Organogels are a class of polymer networks that consist of an organic liquid phase within a three-dimensional crosslinked network.1–3 Currently, organogels are used in applications such as anti-icing,4,5 anti-fouling,6 drug delivery,7–9 and food processing.10,11 Performing chain extension polymerizations on organogel networks provide an opportunity to modify properties such as amphiphilicity,12–14 composition,15–18 shape,19–21 and mechanical properties.22–25 For example, Chen and Gu et al. showed that parent-to-daughter gel network extensions can be used to maintain or modify gel moduli and introduce copolymers to modify solvophilicity.26 However, only a few controlled polymerization techniques result in the retention of chain end functionality required to synthesize block copolymer networks.
Complex architectures including block copolymers and block copolymer networks can be synthesized using reversible-deactivation radical polymerization (RDRP) because the polymer chain ends can be reactivated to perform chain extensions.27 For example, reversible addition–fragmentation chain transfer (RAFT) polymerization is used to synthesize organogels, resulting in more uniform networks compared to free radical polymerization (FRP).28–30 However, using RAFT polymerization to synthesize network polymers leads to a loss in molecular weight control at higher degrees of polymerization (DP) due to mobility issues in viscous media and because degenerative chain transfer is required for molar mass control.28 Photoiniferter and photoinduced electron/energy transfer (PET) initiation pathways are used instead of conventional radical initiators because the added reversible deactivation reaction leads to higher chain end retention,31,32 providing a platform to access structurally tailored and engineered macromolecular (STEM) gel networks.19,20,33–36 However, there is not clarity on how PET-RAFT polymerizations affect both the molecular weight control of STEM organogel chain extensions and the physical property changes that result from these modifications.
We aimed to determine if PET-RAFT polymerization yields well-defined block copolymers and networks with tunable properties by using degradable crosslinkers to characterize the copolymers. The disulfide-based diacrylate (DSDA) crosslinker enables organogel synthesis that is stable under atmospheric conditions but can be cleaved in the presence of a reducing agent.28 A stable yet degradable crosslinker is important to ensure that physical properties are not compromised by prematurely degraded crosslinkers. We hypothesized that PET-RAFT polymerization can be used to chain extend organogels into block copolymer networks with tunable physical properties, while DSDA will provide a degradable crosslinker to analyze the chain extension molecular weight control.
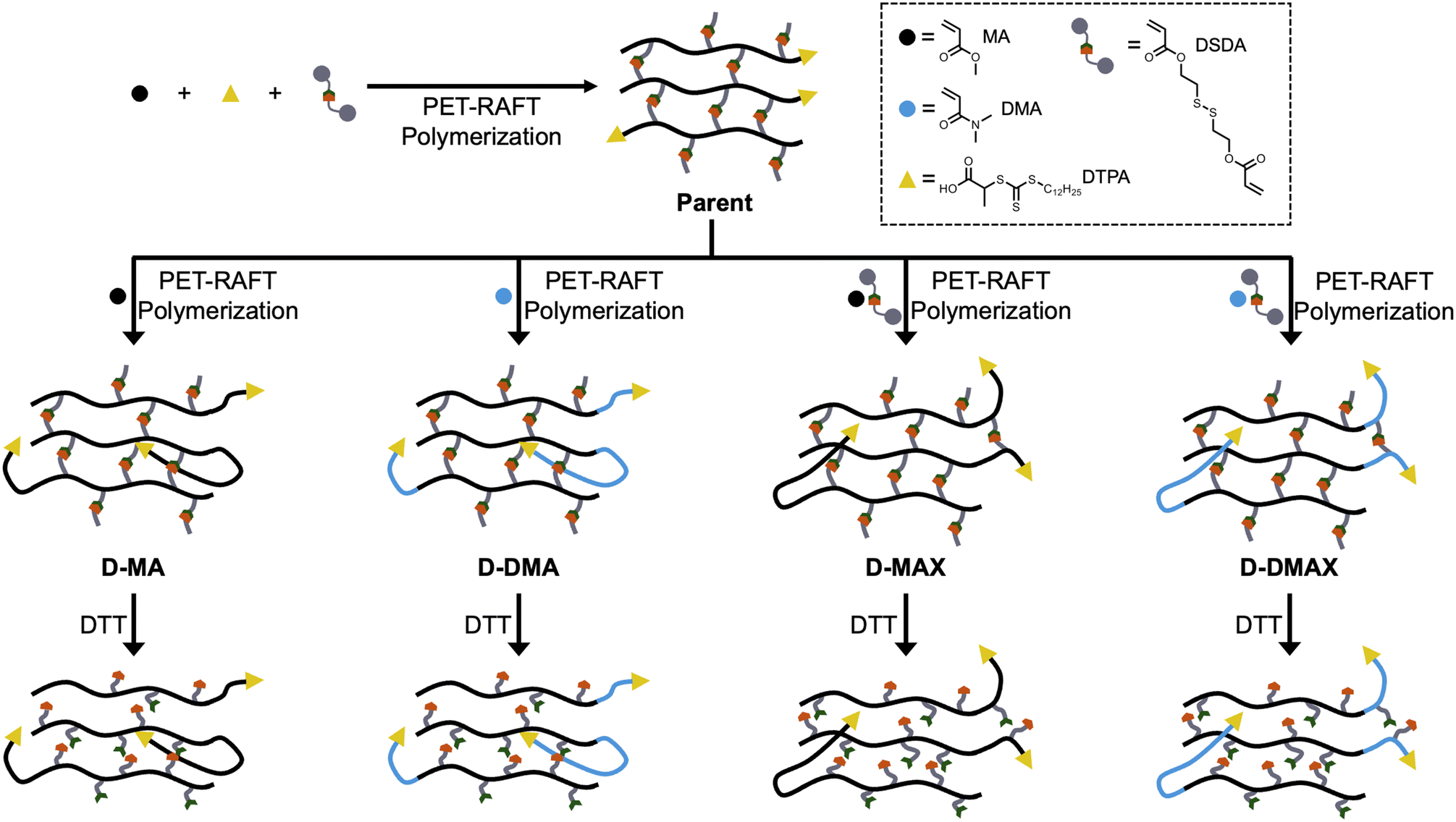
Herein, poly(methyl acrylate-stat-DSDA) (P(MA-s-DSDA)) STEM organogels were synthesized as the first block comprising the parent organogel (Fig. 1). To synthesize block copolymer networks with different properties, methyl acrylate (MA) or N,N-dimethylacrylamide (DMA) were chosen as the second block due to the different physical characteristics of the resulting poly(methyl acrylate) (PMA) or poly(N,N-dimethylacrylamide) (PDMA) polymers. Each chain extension using MA or DMA was performed with or without additional DSDA to determine the effect of a crosslinked versus non-crosslinked second block. The resultant gels were then analyzed via swelling ratio studies and rheology to measure network physical properties. Following physical properties testing, the DSDA crosslinks were reduced to thiols to yield linear copolymers that were analyzed via size exclusion chromatography (SEC). Overall, the primary and chain-extended organogels were characterized to determine network physical property differences and molecular weight control.
Dimethyl sulfoxide (DMSO, Thermo Scientific, 99.7%), N,N-dimethylacetamide (DMAc, Thermo Scientific, 99%), N,N-dimethylformamide (DMF, Acros Organics, 99.8%), acryloyl chloride (Sigma, 97%), bis(2-hydroxyethyl) disulfide (BHEDS, Sigma, technical grade), and triethylamine (Thermo Scientific, 99%) were used as received. Methyl acrylate (MA, Thermo Scientific, 99%) and N,N-dimethylacrylamide (DMA, TCI, 99%) were filtered through basic alumina prior to use. Zinc tetraphenyl porphyrin (ZnTPP) (TCI, 97%) was prepared as a 10 mg mL−1 solution in DMSO prior to use. Dithiothreitol (DTT, Fisher, 99%) was prepared as a 20 mg mL−1 solution in DMAc prior to use. Disulfide-based diacrylate (DSDA) was synthesized from a previous report.28 2-(Dodecylthiocarbonothioylthio)propionic acid (DTPA) was synthesized using an adapted procedure from a previous report.37
For a visible light source, 76.2 cm Supernight Blue LED Light Strips were purchased from Amazon and placed on a Xnrtop Silver Tone Aluminum Radiator Heatsink Heat Sink 150 × 80 × 27 mm from Amazon.
1H nuclear magnetic resonance (NMR) spectroscopy was conducted on an Agilent U4-DD2 400 MHz at 25 °C. DMSO-d6 (Cambridge Isotopes Laboratories, Inc., 99.9%) and CDCl3 (Cambridge Isotopes Laboratories, Inc., 99.8%) were used as received.
Size exclusion chromatography (SEC) was performed in N,N-dimethylacetamide (DMAc) with 50 mM LiCl at 50 °C at a flow rate of 0.5 mL min−1 (Agilent isocratic pump, degasser, and autosampler, columns: TOSOH TSKgel Guard Alpha and TOSOH TSKgel Alpha-4000: exclusion limit of 1 × 106 Da). Detection consisted of an Agilent 1260 Infinity II refractive index detector and an Agilent 1260 Infinity II variable wavelength detector operating at 280 nm. The system was calibrated with poly(methyl methacrylate) standards with molecular weights in the range of 1480 to 1 062 000 g mol−1, which corresponds to a linear calibration region of 12.8 to 21.6 minutes.
Rheological measurements were performed on an HR 20 Discovery Hybrid Rheometer (TA Instruments) equipped with stainless steel 8 mm parallel plates. Temperature was controlled with an environmental test chamber and maintained at 23 ± 1 °C. Frequency sweeps were performed at 0.5% strain from 0.01 to 15 Hz (0.06 to 94 rad per s). Strain (amplitude) sweeps were performed at 1 Hz from 0.01 to 200%. Time sweeps were performed at 0.5% strain and 1 Hz over 300 s. Sample thickness was approximately 2 mm and a small normal force (∼0.1 N) was used to compress the sample between the plates to reduce slippage.
DMSO (2.81 mL), MA (2.45 g, 28.5 mmol), DSDA (75.0 mg, 0.285 mmol), DTPA (20.0 mg, 0.0570 mmol), and ZnTPP (0.193 mg, 0.000285 mmol, 31.4 ppm) were combined in a 20 mL scintillation vial. The mixture was capped and irradiated with a blue LED strip (7.88 mW cm−2) for 20 h at room temperature (23 °C). The reaction was quenched by turning off the LED strip and opening the vial to air. The gel was then washed 3× with methanol to remove unreacted monomer and catalyst. The gel was then dried overnight in vacuo at room temperature (23 °C).
DMSO, MA or DMA (10.0 mmol), parent gel network (disk = ∼10 mm in diameter by 3 mm tall), and ZnTPP (2.71 mg, 0.004 mmol, 271 ppm) were combined in the bottom of a Petri dish with a diameter of 94 mm at 1 M [monomer]. The mixture was covered with the top of the Petri dish, sealed with parafilm and electrical tape, and equilibrated at room temperature (23 °C) for 24 hours. The mixture was then irradiated with a blue LED strip (7.88 mW cm−2) for 20 h at room temperature (23 °C). The reaction was quenched by turning off the LED strip and opening the Petri dish to air. The gel was then washed 3× with methanol to remove unreacted monomer and catalyst. The gel was then dried overnight in vacuo at room temperature (23 °C).
DMSO, MA or DMA (10.0 mmol), DSDA (26.2 mg, 0.100 mmol), parent gel network (disk = ∼10 mm in diameter by 3 mm tall), and ZnTPP (2.71 mg, 0.004 mmol, 271 ppm) were combined in the bottom of a Petri dish with a diameter of 94 mm at 1 M [monomer]. The mixture was covered with the top of the Petri dish and sealed with parafilm and electrical tape and equilibrated at room temperature (23 °C) for 24 hours. The mixture was then irradiated with a blue LED strip (7.88 mW cm−2) for 20 h at room temperature (23 °C). The reaction was quenched by turning off the LED strip and opening the Petri dish to air. The gel was then washed 3× with methanol to remove unreacted monomer and catalyst. The gel was then dried overnight in vacuo at room temperature (23 °C).
The dried gels (20 mg – 250 mg) were weighed (WD) and placed in excess DMAc or DI water for 24 h at room temperature (23 °C) in sealed vials. The swollen gels were removed from the vials and dabbed dry with paper towels and weighed (WS). The swelling ratios were calculated by eqn (1).
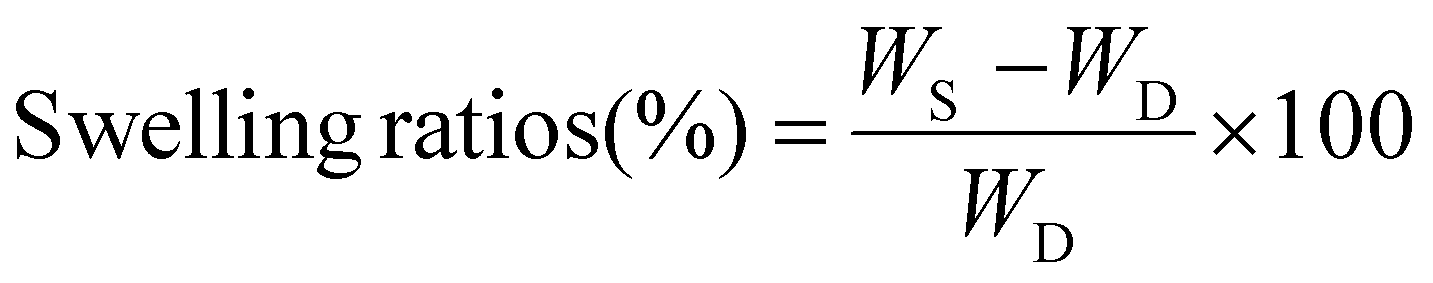 (1)
(1)
A piece of dried gel (∼20 mg) was placed in a 2-dram scintillation vial containing 3 mL of 20 mg mL−1 DTT solution in DMAc. The vial was subsequently capped with a septum and sparged with argon for 5 minutes. The sparged vial was placed in a 65 °C oil bath for 24 hours. The vial was then uncapped and MA (284 mg, 3.3 mmol) was added. The vial was subsequently capped with a septum and sparged with argon for 5 minutes. The sparged vial was placed in a 65 °C oil bath for 24 hours. The resulting solution was diluted by 50% with DMAc containing 50 mM LiCl and passed through a 0.2 μm PTFE syringe filter prior to SEC analysis.
The STEM organogel network was synthesized using oxygen tolerant PET-RAFT polymerization to yield P(MA-s-DSDA) copolymers terminated with a trithiocarbonate (Fig. 2A). The parent gel polymerization displayed a linear relationship in the pseudo-first-order kinetics plot (Fig. 2B and Table S1†) after an inhibition period of 2 hours. This inhibition period can occur in PET-RAFT polymerizations performed without deoxygenation.38 Following blue light irradiation from the bottom of the vial, a layer of the gel was green and showed signs of cracking (noted as bottom of the gel), whereas the majority of the gel was brown and appeared to be homogeneous with no cracking (as top of the gel, Fig. 2C and S1†).
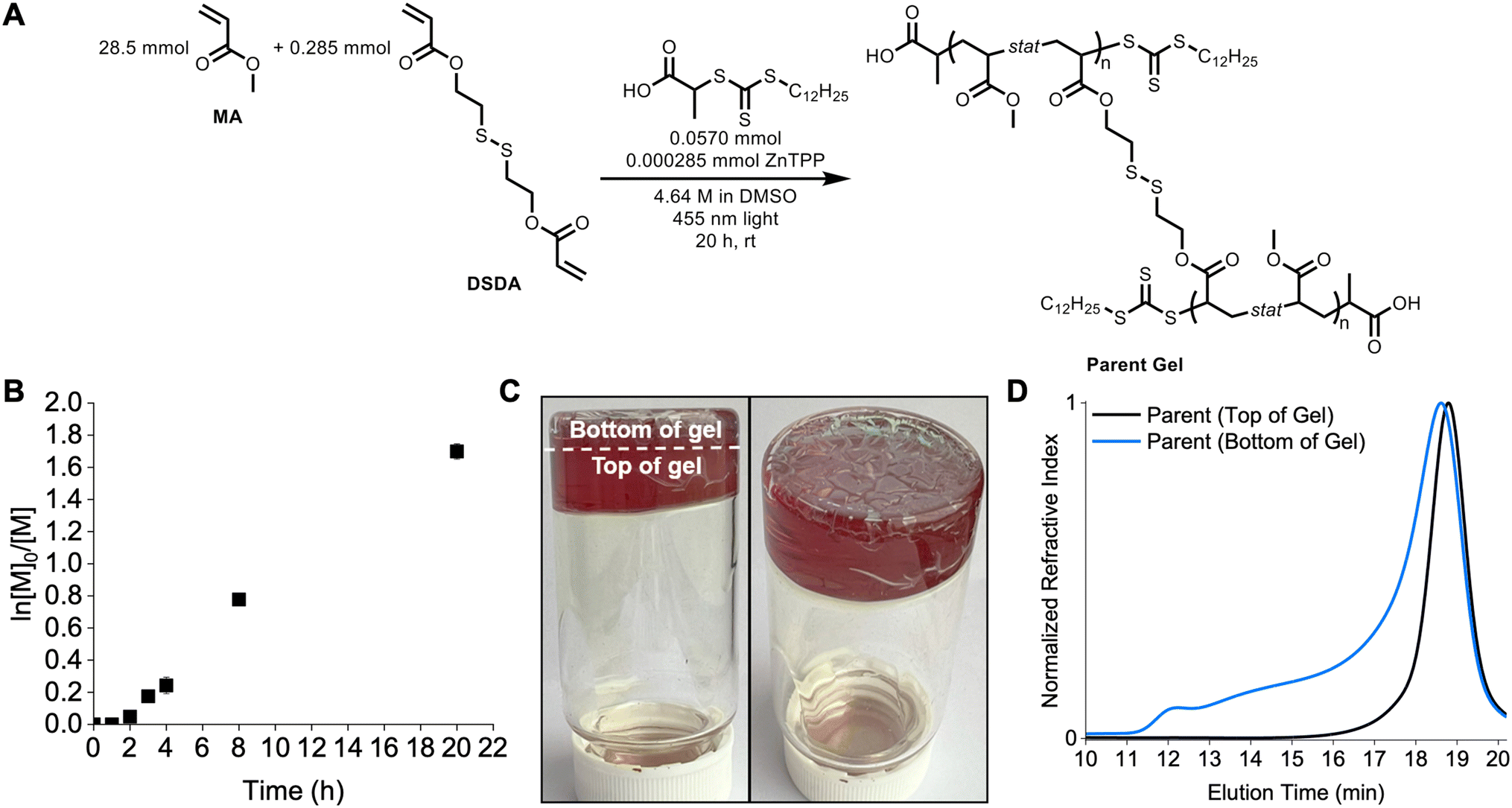
To investigate the differences between the top and bottom of the synthesized STEM gel, SEC was used to analyze both regions separately after degradation of the network polymers into linear chains using DTT. The SEC trace of the green (bottom) portion of the parent gel consisted of a broad distribution spanning the elution volume of the column and a number-average molecular weight (Mn) of 34.6 kg mol−1 with a dispersity (Đ) value of 2.79 (Fig. 2D) according to poly(methyl methacrylate) standards. The SEC trace of the top portion of the degraded gel showed a monomodal trace and Mn = 20.9 kg mol−1 with a low dispersity (Đ = 1.09) (Fig. 2D). The difference between the top and bottom parts of the parent gel is related to the positioning of the light source in the reaction setup. Specifically, the bottom of the vial absorbed the greatest intensity of light, generating a high concentration of radicals, high amounts of termination, and uncontrolled polymerization.26 The top portion of the vial received fewer photons due to scattering, resulting in a lower radical concentration and a controlled PET-RAFT polymerization. Therefore, only the top portion of the gel was used throughout the rest of the study.
Following STEM gel synthesis, we investigated the effect of PET-RAFT polymerization on the chain extension to achieve block copolymer networks. MA and DMA were used as model monomers to investigate the tunability of the physical properties, while DSDA was used as the crosslinker to maintain network degradability for molecular weight analysis. STEM gel networks were swollen in a combination of DMSO, monomers, and ZnTPP and then irradiated with blue light for 20 hours to yield four discrete daughter gels (Fig. 3A). An MA chain extension without DSDA yielded the daughter gel with a polymer structure of P((MA-s-DSDA)-b-MA) (D-MA). An MA chain extension with DSDA yielded the daughter gel with a polymer structure of P((MA-s-DSDA)-b-(MA-s-DSDA)) (D-MAX). A DMA chain extension without DSDA yielded the daughter gel with a polymer structure of P((MA-s-DSDA)-b-DMA) (D-DMA). Lastly, a DMA chain extension with DSDA yielded the daughter gel with a polymer structure of P((MA-s-DSDA)-b-(DMA-s-DSDA)) (D-DMAX). The ratio of monovinyl monomer (i.e., MA or DMA) to the divinyl DSDA monomer was kept constant at 100 : 1.
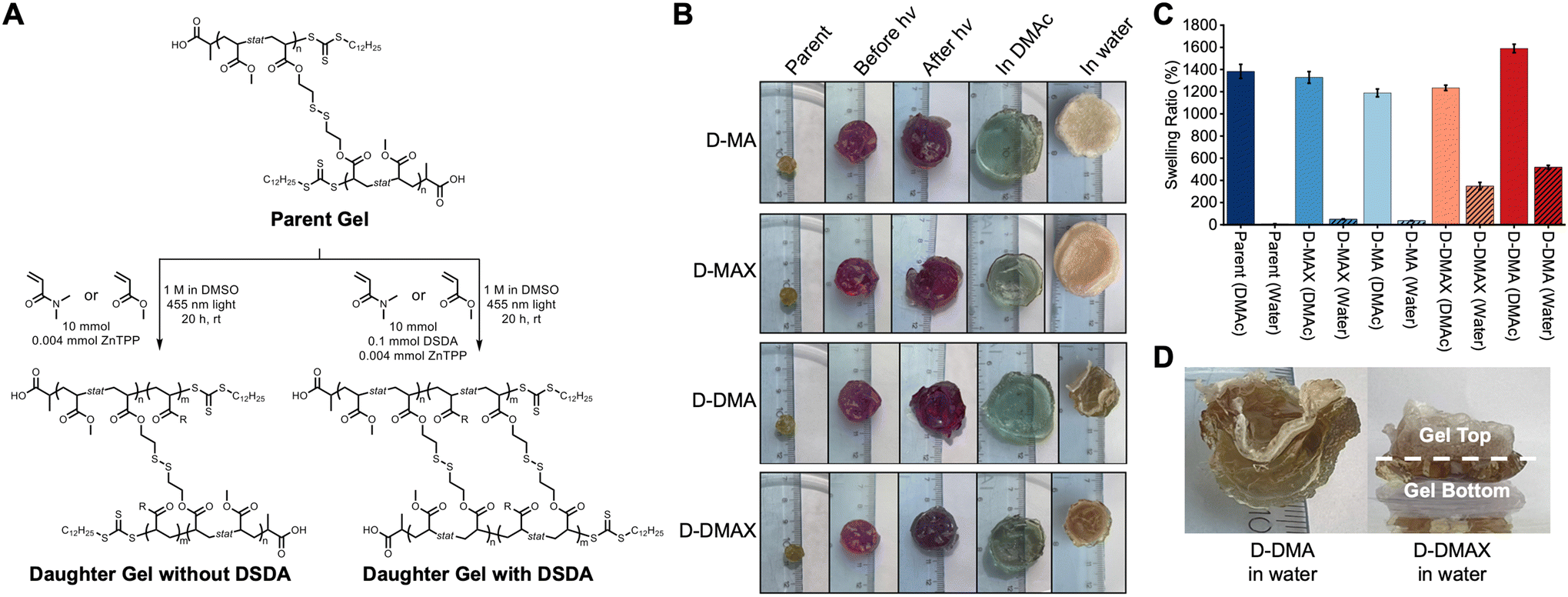
Following the chain extensions, all the daughter gels became larger in size, indicating that network extension had occurred (Fig. 3B). The daughter gels were washed 3× with methanol and dried under vacuum. Swelling ratios were then performed in excess DMAc (a good solvent for both PMA and PDMA). A decrease in swelling ratios was observed when comparing parent gels (14× swelling ratio) to D-MA (12× swelling ratio, 16% decrease compared to parent gel, Fig. S2†), D-MAX (13× swelling ratio, 4.1% decrease compared to parent gel, Fig. S3†), and D-DMAX (12× swelling ratio, 12% decrease compared to parent gel, Fig. S4†), while D-DMA showed an increase in the DMAc swelling ratio of 16× and a 15% increase compared to the parent gel (Fig. 3C, S5, and Table S2†).
Following the DMAc swelling ratio experiments, the gels were placed into excess deionized water for 24 hours (Fig. 3B, C, and Table S3†). The parent organogel swelled to 0.082× of the dry mass. An increase in swelling ratios was observed when comparing parent gels to D-MA (0.37× swelling ratio, 350% increase compared to parent gel, Fig. S6†) and D-MAX (0.50× swelling ratio, 510% increase compared to parent gel, Fig. S7†). When the parent gel was compared to the DMA-containing daughter gels an increase was observed for D-DMA (5.2× swelling ratio, 6200% increase compared to parent gel, Fig. S8†) and D-DMAX (3.5× swelling ratio, 4200% increase compared to parent gel, Fig. S9†). The DMA-containing daughter gels favorable swelling in water was expected since PDMA will impart hydrophilicity to the organogel network, transforming it into an amphiphilic network.
We observed that the chain extensions were spatiotemporally controlled. The polymerizations to synthesize the block copolymer networks were irradiated from the bottom of the Petri dish which resulted in the chain extension polymerization localized to one side of the organogel and asymmetric expansion of the materials (Fig. S10†). For example, the localized chain extensions were observable with D-DMA and D-DMAX block copolymer networks, which showed heterogeneous water swelling. In both examples, the side that was facing the light during the chain extension swelled in water and the side facing away from the light contracted (Fig. 3D).
To further investigate the spatiotemporal control of the block copolymer networks, we synthesized a daughter gel using a checkerboard pattern on the bottom of the Petri dish during an MA without DSDA chain extension polymerization. A lattice structure was imprinted onto the organogel as evidenced by distinct squares on the surface of the gel following the polymerization and swelling in DMAc (Fig. S11†).
The parent and bottoms of the daughter organogels were subjected to oscillatory shear rheometry to measure how the chain extensions modified the moduli (storage modulus (G′), loss modulus (G′′)) and overall viscoelastic behavior of the block copolymer networks compared to the STEM organogels. Amplitude, frequency, and time sweeps were run on the parent and daughter samples after being swelled in DMAc for 24 hours.
All block copolymer networks were within an order of magnitude stiffer compared to the parent organogel due to increased numbers of chain entanglements and/or chemical crosslinks (Fig. 4A, B, S12, and S13†). The materials chain-extended with DMA (D-DMA and D-DMAX) showed a higher modulus than MA chain-extended materials (D-MA and D-MAX), with the samples containing additional crosslinker (D-DMAX and D-MAX) being stiffer than non-crosslinked counterparts (D-DMA and D-MA) (Fig. 4A and S14†).
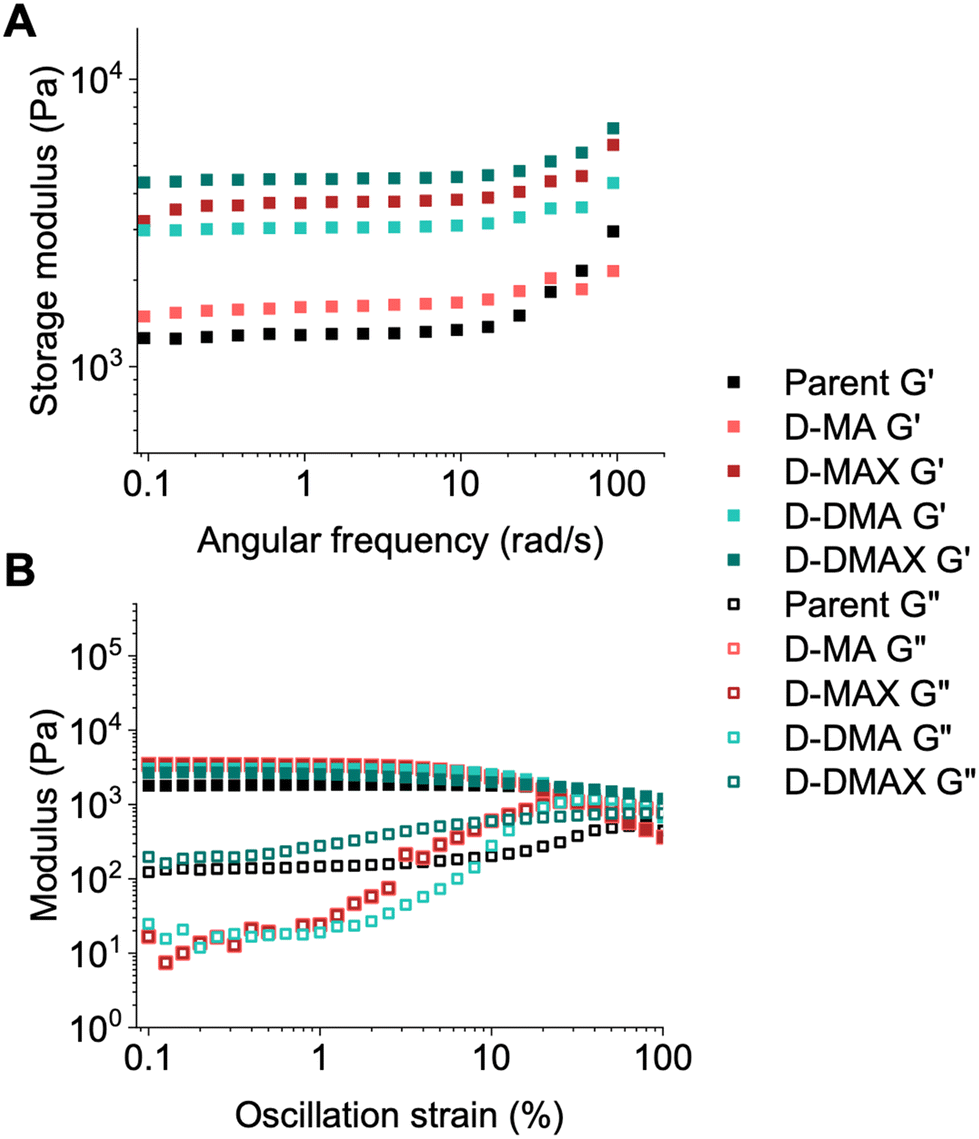
All gels consistently showed a weak frequency dependence up to ∼20 rad s−1, after which, inertial effects are likely present (Fig. 4A). These data suggest that the block copolymer networks behave as robust solid-like (i.e., elastic) materials. The amplitude sweep experiments showed that the linear-viscoelastic range was nearly constant for most of the gels (up to ∼7% strain, Fig. 4B). Interestingly, D-DMAX did not display an observable cross-over point up to 100% strain suggesting a more solid-like structure compared to the other block copolymer networks.
These rheological data indicate that the organogels became stiffer after the block copolymer network synthesis, and G′ can be modulated by a factor of 2–3× via monomer choice. Overall, the swelling ratios and rheological analyses show that the physical properties of organogels can be readily modified via PET-RAFT chain extensions.
Degradation studies were performed on the daughter gels to assess how well the molecular weights were controlled during the organogel network chain extension. Each daughter gel was separated into top and bottom portions and subjected to a solution of dithiothreitol (DTT) at 20 mg mL−1 in DMAc at 65 °C for 24 hours to degrade the network into linear chains. MA was then added to the reaction mixture to perform a thiol-Michael addition to prevent oxidation of the sulfides, which would reform the crosslinks. After 24 h at 65 °C, the mixture was diluted and filtered for characterization by SEC (Fig. 5A).
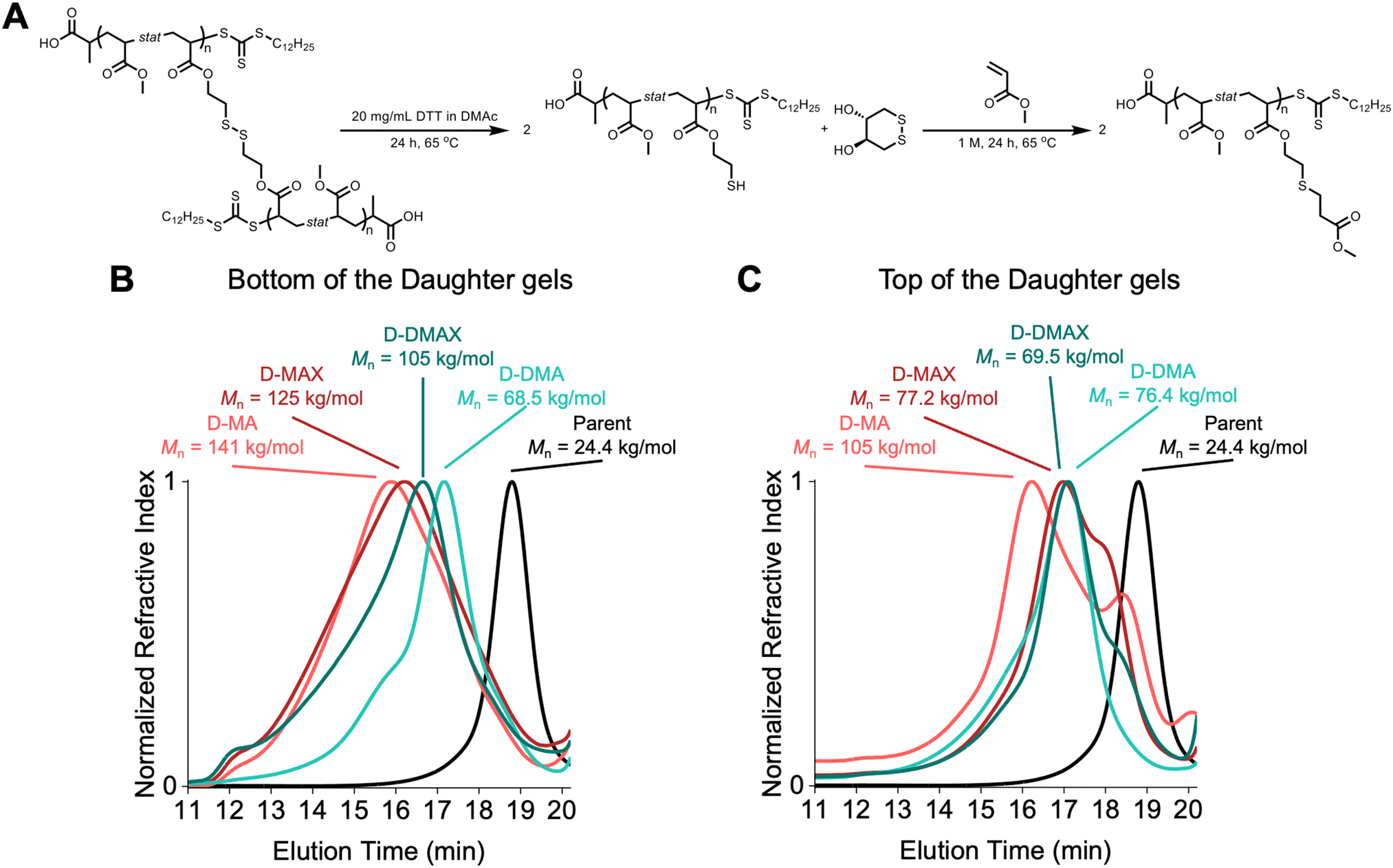
The SEC traces showed the successful network chain extension of the parent gel, indicating that the trithiocarbonate chain-ends were retained following the synthesis of the STEM organogel. However, all the organogel network chain extension block copolymers showed broad SEC traces that spanned most of the entire column volume (∼8 minutes of elution time, Fig. 5B). Two observations can be made about the chain extensions. First, the bottoms of the daughter gels showed higher molecular weights than the top counterparts, except for D-DMA. This observation is attributed to the bottom of the gels being exposed to higher light intensity, which led to a higher radical concentration and resulted in a broader molecular weight distribution, analogous to a free radical polymerization. The chain extensions resulted in heterogeneous networks as the tops and bottoms did not undergo similar chain extension reactions. Second, PMA–PMA block copolymers eluted at lower retention times compared to PMA–PDMA block copolymers. This difference is attributed to MA being a better solvent for the parent networks than DMA. Resultantly, more MA diffused into the parent gel network prior to irradiation, leading to higher local concentrations of MA compared to DMA.
The broad dispersity of molecular weights indicated that the PET-RAFT chain extension did not undergo a controlled polymerization. We expect that the limited chain mobility prevented degenerative chain transfer reactions from occurring, which are needed for a controlled RAFT polymerization.39,40 The limited chain transfer led to free radical polymerization and high amounts of termination. This result disproved our initial hypothesis that using PET-RAFT polymerization would lead to molecular weight control over the parent-to-daughter gel network extensions and suggests that additional degenerative chain transfer reaction pathways are required to maintain control over network block copolymer synthesis.
In this study, degradable organogels consisting of MA, DSDA, and DMA were synthesized via oxygen tolerant PET-RAFT polymerization. PET-RAFT block copolymer network chain extensions did not result in uniform molecular weight control as the daughter polymer chains showed broad and disperse traces by SEC analysis. Although PET-RAFT polymerization was found to result in poor molecular weight control over block copolymer network chain extensions, tunable physical properties (storage moduli, solvophilicity) according to monomer identity were still achieved. Looking forward, introducing additional reversible deactivation pathways to preserve chain-end fidelity could lead to molecular weight control of network chain extensions and better network uniformity, enabling investigations into how dispersity and molecular weight impact physical properties in block copolymer networks.
Z. B. and J. G. B. contributed equally to this manuscript about authorship; Z. B. data curation, formal analysis, visualization, writing – original draft, writing – reviewing & editing; J. G. B. conceptualization, data curation, formal analysis, visualization, writing – original draft, writing – reviewing & editing; M. J. H. data curation, formal analysis; J. D. N. data curation, formal analysis; M. G. data curation, formal analysis; N. T. data curation, formal analysis; J. C. W. data curation, formal analysis, visualization, writing – reviewing & editing; C. A. F. conceptualization, data curation, formal analysis, project administration, supervision, visualization, writing – reviewing & editing.
The data supporting this article have been included as part of the ESI.†
There are no conflicts to declare.
We gratefully acknowledge financial support from startup funds from the Department of Chemistry at Virginia Tech. This work was completed as part of CHEM 4074, so we gratefully acknowledge the academic and administrative support of the Virginia Tech Chemistry Department and College of Science. We thank Claudia Brodkin and the chemistry laboratory stockroom for their assistance with materials.
This journal is © The Royal Society of Chemistry 2024