
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitro and nitritosilanes: do they and can they exist?
Houari Dahmani,
Louis-Philippe Poulin,
Charles-Émile Fecteau,
Lara Harter,
Paul Andrew Johnson and
Guillaume Bélanger-Chabot*
and
Guillaume Bélanger-Chabot*
Department of Chemistry, Centre de recherche sur les matériaux avancés (CERMA) and Centre en Chimie Verte et Catalyse (CCVC), Université Laval, 1045 ave. de la Médecine, Québec, QC, Canada G1V 0A6. E-mail: gbchabot@chm.ulaval.ca
First published on 22nd August 2025
Abstract
Nitrosilanes are unknown, much like the nitro analogues of most metalloids. Their nitrite isomers are better known and even used as reagents. Still, most studies on nitritosilanes focus on reactivity and there remains a doubt on their identity and even their existence. In this contribution, we verify computationally whether nitrosilanes could exist and obtain full confirmation for the existence of nitritosilanes.
Nitro compounds are ubiquitous in organic chemistry. They are industrial precursors (cf.: nitrobenzene),5,6 widely used as energetic materials, both civil and military, and display highly unusual physical properties because of the strong electron-withdrawing effect of the nitro functional group.1–3 The functional group nitro (–NO2) can also be found in the isomeric form nitrite (–ONO), where the group is attached at one of the oxygen positions. Nitrites are also very well known in organic chemistry, as reagents (often as nitrosating agents)4 and pharmaceuticals5 (amyl nitrite, etc.). While the chemistry of nitro and nitrite groups is well developed with carbon, the same cannot be said of the metalloids and most other non-metals of the main group. Indeed, very few nitrito derivatives of boron, silicon, phosphorus, etc. are known and most have not been convincingly characterized, while only two electron-precise B–NO2 compounds have ever been reported.6,7 Nitritosilanes are well-known and invoked as nitrosylating and (de)oximating reagents,8–12 yet their isolation and characterization is the topic of only one report claiming the isolation of triisopropylnitritosilane and tricyclohexylnitritosilane.10 To the best of our knowledge, no nitrosilane has ever been reported. Our group is interested in trends influencing the stability of main-group compounds bearing oxidizing groups like the nitro group. Nitro derivatives of oxophilic elements like silicon would be expected to display interesting performances as energetic materials.13–16 Therefore, the problem of the apparent non-existence of nitrosilanes is of significant fundamental importance and of potential applied relevance. This contribution deals with our computational predictions on the plausibility of the existence of nitrosilanes and with our experimental confirmation of the structure, nature and physical properties of several nitritosilanes.
Our computational estimates in acetonitrile solution (B3LYP/6-311+g(d,p))-SMD17–19 show that nitrosilanes (Table 1) would have expected spectroscopic features for nitro compounds. Indeed, predicted values for νN–O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) asym range from 1457 to 1542 cm−1, those for νN–Osym from 1371 to 1417 cm−1. Predicted 14N chemical shifts are slightly outside of the usual region expected for nitro compounds (typically 0 to −100 ppm), with δ values ranging from 24 to 83 ppm (see SI). Nitrosilanes that bear electronegative substituents like –F (Fig. 1) and –OMe (see SI) have significantly outlying values. Predicted Si–N bond lengths vary between 1.853 (F3SiNO2) and 1.970 Å ((NMe2)3SiNO2), while N–O bond lengths are all ca. 1.23 Å. From inspection, all the nitro compounds studied appear structurally stable.
asym range from 1457 to 1542 cm−1, those for νN–Osym from 1371 to 1417 cm−1. Predicted 14N chemical shifts are slightly outside of the usual region expected for nitro compounds (typically 0 to −100 ppm), with δ values ranging from 24 to 83 ppm (see SI). Nitrosilanes that bear electronegative substituents like –F (Fig. 1) and –OMe (see SI) have significantly outlying values. Predicted Si–N bond lengths vary between 1.853 (F3SiNO2) and 1.970 Å ((NMe2)3SiNO2), while N–O bond lengths are all ca. 1.23 Å. From inspection, all the nitro compounds studied appear structurally stable.
|
||||
|---|---|---|---|---|
| Nitrosilane | ΔHTSisom | ΔGTSisom | ΔHisom | ΔGisom |
| a These nitrites not stable minima in solution. | ||||
| H3SiNO2 | 17.0 | 18.0 | −10.5 | −11.3 |
| Cl3SiNO2 | 12.0 | 11.4 | −20.7a | −20.4a |
| F3SiNO2 | 6.6 | 8.6 | −25.6a | −24.3a |
| (Me3Si)3SiNO2 | 20.5 | 19.9 | −4.9 | −5.9 |
| (MeO)3SiNO2 | 14.6 | 16.7 | −12.9 | −12.7 |
| (NMe2)3SiNO2 | 17.1 | 17.6 | −9.6 | −11.1 |
| Me3SiNO2 | 18.7 | 19.9 | −8.6 | −8.1 |
| Ph3SiNO2 | 17.1 | 17.6 | −10.5 | −11.2 |
| iPr3SiNO2 | 18.3 | 18.2 | −9.0 | −9.8 |
| tBuMe2SiNO2 | 18.4 | 19.1 | −8.6 | −9.1 |
| tBu3SiNO2 | 16.5 | 16.2 | −11.2 | −11.7 |
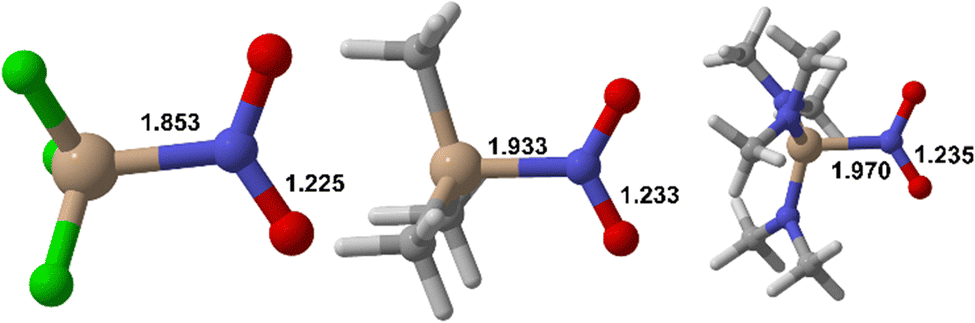 | ||
| Fig. 1 Predicted geometries of selected hypothetical nitrosilanes (B3LYP/6-311+g(d,p))-SMD(acetonitrile). Left: F3SiNO2; center: Me3SiNO2; right: (NMe2)3SiNO2. Bond distances in Å. Beige = silicon, blue = nitrogen, red = oxygen, white = hydrogen, light gray = carbon, green = fluorine. | ||
Our estimates, however, show that the nitrito isomers of these compounds are all significantly thermodynamically favoured, by 5.9 kcal mol−1 for (Me3Si)3SiONO to 24.3 kcal mol−1 for F3SiONO (Table 1, right two columns). This is in stark contrast with the opposite trend in nitro/nitrito alkanes, for which the nitro isomers are typically favored20,21 (we predict by 7 kcal mol−1 for MeNO2). Explanations for this trend are likely to involve the high oxophilicity of silicon. F3SiONO and Cl3SiONO are not stable minima in solution but are weak X3SiO⋯NO adducts. This feature is similar to what has been predicted for the hypothetical molecule dinitroacetylene, which is ca. 15–19 kcal mol−1 higher in Gibbs free energy than its nitrite isomers, both of which are weak CO⋯NO adducts.22
Before any serious synthetic effort at observing nitrosilanes is to be undertaken, it is therefore necessary to verify whether the hypothetical compounds could easily interconvert to their thermodynamically more stable nitrito isomers. We estimated the isomerization kinetic barriers, via a unimolecular process going through a Si–O–N three membered ring, for 11 silane derivatives (Table 1). Our predicted free energy barriers towards isomerization in acetonitrile solution range between 8.6 (F3SiNO2) to 19.9 kcal mol−1 ((Me3Si)3SiNO2). An earlier prediction for the parent nitrosilane H3SiNO2 similarly pointed towards very fast isomerization at ambient temperature.23 All the computed barriers would lead to short half-lives for the nitro isomers at room temperature. This feature is again in stark contrast with the isomerization of nitromethane into nitritomethane, which is known to occur only at high temperatures20,21 (we predict a free energy barrier of 63.2 kcal mol−1 for its unimolecular isomerization). Because other conceivable mechanisms could lead to decomposition, like autooxidation, the observation of nitrosilanes might be a formidable challenge, which would limit their interest to a niche scientific curiosity.
We therefore shifted our attention towards the thermodynamically favoured nitrite isomers. While nitritosilanes are invoked as nitrosating agents, a survey of the literature indicates that only one report deals with the isolation and characterization (by 1H NMR, MS and IR) of triisopropyl- and tricyclohexylnitritosilane.10 Other reports either claim nitritosilanes as intermediates in the formation of nitrosyl chloride,24–27 as nitrosating or (de)oximating agents,8,11,12 or as highly unstable molecules that act as N2O3 sources.8,11 All of these reports use the reactivity of the invoked nitritosilanes, the appearance of decomposition products or even the color of the reaction mixture as a characterization tool. Because these features could all conceivably be explained by some of the decomposition products, the body of evidence for the existence and stability of nitritosilanes remains limited. While there is no doubt that all those reports conclusively indicate the usefulness of “nitritosilanes” (or their decomposition products) as reagents, our genuine interest in the existence of the actual nitritosilane molecules prompted us to revisit these systems computationally and synthetically.
Our predictions indicate that stable nitritosilanes should display an 14N NMR signal at ca. 200 ppm (207 to 227 ppm) and ca. 20 ppm upfield from free nitrite or nitrosyl chloride (see SI). This is in the same region as that of organic nitrite esters. To our knowledge, 14N NMR has never been used to identify nitritosilanes, even though the breadth of chemical shifts (commonly from +300 to −400 ppm) makes it a powerful method to qualitatively identify nitrogen oxide functional groups in diamagnetic molecules.28 We also predict that the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch should be observable between 1729 and 1641 cm−1 as an intense IR band and a medium to weak Raman peak, which is in fair agreement with the bands at 1630 and 1625 cm−1 observed for the claimed Cy3SiONO and iPr3SiONO, respectively.10 Most nitrosilanes from Table 1 display predicted Si–O bond lengths of ca. 1.77 Å, (Si)O–N lengths of ca. 1.38 Å and (SiO)NO bond lengths of ca. 1.19 Å (see SI). Strong deviations from these values are observed for F3SiONO and Cl3SiONO, which display very short Si–O bonds and long (Si)O–N bonds (Fig. 2), again indicating they would not be stable compounds in solution, but rather weak X3SiO⋯NO adducts. We predict that tris(trimethylsilyl)nitritosilane, on the other hand, would display a slightly elongated Si–O bond (Fig. 2).
O stretch should be observable between 1729 and 1641 cm−1 as an intense IR band and a medium to weak Raman peak, which is in fair agreement with the bands at 1630 and 1625 cm−1 observed for the claimed Cy3SiONO and iPr3SiONO, respectively.10 Most nitrosilanes from Table 1 display predicted Si–O bond lengths of ca. 1.77 Å, (Si)O–N lengths of ca. 1.38 Å and (SiO)NO bond lengths of ca. 1.19 Å (see SI). Strong deviations from these values are observed for F3SiONO and Cl3SiONO, which display very short Si–O bonds and long (Si)O–N bonds (Fig. 2), again indicating they would not be stable compounds in solution, but rather weak X3SiO⋯NO adducts. We predict that tris(trimethylsilyl)nitritosilane, on the other hand, would display a slightly elongated Si–O bond (Fig. 2).
 | ||
| Fig. 2 Predicted geometries of selected hypothetical nitritosilanes (B3LYP/6-311+g(d,p))-SMD(acetonitrile). Left: F3SiONO (not a stable minimum); center: Me3SiONO; right: (Me3Si)3SiONO. Bond distances in Å. Beige = silicon, blue = nitrogen, red = oxygen, white = hydrogen, light gray = carbon, green = fluorine. | ||
We chose tert-butyldimethylnitritosilane (1) as our target for isolation. No reference to it could be found in the literature, but its expected volatile nature (and that of its expected decomposition products) would make it a potentially convenient anhydrous nitrosation reagent and therefore a relevant target for isolation. Upon treatment of tert-butyldimethylchlorosilane with silver nitrite in a pentane suspension at room temperature and removal of the solvent under vacuum at ca. −60 °C, a colourless, somewhat volatile liquid was obtained and analysed at room temperature. Raman and IR spectra both showed a band at 1636 cm−1 and the 14N NMR spectrum of an acetonitrile solution showed a broad signal at +196 ppm. These features all agree with the predicted signals for a nitritosilane and fall in the region expected for covalent nitrites. In a similar manner, we re-obtained triisopropylnitritosilane (2) as a colorless liquid that displayed the same characteristic spectroscopic signals, thus strongly supporting its identity as a nitritosilane. This assignment was finally fully confirmed when marginally stable triphenylnitritosilane (6, vide infra) could be observed by X-ray diffraction crystallography (Fig. 3).
 | ||
| Fig. 3 Synthesis and attempted synthesis of various nitritosilanes. Bottom right: solid-state structure of 6 obtained from X-ray diffraction analysis. As expected from its slow formation in DCM, the compound co-crystallized with a small amount of the chloride starting material, which was modeled as a substitutional disorder (refined ratio ONO:Cl 0.85:0.15, not shown, see SI). Hydrogen atoms were omitted for clarity. Beige: silicon; dark grey: carbon; blue: nitrogen; red: oxygen. | ||
Both isolated nitritosilanes 1 and 2 are very moisture-sensitive, and their hydrolysis yields their siloxane derivative and nitrogen oxides, presumably from the disproportionation of somewhat unstable HNO2. Both compounds are relatively stable as neat compounds and can be stored for months at −30 °C. At room temperature, compounds 1 and 2 and their acetonitrile solutions will only slowly decompose over weeks, with 2 being particularly robust. Major decomposition products are the siloxane derivatives, NO and NO2.
Since both these compounds were kinetically stable enough to be isolated at room temperature, we wondered whether true nitritosilanes were responsible for the reactivity described in many reports. To verify this, we examined trimethylnitritosilane (3), which is the species most often invoked in several reports.11,24,25 Treating chlorotrimethylsilane with silver nitrite in acetonitrile, even at −30 °C, led to gas evolution and produced orange nitrogen dioxide fumes and a blue solution, as some reports indicate.11,12 14N NMR analysis of such solutions showed no sign of nitritosilane formation and 1H NMR showed the clear presence of hexamethyldisiloxane as the sole proton-containing product, as one report indicates.11 Gas-phase IR analysis of a portion of the volatiles from the reaction mixture clearly shows the presence of both NO (1877 cm−1, νNO) and NO2 (1616 cm−1, νNO2asym). Their presence in solution is expected to lead to some association, presumably yielding low concentrations of blue N2O3, which is undoubtedly responsible for the blue colour of the solution. Throughout our studies, the appearance of blue solutions was often accompanied by orange vapours of NO2 and could always be correlated with decomposition by 1H NMR spectroscopy. Therefore, claims based on 3 in acetonitrile, especially those relating the blue color to the presence of nitritosilanes, even at low temperature, were most likely incorrect, with the most likely active species being N2O3 and its dissociation products, as proposed by one report.11
We nevertheless sought to demonstrate that 3 indeed exists as an intermediate. In dichloromethane, the formation and decomposition of 3 were substantially slowed, even though the bluish color of N2O3 could still be observed in the cold solutions. Transferring the volatiles of the reaction mixture by vacuum condensation allowed us to observe, even at room temperature, a significant signal at +196 ppm in the 14N NMR spectrum, as well as an intense, sharper signal at ca. +230 ppm, indicative of the presence of ClNO. The presence of ClNO was confirmed by gas-phase IR (NO stretch at 1800 cm−1),29 thereby confirming previous reports that claimed the presence of ClNO from the reaction of excess R3SiCl with R3SiONO, again based on reactivity patterns.24,25,27 1H NMR showed three signals, one for the starting material (which had not finished reacting), one for the nitritosilane and one for the hexamethyldisiloxane decomposition product. Because of slow reactions with insoluble AgNO2 and competing reaction between the starting material and the nitritosilane, we deemed the isolation of pure 3 impractical and did not pursue it further.
To gain a broader sense of the factors affecting the stability of the nitrites, we proceeded to generate several other nitritosilanes. As predicted by our calculations, the treatment of SiCl4 with silver nitrite rapidly leads to vigorous gas formation in acetonitrile, yielding no observable species by NMR spectroscopy, thus supporting our prediction that Cl3SiONO would not be a stable compound (Fig. 2 and Table 1). We could however detect tBuPh2SiONO (4), and (EtO)3SiONO (5) as marginally stable nitritosilanes in acetonitrile solutions. Similarly, Ph3SiONO (6) and PhMe2SiONO (7) were observed but only in dichloromethane solutions. None of species 4–7 could be isolated, although 5 persists in the neat “isolated” mixture. 6 is particularly problematic because its formation is slow in dichloromethane and its decomposition proceeds marginally slower. Still, to our surprise, crystals stored for months at −30 °C were shown to contain the desired nitritosilane. Because the compound does not usually survive more than a few days in solution, we concluded that the compound is considerably more stable in the solid state (Fig. 3). The crystallization of 6 finally demonstrated conclusively the identity of 1–7 as nitrites by relating spectroscopic observations to structural data.
The instability of the aryl derivatives is puzzling and cannot be tied to steric hindrance, since 4 is more crowded than 1 and yet, is less stable. Preliminary results suggest that there is no internal aromatic nitrosation reactions occurring. The mechanisms for the decomposition of nitritosilanes and their reactivity with chlorosilanes is still under investigation. Preliminary predictions indicate that simple Si–O or (Si)O–N bond dissociations are not accessible decomposition pathways for H3SiONO at ambient temperatures, with homolytic bond dissociation enthalpies of 42 to 71 kcal mol−1 in solution. Heterolytic pathways are even less favourable. Intermolecular processes, including solvent involvement (as supported by the observed increased stability in non-coordinating solvent dichloromethane), appear to be more plausible.
This study sheds light onto the existence of nitrosilanes. While these hypothetical compounds would be of great importance to the design of future energetic materials and would likely display exotic properties, akin to their carbon analogues, the low barrier towards isomerization into their nitrite analogues alone suggests that most conceivable nitrosilanes would not exist at room temperature. Other decomposition mechanisms beyond the scope of this work could also likely conspire against their existence. Their nitrite isomers remain highly interesting from fundamental and reactivity perspectives. For the first time, we provide direct spectroscopic evidence for their existence with strong X-ray diffraction structural support. We isolated two storable nitritosilanes, while showing the transient existence of several more. We conclusively showed that the greenish-blue colour observed in some reports does not correlate with the presence of nitritosilanes, but rather with their decomposition into NO and NO2. These results will clarify the interpretation of the reactivity of these useful oxidizing agents in future uses. Our group is exploring further reactivity and the extension of the use of nitritosilanes in an anhydrous, main-group synthesis setting, particularly that of 1, a conveniently volatile reagent.
The authors acknowledge the Natural Sciences and Engineering Research Council of Canada for a Discovery grant and an RTI grant, the Fonds de recherche du Québec Nature et Technologies for a Research Support for New Academics grant and the Canada Foundation for Innovation for a John R. Evans Leaders Fund grant.
Conflicts of interest
There are no conflicts to declare.Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its SI. See DOI: https://doi.org/10.1039/d5cc03923aGaussian output (.log) files were deposited on the Borealis data repository and are available at: https://doi.org/10.5683/SP3/LQ7JKU.
CCDC 2469460 contains the supplementary crystallographic data for this paper.30
Notes and references
- A. W. Knudsen, Am. J. Phys., 1975, 43, 888–894 CrossRef CAS.
- T. Ueno, Y. Urano, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2006, 128, 10640–10641 Search PubMed.
- M. R. Niazi, E. Hamzehpoor, P. Ghamari, I. F. Perepichka and D. F. Perepichka, Chem. Commun., 2020, 56, 6432–6435 RSC.
- A. Hashidoko, T. Kitanosono, Y. Yamashita and S. Kobayashi, Org. Lett., 2024, 26, 5517–5521 CrossRef CAS PubMed.
- D. T. Mason, E. Braunwald, F. A. Bullock and C. V. King, Circulation, 1965, 32, 755–766 CrossRef CAS PubMed.
- D. J. Brauer, H. Bürger, Y. Chebude and G. Pawelke, Eur. J. Inorg. Chem., 1999, 247–253 Search PubMed.
- A. Solovyev, Q. Chu, S. J. Geib, L. Fensterbank, M. Malacria, E. Lacôte and D. P. Curran, J. Am. Chem. Soc., 2010, 132, 15072–15080 Search PubMed.
- M. Baidya, Nitrous acid, tris(1-methylethyl)silyl Ester, Encyclopedia of Reagents for Organic Synthesis, 2013 DOI:10.1002/047084289X.rn01624.
- C. Eaborn, J. Chem. Soc., 1950, 3077–3089 RSC.
- M. Weidenbruch and F. Sabeti, Z. Naturforsch., B: J. Chem. Sci., 1976, 31, 1212 Search PubMed.
- A. S. Demir and H. Findik, Lett. Org. Chem., 2005, 2, 602–604 Search PubMed.
- M. Baidya and H. Yamamoto, J. Am. Chem. Soc., 2011, 133, 13880–13882 CrossRef CAS PubMed.
- P. Portius, B. Peerless, M. Davis and R. Campbell, Inorg. Chem., 2016, 55, 8976–8984 Search PubMed.
- E.-C. Koch and T. M. Klapötke, Propellants, Explos., Pyrotech., 2012, 37, 335–344 CrossRef CAS.
- T. M. Klapötke, B. Krumm, R. Ilg, D. Troegel and R. Tacke, J. Am. Chem. Soc., 2007, 129, 6908–6915 CrossRef PubMed.
- T. M. Klapötke, B. Krumm, A. Nieder, O. Richter, D. Troegel and R. Tacke, Z. Anorg. Allg. Chem., 2012, 638, 1075–1079 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- G. N. Spokes and S. W. Benson, J. Am. Chem. Soc., 1967, 89, 6030–6035 CrossRef CAS.
- M. J. S. Dewar, J. P. Ritchie and J. Alster, J. Org. Chem., 1985, 50, 1031–1036 CrossRef CAS.
- L. Harter, G. Bélanger-Chabot and M. Rahm, Propellants, Explos., Pyrotech., 2025, 50, e12019 CrossRef CAS.
- Z.-H. Tang, J.-S. Chen, Z.-M. Sun, A.-M. Tian and G.-S. Yan, Gaodeng Xuexiao Huaxue Xuebao, 1995, 16, 761–764 CAS.
- G. L. Jong, H. K. Ki and P. H. Je, Tetrahedron Lett., 1990, 31, 6677–6680 CrossRef.
- J. G. Lee and H. T. Cha, Tetrahedron Lett., 1992, 33, 3167–3168 CrossRef CAS.
- G. A. Olah, P. Ramaiah, G. Sandford, A. Orlinkov and G. K. Surya Prakash, Synthesis, 1994, 468–469 CrossRef CAS.
- M. Narendra Mallya, G. Nagendrappa, J. Shashidhara Prasad, M. A. Sridhar, N. K. Lokanath and N. S. Begum, Tetrahedron Lett., 2001, 42, 2565–2568 CrossRef CAS.
- J. Mason, in Multinuclear NMR, ed. J. Mason, Plenum Press, New York, 1987, p. 227 Search PubMed.
- T. Shimanouchi, J. Phys. Chem. Ref. Data, 1972, 6, 993–1102 CrossRef.
- H. Dahmani, L.-P. Poulin, C.-É. Fecteau, L. Harter, P. A. Johnson and G. Bélanger-Chabot, CCDC 2000643: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2nwp08.
| This journal is © The Royal Society of Chemistry 2025 |