
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
δ-Bonding modulates the electronic structure of formally divalent nd1 rare earth arene complexes†
Ross E. MacKenzie ab,
Tomáš Hajdubd,
John A. Seedab,
George F. S. Whiteheadb,
Ralph W. Adamsb,
Nicholas F. Chiltonbc,
David Collisonbd,
Eric J. L. McInnesbd and
Conrad A. P. Goodwin*ab
ab,
Tomáš Hajdubd,
John A. Seedab,
George F. S. Whiteheadb,
Ralph W. Adamsb,
Nicholas F. Chiltonbc,
David Collisonbd,
Eric J. L. McInnesbd and
Conrad A. P. Goodwin*ab
aCentre for Radiochemistry Research, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
bDepartment of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
cResearch School of Chemistry, The Australian National University, Sullivans Creek Road, Canberra, 2601, Australia
dPhoton Science Institute, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
First published on 5th August 2024
Abstract
Landmark advances in rare earth (RE) chemistry have shown that divalent complexes can be isolated with non-Aufbau 4fn{5d/6s}1 electron configurations, facilitating remarkable bonding motifs and magnetic properties. We report a series of divalent bis-tethered arene complexes, [RE(NHAriPr6)2] (2RE; RE = Sc, Y, La, Sm, Eu, Tm, Yb; NHAriPr6 = {N(H)C6H3-2,6-(C6H2-2,4,6-iPr3)2}). Fluid solution EPR spectroscopy gives giso < 2.002 for 2Sc, 2Y, and 2La, consistent with formal nd1 configurations, calculations reveal metal–arene δ-bonding via mixing of nd(x2−y2) valence electrons into arene π* orbitals. Experimental and calculated EPR and UV-Vis-NIR spectroscopic properties for 2Y show that minor structural changes markedly alter the metal d(x2−y2) contribution to the SOMO. This contrasts 4fn{5d/6s}1 complexes where the valence d-based electron resides in a non-bonding orbital. Complexes 2Sm, 2Eu, 2Tm, and 2Yb contain highly-localised 4fn+1 ions with no appreciable metal–arene bonding by density functional calculations. These results show that the physicochemical properties of divalent rare earth arene complexes with both formal nd1 and 4fn+1 configurations are nuanced, may be controlled through ligand modification, and require a multi-pronged experimental and theoretical approach to fully rationalise.
Introduction
A hallmark of trivalent lanthanide ions is the relative insensitivity of valence 4f-orbitals to the coordination environment. While this gives rise to useful physical and optical properties, such as their narrow optical emission profiles and large magnetic moments from their unquenched orbital angular momentum, the weakness of this interaction also inhibits the extent to which their properties may be tailored through molecular design. Advancements in divalent molecular rare earth and lanthanide (Sc, Y, La–Lu; collectively Ln henceforth) chemistry have shown that in coordination environments such as [Ln(CpR)3]− or [Ln(CpR)2] (CpR = substituted cyclopentadienide ligands), Ln(II) complexes can be isolated with 4fn5d1 or 4fn{5d/6s}1 valence electron configurations, at least for La–Lu, while Sc and Y necessarily give 3d1 or 4d1 ions.1–23 Further work shows these non-Aufbau ions can be exploited as potential qubit candidates,15–17 exhibit record-setting magnetic properties,18–20 and have produced the first examples of molecular Ln–Ln bonding outside of endohedral fullerenes.20–22 Similar advancements have also been made with actinide elements.24 However, with few exceptions, design strategies employed to isolate examples of these ions use geometries (e.g. C3h or D5h) that minimise or forbid, by symmetry, the mixing of 5d (or 5d/6s hybridised) valence electrons with ligand orbitals. This is useful in some applications,15–17 but in analogy to the poor tunability of 4f-orbitals in Ln(III) ions, it limits the extent to which the electronic structures of 4fn5d1 or 4fn{5d/6s}1 Ln(II) complexes may be controlled through ligand design.Using ligand field principles commonplace in the d-block that also apply to 4fn5d1 Ln(II) ions, we may instead target molecular designs where the electronic structure and physicochemical properties are more sensitive to changes in the coordination environment. Arene ligands provide a promising route towards these goals due to their ability to act as symmetry-allowed donors into vacant d-orbitals, and further stabilise lower oxidation states through back donation.25–32 Indeed, the only examples of formally zero-valent rare earth complexes are within [M(η6-C6R6)2] frameworks.33–37
Herein, we present a series of structurally analogous bis-tethered arene divalent rare earth complexes of the form [M(NHAriPr6)2] (2M, M = Sc, Y, La, Sm, Eu, Tm, Yb; NHAriPr6 = {N(H)C6H3-2,6-(C6H2-2,4,6-iPr3)2}) – the synthesis and some properties of 2Y have been reported previously.38 In all cases, characterisation using SQUID magnetometry, solid and solution phase EPR spectroscopy, UV-Vis-NIR spectroscopy, density functional theory (DFT), and complete active space self-consistent field (CASSCF) calculations support the description of these as formal Ln(II) complexes, demonstrating that this framework is robust across a range of Ln(II) ion sizes and (formal) reduction potentials. While all seven divalent complexes display close metal–arene contacts, only 2Sc, 2Y, and 2La show mixing of a metal valence nd(x2−y2) orbital with the arene π* to give δ-bonding interactions. Complexes 2Sm, 2Eu, 2Tm, and 2Yb instead adopt metal-localised 4fn+1 configurations in accordance with their large Ln(II) 4fn+1 → 4fn5d1 promotional energies.39,40 Solid and solution phase UV-Vis-NIR and EPR for 2Y, combined with DFT calculations, reveals that the balance of metal vs. arene-centred spin-density is sensitive to small structural changes between these phases, suggesting the properties of divalent rare earth complexes with nd1 may be tuned through ligand design.
Results and discussion
Synthesis
A reductive route was pursued for the synthesis of 2M (M = Sc, Y, La, and Tm; Scheme 1A), which began with the synthesis of trivalent [M(NHAriPr6)2(I)] (1M, M = Sc, Y, La, Tm) precursors. Complexes 1M were synthesised by salt elimination between KNHAriPr6 (ref. 41 and 42) and the relevant rare earth tri-iodide salt [MI3(THF)n] (n = 4, M = La; n = 3.5, M = Y, Tm; n = 3, M = Sc) in Et2O.43 Workup and crystallisation from toluene gave fair to good crystalline yields (47–62%). 1H NMR spectroscopy of diamagnetic 1Sc, 1Y, and 1La in d6-benzene show the M⋯C6-arene interaction is dynamic in solution and that all four Tripp groups are in exchange (see ESI† for detailed assignments).38 | ||
| Scheme 1 Synthesis of (Route A) [M(NHAriPr6)2(I)] (1M, M = Sc, Y, La, Tm), and (Route B) [M(NHAriPr6)2] (M = Sc, Y, La, Sm, Eu, Tm, Yb). Tripp = {C6H2-2,4,6-iPr3}. | ||
Reduction (KC8, 1.2 equiv.) of 1M (M = Sc, Y, La, Tm) in Et2O gave, after workup and crystallization from Et2O, dark crystals of 2M in fair crystalline yields (shown as % yield ahead): 2Sc (red, 53%), 2Y (red/green, 55%), 2La (red/brown, 45%), and 2Tm (red/brown, 65%). Salt elimination between KNHAriPr6 and MI2(THF)2 (M = Sm, Eu, Yb)44 in Et2O gave poor to excellent isolated crystalline yields of 2Sm (69%), 2Eu (20%), and 2Yb (82%) (Scheme 1B). Attempts to synthesise 1Sm and 1Yb using [MI3(THF)n] precursors were unsuccessful, leading to intractable mixtures (see ESI†), and we did not attempt the synthesis of 1Eu.
While the 1H NMR spectra of 1Sc, 1Y, and 1La showed the metal-bound and “terminal” {C6H2-2,4,6-iPr3} (Tripp) groups were in exchange at room temperature, the spectrum of diamagnetic 2Yb in d6-benzene shows six doublets for the CH3-iPr groups, along with a single N(H) resonance. Thus, 2Yb is C2 symmetric in solution and the Tripp groups do not appreciably exchange at room temperature. This does not appear to be due to steric hindrance as the Sc(III) in 1Sc is smaller than Yb(II) (6-coordinate radii: Yb(II), 0.868 Å; Sc(III), 0.745 Å). A variable temperature 1H NMR study in d8-toluene shows the CH3-iPr peaks begin to coalesce at 308 K, but full equilibrium is not reached until ca. 358 K (Fig. S40 and S41†). The 171Yb–1H HMBC NMR spectrum gave two cross-peaks at δ171Yb = −83 ppm (δ1H = 3.53 and 7.02 ppm), showing the 171Yb couples to both the anilido proton and the Tripp 3,5-CH groups. The 1H NMR spectra of paramagnetic 2M complexes were uninformative, see the ESI† for all NMR spectra.
Molecular structures
Single crystal X-ray diffraction studies on 1M (M = Sc, Y, La, Tm) show all crystallize in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (Z′ = 1) and are pseudo three-coordinate – see Fig. 1. In 1La, the metal is sandwiched almost equally between two Tripp groups (La⋯C6-centroid = 2.8714(12) and 2.8783(12) Å; La⋯N2I plane deviation = 0.0042(15) Å). Conversely, 1Sc and 1Y have a single Tripp group close to the metal (M⋯C6-centroid: 1Sc, 2.3391(8) Å; and 1Y, 2.4949(9) Å) and hence are trigonal pyramidal. The structure of 1Y is comparable to recently reported [Y(NHAriPr6)2(Cl)].38 See the ESI† for the structure of 1Tm, and a comparison of 1M complexes. In the following sections, we shall continue to use the term “C6-centroid”, noting it is not strictly appropriate to define a centroid for non-planar groups.
(Z′ = 1) and are pseudo three-coordinate – see Fig. 1. In 1La, the metal is sandwiched almost equally between two Tripp groups (La⋯C6-centroid = 2.8714(12) and 2.8783(12) Å; La⋯N2I plane deviation = 0.0042(15) Å). Conversely, 1Sc and 1Y have a single Tripp group close to the metal (M⋯C6-centroid: 1Sc, 2.3391(8) Å; and 1Y, 2.4949(9) Å) and hence are trigonal pyramidal. The structure of 1Y is comparable to recently reported [Y(NHAriPr6)2(Cl)].38 See the ESI† for the structure of 1Tm, and a comparison of 1M complexes. In the following sections, we shall continue to use the term “C6-centroid”, noting it is not strictly appropriate to define a centroid for non-planar groups.
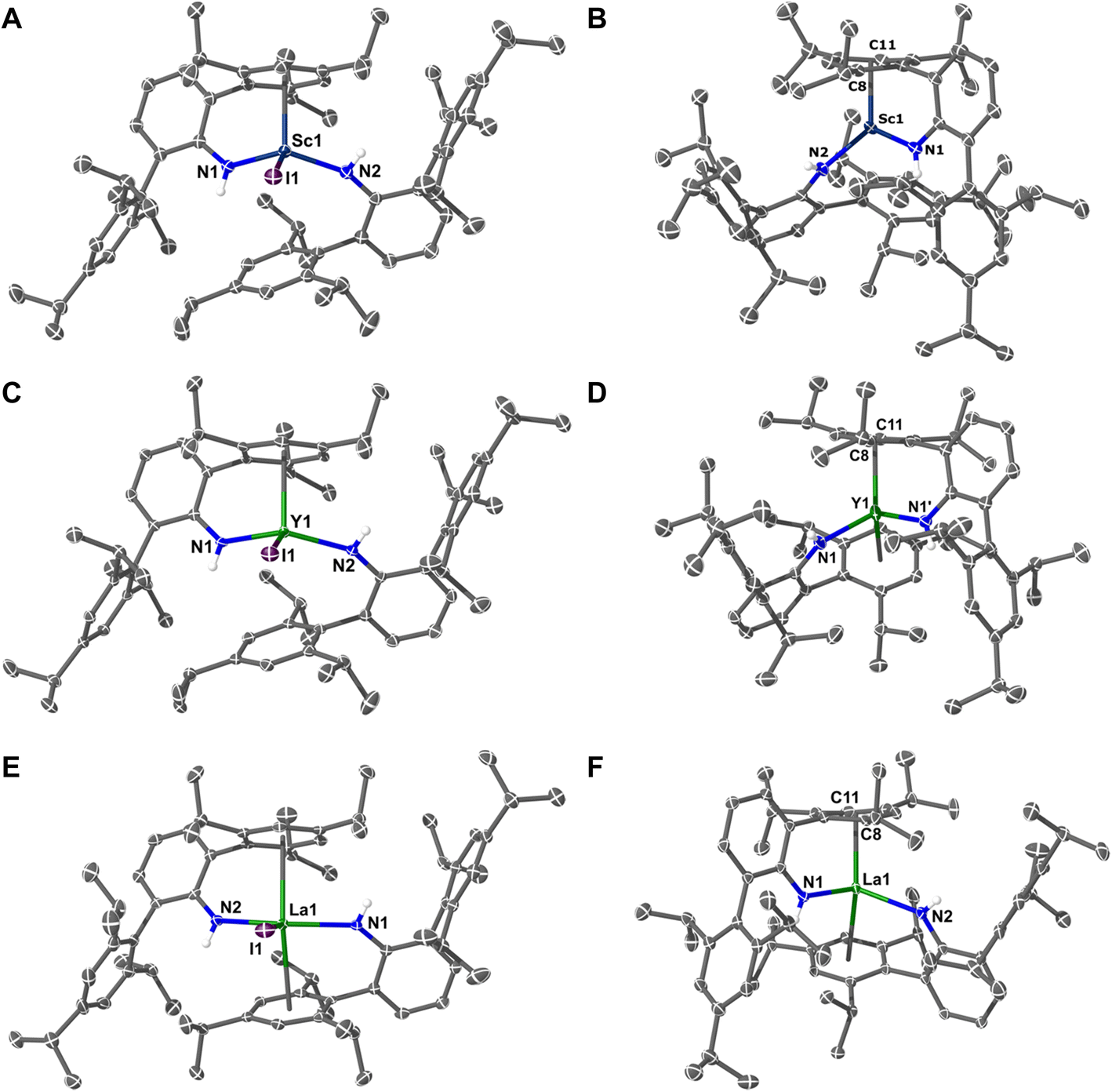 | ||
| Fig. 1 Molecular structures: (A) 1Sc; (B) 2Sc; (C) 1Y; (D) 2Y; (E) 1La; (F) 2La. Thermal ellipsoids have been set at 40% probability. H-atoms except those on N–H groups, solvents of crystallization, and disordered components have been removed for clarity. | ||
The molecular structures of 2Sc, 2Y, and 2La are also shown in Fig. 1 (see ESI† for 2Sm, 2Eu, 2Tm, and 2Yb). With the exception of 2Sc, all 2M complexes (M = Y, La, Sm, Eu, Tm, Yb) show two Tripp groups closely approaching the metal. When viewed along the C6-centroid⋯C6-centroid axis these groups are either fully eclipsed due to crystallographic C2 symmetry (2Y, 2Eu, and 2Yb), or are pseudo-eclipsed (2Sc, 2La, 2Sm, and 2Tm). In 2Sc, only one Tripp group is close to the metal. The nature of this metal–arene interaction provides insight into the electronic structure vis-à-vis metal- or ligand-centred reduction.45,46 Complexes 2Sc and 2La crystallize with a whole molecule in the asymmetric unit, and in both one metal-bound Tripp group is non-planar, showing an “open book” deformation for which a “hinge angle” (∠arene) can be calculated – 11.43(11)° for 2Sc, and 12.9(3)° for 2La. In 2Sc, the next shortest M⋯C6-centroid distance (3.8304(7) Å) is too long to constitute a strong interaction; but, in 2La the equivalent group is only ca. 0.4 Å further (M⋯C6-centroid = 2.8348(12) Å) than the deformed arene ring, but is planar. Complex 2Sc is similar to the Ti(IV) analogue 2Ti,46 but the latter exhibits ∠arene of 24.19(18)° and is diamagnetic by SQUID magnetometry, indicating the presence of a dianionic Tripp ring.
Complex 2Y is different to 2Sc and 2La as only half the molecule is present in the asymmetric unit (Z′ = 0.5), as previously reported;38 and also to that of 2U.47 The ∠arene angle for both ligands in 2Y is 7.27(12)°, which compares well to 9.5(1)° in 2U.47 There is no clear trend in ∠arene values (2Y < 2U < 2La < 2Sc) except that C2 symmetric complexes (2Y and 2U) have smaller ∠arene angles,38,47 and that larger values (with a single deformed arene) correlate with a greater degree of metal electron localisation (see below). The Gd(0) complex, [Gd(C6H3-1,3,5-tBu)2], also displays two symmetry-equivalent distorted arene rings (∠arene = 3.1(3)°).33 The remaining divalent complexes, 2Sm, 2Eu, 2Tm, and 2Yb, are analogous to 2La, except only the latter shows arene deformation. Table 1 summarizes all 2M complexes now reported (M = Sc, Y, La, Sm, Eu, Tm, Yb, U).38,47
| (Å or °) | 2Sc | 2Ya | 2La | 2Sm | 2Eub | 2Tm | 2Ybb | 2U | |
|---|---|---|---|---|---|---|---|---|---|
| a The solid-state structure is C2-symmetric so there is a single metal and ligand per asymmetric unit.b The solid-state structures show two half-molecules in the asymmetric unit, so M(1) and M(2) each have only one unique ligand. | |||||||||
| M–N | N(1) | 2.0884(11) | 2.2600(12) | 2.395(3) | 2.412(2) | 2.411(4) | 2.3060(17) | 2.310(6) | 2.330(2) |
| 2.414(5)b | 2.294(6)b | ||||||||
| N(2) | 2.0678(10) | —a | 2.434(3) | 2.425(2) | —b | 2.3169(18) | —b | —a | |
| M–C6-range | Ring(1) | 2.3913(12)–2.6304(14) | 2.7276(14)–2.9273(15) | 2.778(16)–2.971(9) | 2.955(3)–3.160(3) | 2.972(4)–3.176(5) | 2.8015(19)–2.971(2) | 2.840(6)–3.047(6) | 2.723(3)–2.870(3) |
| Ring(2) | — | —a | 3.047(3)–3.240(3) | 2.953(3)–3.201(3) | —b | 2.819(2)–3.118(2) | —b | —a | |
| M–C | C(8) | 2.3913(12) | 2.7684(14) | 2.843(13) | — | — | — | — | 2.731(3) |
| C(11) | 2.5418(13) | 2.7859(15) | 2.903(12) | — | — | — | — | 2.723(3) | |
| Arene fold angle | 11.43(11) | 7.27(12) | 12.9(9) | — | — | — | — | 9.3(2) | |
UV-Vis-NIR spectroscopy
UV-Vis-NIR spectra were collected for 1M (M = Sc, Y, La, Tm) and 2M (M = Sc, Y, La, Sm, Eu, Tm, Yb) at ambient temperature as 1 mM solutions in Et2O (Fig. 2, except for 1Tm). The spectra of 1Sc, 1Y, and 1La are uninformative, showing only a broad ligand-to-metal charge transfer (LMCT) process from ca. 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1 (500 nm) to well into the UV region, accounting for the intense yellow colour of all three in solution. The colours and spectra of 1Tm and 2Sm, 2Eu, 2Tm, and 2Yb are typical for these elements in their respective oxidation states (see ESI†).13,14,48–51
000 cm−1 (500 nm) to well into the UV region, accounting for the intense yellow colour of all three in solution. The colours and spectra of 1Tm and 2Sm, 2Eu, 2Tm, and 2Yb are typical for these elements in their respective oxidation states (see ESI†).13,14,48–51
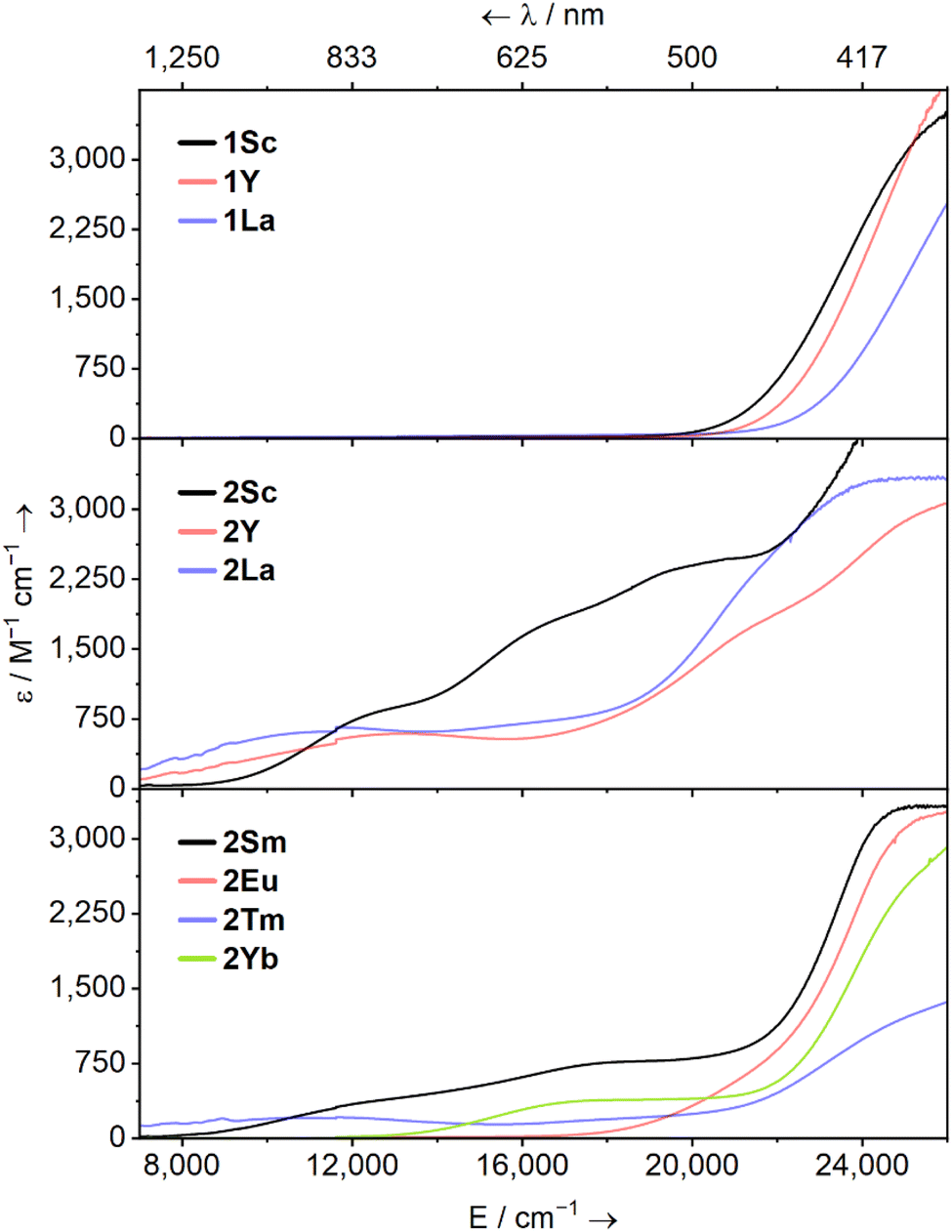 | ||
| Fig. 2 Solution UV-Vis-NIR spectra of [M(NHAriPr6)(I)] (1M, M = Sc, Y, La; top) and [M(NHAriPr6)] (2M, middle, M = Sc, Y, La; bottom, M = Sm, Eu, Tm, Yb) – as 1 mM solutions in Et2O between 7000–26000 cm−1 (1429–385 nm) at ambient temperature. | ||
The spectrum of 2Sc shows three well-resolved absorptions at 13400 cm−1 (756 nm, 805 M−1 cm−1), 17400 cm−1 (575 nm, 1727 M−1 cm−1), and 21300 cm−1 (469 nm, 2471 M−1 cm−1). The modest intensity of these peaks coupled with their energies suggests 3d → 3d transitions and/or metal-to-ligand charge transfer (MLCT) bands. For 2Y, two peaks are resolved at 13700 cm−1 (729 nm, 594 M−1 cm−1) and 21600 cm−1 (463 nm, 1787 M−1 cm−1), while a third tails in from above 26000 cm−1 (385 nm), in agreement with the previous report.38 Finally, in 2La, a single clear peak is resolved at 12300 cm−1 (816 nm, 650 M−1 cm−1), which we suggest is a 5d → 5d transition. A broad feature at ca. 16000 cm−1 and a peak with a maximum at ca. 24000 cm−1 (417 nm) can also be seen, but background absorption precludes accurately describing these.
SQUID magnetometry
Direct current (DC) magnetic susceptibility data were collected for 1Tm, and 2M (M = Sc, Y, La, Sm, Eu, Tm) from 1.8 to 300 K under an applied field of 1 kOe. At 300 K, the χMT (χM is the molar magnetic susceptibility) values for 1Tm (6.61 cm3 mol−1 K), 2Eu (7.27 cm3 mol−1 K), and 2Tm (2.47 cm3 mol−1 K) closely match theoretical values for 4fn+1 configurations for Tm(III), 4f12 (3H6, 7.15 cm3 mol−1 K), Eu(II), 4f7 (8S7/2, 7.88 cm3 mol−1 K), and Tm(II), 4f13 (2F7/2, 2.57 cm3 mol−1 K). For 2Sm (4f6) the value at 300 K is 1.43 cm3 mol−1 K, which is in the range observed for Eu(III) (4f6) complexes (1.3 to 1.5 cm3 mol−1 K),52 and is non-zero due to population of excited 7FJ states.48–50 Upon cooling to 1.8 K, χMT for 2Sm lowers to 0.02 cm3 mol−1 K which is consistent with a 7F0 ground state; thus, a 4f6 configuration.Complexes 2Sc, 2Y and 2La exhibit χMT values at 300 K (0.31, 0.36, and 0.29 cm3 mol−1 K respectively), which are in reasonable agreement with the spin-only value for an S = 1/2 system (0.375 cm3 mol−1 K for g = 2.00), and hence with a formal d1 configuration. In each case, the magnetic moment is essentially invariant with temperature down to 8–10 K, where a sudden drop can be seen, though this varies across independently synthesised samples (see ESI† for more details).
Electronic structure calculations
Unrestricted Kohn–Sham density-functional theory (DFT) calculations were performed on 2Sc, 2Y, 2La, 2Tm (S = 1/2), 2Sm (S = 3), 2Eu (S = 7/2), and 2Yb (S = 0) using partially geometry-optimised structures (see ESI† for full details). Löwdin population and spin analyses of 2Sm, 2Eu, 2Yb, and 2Tm are consistent with experimental data and describe all four as 4fn+1 Ln(II) ions with metal-localised valence electrons. Fig. 3A and C shows the SOMOs of 2Sc and 2La, which depict M–arene δ-bonds comprised of 36% 3d (with 4% 4s) and 14% 5d (with 10% 4f and 1% 6s) metal character, respectively, with the bound Tripp ring making up 41% (2Sc) and 56% (2La) – the remainder is diffused over the rest of the molecule. Defining the M⋯Tripp direction as z, the d-orbital contribution is described as d(xy/x2−y2) (the superficial resemblance to dx2 or dy2 is due to a small degree of dz2 mixing in this axis system) – this is comparable to the e2g MO of bis-benzene transition metal complexes in D6h symmetry.53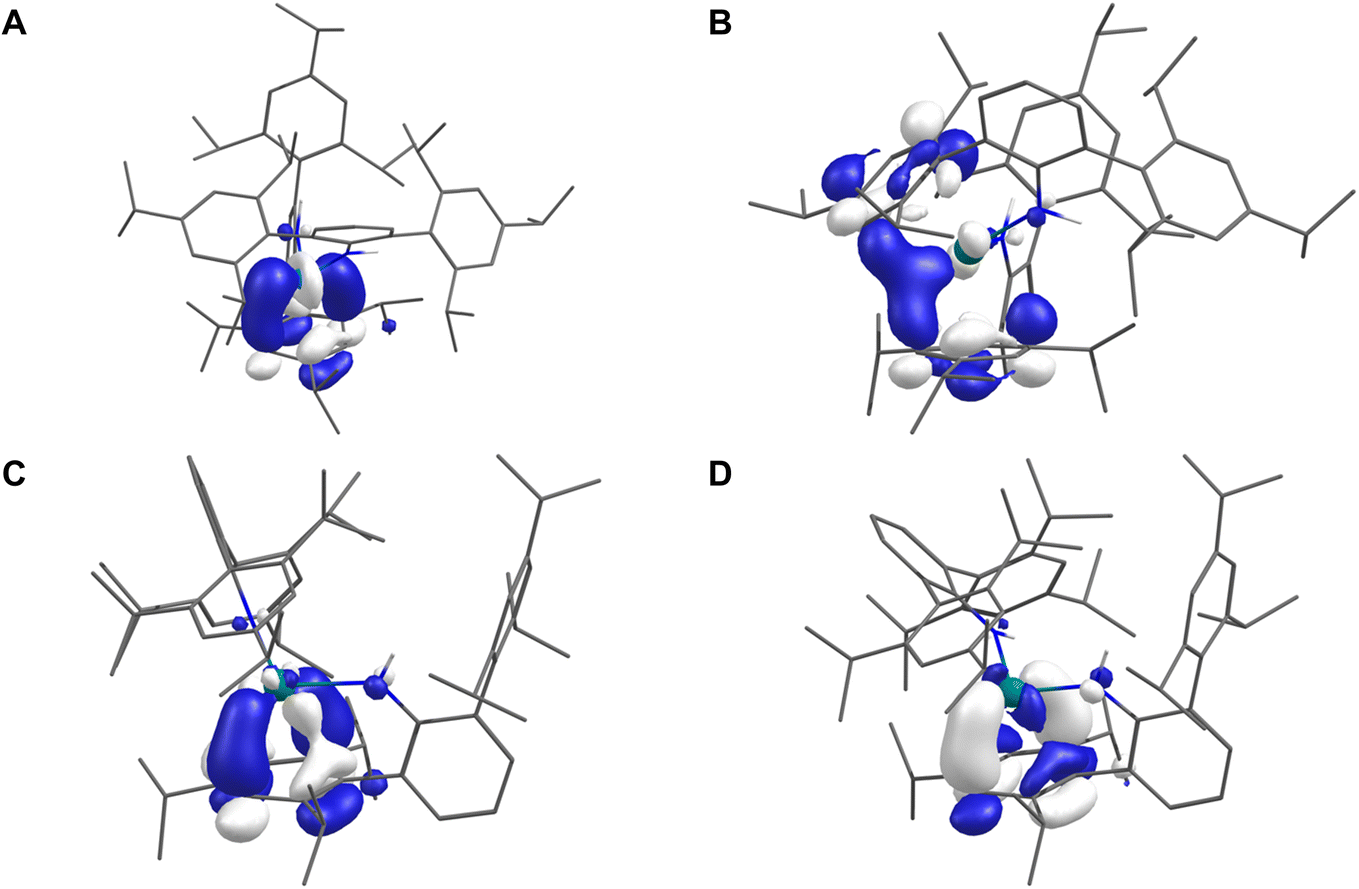 | ||
| Fig. 3 SOMOs of: (A) 2Sc; (B) 2Y; (C) 2La; (D) 2Y-Et2O (isovalues = 0.05) using geometries derived from single crystal X-ray diffraction with H-atoms optimised (2Sc, 2Y), select disordered C-atoms and all H-atoms optimised (2La), or all atom positions optimised (2Y-Et2O). H-atoms except those of the N(H) group omitted for clarity. | ||
In the case of 2Y (Fig. 3B), the SOMO is delocalised across the symmetry equivalent metal-bound Tripp groups such that the SOMO composition is 14% Y (12% 4d, 0.5% 5s), while the bound Tripp rings sum to 64.6% – the SOMO resembles a delocalised δ-bonding interaction. These results agree with previous work on 2Y,38 but it is an outlier compared to 2Sc and 2La. Full geometry optimisation of 2Y using the lower-symmetry structure of 2La as the starting point was performed in the gas phase and using an Et2O solvent model (2Y-Et2O henceforth). Both calculations produced geometries that have only one metal-bound Tripp group deformed (i.e. like 2Sc and 2La) and are true local minima on the potential energy surface. Fig. 3D shows the SOMO of 2Y-Et2O, and Löwdin population analysis shows it to be more metal-localised (22% 4d, 2% 5s, and 1% 4f – total 25%) than in the C2-symmetric 2Y (14%), which is accompanied by a corresponding decrease in Tripp contributions to the SOMO – 65% in 2Y (over two Tripp groups) and 56% in 2Y-Et2O (over a single Tripp group). The Löwdin spin populations at the metal in 2Y-Et2O (0.245) and 2Y (0.142) reflect these differences.
Time-dependent DFT (TD-DFT) and simplified TD-DFT (sTD-DFT) calculations were employed to model the UV-Vis-NIR spectra of 2Sc, 2Y, 2La, and 2Y-Et2O; here we focus on sTD-DFT with TPSSh for consistency with prior art (Fig. 4),11,12,16,23,54–56 see ESI (Fig. S102–S111†) for more details. Experimental features of 2Sc and 2La are well represented, and the Natural Transition Orbitals (NTOs) suggests the broad features in the spectrum of 2Sc are 3d → 3d transitions, the lowest energy of which resembles a 3d(xy/x2−y2) → 3d(x2−y2/xy) transition maintaining the δ-bonding interaction. For 2La, the lowest energy feature is a 5d(xy/x2−y2) → 5d(x2−y2/xy) transition, and the next two lowest energy features are a combination of MLCT and 5d → 5d transitions. There is poor agreement with all methods for 2Y (Fig. 4 middle panel, red line), however, calculations for 2Y-Et2O are substantially better (Fig. 4 middle panel, blue line); this suggests that the structure of 2Y in Et2O solution is similar to the solid-state structures of 2Sc and 2La. The lowest energy feature in 2Y-Et2O is comprised of two components, a 4d(xy/x2−y2) → 4d(x2−y2/xy) transition and an MLCT process.
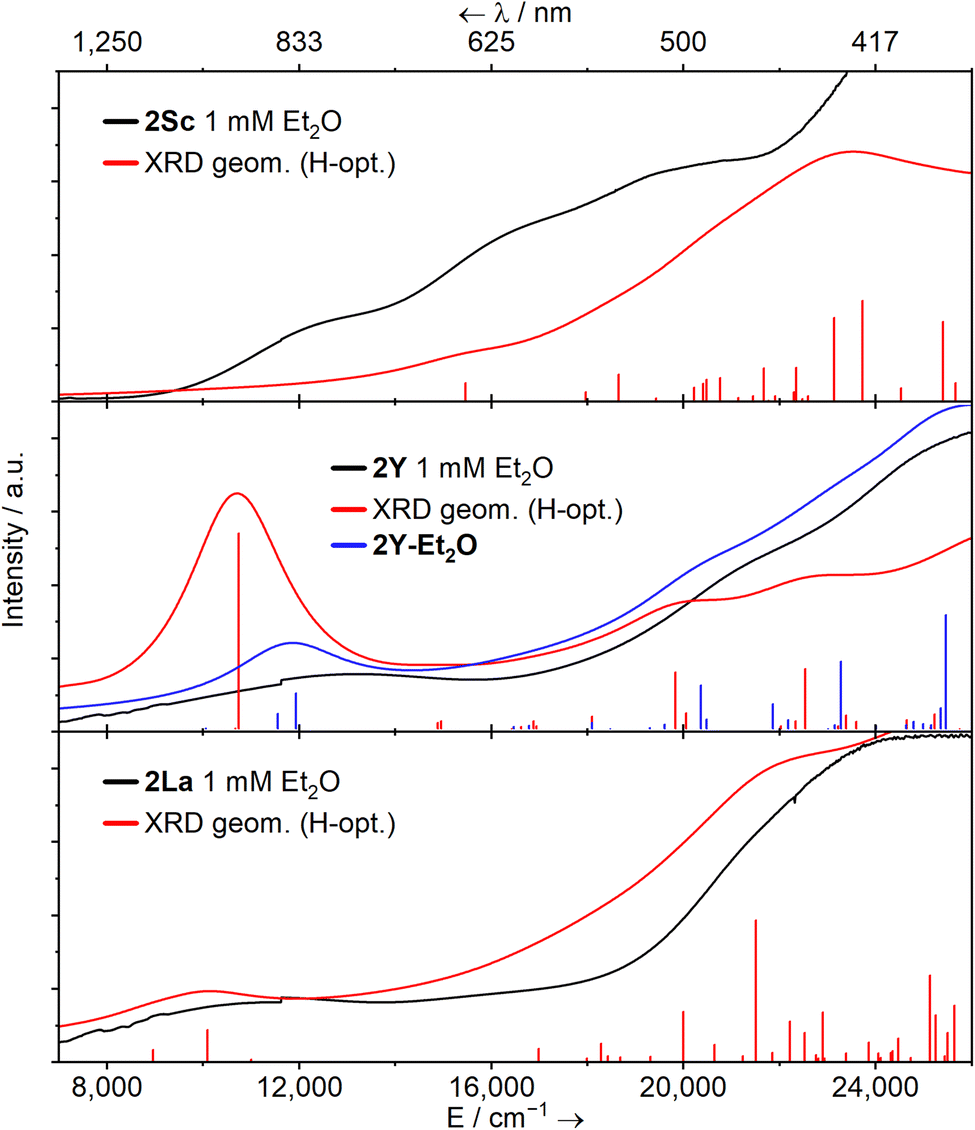 | ||
Fig. 4 Experimental (1 mM, Et2O, black), calculated sTD-DFT transitions (red and blue vertical lines), and simulated spectra (red and blue solid lines, with Gaussian broadening and a linewidth factor of 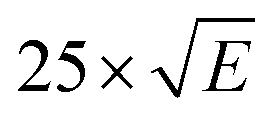 ) UV-Vis-NIR spectra for complexes 2Sc, 2Y (and 2Y-Et2O) and 2La calculated transitions were performed using a solvent model accounting for the dielectric constant (ε) and refractive index (nD) of Et2O. ) UV-Vis-NIR spectra for complexes 2Sc, 2Y (and 2Y-Et2O) and 2La calculated transitions were performed using a solvent model accounting for the dielectric constant (ε) and refractive index (nD) of Et2O. | ||
Complete active space self-consistent field (CASSCF) calculations (see ESI†) on 2M confirm the DFT and experimental results: 2Sc, 2Y and 2La exhibit nd1 ground states with significant orbital mixing with the arene ligand(s), and 2Sm, 2Eu, 2Tm and 2Yb have 4fn+1 ground states. Calculations for the lowest-lying excitations in 2Sc, 2Y and 2La including multi-configurational pair-density functional theory (MC-PDFT) corrections for dynamic correlation show nd/arene → nd/arene excitations in the UV-Vis-NIR range, in good agreement with the experimental spectra. The character of these excitations is broadly in line with that found using (s)TD-DFT, where the lowest-lying excitations for 2Sc are mostly localised to one side of the molecule and resemble d → d transitions, while some for 2La and 2Y-Et2O are combined MLCT and 5d → 5d transitions to the opposite Tripp ring.
EPR spectroscopy
Continuous wave (c. w.) EPR spectroscopy was used to study the orbitally non-degenerate species 2Sc, 2Y and 2La. All three are EPR active as polycrystalline solids and in solution (1 mM Et2O or nPr2O); spectra of 2Y in Et2O have been reported previously.38 We find better resolved frozen solution spectra in nPr2O than in Et2O (although the spectra are consistent; see ESI†).X-band spectra of powders at room temperature show features around g = 2, consistent with the formal M(II) oxidation states. There is partial resolution of the metal hyperfine for 2Sc (45Sc, I = 7/2, 100% abundant) and 2Y (89Y, I = 1/2, 100%) but is unresolved for 2La (139La, I = 7/2, 100%). For 2Sc, a hyperfine octet is observed, with g = 2.000 and A = 145 MHz, for 2Y we observe a hyperfine doublet (estimated A = 14 MHz) with g⊥ = 2.005 and g‖ = 1.995, and for 2La, we observe g⊥ = 2.019 and g‖ = 1.958. There are small changes in these parameters upon cooling to 5 K, without any improvement in resolution; the limited resolution of the powder spectra is indicative of intermolecular magnetic interactions.
Fluid solution spectra of 2Y and 2La (Fig. 5 and Table 2) give a hyperfine doublet (Aiso = 46 MHz, giso = 1.9995) and octet (Aiso = 112 MHz, giso = 1.998), respectively. For 2Sc (Fig. 5 and Table 2) we also obtain an octet (Aiso = 205 MHz, giso = 1.989), but there is a second minor octet spectrum which differs subtly in the magnitude of the hyperfine (Aiso = 186 MHz), suggesting two Sc(II) species in solution with a relative abundance of ca. 12:1 (similar features have been observed recently in a different Sc(II) system23). In each case giso < ge, consistent with the formal d1 configuration. The isotropic part of the hyperfine interaction derives from s-orbital spin density, and from theoretical values of the hyperfine interaction for unit population of the valence s-orbitals57 we estimate 7.3% (2Sc), 3.7% (2Y) and 2.0% (2La) s-orbital character of the SOMO; these are in good agreement with DFT calculations (3.6%, 1.8%, and 1.0% s-orbital character, or 3.8%, 2.0%, 1.1% Löwdin s-orbital spin populations). In summary, 2Sc has the largest metal valence s-orbital spin density, then 2Y > 2La.
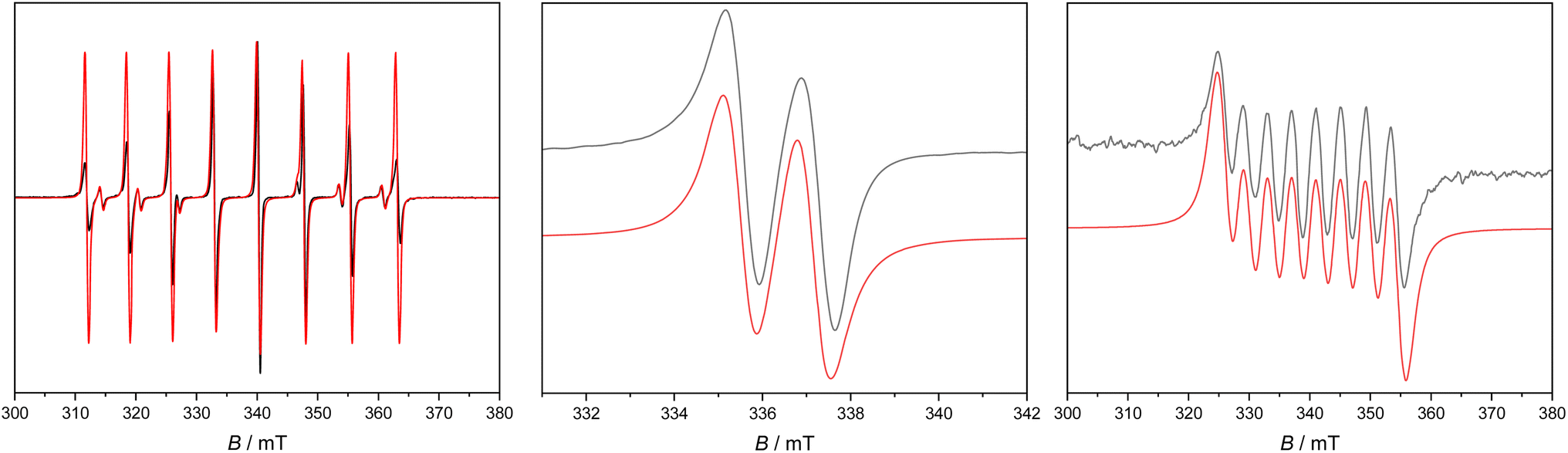 | ||
| Fig. 5 X-band c. w. EPR spectrum of 1 mM 2Sc in Et2O at 250 K (left), 2Y in nPr2O at 180 K (middle), and 2La in Et2O at 200 K (right). Black, experimental; red, simulations with parameters in the text. | ||
| g1 | g2 | g3 | A1 | A2 | A3 | Aiso | ||
|---|---|---|---|---|---|---|---|---|
| 2Sc | Exp. | 1.990 | 2.002 | 195 | 210 | 205 | ||
| Calc. | 1.990 | 2.009 | 2.015 | 159 | 171 | 184 | 171 | |
| 2Y | Exp. | 1.986 | 2.004 | −36 | −39 | −38 | ||
| 2Y-Et2O | Calc. | 1.983 | 2.004 | 2.006 | −47 | −49 | −50 | −48 |
| 2La | Exp. | 1.952 | 2.005 | 100 | 110 | 107 | ||
| Calc. | 1.954 | 1.971 | 1.993 | 101 | 106 | 108 | 105 | |
Frozen solutions gave well-resolved spectra in each case (Fig. 6 and Table 2). For 2Sc there is a dominant perpendicular hyperfine coupling A⊥ ≈ 210 MHz (g⊥ = 2.002), from which we can determine A‖ ≈ 195 MHz (using Aiso = 205 MHz from the fluid spectra), and by simulation we find g‖ = 1.99. However, these parameters are not well defined as there is evidence of a second species. For 2Y and 2La the spectra appear axially symmetric giving g⊥ = 2.004, g‖ = 1.986, with a near isotropic metal hyperfine of A⊥ = −39, A‖ = −36 MHz for 2Y, while for 2La we find g⊥ = 2.005, g‖ = 1.952 with A⊥ = 110, A‖ = 100 MHz.
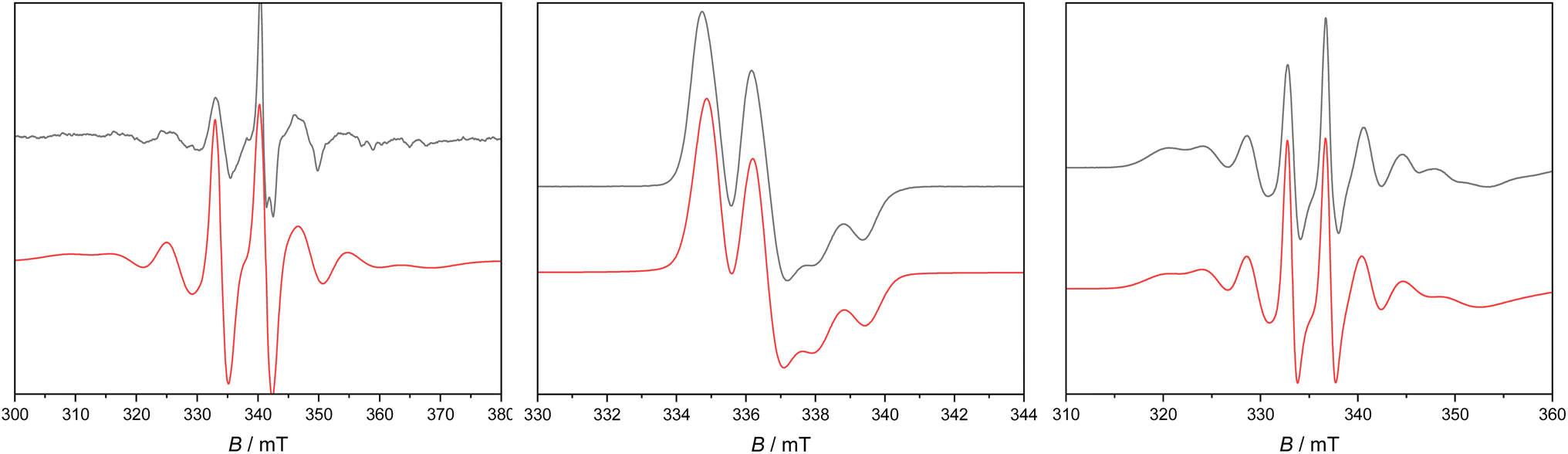 | ||
| Fig. 6 X-band c. w. EPR spectrum of 1 mM 2Sc (left) in Et2O at 130 K, 2Y (middle) and 2La (right) in nPr2O at 60 K. Black experimental, red simulations. | ||
The anisotropy of the g-values for 2Sc, 2Y and 2La is due to the significant d-orbital character of the SOMO. The g⊥ > g‖ pattern indicates a dominant dx2−y2 contribution (where z is the M-bound arene direction), while the greater deviation of g‖ from ge in the series 2La > 2Y > 2Sc is in keeping with greater spin–orbit coupling for the heavier elements,58 and also the trend in the lowest energy excited states (see above). A simple analysis of the anisotropic metal hyperfine interaction to give the metal dx2−y2 contribution to the SOMO [A‖ − A⊥ = (−6/7)a2Pd, where a is the dx2−y2 coefficient of the SOMO and values of Pd are tabulated in ref. 57] gives 7.3% for 2Sc, 5.6% for 2Y and 4.9% for 2La. DFT calculations give much larger d-orbital contributions of 36%, 20%, and 14%, respectively (see above), despite also showing reasonable agreement with the measured hyperfine coupling constants (Table 2), implying an inadequacy in the simple analysis above; we have previously noted similar discrepancies in related [MIIL3]− systems (M = Sc, Y, La, Lu).17
Indeed, the EPR parameters and electronic structures of 2Sc, 2Y and 2La contrast to those related d1 [MIIL3]− species where the trigonal crystal field instead stabilises the dz2 (defined by the C3 axis) orbital, or a d/s hybrid giving rise to characteristic g⊥ < g‖ (≈ge) patterns.2,16,17 The electronic structures of the present compounds have more in common with [Sc(Cpttt)2],23 where DFT calculations give a dx2−y2-dominated SOMO, and a similar g⊥ > g‖ pattern can be observed from the frozen solution EPR data. The hyperfine coupling in 2Y and 2La is much weaker than in the trigonal M(II) cyclopentadienyl species: for example, [Y(CpR)3]− with various substituents [e.g. (CpR)3 = Cp′3, Cpt3, {Cp′′2(C5H5)}] have |Aiso| = 98–130 MHz;17,59,60 and [La(CpR)3]− (CpR = Cp′, Cp′′, Cptt) have |Aiso| = 390–640 MHz.2,17,61 Hence, there is greater metal character in the SOMOs of [M(CpR)3]− than in 2M. Comparing the present compounds with more symmetric sandwich compounds, [Y(CpiPr5)2] and [La(CpiPr5)2] have larger magnitude |Aiso| = 505 and 2000 MHz, respectively,14 while curiously [Sc(Cpttt)2] has smaller magnitude |Aiso| = 83 MHz,23 although it has been reported that [Sc(Cp*)2] (not structurally authenticated) has |Aiso| = 824 MHz.23
Conclusions
In summary, we have reported a series of room-temperature stable crystalline divalent rare earth bis-tethered arene complexes of the form [M(NHAriPr6)2] (2M; M = Sc, Y, La, Sm, Eu, Tm, Yb). In the case of Sc and La, these represent the second examples of neutral divalent complexes with these elements and are amongst just a few in any charge state. All 2M complexes feature close metal–arene contacts, which in 2Sc, 2Y, and 2La, results in an “open book” arene deformation suggestive of charge transfer, whereas in 2Sm, 2Eu, 2Tm, and 2Yb the equivalent arene groups remain planar. SQUID magnetometry and UV-Vis-NIR spectroscopy demonstrate 2Sm, 2Eu, 2Tm, and 2Yb are examples of 4fn+1 ions, and quantum chemical calculations show that the metal-localised 4f orbitals do not interact with the arene π-orbitals.Solution-phase c. w. EPR spectroscopy of 2Sc, 2Y, and 2La are consistent with formal nd1 ions, where the SOMO has nd(x2−y2) character with delocalisation of the spin onto the Tripp groups. Quantum chemical and ab initio calculations further support this description and reveal mixing between metal nd(x2−y2) and arene–π orbitals to give δ-bonds, which explain the arene deformation in their structures.
The electronic structures of nd1 2Sc, 2Y, and 2La closely resemble those of bis-benzene transition metal complexes; and, going forward, we posit that these rare nd(x2−y2) configurations afford as-yet unexplored opportunities to tune the physicochemical properties of divalent rare earth ions with formal {5d/6s}1 valence electron configurations. This, along with work to probe the limits of the bis-{NHAriPr6} framework to stabilise other divalent f-block ions, remains an active area of research in our laboratory.
Data availability
The following CCDC references contain the ESI† crystal data for this article: 1Sc (2266263), 1Y (2266261), 1La (2266235), 1Tm (2266255), 1Yb (2266257), 1Yβ (2295306), 1Laβ (2295307), 2Sc (2266264), 2Y (2266262), 2La (2266236), 2Sm (2266243), 2Eu (2266244), 2Tm (2266256), 2Yb (2266258), and 3 (2282041). Raw experimental data (NMR, ATR-IR, UV-Vis-NIR, SQUID Magnetometry) and computational inputs/outputs can be found freely at DOI: https://doi.org/10.48420/25245760.Author contributions
C. A. P. G. provided the original concept. R. E. M. synthesised and characterised the complexes. R. E. M. and C. A. P. G. collected, solved, and refined the single crystal XRD data. G. F. S. W. performed final refinement and validation of XRD data. J. A. S. performed SQUID magnetometry measurements and interpreted the data. T. H. performed EPR spectroscopy measurements, E. J. L. M. and D. C. supervised the EPR measurements and data interpretation. C. A. P. G. performed DFT calculations and interpretation, N. F. C. performed CASSCF and MC-PDFT calculations and interpreted the data. C. A. P. G. wrote the manuscript with contributions from all other authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Royal Society for a University Research Fellowship (URF\211271 to C. A. P. G., and URF\191320 to N. F. C.); and the UoM Department of Chemistry for the funding of a PhD studentship under EPSRC DTP (EP/W524347/1 to R. E. M., and EP/T517823/1 to T. H.). We acknowledge funding from the EPSRC (EP/K039547/1, EP/V007580/1, and EP/P001386/1 for NMR spectroscopy and X-ray diffraction). We also thank the EPSRC UK EPR National Research Facility including for access to the SQUID magnetometer (EP/W014521/1, EP/V035231/1, EPS033181/1). C. A. P. G. and N. F. C. also thank the Computational Shared Facility at the University of Manchester for support. We thank Prof. Stephen Liddle for support (J. A. S.). Elemental analyses were performed at the UoM by Mr Martin Jennings and Ms Anne Davies. We are also grateful to Prof. Aaron Odom and Prof. Selvan Demir for scientific discussions and encouragement.References
- K. R. Meihaus, M. E. Fieser, J. F. Corbey, W. J. Evans and J. R. Long, J. Am. Chem. Soc., 2015, 137, 9855–9860 CrossRef CAS PubMed.
- J. Liu, L. E. Nodaraki, D. O. T. A. Martins, M. J. Giansiracusa, P. J. Cobb, J. Emerson-King, F. Ortu, G. F. S. Whitehead, G. K. Gransbury, E. J. L. Mclnnes, F. Tuna and D. P. Mills, Eur. J. Inorg. Chem., 2023, 26, e202300552 CrossRef CAS.
- M. R. MacDonald, J. W. Ziller and W. J. Evans, J. Am. Chem. Soc., 2011, 133, 15914–15917 CrossRef CAS PubMed.
- M. R. MacDonald, J. E. Bates, M. E. Fieser, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2012, 134, 8420–8423 CrossRef CAS PubMed.
- M. R. MacDonald, J. E. Bates, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2013, 135, 9857–9868 CrossRef CAS PubMed.
- M. E. Fieser, M. G. Ferrier, J. Su, E. Batista, S. K. Cary, J. W. Engle, W. J. Evans, J. S. Lezama Pacheco, S. A. Kozimor, A. C. Olson, A. J. Ryan, B. W. Stein, G. L. Wagner, D. H. Woen, T. Vitova and P. Yang, Chem. Sci., 2017, 8, 6076–6091 RSC.
- M. T. Trinh, J. C. Wedal and W. J. Evans, Dalton Trans., 2021, 50, 14384–14389 RSC.
- T. F. Jenkins, D. H. Woen, L. N. Mohanam, J. W. Ziller, F. Furche and W. J. Evans, Organometallics, 2018, 37, 3863–3873 CrossRef CAS.
- A. J. Ryan, L. E. Darago, S. G. Balasubramani, G. P. Chen, J. W. Ziller, F. Furche, J. R. Long and W. J. Evans, Chem.–Eur. J., 2018, 24, 7702–7709 CrossRef CAS PubMed.
- S. A. Moehring, M. Miehlich, C. J. Hoerger, K. Meyer, J. W. Ziller and W. J. Evans, Inorg. Chem., 2020, 59, 3207–3214 CrossRef CAS PubMed.
- D. N. Huh, S. R. Ciccone, S. Bekoe, S. Roy, J. W. Ziller, F. Furche and W. J. Evans, Angew. Chem., Int. Ed., 2020, 59, 16141–16146 CrossRef CAS PubMed.
- L. M. Anderson-Sanchez, J. M. Yu, J. W. Ziller, F. Furche and W. J. Evans, Inorg. Chem., 2023, 62, 706–714 CrossRef CAS PubMed.
- M. E. Fieser, M. R. MacDonald, B. T. Krull, J. E. Bates, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2015, 137, 369–382 CrossRef CAS PubMed.
- K. R. McClain, C. A. Gould, D. A. Marchiori, H. Kwon, T. T. Nguyen, K. E. Rosenkoetter, D. Kuzmina, F. Tuna, R. D. Britt, J. R. Long and B. G. Harvey, J. Am. Chem. Soc., 2022, 144, 22193–22201 CrossRef CAS PubMed.
- P. W. Smith, J. Hrubý, W. J. Evans, S. Hill and S. G. Minasian, J. Am. Chem. Soc., 2024, 146, 5781–5785 CrossRef CAS PubMed.
- K. Kundu, J. R. K. White, S. A. Moehring, J. M. Yu, J. W. Ziller, F. Furche, W. J. Evans and S. Hill, Nat. Chem., 2022, 14, 392–397 CrossRef CAS PubMed.
- A.-M. Ariciu, D. H. Woen, D. N. Huh, L. E. Nodaraki, A. K. Kostopoulos, C. A. P. Goodwin, N. F. Chilton, E. J. L. McInnes, R. E. P. Winpenny, W. J. Evans and F. Tuna, Nat. Commun., 2019, 10, 3330 CrossRef PubMed.
- C. A. Gould, K. R. McClain, J. M. Yu, T. J. Groshens, F. Furche, B. G. Harvey and J. R. Long, J. Am. Chem. Soc., 2019, 141, 12967–12973 CrossRef CAS PubMed.
- P.-B. Jin, Q.-C. Luo, G. K. Gransbury, I. J. Vitorica-Yrezabal, T. Hajdu, I. Strashnov, E. J. L. McInnes, R. E. P. Winpenny, N. F. Chilton, D. P. Mills and Y.-Z. Zheng, J. Am. Chem. Soc., 2023, 145, 27993–28009 CrossRef CAS PubMed.
- C. A. Gould, K. R. McClain, D. Reta, J. G. C. Kragskow, D. A. Marchiori, E. Lachman, E. S. Choi, J. G. Analytis, R. D. Britt, N. F. Chilton, B. G. Harvey and J. R. Long, Science, 2022, 375, 198–202 CrossRef CAS PubMed.
- J. C. Wedal, L. M. Anderson-Sanchez, M. T. Dumas, C. A. Gould, M. J. Beltran-Leiva, C. Celis-Barros, D. Paez-Hernandez, J. W. Ziller, J. R. Long and W. J. Evans, J. Am. Chem. Soc., 2023, 145, 10730–10742 CrossRef CAS PubMed.
- K. R. McClain, H. Kwon, K. Chakarawet, R. Nabi, J. G. C. Kragskow, N. F. Chilton, R. D. Britt, J. R. Long and B. G. Harvey, J. Am. Chem. Soc., 2023, 145, 8996–9002 CrossRef CAS PubMed.
- J. D. Queen, L. M. Anderson-Sanchez, C. R. Stennett, A. Rajabi, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2024, 146, 3279–3292 CrossRef CAS PubMed.
- J. T. Boronski, J. A. Seed, D. Hunger, A. W. Woodward, J. van Slageren, A. J. Wooles, L. S. Natrajan, N. Kaltsoyannis and S. T. Liddle, Nature, 2021, 598, 72–75 CrossRef CAS PubMed.
- M. D. Straub, E. T. Ouellette, M. A. Boreen, R. D. Britt, K. Chakarawet, I. Douair, C. A. Gould, L. Maron, I. Del Rosal, D. Villarreal, S. G. Minasian and J. Arnold, J. Am. Chem. Soc., 2021, 143, 19748–19760 CrossRef CAS PubMed.
- J. D. Cryer and S. T. Liddle, in Comprehensive Organometallic Chemistry IV, ed. G. Parkin, K. Meyer and D. O'Hare, Elsevier, Oxford, 2022, pp. 460–501 Search PubMed.
- J. Murillo, R. Bhowmick, K. L. M. Harriman, A. Gomez-Torres, J. Wright, R. W. Meulenberg, P. Miro, A. Metta-Magana, M. Murugesu, B. Vlaisavljevich and S. Fortier, Chem. Sci., 2021, 12, 13360–13372 RSC.
- R. A. Keerthi Shivaraam, M. Keener, D. K. Modder, T. Rajeshkumar, I. Zivkovic, R. Scopelliti, L. Maron and M. Mazzanti, Angew. Chem., Int. Ed., 2023, 62, e202304051 CrossRef CAS PubMed.
- M. Keener, R. A. K. Shivaraam, T. Rajeshkumar, M. Tricoire, R. Scopelliti, I. Zivkovic, A. S. Chauvin, L. Maron and M. Mazzanti, J. Am. Chem. Soc., 2023, 145, 16271–16283 CrossRef CAS PubMed.
- F.-C. Hsueh, T. Rajeshkumar, L. Maron, R. Scopelliti, A. Sienkiewicz and M. Mazzanti, Chem. Sci., 2023, 14, 6011–6021 RSC.
- Y. Wang, J. Liang, C. Deng, R. Sun, P.-X. Fu, B.-W. Wang, S. Gao and W. Huang, J. Am. Chem. Soc., 2023, 145, 22466–22474 CrossRef CAS PubMed.
- H. S. La Pierre, A. Scheurer, F. W. Heinemann, W. Hieringer and K. Meyer, Angew. Chem., Int. Ed., 2014, 53, 7158–71562 CrossRef CAS PubMed.
- J. G. Brennan, F. G. N. Cloke, A. A. Sameh and A. Zalkin, J. Chem. Soc., Chem. Commun., 1987, 1668–1669 RSC.
- D. M. Anderson, F. G. N. Cloke, P. A. Cox, N. Edelstein, J. C. Green, T. Pang, A. A. Sameh and G. Shalimoff, J. Chem. Soc., Chem. Commun., 1989, 53–55 RSC.
- W. A. King, T. J. Marks, D. M. Anderson, D. J. Duncalf and F. G. N. Cloke, J. Am. Chem. Soc., 1992, 114, 9221–9223 CrossRef CAS.
- F. G. N. Cloke, Chem. Soc. Rev., 1993, 22, 17–24 RSC.
- W. A. King, S. Di Bella, G. Lanza, K. Khan, D. J. Duncalf, F. G. N. Cloke, I. L. Fragala and T. J. Marks, J. Am. Chem. Soc., 1996, 118, 627–635 CrossRef CAS.
- R. Jena, F. Benner, F. Delano, D. Holmes, J. McCracken, S. Demir and A. L. Odom, Chem. Sci., 2023, 14, 4257–4264 RSC.
- P. Dorenbos, J. Condens. Matter Phys., 2003, 15, 575–594 CrossRef CAS.
- P. Dorenbos, J. Lumin., 2000, 91, 91–106 CrossRef CAS.
- B. R. Barnett, C. C. Mokhtarzadeh, P. Lummis, S. Wang, J. D. Queen, J. Gavenonis, N. Schüwer, T. D. Tilley, J. N. Boynton, N. Weidemann, D. W. Agnew, P. W. Smith, T. B. Ditri, A. E. Carpenter, J. K. Pratt, N. D. Mendelson, J. S. Figueroa and P. P. Power, in Inorganic Syntheses, 2018, pp. 85–122 Search PubMed.
- B. Twamley, C.-S. Hwang, N. J. Hardman and P. P. Power, J. Organomet. Chem., 2000, 609, 152–160 CrossRef CAS.
- K. Izod, S. T. Liddle and W. Clegg, Inorg. Chem., 2004, 43, 214–218 CrossRef CAS PubMed.
- P. Girard, J. L. Namy and H. B. Kagan, J. Am. Chem. Soc., 1980, 102, 2693–2698 CrossRef CAS.
- S. R. Chowdhury, C. A. P. Goodwin and B. Vlaisavljevich, Chem. Sci., 2024, 15, 1810–1819 RSC.
- J. N. Boynton, J. D. Guo, F. Grandjean, J. C. Fettinger, S. Nagase, G. J. Long and P. P. Power, Inorg. Chem., 2013, 52, 14216–14223 CrossRef CAS PubMed.
- B. S. Billow, B. N. Livesay, C. C. Mokhtarzadeh, J. McCracken, M. P. Shores, J. M. Boncella and A. L. Odom, J. Am. Chem. Soc., 2018, 140, 17369–17373 CrossRef CAS PubMed.
- N. F. Chilton, C. A. P. Goodwin, D. P. Mills and R. E. P. Winpenny, Chem. Commun., 2015, 51, 101–103 RSC.
- C. A. P. Goodwin, K. C. Joslin, S. J. Lockyer, A. Formanuik, G. A. Morris, F. Ortu, I. J. Vitorica-Yrezabal and D. P. Mills, Organometallics, 2015, 34, 2314–2325 CrossRef CAS.
- C. A. P. Goodwin, N. F. Chilton, G. F. Vettese, E. Moreno Pineda, I. F. Crowe, J. W. Ziller, R. E. P. Winpenny, W. J. Evans and D. P. Mills, Inorg. Chem., 2016, 55, 10057–10067 CrossRef CAS PubMed.
- C. A. P. Goodwin, N. F. Chilton, L. S. Natrajan, M.-E. Boulon, J. W. Ziller, W. J. Evans and D. P. Mills, Inorg. Chem., 2017, 56, 5959–5970 CrossRef CAS PubMed.
- Y. Takikawa, S. Ebisu and S. Nagata, J. Phys. Chem. Solids, 2010, 71, 1592–1598 CrossRef CAS.
- V. M. Rayón and G. Frenking, Organometallics, 2003, 22, 3304–3308 CrossRef.
- A. Rajabi, R. Grotjahn, D. Rappoport and F. Furche, Dalton Trans., 2024, 53, 410–417 RSC.
- M. E. Fieser, C. T. Palumbo, H. S. La Pierre, D. P. Halter, V. K. Voora, J. W. Ziller, F. Furche, K. Meyer and W. J. Evans, Chem. Sci., 2017, 8, 7424–7433 RSC.
- C. T. Palumbo, D. P. Halter, V. K. Voora, G. P. Chen, A. K. Chan, M. E. Fieser, J. W. Ziller, W. Hieringer, F. Furche, K. Meyer and W. J. Evans, Inorg. Chem., 2018, 57, 2823–2833 CrossRef CAS PubMed.
- J. R. Morton and K. F. Preston, J. Magn. Reson., 1978, 30, 577–582 CAS.
- S. Koseki, N. Matsunaga, T. Asada, M. W. Schmidt and M. S. Gordon, J. Phys. Chem. A, 2019, 123, 2325–2339 CrossRef CAS PubMed.
- J. F. Corbey, D. H. Woen, C. T. Palumbo, M. E. Fieser, J. W. Ziller, F. Furche and W. J. Evans, Organometallics, 2015, 34, 3909–3921 CrossRef CAS.
- M. A. Angadol, D. H. Woen, C. J. Windorff, J. W. Ziller and W. J. Evans, Organometallics, 2019, 38, 1151–1158 CrossRef CAS.
- S. A. Moehring and W. J. Evans, Organometallics, 2020, 39, 1187–1194 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the starting materials used, descriptions of sample preparation for spectroscopic measurements, and methodologies and coordinates for the DFT and CASSCF calculations. CCDC 2266235, 2266236, 2266243, 2266244, 2266255–2266258, 2266261–2266264, 2282041, 2295306 and 2295307. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc03005b. |
| This journal is © The Royal Society of Chemistry 2024 |