The effect of γ-Al2O3 surface hydroxylation on the stability and nucleation of Ni in Ni/γ-Al2O3 catalyst: a theoretical study†
Zhixue Liua,
Yuhan Wangb,
Jingrui Lia and
Riguang Zhang*a
aKey Laboratory of Coal Science and Technology of Ministry of Education and Shanxi Province, Taiyuan University of Technology, No. 79 Yingze West Street, Taiyuan 030024, Shanxi, China. E-mail: zhangriguang@tyut.edu.cn; zhangriguang1981@163.com; Fax: +86 351 6041237; Tel: +86 351 6018239
bSchool of Chemical Engineering and Environment, Beijing Institute of Technology, Beijing 100081, China
First published on 15th January 2014
Abstract
Using recent well-defined models of γ-Al2O3 surfaces, the interactions of Nin(n = 1–7) clusters with different γ-Al2O3 surfaces have been investigated in order to illustrate, by density functional theory periodic calculations, the effect of γ-Al2O3 surface hydroxylation on the stability and nucleation of Ni in Ni/γ-Al2O3 catalyst. Three types of γ-Al2O3 surfaces, dehydrated γ-Al2O3(100), dehydrated γ-Al2O3(110) and hydrated γ-Al2O3(110) were considered. Our results show that for the adsorption of Nin(n = 3–7) clusters, the γ-Al2O3(110) surface is more favorable than the γ-Al2O3(100) surface, however, for single Ni atoms and Ni2 clusters, the reverse becomes true. Meanwhile, for the adsorption of Nin(n = 2–7) clusters, the hydrated (110) surface is not favorable compared to the dehydrated (110) surface, due to the presence of surface hydroxyls on the former. The reverse is true for single Ni atoms due to weaker surface deformation. Further, the support stabilizes Nin(n = 2–7) clusters well in the supported state, in which the presence of surface hydroxyls reduces the stability of the supported Nin clusters. On the other hand, the nucleation ability of Nin clusters on different γ-Al2O3 surfaces, is more favorable on the γ-Al2O3(110) surface than on the γ-Al2O3(100) surface, and the dehydrated (110) surface is more favorable than the hydrated (110) surface due to the presence of surface hydroxyls, namely, surface hydroxylation reduces the nucleation ability of Nin clusters on the γ-Al2O3 surface. More importantly, the exothermicity of supported Nin(n = 2–7) clusters on different γ-Al2O3 surfaces is lower than that of isolated Nin clusters, indicating that the support is not favorable for the nucleation of Nin(n = 2–7) clusters, as a result, the support can inhibit the aggregation of clusters, and favors the formation of small clusters.
1. Introduction
Oxide-supported metal catalysts play a pivotal role in a lot of technologically important applications, including sensors, solid oxide fuel cells, and heterogeneous catalysts.1 Among the oxide-supported metal catalysts, Ni-based catalysts are often used in industrial processes such as the hydrogenation, hydrotreatment and hydrogenolysis of hydrocarbons, steam reforming of hydrocarbons, and methanation.2–5 Different supports are widely used to disperse Ni particles, such as γ-Al2O3,6,7 TiO2,8 CeO2,9 SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10,11 or La2O3,7 among them, γ-Al2O3 is one of the most common supports as its large surface area and high degree of porosity favors the good dispersion of active species.12
10,11 or La2O3,7 among them, γ-Al2O3 is one of the most common supports as its large surface area and high degree of porosity favors the good dispersion of active species.12
Hydroxylation is a common process that occurs in many processes.13–18 For the oxide-supported metal catalysts, the hydroxylation of the oxide support has an effect on the metal–support interaction, and further on the reactions taking place on the catalyst.19–24 For example, Cu(111) and Cu/TiO2(110) are both active towards the water–gas shift (WGS) reaction, however, due to the surface hydroxylation of the TiO2 support, the Cu/TiO2(110) catalyst has a much higher catalytic activity in the WGS reaction than the Cu(111) catalyst.19 For γ-Al2O3-supported metal catalysts, under normal reaction conditions, the surface of the γ-Al2O3 support inevitably becomes hydrated/hydroxylated; subsequently, the surface nature of the support changes,22–29 this change affects both the metal–support interaction and the reactions taking place over the catalyst. For example, Valero et al.30 have investigated the effect of γ-Al2O3 surface hydroxylation on the stability and diffusion of a single Pd atom, and found that the binding energy of the Pd atom decreases as the OH coverage of the γ-Al2O3 surface increases. In addition, previous studies have shown the effect of γ-Al2O3 surface hydroxylation on the selectivity of CO2 hydrogenation over different Ni/γ-Al2O3,27 Pd/γ-Al2O331 and Cu/γ-Al2O332 catalysts, suggesting that the hydroxylation of the γ-Al2O3 surface can alter the reaction pathway and the selectivity of CO2 hydrogenation.
On the other hand, the activity and selectivity of metal-supported catalysts are strongly influenced by the size of the dispersed metal particles, and the metal–support interaction.33–38 Small metal clusters or particles have interesting properties for catalytic processes due to the presence of low-coordination atoms and electron confinement effects,39 which is different from those of bulk metals. For example, Zhang et al.31 have suggested that the presence and number of low-coordinated Pd particles on a Pd/γ-Al2O3 catalyst is of great importance to improve the overall activity and selectivity of CO2 hydrogenation. The quasi in situ X-ray photoelectron studies of Loviat et al.6 have shown that the reactivity and the observed reaction mechanisms for methane production from syngas using a Ni/γ-Al2O3 catalyst are directly influenced by both the size and the composition of the Ni particle on the γ-Al2O3 support. Moreover, on the basis of EXAFS and IR spectra studies using an Ir/γ-Al2O3 catalyst, Argo et al.35 have pointed out that the rate of ethene hydrogenation on Ir4 clusters is typically several times greater than that on Ir6 clusters. Further, Prasad and co-authors37,38 and Meier et al.40 also pointed out that the metal cluster size, the support material, or both, are critical in determining the catalytic activity.
Nowadays, Ni/γ-Al2O3 catalyst is widely used in many chemical processes such as hydrogenation,27 dehydrogenation, reformation,41 methanation, desulfurization, methane oxidation, and in the production of many bulk and fine chemicals.42–47 Unfortunately, it is often difficult to obtain pertinent information from experiments on the metal–support interaction in Ni/γ-Al2O3 catalyst, as well as the stability and nucleation of metals on supports with different properties. Theoretical calculations are a valuable tool to complement experimental results, and a detailed investigation of metal–support interactions, as well as the study of the stability and nucleation of metals on the support at the molecular level not only help to increase the understanding of the underlying interaction mechanisms, but also serve as the basis for the selective design of Ni-based catalysts to improve the catalytic performance towards the desired reaction. A number of theoretical studies have examined the stability and nucleation of metals on an Al2O3 support, including Pd on γ-Al2O3,48 Rh on γ-Al2O3,49 Cu on α-Al2O350 and γ-Al2O3,51 as well as Ag on α-Al2O3(0001).52 For example, Valero et al.48 have shown that with a very low metal coverage, the surface hydroxylation of hydrated γ-Al2O3(110) surfaces is favorable for the nucleation of Pd, whereas, with a higher metal coverage, the reverse is true for dehydrated γ-Al2O3(100) . Further, Shi et al.49 have shown that the nucleation of Rh is better on hydrated γ-Al2O3 surfaces than on dehydrated surfaces. Hu et al.53 have found that Pd13 and Pt13 clusters are more stable on a dehydrated γ-Al2O3(100) surface than on a hydrated γ-Al2O3(110) surface. However, to our knowledge, little attention has been paid to the effect of γ-Al2O3 surface hydroxylation on the stability and nucleation of Ni in a Ni/γ-Al2O3 catalyst.
In this study, by using density functional theory (DFT) method, we present a detailed and systematic theoretical investigation of the stability and nucleation of small Nin(n = 1–7) clusters on three different γ-Al2O3 surfaces including the dehydrated (100), dehydrated and hydrated (110) surfaces. The objective was twofold: (i) to identify the preferred adsorption site, and how the isolated Nin clusters were modified after adsorption on the three different γ-Al2O3 surfaces, and (ii) to explore the effect of γ-Al2O3 surface hydroxylation on the stability and nucleation of Ni in Ni/γ-Al2O3 catalyst.
2. Models and methods
2.1 Computational models
Both spinel and non-spinel structure models have been proposed for γ-Al2O3.54–63 The non-spinel model has been well characterized both experimentally and computationally.54–63 Moreover, the non-spinel model can produce diffraction patterns that are close to the characteristic diffraction patterns of γ-Al2O3.58–60 Further, the non-spinel γ-Al2O3 model has been widely used to construct surfaces in previous studies,62,64,65 thus, we employed the non-spinel-γ-Al2O3 as presented in previous studies to model the γ-Al2O3 surface in this study. The corresponding bulk model of the non-spinel-γ-Al2O3 used was taken from a previous theoretical investigation62 of the calcination of boehmite, the hydrated precursor of γ-Al2O3, in which a wide range of aluminum atom distributions in this oxygen atom matrix was simulated, and the most stable structure with 25% of tetrahedral aluminum was retained as the best model for γ-Al2O3, as shown in ref. 55.On the other hand, it has been reported in ref. 55 and 63 that by using the neutron diffraction analysis method of Beaufils and Barbaux,66 the (110) surface was estimated to be 83% of the total surface area, whereas the (100) surface was 17% of the total surface area. In addition, the electron microscopy study carried out by Nortier et al.,67 confirmed that the (110) surface is the predominant facet, covering about 70% of the total surface area, the remaining 30% corresponding to the (100) and (111) surfaces. Further, the (100) surface was found to be poorly hydrated under a broad range of experimental conditions.68 Thus, in this study, we chose the (110) and (100) surfaces to study the stability and nucleation of Ni using DFT slab calculations. The (100) surface was modeled by a (2 × 1) supercell with a dimension of 11.17 × 8.41 × 20.44 Å, and included ten atomic layers, as shown in Fig. 1(a). The vacuum region was set to 12 Å in order to separate the slabs in the direction perpendicular to the surface. During geometry optimization, the bottom four layers were frozen in their bulk position, whereas, the remaining six layers together with the adsorbed Nin(n = 1–7) clusters were allowed to relax. For the γ-Al2O3(110) surface, the dehydrated and hydrated surfaces were considered. The supercell for the (110) surface included six layers with a vacuum region of 12 Å, resulting in a supercell size of 8.41 × 8.07 × 19.17 Å. In this case, the adsorbed Nin(n = 1–7) clusters and/or hydroxyls together with the Al and O atoms in the top four layers were allowed to relax.
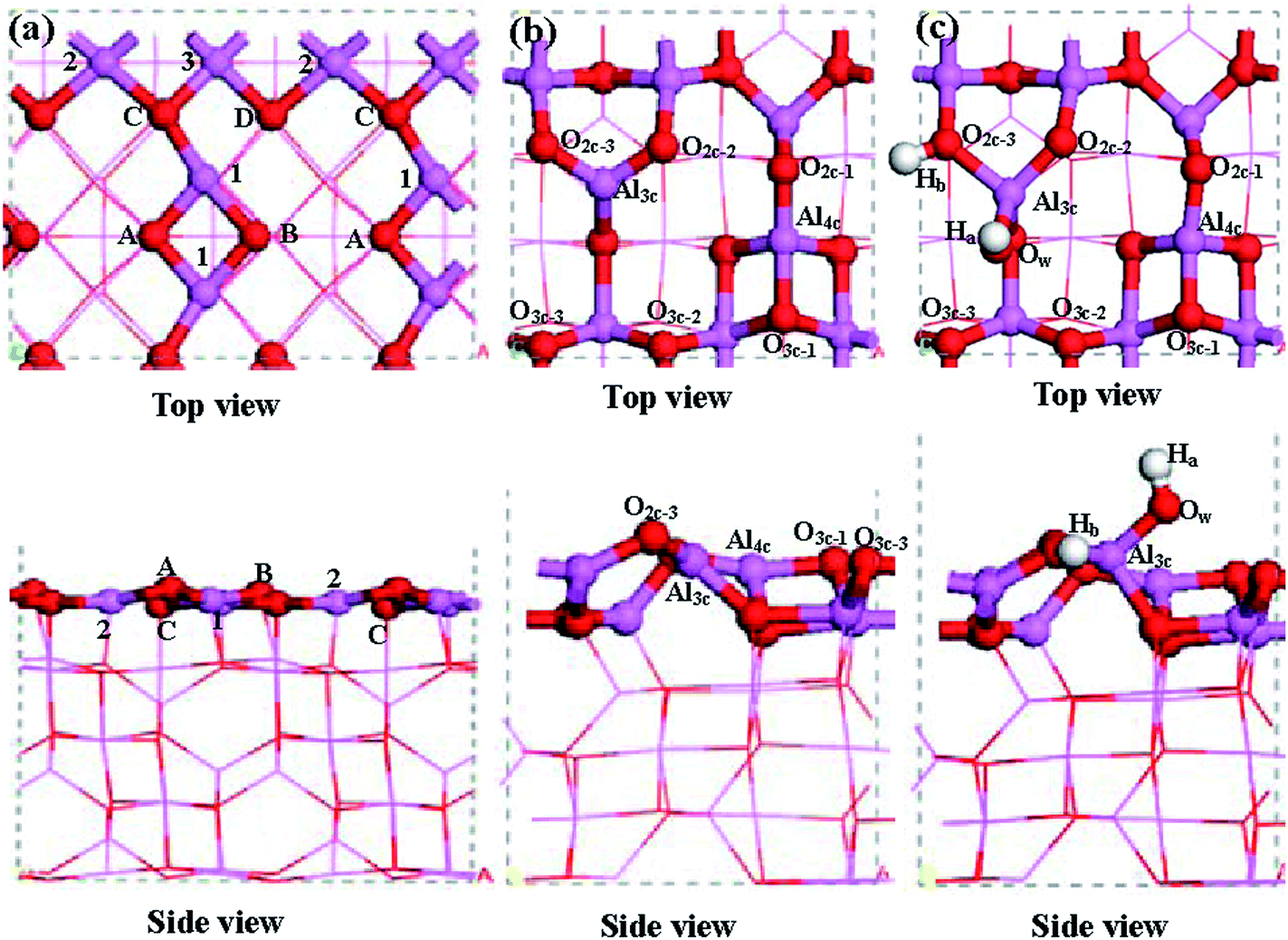 | ||
| Fig. 1 The top and side views of (a) the dehydrated γ-Al2O3(100) surface, (b) the dehydrated γ-Al2O3(110) surface, and (c) the hydrated γ-Al2O3(110) surface. Pink, red and white balls stand for Al, O and H atoms, respectively. | ||
In the dehydrated γ-Al2O3(100) surface presented in Fig. 1(a), 5-fold-coordinated aluminum Al(1), Al(2) and Al(3), and 3-fold-coordinated oxygen O(A), O(B), O(C) and O(D) are exposed. For the dehydrated γ-Al2O3(110) surface, as shown in Fig. 1(b), the 3-fold-coordinated aluminum (Al3c), 4-fold-coordinated aluminum (Al4c), 6-fold-coordinated aluminum (Al6c), 2-fold-coordinated oxygen (O2c), and 3-fold-coordinated oxygen (O3c) atoms are exposed, of which Al3c, Al4c, O2 are coordinately unsaturated.
For the hydrated γ-Al2O3(110) surface, under normal reaction conditions as demonstrated by Digne et al.,55 the (110) surface inevitably becomes hydrated/hydroxylated, thus, a partially hydrated γ-Al2O3(110) surface was employed to characterize the effect of surface hydroxylation on the stability and nucleation of Ni in the Ni/γ-Al2O3 catalyst. The hydroxylated surfaces are formed by dissociative adsorption of water on the dry surface. The thermodynamics of hydroxylation with various OH coverages were studied by Digne et al.55 However, our aim in this work was to study the qualitative effect of γ-Al2O3 surface hydroxylation on the stability and nucleation of Ni in the Ni/γ-Al2O3 catalyst, not to describe the specific hydroxyl coverage, which can vary under the given experimental conditions. As a result, only one H2O molecule was considered for the hydrated γ-Al2O3(110) surface in this study. The previous DFT studies carried out by Pan et al.29 investigated the hydroxylation process by the dissociation of one H2O molecule on the dry γ-Al2O3(110) surface, which is the same as our model of the dry γ-Al2O3(110) surface, as shown in Fig. 1(b), their results showed that water dissociation across the O2c–Al3c bridge site is favorable both thermodynamically and kinetically, and the hydroxylation of the dry γ-Al2O3(110) surface is an activated process with only a very small barrier of 40.5 kJ mol−1. Fig. 1(c) presents the side and top views of the partially hydroxylated γ-Al2O3(110) surface, in which one H2O molecule dissociates into an hydroxyl (OwHa) with a bond length of 0.967 Å, which occupies the Al3c site through an Ow–Al3c bond (1.726 Å). Meanwhile, a proton (Hb) binds with a neighboring O2c–3 site through the Hb–O2c–3 bond (1.027 Å). Thus, two hydroxyls are formed in each surface unit cell.
2.2 Computational methods
All the calculations were carried out in the framework of DFT using the Dmol3 program in the Materials Studio 4.4 package.69,70 The generalized gradient approximation (GGA) corrected exchange-correlation functional proposed by the Perdew–Burke–Ernzerhof (PBE) functional71 was employed with a double numerical basis set with polarization (DNP).72 The inner electrons of Ni and Al atoms were kept frozen and replaced by an effective core potential (ECP),73,74 and other atoms were treated with an all electron basis set. For optimization of the geometry, the forces imposed on each atom were converged to be less than 0.0004 Ha Å−1 (1 Ha = 2625.5 kJ mol−1), the total energy was converged to be less than 2.0 × 10−5 Ha and the displacement convergence was less than 5 × 10−3 Å. A Fermi smearing of 0.005 Ha for orbital occupancy was used to improve the computational performance. In addition, the properties of isolated Nin clusters were calculated using a 15 × 15 × 15 Å cubic unit cell, the charge transfer was calculated using Mulliken charge, and the dipolar correction was not considered in this study.Different k-point grid samplings ranging from (1 × 1 × 1) to (4 × 4 × 1) were tested for both the bare (100) and (110) surface slabs, as well as the surfaces containing adsorbed Ni4 clusters. The differences between the surface energies and Ni4 cluster adsorption energies were found to be negligible using the (2 × 2 × 1), (3 × 3 × 1), and (4 × 4 × 1) k-point grids for both surfaces. As a result, for a reasonable CPU cost, the (2 × 2 × 1) k-point sampling scheme was employed in this work, and gave good converged results.
3. Results and discussion
In this section, we firstly investigate the stable configuration of isolated Nin(n = 2–7) clusters in section 3.1, then, the adsorption of the most stable Nin clusters on three different γ-Al2O3 surfaces, and the stability of the Nin clusters are discussed in section 3.2, Further, the nucleation of Nin clusters on the three different γ-Al2O3 surfaces are studied in section 3.3. Finally, a comparison of the stability and nucleation between Ni and other metals is provided in section 3.4.3.1 Structures of isolated Nin(n = 2–7) clusters
Isolated Ni clusters have been widely studied previously.6,75 In this section, details are given of the investigation of the geometries of isolated Nin clusters. To study the configuration of Nin(n = 2–7) clusters, one-dimensional (1D), two-dimensional (2D), and three-dimensional (3D) structures were considered. And only the most stable geometries and key parameters of the Nin(n = 2–7) clusters were considered, as listed in Table 1.| n | Geometry | ![[d with combining macron]](https://www.rsc.org/images/entities/i_char_0064_0304.gif) (Ni–Ni)/Å (Ni–Ni)/Å |
Ebind(Nin)/kJ mol−1/atom |
|---|---|---|---|
| 2 |  |
2.163 | −98.6 |
| 3 | 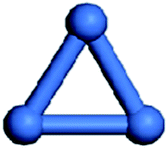 |
2.277 | −144.5 |
| 4 |  |
2.354 | −171.1 |
| 5 |  |
2.361 | −198.9 |
| 6 | 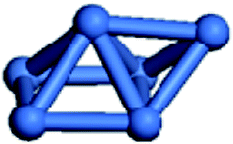 |
2.413 | −211.9 |
| 7 |  |
2.411 | −227.7 |
In order to evaluate the stability of isolated Nin(n = 2–7) clusters, the average binding energy of isolated Nin(n = 2–7) clusters, Ebind(Nin), was calculated as follows:51–53
| Ebind(Nin) = [E(Nin) − n × E(Ni)]/n |
For Ni2 clusters, the Ni–Ni bond length and the average binding energy were 2.163 Å and −98.6 kJ mol−1, respectively, which is in accordance with the experimental results.76,77 For Ni3 clusters, the most stable configuration is a 2D planar one, the average Ni–Ni bond length and the average binding energy were 2.277 Å and −144.5 kJ mol−1, respectively. For Ni4 clusters, the isolated 3D tetrahedral configuration is more stable than the isolated 2D planar one, which is in accordance with the corresponding Pd4 clusters,78 the average Ni–Ni bond length and the binding energy of Ni4 clusters were 2.354 Å and −171.1 kJ mol−1, respectively. For Ni5 clusters, the most stable configuration is a trigonal bipyramid with an average Ni–Ni bond length of 2.361 Å, which is in agreement with the previous results of Reuse et al.,75 the average binding energy of the Ni5 clusters was −198.9 kJ mol−1. For Ni6 clusters, the average Ni–Ni bond length and the binding energy were 2.413 Å and −211.9 kJ mol−1, respectively. Finally, the most stable configuration of Ni7 clusters is a pentagonal bipyramid, which is similar to that of Pd779 and Ag752 clusters, the average bond length and the binding energy for Ni7 clusters were 2.411 Å and −227.7 kJ mol−1, respectively.
On the basis of the above results, we can see from Table 1 that the average Ni–Ni bond length in Nin(n = 2–7) clusters was shorter than that in the bulk structure (d(Ni–Ni) = 2.492 Å), and the average Ni–Ni bond length increases with an increase of the cluster size, however, it cannot reach the corresponding bulk value. Meanwhile, with an increase of atomic coordination in the Nin(n = 2–7) clusters, the value of the Ebind(Nin) decreases.
3.2 Nin clusters adsorbed on different γ-Al2O3 surfaces
Nin(n = 1–7) clusters were adsorbed on all possible positions of the γ-Al2O3 surfaces, here, only the most stable adsorption configurations are reported according to the adsorption energy, which is calculated as follows:25,51| Eads = E(Nin/γ-Al2O3) − E(γ-Al2O3) − E(Nin) |
Distortion of the surface or the metal cluster alone towards the geometry needed to overcome some energy is defined as the surface or metal cluster deformation energy. The surface deformation energy is calculated as follows:25,51
| Edef,surface = E(γ-Al2O3′) − E(γ-Al2O3) |
where E(γ-Al2O3′) is the total energy of the surface with deformed geometry obtained after Nin cluster adsorption. With this definition, lower the Edef,surface results in weaker surfaces.
The deformation energy of the Nin(n = 1–7) clusters is calculated in a similar way:25,51
| Edef,Nin = E(Nin′) − E(Nin) |
The interaction between the Nin clusters and the support is another key parameter. The metal–support interaction energy, EMSI, is calculated as follows:25,51
| EMSI = E(Nin/γ-Al2O3) − E(Nin′) − E(γ-Al2O3′) |
EMSI is simply the adsorption energy between the already deformed partners, i.e., without the energy contributions of geometry deformation in either fragment of the Ni/γ-Al2O3 system.
Therefore, the adsorption energy can be decomposed according to the following equation:25,51
| Eads = EMSI + Edef,Nin + Edef,surface |
This energy decomposition scheme clearly shows that adsorption energy is a trade-off between two antagonistic effects: metal–support interaction energy and deformation energy.
 | ||
| Fig. 2 The most stable adsorption configuration of Nin(n = 1–7) clusters on the dehydrated γ-Al2O3(100) surface. Bond lengths are in Å. The blue balls represent Ni atoms, and the others are the same as in Fig. 1. | ||
(Ni–Ni) for the most stable adsorption configuration of Nin(n = 1–7) clusters on three different γ-Al2O3 surfaces
| n | Eads/kJ mol−1 | EMSI/kJ mol−1 | Edef,Nin/kJ mol−1 | Edef,surface/kJ mol−1 | (Ni–Ni)/Å |
|---|---|---|---|---|---|
| Dehydrated γ-Al2O3(100) surface | |||||
| 1 | −352.2 | −476.2 | — | 124.0 | — |
| 2 | −480.3 | −750.4 | 50.9 | 219.2 | 2.603 |
| 3 | −374.4 | −569.1 | 41.6 | 153.1 | 2.501 |
| 4 | −442.8 | −567.3 | 3.9 | 120.6 | 2.377 |
| 5 | −431.7 | −600.1 | 40.2 | 128.2 | 2.362 |
| 6 | −464.7 | −706.0 | 36.4 | 204.9 | 2.434 |
| 7 | −493.5 | −658.0 | 42.6 | 121.9 | 2.427 |
| Dehydrated γ-Al2O3(110) surface | |||||
| 1 | −269.9 | −359.4 | — | 89.5 | — |
| 2 | −408.8 | −540.7 | 3.5 | 128.4 | 2.251 |
| 3 | −483.6 | −623.2 | 6.7 | 132.9 | 2.366 |
| 4 | −578.9 | −730.3 | 6.0 | 145.4 | 2.377 |
| 5 | −571.2 | −859.6 | 95.4 | 193.0 | 2.434 |
| 6 | −602.9 | −870.1 | 64.5 | 202.7 | 2.477 |
| 7 | −563.1 | −813.8 | 52.6 | 198.1 | 2.463 |
| Hydrated γ-Al2O3(110) surface | |||||
| 1 | −277.6 | −331.6 | — | 54.0 | — |
| 2 | −349.9 | −449.6 | 0.8 | 98.9 | 2.204 |
| 3 | −470.3 | −572.0 | 1.4 | 100.3 | 2.281 |
| 4 | −524.2 | −625.5 | 9.3 | 92.0 | 2.387 |
| 5 | −504.1 | −656.9 | 29.4 | 123.4 | 2.426 |
| 6 | −524.0 | −702.0 | 41.8 | 136.2 | 2.452 |
| 7 | −500.5 | −652.0 | 40.1 | 111.4 | 2.404 |
For single Ni atoms, it is preferrable to occupy the bridge site along O(C)–O(D) with an adsorption energy of −352.2 kJ mol−1, as shown in Fig. 2(a), and 0.027e was transferred to support the surface from single Ni atoms. The adsorption of a single Ni atom can lead to strong surface deformation of the dehydrated γ-Al2O3(100) surface with a surface deformation energy of 124.0 kJ mol−1, this has also been observed in previous studies of the adsorption of single Rh atoms,49 Pd atoms48 and Cu atoms.51 The bond lengths of Ni–O(C) and Ni–O(D) were 1.866 and 1.905 Å, respectively. The Ni1–support interaction energy was −476.2 kJ mol−1, which makes a major contribution to the adsorption energy.
For Ni2 clusters, the most stable configuration is shown in Fig. 2(b), which is different from that of Cu2 clusters.51 In this configuration, two Ni atoms interact directly with the surface sites: the Ni1 atom occupies the O(C)–O(D) bridge site with Ni1–O(C), Ni1–O(D) and Ni1–Al(2) bond lengths of 1.895, 1.905 and 2.267 Å, respectively, while the Ni2 atom is located at the O(A)–O(B) bridge site with Ni2–O(A) and Ni2–O(B) bond lengths of 1.925 and 1.858 Å, respectively. The adsorption of Ni2 clusters introduces strong surface deformation (219.2 kJ mol−1). The Ni1–Ni2 bond length of the adsorbed Ni2 clusters was stretched to 2.603 Å from 2.163 Å in the isolated Ni2 clusters with a cluster deformation energy of 50.9 kJ mol−1. The adsorption energy and the Ni2–support interaction energy were −480.3 and −750.4 kJ mol−1, respectively, which are lower than the corresponding energies for single Ni atoms (−352.2 and −476.2 kJ mol−1). Meanwhile, 0.038e was transferred to the Ni2 clusters from the support surface. Thus, the decrease of adsorption energy is mainly due to the decrease of the Ni2–support interaction.
As shown in Fig. 2(c), the most stable adsorption configuration of Ni3 clusters resembles that of the Cu3 clusters obtained in our previous work.51 In this configuration, the plane of the Ni3 clusters lies aslant relative to the (100) surface forming Ni1–Al(2) (2.373 Å), Ni1–O(C) (1.993 Å), Ni1–O(D) (1.995 Å), Ni2–O(A) (1.915 Å), Ni2–O(B) (1.868 Å) and Ni3–Al(1) (2.414 Å) bonds. The adsorption energy of the Ni3 clusters was −374.4 kJ mol−1, which is higher than that of the Ni2 clusters (−480.3 kJ mol−1). 0.145e was transferred to the Ni3 clusters from the support surface. Meanwhile, the Ni3–support interaction energy obviously increased to −569.1 kJ mol−1 from −750.4 kJ mol−1 for the Ni2 clusters, which is mainly due to the increase of the adsorption energy of the Ni3 clusters. Compared to the Ni2 clusters (50.9 and 219.2 kJ mol−1), the adsorption of Ni3 clusters is accompanied by relatively weak cluster and surface deformations with corresponding energies of 41.6 and 153.1 kJ mol−1.
As shown in Fig. 2(d), Ni4 clusters interact with the surface via three Ni atoms, the Ni4–support interaction energy was −567.3 kJ mol−1, which is close to that of the Ni3 clusters (−569.1 kJ mol−1). The lengths of Ni1–Al(2), Ni1–O(D), Ni2–O(A), Ni2–O(B), Ni3–O(C) and Ni3–Al(1) bonds were 2.449, 1.879, 2.029, 2.023, 1.916 and 2.463 Å, respectively. The adsorption energy of the Ni4 clusters (−442.8 kJ mol−1) was lower than that of the Ni3 clusters (−374.4 kJ mol−1), meanwhile, the adsorption of Ni4 clusters can lead to weaker cluster and surface deformations (3.9 and 120.6 kJ mol−1) relative to those for Ni3 clusters (41.6 and 153.1 kJ mol−1), which is the main reason for the decrease of adsorption energy, and leads to 0.102e being transferred to the Ni4 clusters from the support surface.
For the Ni5 clusters, as shown in Fig. 2(e), the most stable configuration is a trigonal bipyramid with one Ni–Ni bond cleavage, and Ni5 clusters interact with the surface via four Ni atoms. The adsorption energy (−431.7 kJ mol−1) was slightly higher than that for Ni4 clusters (−442.8 kJ mol−1), and 0.204e was transferred to the Ni5 clusters from the support surface. The Ni5–support interaction energy (−600.1 kJ mol−1) was lower than that of the Ni4 clusters (−567.3 kJ mol−1), which makes a major contribution to the adsorption energy for Ni5 clusters. The cluster and surface deformation energies were 40.2 and 128.2 kJ mol−1, respectively, which are higher than those for Ni4 clusters (3.9 and 120.6 kJ mol−1). The stronger cluster deformation of Ni5 clusters is mainly due to the increase of adsorption energy. In the adsorption configuration, the bond lengths of Ni1–Al(2), Ni2–O(D), Ni2–Al(1), Ni3–O(B), Ni3–O(A), Ni4–O(C) and Ni4–Al(1) were 2.411, 1.939, 2.598, 2.001, 2.014, 1.930 and 2.611 Å, respectively.
As shown in Fig. 2(f), five Ni atoms in the most stable adsorption configuration of Ni6 clusters prefer to interact with the surface, and form two Ni–Al(2) bonds (2.545 and 2.630 Å), one Ni1–O(D) bond (1.900 Å), three Ni–Al(1) bonds (2.519, 2.607 and 2.614 Å), one Ni2–O(B) bond (1.881 Å), one Ni3–O(A) bond (1.971 Å) and one Ni5–O(C) bond (1.897 Å). The adsorption energy and the Ni6–support interaction energy (−464.7 and −706.0 kJ mol−1) were lower than those for the Ni5 cluster (−431.7 and −600.1 kJ mol−1), and 0.289e was transferred to the Ni6 cluster from the support surface. The decrease of Ni6–support interaction energy has a major effect on the adsorption energy. The adsorption of Ni6 clusters on the surface leads to a stronger surface deformation (204.9 kJ mol−1) than that of Ni5 clusters (128.2 kJ mol−1). The adsorption also leads to Ni6 cluster deformation (36.4 kJ mol−1), whereas, it is slightly weaker than that of the Ni5 clusters (40.2 kJ mol−1).
The most stable adsorption configuration of Ni7 clusters is presented in Fig. 2(g), and is characterized by a strong adsorption (−493.5 kJ mol−1) and a weak Ni7–support interaction (−658.0 kJ mol−1) in comparison with the adsorption of Ni6 cluster (−464.7 and −706.0 kJ mol−1). Upon adsorption to the dehydrated (100) surface, the pentagonal bipyramid structure of the Ni7 clusters is distorted. The adsorption of Ni7 clusters introduces both cluster and surface deformations with deformation energies of 42.6 and 121.9 kJ mol−1, respectively, 0.232e was transferred from the support surface to the Ni7 clusters. The decrease of the adsorption energy for the Ni7 clusters was mainly due to the weaker surface deformation compared to that for the Ni6 clusters (204.9 kJ mol−1). In this stable configuration, four Ni atoms directly interact with the surface, and form two Ni–Al(2) bonds (2.468 and 2.655 Å), Ni2–O(D) bond (1.929 Å), Ni3–O(B) bond (1.938 Å), Ni3–O(A) bond (2.008 Å) and Ni4–O(C) bond (1.940 Å).
From the obtained energy values listed in Table 2, we can obtain that on dehydrated (100) surface, for the adsorption of Nin cluster, Nin cluster deformation, surface deformation and Nin–support interaction, these four parameters have the strong correlation. The adsorption of Nin clusters on the support is not only affected by the Nin–support interaction, but is also affected by the Nin cluster and surface deformations, moreover, the Nin cluster deformation cannot be neglected except for the Ni4 cluster due to the high stability of the tetrahedron. The results we obtained in the study of Nin clusters are different from the results obtained in previous studies of Cun clusters,51 which show that the deformation of Cun(n = 1–4) clusters is negligible.
We found that the Ni–Ni bond length of adsorbed Nin(n = 2–7) clusters is longer than that of the isolated clusters, and increases with an increase of Nin cluster size, as a result, the Ni–Ni bond length gradually approaches the value in the bulk structure (2.492 Å). For adsorbed Nin(n = 2–4) clusters, the corresponding cluster deformation is very weak, and makes a negligible contribution to the adsorption energy. However, beginning with Ni5 clusters, the cluster deformation increases sharply, and cannot be neglected. On the other hand, for Nin(n ≥ 5) clusters, the surface deformation changes slightly. The results presented above indicate that when the number of Ni atoms in Nin(n = 1–7) clusters is 5 or more, the adsorption can lead to a strong Nin cluster deformation, however, the adsorption cannot lead to a large change of surface deformation.
On the other hand, compared to the dehydrated (110) surface, both the adsorption energy of Nin(n = 2–7) clusters and the Nin–support interaction energy increase due to the presence of surface hydroxyls, whereas, for single Ni atoms, the adsorption energy decreases because of the weaker surface deformation. For Nin(n = 1–7) clusters, the surface deformation of the hydrated (110) surface is weaker than that of the dehydrated (110) surface, namely, the presence of surface hydroxyls can make the (110) surface more stable. Similarly, the adsorption of Rhn(n = 1–5) on both dehydrated and hydrated γ-Al2O3(100) surfaces also shows that the presence of surface hydroxyls is favorable for the stability of the γ-Al2O3 surface.49 Meanwhile, for Nin(n ≥ 5) clusters, due to the presence of surface hydroxyls, cluster deformation on the hydrated γ-Al2O3(110) surface is weaker than that on the dehydrated γ-Al2O3(110) surface, which means that the surface hydroxyls can reduce the effect of the support on the adsorbed cluster.
Secondly, for the γ-Al2O3(110) surface, due to the weaker surface deformation, the adsorption stability of single Ni atoms on the hydrated surface is stronger than that on the dehydrated surface. Whereas, for Nin(n = 2–7) clusters, both the adsorption energy and the Nin–support interaction energy for the hydrated surface are higher than those for the dehydrated surface, indicating that surface hydroxyls have a negative effect on the adsorption stability of Nin(n = 2–7) clusters, and reduce the interaction between the Nin clusters and the γ-Al2O3 surface, which is in accordance with the previous studies.32,49 Meanwhile, the surface deformation of the hydrated surface is weaker than that of the dehydrated surface, namely, the presence of surface hydroxyls can improve the stability of the (110) surface. Moreover, for Nin(n ≥ 5) clusters, the cluster deformation is much larger than that of the Nin(n = 2–4) clusters, and the cluster deformation of Nin(n ≥ 5) clusters on the hydrated surface is weaker than that on the dehydrated surface, which means that the surface hydroxyls can reduce the effect of the support on the adsorbed cluster.
On the other hand, to probe into the effect of the support and surface hydroxyls on the stability of Nin(n = 2–7) clusters, the average binding energy of supported Nin(n = 2–7) clusters on three different γ-Al2O3 surfaces, Ebind(Nin/γ-Al2O3), was calculated, as listed in Table 3.
| n | Ebind(Nin)/kJ per mol per atom | Ebind(Nin/γ-Al2O3)/kJ per mol per atom | ||
|---|---|---|---|---|
| Isolated | Dehydrated (100) | Dehydrated (110) | Hydrated (110) | |
| 2 | −98.6 | −338.8 | −303.0 | −273.5 |
| 3 | −144.5 | −269.3 | −305.7 | −301.2 |
| 4 | −171.1 | −281.8 | −315.8 | −302.1 |
| 5 | −198.9 | −285.2 | −313.1 | −297.1 |
| 6 | −211.9 | −289.4 | −312.4 | −295.6 |
| 7 | −227.7 | −298.2 | −308.1 | −299.2 |
Ebind(Nin/γ-Al2O3) is given by the following equation:51,53
| Ebind(Nin/γ-Al2O3) = [E(Nin/γ-Al2O3) − n × E(Ni) −E(γ-Al2O3)]/n |
Our results show that the average binding energy of Nin(n = 2–7) clusters in the supported state is higher than that in the corresponding isolated state, which means that the support can stabilize the Nin(n = 2–7) clusters well. Meanwhile, the stability of the supported Nin(n = 3–7) clusters on the γ-Al2O3(110) surface is better than that on the γ-Al2O3(100) surface, however, for Ni2 clusters, the reverse becomes true. For the γ-Al2O3(110) surface, the stability of the supported Nin(n = 2–7) clusters on the dehydrated surface is better than that on the hydrated surface, namely, surface hydroxyls reduce the stability of the supported Nin clusters.
3.3 Nucleation of Nin cluster on the different γ-Al2O3 surfaces
Based on the above stable adsorption configurations of Nin(n = 1–7) clusters on the different γ-Al2O3 surfaces, the nucleation of Nin clusters on the different γ-Al2O3 surfaces has been further discussed. The nucleation is defined as the chemical reaction of Nin−1/γ-Al2O3 + Ni1/γ-Al2O3 = Nin/γ-Al2O3, and the corresponding nucleation energy is calculated as follows:51| Enuc = E(Nin/γ-Al2O3) + E(γ-Al2O3) − E(Nin−1/γ-Al2O3) − E(Ni1/γ-Al2O3) |
Table 4 lists the calculated nucleation energies on different surfaces together with the corresponding isolated Nin cluster. We can see that on the dehydrated (100) surface, the nucleation of Nin(n = 2–7) clusters is not thermodynamically favored, namely, the dehydrated (100) surface is unfavorable for the nucleation of Ni clusters. On the dehydrated (110) surface, the nucleation of all the Ni clusters is exothermic, whereas, on the hydrated (110) surface, the nucleation of Ni2 clusters is thermodynamically unfavorable; however, for the Nin clusters with 3 or more Ni atoms, the nucleation becomes favorable, which means that the critical cluster size for Ni cluster nucleation on the hydrated (110) surface is 3. More importantly, the exothermicity of supported Nin(n = 2–7) clusters on different γ-Al2O3 surfaces is lower than that of isolated Nin clusters, which means that the support can reduce the nucleation ability of Nin(n = 2–7) clusters, therefore, the support can inhibit the aggregation of clusters, and favors the formation of highly dispersed Ni particles to prevent their sintering into supranano ones. This result is in agreement with previous studies on the nucleation of Pd,48 Rh,49 Cu51 and Pt80 on γ-Al2O3 surfaces.
| n | Enuc/kJ mol−1 | |||
|---|---|---|---|---|
| Dehydrated (100) | Dehydrated (110) | Hydrated (110) | Isolated | |
| 2 | 26.8 | −66.2 | 8.1 | −197.2 |
| 3 | 221.8 | −41.1 | −79.1 | −236.3 |
| 4 | 32.8 | −76.3 | −27.2 | −250.9 |
| 5 | 53.4 | −32.3 | −12.2 | −309.9 |
| 6 | 41.9 | −39.1 | −10.3 | −277.2 |
| 7 | 1.0 | −12.6 | −21.2 | −322.4 |
On the other hand, for the nucleation energy of Nin(n = 2–7) clusters, the γ-Al2O3(110) surface is weaker than the γ-Al2O3(100) surface, namely, the nucleation of Nin(n = 2–7) clusters is preferred on the γ-Al2O3(110) surface compared to the γ-Al2O3(100) surface. For the γ-Al2O3(110) surface, the nucleation energy of Nin(n = 2–7) clusters on the dehydrated surface is lower than that on the hydrated surface except for Ni3 clusters, suggesting that the nucleation of Nin(n = 2–7) clusters is preferred on the dehydrated (110) surface, namely, the presence of surface hydroxyls reduces the nucleation ability of Nin(n = 2–7) clusters.
Further, although the Ni/γ-Al2O3 catalyst has been widely studied both experimentally and theoretically,27,81–89 these studies have all focussed on the effect of Ni particle size on the target reaction or products, for example, the studies by Chen et al.83 have reported CH4 decomposition over supported Ni catalysts with different Ni particle sizes, suggesting that the smaller sizes of Ni particles give lower carbon yield. Meanwhile, the support plays a key role in the particle size, metal–support interactions, as well as Ni particle crystallographic orientations. Thus, the support has a significant effect on the structure of the target product nanofibers and carbon yield. The effect of various supports has been investigated.86–89 Up to now, to our knowledge, little attention has been paid to the effect of γ-Al2O3 surface hydroxylation on the stability and nucleation of Ni in a for Ni/γ-Al2O3 catalyst, meanwhile, the effect of the support surface properties on Ni particle sizes, Ni-support interactions, and Ni morphology has not been systematically well-studied. Recently, only Briquet et al.90–92 have investigated the adsorption of the single Ni atom on the clean and hydrated (0001) surface of α-Al2O3, as well as the adsorption of Ni6 cluster on the clean (0001) surface, suggesting that the adsorption on hydrated surfaces is different from that on the clean surface, strong stabilization of the single Ni atom by the hydroxyl group was observed, which agrees with our calculated result. The studies by Li et al.93 have investigated the adsorption of the tetrahedrally configured Ni4 cluster on a γ-Al2O3(100) surface, the optimized stable configuration was different, the corresponding adsorption energy in our study was much higher than that obtained by Li et al., thus, the optimized Ni4/γ-Al2O3(100) configuration in our study is thought to be the most stable.
On the basis of the above reported results, we obtained little information on the systematic investigation of the effect of γ-Al2O3 surface hydroxylation on the stability and nucleation of different sized Nin(n = 2–7) cluster in Ni/γ-Al2O3 catalysts, to carry out a comparison between our results and previously reported results. However, a large number of studies have focussed on the effect of γ-Al2O3 surface hydroxylation on the stability and nucleation of Pd,48 Rh,49 Cu51 and Pt80 on the γ-Al2O3 surface, thus, a comparison of the stability and nucleation of Ni with that of other metals Pd, Rh, Cu and Pt was carried out and is presented in the following section.
3.4 Comparison of the stability and nucleation of Ni with that of other metals Pt, Rh, Pd and Cu
On the basis of the nucleation of Ni, in this section, we made a simple comparison with previous studies on the nucleation of other metals Pt, Rh, Pd and Cu. Our results show that the most stable configurations of Pd,48 Rh,49 Cu,51 Pt80 and Ni on the γ-Al2O3 surface are quite different, only on the γ-Al2O3(100) surface does the adsorption of single Pt atoms have a similar stable configuration to that of single Ni atoms, indicating that when different metals with the same size are adsorbed on the same γ-Al2O3 surface, they correspond to different adsorption sites and the most stable configurations. Meanwhile, the studies by Briquet et al.90–92 have shown that the interaction of single Ni atoms on the α-Al2O3(0001) surface results in less exothermic values (close to 170 kJ mol−1) compared to those on different γ-Al2O3 surfaces, this may be explained by the higher coordination of Al and O surface atoms of the α-Al2O3(0001) surface compared to the γ-Al2O3 surface, thus, the γ-Al2O3 surface has a higher intrinsic surface reactivity than the α-Al2O3 surface. Further, the nucleation of Pd,48 Rh,49 Cu51 and Ni also differs, firstly, on the dehydrated (100) surface, the nucleation energy of Pdn(n = 1–5) clusters is positive until n = 4, namely, the critical cluster size of Pd is 4; for Rhn(n = 1–5) and Cun(n = 1–4) clusters, it is 3; however, for the Nin(n = 1–7) cluster in this study, the nucleation energy was always positive, which means that the critical cluster size for Ni is greater than 7. Secondly, on the hydrated (110) surface, the critical cluster size of Pd is 3; for Rh and Cu, it is 2; while for Ni in this study, it was 3. Thirdly, on the dehydrated (110) surface, the critical cluster size of Cu and Ni is 2.
On the other hand, the nucleation of Rhn(n = 1–5) clusters is preferred on the hydrated γ-Al2O3 surface relative to that on the dehydrated γ-Al2O3 surface. For Pdn(n = 1–5) clusters, nucleation at very low metal coverage on an hydrated surface is more favorable than that on a dehydrated surface, whereas, at a higher metal coverage, the reverse becomes true. For Cu and Ni clusters, compared to the γ-Al2O3(100) surface, the γ-Al2O3(110) surface is more favorable for the nucleation of Cu and Ni clusters, in which surface hydroxyls reduce the nucleation ability of Cu and Ni clusters. In addition, Mager-Maury et al.80 carried out studies on the adsorption of Ptn clusters (1 ≤ n ≤ 5 and n = 13) on four γ-Al2O3 surfaces to investigate their stability as a function of particle's size, suggesting that the migration of H and Cl on γ-Al2O3 surfaces induces a stronger interaction for the Pt3 cluster, which is thus proposed to be at the origin of the formation of highly dispersed platinum particles.
4. Conclusions
The effect of γ-Al2O3 surface hydroxylation on the stability and nucleation of Nin(n = 1–7) clusters over dehydrated γ-Al2O3(100), dehydrated γ-Al2O3(110) and hydrated γ-Al2O3(110) surfaces has been systematically investigated by the DFT method. The results show that the Nin–support interaction mainly contributes to the stability of Nin(n = 1–7) clusters on the three different γ-Al2O3 surfaces, Nin(n = 3–7) clusters on the γ-Al2O3(110) surface are much more stable than the corresponding clusters on the γ-Al2O3(100) surface, whereas, for single Ni atoms and Ni2 clusters, the reverse becomes true. For the γ-Al2O3(110) surface, surface hydroxyls do not favor the adsorption stability of Nin(n = 2–7) clusters, and lower the interaction between the Nin clusters and the γ-Al2O3 surface, however, surface hydroxyls increase the surface stability of γ-Al2O3(110).The critical cluster size for the nucleation of Nin(n = 2–7) clusters on the hydrated γ-Al2O3(110) surface is 3, while on the dehydrated (100) surface, the critical cluster size should be greater than 7. Meanwhile, from the viewpoint of thermodynamics, the nucleation of Nin clusters on different γ-Al2O3 surfaces show different trends with an increase of the Nin(n = 2–7) cluster size. For the exothermicity of the nucleation, the supported Nin(n = 2–7) clusters are smaller than the isolated Nin(n = 2–7) clusters, suggesting that the support can reduce the nucleation ability of Nin(n = 2–7) clusters. Further, for the nucleation of Nin(n = 2–7) clusters on the support, the γ-Al2O3(110) surface is more favorable than the γ-Al2O3(100) surface. Finally, for the γ-Al2O3(110) surface, the nucleation of Nin(n = 2–7) clusters preferably occurs on a dehydrated surface, indicating that surface hydroxyls can lower the nucleation ability of Nin clusters. Therefore, the γ-Al2O3 support and its surface hydroxyls can inhibit the aggregation of clusters, and favor the formation of highly dispersed Ni particles preventing their sintering into supranano ones.
Acknowledgements
This works is supported by the National Natural Science Foundation of China (21276003, 20906066 and 21276171). The authors especially thank the two anonymous reviewers for their helpful suggestions on the quality improvement of our present paper.References
- V. E. Henrich and P. A. Cox, The Surface Science of Metal Oxides, Cambridge University Press, Cambridge, U.K, 1994 Search PubMed.
- G. Ertl, H. Knözinger and J. Weitkamp, Handbook of Heterogeneous Catalysis, Wiley Company, Weinheim, 1997 Search PubMed.
- D. Nazimek, A. Machocki and T. Borowiecki, The Influence of Nickel Dispersion in Ni/Al2O3 Catalysts on Their Properties in the Reaction with Hydrogen, Hydrocarbons and Steam, Adsorpt. Sci. Technol., 1998, 16, 747–757 CAS.
- J. P. Jacobs, L. P. Lindfors, J. G. H. Reintjes, O. Jylhä and H. H. Brongersma, The Growth Mechanism of Nickel in the Preparation of Ni/Al2O3 Catalysts Studied by LEIS, XPS and Catalytic Activity, Catal. Lett., 1994, 25, 315–324 CAS.
- T. J. Šarapatka, XPS-XAES Study of Charge Transfers at Ni/Al2O3/Al Systems, Chem. Phys. Lett., 1993, 212, 37–42 Search PubMed.
- F. Loviat, I. Czekaj, J. Wambach and A. Wokaun, Nickel Deposition on γ-Al2O3 Model Catalysts: An Experimental and Theoretical Investigation, Surf. Sci., 2009, 603, 2210–2217 CrossRef CAS PubMed.
- A. N. Fatsikostas and X. E. Verykios, Reaction Network of Steam Reforming of Ethanol over Ni-based Catalysts, J. Catal., 2004, 225, 439–452 CrossRef CAS PubMed.
- J. Zhou, Y. C. Kang and D. A. Chen, Controlling Island Size Distributions: A Comparison of Nickel and Copper Growth on TiO2(110), Surf. Sci., 2003, 537, L429–L434 CrossRef CAS.
- Y. W. Zhang, Z. H. Wang, J. H. Zhou, J. Z. Liu and K. Cen, Catalytic Decomposition of Hydrogen Iodide over Pre-treated Ni/CeO2 Catalysts for Hydrogen Production in the Sulfur-iodine Cycle, Int. J. Hydrogen Energy, 2009, 34, 8792–8798 CAS.
- M. A. Keane, G. Jacobs and P. M. Patterson, Ni/SiO2 Promoted Growth of Carbon Nanofibers from Chlorobenzene: Characterization of the Active Metal Sites, J. Colloid Interface Sci., 2006, 302, 576–588 CrossRef CAS PubMed.
- A. Venugopal, S. N. Kumar, J. Ashok, H. D. Prasad, V. D. Kumari, K. B. S. Prasad and M. Subrahmanyam, Hydrogen Production by Catalytic Decomposition of Methane over Ni/SiO2, Int. J. Hydrogen Energy, 2007, 32, 1782–1788 CrossRef CAS PubMed.
- P. Euzen, P. Raybaud, X. Krokidis, H. Toulhoat, J. L. Le Loarer, J. P. Jolivet and C. Froidefond, in Handbook of Porous Solids, ed. F. Schüth, K. S. W. Sing and J. Weitkamp, Wiley–VCH, Weinheim, 2002, pp. 1591–1677 Search PubMed.
- A. Dey, Y. I. Jiang, P. O. de Montellano, K. O. Hodgson, B. Hedman and E. I. Solomon, S K-edge XAS and DFT Calculations on Cytochrome P450: Covalent and Ionic Contributions to the Cysteine-Fe Bond and Their Contribution to Reactivity, J. Am. Chem. Soc., 2009, 131, 7869–7878 CrossRef CAS PubMed.
- S. P. de Visser and L. S. Tan, Is the Bound Substrate in Nitric Oxide Synthase Protonated or Neutral and What Is the Active Oxidant that Performs Substrate Hydroxylation?, J. Am. Chem. Soc., 2008, 130, 12961–12974 CAS.
- M. Digne, P. Raybaud, P. Sautet, D. Guillaume and H. Toulhoat, Atomic Scale Insights on Chlorinated γ-Alumina Surfaces, J. Am. Chem. Soc., 2008, 130, 11030–11039 CrossRef CAS PubMed.
- X. D. Xu and P. Servati, Facet-Dependent Electronic Properties of Hexagonal Silicon Nanowires under Progressive Hydroxylation and Surface Reconstruction, Nano Lett., 2009, 9, 1999–2004 CrossRef CAS PubMed.
- Z. L. Hong, L. F. Cheng, L. T. Zhang and Y. G. Wang, Water Vapor Corrosion Behavior of Scandium Silicates at 1400 °C, J. Am. Ceram. Soc., 2009, 92, 193–196 CrossRef CAS.
- A. Arranz, C. Palacio, D. Garcia-Fresnadillo, G. Orellana, A. Navarro and E. Muñoz, Influence of Surface Hydroxylation on 3-Aminopropyltriethoxysilane Growth Mode during Chemical Functionalization of GaN Surfaces: An Angle-Resolved X-ray Photoelectron Spectroscopy Study, Langmuir, 2008, 24, 8667–8671 CAS.
- J. A. Rodriguez, J. Evans, J. Graciani, J.-B. Park, P. Liu, J. Hrbek and J. F. Sanz, High Water–Gas Shift Activity in TiO2(110) Supported Cu and Au Nanoparticles: Role of the Oxide and Metal Particle Size, J. Phys. Chem. C, 2009, 113, 7364–7370 CAS.
- Z. P. Qu, W. X. Huang, S. T. Zhou, H. Zheng, X. M. Liu, M. J. Cheng and X. H. Bao, Enhancement of the Catalytic Performance of Supported-Metal Catalysts by Pretreatment of the Support, J. Catal., 2005, 234, 33–36 CrossRef CAS PubMed.
- M. Daturi and L. G. Appel, Infrared Spectroscopic Studies of Surface Properties of Mo/SnO2 Catalyst, J. Catal., 2002, 209, 427–432 CrossRef CAS.
- D. Lomot and Z. Karpinski, The effect of Pd/Al2O3 Pretreatment on Catalytic Activity in Cyclopentane/Deuterium Exchange, Catal. Lett., 2000, 69, 133–138 CAS.
- C. J. Bertole, C. A. Mims and G. Kiss, The Effect of Water on the Cobalt-Catalyzed Fischer–Tropsch Synthesis, J. Catal., 2002, 210, 84–96 CrossRef CAS.
- F. G. Botes, The Effects of Water and CO2 on the Reaction Kinetics in the Iron-Based Low-Temperature Fischer-Tropsch Synthesis: A Literature Review, Catal. Rev. Sci. Eng., 2008, 50, 471–491 CAS.
- M. C. Valero, P. Raybaud and P. Sautet, Interplay between Molecular Adsorption and Metal–Support Interaction for Small Supported Metal Clusters: CO and C2H4 Adsorption on Pd4/γ-Al2O3, J. Catal., 2007, 247, 339–355 CAS.
- S. X. Yin, T. Swift and Q. F. Ge, Adsorption and Activation of CO2 over the Cu-Co Catalyst Supported on Partially Hydroxylated γ-Al2O3, Catal. Today, 2011, 165, 10–18 CAS.
- Y. X. Pan, C. J. Liu and Q. F. Ge, Effect of Surface Hydroxyls on Selective CO2 Hydrogenation Over Ni4/γ-Al2O3: A Density Functional Theory Study, J. Catal., 2010, 272, 227–234 CrossRef CAS PubMed.
- Y. X. Pan, C. J. Liu, T. S. Wiltowski and Q. F. Ge, CO2 Adsorption and Activation Over γ-Al2O3-Supported Transition Metal Dimers: A Density Functional Study, Catal. Today, 2009, 147, 68–76 CrossRef CAS PubMed.
- Y. X. Pan, C. J. Liu and Q. F. Ge, Adsorption and Protonation of CO2 on Partially Hydroxylated γ-Al2O3 Surfaces: A Density Functional Theory Study, Langmuir, 2008, 24, 12410–12419 CrossRef CAS PubMed.
- M. C. Valero, P. Raybaud and P. Sautet, Influence of the Hydroxylation of γ-Al2O3 Surfaces on the Stability and Diffusion of Single Pd Atoms: A DFT Study, J. Phys. Chem. B, 2006, 110, 1759–1767 CAS.
- R. G. Zhang, H. Y. Liu, B. J. Wang and L. X. Ling, Insights into the Effect of Surface Hydroxyls on CO2 Hydrogenation Over Pd/γ-Al2O3 Catalyst: A Computational Study, Appl. Catal., B, 2012, 126, 108–120 CAS.
- R. G. Zhang, B. J. Wang, H. Y. Liu and L. X. Ling, Effect of Surface Hydroxyls on CO2 Hydrogenation Over Cu/γ-Al2O3 Catalyst: A Theoretical Study, J. Phys. Chem. C, 2011, 115, 19811–19818 CAS.
- B. Hvolbæk, T. V. W. Janssens, B. S. Clausen, H. Falsig, C. H. Christensen and J. K. Nøskov, Catalytic Activity of Au Nanoparticles, Nano Today, 2007, 2, 14–18 CrossRef.
- F. Li and B. C. Gates, Size-Dependent Catalytic Activity of Zeolite-Supported Iridium Clusters, J. Phys. Chem. C, 2007, 111, 262–267 CAS.
- A. M. Argo, J. F. Odzak and B. C. Gates, Role of Cluster Size in Catalysis: Spectroscopic Investigation of γ-Al2O3-Supported Ir4 and Ir6 during Ethene Hydrogenation, J. Am. Chem. Soc., 2003, 125, 7107–7115 CrossRef CAS PubMed.
- M. Montano, K. Bratlie, M. Salmeron and G. A. Somorjai, Hydrogen and Deuterium Exchange on Pt(111) and Its Poisoning by Carbon Monoxide Studied by Surface Sensitive High-Pressure Techniques, J. Am. Chem. Soc., 2006, 128, 13229–13234 CrossRef CAS PubMed.
- N. S. Babu, N. Lingaiah, R. Gopinath, P. S. S. Reddy and P. S. S. Prasad, Characterization and Reactivity of Alumina-Supported Pd Catalysts for the Room-Temperature Hydrodechlorination of Chlorobenzene, J. Phys. Chem. C, 2007, 111, 6447–6453 CAS.
- R. Gopinath, N. S. Babu, J. V. Kumar, N. Lingaiah and P. S. S. Prasad, Influence of Pd Precursor and Method of Preparation on Hydrodechlorination Activity of Alumina Supported Palladium Catalysts, Catal. Lett., 2007, 120, 312–319 CrossRef.
- B. C. Gates, Supported Metal Cluster Catalysts, J. Mol. Catal. A: Chem., 2000, 163, 55–65 CAS.
- D. C. Meier and D. W. Goodman, The Influence of Metal Cluster Size on Adsorption Energies: CO Adsorbed on Au Clusters Supported on TiO2, J. Am. Chem. Soc., 2004, 126, 1892–1899 CrossRef CAS PubMed.
- Z. X. Cheng, X. G. Zhao, J. L. Li and Q. M. Zhu, Role of Support in CO2 Reforming of CH4 over a Ni/γ-Al2O3 Catalyst, Appl. Catal., A, 2001, 205, 31–36 CrossRef CAS.
- P. G. Savva, K. Goundani, J. Vakros, K. Bourikas, C. Fountzoula, D. Vattis, A. Lycourghiotis and C. Kordulis, Benzene Hydrogenation over Ni/Al2O3 Catalysts Prepared by Conventional and Sol–Gel Techniques, Appl. Catal., B, 2008, 79, 199–207 CAS.
- S. Yolcular and Ö. Olgun, Ni/Al2O3 Catalysts and Their Activity in Dehydrogenation of Methylcyclohexane for Hydrogen Production, Catal. Today, 2008, 138, 198–202 CrossRef CAS PubMed.
- A. S. A. Al-Fatish, A. A. Ibrahim, A. H. Fakeeha, M. A. Soliman, M. R. H. Siddiqui and A. E. Abasaeed, Coke Formation During CO2 Reforming of CH4 over Alumina-Supported Nickel Catalysts, Appl. Catal., A, 2009, 364, 150–155 CAS.
- A. E. Aksoylu and Z. İlsenÖnsan, Hydrogenation of Carbon Oxides Using Coprecipitated and Impregnated Ni/Al2O3 Catalysts, Appl. Catal., A, 1997, 164, 1–11 CrossRef.
- A. R. Suzdorf, S. V. Morozov, N. N. Anshits, S. I. Tsignova and A. G. Anshits, Gas Phase Hydrodechlorination of Chlorinated Aromatic Compounds on Nickel Catalysts, Catal. Lett., 1994, 29, 49–55 CrossRef CAS.
- S. Hernández, L. Solarino, G. Orsello, N. Russo, D. Fino, G. Saracco and V. Specchia, Desulfurization Processes for Fuel Cells Systems, Int. J. Hydrogen Energy, 2008, 33, 3209–3214 Search PubMed.
- M. C. Valero, P. Raybaud and P. Sautet, Nucleation of Pdn(n = 1–5) Clusters and Wetting of Pd Particles on γ-Al2O3 Surfaces: A Density Functional Theory Study, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75 CAS.
- X. R. Shi and D. S. Sholl, Nucleation of Rhn(n = 1–5) Clusters on γ-Al2O3 Surfaces: A Density Functional Theory Study, J. Phys. Chem. C, 2012, 116, 10623–10631 CAS.
- N. C. Hernández, J. Graciani, A. Márquez and J. F. Sanz, Cu, Ag and Au Atoms Deposited on the α-Al2O3(0001) Surface: A Comparative Density Functional Study, Surf. Sci., 2005, 575, 189–196 Search PubMed.
- J. R. Li, R. G. Zhang and B. J. Wang, Influence of the Hydroxylation of γ-Al2O3 Surfaces on the Stability and Growth of Cu for Cu/γ-Al2O3 Catalyst: A DFT Study, Appl. Surf. Sci., 2013, 270, 728–736 CAS.
- S. Nigam and C. Majumder, Growth Pattern of Agn(n = 1–8) Clusters on the α-Al2O3(0001) Surface: A First Principles Study, Langmuir, 2010, 26, 18776–18787 CAS.
- C. H. Hu, C. Chizallet, C. Mager-Maury, M. Corral-Valero, P. Sautet, H. Toulhoat and P. Raybaud, Modulation of Catalyst Particle Structure upon Support Hydroxylation: Ab Initio Insights into Pd13 and Pt13/γ-Al2O3, J. Catal., 2010, 274, 99–110 CrossRef CAS PubMed.
- M. C. Valero, M. Digne, P. Sautet and P. Raybaud, DFT Study of the Interaction of a Single Palladium Atom with γ-Alumina Surfaces: the Role of Hydroxylation, Oil Gas Sci. Technol., 2006, 61, 535–545 CAS.
- M. Digne, P. Sautet, P. Raybaud, P. Euzen and H. Toulhoat, Use of DFT to Achieve a Rational Understanding of Acid–Basic Properties of γ-Alumina Surfaces, J. Catal., 2004, 226, 54–68 CrossRef CAS PubMed.
- G. Paglia, C. E. Buckley, A. L. Rohl, B. A. Hunter, R. D. Hart, J. V. Hanna and L. T. Byrne, Tetragonal Structure Model for Boehmite-Derived γ-Alumina, Phys. Rev. B: Condens. Matter, 2003, 68 CrossRef CAS.
- G. Paglia, C. E. Buckley, A. L. Rohl and L. T. Byrne, Towards the Determination of the Structure of γ-Alumina, J. Australas. Ceram. Soc., 2002, 38, 92–98 CAS.
- E. Menendez-Proupin and G. Gutierrez, Electronic Properties of Bulk γ-Al2O3, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72 Search PubMed.
- G. Paglia, E. S. Bozǐn and S. J. L. Billinge, Fine-Scale Nanostructure in γ-Al2O3, Chem. Mater., 2006, 18, 3242–3248 CAS.
- G. Paglia, C. E. Buckley and A. L. Rohl, Comment on “Examination of Spinel and Nonspinel Structural Models for γ-Al2O3 by DFT and Rietveld Refinement Simulations”, J. Phys. Chem. B, 2006, 110, 20721–20723 CrossRef CAS PubMed.
- M. Digne, P. Raybaud, P. Sautet, B. Rebours and H. Toulhoat, Comment on “Examination of Spinel and Nonspinel Structural Models for γ-Al2O3 by DFT and Rietveld Refinement Simulations”, J. Phys. Chem. B, 2006, 110, 20719–20726 CrossRef CAS PubMed.
- X. Krokidis, P. Raybaud, A. E. Gobichon, B. Rebours, P. Euzen and H. Toulhoat, Theoretical Study of the Dehydration Process of Boehmite to γ-Alumina, J. Phys. Chem. B, 2001, 105, 5121–5130 CrossRef CAS.
- M. Digne, P. Sautet, P. Raybaud, P. Euzen and H. Toulhoat, Hydroxyl Groups on γ-Alumina Surfaces: A DFT Study, J. Catal., 2002, 211, 1–5 CAS.
- C. Wolverton and K. C. Hass, Phase Stability and Structure of Spinel-Based Transition Aluminas, Phys. Rev. B: Condens. Matter, 2000, 63 CrossRef.
- G. Paglia, A. L. Rohl, C. E. Buckley and J. D. Gale, Determination of the Structure of γ-Alumina from Interatomic Potential and First-Principles Calculations: The Requirement of Significant Numbers of Nonspinel Positions to Achieve an Accurate Structural Model, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71 CrossRef CAS.
- J. P. Beaufils and Y. Barbaux, Détermination, Par Diffraction Différentielle de Neutrons, des Faces Cristallines Exposées par des Supports de Catalyseurs en Poudre, J. Chim. Phys. Rev. Gen. Colloides, 1981, 78, 347–352 CAS.
- P. Nortier and P. Fourre, Mohammed Saad, A. B.; Saur, O.; Lavalley, J. C. Effects of Crystallinity and Morphology on the Surface Properties of Alumina, Appl. Catal., 1990, 61, 141–160 CAS.
- J. Libuda, M. Frank, A. Sandell, S. Andersson, P. A. Bruhwiler, M. Bäumer, N. Martensson and H. J. Freund, Interaction of Rhodium with Hydroxylated Alumina Model Substrates, Surf. Sci., 1997, 384, 106–119 CrossRef CAS.
- B. Delley, An All-Electron Numerical Method for Solving the Local Density Functional for Polyatomic Molecules, J. Chem. Phys., 1990, 92, 508–517 CAS.
- B. Delley, From Molecules to Solids with the DMol3 Approach, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- P. Hohenberg and W. Kohn, Inhomogeneous Electron Gas, Phys. Rev., 1964, 136, B864–B871 Search PubMed.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, Energy-Adjusted Ab Initio Pseudopotentials for the First Row Transition Elements, J. Chem. Phys., 1987, 86, 866–872 CAS.
- A. Bergner, M. Dolg, W. Küchle, H. Stoll and H. Preuss, Ab Initio Energy-Adjusted Pseudopotentials for Elements of Groups 13–17, Mol. Phys., 1993, 80, 1431–1441 CAS.
- F. A. Reuse and S. N. Khanna, Geometry, Electronic Structure, and Magnetism of Small Nin(n = 2–6, 8, 13) Clusters, Chem. Phys. Lett., 1995, 234, 77–81 CrossRef CAS.
- M. D. Morse, G. P. Hansen, P. R. R. Langridge-Smith, L. S. Zheng, M. E. Gensic, D. L. Michalopoulos and R. E. Smalley, Spectroscopic Studies of the Jet-cooled Nickel Dimer, J. Chem. Phys., 1984, 80, 5400–5405 CAS.
- J. Ho, M. L. Polak, K. M. Ervin and W. C. Lineberger, Photoelectron Spectroscopy of Nickel Group Dimmers: Ni2−, Pd2−, and Pt2−, J. Chem. Phys., 1993, 99, 8542–8551 CAS.
- S. Nigam and C. Majumder, Adsorption of Small Palladium Clusters on the α-Al2O3(0001) Surface: A First Principles Study, J. Phys. Chem. C, 2012, 116, 2863–2871 CAS.
- P. Liu, Understanding the Behavior of TiO2(110)-Supported Pd7 Cluster: A Density Functional Study, J. Phys. Chem. C, 2012, 116, 25337–25343 CAS.
- C. Mager-Maury, C. Chizallet, P. Sautet and P. Raybaud, Platinum Nanoclusters Stabilized on γ-Alumina by Chlorine Used as a Capping Surface Ligand: A Density Functional Theory Study, ACS Catal., 2012, 2, 1346–1357 CAS.
- X. L. Zhu, D. G. Cheng and P. Kuai, Catalytic Decomposition of Methane over Ni/Al2O3 Catalysts: Effect of Plasma Treatment on Carbon Formation, Energy Fuels, 2008, 22, 1480–1484 CrossRef CAS.
- L. B. Avdeeva, T. V. Reshetenko, Z. R. Ismagilov and V. A. Likholov, Iron-Containing Catalysts of Methane Decomposition: Accumulation of Filamentous Carbon, Appl. Catal., A, 2002, 228, 53–63 CrossRef CAS.
- D. Chen, K. O. Christensen, E. Ochoa-Fernández, Z. X. Yu, B. Tøtdal, N. Latorre, A. Monzón and A. Holmen, Synthesis of Carbon Nanofibers: Effects of Ni Crystal Size During Methane Decomposition, J. Catal., 2005, 229, 82–96 CAS.
- H. Dai, A. G. Rinzler, P. Nikolaev, A. Thess, D. T. Colbert and R. E. Smalley, Single-Wall Nanotubes Produced by Metal-Catalyzed Disproportionation of Carbon Monoxide, Chem. Phys. Lett., 1996, 260, 471–475 CAS.
- C. L. Cheung, A. Kurtz, H. Park and C. M. Lieber, Diameter-Controlled Synthesis of Carbon Nanotubes, J. Phys. Chem. B, 2002, 106, 2429–2433 CrossRef CAS.
- C. Park and M. A. Keane, Catalyst Support Effects in the Growth of Structured Carbon from the Decomposition of Ethylene over Nickel, J. Catal., 2004, 221, 386–399 CAS.
- S. Takenaka, Y. Shigeta, E. Tanabe and K. Otuska, Methane Decomposition into Hydrogen and Carbon Nanofibers over Supported Pd–Ni Catalysts: Characterization of the Catalysts during the Reaction, J. Phys. Chem. B, 2004, 108, 7656 CAS.
- R. L. Van Wal, T. M. Ticich and V. E. Curtis, Substrate-Support Interactions in Metal-Catalyzed Carbon Nanofiber Growth, Carbon, 2001, 39, 2277–2289 CrossRef.
- X. N. Li, Y. Zhang and K. J. Smith, Metal–Support Interaction Effects on the Growth of Filamentous Carbon over Co/SiO2 Catalysts, Appl. Catal., A, 2004, 264, 81–91 CAS.
- L. G. V. Briquet, C. R. A. Catlow and S. A. French, Comparison of the Adsorption of Ni, Pd, and Pt on the (0001) Surface of α-Alumina, J. Phys. Chem. C, 2008, 112, 18948–18954 CAS.
- L. G. V. Briquet, C. R. A. Catlow and S. A. French, Platinum Group Metal Adsorption on Clean and Hydroxylated Corundum Surfaces, J. Phys. Chem. C, 2009, 113, 16747–16756 CAS.
- L. G. V. Briquet, C. R. A. Catlow and S. A. French, Structure and Reactivity of Aluminum Oxide Supported Nickel Clusters, J. Phys. Chem. C, 2010, 114, 22155–22158 CAS.
- J. Li, E. Croiset and L. Ricardez-Sandoval, Effect of Metal–Support Interface During CH4 and H2 Dissociation on Ni/γ-Al2O3: A Density Functional Theory Study, J. Phys. Chem. C, 2013, 117, 16907–16920 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: details of the adsorption of Nin clusters on dehydrated and hydrated γ-Al2O3(110) surfaces, as well as the corresponding most stable configurations of Nin clusters. See DOI: 10.1039/c3ra46352d |
| This journal is © The Royal Society of Chemistry 2014 |