DOI:
10.1039/D4NR02692F
(Paper)
Nanoscale, 2024,
16, 19743-19755
CD56-targeted in vivo genetic engineering of natural killer cells mediates immunotherapy for acute myeloid leukemia†
Received
29th June 2024
, Accepted 15th September 2024
First published on 4th October 2024
Abstract
Acute myeloid leukemia (AML) is a heterogeneous hematological malignancy that starts from bone marrow and spreads to other organs. At the time of diagnosis, both innate and defective natural killer (NK) cells are present in AML patients. The dysfunction of the NK cells is due to the absence of NK cell receptors such as NKG2D on tumor cells that help with tumor immune escape, and also the polycomb protein, EzH2, which plays an important role in the commitment and differentiation of NK cells. The inhibition of EzH2 activates NK cells towards enhanced lytic activity. However, the adoptive transfer of NK cells for cancer treatment is still under scrutiny due to limitations like production cost, vein-to-vein time, and complicated experimental procedures. In order to circumvent these issues, here, in vivo CD56+ NK cell genetic engineering is hypothesized through the CD56-directed delivery of the pSMP-EzH2 shRNA plasmid encapsulated in chitosan nanoparticles (pEzH2@CSNPs@CD56). The pSMP-EzH2 shRNA plasmid was encapsulated in chitosan nanoparticles followed by CD56 antibody conjugation through EDC–NHS chemistry. CD56 antibody-conjugated nanoparticles selectively target CD56+ NK cells and downregulate EzH2 expression in CD56+ NK cells of human PBMCs. The in vitro CD56+ CD3− NK cells were enriched and stably suppressed EzH2 expression to prepare adoptive CD56+ CD3− NK (EzH2−) cells for anti-AML immunotherapy. The in vitro NK (EzH2−) cells and pEzH2@CSNPs@CD56 reduced splenomegaly while immunophenotyping revealed in vivo downregulation of the c-Kit+ leukemia stem cell population along with upregulation of the differentiation markers CD11b and Gr-1 in the peripheral blood and bone marrow of AML1-ETO9a-induced xenograft nude mice. CD56+CD3− and CD56+CD38+ cell populations were significantly increased in the peripheral blood and bone marrow, which indicated NK cell-mediated AML cell killing took place suggesting that use of pEzH2@CSNPs@CD56 is a safe and viable strategy for NK cell-mediated anti-AML immunotherapy.
Introduction
Acute myeloid leukemia (AML) is a heterogeneous hematological malignancy of the myeloid lineage of white blood cells that manifests a clonal propagation of immature blasts with complex genetic and epigenetic landscapes.1,2 Genetically, chromosomal translocation t(8;21) creates the genetic fusion of AML1 and ETO to generate AML1-ETO with the ability to transform normal hematopoietic stem cells. AML1-ETO regulates several downstream targets involved in cellular progression.3 The t(8;21) anomaly is found in ∼15% of AML cases.4 AML originates in the bone marrow and further infiltrates into other organs such as the spleen.5 AML events are reported irrespective of age, race or gender with the majority of occurrences in children and in older patients’ groups but the overall survival rate is yet to be improved.6 In addition, several therapeutic regimens have been developed for AML treatment7 including immunotherapy based on chimeric antigen receptor (CAR) T cells8 and natural killer (NK) cells.9 Even though CAR T-cell therapies are under clinical trials, they still possess certain major limitations like cytokine release syndrome and neurotoxicity.10,11 In order to mitigate the risk of graft versus host disease, a complex genetic editing platform is required to remove the T cell receptor (TCR) in T cells that makes NK cells better candidates for adoptive transfer. In addition, NK cells are genetically engineered to perform an enhanced anti-tumor function by utilizing their interactions with the cancer microenvironment.12,13 Cytotoxic T cells execute tumor cell killing after priming by antigen-presenting cells14 but NK cells kill the tumor cells without any priming or prior activation.13 NK cells function with a balance of inhibitory and activation receptors, which becomes skewed towards immune escape in cancer. The inhibitory killer cell immunoglobin-like receptors (KIRs) bind to major histocompatibility complex (MHC) class I molecules and spare the healthy cells. To implement immune escape, the tumor cells suppress MHC class I and imperfect recognition activates killing signals.15,16 Afterwards, the activated NK cells secrete cytotoxic granules into the intercellular region between tumor cells and NK cells, which help execute the killing of tumor cells.17 The cytotoxic granules are complex cell organelles, mainly consisting of granzymes, perforin, and granulysin that implement pore formation on the target cell surface and finally activate apoptosis to eliminate the target cells.18 The cytolytic activity of NK cells is reduced under the prohibited expression of the CD56 NK cell surface marker, which impairs granule secretion.19 In humans, the CD56+CD3− cell population is considered a subset of NK cells, which contributes to 5–10% of all lymphocytes.20 When activated, CD56+ NK cells produce adenosine through the constitutive expression of CD38, which promotes the secretion of cytokines and granules for the effector function of NK cells.21,22 In addition, CD38 is known to be regulated by the polycomb protein EzH2 through the presence of the epigenetic marker, histone protein H3, which is trimethylated at the lysine 27 position (H3K27me3) in the CD38 promoter region.23 As per recent literature, under EzH2 inhibition in NK cells, its lineage commitment and cytotoxicity towards tumor cells are reported to have been enhanced with increased degranulation.24,25 The inhibition of EzH2 relates to the suppression of the epigenetic marker, H3K27me3, which in turn promotes the expression of the IL-15 receptor (CD122) and the NKG2D-activating receptor to increase the cytolytic function of NK cells.24 These findings enlighten the contribution of the epigenetic regulation of NK cell genesis and counsel that EzH2 inhibitors inhibit tumor growth indirectly through the adoptive transfer of NK cells. The EzH2 shRNA plasmid, pSMP-EzH2, is known to knockdown EzH2 expression in mammalian systems26 and can be utilized for genetic manipulation of NK cells with retroviral systems to prepare retrovirally in vitro EzH2− knockdown-NK cells (NK(EzH2−)) to induce anti-AML effects. However, the adoptive transfer of such NK cells for anti-tumor activity requires a robust and sophisticated cell culture facility along with trained professionals, which increases the vein-to-vein time along with the production cost.27 So, alternative strategies are required to circumvent the current limitations and risks of adoptive transfer of engineered NK cells.
Thus, we hypothesize the genetic engineering of NK cells through the CD56-targeted delivery of the EzH2 shRNA plasmid (pSMP-EzH2) encapsulated in cationic nanoparticles under heterogeneous and in situ conditions along with its comparison with in vitro retrovirally prepared EzH2− knockdown-NK cells (NK(EzH2−)). Chitosan is a cationic polymer that forms nanoparticles through electrostatic interactions with sodium tripolyphosphate (sTPP) and provides suitable non-viral vehicles for nucleic acid and gene delivery because the amine groups of chitosan electrostatically interact with the phosphate groups of plasmid DNA. The amine groups of chitosan nanoparticles protract its surface functionalization with carboxylic groups of antibodies.28,29 Here, we have prepared pSMP-EzH2-encapsulated and anti-CD56 antibody-conjugated chitosan nanoparticles (pEzH2@CsNPs@CD56) to genetically engineer in vivo NK cells and compare their therapeutic efficacy with that of NK(EzH2−) cells in AML1-ETO9a-induced AML xenografts. The surfaces of CsNPs and pSMP-EzH2-encapsulated CsNPs (pEzH2@CsNPs) were conjugated with anti-CD56 antibodies to synthesize, CsNPs@CD56 and pEzH2@CsNPs@CD56 followed by their characterization using a Zetasizer instrument, transmission electron microscopy and fluorescence analysis. The CsNPs and CsNPs@CD56 exhibited biocompatibility and excretion through a reticuloendothelial system. CsNPs@CD56 internalized in CD56+ cells compared to CD56− cells and pEzH2@CsNPs@CD56 exhibited the downregulation of EzH2 in CD56+ cells compared to CD56− cells under in vitro conditions. Furthermore, splenomegaly was reduced in the pEzH2@CsNPs@CD56-treated xenograft compared to control xenograft mice. The leukemia stem cell marker, c-Kit, was reduced and myeloid differentiation markers, CD11b and Gr-1, were upregulated in the peripheral blood and bone marrow of the pEzH2@CsNPs@CD56-treated xenografts. The CD56+ and CD38+ populations were increased in the peripheral blood and bone marrow of the pEzH2@CsNPs@CD56-treated xenografts. Also, the expression of EzH2, H3K27me3 and NPM1 were downregulated in the spleen tissues of pEzH2@CsNPs@CD56-treated xenografts, which suggested EzH2-specific immunotherapy (Scheme 1). These in vivo analyses exhibited proof of pEzH2@CsNPs@CD56 targeting CD56+ cells to activate the NK cells for the expression of CD38, which in turn executes NK cell immunotherapy towards the AML1-ETO9a-induced AML xenograft model.
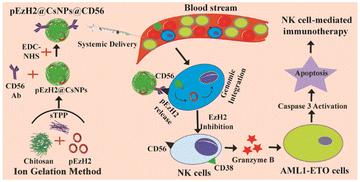 |
| | Scheme 1 Schematic representation of pEzH2@CsNPs@CD56 synthesis and its anti-AML efficacy. | |
Materials and methods
Materials
Chitosan, sodium tripolyphosphate (sTPP), ethyl(dimethylamino propyl)carbodiimide (EDC), N-hydroxysuccinimide (NHS), rhodamine B and Indocyanine Green (ICG) were purchased from TCI, Japan. Gag-Pol and VSV-G plasmids were gift samples from Dr. Johannes Zuber.30 MSCV-AML1/ETO-IRES-GFP (Addgene #60832)30 and pSMP-EzH2_1 (Addgene #36387)26 plasmids were purchased from Addgene, USA. The antibodies were purchased from Santa Cruz Biotechnology, USA.
Synthesis, modification, and characterization of chitosan nanoparticles
Chitosan nanoparticles were synthesized by the ionic gelation method using sTPP as an anionic agent having a negatively charged phosphate group that interacts with the positively charged amino groups (–NH2) of chitosan. The working chitosan stock of 1.5 mg ml−1 in 20 mM sodium acetate (NaOAc) buffer (pH 4.5) was added to 3 mg ml−1 sTPP stock at different weight/weight ratios. The optimized ratio of chitosan and sTPP was used to encapsulate the pSMP-EzH2 plasmid DNA into chitosan nanoparticles with an optimized ratio of DNA and chitosan (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000). The synthesized nanoparticles were centrifuged at 15000 rpm for 15 min and the pellets were resuspended in NaOAc buffer for dynamic light scattering (DLS) and zeta potential analysis using a Zetasizer (Malvern Instruments, UK). The DNA loading was confirmed from 0.8% agarose gel electrophoresis at 80 mV for 20 min and the intensity of stuck bands in the wells was quantified using Image Lab software (Bio-Rad) to calculate the DNA encapsulation efficiency. Furthermore, the conjugation of CD56 antibodies with blank and DNA-loaded CsNPs was performed using EDC–NHS coupling wherein –COOH groups from CD56 antibodies were activated with EDC and NHS followed by amide bond conjugation with the –NH2 group from CsNPs. Different ratios of antibody and chitosan (1:100, 1:500 and 1:1000) were used to optimize the conditions for EDC–NHS coupling. At first, the antibody was incubated with 0.5 mM EDC for 30 min and then 0.25 mM NHS was added, followed by incubation for 2 h at room temperature. Different amounts of CsNPs were mixed with EDC–NHS-activated antibodies and the whole mixture was incubated under constant stirring (200 rpm) at 4 °C for 12 h. The CD56 antibody-conjugated CsNPs (CsNPs@CD56) were settled down with centrifugation at 15000 rpm for 15 min and the pellets were resuspended in 20 mM NaOAc buffer for the DLS and zeta potential analysis. The antibody conjugation to CsNPs was confirmed by the presence of tryptophan fluorescence from the antibody (excitation at 280 nm) detected using a multiplate reader (Infinite Mplex, Tecan, Switzerland), and the antibody conjugation efficiency was quantified. An optimized ratio of the antibody and CsNPs was used to synthesize CD56 antibody-conjugated blank and DNA-loaded CsNPs followed by DLS and zeta potential analysis. Furthermore, these nanoparticles were characterized using transmission electron microscopy (TEM) analysis.
:1000). The synthesized nanoparticles were centrifuged at 15000 rpm for 15 min and the pellets were resuspended in NaOAc buffer for dynamic light scattering (DLS) and zeta potential analysis using a Zetasizer (Malvern Instruments, UK). The DNA loading was confirmed from 0.8% agarose gel electrophoresis at 80 mV for 20 min and the intensity of stuck bands in the wells was quantified using Image Lab software (Bio-Rad) to calculate the DNA encapsulation efficiency. Furthermore, the conjugation of CD56 antibodies with blank and DNA-loaded CsNPs was performed using EDC–NHS coupling wherein –COOH groups from CD56 antibodies were activated with EDC and NHS followed by amide bond conjugation with the –NH2 group from CsNPs. Different ratios of antibody and chitosan (1:100, 1:500 and 1:1000) were used to optimize the conditions for EDC–NHS coupling. At first, the antibody was incubated with 0.5 mM EDC for 30 min and then 0.25 mM NHS was added, followed by incubation for 2 h at room temperature. Different amounts of CsNPs were mixed with EDC–NHS-activated antibodies and the whole mixture was incubated under constant stirring (200 rpm) at 4 °C for 12 h. The CD56 antibody-conjugated CsNPs (CsNPs@CD56) were settled down with centrifugation at 15000 rpm for 15 min and the pellets were resuspended in 20 mM NaOAc buffer for the DLS and zeta potential analysis. The antibody conjugation to CsNPs was confirmed by the presence of tryptophan fluorescence from the antibody (excitation at 280 nm) detected using a multiplate reader (Infinite Mplex, Tecan, Switzerland), and the antibody conjugation efficiency was quantified. An optimized ratio of the antibody and CsNPs was used to synthesize CD56 antibody-conjugated blank and DNA-loaded CsNPs followed by DLS and zeta potential analysis. Furthermore, these nanoparticles were characterized using transmission electron microscopy (TEM) analysis.
CsNPs tagging with rhodamine B and Indocyanine Green (ICG)
Chitosan nanoparticles and modified chitosan nanoparticles were incubated overnight with 100 μM rhodamine B and ICG with constant shaking followed by centrifugation at 15000 rpm for 15 min. The pellets were resuspended in 20 mM sodium acetate buffer and UV-sterilized for 1 h before treating the cells and animals.
In vivo biodistribution of nanoparticles
All animal experiments used in the present study complied with the guidelines of the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA) and were approved by the Institutional Animal Ethical Committee (IAEC) of the Indian Institute of Science Education and Research, Mohali (IISER, Mohali) and National Agri-Food Biotechnology Institute (NABI, Mohali). Concentrations of 1 mg kg−1 ICG-tagged CsNPs and CsNPs@CD56 were administered to female Balb/c mice through the tail vein and time-dependent ICG photoluminescence was recorded as a function of nanoparticle accumulation in different organs. After 24 h, the ICG photoluminescence was monitored under ex vivo conditions using an In Vivo Imaging System (IVIS) (PerkinElmer).
In vitro plasmid DNA release assay
2 μg equivalents of pEzH2@CsNPs and pEzH2@CsNPs@CD56 were resuspended in 10 mM phosphate buffered saline (PBS) (pH 7.4) and kept under orbital shaking. At each time point, the nanoparticles were centrifuged at 15000 rpm for 15 min and 10 μl of the supernatant was collected followed by the replenishment of 10 μl of fresh PBS. The aliquots of supernatant samples were read on a NanoQuant Plate (Mplex Infinite, Tecan, Switzerland) and the percentage of cumulative release DNA was plotted against the incubation time.
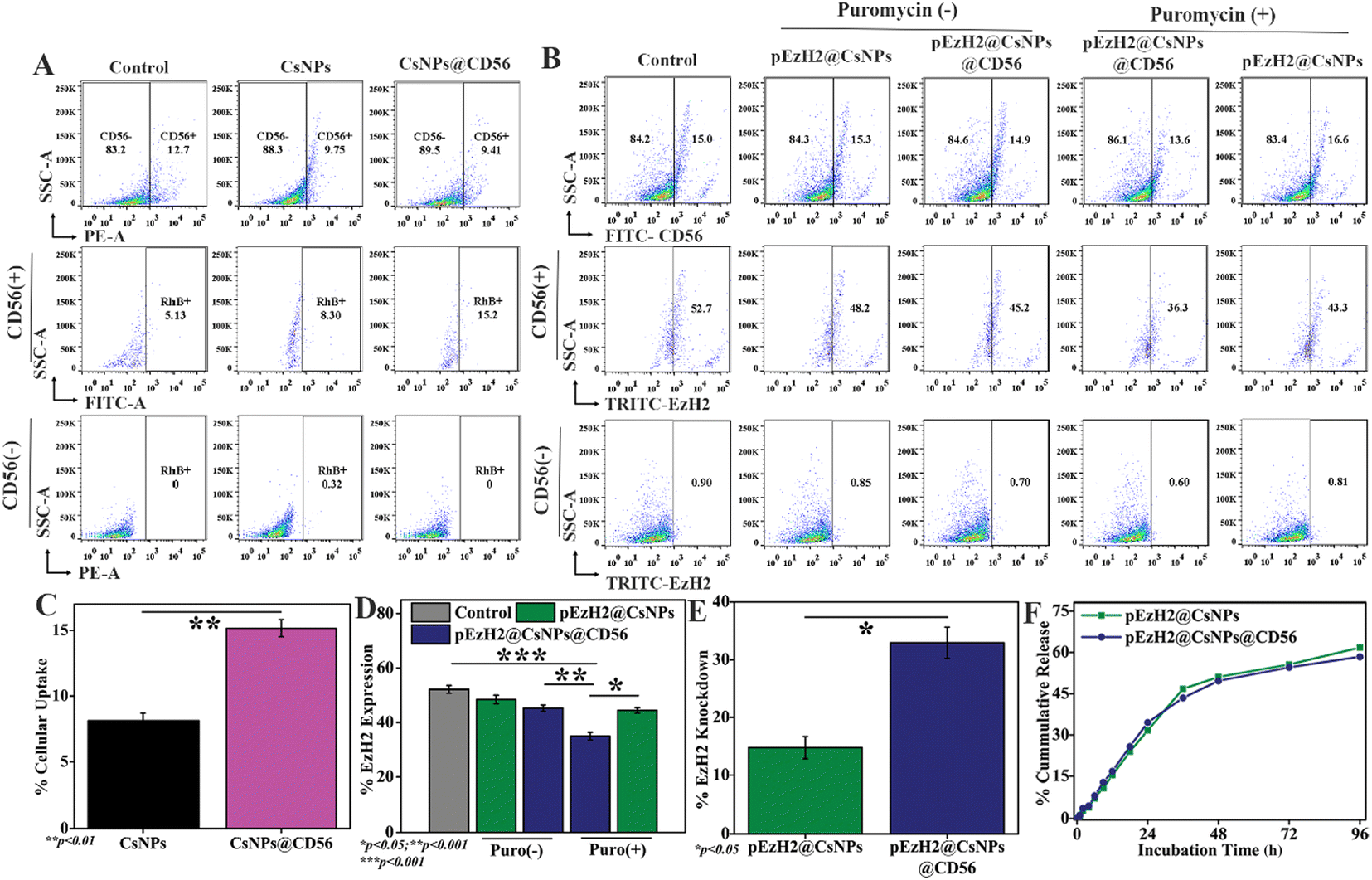
In vitro evaluation of the CD56-specific downregulation of EzH2 by pEzH2@CsNPs@CD56
U937 and HEK293T cells were procured from NCCS, Pune and maintained in RPMI-1640 and DMEM, respectively, supplemented with 10% fetal bovine serum (FBS) and 1% antibiotics. MSCV-AML1/ETO-IRES-GFP and pSMP-EzH2_1 lipofectamine-based transfection in HEK293T cells using Gag-Pol and VSV-G plasmids was carried out to produce AML1-ETO9a retroviral particles with a titre value of 3.5 × 105 transduction units per ml when the GFP expression was estimated using a multiplate reader. The following formula was used to calculate the retroviral titre:
Furthermore, AML1-ETO9a cells were prepared by infecting U937 cells with the MSCV-AML1/ETO-IRES-GFP retrovirus and the polybrene reagent on fibronectin-coated culture flasks. After infection, the green fluorescent protein (GFP) as a function of AML1-ETO9a expression was checked by confocal laser scanning microscopy (CLSM) to confirm the formation of AML1-ETO9a expressing cells.
In addition, the peripheral blood samples were collected from AML patients at the Post-Graduate Institute of Medical Education and Research (PGIMER), Chandigarh as per ethical guidelines and approval (IEC no.: IEC-11/2020-1841). The blood samples were treated with Ficoll-Paque to isolate the peripheral blood mononuclear cells (PBMCs). The PBMCs were cultured under in vitro conditions of RPMI-1640 supplemented with 10% fetal bovine serum (FBS), and 1% antibiotics. Afterwards, CD56+CD3− NK cells were analyzed in PBMCs using a flow cytometer (Aria Fusion, BD). The PBMCs were treated with rhodamine B-tagged CsNPs and CsNPs@CD56 for 4 h to perform the internalization of nanoparticles under heterogeneous cell-mimicking conditions. Afterwards, the cells were harvested and stained with the FITC-tagged CD56 antibody (mouse produced) and flow cytometry analysis was carried out for the cellular internalization of CsNPs and CsNPs@CD56 in CD56+ and CD56− populations. Furthermore, PBMCs were treated with pEzH2@CsNPs and pEzH2@CsNPs@CD56 for 48 h to inhibit EzH2 expression through nanoparticles under heterogeneous cell-mimicking conditions. These cells were treated with puromycin (10 μg ml−1) for an additional 24 h and were harvested, washed with 10 mM PBS, and stained with the FITC-tagged CD56 antibodies for 1 h. These stained cells were washed, fixed using formaldehyde, permeabilized with 90% methanol and blocked using 2.5% albumin. The expression of EzH2 was examined in CD56+ and CD56− populations using a flow cytometer after staining with anti-EzH2 primary antibodies (rabbit produced) and counterstaining with TRITC-conjugated anti-rabbit secondary antibodies. Furthermore, T cells were depleted in human PBMCs using anti-CD3 antibodies and interleukin-2 (IL-2) to enrich the CD56+ cell population.31 CD56+CD3− cells were sorted from enriched PBMCs using a flow cytometer and maintained under in vitro conditions. The sorted CD56+CD3− cells were treated with different amounts of pEzH2@CsNPs@CD56 for 48 h and the expression of EzH2 was estimated as above using flow cytometry. The percentage EzH2 knockdown efficiency was calculated as per the control. Additionally, CD56+CD3− cells were infected with pSMP-EzH2 retroviral particles with the polybrene reagent as above. The infection-positive cells were stably selected with puromycin for 7 days and EzH2 expression was monitored using a flow cytometer as described above and the percentage EzH2 knockdown efficiency was calculated as per the control. These retrovirally produced CD56+CD3−EzH2− (NK(EzH2−)) cells were propagated under in vitro conditions for in vivo experimentations.
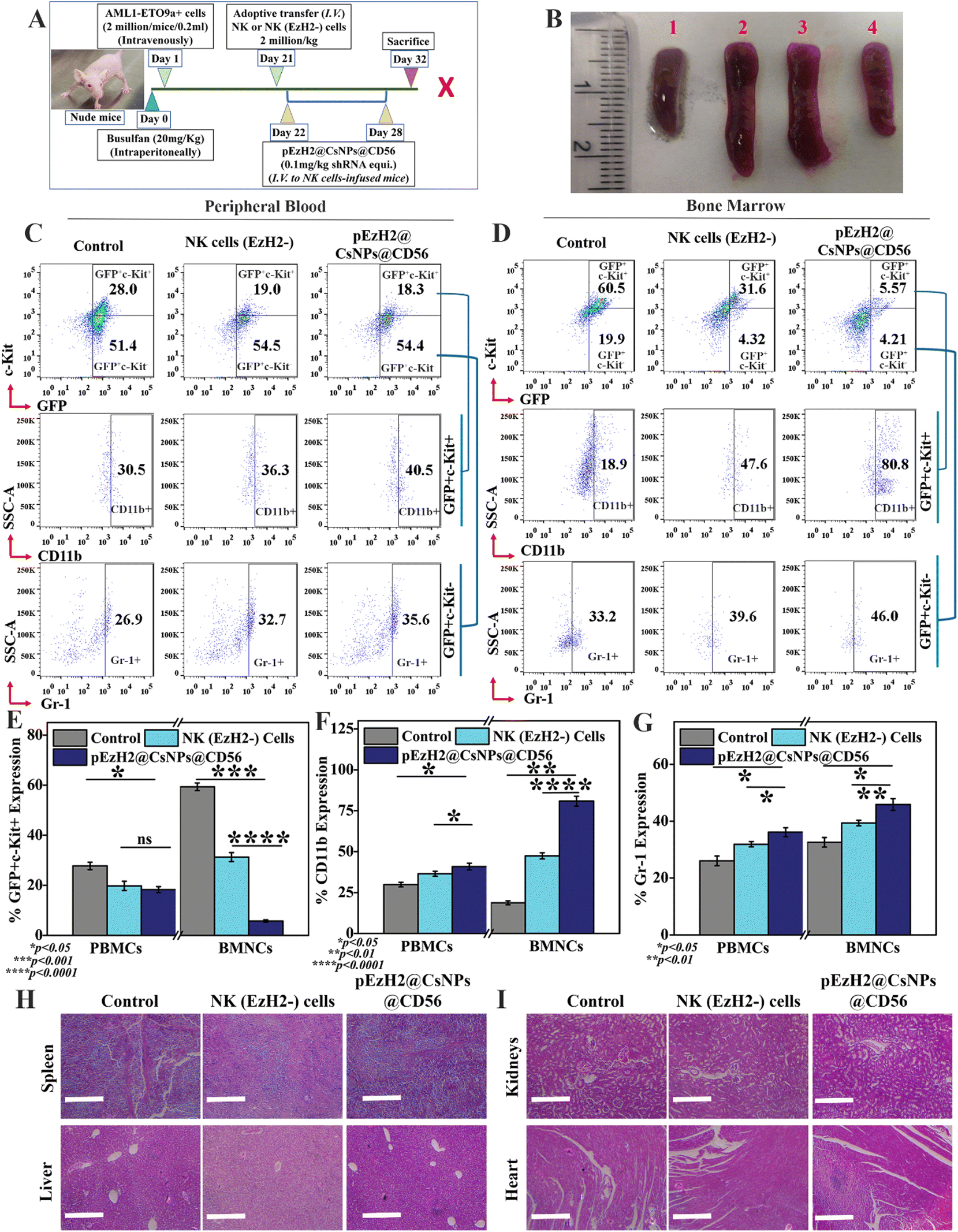
NK cell immunotherapeutic evaluation of pEzH2@CsNPs@CD56 in AML1-ETO9a-induced AML athymic xenograft
All animal experiments used in the present study complied with the guidelines of the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA) and were approved by the Institutional Animal Ethical Committee (IAEC) of the Indian Institute of Science Education and Research, Mohali (IISER, Mohali). 2 million AML1-ETO9a cells were intravenously infused in busulfan-treated athymic mice and, at day 21, GFP signals were evaluated in peripheral blood samples using a flow cytometer. Afterwards, 2 million CD56+CD3− NK and CD56+CD3−EzH2− NK (EzH2−) cells per kg were adoptively transferred to animals and monitored. The xenograft mice infused with CD56+CD3− cells were further intravenously administered with pEzH2@CsNPs@CD56 (0.1 mg shRNA plasmid per kg equivalent) that had been resuspended in puromycin solution (10 μg ml−1) for 5 days.32,33 Post-treatment, splenomegaly was documented followed by GFP signal monitoring and immunophenotyping with C-Kit, CD11b and Gr-1 antibodies in the peripheral blood and bone marrow samples. Furthermore, major organs like the spleen, liver, kidneys, and heart were isolated, fixed and sectioned into 10 μm slices for hematoxylin and eosin (H & E) staining to study the toxicity of the treatment regimen. In addition, the peripheral blood and bone marrow-resident CD56+CD3− NK cell population was monitored through flow cytometry. Also, the peripheral blood and bone marrow-resident CD56+CD38+ cell population was monitored through flow cytometry. The protein was isolated from spleen tissues of control and treated xenografts and transferred to a PVDF membrane for western blot analysis using β-actin, EzH2, H3K27me3, NPM1, caspase 3 and CD38 expression profiling. Spleen tissues were homogenized and cross-linked for anti-EzH2 chromatin immunoprecipitation (ChIP) assay to analyse the presence of EzH2 on the promoter regions of NPM1. ChIP DNA was used to perform the qPCR analysis using NPM1 promoter region-specific primers as listed in ESI Table S2.†
Statistics analysis
All experiments were performed with n = 3, where n is the number of experimental repeats. Bonferroni and Tuckey tests were performed at a significance level of *p < 0.05.
Results and discussion
CD56 antibody conjugates to chitosan nanoparticles through EDC–NHS coupling
The CD56+ NK cell-based immunotherapy follows through the adoptive transfer of in vitro activated CD56+ NK cells wherein EzH2 downregulated NK cells exhibit enhanced activity and can be achieved through in vitro retroviral-based infection followed by infusion.24,25 The high-end requirement of this method demands an innovative non-viral method wherein we hypothesize a CD56-targeted delivery of EzH2 shRNA to specifically downregulate the EzH2 expression in NK cells for enhancing anti-AML immunotherapy in AML1-ETO9a-induced AML xenografts. The delivery of the EzH2 shRNA plasmid through chitosan nanoparticles (CsNPs) as vehicles has been reported in earlier literature and these spatially provide CD56 antibody conjugation sites.29,34 Here, chitosan nanoparticles (CsNPs) were synthesized using different ratios of chitosan and sTPP as the cross-linker. Here, the 2:0.9 ratio of chitosan and sTPP, the nanoparticle size of 142 nm and zeta potential of 25.3 ± 2.7 mV were found to be the optimum conditions that were used for further experimentation (Fig. S1†). The amines from the chitosan structure interact with the phosphates from sodium tripolyphosphate (sTPP) and the optimized ratio between the two provides a stable assembly of chitosan and sTPP in the form of nanoparticles.28 Chitosan also provides an electrostatic platform for phosphate groups from nucleic acids and encapsulates them during the ion gelation process with a slight change in the dynamics of electrostatic interaction. The agarose gel retardation assay demonstrated that chitosan held the plasmid DNA in the wells of the gel as indicated in Fig. 1C. The movement of free plasmid DNA was as per the electrophoretic mobility suggesting that the DNA and chitosan interaction was strong and became stronger as the ratio of DNA to chitosan increased (Fig. 1C). The DNA was loaded onto CsNPs with a 1:1000 ratio of DNA and chitosan, and the size of EzH2 plasmid DNA-loaded CsNPs (pEzH2@CsNPs) was found to be similar to that of blank CsNPs (Fig. 1A) but the zeta potential of pEzH2@CsNPs was reduced to 18.7 ± 1.5 mV (p value: 0.038) (Fig. 1B), which suggested DNA loading to CsNPs. CD56 antibody–CsNP conjugation using EDC–NHS chemistry was optimized based on the ratio of the antibody and CsNPs. Here, the 1:500 ratio of the CD56 antibody and CsNPs was found to be optimum as is evident from the significantly increased hydrodynamic size of 285 nm (Fig. 1E), and reduced zeta potential of 16.8 ± 0.9 mV (Fig. 1F). However, the size of this ratio was found to be similar and overlapping to that of other ratios of CD56 antibody and CsNPs (Fig. 1E). The CD56 antibody functionalization of CsNPs changes the surface characteristics of the nanoparticles as is evident from the reduced zeta potential and supported by an earlier study on the functionalization of CsNPs with CD7 antibodies.35 Furthermore, the percentage of CD56 antibody conjugation to CsNPs was calculated through the standard graph of CD56 antibody intrinsic fluorescence due to tryptophan residues (Fig. S2†) and found to be at maximum (70.9%) for a 1:500 ratio of antibody and CsNPs (ESI Table S1†). The surface of CsNPs was conjugated with CD56 antibody conjugation as is evident from the presence of tryptophan fluorescence in the pellets of the nanoparticles. The maximum antibody conjugation was found with a 1:500 ratio of the CD56 antibody and CsNPs (Fig. 1G). In general, tryptophan residues are found in the antibody structure, which exhibits fluorescence characteristics with the maximum emission at ∼320 nm36 and when the antibody is surface-functionalized to the nanoparticles, the presence of tryptophan fluorescence of the antibody in CsNPs establishes a proof of antibody conjugation of nanoparticles. An earlier report on gold nanoparticles and antibody conjugation highlights the presence of tryptophan fluorescence of the antibody after the conjugation, which led to an efficient detection of the C-reactive protein.37 The hydrodynamic size and zeta potential of pEzH2@CsNPs@CD56 were found to be 301 nm (Fig. 1E) and 13.33 ± 0.7 mV (p value: 0.0163) (Fig. 1F), respectively, along with the CD56 conjugation of 70.1% (Fig. 1G and ESI Table S1†). The negative charge of DNA contributes to the reduction of the zeta potential of CsNPs. Also, the DNA loading was confirmed from agarose gel electrophoresis as indicated by the presence of DNA in the well of the pellets of pEzH2@CsNPs and pEzH2@CsNPs@CD56 (Fig. 1D). The DNA encapsulation efficiency was found to be ∼83.33% and ∼82.3% for pEzH2@CsNPs and pEzH2@CsNPs@CD56, respectively. The chitosan encapsulates plasmid DNA utilizing the electrostatic interaction between amine and phosphate groups wherein sTPP cross-links the structure to hold the nanoparticulates in a stable form as supported by earlier literature.38 Transmission electron microscopy (TEM) revealed the spherical shape and even distribution of nanoparticles with small aggregates (Fig. 1H–K). The diameters of CsNPs, pEzH2@CsNPs, CsNPs@CD56 and pEzH2@CsNPs@CD56 were found to be 71.5 nm, 77.8 nm, 102.1 nm and 110.1 nm, respectively. The size distribution based on the TEM imaging exhibited a similar pattern to that in DLS analysis (Fig. S3†). Here, the pEzH2 plasmid encapsulation did not change the overall size distribution of nanoparticles whereas the surface conjugation of CD56 antibodies altered the size distribution of nanoparticles. This analysis is supported by an earlier study on sTPP-stabilized chitosan nanoparticles, which exhibit a spherical nanoparticle shape.39
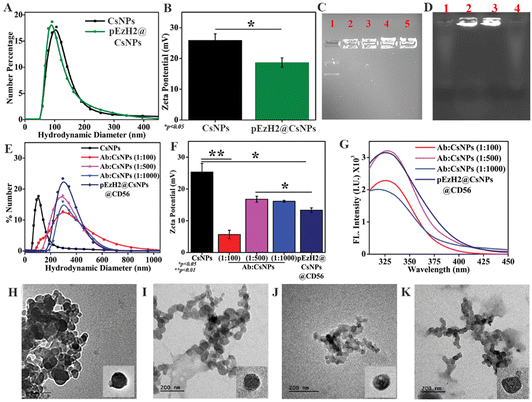 |
| | Fig. 1 Synthesis, modification, and characterization of chitosan nanoparticles; (A) dynamic light scattering and (B) zeta potential analysis of CsNPs and pEzH2@CsNPs; 0.8% (w/v) agarose gel electrophoresis for (C) pEzH2 and chitosan interaction analysis (Lane 1- pSMP-EzH2 free, plasmid: chitosan ratio of Lane 2–1:100, Lane 3–1:250, Lane 4–1:500, and Lane 5: 1:1000), and (D) pEzH2 encapsulation (Lane 1: supernatant and Lane 2: pellets of pEzH2@CsNPs; and Lane 3: pellets and Lane 4: supernatant of pEzH2@CsNPs@CD56); conjugation and characterization of the CD56 antibody to CsNPs, (E) dynamic light scattering, (F) zeta potential analysis, and (G) intrinsic tryptophan fluorescence analysis. Transmission electron microscopy (TEM) analysis of (H) CsNPs, (I) pEzH2@CsNPs, (J) CsNPs@CD56, and (K) pEzH2@CsNPs@CD56. All experiments were performed with n = 3, where n is the number of experimental repeats. Bonferroni and Tuckey tests were performed at a significance level of *p < 0.05 and **p < 0.01. | |
Chitosan nanoparticles exhibit renal clearance
The time-dependent ICG photoluminescence as a function of in vivo biodistribution of CsNPs and CsNPs@CD56 revealed the in vivo localization and excretion of nanoparticles. The ICG photoluminescence was depleted within 24 h of intravenous administration suggesting the clearance of nanoparticles (Fig. 2A). The ex vivo ICG photoluminescence revealed the presence of traces of nanoparticles in kidneys suggesting the renal clearance of the chitosan nanoparticles (Fig. 2B–E). This is in lieu of an earlier study on tissue accumulation (as indicated in low renal retention) and biocompatibility patterns of CsNPs, which followed the excretion via the reticuloendothelial system after intravenous administration.40 The intravenous administration of chitosan nanoparticles (1 mg kg−1) is proven safe and biocompatible for mice as the earlier article explained the absence of the systemic hemolysis up to 4 mg kg−1 dosage.40
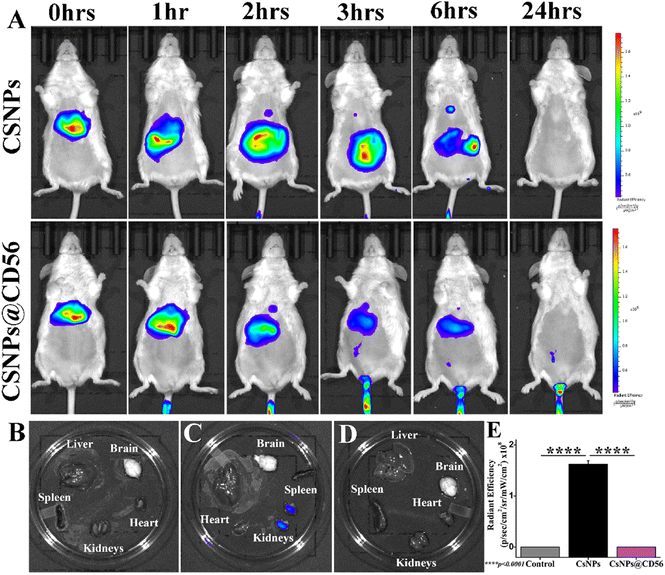 |
| | Fig. 2
In vivo biodistribution of ICG-tagged CsNPs and CsNPs@CD56; (A) time-dependent ICG photoluminescence analysis as a function of nanoparticles’ localization and accumulation, ICG photoluminescence analysis under ex vivo conditions of (B) control, (C) CsNPs, and (D) CsNPs@@CD56-administered mice; and (E) quantification of photoluminescence of ICG as a function of accumulation of CsNPs in mice kidneys. All experiments were performed with n = 3, where n is the number of experimental repeats. Bonferroni and Tuckey tests were performed at a significance level of ****p < 0.0001. | |
pEzH2@CsNPs@CD56 specifically downregulates EzH2 in CD56+ NK cells of human PBMCs
To exhibit the CD56-specific downregulation of EzH2, pSMP-EzH2 shRNA must specifically internalize into NK cells and release into the intracellular space for genomic integration where the systemic presence of pSMP-EzH2 shRNA downregulates EzH2.26 The pSMP-EzH2 shRNA plasmid DNA was released from pEzH2@CsNPs and pEzH2@CsNPs@CD56 through a sustained release pattern with maximum release of ∼50% in 48 h. The release patterns from both nanoparticles were found to be similar under physiological-mimicking conditions of pH 7.4 provided by 10 mM PBS (Fig. 3F). Chitosan nanoparticles are known to exhibit the sustained release pattern of the encapsulated pSMP-EzH2 shRNA plasmid and enhanced the transfection efficiency.41 This released DNA can be incorporated into the genomic DNA of target cells and express the EzH2 shRNA to suppress the expression of EzH2 in the presence of puromycin as explained by Onder et al.26 The CD56 antibody conjugation was performed to selectively inhibit the EzH2 expression in CD56+ NK cells for enhanced immunotherapy. Furthermore, the CD56+ NK cells were analyzed using a flow cytometer and found to be ∼10.1% in human peripheral blood mononuclear cells (PBMCs) from a healthy donor (Fig. S4†), which is supported by earlier reported data.20 To evaluate the CD56 targeting ability, human PBMCs were treated with nanoparticles to mimic in vivo heterogeneous conditions. CsNPs@CD56 were 1.83-fold and 2.96-fold uptaken by CD56+ NK cells of human PBMCs compared to CsNPs and the control, respectively, as analyzed by flow cytometry (Fig. 3A). The rhodamine B fluorescence intensity as a functional validation of uptake by nanoparticles was also found to be increased ∼1.9-fold by CsNPs@CD56 compared with CsNPs in CD56+ cells (p value: 0.0097) (Fig. 3C). The presence of CD56 antibodies on CsNPs@CD56 promotes their internalization into the CD56+ NK cells as indicated by the presence of rhodamine B fluorescence. This follows the receptor-mediated endocytosis of CD56 antibodies, which reaches the intracellular compartments as explained earlier by the internalization of CD56 antibody–drug conjugates and their therapeutic effects on neuroblastoma cell lines.42 This CD56-specific and higher internalization of CsNPs@CD56 makes them suitable vehicles for the pSMP-EzH2 shRNA plasmid for genetic engineering of NK cells. The EzH2 expression was downregulated by ∼1.7-fold after pEzH2@CsNPs@CD56 treatment compared to that of pEzH2@CsNPs treatment in CD56+ NK cells from human PBMCs (p value: 0.0048). The CD56 antibody conjugation effectively escorts the pSMP-EzH2 plasmid DNA to CD56+ cells wherein pSMP-EzH2 downregulates the EzH2 expression. In addition, the EzH2 expression was downregulated by ∼16.4% and ∼9.4% in CD56+ NK cells after treatment with pEzH2@CsNPs@CD56 and pEzH2@CsNPs in the presence of puromycin (Fig. 3B & D). The percentage EzH2 knockdown efficiency in CD56+ NK cells was 14.8% and 32.9% by pEzH2@CsNPs and pEzH2@CsNPs@CD56, respectively (p value: 0.0201) (Fig. 3E). The plasmid expresses EzH2 shRNA, which suppresses EzH2 expression in the presence of puromycin.26 These analyses establish the CD56-selectivity of pEzH2@CsNPs@CD56 in a heterogeneous cell population of PBMCs and can be used for in vivo genetic engineering of CD56+ NK cells for anti-AML immunotherapy in AML1-ETO9a-induced AML xenografts. To compare the pEzH2@CsNPs@CD56 efficacy with the adoptive transfer of CD56+ NK cells, the CD56+CD3− cell population was enriched in human PBMCs with anti-CD3 antibodies and IL-2, which was found to be ∼27.8% (Fig. S5†). Anti-CD3 antibodies deplete the CD3+ T cells from PBMCs and help increase the CD56+ NK cells, which can affect the adoptive NK cell activity while IL-2 promotes the CD56 activation.31 The sorted CD56+CD3− population increased the CD56 expression by ∼62.6% when maintained under in vitro conditions as supported by an earlier study.31 When the sorted CD56+CD3− cells were treated with pEzH2@CsNPs@CD56, the EzH2 expression was downregulated with increasing numbers of nanoparticles (Fig. S6A and B†). The percentage of the EzH2 knockdown efficiency was found to be at a maximum for 50 ng of nanoparticles, which was 29.4% in CD56+ NK cells (Fig. S6C†). The exogenous plasmid shRNA plasmid DNA reaches the cytoplasm through cellular interactions of nanoparticles and releases the DNA into the cytoplasm. These shRNA DNA strands enter the nucleus by wrapping the nuclear envelope-like membrane at the telophase to exert their effect as explained by an earlier study of nuclear envelope formation at the telophase of mitosis.43 In addition, EzH2 expression was suppressed by ∼29.3% in enriched CD56+CD3− cells when infected with the pSMP-EzH2 retroviral particle in the presence of puromycin (Fig. S7†). The stable integration of EzH2 shRNA suppresses the EzH2 expression through puromycin selection as explained earlier.26 These enriched CD56+CD3−EzH2− NK cells (NK(EzH2−) cells) can be utilized for NK cell-mediated anti-AML immunotherapy. The EzH2 downregulation by in vitro NK(EzH2−) cells and pEzH2@CsNPs@CD56 in NK cells under in vitro conditions explain the enhanced targetability of pEzH2@CsNPs@CD56 and argues for the superior efficiency and safety of the non-viral preparation44 of genetically engineered NK cells for anti-AML therapeutics. In order to study NK(EzH2−) cells and pEzH2@CsNPs@CD56-mediated anti-AML therapeutics, the genetic fusion oncogene, AML1-ETO9a-induced AML model was developed under in vitro conditions through retroviral transduction with a titre value of 3.5 × 105 transduction units per ml, and the presence of green fluorescent protein (GFP) as a function of AML1-ETO9a (an alternatively spliced variant of AML1-ETO) expression was monitored (Fig. S8†). The GFP expression correlates with the AML1-ETO9a expression, which efficiently generates AML pathology as supported by an earlier study of Zuber et al.,30 and these AML1-ETO9a cells can be used for xenograft development followed by the evaluation and comparison of NK(EzH2−) cells and pEzH2@CsNPs@CD56-mediated anti-AML therapeutics.
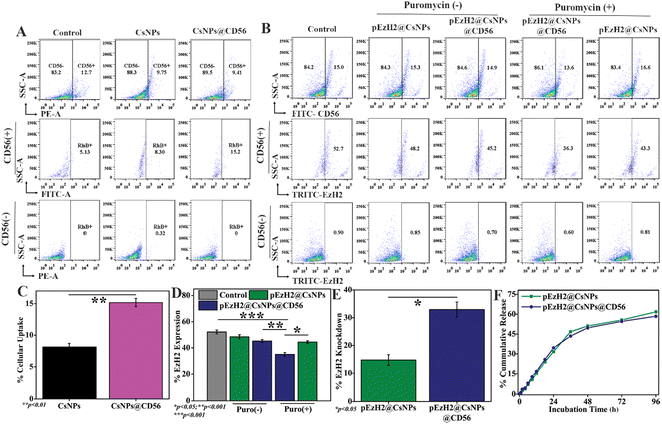 |
| | Fig. 3
In vitro studies to evaluate the targetability of CD56 conjugated chitosan nanoparticles; (A) flow cytometry of cellular internalization of rhodamine B-tagged CsNPs and CsNPs@CD56 under heterogeneous-mimicking conditions of peripheral blood mononuclear cells, and (C) quantification of rhodamine B fluorescence in CD56+ cells; (B) flow cytometry of the EzH2 expression under heterogeneous-mimicking conditions of peripheral blood mononuclear cells, and (D) its quantification in CD56+ NK cells after treatment of pEzH2@CsNPs and pEzH2@CsNPs@CD56, in the presence and absence of puromycin along with (E) calculation of the EzH2 knockdown efficiency of pEzH2@CsNPs and pEzH2@CsNPs@CD56 in CD56+ NK cells; (F) In vitro plasmid DNA release assay for pEzH2@CsNPs and pEzH2@CsNPs@CD56 in 10 mM PBS (pH7.4). All experiments were performed with n = 3, where n is the number of experimental repeats. Bonferroni and Tuckey tests were performed at a significance level of *p < 0.05, **p < 0.01 and ***p < 0.001. | |
pEzH2@CsNPs@CD56 induce superior immunotherapeutics in AML1-ETO9a-induced AML
NK cells exhibit cytotoxic effects through the secretion of granzyme B and activation of apoptosis of target cells without any priming or prior activation13 wherein the EzH2 downregulation increases the CD38 expression, which is related to the production of granzyme B and activation of caspase 3-dependent apoptosis of target cells.21,23 The downregulation of EzH2 by NK(EzH2−) cells and pEzH2@CsNPs@CD56 was achieved under in vitro conditions and to evaluate the in vivo anti-AML therapeutics implication of NK(EzH2−) cells and pEzH2@CsNPs@CD56, the genetic fusion oncogene, AML1-ETO9a-induced AML xenograft model was developed. Fig. 4A schematically represents the study plan of the in vivo therapeutics. After the administration of AML1-ETO9a cells, the GFP expression was found to be ∼37.2% in the peripheral blood of xenografts compared to that of non-xenograft mice (Fig. S9†). The presence of GFP expression in the peripheral blood of xenograft mice entails the propagation of AML1-ETO9a cells under in vivo conditions, which validates the xenotransplantation as supported by an earlier study.45 As AML1-ETO9a cells propagate to the peripheral blood, they infiltrate the spleen causing an increase in spleen size, known as splenomegaly.46 The adoptive transfer of NK(EzH2−) cells exhibited the reduction in splenomegaly compared to the control xenograft wherein pEzH2-CsNPs@CD56-treated xenograft exhibited a further reduction in splenomegaly (Fig. 4B), which is related to superior anti-AML therapeutics. The NK cells can recognize the AML1-ETO9a cells as foreign entities through MHC I mismatch, which block the spleen infiltration of AML1-ETO9a cells and reduce splenomegaly by activating the killing mechanism.47 As postulated, the peripheral blood immunophenotyping of NK(EzH2−) cell-treated and pEzH2-CsNPs@CD56-treated xenografts exhibited the repression of leukemia stem cells, GFP+c-Kit+ by ∼9.0% and ∼9.7%, respectively (p value: 0.0415). Also, NK(EzH2−) cell-treated and pEzH2-CsNPs@CD56-treated xenografts exhibited the increment in the myeloid differentiation marker CD11b by ∼5.8% and ∼10.0%, respectively (p value: 0.0295), in the GFP+c-Kit− cell population and enhanced Gr-1 expression by ∼5.8% and ∼8.7% (p value: 0.0342) when analyzed in the GFP+c-Kit− cell population (Fig. 4C & E–G). Furthermore, the bone marrow immunophenotyping of NK(EzH2−) cell-treated and pEzH2-CsNPs@CD56-treated xenografts exhibited the repression of leukemia stem cells GFP+c-Kit+ by ∼28.9% and ∼54.9%, respectively (p value: 0.0004). Also, NK(EzH2−) cell-treated and pEzH2@CsNPs@CD56-treated xenografts exhibited an increment in the myeloid differentiation marker CD11b by ∼28.7% and ∼61.9%, respectively (p value: 0.0016) when analyzed for the GFP+c-Kit+ cell population and Gr-1 expression was increased by ∼6.4% and ∼12.8% (p value: 0.0272) when analyzed for the GFP+c-Kit− cell population in BMNCs (Fig. 4D & E–G). This therapeutics efficacy analysis establishes the modified-NK cell-based immunotherapy since the presence of activated NK cells secrete killing signals to induce the apoptosis of AML1-ETO9a cells.48 AML exhibits a halted differentiation mechanism where the immunotherapy of NK cells induces the differentiation of AML1-ETO9a cells and provides a viable solution for anti-AML therapy.49 The histopathological analysis revealed intact and healthy tissue structures in the spleen, liver, kidneys, and heart of xenograft mice after the treatment of NK(EzH2−) cells and pEzH2@CsNPs@CD56 when compared to the control xenograft mice (Fig. 4H and I). AML creates a damaged pathology in the spleen and liver that is corrected by the treatment regimen as supported by an earlier article.50 The mechanism behind this anti-AML therapeutic efficacy by NK(EzH2−) cells and pEzH2@CsNPs@CD56 is a necessary factor to be analyzed to establish NK cell-mediated immunotherapy.
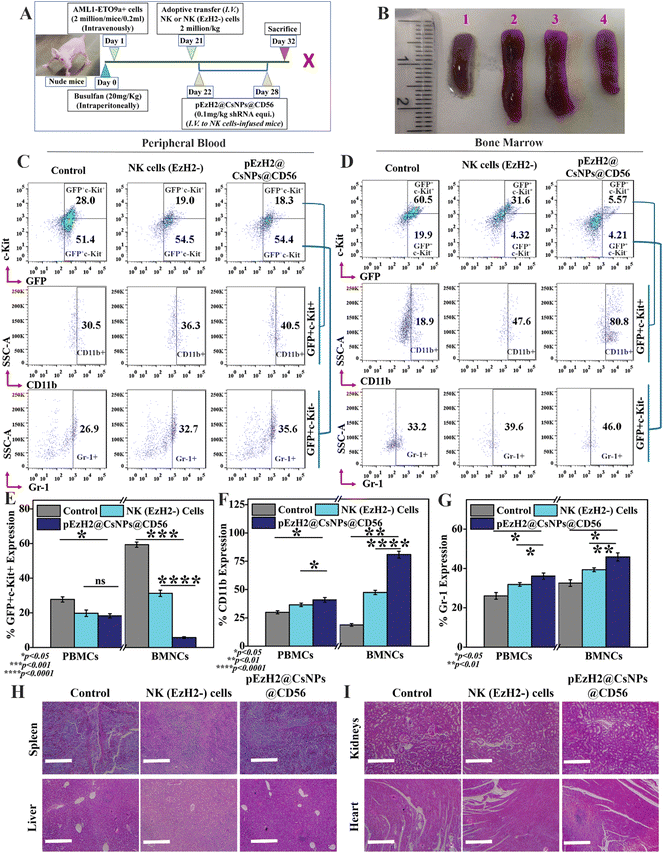 |
| | Fig. 4
In vivo therapeutics of CD56 conjugated and EzH2 shRNA encapsulated chitosan nanoparticles in an athymic nude mice xenograft model; (A) a schematic representation of the in vivo study plan, (B) splenomegaly imaging (1: Non-xenograft mice, 2: Xenograft mice, 3: NK (EzH2-) cells-treated mice, and 4: pEzH2@CsNPs@CD56-treated mice); immunophenotyping for c-Kit, CD11b and Gr-1 markers along with GFP (as a function of AML1-ETO9a expression) analysis in (C) peripheral blood, and (D) bone marrow of NK cells (EzH2−) and pEzH2@CsNPs@CD56. Quantification of (E) GFP+c-Kit+ cell population, (F) CD11b cell population and (G) Gr-1 cell population in peripheral blood and bone marrow of NK (EzH2−) cells and pEzH2@CsNPs@CD56-treated mice. (H and I) Hematoxylin and eosin staining of spleen, liver, kidneys, and heart tissues after the treatment of NK (EzH2−) cells and pEzH2@CsNPs@CD56 (Scale: 400 μm). All experiments were performed with n = 3, where n is the number of experimental repeats. Bonferroni and Tuckey tests were performed at a significance level of *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001; and ‘ns’ stands for ‘not significant’. | |
Increased CD38 expression activates caspase 3-dependent apoptosis and inhibits NPM1 in AML1-ETO9a-induced AML
EzH2 downregulation in NK cells mediates CD56 activation and CD38 expression, which in turn enhances NK cell immunotherapy. The CD56+ NK cell population in peripheral blood was increased by ∼7.6% and ∼15.8%, respectively, in NK(EzH2−) cell-treated and pEzH2@CsNPs@CD56-treated xenografts (p value: 0.0099) (Fig. 5A, E and F). The bone marrow exhibited an increase in the CD56+ cell population by ∼8.1% and ∼14.4%, respectively, in NK(EzH2−) cell-treated and pEzH2@CsNPs@CD56-treated xenograft mice (p value: 0.0161) (Fig. 5B, E and F). The EzH2 downregulation by pEzH2@CsNPs@CD56 under in vitro conditions (Fig. 3 & S7†) and an enhanced in vivo anti-AML therapeutics in AML1-ETO9a-induced AML (Fig. 4) as discussed above infer that pEzH2@CsNPs@CD56 efficiently targets CD56+ NK cells. The downregulated EzH2 exhibits compromised catalytic activity in histone H3 trimethylation, and promotes differentiation of NK cells and their cytolytic function by increasing the degranulation24,25 wherein CD38 activation leads to granzyme B production.22 This promotion of NK cells by pEzH2@CsNPs@CD56 is perceived as superior anti-AML NK cell immunotherapeutics, which overcomes the limitations of conventional and genetically engineered NK cells in terms of effectiveness, viral safety, and involvement of robust processes. Furthermore, CD56-associated CD38+ cells were found to be increased by ∼3.4% and ∼23.0% in the peripheral blood of NK(EzH2−) cell-treated and pEzH2@CsNPs@CD56-treated xenografts (p value: 0.0061) (Fig. 5C & G). The bone marrow exhibited an increment in CD56-associated CD38+ cells by ∼8.6% and ∼22.9%, respectively, in NK(EzH2−) cell-treated and pEzH2@CsNPs@CD56-treated xenografts (p value: 0.0107) (Fig. 5D & G). The downregulation of EzH2 in NK cells promotes CD38 expression through epigenetic regulation at the promoter site,23 which indicates that EzH2 downregulation in NK cells promotes CD56 differentiation and CD38 upregulation and, in turn, activates the caspase 3-dependent apoptosis of target cells. Furthermore, western blotting analysis revealed that CD38 expression increased by ∼1.3-fold and ∼1.5-fold along with cleaved caspase 3 expression by ∼1.9-fold and ∼2.1-fold in the spleen tissues of NK(EzH2−) cell-treated and pEzH2@CsNPs@CD56-treated xenografts, respectively (Fig. 5H & S10†). When NK cells identify their target cells, the CD56+ NK cells activate granzyme B production, which is regulated by CD38.21,22 As NK-secreted granzyme B reaches the cytoplasm,51 it induces caspase 3-dependent apoptosis of target cells as supported by western blot analysis and earlier literature studies.52 As per the earlier reports, an inverse relation of CD38 and EzH2 has been observed wherein EzH2 negatively and transcriptionally regulates CD38 expression.23 The EzH2 was downregulated by ∼2.8-fold and ∼3.0-fold, respectively, in NK(EzH2−) cell-treated and pEzH2@CsNPs@CD56-treated xenografts compared to the control wherein the EzH2-associated epigenetic marker, H3K27me3, was downregulated by ∼1.3-fold and ∼2.9-fold, respectively, in NK(EzH2−) cell-treated and pEzH2@CsNPs@CD56-treated xenografts. The EzH2 downregulation promotes the commitment of NK cells through epigenetic regulation, which activates enhanced and superior cytolytic activity24 as supported by increased CD56+ NK cell populations and enhanced therapeutic efficacy (Fig. 4, 5A and B). This is the first time CD56-targeted NK-cell genetic engineering has been performed under in situ conditions through a nanoparticle-based EzH2 shRNA mechanism that promotes NK cell immunotherapy under AML1-ETO9a-induced AML conditions. In addition, NPM1 was downregulated by ∼2.9-fold and ∼3.2-fold in NK (EzH2−) cell-treated and pEzH2@CsNPs@CD56-treated xenografts compared to the control (Fig. 5H & S10†). EzH2 shRNA exhibits the downregulation of EzH2, which further downregulates H3K27me3.26 The anti-EzH2 chromatin immunoprecipitation (ChIP)-qPCR analysis revealed the binding of EzH2 to NPM1 promoter regions at (−273/+48 bp) and (+561/+913 bp) in AML1-ETO9a-induced xenografts wherein NK(EzH2−) cell-treated and pEzH2@CsNPs@CD56-treated xenografts exhibit the abrogation of this relationship (Fig. 5I). The ChIP DNA amplification of (−273/+48 bp) and (+561/+913 bp) regions was reduced by treatment with NK(EzH2−) cells and pEzH2-CsNPs@CD56 in AML1-ETO9a-induced xenografts (Fig. 5J) and explained the binding of EzH2 to the NPM1 promoter as depicted in Fig. 5K. Here, the NPM1–EzH2 relationship, a known molecular interaction that regulates AML pathogenesis and progression, was abrogated by EzH2-specific NK cell immunotherapy,53 which suggests superior and efficient EzH2-mediated epigenetic regulation. Together, these analyses indicate that pEzH2@CsNPs@CD56 targeted and downregulated EzH2 expression in CD56+ NK cells, which increases NK cell immunotherapy towards AML1-ETO9a-induced AML. This approach can effectively minimize the vein-to-vein time along with the production cost and provide an alternative regimen of adoptive NK cell-transfer for anti-AML immunotherapy. The therapeutic effectiveness and safety of non-viral anti-AML NK cell-based immunotherapeutics by pEzH2@CsNPs@CD56 extenuates further translational development and promises viable anti-AML therapeutics in the future.
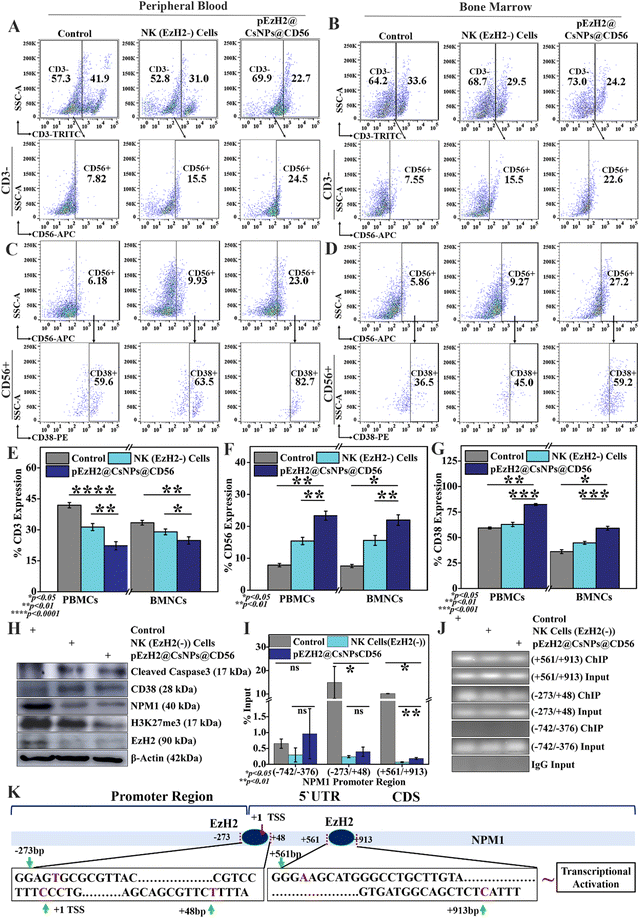 |
| | Fig. 5 Evaluation of anti-AML immunotherapeutic effect of natural killer cells; immunophenotyping for CD56 and CD38 analysis in (A and C) peripheral blood and (B and D) bone marrow samples; quantification of (E) the CD3 cell population, (F) CD56 cell population and (G) CD38 cell population in peripheral blood and bone marrow of NK (EzH2−) cells and pEzH2@CsNPs@CD56-treated mice. Molecular analysis of the spleen tissue with (H) western blotting, (I) anti-EzH2 chromatin immunoprecipitation (ChIP)-qPCR analysis on the NPM1 promoter, (J) agarose gel electrophoresis of amplified NPM1 promoter regions, and (K) schematic representation of EzH2 binding to NPM1 promoter regions. All experiments were performed with n = 3, where n is the number of experimental repeats. Bonferroni and Tuckey tests were performed at a significance level of *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001; and ‘ns’ stands for ‘not significant’. | |
Conclusion
Here, the pSMP-pEzH2 shRNA plasmid was encapsulated in chitosan nanoparticles that were further conjugated with CD56 antibodies and characterized. The activity of NK cells is promoted by the CD56-directed delivery of the pSMP-pEzH2 shRNA plasmid, which can knockdown the expression of EzH2 in NK cells and promote the activation of CD56+ NK cells and CD38+ NK cells. This in turn promotes the killing mechanism of AML1-ETO9a cells in the peripheral blood and bone marrow of xenograft mice. The c-Kit+ leukemia stem cell population was deregulated by NK cell immunotherapy wherein myeloid differentiation markers, CD11b and Gr-1, were upregulated in the peripheral blood and bone marrow of xenograft mice. These analyses exhibited proof of pEzH2@CsNPs@CD56-based in vivo genetic engineering of CD56 cells to enhance NK cell-mediated anti-AML immunotherapy and provide low production costs along with safety from virus-based genetic editing of NK cells for adoptive transfer. pEzH2@CsNPs@CD56 presents itself as a non-viral method to improve NK cell-mediated immunotherapy, which provides a potential future in translational development.
Author contributions
ACK: conception of idea, design and conduct of the experiments and manuscript writing; MB: experimental conduct; PM: guidance and manuscript correction; SRC: experimental design, guidance and manuscript correction.
Data availability
All data have been incorporated into the article and the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was partly supported by funding from the ICMR (35/4/2020-Nano/BMS) and DST Nano Mission (DST/NM/TBI-02/2020). The facilities of the Indian Institute of Science Education and Research, Mohali (IISER, Mohali), the National Agri-Food Biotechnology Institute (NABI, Mohali), and the Institute of Nano Science and Technology (INST, Mohali) are duly acknowledged. We are also thankful to Prof. Surajit Karmakar of the Institute of Nano Science and Technology (INST, Mohali) for his scientific input for the development of the PDX model. We are also thankful to Dr Nitin Singhal of the National Agri-Food Biotechnology Institute (NABI, Mohali) for providing support in the use of the in vivo imaging facility of NABI, Mohali.
References
- R. J. Austin,
et al., Oncogenic drivers dictate immune control of acute myeloid leukemia, Nat. Commun., 2023, 14, 2155 CrossRef CAS PubMed.
- H. Döhner, D. J. Weisdorf and C. D. Bloomfield, Acute Myeloid Leukemia, 2015, 373, 1136–1152 Search PubMed.
- K. Rejeski, J. Duque-Afonso and M. Lübbert, AML1/ETO and its function as a regulator of gene transcription via epigenetic mechanisms, Oncogene, 2021, 40, 5665–5676 CrossRef CAS PubMed.
- K. E. Elagib and A. N. Goldfarb, Oncogenic pathways of AML1-ETO in acute myeloid leukemia: Multifaceted manipulation of marrow maturation, Cancer Lett., 2007, 251, 179–186 CrossRef CAS PubMed.
- B. Kumar,
et al., Acute myeloid leukemia transforms the bone marrow niche into a leukemia-permissive microenvironment through exosome secretion, Leukemia, 2018, 32, 575–587 CrossRef CAS PubMed.
- B. Deschler and M. Lübbert, Acute myeloid leukemia: Epidemiology and etiology, Cancer, 2006, 107, 2099–2107 CrossRef PubMed.
- C. D. DiNardo and A. H. Wei, How I treat acute myeloid leukemia in the era of new drugs, Blood, 2020, 135, 85–96 CrossRef PubMed.
- M. Sugita,
et al., Allogeneic TCRαβ deficient CAR T-cells targeting CD123 in acute myeloid leukemia, Nat. Commun., 2022, 13, 2227 CrossRef CAS PubMed.
- N. Albinger,
et al., Primary CD33-targeting CAR-NK cells for the treatment of acute myeloid leukemia, Blood Cancer J., 2022, 12, 61 CrossRef PubMed.
- J. N. Brudno and J. N. Kochenderfer, Toxicities of chimeric antigen receptor T cells: recognition and management, Blood, 2016, 127, 3321–3330 CrossRef CAS PubMed.
- X. Zhang,
et al., Cytokine Release Syndrome After Modified CAR-NK Therapy in an Advanced Non-small Cell Lung Cancer Patient: A Case Report, Cell Transplant., 2022, 31, 9636897221094244 Search PubMed.
- J. Daly, M. Carlsten and M. O’Dwyer, Sugar Free: Novel Immunotherapeutic Approaches Targeting Siglecs and Sialic Acids to Enhance Natural Killer Cell Cytotoxicity Against Cancer, Front. Immunol., 2019, 10, 1047 CrossRef CAS PubMed.
- T. Bald, M. F. Krummel, M. J. Smyth and K. C. Barry, The NK cell–cancer cycle: advances and new challenges in NK cell–based immunotherapies, Nat. Immunol., 2020, 21, 835–847 CrossRef CAS PubMed.
- H. Raskov, A. Orhan, J. P. Christensen and I. Gögenur, Cytotoxic CD8+ T cells in cancer and cancer immunotherapy, Br. J. Cancer, 2021, 124, 359–367 CrossRef CAS PubMed.
- T. J. Laskowski, A. Biederstadt and K. Rezvani, Natural killer cells in antitumour adoptive cell immunotherapy, Nat. Rev. Cancer, 2022, 22, 557–575 CrossRef CAS PubMed.
- S. Paul and G. Lal, The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy, Front. Immunol., 2017, 8, 1124 CrossRef PubMed.
- I. Prager and C. Watzl, Mechanisms of natural killer cell-mediated cellular cytotoxicity, J. Leukocyte Biol., 2019, 105, 1319–1329 CrossRef CAS PubMed.
- M. J. Smyth,
et al., Activation of NK cell cytotoxicity, Mol. Immunol., 2005, 42, 501–510 CrossRef CAS PubMed.
- J. T. Gunesch,
et al., CD56 regulates human NK cell cytotoxicity through Pyk2, eLife, 2020, 9, e57346 CrossRef CAS PubMed.
- E. L. Azeredo,
et al., NK cells, displaying early activation, cytotoxicity and adhesion molecules, are associated with mild dengue disease, Clin. Exp. Immunol., 2005, 143, 345–356 CrossRef PubMed.
- S. Dwivedi, E. P. Rendón-Huerta, V. Ortiz-Navarrete and L. F. Montaño, CD38 and Regulation of the Immune Response Cells in Cancer, J. Oncol., 2021, 2021, 6630295 Search PubMed.
- R. Zambello, G. Barilà, S. Manni, F. Piazza and G. Semenzato, NK cells and CD38: Implication for (Immuno)Therapy in Plasma Cell Dyscrasias, Cells, 2020, 9, 768 CrossRef CAS PubMed.
- D. Chemlal,
et al., EZH2 Targeting Induces CD38 Upregulation and Response to Anti-CD38 Antibodies in Multiple Myeloma, Blood, 2022, 140, 4220–4221 CrossRef.
- J. Yin,
et al., Ezh2 regulates differentiation and function of natural killer cells through histone methyltransferase activity, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 201521740 Search PubMed.
- M. Yu,
et al., Histone methyltransferase Ezh2 negatively regulates NK cell terminal maturation and function, J. Leukocyte Biol., 2021, 110, 1033–1045 CrossRef CAS PubMed.
- T. T. Onder,
et al., Chromatin-modifying enzymes as modulators of reprogramming, Nature, 2012, 483, 598–602 CrossRef CAS PubMed.
- S. Kundu, M. Gurney and M. O'Dwyer, Generating natural killer cells for adoptive transfer: expanding horizons, Cytotherapy, 2021, 23, 559–566 CrossRef CAS PubMed.
- F. G. de Carvalho,
et al., Synthesis and characterization of TPP/chitosan nanoparticles: Colloidal mechanism of reaction and antifungal effect on C. albicans biofilm formation, Mater. Sci. Eng., C, 2019, 104, 109885 CrossRef CAS PubMed.
- C. Carrillo,
et al., Chitosan nanoparticles as non-viral gene delivery systems: Determination of loading efficiency, Biomed. Pharmacother., 2014, 68, 775–783 CrossRef CAS PubMed.
- J. Zuber,
et al., Mouse models of human AML accurately predict chemotherapy response, Genes Dev., 2009, 23, 877–889 CrossRef CAS PubMed.
- S. Carlens,
et al., A new method for in vitro expansion of cytotoxic human CD3−CD56+ natural killer cells, Hum. Immunol., 2001, 62, 1092–1098 CrossRef CAS PubMed.
- Y. Qazi,
et al., Nanoparticle-mediated delivery of shRNA.VEGF-a plasmids regresses corneal neovascularization, Invest. Ophthalmol. Visual Sci., 2012, 53, 2837–2844 CrossRef CAS PubMed.
- D. Hattab, A. M. Gazzali and A. Bakhtiar, Clinical Advances of siRNA-Based Nanotherapeutics for Cancer Treatment, Pharmaceutics, 2021, 13, 1009 CrossRef CAS PubMed.
- K. Bowman and K. W. Leong, Chitosan nanoparticles for oral drug and gene delivery, Int. J. Nanomed., 2006, 1, 117–128 CrossRef CAS PubMed.
- J. Lee,
et al., T Cell-Specific siRNA Delivery Using Antibody-Conjugated Chitosan Nanoparticles, Bioconjugate Chem., 2012, 23, 1174–1180 CrossRef CAS PubMed.
- P. Garidel, M. Hegyi, S. Bassarab and M. Weichel, A rapid, sensitive and economical assessment of monoclonal antibody conformational stability by intrinsic tryptophan fluorescence spectroscopy, Biotechnol. J., 2008, 3, 1201–1211 CrossRef CAS PubMed.
- D. V. Sotnikov, N. A. Byzova, A. V. Zherdev and B. B. Dzantiev, Retention of Activity by Antibodies Immobilized on Gold Nanoparticles of Different Sizes: Fluorometric Method of Determination and Comparative Evaluation, Nanomaterials, 2021, 11, 3117 CrossRef CAS PubMed.
- R. Jha and R. A. Mayanovic, A Review of the Preparation, Characterization, and Applications of Chitosan Nanoparticles in Nanomedicine, Nanomaterials, 2023, 13, 1302 CrossRef CAS PubMed.
- S. Sarangapani,
et al., Chitosan nanoparticles’ functionality as redox active drugs through cytotoxicity, radical scavenging and cellular behaviour, Integr. Biol., 2018, 10, 313–324 CrossRef CAS PubMed.
- D. Sonin,
et al., Biological Safety and Biodistribution of Chitosan Nanoparticles, Nanomaterials, 2020, 10, 810 CrossRef CAS PubMed.
- T. Huang,
et al., Chitosan-DNA nanoparticles enhanced the immunogenicity of multivalent DNA vaccination on mice against Trueperella pyogenes infection, J. Nanobiotechnol., 2018, 16, 8 CrossRef PubMed.
- Y. Feng,
et al., Differential killing of CD56-expressing cells by drug-conjugated human antibodies targeting membrane-distal and membrane-proximal non-overlapping epitopes, mAbs, 2016, 8, 799–810 CrossRef CAS PubMed.
- T. Haraguchi,
et al., Transfected plasmid DNA is incorporated into the nucleus via nuclear envelope reformation at telophase, Commun. Biol., 2022, 5, 78 CrossRef CAS PubMed.
- B. Santos-Carballal, E. Fernández Fernández and F. M. Goycoolea, Chitosan in Non-Viral Gene Delivery: Role of Structure, Characterization Methods, and Insights in Cancer and Rare Diseases Therapies, Polymers, 2018, 10, 444 CrossRef PubMed.
- E. Sosa,
et al., Differentiation of primate primordial germ cell-like cells following transplantation into the adult gonadal niche, Nat. Commun., 2018, 9, 5339 CrossRef PubMed.
- B. Zhou,
et al., Targeting miR-193a-AML1-ETO-β-catenin axis by melatonin suppresses the self-renewal of leukaemia stem cells in leukaemia with t (8;21) translocation, J. Cell. Mol. Med., 2019, 23, 5246–5258 CrossRef CAS PubMed.
- C. King, CAR NK Cell Therapy for T Follicular Helper Cells, Cell Rep. Med., 2020, 1, 100009 CrossRef CAS PubMed.
- E. Fathi, R. Farahzadi and B. Valipour, Alginate/gelatin encapsulation promotes NK cells differentiation potential of bone marrow resident C-kit+ hematopoietic stem cells, Int. J. Biol. Macromol., 2021, 177, 317–327 CrossRef CAS PubMed.
- J. Xu and T. Niu, Natural killer cell-based immunotherapy for acute myeloid
leukemia, J. Hematol. Oncol., 2020, 13, 167 CrossRef CAS PubMed.
- Z. Her,
et al., An improved pre-clinical patient-derived liquid xenograft mouse model for acute myeloid leukemia, J. Hematol. Oncol., 2017, 10, 162 CrossRef PubMed.
- I. Prager,
et al., NK cells switch from granzyme B to death receptor-mediated cytotoxicity during serial killing, J. Exp. Med., 2019, 216, 2113–2127 CrossRef CAS PubMed.
- I. Rousalova and E. Krepela, Granzyme B-induced apoptosis in cancer cells and its regulation (review), Int. J. Oncol., 2010, 37, 1361–1378 CAS.
- J. Stomper,
et al., Integrative study of EZH2 mutational status, copy number, protein expression and H3K27 trimethylation in AML/MDS patients, Clin. Epigenet., 2021, 13, 77 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a
*a