
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cell-penetrating peptide-conjugated, splice-switching oligonucleotides mitigate the phenotype in BTK/Tec double deficient X-linked agammaglobulinemia model†
Burcu
Bestas
 a,
H. Yesid
Estupiñán
am,
Qing
Wang
a,
Shabnam
Kharazi
a,
Chenfei
He
b,
Dara
K. Mohammad
a,
Dhanu
Gupta
af,
Oscar P. B.
Wiklander
an,
Taavi
Lehto
acp,
Karin E.
Lundin
a,
Anna
Berglöf
a,
Mikael C. I.
Karlsson
b,
Frank
Abendroth
do,
Samir
El Andaloussi
ae,
Michael J.
Gait
d,
Matthew J. A.
Wood
f,
Christian J.
Leumann
g,
Dmitry A.
Stetsenko
ij,
Robert
Månsson
ah,
Jesper
Wengel
k,
Rula
Zain
ael and
C. I. Edvard
Smith
*ae
a,
H. Yesid
Estupiñán
am,
Qing
Wang
a,
Shabnam
Kharazi
a,
Chenfei
He
b,
Dara
K. Mohammad
a,
Dhanu
Gupta
af,
Oscar P. B.
Wiklander
an,
Taavi
Lehto
acp,
Karin E.
Lundin
a,
Anna
Berglöf
a,
Mikael C. I.
Karlsson
b,
Frank
Abendroth
do,
Samir
El Andaloussi
ae,
Michael J.
Gait
d,
Matthew J. A.
Wood
f,
Christian J.
Leumann
g,
Dmitry A.
Stetsenko
ij,
Robert
Månsson
ah,
Jesper
Wengel
k,
Rula
Zain
ael and
C. I. Edvard
Smith
*ae
aDepartment of Laboratory Medicine, Karolinska Institutet, ANA Futura, Alfred Nobels Allé 8 Floor 8, SE-141 52 Huddinge, Sweden. E-mail: edvard.smith@ki.se
bDepartment of Microbiology, Tumor and Cell Biology, Karolinska Institutet, Stockholm, Sweden
cInstitute of Techology, University of Tartu, Tartu, Estonia
dMedical Research Council Laboratory of Molecular Biology, Cambridge CB2 0QH, UK
eKarolinska ATMP Center, Karolinska Institutet, Karolinska University Hospital, SE-171 76 Stockholm, Sweden
fDepartment of Paediatrics, University of Oxford, Oxford OX3 7TY, UK
gDepartment of Chemistry and Biochemistry, University of Bern, Freiestrasse 3, CH-3012 Bern, Switzerland
hDepartment of Clinical Immunology and Transfusion Medicine, Karolinska University Hospital, Stockholm, Sweden
iInstitute of Cytology and Genetics, Siberian Branch of the Russian Academy of Sciences, 10 Lavrentiev Ave., Novosibirsk 630090, Russia
jDepartment of Physics, Novosibirsk State University, 2 Pirogov Str., Novosibirsk 630090, Russia
kDepartment of Physics, Chemistry and Pharmacy, Biomolecular Nanoscale Engineering Center, University of Southern Denmark, Odense, Denmark
lCentre for Rare Diseases, Department of Clinical Genetics and Genomics, Karolinska University Hospital, SE-171 76 Stockholm, Sweden
mDepartamento de Ciencias Básicas, Universidad Industrial de Santander, Bucaramanga, Colombia
nBreast Center, Karolinska Comprehensive Cancer Center, Karolinska University Hospital, Stockholm, Sweden
oDepartment of Chemistry, University of Marburg, Marburg, D-35043, Germany
pInstitute of Pharmacy, Faculty of Medicine, University of Tartu, Nooruse 1, 50411 Tartu, Estonia
First published on 31st March 2025
Abstract
Splice-switching oligonucleotides (SSOs) have been developed as a treatment for various disorders, including Duchenne muscular dystrophy and spinal muscular atrophy. Here, the activity of several different SSOs was investigated as potential treatments for B lymphocyte disorders with a focus on X-linked agammaglobulinemia (XLA), caused by defects in the gene encoding Bruton's tyrosine kinase (BTK). In this study, the activity of locked nucleic acid (LNA), tricyclo-DNA (tcDNA), phosphoryl guanidine oligonucleotides (PGO) and phosphorodiamidate morpholino oligomers (PMO) were compared, targeting the pseudoexon region of BTK pre-mRNA. We further investigated the effect of conjugating cell-penetrating peptides, including Pip6a, to the SSOs. The effect was measured as splice-switching in vitro as well as in a further developed, bacterial artificial chromosome transgenic mouse model of XLA. Therapy in the form of intravenous infusions 2 times a week during 3 weeks of PMO oligomers conjugated to Pip6a was sufficient to partly restore the in vivo B lineage phenotype. SSOs treatment also provides a unique opportunity to get insights into a restoration process, when B lymphocytes of different maturation stages are simultaneously splice-corrected.
Introduction
Splice-switching oligonucleotides (SSOs) interfering with pre-mRNA processing, first developed in 1993,1 have increasingly impacted the treatment of a number of genetic diseases. Consequently, SSOs have been approved as therapy in Duchenne muscular dystrophy,2,3 and such a strategy was recently used for the innovative treatment of a newly discovered neurodegenerative disorder, with only a single known, affected patient.4,5 This demonstrates the versatility and short time from the identification of a mutation to the actual development of a novel treatment modality. Furthermore, the perhaps most successful treatment to date is in spinal muscular atrophy. By administrating an SSO-based drug, spinraza, into the cerebrospinal fluid, repression of a splice silencer in the survival motor neuron gene 2 (SMN2) leads to the inclusion of exon 7 into the pre-mRNA with great clinical benefit.6As with other forms of oligonucleotide (ON) therapies, the challenge is to successfully transfer sufficient numbers of SSOs into relevant cells in the disease-affected tissue. Hepatocytes and cells in the central nervous system can be readily targeted, but for many other organs uptake remains a hurdle.7 To this end, we have previously developed an experimental treatment strategy for X-linked agammaglobulinemia (XLA).8 In this B-lymphocyte disorder, a developmental block occurs at the pro-B to pre-B transition9 owing to that the BTK cytoplasmic protein-tyrosine kinase is lacking or functionally disabled.10 The blocked maturation results in an increased susceptibility to bacterial and enteroviral infections.10,11 By generating a BTK-transgenic mouse, which carries a mutated human bacterial artificial chromosome (BAC), and, simultaneously lacks the endogenous BTK kinase, it was possible to obtain an XLA mouse model to study the effect of SSOs.8 We observed that when targeting B-lineage cells, only SSOs carrying a cell-penetrating peptide were efficient for in vivo correction. In these studies, BTK protein synthesis was restored through the removal of a pseudo-exon erroneously introduced by a single point mutation. However, while our previous work demonstrated protein restoration, it did not assess the phenotypic and functional consequences of this correction in vivo.
We here analyze a set of chemistries for efficient pseudo-exon removal in B-lymphocytes and demonstrate for the first time that by repeated administration of cell-penetrating peptide SSO conjugates, it is possible to partly restore the phenotype of the affected lymphocytes in vivo. Since BTK deficiency alone only results in a mild B-lineage defect in mice,12–14 we have used a BTK/Tec double knockout mouse strain lacking both of these kinases. This animal model manifests a more severe phenotype resembling XLA.15 Prior to treatment, spleen and lymph nodes of Tec/BTK double-deficient mice displayed a dramatic reduction of B cells compared with BTK single knockout mice. Similar to human settings, a relative decrease of the mature IgMloIgDhi population and accumulation of CD43+ (Pro-B) B cell precursors were noted, indicating a partial developmental block at the large pre-B cell stage.15 Thus, this study goes beyond protein restoration to assess phenotypic recovery in a BTK/Tec double knockout model, which more closely mimics human XLA, allowing us to evaluate functional B cell development after sustained SSO treatment.
Disorders affecting B lineage cells comprise a large part of human disease including autoimmunity, autoinflammation, allergies, leukemias&lymphomas, primary immunodeficiencies and infectious disease. Our study not only confirms the feasibility of targeting pseudoexon 4a with SSO-CPP conjugates but also provides the first in vivo evidence of functional restoration in a severe B-cell deficiency model. These findings broaden the applicability of ON-based therapeutics for genetic immunodeficiencies, highlighting their potential to address complex hematopoietic disorders beyond XLA. Hence, apart from addressing inherited loss-of-function disorders affecting the B-cell lineage, these studies may also be of interest for the general development of ON therapeutic strategies targeting B-lymphocyte-derived leukemias and lymphomas.
Results and discussion
In vitro comparison of oligonucleotides with different chemistries in a reporter cell line
The strategy for pseudo-exon (exon 4a) exclusion is depicted in Fig. 1a. We previously screened and optimized a large number of SSOs based on locked nucleic acid (LNA) and phosphorodiamidate morpholino oligomer (PMO) chemistries spanning the intron 4 of the human BTK gene.8 The most efficient SSO, named 186, targeting the predicted exon splice enhancer (ESE) and another SSO targeting the 5′-splice site (named 187) were chosen for the current study. Owing to that our previous studies were restricted to LNA-DNA mixmers and PMOs,8 we here wanted to extend the analysis to other types of nucleotides. Different chemistries of both the 186- and the 187-series of SSOs were first tested in a U2OS reporter cell line carrying an interrupted luciferase gene with the mutated patient-derived BTK intron 4. The first sets of SSOs were synthesized with tricyclo-DNA (tcDNA) chemistry, which has been shown to have a potent activity in a dystrophic mouse model.16 A second set of SSOs was synthesized as novel phosphoryl guanidine oligonucleotides (PGO) as charge-neutral phosphate mimics. The PGOs represent a third class of charge-neutral oligonucleotide derivatives, alongside the previously known peptide nucleic acids (PNA) and morpholino oligomers (PMO).17 This chemistry has not been tested in the context of splice-switching in B lymphocytes up to date but for antisense properties including gapmers18 and as well as G-quadruplex-forming agents.19 The structures of these SSOs are shown in Fig. 1b. The SSOs were transfected into reporter cells via gymnosis20 and compared to the previously described LNA-modified SSOs, with the splice-switching activity calculated based on the luminescence as well as RT-PCR. Treatment of the reporter cells showed that the LNA-DNA mixmer SSO 186 and tcDNA modified SSO 187 display the highest activity (Fig. 1c). Although the unconjugated phosphoryl guanidine (PGO) SSOs demonstrated a dose response, the overall in vitro activity was much lower compared to other chemistries (Fig. 1c). | ||
| Fig. 1 Gymnosis activity of SSOs with different chemistries in a reporter cell line model. (a) Outcome of predicted splicing in the reporter luciferase U2OS cell line model with a pseudo-exon-interrupted (exon 4a) mutated BTK intron 4, SSO treatment leads to restored splicing and generation of functional luciferase protein. (b) Structures of oligonucleotide analogues used in the study: (1) locked nucleic acid (LNA) phosphorothioate; (2) tricyclo-DNA (tcDNA) phosphodiester; (3) phosphorodiamidate morpholino oligomer (PMO); (4) phosphoryl guanidine oligo-2′-O-methylribonucleotide (2′-OMe PGO). (c) The upper panel shows the luciferase activity 24 h after SSO transfection. Tricyclo-DNA (tcDNA), locked nucleic acid (LNA) and PGO modified SSOs were tested. Data represent mean + SD of 2 independent experiments, each with 2 replicates. The lower panel shows a representative RT-PCR result of the same experiment. 389-bp and 271-bp bands correspond to aberrant (mut) and corrected (with BTK intron 4 excised) mRNA bands, respectively. The lower, 18S ribosomal RNA band served as control. The percentage of WT BTK RNA was calculated as WT RNA fraction × 100/(mis-spliced + WT RNA fractions). PMO = phosphorodiamidate morpholino oligomer and UT = untreated control. The corrected peak area was used for the calculation. | ||
CPP-mediated delivery of the SSOs in a reporter cell line
We previously studied the effect of PMO-based ONs conjugated to a cell-penetrating peptide.8 In order to determine whether the cell-penetrating peptide (CPP) conjugates could further improve the splice-switching activity, we decided to test Pip6a-PMO, an improved derivative of Pip5e-PMO with alterations in the hydrophobic core, which has shown the ability to restore dystrophin function in mdx mouse model.21,22 A more recent study using Pip6a-PMO targeting the pathologic CAG repeats in myotonic dystrophy type 1 (DM1) mice showed normalization of the disease transcriptome and phenotype.23 To this end, we used the B-PMO 186 as the most efficient conjugate from our previous study8 and compared it to Pip6a-PMO 186. Pip6a was previously never tested for SSO transfer into B lymphocytes. The comparison between the two derivatives showed a striking difference as Pip6a-PMO 186 demonstrated superior activity over B-PMO 186, even at lower concentration ranges both on protein and RNA levels (Fig. 2a). This prompted us to also test the novel phosphoryl guanidine (PGO) modification with the same conjugation, which showed several fold increase in activity with the overall efficiency being similar to the Pip6a-PMO conjugates. Moreover, Pip6a-Control targeting the dystrophin locus as a control and PMO-BTK 186 without CPP conjugation did not show any effect (Fig. 2a and b). Due to the uncharged backbone of PMO and PGO SSOs, they were chosen to be conjugated to positively charged Pip6a cell-penetrating peptide. | ||
| Fig. 2 CPP-mediated delivery in a reporter cell line. (a) Comparison of Pip6a-PMO 186 with B-PMO 186 as measured by luciferase activity 24 h after transfection. (b) Increase in luciferase activity by Pip6a-PMO and by unconjugated and Pip6a-conjugated phosphoryl guanidine (PGO) SSOs. The lower panels (a) and (b) show RT-PCR results for aberrant and corrected mRNA. PMO-BTK is the unconjugated 186.258 and Pip6a-Control is dystrophin targeting control oligonucleotide. Data represent mean + SD of 2 independent experiments, each with 2 replicates. A dosage of 15 μM was only used for PGO-186 and PGO-187 and a dosage of 5 μM was used for Pip6a-Control. 389-bp and 271-bp bands correspond to aberrant and corrected (with BTK intron 4 excised) mRNA bands, respectively. The 18S serves as an RNA quality control. The percentage of WT BTK RNA was calculated as WT RNA fraction × 100/(mis-spliced + WT RNA fractions). The corrected peak area was used for the calculation. Two-tailed Mann–Whitney test was used to calculate significance, *P ≤ 0.05. | ||
CPP-conjugated SSOs induce splice-correction ex vivo in primary B cells
In order to test the activity of the SSOs in a physiological, genomic setting, we first isolated primary B cells from mice expressing the mutant BTK transcript. We subsequently treated cells with the CPP-conjugated SSOs (Pip6a-PMO 186, B-PMO 186, Pip6a-PGO-187 and Pip6a-PGO-186) using two different doses (0.3 μM and 5 μM) and incubated cells for 48 h. Following the incubation, primary B cells were collected and BTK expression was evaluated by western blot. As shown in Fig. 3, the two Pip6a 186 conjugates with the different chemistries demonstrated the most efficient restoration of BTK protein. | ||
| Fig. 3 Ex vivo treatment of primary B cells with 0.3 μM and 5 μM of the splice-switching oligonucleotides (SSOs) Pip6a-PMO 186, B-PMO 186, Pip6a-PGO-187 and Pip6a-PGO-186 results in detectable BTK protein. Western blot shows a representative experiment of BTK protein after 48 h from 2 independent tests. Actin served as loading control. | ||
Pip6a-PMO restores BTK in vivo upon systemic delivery
The most efficient Pip6a-PMO 186 conjugate was subsequently injected intravenously into our previously described BAC transgenic mice,8 carrying the entire human mutant BTK locus in its germline and with an inactivated mouse BTK gene thereby preventing expression of endogenous mouse BTK protein.The correction of the mutant, human BTK mRNA in this mouse model generates a functional human BTK protein,8 which is almost identical to the mouse protein.24 In order to find the most optimal dose, different concentrations were intravenously injected and after 48 h, B cells from the spleen and the bone marrow of the mice were isolated. As depicted in Fig. 4, efficient BTK restoration was achieved already at 4 mg kg−1, which was also confirmed by the RT-PCR results (lower panel). This data overall suggests that Pip6a-PMO 186 is efficient already at lower doses. The biodistribution of Pip-series has been studied before;25,26 however, this is the first time it has been shown to be effective for the systemic treatment of B cells in vivo.
 | ||
| Fig. 4 In vivo, injection of Pip-6a PMO 186 into the BAC transgenic mice restores the BTK expression. The upper panel shows the BTK expression in splenic B cells by western blot. The lower panel shows the restoration of the WT mRNA in B cells isolated from the spleen (left) and the bone marrow (right) of the transgenic mouse after the indicated treatments. The percentage of WT BTK RNA was calculated as WT RNA fraction × 100/(mis-spliced + WT RNA fractions). The corrected peak area was used for the calculation. Two mice were injected for each dose. | ||
Pip6a-PMOs rescue the phenotype in the BTK/Tec double knock-out, BAC-transgenic mouse model
In order to assess the functionality of the most efficient conjugate, we injected 10 mg kg−1 Pip6a-PMO 186 as intravenous infusions 2 times a week during 3 weeks into BTK/Tec double-deficient mice. Three weeks later, cells from the spleen and bone marrow as well as sections of the spleen were collected for FACS and immunohistochemistry analysis.We observed significant changes in switched and marginal zone (MZ) B-cells in the spleen (Fig. 5A and B). Interestingly, the frequency of MZ B-cells was reduced in treated animals and the frequency of these cells was higher in the BTK/Tec double-deficient animals compared to WT. This is in accordance with previously published data on untreated mice27 and suggests normalization of the phenotype after therapy. We also observed significantly increased numbers of switched B-cells (Fig. 5A and B) and an increased tendency to develop a higher level of the T1 B-cell subset (Fig. 5A and B). To this end, Follicular (FoB) cells and the T2 B-cell subset did not show significant changes. BTK expression as measured by WB was confirmed in treated mice (C).
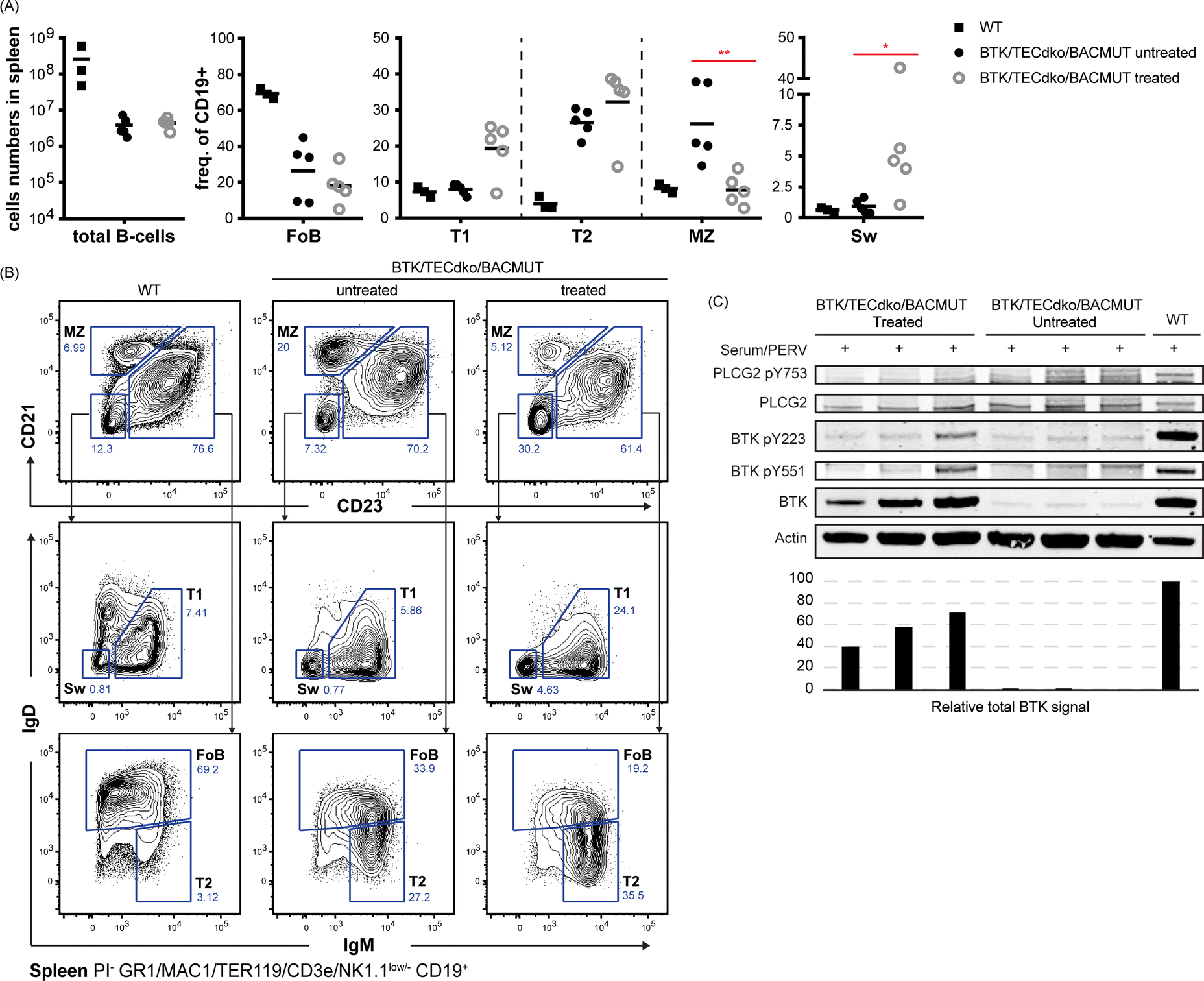 | ||
| Fig. 5 Characterization of the spleen B cell compartment following treatment with Pip6a-conjugated splice-switching PMOs. Panel (A), total (B) cells and B-cell subpopulations, Follicular (FoB), transitional type 1 (T1), transitional type 2 (T2) marginal zone (MZ), and switched (sw) cells. Panel B, Gating strategy for identification of indicated B-cell subsets. Prior gating was performed as indicated below the fluorescence-activated cell sorting plots. Propidium iodide (PI) was used for dead cell discrimination. Panel (C), western blot showing expression of total BTK, PLCG2 and actin (loading control) as well as activated forms as detected by phosphorylation-specific antibodies for BTK pY223 and pY551 and PLCG2 pY753. The total BTK levels are presented in the bar graph. | ||
To investigate the effect of the treatment on the follicular structure of the spleen we performed immunohistochemistry analysis (Fig. 6). We stained for B cells (B220), MZ B cells (B220/CD1d), metallophilic macrophages (MOMA-1), T lymphocytes (TCRβ) and MZ macrophages (MARCO). The data show that there is a normalization of the splenic follicular structure with a more organized follicle where the specialized macrophages of the marginal zone surrounded the B cell areas and with the marginal zone B cells being localized to the correct position within the marginal zone and next to the metallophilic macrophages.
 | ||
| Fig. 6 Characterization of the spleen architecture following treatment with Pip6a-conjugated splice-switching PMOs. Cells were detected using specific monoclonal antibodies: B cells (B220), marginal zone B cells (B220/CD1d), T lymphocytes (TCRβ), metallophilic macrophages (MOMA-1) and marginal zone macrophages (MARCO). | ||
More specifically we observed that the white pulp in BTK/Tec double-deficient mice was altered and only a few clearly defined, but small follicular structures were observed. The majority of the B220+ B cell areas appeared to be loosely associated with T cell areas and no clear and distinct surrounding anti–MOMA-1 staining was observed, indicating an increased discontinuity of the marginal zone compared with BTK/Tec null mice. Germinal centers were also detected in BTK/Tec double-deficient mice. Interestingly, a relative increase in CD21+ CD23lo MZ B cells was observed in BTK/Tec double-deficient mice when compared to WT, Tec ko or BTK ko animals.27
We also studied the B cell compartment in the bone marrow (Fig. 7). As can be seen, both the frequency of ProB cKit+ and pre-B increased in treated animals, and this alteration is statistically significant (Panel A).
 | ||
| Fig. 7 Characterization of the bone marrow B cell compartment following treatment with Pip6a-conjugated splice-switching PMOs. Panel (A), total (B) cells and B-lineage subpopulations, c-kit+ pro-B cells, c-kit− pro-B cells, pre-B cells, immature B cells and mature B cells. Panel B, Gating strategy for identification of indicated B-cell subsets. Prior gating was performed as indicated below the fluorescence-activated cell sorting plots. Propidium iodide (PI) was used for dead cell discrimination. Panel (C), western blot showing expression of total BTK, PLCG2 and actin (loading control) as well as activated forms as detected by phosphorylation-specific antibodies for BTK pY223 and pY551 and PLCG2 pY753 in individual mice. The total BTK levels are presented in the bar graph. | ||
Our results demonstrate that different chemistries can be used for splice-switching in B lymphocytes. A major obstacle in the antisense field is the targeted delivery, especially to extrahepatic sites. Numerous modifications have been tested involving the sugar, nucleobase and internucleotide linkage to improve stability, cellular uptake and in vivo distribution.7,28 The most promising chemical modifications, phosphorothioate (PS) and phosphorodiamidate morpholino oligomer (PMO), as well as the 2′-substitution on the ribose sugar, have been tested in several clinical studies and approved as drugs by FDA.29 Our previous study showed for the first time the successful correction of an intronic mutation in the BTK gene by steric blocking of a cryptic splice-site both in vitro and in vivo, achieving efficient splice-switching into hard-to-transfect primary B cells.8 However, the functional impact of this correction has never been assessed in vivo.
In this current study, we decided to screen several new chemistries in vitro and, moreover, to select the best compounds and evaluate their efficacy using the BAC-transgenic model. Since humoral immunity in BTK-deficient mice is much less impaired as compared to humans with XLA, we now bred the mice onto a BTK/Tec double knock-out background, which resembles human XLA in terms of the humoral deficiency.15
We first selected the most efficient SSOs from our previous study (SSO 186 and SSO 187) targeting predicted ESE and 5′-cryptic splice-site proximal to pseudo-exon respectively. We subsequently compared the efficacy of SSOs made from LNA, tcDNA and the novel phosphoryl guanidine (PGO) chemistry. The neutral chemistries were thereafter combined with cell-penetrating peptide Pip6a and compared to the B-PMO conjugates that we studied previously.8
To achieve efficient targeting in our double knock-out mouse model we first tested CPP-conjugated SSOs in our in vitro reporter model. We here used CPP-PMO conjugates. PMO, an uncharged nucleic acid with a morpholino group and phosphorodiamidate linkages,30 has been approved by the FDA for the treatment of Duchenne's muscular dystrophy, e.g. via exon 51 skipping.2 In a number of studies, the bioavailability of PMOs has been improved by direct conjugation to CPPs and such peptide-PMOs have been successfully exploited in preclinical studies of DMD by using exon skipping strategy for dystrophin rescue.31–33 Especially the optimized CPP platform named Pip-peptides with a designed hydrophobic core has been shown to be potent in dystrophic mouse models and reach hard to target sites such as cardiac muscle and CNS.25,34,35 We compared our previously tested B-PMO conjugate to the more efficient variant Pip6a-PMO.20,21 Strikingly, Pip6a conjugated SSO 186 PMO demonstrated two-to-three-fold more activity in a concentration range of 0.625–5 μM which is in line with the previous reports.36 Pip6a is a highly cationic charged peptide with a hydrophobic core, which has been shown, in conjugation with charged neutral oligonucleotides (PMO, PNA), to be efficiently taken up by cells.37 This property likely contributes to the enhanced efficacy observed when Pip6a is conjugated with SSOs, as seen in our comparative analyses with B-PMO conjugates. This prompted us to test the novel phosphoryl guanidine (PGO) modification in conjugation with Pip6a. Even though unconjugated PGO SSOs 186 and 187 showed very minor activity in vitro, Pip6a conjugation enhanced their efficacy to a similar level as with Pip6a-186 PMO. In order to evaluate the efficacy of Pip6a conjugated to PMO or PGO SSOs, we compared the efficiency of splice-switching in primary B cells derived from our previously described human BAC transgenic mouse model with a full-length mutated human BTK pre-mRNA. Interestingly, evaluation of BTK protein levels after 48 h showed similar efficiency for SSOs Pip6a PMO 186 and Pip6a-PGO-186, whereas B-PMO 186 and Pip6a-PGO-187 had minor restoration. While the PGO-modification has been previously reported to efficiently restore the sensitivity to vinblastine in tumor cells by targeting MDR1 RNA,18 this is the first time that their efficiency has been demonstrated in a primary B cell setting. Although LNA and tcDNA SSOs showed high activity in a reporter cell line setting, we have chosen CPP-conjugates of the neutral chemistries for in vivo testing owing to the nature of hard to target B cells. Further studies are needed to investigate LNA and tcDNA modified SSOs in an in vivo setting for targeting B cells.
Next, we tested the Pip6a PMO conjugate in the aforementioned double knock-out mouse model in order to study the functional activity of restored BTK protein after systemic administration. We used the Pip6a 186 PMO owing to its efficiency as well as having enough material for a long-term repeated dosing study. Initially, we tested the optimal dose for BTK restoration in the BAC transgenic mouse model. Intravenous injection of Pip6a 186 PMO revealed efficient BTK restoration at doses of 4 mg kg−1 and 12 mg kg−1 both in the spleen and bone-marrow cells isolated after 48 h. In order to monitor relevant functional change, we injected the BTK/Tec double-deficient mice with 10 mg kg−1 Pip6a-PMO 186 2 times a week during 3 weeks and investigated restoration of the B cell lineage by FACS and immunohistochemistry of the bone marrow and spleen. Analysis of the spleen compartment revealed that MZ B cells were normalized in comparison to non-treated animals. Moreover, we observed a tendency for an increase in the T1 B-cell subset, whereas follicular mature B cells did not show any augmentation. We further analyzed the BTK expression and the phosphorylation status upon activation as an indicator for a functional protein, as outlined before,8 and found clear signs of activation upon stimulation suggesting that the molecule is intact. To this end, phosphorylation of BTK at Y551 enhances its catalytic activity and results in its autophosphorylation at position Y223, although Y223 phosphorylation is functionally dispensable.38 Moreover, a fully active BTK phosphorylates PLCγ2 at position Y753. Although we could detect BTK in both splenic and bone marrow B cells, only the treated animal with the highest BTK expression in splenic B cells showed significant Y551 and Y223 phosphorylation. This suggests that treatment more often than intravenous infusions 2 times a week during 3 weeks is necessary. In our previous study using a B-PMO construct, treatment 3 times per week yielded higher expression.8
We also investigated the spleen architecture by immunohistochemistry in order to see the early stage changes in the lymphoid follicle. Staining of the spleen tissue sections for B cells (B220), Marginal zone B cells (B220/CD1d), metallophilic macrophages (MOMA-1) and Marginal zone macrophages (MARCO) revealed a normalization in the splenic follicular compartment. Moreover, we have also checked for the B cells in the bone marrow compartment. FACS analysis showed that treated animals have increased ProB cKit+ and pre-B cell numbers indicating developing B cells.
To this end, earlier studies have utilized lentiviral-based gene delivery into hematopoietic stem cells (HSCs) following transplantation in BTK/Tec double knockout mice, leading to significant restoration of major splenic B cell subsets and bone marrow B cell populations.39,40 In contrast, our non-viral approach partially restored the B cell phenotype, primarily normalizing splenic MZ B cells and increasing the proportion of ProB cKit+ and pre-B cells in the bone marrow. However, the transient SSO treatment seems to have been insufficient for driving all B cell subsets to full maturation. Targeting cells over a longer period could potentially lead to complete restoration of the B cell phenotype. Interestingly, while the phenotype was partly reverted in the Btk/Tec double-deficient mice, there were also changes that were unexpected. Thus, the increase in the transitional T1 subset differed from both normal and untreated mice and is likely caused by the instant loss of the developmental block, when BTK expression is turned on. It has been also shown that the differentiation into either follicular B cells (FoB) or marginal zone B cells (MZ B) is partially influenced by the strength of B-cell receptor (BCR) signaling.41 Notably, our ability to normalize only the MZ B cell population provides valuable insights into the BCR signaling threshold required for the maturation of distinct B cell subsets.
Hence, continued studies may give unique insights into the time course of B lineage subpopulation development during SSO-mediated phenotypic restoration. The SSO therapy here differs from the lentivirus-mediated treatment where all B cells develop from the HSC population. In contrast, the SSO therapy likely immediately affects all B lineage cells and not only the early progenitors derived from the HSCs. Thus, with SSOs, defined, developmental windows can be readily investigated by turning on and off the administration.
Conclusions
This study provides evidence that SSOs made from a rather unique collection of different chemistries are able to correct aberrantly spliced BTK in B lymphocytes using a mouse model of X-linked agammaglobulinemia. To obtain sufficient restoration in vivo it was necessary to conjugate the ONs to cell-penetrating peptides. The effect was studied by a battery of different methods. Treatment with intravenous infusions 2 times a week during 3 weeks clearly reverted the phenotype, but longer treatment periods will be needed in order to restore the phenotype into normality. Owing to that the most mature B lineage cells, the plasma cells, do not need BTK, it may be possible, as a first step, to correct splicing in mature B lymphocytes, followed by immunization against infectious pathogens. Subsequently, the responding B cells would undergo maturation into plasma cells and, since human plasma cells can survive for decades, a transient treatment could have long-lasting clinical benefit. Hence our study provides the first evidence that repeated administration of SSO-CPP conjugates can lead to a partial restoration of B-cell development in vivo, demonstrating a functional phenotypic rescue and reinforcing the potential of SSO-based therapies for primary immunodeficiencies.Ethical statement
All animal work was performed according to the protocols approved by the Ethical Committee on Animal Experiments, Stockholm South, Sweden, S175-12, S7-11, 20091309-31/2. The animals were bred and maintained in accordance with Karolinska Institute's guidelines for animal welfare.Author contributions
The project was conceived by CIES and further developed by BB, YEH and RZ. The experiments were mainly carried out by BB and YHE with the help of QW, CH, DKM, DG, OPBW, TL, KEL, AB, MCIK, RM. SEA and MAW were involved in discussions related to the chemistry of oligonucleotides and FA, MJG, CL, DAS and JW synthesized and provided the oligonucleotides. BB and CIES wrote the manuscript and all authors were provided with the possibility to comment on the manuscript.Data availability
The data that support the findings of this study are available from the corresponding author, CIES, upon request. The relevant data supporting this article will be uploaded as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Swedish Cancer Society and the Swedish Medical Research Council. T. L. is supported by Estonian Research Council (PUTJD71 and PRG1882). D. A. S. is supported by the Ministry of Science and Higher Education of the Russian Federation (project of Novosibirsk state University no. FSUS-2025-0012). We thank Wilfried Ellmeier for providing us the BTK/Tec double deficient mouse strain.References
- Z. Dominski and R. Kole, Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides, Proc. Natl. Acad. Sci. U. S. A., 1993, 90(18), 8673–8677 CAS.
- J. S. Charleston, F. J. Schnell, J. Dworzak, C. Donoghue, S. Lewis and L. Chen, et al., Eteplirsen treatment for Duchenne muscular dystrophy: Exon skipping and dystrophin production, Neurology, 2018, 90(24), e2146–e2154 CAS.
- A. Aartsma-Rus and D. R. Corey, The 10th Oligonucleotide Therapy Approved: Golodirsen for Duchenne Muscular Dystrophy, Nucleic Acid Ther., 2020, 30(2), 67–70 CAS.
- J. Kim, C. Hu, C. Moufawad El Achkar, L. E. Black, J. Douville and A. Larson, et al., Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease, N. Engl. J. Med., 2019, 381(17), 1644–1652 CAS.
- A. H. Jonker, E. A. Tataru, H. Graessner, D. Dimmock, A. Jaffe and G. Baynam, et al., The state-of-the-art of N-of-1 therapies and the IRDiRC N-of-1, Nat. Rev. Drug Discovery, 2024, 24, 40–56 Search PubMed.
- R. S. Finkel, C. A. Chiriboga, J. Vajsar, J. W. Day, J. Montes and D. C. De Vivo, et al., Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study, Lancet, 2016, 388(10063), 3017–3026 CAS.
- C. I. E. Smith and R. Zain, Therapeutic Oligonucleotides: State of the Art, Annu. Rev. Pharmacol. Toxicol., 2019, 59, 605–630 CAS.
- B. Bestas, P. M. Moreno, K. E. Blomberg, D. K. Mohammad, A. F. Saleh and T. Sutlu, et al., Splice-correcting oligonucleotides restore BTK function in X-linked agammaglobulinemia model, J. Clin. Invest., 2014, 124(9), 4067–4081 CrossRef CAS PubMed.
- L. Del Pino Molina, M. Wentink, M. van Deuren, P. M. van Hagen, C. I. E. Smith and M. van der Burg, Precursor B-cell development in bone marrow of Good syndrome patients, Clin. Immunol., 2019, 200, 39–42 CrossRef CAS PubMed.
- D. Vetrie, I. Vorechovský, P. Sideras, J. Holland, A. Davies and F. Flinter, et al., The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases, Nature, 1993, 361(6409), 226–233 CrossRef CAS PubMed.
- D. Bearden, M. Collett, P. L. Quan, B. T. Costa-Carvalho and K. E. Sullivan, Enteroviruses in X-Linked Agammaglobulinemia: Update on Epidemiology and Therapy, J. Allergy Clin. Immunol.:Pract., 2016, 4(6), 1059–1065 Search PubMed.
- C. I. E. Smith and A. Berglöf, GeneReviews®, University of Washington, Seattle (WA), 2001 Search PubMed.
- J. D. Thomas, P. Sideras, C. I. Smith, V. Chapman and W. E. Paul, Colocalization of X-linked agammaglobulinemia and X-linked immunodeficiency genes, Science, 1993, 5119, 355–358 CrossRef PubMed.
- D. J. Rawlings, D. C. Saffran, S. Tsukada, D. A. Largaespada, J. C. Grimaldi and L. Cohen, et al., Mutation of unique region of Bruton's tyrosine kinase in immunodeficient XID mice, Science, 1993, 261(5119), 358–361 CrossRef CAS PubMed.
- W. Ellmeier, S. Jung, M. J. Sunshine, F. Hatam, Y. Xu and D. Baltimore, et al., Severe B cell deficiency in mice lacking the tec kinase family members Tec and Btk, J. Exp. Med., 2000, 192(11), 1611–1624 CrossRef CAS PubMed.
- A. Goyenvalle, G. Griffith, A. Babbs, S. El Andaloussi, K. Ezzat and A. Avril, et al., Functional correction in mouse models of muscular dystrophy using exon-skipping tricyclo-DNA oligomers, Nat. Med., 2015, 21(3), 270–275 CrossRef CAS PubMed.
- M. S. Kupryushkin, D. V. Pyshnyi and D. A. Stetsenko, Phosphoryl guanidines: A new type of nucleic acid analogues, Acta Nat., 2014, 6(4), 116–118 CrossRef CAS.
- M. S. Kupryushkin, A. V. Filatov, N. L. Mironova, O. A. Patutina, I. V. Chernikov and E. L. Chernolovskaya, et al., Antisense oligonucleotide gapmers containing phosphoryl guanidine groups reverse MDR1-mediated multiple drug resistance of tumor cells, Mol. Ther.–Nucleic Acids, 2022, 27, 211–226 CrossRef CAS PubMed.
- Y. Su, H. Fujii, E. A. Burakova, B. P. Chelobanov, M. Fujii and D. A. Stetsenko, et al., Neutral and Negatively Charged Phosphate Modifications Altering Thermal Stability, Kinetics of Formation and Monovalent Ion Dependence of DNA G-Quadruplexes, Chem. – Asian J., 2019, 14(8), 1212–1220 CrossRef CAS.
- C. A. Stein, J. B. Hansen, J. Lai, S. Wu, A. Voskresenskiy and A. Høg, et al., Efficient gene silencing by delivery of locked nucleic acid antisense oligonucleotides, unassisted by transfection reagents, Nucleic Acids Res., 2010, 38(1), e3 CrossRef CAS.
- C. A. Betts, S. M. Hammond, H. F. Yin and M. J. Wood, Optimizing tissue-specific antisense oligonucleotide-peptide conjugates, Methods Mol. Biol., 2012, 867, 415–435 CrossRef CAS PubMed.
- T. Lehto, A. Castillo Alvarez, S. Gauck, M. J. Gait, T. Coursindel and M. J. Wood, et al., Cellular trafficking determines the exon skipping activity of Pip6a-PMO in mdx skeletal and cardiac muscle cells, Nucleic Acids Res., 2014, 42(5), 3207–3217 CrossRef CAS.
- A. F. Klein, M. A. Varela, L. Arandel, A. Holland, N. Naouar and A. Arzumanov, et al., Peptide-conjugated oligonucleotides evoke long-lasting myotonic dystrophy correction in patient-derived cells and mice, J. Clin. Invest., 2019, 129(11), 4739–4744 CrossRef CAS PubMed.
- J. M. Lindvall, K. E. Blomberg, J. Väliaho, L. Vargas, J. E. Heinonen and A. Berglöf, et al., Bruton's tyrosine kinase: cell biology, sequence conservation, mutation spectrum, siRNA modifications, and expression profiling, Immunol. Rev., 2005, 203, 200–215 CAS.
- S. M. Hammond, G. Hazell, F. Shabanpoor, A. F. Saleh, M. Bowerman and J. N. Sleigh, et al., Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy, Proc. Natl. Acad. Sci. U. S. A., 2016, 113(39), 10962–10967 CAS.
- F. Shabanpoor, S. M. Hammond, F. Abendroth, G. Hazell, M. J. A. Wood and M. J. Gait, Identification of a Peptide for Systemic Brain Delivery of a Morpholino Oligonucleotide in Mouse Models of Spinal Muscular Atrophy, Nucleic Acid Ther., 2017, 27(3), 130–143 CAS.
- M. J. de Bruijn, J. Rip, E. K. van der Ploeg, L. W. van Greuningen, V. T. Ta and L. P. Kil, et al., Distinct and Overlapping Functions of TEC Kinase and BTK in B Cell Receptor Signaling, J. Immunol., 2017, 198(8), 3058–3068 CAS.
- T. C. Roberts, R. Langer and M. J. A. Wood, Advances in oligonucleotide drug delivery, Nat. Rev. Drug Discovery, 2020, 19(10), 673–694 CAS.
- S. T. Crooke, X. H. Liang, B. F. Baker and R. M. Crooke, Antisense technology: A review, J. Biol. Chem., 2021, 296, 100416 CAS.
- J. Summerton and D. Weller, Morpholino antisense oligomers: design, preparation, and properties, Antisense Nucleic Acid Drug Dev., 1997, 7(3), 187–195 CAS.
- H. Yin, H. M. Moulton, Y. Seow, C. Boyd, J. Boutilier and P. Iverson, et al., Cell-penetrating peptide-conjugated antisense oligonucleotides restore systemic muscle and cardiac dystrophin expression and function, Hum. Mol. Genet., 2008, 17(24), 3909–3918 CAS.
- G. McClorey, H. M. Moulton, P. L. Iversen, S. Fletcher and S. D. Wilton, Antisense oligonucleotide-induced exon skipping restores dystrophin expression in vitro in a canine model of DMD, Gene Ther., 2006, 13(19), 1373–1381 CAS.
- B. Wu, H. M. Moulton, P. L. Iversen, J. Jiang, J. Li and J. Li, et al., Effective rescue of dystrophin improves cardiac function in dystrophin-deficient mice by a modified morpholino oligomer, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(39), 14814–14819 CAS.
- H. Yin, A. F. Saleh, C. Betts, P. Camelliti, Y. Seow and S. Ashraf, et al., Pip5 transduction peptides direct high efficiency oligonucleotide-mediated dystrophin exon skipping in heart and phenotypic correction in mdx mice, Mol. Ther., 2011, 19(7), 1295–1303 CAS.
- C. A. Betts, A. F. Saleh, C. A. Carr, S. M. Hammond, A. M. Coenen-Stass and C. Godfrey, et al., Prevention of exercised induced cardiomyopathy following Pip-PMO treatment in dystrophic mdx mice, Sci. Rep., 2015, 5, 8986 CAS.
- O. A. Patutina, S. K. Gaponova Miroshnichenko, A. V. Senkova, I. A. Savin, D. V. Gladkikh and E. A. Burakova, et al., Mesyl phosphoramidate backbone modified antisense oligonucleotides targeting miR-21 with enhanced in vivo therapeutic potency, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(51), 32370–32379 CAS.
- C. Betts, A. F. Saleh, A. A. Arzumanov, S. M. Hammond, C. Godfrey and T. Coursindel, et al., Pip6-PMO, a new generation of peptide-oligonucleotide conjugates with improved cardiac exon skipping activity for DMD treatment, Mol. Ther.–Nucleic Acids, 2016, 5(9), e273 Search PubMed.
- H. Y. Estupiñán, T. Bouderlique, C. He, A. Berglöf, A. Cappelleri and N. Frengen, et al., In BTK, phosphorylated Y223 in the SH3 domain mirrors catalytic activity, but does not influence biological function, Blood Adv., 2024, 8(8), 1981–1990 Search PubMed.
- B. J. Seymour, S. Singh, H. M. Certo, K. Sommer, B. D. Sather and S. Khim, et al., Effective, safe, and sustained correction of murine XLA using a UCOE-BTK promoter-based lentiviral vector, Nat. Commun., 2024, 15(1), 1243 Search PubMed.
- H. M. Kerns, B. Y. Ryu, B. V. Stirling, B. D. Sather, A. Astrakhan and S. Humblet-Baron, et al., B cell-specific lentiviral gene therapy leads to sustained B-cell functional recovery in a murine model of X-linked agammaglobulinemia, Blood, 2010, 115(11), 2146–2155 CAS.
- T. Lopes-Carvalho and J. F. Kearney, Development and selection of marginal zone B cells, Immunol. Rev., 2004, 197(1), 192–205 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cb00312h |
| This journal is © The Royal Society of Chemistry 2025 |