Cotton effects remained nearly zero (Fig. S2 in ESI†), which supports the fact that the small conjugate was composed of 16RD and sixteen AB-LPs and that 16RD cannot accommodate more than 16 mol equivalents of the guest peptides.
The hydrodynamic diameter of 49 nm, however, is significantly larger than the estimated value of ca. 25–30 nm for the guest–host conjugate. TEM observations revealed that the guest–host conjugates aggregated to some extent in a time-dependent manner. It is therefore considered that the surface property of the AB-LP/16RD conjugate was not hydrophilic enough to shield the hydrophobic helix layer of the conjugate. Indeed, the morphology of the conjugate was found to be disk-like, which has been frequently observed with lower generation dendrimers due to the low density of the dendritic chains inside. Therefore, in order to suppress the aggregation of the conjugate, we designed A3B-type guests.
The hydrodynamic diameters of the A3B-type guest peptides/host (16RD) conjugate were measured by varying the ratios of the guest against the host. The minimum hydrodynamic diameters were obtained with the additions of 16 mol equivalent A3B-LP and 14 mol equivalent A3B-apLP to be 32 nm and 27 nm, respectively (Fig. S3 in ESI†). Thus, these guest peptides also associated stoichiometrically with the host dendrimer on the basis of the stereo-complex formation between the right-handed and the left-handed helices. Furthermore, at mixing ratios of 8:1, 12:1 and 14:1, the guest–host associates also maintained a similar size (Fig. S3†), suggesting that the dendrimer core should be a primary determinant of the associate sizes. TEM observation revealed that the conjugates still maintained the disk-shaped morphology based on the wormlike-shaped side-view and the sphere-shaped top-view (Fig. 3), but no aggregation was detected at room temperature for 24 h. The local high density of poly(sarcosine) chains of the A3B-type peptides should provide sufficient hydrophilicity. Not sphere-shaped, but a disk-shaped assembly may be obtained because the 3G dendrimer was not sphere-shaped but ellipsoid-shaped.20
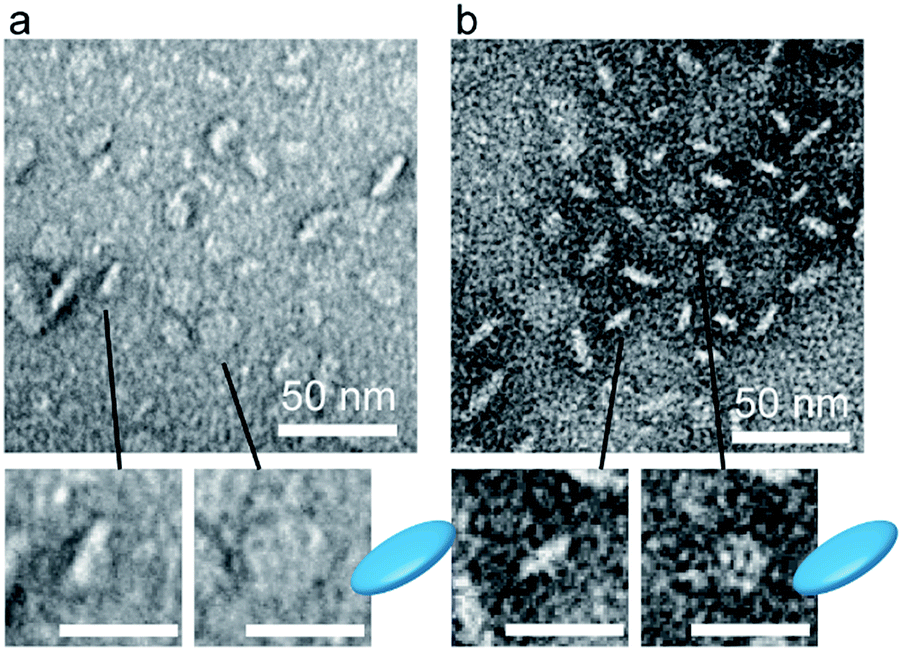 | ||
| Fig. 3 TEM images of molecular assemblies prepared from a mixture of A3B-LP/16RD (a) and A3B-apLP/16RD (b) at the molar ratio of 16:1. Magnified figures are shown at the bottom. Scale bars in the magnification are 25 nm. | ||
The hydrodynamic diameter of 27 nm for the A3B-apLP/16RD conjugate corresponds just to the estimated diameter where the guest helices and the host helices take on an interdigitated side-by-side arrangement with antiparallel dipole orientation (Fig. 4). On the other hand, the A3B-LP/16RD conjugate showed a slightly larger diameter of 32 nm, which may be explained by the dipole–dipole interaction, which works to thicken the helical peptide layer in the conjugate as follows. When A3B-LP is inserted into the surface peptide region of 16RD, A3B-LP should take an orientation to keep the bulky A3-block outside of 16RD. The helix block of A3B-LP then prefers a head-to-tail arrangement with the helix block of 16RD in order to avoid a side-by-side arrangement, where the parallel arrangement of the helix dipoles should destabilize the helix packing in the conjugate. It is therefore considered that the steric effect in the association of the guest peptide with the host dendrimer should take priority, and the following dipole–dipole interaction should decide the helix packing of the side-by-side or the head-to-tail arrangement in the conjugates (Fig. 4).21 Accordingly, as far as the 2nd generation12 and the 3rd generation dendrimers are concerned, there is no difference in the host–guest chemistry in the present molecular systems.
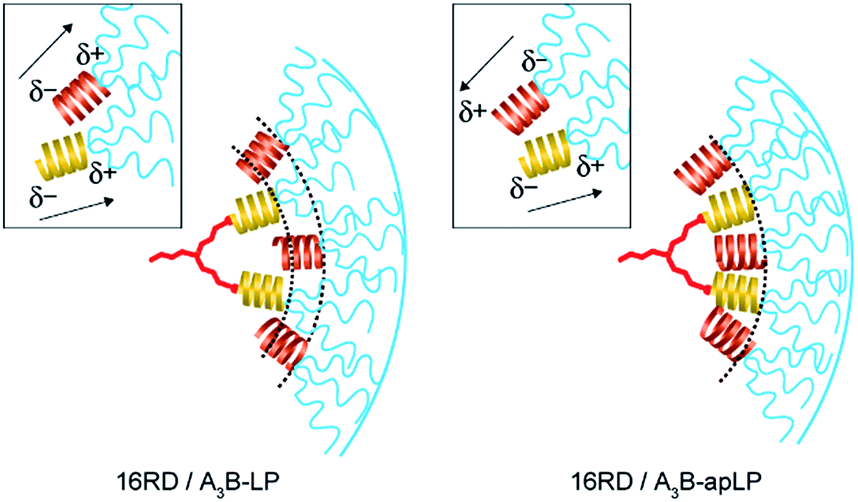 | ||
| Fig. 4 Illustrations of molecular packing in the hydrophobic layer in the case of A3B-LP/16RD (left) and A3B-apLP/16RD (right). Arrows mean the dipole moments along hydrophobic α-helices. | ||
In vivo disposition
The A3B-LP/16RD conjugate and the A3B-apLP/16RD conjugate have the same constituents except for the direction of the helix dipoles of the guest being parallel or antiparallel to that of the host. The structural difference makes the size of the A3B-LP/16RD conjugate slightly larger than the A3B-apLP/16RD conjugate. These two types of conjugates were labeled with ICG and examined for tumor imaging of mice. Even though the surface modification between these two conjugates looks very similar, they showed a different behavior for in vivo pharmacokinetics.A mixture of 16RD, A3B-apLP and ICG-LP (Fig. 5a) at a ratio of 1:14:0.16 generated the ICG-labeled conjugate with a diameter of 27 nm, which was the same size of the A3B-apLP/16RD conjugate at a mixing ratio of 1:14 (Fig. 5b). This observation is a good evidence for the present guest–host chemistry in the preparation of functionalized nanoparticles, as it is very useful for size control. The buffered solutions of the ICG-labeled conjugates were injected to tumor bearing mice from the tail vein and NIRF images were taken by a Shimadzu Clairvivo OPT (Fig. 5). When the images that were taken 9 h after the injection were compared, the whole body fluorescence intensity was significantly larger with the A3B-apLP/16RD conjugate than the A3B-LP/16RD conjugate, reflecting that the circulating amount of the A3B-apLP/16RD conjugate in the blood stream was larger than that of the A3B-LP/16RD conjugate (Fig. 5c and d). This is because the A3B-LP/16RD conjugate is initially more easily captured by the liver to reduce its concentration in the blood stream, as shown in Fig. 5e. As a result, the amount of accumulation of the A3B-apLP/16RD conjugate in the tumor was nearly two-times higher than that of the A3B-LP/16RD conjugate. The tumor/liver signal ratio is also better with the A3B-apLP/16RD conjugate than the A3B-LP/16RD conjugate (Fig. 5e). The stability of the A3B-apLP/16RD conjugate in the blood stream may be attributable to the 1.3-times higher surface density of the poly(sarcosine) chains with the A3B-apLP/16RD conjugate than the A3B-LP/16RD conjugate on the basis of a simple calculation of the surface areas of the diameters of 27 nm and 32 nm.
 | ||
| Fig. 5 Chemical structure of ICG-labeled amphiphiles, ICG-LP (a), DLS profiles of molecular assemblies prepared from a mixture of A3B-apLP and 16RD at the mixing ratio of 14:1 with and without ICG-LP at 0.16 eq. (b) In vivo NIRF imaging results (c–e). NIRF images of nanocarriers from A3B-LP/16RD/ICG-LP (c) and A3B-apLP/16RD/ICG-LP (d) after 9 h from administration. The region of interest (ROI) at the tumor site, liver site and tumor/liver NIRF intensity ratio (e). | ||
Furthermore, the antiparallel packing of the helix dipoles in the A3B-apLP/16RD conjugate may contribute to the physical stability in the blood stream. The physical stability is indeed supported by the observation that the amount of accumulation in the tumor site increased with time up to 20 h, which means that the ICG-LP stayed stably in the conjugate in the blood stream.
The A3B-apLP/16RD conjugate was examined for in vivo pharmacokinetics upon repeated administrations. It has been reported that the PEGylated liposome and the polymeric micelles at the second dose were immediately captured by liver even though they showed a long life time in the blood stream at the first dose.6–9 This type of alteration in pharmacokinetics is called the accelerated blood clearance (ABC) phenomenon. When the A3B-apLP/16RD conjugate was dosed at 7 days after the first injection, the time-profiles of the accumulation in the tumor, liver, and background coincided just with those at the first dose (Fig. 6). The reason for no ABC phenomenon with the A3B-apLP/16RD conjugate is yet to be solved, but this type of the conjugate should be useful as a nanocarrier platform for clinical tumor diagnostics and therapy, which can be attained by loading the diagnostic or therapeutic agent on the guest peptide. With this platform, nanoparticles can keep their size upon functionalization without an immune response on multiple administrations.
 | ||
| Fig. 6 NIRF images of nanocarrier from A3B-apLP/16RD/ICG-LP after 9 h from 1st and 2nd administrations (a) and the time profiles of ROI at the tumor site, liver site and background (left-leg site) (b). | ||
Conclusions
We demonstrated a useful nanocarrier whose diameter is less than 30 nm. Various chemical agents can be loaded on the nanocarrier with a defined concentration and keeping the size unchanged. The nanocarrier showed a long life time in the blood stream, and no ABC phenomenon was triggered. Tumor imaging is available with the nanocarrier due to the enhanced permeability and retention effect. The chemistry of the nanocarrier is based on the stereocomplex formation between the right-handed and the left-handed helices. The 16 guest peptides can be accommodated in the 16-helix terminated dendrimer due to the stereo-complex formation. The surface of the conjugate is densely covered with poly(sarcosine) chains due to its A3B-type molecular architecture. The conjugate was highly stable in the blood stream because of the tight helix packing and antiparallel arrangement of the helix dipoles in the conjugate. We tried functionalizing the conjugate using an ICG-modified guest peptide for tumor imaging. We think various functionalization will be available with just changing the ICG moiety with other agents, by which the nanoparticle size and the long life time in the blood stream will be kept unchanged, and noticeably no ABC phenomenon of the conjugate nanoparticle will be induced.Experimental section
Materials and methods
:1, 1:1, 2:1, 4:1, 8:1, 16:1, 24:1, and 32:1 was injected into a buffer (0.5 mL, 10 mM Tris–HCl, pH 7.4) with stirring at 4 °C.
:14:0.16 was injected into saline (0.5 mL) kept in a sample vial at 4 °C. This dispersion was kept stirring at 4 °C for 30 min, allowed to reach room temperature, filtered through a membrane filter (polyethersulfone, 100 nm), and then used for the fluorescent analysis and in vivo imaging experiment.
Acknowledgements
This study is a part of translational research network program by MEXT, a part of S-innovation by JST, and a part of the innovative techno-hub for integrated medical bio-imaging of the project for developing innovation systems by MEXT.Notes and references
- J. Xie, S. Lee and X. Y. Chen, Adv. Drug Delivery Rev., 2010, 62, 1064 CrossRef CAS PubMed.
- Z. Wang, G. Niu and X. Y. Chen, Pharm. Res., 2014, 31, 1358 CrossRef CAS PubMed.
- T. Moore, H. Y. Chen, R. Morrison, F. L. Wang, J. N. Anker and F. Alexis, Mol. Pharmaceutics, 2014, 11, 24 CrossRef CAS PubMed.
- K. Y. Choi, G. Liu, S. Lee and X. Y. Chen, Nanoscale, 2012, 4, 330 RSC.
- N. Ahmed, H. Fessi and A. Elaissari, Drug Discovery Today, 2012, 17, 928 CrossRef CAS PubMed.
- M. Yokoyama, J. Drug Targeting, 2014, 22, 576 CrossRef CAS PubMed.
- E. Hara, M. Ueda, C. J. Kim, A. Makino, I. Hara, E. Ozeki and S. Kimura, J. Pept. Sci., 2014, 20, 570 CrossRef CAS PubMed.
- H. Koide, T. Asai, K. Hatanaka, T. Urakami, T. Ishii, E. Kenjo, M. Nishihara, M. Yokoyama, T. Ishida, H. Kiwada and N. Oku, Int. J. Pharm., 2008, 362, 197 CrossRef CAS PubMed.
- T. Ishida, M. Harada, X. Y. Wang, M. Ichihara, K. Irimura and H. Kiwada, J. Controlled Release, 2005, 105, 305 CrossRef CAS PubMed.
- B. F. Lin, R. S. Marullo, J. F. Robb, D. V. Krogstad, P. Antoni, D. J. Hawker, L. M. Campos and M. V. Tirrell, Nano Lett., 2011, 11, 3946 CrossRef CAS PubMed.
- A. Uesaka, I. Hara, T. Imai, J. Sugiyama and S. Kimura, RSC Adv., 2015, 5, 14697 RSC.
- H. Matsui, M. Ueda, A. Makino and S. Kimura, Chem. Commun., 2012, 48, 6181 RSC.
- M. Ueda, A. Makino, T. Imai, J. Sugiyama and S. Kimura, Soft Matter, 2011, 7, 4143 RSC.
- M. Ueda, A. Makino, T. Imai, J. Sugiyama and S. Kimura, Langmuir, 2011, 27, 4300 CrossRef CAS PubMed.
- M. Ueda, A. Makino, T. Imai, J. Sugiyama and S. Kimura, Polym. J., 2013, 45, 509 CrossRef CAS.
- A. Uesaka, M. Ueda, A. Makino, T. Imai, J. Sugiyama and S. Kimura, Langmuir, 2014, 30, 1022 CrossRef CAS PubMed.
- T. Kanzaki, Y. Horikawa, A. Makino, J. Sugiyama and S. Kimura, Macromol. Biosci., 2008, 8, 1026 CrossRef CAS PubMed.
- M. Ueda, A. Makino, T. Imai, J. Sugiyama and S. Kimura, J. Pept. Sci., 2011, 17, 94 CrossRef CAS PubMed.
- M. Ueda, A. Makino, T. Imai, J. Sugiyama and S. Kimura, Chem. Commun., 2011, 47, 3204 RSC.
- P. K. Maiti, T. Cagin, G. F. Wang and W. A. Goddard, Macromolecules, 2004, 37, 6236 CrossRef CAS.
- S. Kimura, Org. Biomol. Chem., 2008, 6, 1143 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra01685a |
| This journal is © The Royal Society of Chemistry 2015 |