
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Drug delivery via a 3D electro-swellable conjugated polymer hydrogel†
Ilaria
Abdel Aziz
 ab,
Johannes
Gladisch
a,
Sophie
Griggs
c,
Maximilian
Moser
c,
Hanne
Biesmans
a,
Ana
Beloqui
de,
Iain
McCulloch
c,
Magnus
Berggren
a and
Eleni
Stavrinidou
*a
ab,
Johannes
Gladisch
a,
Sophie
Griggs
c,
Maximilian
Moser
c,
Hanne
Biesmans
a,
Ana
Beloqui
de,
Iain
McCulloch
c,
Magnus
Berggren
a and
Eleni
Stavrinidou
*a
aLaboratory of Organic Electronics, Department of Science and Technology, Linköping University, 601 74 Norrköping, Sweden. E-mail: eleni.stavrinidou@liu.se
bPOLYMAT, University of the Basque Country UPV/EHU, Avenida Tolosa 72, Donostia-San Sebastian, 20018, Gipuzkoa, Spain
cDepartment of Chemistry, Oxford University, Oxford, UK
dPOLYMAT, Applied Chemistry Department, Faculty of Chemistry, University of the Basque Country UPV/EHU, Paseo Manuel de Lardizabal 3, 20018, Donostia-San Sebastian, Spain
eIKERBASQUE, Basque Foundation for Science, Plaza Euskadi 5, Bilbao, 48009, Spain
First published on 26th March 2024
Abstract
Spatiotemporal controlled drug delivery minimizes side-effects and enables therapies that require specific dosing patterns. Conjugated polymers (CP) can be used for electrically controlled drug delivery; however so far, most demonstrations were limited to molecules up to 500 Da. Larger molecules could be incorporated only during the CP polymerization and thus limited to a single delivery. This work harnesses the record volume changes of a glycolated polythiophene p(g3T2) for controlled drug delivery. p(g3T2) undergoes reversible volumetric changes of up to 300% during electrochemical doping, forming pores in the nm-size range, resulting in a conducting hydrogel. p(g3T2)-coated 3D carbon sponges enable controlled loading and release of molecules spanning molecular weights of 800–6000 Da, from simple dyes up to the hormone insulin. Molecules are loaded as a combination of electrostatic interactions with the charged polymer backbone and physical entrapment in the porous matrix. Smaller molecules leak out of the polymer while larger ones could not be loaded effectively. Finally, this work shows the temporally patterned release of molecules with molecular weight of 1300 Da and multiple reloading and release cycles without affecting the on/off ratio.
 Eleni Stavrinidou | Eleni Stavrinidou is Senior Associate Professor of Bioengineering and leader of the Electronic Plants group at Linköping University. She received a PhD in Microelectronics from EMSE (France) in 2014. After a postdoc at Linköping University, she became Assistant Professor and established the Electronic Plants group in 2017. In 2020 she became Associate Professor and Docent in Applied Physics. She received several grants including the Future Research Leaders grant (SSF-Sweden) and the ERC-Staring Grant. Stavrinidou is recipient of the L’ORÉAL-UNESCO FWIS prize in Sweden (2019) and the Tage Erlander Prize from the Royal Swedish Academy of Sciences (2023). Her research interests focus on plant bioelectronics and plant-based biohybrid living materials and devices. |
Introduction
Precision medicine envisages the development of miniaturized drug delivery tools that overcome the limitations of systemic administration enabling targeted and even personalized therapy. In conventional drug delivery systems, drugs are orally administered or injected to the blood stream and usually a high dose is required, as the drugs must cross various physiological barriers before reaching the target tissue.1 If the drugs do not have high specificity for the target tissue, they might cause side effects, which becomes particularly relevant when these substances are toxic. Moreover, many diseases affecting high percentages of population require a specific temporal pattern of drug dosing, such as hormonal regulation, pain relief control or chemotherapy (Table ST1, ESI†). However, conventional system do not provide the required specific temporal pattern for the drug dosing,2–4 All these reasons motivate the need for local drug delivery, in a temporally and spatially controlled manner.To achieve on-demand release, stimuli responsive systems can be used. Temperature, pH or chemical inputs have been widely explored as triggers; however, these stimuli can occur endogenously and therefore hinder high control. Exogenous stimulus on the other hand, such as electric field, magnetic field or light, are promising for highly controlled drug delivery, as they do not interfere with physiological inputs. Electrical stimulation is particularly attractive as it can be easily applied with miniaturized, wearable controllers and can be coupled to sensors for close loop, feedback regulated therapy.1 Electric field-responsive bio-derived hydrogels, such as chitosan, agarose or xanthan gum, have been extensively used for releasing various molecules, drugs, and proteins.5 However, they usually require high operation voltages, far above clinically acceptable values.6 Conjugated polymers (CP) overcome this limitation, as their properties can change electrochemically with low applied voltage (±1 V). CP are organic macromolecules, characterized by a conjugated backbone of alternating single and double bonds, which allows for the formation of overlapping p-orbitals and delocalized π-electrons. The presence of such electrons results in good electronic conduction. Additionally, CPs are mostly biocompatible, and they can be easily processed and integrated into microfabricated circuitry for close loop systems.7 Drug delivery via CPs is based on the electrochemical doping mechanism. When a voltage is applied between the CP and an electrolyte, electronic charges injected in the polymer backbone are compensated by ions from the electrolyte that migrate into the polymer matrix. This results in the increase of the polymer volume due to the incorporation of ions and water molecules as well as changes of polymer chains conformation. When a de-doping potential is applied, charges are extracted from the backbone resulting in the expulsion of the ions and water and consequently volume contraction. The drug therefore can be loaded in the polymer matrix as the dopant or simply by physical entrapment due to the volume changes.
Depending on the polymer composition, the volumetric change that can be achieved varies from a few to hundreds percent, which consequently affects the loaded amount of drug. Polypyrrole (PPY) and poly(3,4-ethylenedioxythiophene)polystyrene sulfonate (PEDOT:PSS) for example, showed a volumetric change up to 35%.8,9 2D thin films resulted in good electrical control of incorporated drugs, showing temporally patterned release. However, this geometry usually enables incorporating a non-clinically relevant drug amount.,10,11 limited by the low volumetric expansion.12 Changing the geometry of the device, i.e., moving from a thin film to a 3D structure, and engineering the micro and nanostructure of the active material increases the charging capacity.13–22 Yet, demonstrations so far are mostly limited to drugs with a low molecular weight, up to 500 Da.23–28 Few reports show the potential of PPY and PEDOT for releasing proteins, such as neutrophin-3 and brain derived growth factors (13 and 30 kDa, respectively).29–32 These drugs were incorporated during the polymerization step and possible reloading in situ was not possible.29,33,34
Recently, we demonstrated that a glycolated polythiophene (p(g3T2)) converts from the solid phase to gel phase when it is electrochemically oxidized.35,36 The glycolated side chains of the polymer chelate and retain water, conferring to this material a stable, soft hydrated gel state once doped. The intercalation of water, together with ions, drives volumetric changes of the bulk polymeric matrix, reaching more than 1000% expansion during the first switching and a reversible volumetric change of 300% in subsequent cycles.35 Molecular dynamics suggested that the large volume change arises from the reorganization of the polymer matrix that forms a porous network to accommodate the water between the hydrophilic side chains, still maintaining the charge percolation path. Indeed, the polymer maintains the π–π stacking also after the pore formation, which ensures a conductive network over the whole extension of the sample. The pore dimension has been estimated to be lower than tens of nanometres and it depends on the oxidation level of the polymer. Additionally, the glycolated side chains increase the stability of the polymer upon charging/discharging cycling, as the polymer maintains its expansion capacity over 300 cycles (ca. 5 hours continuously switched).36,37 While the p(g3T2) has been used as an active layer in tuneable filters38 and in organic electrochemical transistors,37 it has not been explored in drug delivery, despite the unique volumetric expansion and the soft gel transition achieved upon electrochemical doping.
In this work, we harness the large volume changes of p(g3T2) for controlled drug delivery. By coating carbon sponges with p(g3T2) we developed a 3D active matrix that can be loaded with anionic drugs during electrochemical doping and released on-demand during de-doping. Controlled delivery can be achieved for molecules in the 800–6000 Da range, whereas smaller molecules leak out and larger ones cannot be loaded effectively. Finally, we show that the polymer can be reloaded multiple times without affecting the on/off ratio and that it is possible to achieve temporally patterned release on demand.
Results and discussion
We hypothesize that drugs can be loaded in the p(g3T2)-coated sponge during the oxidation cycle, when the material expands, and be released during reduction, when the material contracts (Fig. 1(A)). The loading capacity will be therefore correlated with the amount of available polymer, so we coated substrates with three different geometries with increasingly exposed area, namely a carbon fibre, a metal mesh, and a glassy-carbon sponge. As expected, the 3D geometry results in higher capacitance in comparison with the 1D carbon fibre or the 2D mesh (Fig. 1(B)). For all the measurements, we thus selected the 3D sponge. We previously found that for p(g3T2) coated carbon fibres, the volume reversibly changes by 300% in the [−0.2, 0.5] V window for over 300 cycles, whereas in the [−0.5, −0.2] V window no additional volume changes were observed.35,36 We therefore decided to use the same electrochemical window, selecting 0.5 V as the loading potential and −0.2 V as the releasing potential. Indeed, we observe that at 0.5 V, the polymer volume on the carbon sponge expands, which closes the substrate pores, and at −0.2 V it contracts; however, it does not recover the initial volume, in agreement with previous studies35 (Fig. 1(C)). The same voltage responsive behaviour was recorded on the entire sponge (Fig. S1, ESI†).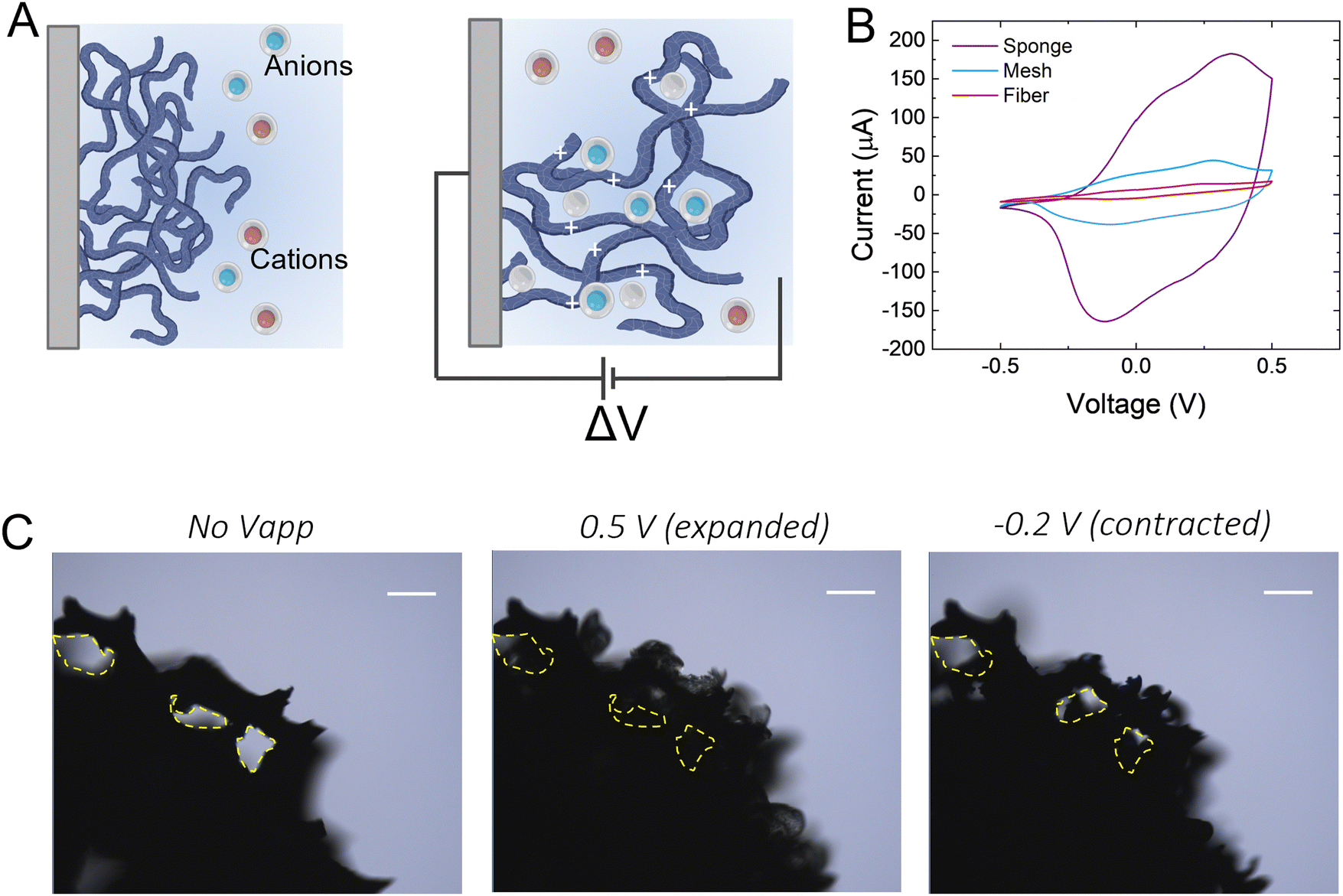 | ||
| Fig. 1 (A) Schematic of the polymer doping/de-doping process in electrolytic environment. Left: Pristine state. Right: Doped state. (B) Cyclic voltammetry of different coated substrates, i.e., a carbon fibre (red line), a metal mesh (blue line) and a carbon sponge (violet line). (C) Representative images of coated p(g3T2)-coated sponge in the pristine state (left figure), expanded state (central figure) and contracted state (right image). Scale bar: 500 μm. Dotted lines highlight the visible pores, which are closed and re-opened when expanding and contracting, respectively. | ||
To investigate whether it is possible to load drugs during the charging/expansion and release it on demand during the discharging/contraction we selected the molecule methyl blue (MB, 799 g mol−1), for its anionic charge, water solubility, and easy colorimetric readout. With microscopy, we qualitatively evaluated the temporal release of the p(g3T2)-coated sponges in two different conditions, namely leaking (no applied bias) and active release (applying −0.2 V), for 10 minutes each (Fig. 2(A)). The first frame during leaking shows that the sponge was loaded with MB, as highlighted by the blue corona around the sponge surface. The latter disappears after 10 minutes of leaking, indicating that part of the loaded drug is not electrostatically bound to the polymer backbone, and it diffuses away from the sponge over time. Additionally, the sponge volume is slightly reduced during the leaking, indicating that in the absence of the doping potential, the polymer does not retain completely the doped state. The first frame of the active release shows the reappearance of the MB at the interface between the p(g3T2)-coated sponge and the electrolyte. After 10 min of active release, the surrounding electrolyte turns to dark blue, a clear indication that the MB is successfully released. This is in contrast with the leaking phase where the electrolyte remained clear and transparent, indicating that very little amount of MB has leaked. The qualitative temporal evolution, evaluated as the number of blue pixels in the image over time, shows that upon application of −0.2 V a clear release peak appears (Fig. 2(B)). This result is further confirmed by quantitatively evaluating the concentration (C) of the dye in the electrolyte spectroscopically with a plate reader at the different time steps (Fig. 1(C)). We define then the active release as the ON state and the leakage as the off state of the polymer. The on/off ratio percentage, defined as (Crelease − Cleaking)/(Cleaking), is 4.6 ± 0.43 for the MB. To test whether the drug release is indeed charge selective, and that the molecules are loaded as dopants, we repeated the same experimental procedure using propidium iodide (PI, 699 g mol−1), a cationic, water soluble, fluorescent dye that has a similar size with respect to the MB, but opposite charge. Microscopy revealed that the PI is loaded into the p(g3T2)-coated sponge, possibly passively driven by the water movement, but cannot be released in a controlled manner. Indeed, we cannot identify a peak in the release dynamics (Fig. 2(E)), nor a statistical difference between the leaking and the active release in the quantitative evaluation (Fig. 2(F)). Interestingly, the concentration of PI is comparable to that of the MB, whereas the on/off ratio for the MB (4.6 ± 0.43) outperforms the one of the PI (0.5 ± 0.12). We speculate that MB is loaded in the polymer matrix both due to electrostatic interactions with the charged backbone, acting as the dopant, and to physical entrapment during volume change facilitated by the water molecules diffusion. For the PI, as it is positively charged, the loading will depend only on the water diffusion, thus controlled release is hindered. Indeed, the fact that the on/off ratio of the MB surpasses the one of the PI supports this interpretation. Therefore, we can conclude that it is possible to load the p(g3T2)-coated sponges both through the doping process and water movements, and that the controlled release is charge selective.
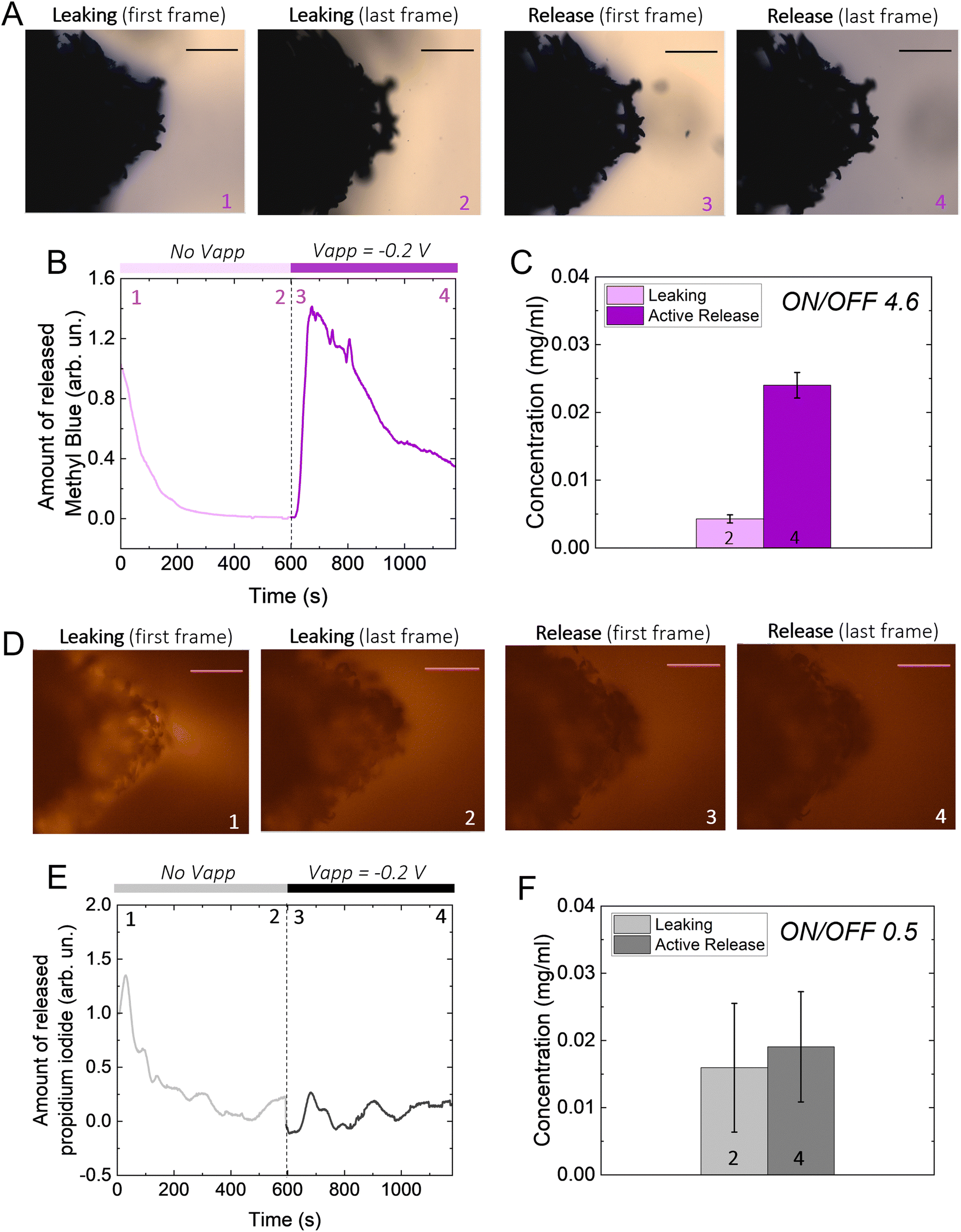 | ||
| Fig. 2 (A), (D) Representative images of release during leaking (1–2) and active release (3–4) for methyl blue (MB) and propidium iodide (PI), respectively (scale bar 500 μm). (B) and (E) Representative analysis of temporal release during leaking and active release for MB and PI, respectively. (C) and (F) Released concentration of MB and PI after 10 minutes of leaking (light magenta and light grey, respectively) and after 10 minutes of active release (magenta and grey, respectively), reported as average ± SD. The on/off ratio is defined as (Crelease − Cleaking)/(Cleaking). The significancy between the bars in panels C and F was evaluated using a t-Student test (0.05 level). C: the means are statistically different, F: the means are not statistically different. | ||
Next, we investigated a range of molecules with different molecular weight, to evaluate whether they can be released in a controlled manner with the p(g3T2)-coated sponges. We studied a wide range of molecules and proteins, namely fluorescein, Direct Red 80, insulin-FITC (conjugated to fluorescein isothiocyanate, FITC) and cyan fluorescent protein (CFP) with 300, 1300, 5800 and 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da, respectively, Fig. 3(A).
000 Da, respectively, Fig. 3(A).
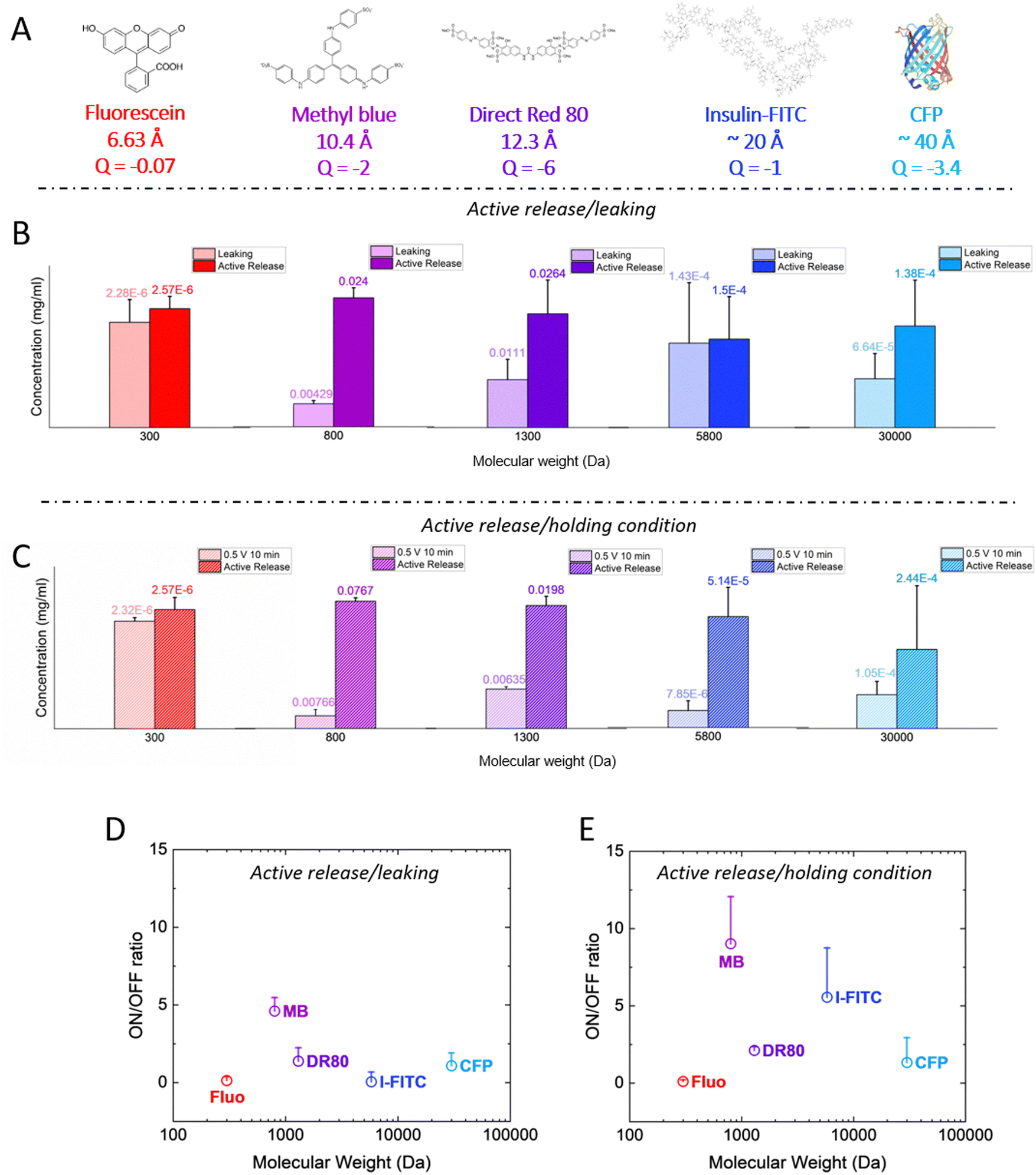 | ||
| Fig. 3 (A) Molecular structure of the different dyes and proteins with increasing size, calculated from chemicalize (https://chemicalize.com/, developed by ChemAxon) for fluorescein, MB and DR. Since the size for the insulin-FITC and the CFP are not reported on the manufacturer's specification sheet, we reported the one of insulin from ref. 39 and we assumed the CFP to be comparable to the GFP.40 We also reported the net charge of the molecules at neutral pH as calculated from Marvin Sketch for fluorescein, MB and DR, from ref. 41 for insulin-FITC and from ProtPi database for the CFP. (B) Concentration of released dye/protein after 10 minutes of leaking (light colours) and after 10 minutes of release (dark colours) for all the different dyes/proteins. The release or leaking values are reported on top of each bar. (C) Concentration of released dye/proteins after 10 minutes of positive bias (holding condition, light colours) and after 10 minutes of release (dark colours). Results are reported as average + standard error. (D) and (E) On/off ratios for the different dyes/proteins for the (D) leaking/active release and for the (E) positive bias/active release cases, respectively. For all panels, the results are reported as average + standard error. | ||
The p(g3T2)-coated sponges were loaded for 10 minutes at +0.5 V, then no bias was applied for 10 minutes to evaluate the leaking and after that, the active release was evaluated by applying −0.2 V for 10 minutes for all the molecules reported except for the insulin-FITC. In that case, we evaluated the release after 20 minutes, as the microscopy images revealed that the protein took longer to be released from the polymer (Fig. S2 and S3, ESI†). The molecule concentration in the solution after leaking and active release showed that only methyl blue and Direct Red 80 could be released in a controlled manner (Fig. 3(B)). For all the other cases, there was no significant difference between leaking and release phase. Since the employed dyes and protein are anionic we hypothesized that by applying a positive bias on the sponge prior the release, the diffusion of the anionic molecules out of the polymer matrix will be prevented,42–45 as their electrostatic interaction with the p(g3T2)-coated sponge will increase. After the loading phase, we therefore kept applying the positive bias across the sponge for 10 minutes and then we applied the release potential. We will refer from now on to the positive bias application as the holding condition. We observed an improvement on the on/off ratio of insulin-FITC, with a statistically significant difference of the active release with respect to the holding condition. For the CFP and the fluorescein, there was no significant difference between the active release and the holding condition (Fig. 3(C)). For the methyl blue and the Direct Red 80, there was no significant improvement, with on/off ratio comparable to the leaking condition. In contrast, for the bigger molecules, the on/off ratio increased up to two-fold with the holding potential with respect to the leaking condition (Fig. 3(D) and (E)). Additionally, the structure of insulin after release is not impaired as evaluated with an insulin-antibody ELISA assay (Fig. S4, ESI†). This indicates that neither the interaction with the sponge nor the application of the voltage impacts the protein structure and integrity.
These results indicate that the combination of electrostatic interactions, water affinity and ion diffusion highlight a sweet spot in terms of controllable drug size. The polymer forms pores where water, Cl anions and drugs are located. Both Cl anions and the anionic drug will compete to compensate for the injected holes, depending on their diffusion coefficient in the polymer matrix, which depends on their size, structure and charge. Additional drug and anions will be loaded in the polymer matrix through entrapment facilitated by water movement. From molecular dynamics simulations,36 we found that the diameter of the pores of p(g3T2) in the expanded state is on average 4 nm, reaching up to 7 nm. As the fluorescein steric hindrance is one order of magnitude lower than the polymer's pores, and its charge is weak, we speculate that it is loaded by expansion, not acting as dopant, and it diffuses from the polymer to the electrolyte because the size of the p(g3T2) pores is bigger compared to the fluorescein dimension. The holding potential does not increase the on/off ratio either, possibly because Fluorescein exhibits a lower negative charge compared to the other molecules, and it is therefore less responsive to the application of a positive bias. The pores size possibly hinders the loading of bigger proteins in the polymer bulk in the employed sponge configuration, which remain confined to the surface of the sponge. Furthermore, we extended the size range up to 66 kDa by evaluating the loading and release of the Bovine serum albumin FITC conjugated protein (BSA-FITC), following the same protocols hereby described (Fig. S6, ESI†). The BSA-FITC mostly leaked out when no bias was applied, while applying the holding potential partially retained the protein. However, no significant on/off ratios were observed. The BSA-FITC is possibly too big with many potential intermolecular interactions,29 which prevents entering the p(g3T2)-coated sponge pores, therefore we speculate that most of the protein was held on the p(g3T2)-coated sponge surface. For this reason, the protein diffused to the electrolyte during both leaking and holding conditions.
We then evaluated the efficiency of the delivery with respect to the loaded amount of drug/protein for those exhibiting the high on/off ratio, i.e. MB, DR80 and insulin-FITC (eff = molreleased/molloaded). The moles of released molecules (molreleased) have been quantified from the absorption or fluorescent measurements. However, it is not possible to quantify the moles of loaded molecules due to the complex geometry of the polymer on the carbon sponge and the presence of competing anions (Cl). From the current we calculated the moles of molecules contributing to the current during release and then we calculated the ratio that is due to the released drugs. Then assuming that this ratio is the same for the loading we estimated the moles of loaded molecules (the details of the calculations are reported in the ESI†).
The release efficiency is quite low for all tested molecules (Table 1). We hypothesize that during release in a single step, with a constant voltage, the loaded drugs compete with the smaller Cl anions (which have a higher diffusion coefficient in water compared to the other molecules, Fig. S5, ESI†) and therefore not all incorporated drug is released. The efficiency for the holding condition is comparable or even smaller than the one of the leaking condition. In the holding condition, after loading of the sponge, we maintain the positive bias with the sponge exposed to a fresh KCl solution that may result into more Cl anions entering the sponge to maintain the oxidized state that could reduce even further the efficiency.
| Efficiency (leaking condition)% | Efficiency (holding condition)% | |
|---|---|---|
| Methyl blue | 37.78 | 25.46 |
| Direct Red 80 | 10.34 | 16.97 |
| Insulin FITC | 45.06 | 35.77 |
One of the main advantages of voltage-responsive materials is the possibility to spatio-temporally pattern the drug release. We therefore explored our system for temporally patterned release using pulsed addressing. Voltage pulses of 5 or 30 s were applied for the release while no bias was applied in between. We used the Direct Red 80 to characterize the pulsed release, for its higher molecular weight in the window of the controlled molecules. For each pulse, the voltage-controlled release is higher than the passive leakage (Fig. 4(A) and (B)). Furthermore, the released amount decreased with increasing pulse number (Fig. 4(C)). Interestingly, the final concentration achieved after 5 pulses is comparable between the two pulses lengths. These observations can be explained by considering the charge/discharge curves of the p(g3T2) (Fig. S7, ESI†). The discharging of the polymer has a time constant of ≈12 s, thus during the first pulse of 30 s, the polymer has mostly discharged and most of the release occurred during this first pulse. In the consecutive pulses a smaller amount of Direct Red 80 is released until the polymer is completely discharged. For the 5 s pulses, most of the release occurs during the first two pulses, according to the time constant of the polymer (Fig. 4(C) and Fig. S7, ESI†).
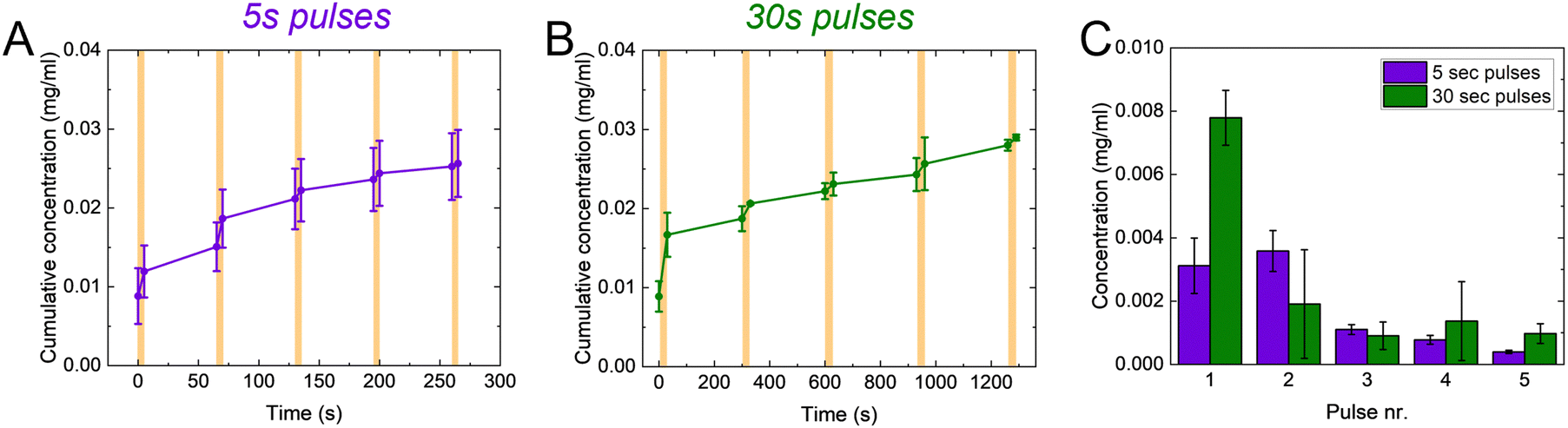 | ||
| Fig. 4 (A) and (B) cumulative concentration during pulsed release from p(g3T2)-coated sponges for 5 seconds and 30 pulses length, respectively. (C) Concentration released during each of the 5 pulses. The results are shown as average ± SE. | ||
When targeting the control of relatively large molecules (MW > 500 Da), conjugated polymers are usually loaded with the molecule of interest during the polymerization process. This strongly limits their applicability since it is not possible to reload the polymer through the same process (as the polymerization already occurred), and loading through electrochemical doping is limited by low delivery doses, as discussed in the Introduction. The p(g3T2)-coated sponge, instead, is loaded through the charging and releases the drug during the discharging, potentially enabling on site reloading. Indeed, the p(g3T2)-coated sponges can be reloaded multiple times from a fresh solution, with no significant difference between successive loadings (Fig. 5(A)). The release performances of the p(g3T2)-coated sponge are not affected by the reloading, as there are no differences between the holding condition and the release, nor between the pulsed release (Fig. 5(B), (C) and Fig. S8, ESI†).
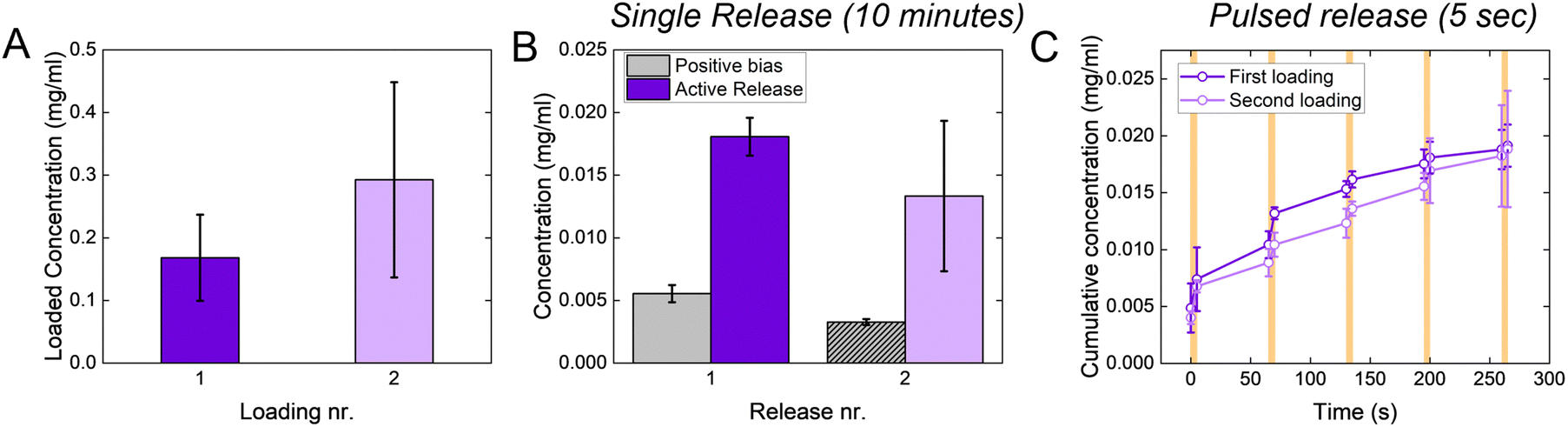 | ||
| Fig. 5 (A) Loaded amount of Direct Red 80 in the first and the second loading events. (B) Concentration released after the first and the second loading event, as a single release. (C) Cumulative concentration released during 5 pulses of 5 seconds each, after the first and the second loading event. In all the panels, the dark purple refers to the first loading event and the light purple refers to the second. The results are shown as average ± SD. | ||
Conclusions
In conclusion we demonstrated that the p(g3T2)-coated sponges can be used for voltage-controlled drug delivery of molecules/proteins in the size range of 800–6000 Da. p(g3T2) becomes gelled in the oxidized state forming nm-sized pores,35,36 facilitating the molecule entrapment. The loading process is therefore limited by the size of the molecule, but not by its charge, as it relies on a combination of electrostatic interactions between molecules and polymer as well as electrolyte intercalation into the polymer matrix. Controlled release on the other hand is achieved only for negatively charged molecules within a specific size range, as positively charged molecules leak out from the matrix. We also demonstrated patterned release of molecules with molecular weight of 1300 Da by applying voltage pulses. The p(g3T2)-coated sponges can be easily re-loaded maintaining their controlled release performance. The possibility to re-load the material through electrochemical cycling provides a key to long-term implantable devices, where a small, exchangeable reservoir can be integrated. To the best of our knowledge, p(g3T2) is one of the few single component conjugated polymers used for drug delivery enabling controlled release of molecules in the size range of 800–6000 g mol−1. Furthermore thanks to its affinity with water, the p(g3T2) exhibits a lower elastic modulus (0.5–80 MPa46) compared to PEDOT:PSS (1–5 GPa47) and PPY (0.5–1 GPa48), reducing the mechanical mismatch between the polymer and the biotic interface without the need of further fabrication steps. Finally, the polymer biocompatibility has been verified by means of live/dead cell stain and alamar blue test (Fig. S9, ESI†), highlighting the potential of the material for drug delivery applications. Overall, the size range of the molecules that can be released, together with the possibility to control the release remotely and on demand through the application of an external bias opens the way to temporally pattern the delivery of payloads, enabling therapies such as those based on hormones and growth factors.Materials and methods
Synthesis of glycolated polymers
The p(g3T2) polymer was synthesized according to previously reported procedures.35–37Electrochemical measurements
Glassy carbon substrates were purchased from Redox.me. The p(g3T2) was drop casted on the substrates from a 10 mg ml−1 Chloroform (Sigma Aldrich) solution. The coated samples were then connected to an Autolab PGSTAT101 in a standard three electrodes electrochemical cell as working electrode (WE), together with a metal mesh (screen printing meshes 80μm pore size, coated screens Scandinavia AB) as a counter electrode (CE) and a quasi-reference Ag/AgCl pellet (RE). The three electrodes were immersed in a KCl (Sigma Aldrich) 0.01 M solution, assembled in a poly(dimethylsiloxane) (PDMS) well and mounted under a fluorescence microscope Nikon Ni-E fluorescence microscope. Reduction/oxidation switches of 30 s −0.2 V/0.5 V for a complete duty cycle of 1 minute were carried out for all the samples as first measurement. To load the dyes and proteins in the coated sponges, we employed a 900 s long chronoamperometry applying a constant voltage of 0.5 V (oxidation potential). The dyes release was carried out applying −0.2 V for 600 s (reduction potential). For both chronoamperometries the sampling rate was 1 s. Carbon monofilaments (Specialty Materials, Lowell, MA, USA, diameter 34.5 ± 2.5 μm) were coated according to the following methodology. A glass micropipette was filled with a chloroform solution of p(g3T2). A carbon fibre was fixed on a rotating motor and dipped in the solution and slowly extracted, allowing the evaporation of the solvent and the casting of the material. A metal mesh (screen printing meshes 80μm pore size, coated screens Scandinavia AB) was dip coated in a chloroform solution of p(g3T2) and then shaken into a vial to remove the excess material. The cyclic voltammetries were carried out at a scan rate of 5 mV s−1 for 3 cycles.
Release − CLeaking)/CLeaking.
Cell culture
PC-12 (ECACC Cat# 88022401, RRID:CVCL 0481) cells were purchased from Sigma Aldrich. PC-12 cells are rat pheochromocytoma cells, that are extensively studied and therefore well characterized as a neuronal model. They were cultured in Gibco's RPMI 1640 with L-glutamine (Thermo Fisher Scientific, cat. no. 21875034), supplemented with 10% Gibco's horse serum (HS, Thermo Fisher Scientific, cat. no. 16050122) and 5% Gibco's fetal bovine serum (FBS, Thermo Fisher Scientific, cat. no. 26140079). The cells were grown at 37 °C in a humidified atmosphere with 5% CO2 and were passaged every other day.Toxicity tests
Two different toxicity tests were used in this manuscript. First, a fluorescent live/dead assay was performed. Three p(g3T2) coated wells and three untreated wells were coated at 37 °C with Collagen IV. After, 200000 cells were seeded. After 24 hours, the cells were tested with a fluorescent live/dead viability kit for mammalian cells (Thermo Fisher Scientific, cat. no. L3224) according to manufacturer's instructions. The cells were imaged using a Zoe fluorescent cell imager (BioRad Laboratories). Statistical analysis (i.e., Mann Whitney statistic) was performed using GraphPad Prism. We also performed an Alamar Blue test. For this test, 200000 cells per ml were seeded to both p(g3T2) coated wells and untreated control ones. After 24 hours, an alamarBlue™ Cell Viability test (Thermo Fisher, cat. no. DAL1025) was performed according to manufacturer's instructions, and a BioTek Synergy H1m plate reader was used for the fluorescence readout. For both tests, two biological replicates with three technical replicates each were employed.
Conflicts of interest
The authors declare that there are no conflicts of interest related to the content of this manuscript.References
- D. Liu, F. Yang, F. Xiong and N. Gu, The Smart Drug Delivery System and Its Clinical Potential, Theranostics, 2016, 6(9), 1306–1323, DOI:10.7150/thno.14858.
- G. Jeon, Y. Yang, J. Byun and J. K. Kim, Electrically Actuatable Smart Nanoporous Membrane for Pulsatile Drug Release, Nano Lett., 2011, 11, 1284–1288, DOI:10.1021/nl104329y.
- G. Adilene, et al., Chondroitin/polypyrrole nanocomposite hydrogels for the accurate release of 5-fluorouracil by electrical stimulation, Polym. Adv. Technol., 2022, 621–633, DOI:10.1002/pat.5915.
- A. Z. Khalifa, et al., Recent advances in remotely controlled pulsatile drug delivery systems, J. Adv. Pharm. Technol. Res., 2022, 13(2), 77, DOI:10.4103/JAPTR.JAPTR_330_21.
- S. Ramanathan and L. H. Block, The use of chitosan gels as matrices for electrically-modulated drug delivery, J. Controlled Release, 2001, 70(1–2), 109–123, DOI:10.1016/S0168-3659(00)00333-3.
- B. C. Thompson, et al., Conducting polymers, dual neurotrophins and pulsed electrical stimulation - Dramatic effects on neurite outgrowth, J. Controlled Release, 2010, 141(2), 161–167, DOI:10.1016/J.JCONREL.2009.09.016.
- J. Rivnay, R. M. Owens and G. G. Malliaras, The rise of organic bioelectronics, Chem. Mater., 2014, 26(1), 679–685, DOI:10.1021/CM4022003/ASSET/IMAGES/MEDIUM/CM-2013-022003_0004.GIF.
- B. Tandon, A. Magaz, R. Balint, J. J. Blaker and S. H. Cartmell, Electroactive biomaterials: Vehicles for controlled delivery of therapeutic agents for drug delivery and tissue regeneration, Adv. Drug Delivery Rev., 2018, 129, 148–168, DOI:10.1016/J.ADDR.2017.12.012.
- A. Puiggalí-Jou, L. J. del Valle and C. Alemán, Drug delivery systems based on intrinsically conducting polymers, J. Controlled Release, 2019, 309, 244–264, DOI:10.1016/J.JCONREL.2019.07.035.
- T. A. Desai, et al., Nanoporous Implants for Controlled Drug Delivery, BioMEMS Biomed. Nanotechnol., 2006, 263–286, DOI:10.1007/978-0-387-25844-7_15.
- C. A. R. Chapman, E. A. Cuttaz, J. A. Goding and R. A. Green, Actively controlled local drug delivery using conductive polymer-based devices, Appl. Phys. Lett., 2020, 116(1), 10501, DOI:10.1063/1.5138587/38172.
- R. Green, M. R. Abidian, R. Green and M. R. Abidian, Conducting Polymers for Neural Prosthetic and Neural Interface Applications, Adv. Mater., 2015, 27(46), 7620–7637, DOI:10.1002/ADMA.201501810.
- J. Pokki, et al., Electroplated porous polypyrrole nanostructures patterned by colloidal lithography for drug-delivery applications, Nanoscale, 2012, 4(10), 3083–3088, 10.1039/C2NR30192J.
- X. Luo and X. T. Cui, Sponge-like nanostructured conducting polymers for electrically controlled drug release, Electrochem. Commun., 2009, 11(10), 1956–1959, DOI:10.1016/J.ELECOM.2009.08.027.
- A. Seyfoddin, A. Chan, W. T. Chen, I. D. Rupenthal, G. I. N. Waterhouse and D. Svirskis, Electro-responsive macroporous polypyrrole scaffolds for triggered dexamethasone delivery, Eur. J. Pharm. Biopharm., 2015, 94, 419–426, DOI:10.1016/J.EJPB.2015.06.018.
- X. Luo and X. T. Cui, Electrochemically controlled release based on nanoporous conducting polymers, Electrochem. Commun., 2009, 11(2), 402–404, DOI:10.1016/J.ELECOM.2008.11.052.
- E. Shamaeli and N. Alizadeh, Nanostructured biocompatible thermal/electrical stimuli-responsive biopolymer-doped polypyrrole for controlled release of chlorpromazine: Kinetics studies, Int. J. Pharm., 2014, 472(1–2), 327–338, DOI:10.1016/J.IJPHARM.2014.06.036.
- M. Sharma, G. I. N. Waterhouse, S. W. C. Loader, S. Garg and D. Svirskis, High surface area polypyrrole scaffolds for tunable drug delivery, Int. J. Pharm., 2013, 443(1–2), 163–168, DOI:10.1016/J.IJPHARM.2013.01.006.
- C. Kleber, K. Lienkamp, J. Rühe and M. Asplund, Electrochemically Controlled Drug Release from a Conducting Polymer Hydrogel (PDMAAp/PEDOT) for Local Therapy and Bioelectronics, Adv. Healthcare Mater., 2019, 8(10), 1801488, DOI:10.1002/ADHM.201801488.
- S. Jiang, et al., Enhanced drug loading capacity of polypyrrole nanowire network for controlled drug release, Synth. Met., 2013, 163(1), 19–23, DOI:10.1016/J.SYNTHMET.2012.12.010.
- R. A. Green, N. H. Lovell, G. G. Wallace and L. A. Poole-Warren, Conducting polymers for neural interfaces: challenges in developing an effective long-term implant, Biomaterials, 2008, 29(24–25), 3393–3399, DOI:10.1016/J.BIOMATERIALS.2008.04.047.
- Y. Yang, et al., Self-Powered Controllable Transdermal Drug Delivery System, Adv. Funct. Mater., 2021, 31(36), 2104092, DOI:10.1002/adfm.202104092.
- M. R. Abidian, D. H. Kim and D. C. Martin, Conducting-polymer nanotubes for controlled drug release, Adv. Mater., 2006, 18(4), 405–409, DOI:10.1002/ADMA.200501726.
- M. Antensteiner, et al., Conducting Polymer Microcups for Organic Bioelectronics and Drug Delivery Applications, Adv. Mater., 2017, 29(39), 1702576, DOI:10.1002/ADMA.201702576.
- D. Svirskis, B. E. Wright, J. Travas-Sejdic, A. Rodgers and S. Garg, Development of a Controlled Release System for Risperidone Using Polypyrrole: Mechanistic Studies, Electroanalysis, 2010, 22(4), 439–444, DOI:10.1002/ELAN.200900401.
- D. Uppalapati, B. J. Boyd, S. Garg, J. Travas-Sejdic and D. Svirskis, Conducting polymers with defined micro- or nanostructures for drug delivery, Biomaterials, 2016, 111, 149–162, DOI:10.1016/J.BIOMATERIALS.2016.09.021.
- D. Svirskis, B. E. Wright, J. Travas-Sejdic, A. Rodgers and S. Garg, Evaluation of physical properties and performance over time of an actuating polypyrrole based drug delivery system, Sens. Actuators, B, 2010, 151(1), 97–102, DOI:10.1016/J.SNB.2010.09.042.
- B. Yao, et al., CommuniCation 1700974 (1 of 7) Ultrahigh-Conductivity Polymer Hydrogels with Arbitrary Structures, Adv. Mater., 2017, 29, 1700974, DOI:10.1002/adma.201700974.
- D. H. Kim, S. M. Richardson-Burns, J. L. Hendricks, C. Sequera and D. C. Martin, Effect of Immobilized Nerve Growth Factor on Conductive Polymers: Electrical Properties and Cellular Response, Adv. Funct. Mater., 2007, 17(1), 79–86, DOI:10.1002/ADFM.200500594.
- B. C. Thompson, et al., Optimising the incorporation and release of a neurotrophic factor using conducting polypyrrole, J. Controlled Release, 2006, 116(3), 285–294, DOI:10.1016/J.JCONREL.2006.09.004.
- R. T. Richardson, et al., The effect of polypyrrole with incorporated neurotrophin-3 on the promotion of neurite outgrowth from auditory neurons, Biomaterials, 2007, 28(3), 513–523, DOI:10.1016/J.BIOMATERIALS.2006.09.008.
- R. T. Richardson, et al., Polypyrrole-coated electrodes for the delivery of charge and neurotrophins to cochlear neurons, Biomaterials, 2009, 30(13), 2614–2624, DOI:10.1016/J.BIOMATERIALS.2009.01.015.
- C. Boehler, et al., Actively controlled release of Dexamethasone from neural microelectrodes in a chronic in vivo study, Biomaterials, 2017, 129, 176–187, DOI:10.1016/J.BIOMATERIALS.2017.03.019.
- C. Boehler, F. Oberueber and M. Asplund, Tuning drug delivery from conducting polymer films for accurately controlled release of charged molecules, J. Controlled Release, 2019, 304, 173–180, DOI:10.1016/J.JCONREL.2019.05.017.
- J. Gladisch, et al., Reversible Electronic Solid–Gel Switching of a Conjugated Polymer, Adv. Sci., 2020, 7(2), 1901144, DOI:10.1002/advs.201901144.
- M. Moser, et al., Controlling Electrochemically Induced Volume Changes in onjugated Polymers by Chemical Design: from Theory to Devices, Adv. Funct. Mater., 2021, 31(26), 2100723, DOI:10.1002/ADFM.202100723.
- M. Moser, et al., Side Chain Redistribution as a Strategy to Boost Organic Electrochemical Transistor Performance and Stability, Adv. Mater., 2020, 32(37), 2002748, DOI:10.1002/adma.202002748.
- J. Gladisch, et al., An Electroactive Filter with Tunable Porosity Based on Glycolated Polythiophene, Small Science, 2022, 2(4), 2100113, DOI:10.1002/SMSC.202100113.
- P. R. Shorten, C. D. McMahon and T. K. Soboleva, Insulin Transport within Skeletal Muscle Transverse Tubule Networks, Biophys. J., 2007, 93(9), 3001, DOI:10.1529/BIOPHYSJ.107.107888.
- M. A. Hink, et al., Structural dynamics of green fluorescent protein alone and fused with a single chain Fv protein, J. Biol. Chem., 2000, 275(23), 17556–17560, DOI:10.1074/jbc.M001348200.
- O. Wintersteiner and H. A. Abramson, The Isoelectric Point Of Insulin Electrical Properties Of Adsorbed And Crystalline Insulin, J. Biol. Chem., 1933, 99(3), 741–753, DOI:10.1016/S0021-9258(18)76023-7.
- N. Alizadeh and E. Shamaeli, Electrochemically controlled release of anticancer drug methotrexate using nanostructured polypyrrole modified with cetylpyridinium: Release kinetics investigation, Electrochim. Acta, 2014, 130, 488–496, DOI:10.1016/J.ELECTACTA.2014.03.055.
- E. Shamaeli and N. Alizadeh, Functionalized gold nanoparticle-polypyrrole nanobiocomposite with high effective surface area for electrochemical/pH dual stimuli-responsive smart release of insulin, Colloids Surf., B, 2015, 126, 502–509, DOI:10.1016/J.COLSURFB.2015.01.003.
- D. Esrafilzadeh, J. M. Razal, S. E. Moulton, E. M. Stewart and G. G. Wallace, Multifunctional conducting fibres with electrically controlled release of ciprofloxacin, J. Controlled Release, 2013, 169(3), 313–320, DOI:10.1016/J.JCONREL.2013.01.022.
- D. Svirskis, B. E. Wright, J. Travas-Sejdic, A. Rodgers and S. Garg, Development of a Controlled Release System for Risperidone Using Polypyrrole: Mechanistic Studies, Electroanalysis, 2010, 22(4), 439–444, DOI:10.1002/ELAN.200900401.
- I. A. Aziz, et al., Electrochemical modulation of mechanical properties of glycolated polythiophenes, Mater. Horiz., 2024, 5 10.1039/D3MH01827J.
- S. Benaglia, S. Drakopoulou, F. Biscarini and R. Garcia, In operando nanomechanical mapping of PEDOT:PSS thin films in electrolyte solutions with bimodal AFM, Nanoscale, 2022, 14(38), 14146–14154, 10.1039/D2NR02177C.
- A. Kaynak, L. Rintoul and G. A. George, Change of mechanical and electrical properties of polypyrrole films with dopant concentration and oxidative aging, Mater. Res. Bull., 2000, 35(6), 813–824, DOI:10.1016/S0025-5408(00)00280-4.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tb02592f |
| This journal is © The Royal Society of Chemistry 2024 |