
Self-assembled nanomicelles for oral delivery of luteolin utilizing the intestinal lymphatic pathway to target pancreatic cancer†
Archana
Karole
 a,
Yirivinti Hayagreeva
Dinakar
a,
Poonam
Sagar
c and
Shyam Lal
Mudavath
*ab
a,
Yirivinti Hayagreeva
Dinakar
a,
Poonam
Sagar
c and
Shyam Lal
Mudavath
*ab
aInfectious Disease Biology Laboratory, Chemical Biology Unit, Institute of Nano Science and Technology, Knowledge City, Sector-81, Mohali-140306, Punjab, India
bDepartment of Animal Biology, School of Life Sciences, University of Hyderabad, Prof. C.R. Rao Road, Gachibowli, Hyderabad, 500046 Telangana, India. E-mail: shyamlal_absls@uohyd.ac.in; shavs0502@gmail.com
cDepartment of Food and Nutrition, National Agri-Food Biotechnology Institute, Knowledge City, Sector 81, Mohali, Punjab, India
First published on 8th March 2024
Abstract
Pancreatic cancer is one of the major cause of cancer-related deaths worldwide, and is mainly associated with carcinomas of the pancreatic tissue. Current therapies for treating pancreatic cancer have a major drawback related to their low bioavailability and non-specificity, which leads to low therapeutic efficacy and side effects. Luteolin (LUT) has been clinically used for treatment of various types of cancer, although its clinical use has declined owing to its low oral bioavailability. In this work, we prepared an effervescent-based nanocarrier (NG) that rapidly triggers an effervescent reaction and transforms into nanomicelles to modulate the oral bioavailability of the hydrophobic drug Luteolin (LUT). Furthermore, we performed tests to assess its in vitro epithelial cell permeability and cellular internalization on a Caco-2 monolayer. We also performed in vivo toxicity assessment using animal models. Further, we evaluated the nanocarrier system's in vivo efficacy in tumor xenograft pancreatic cancer models. We validated that being pH responsive, our effervescent carrier disassembles at intestinal pH and is absorbed through the intestinal lymphatic system (ILS) to further site-specifically invade the pancreatic cancer cells. Furthermore, the negative surface charge and particle size (450 ± 100 nm) of the nanomicelles helped to internalize LUT through the ILS. We observed that LUT-loaded nanomicelles have significant antipancreatic cancer efficacy by activating caspase-3 activity and downregulating VEGF-A, FAK, TNF-α, and Ki-67. Unlike other drug-delivery systems, we developed noninvasive nanocarrier system has the capability of transporting the hydrophobic drug LUT from the intestine to the tumor site by utilizing the ILS.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the predominant types of pancreatic cancer, and involves highly aggressive tumor formation in the pancreatic duct. The incidence of this disease is higher in males (5.7 per 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, with 262865 cases) compared to females (4.1 per 100000, with 232908 overall cases) and accounts for more than 90% of pancreatic cancer cases.1,2 PDAC arises from exocrine epithelial cells of the pancreatic duct.3 However, current treatment options, such as the standard chemotherapy drug gemcitabine, fall short for extreme cases, with 5-year expected survival in 5% of patients.4 This is primarily due to the fact that PDAC exhibits substantial stromal components, comprising more than half of the tumor mass, along with complex tumor blood vessel structures.5 These factors collectively exert significant barriers for the effective treatment and delivery of drugs.6 Addressing these challenges of pancreatic cancer chemotherapy involves effective targeting of metabolic pathways, the acidic tumor microenvironment, and neighboring cancer stem cells.7 These obstacles must be overcome to improve the outcomes for individuals battling pancreatic cancer. Currently, the available treatments include radiation therapy, surgical resection, and chemotherapy.8 Delivering antitumor agents specifically to tumors in pancreatic cancer is crucial to enhance treatment effectiveness while minimizing damage to healthy tissues.
000, with 262865 cases) compared to females (4.1 per 100000, with 232908 overall cases) and accounts for more than 90% of pancreatic cancer cases.1,2 PDAC arises from exocrine epithelial cells of the pancreatic duct.3 However, current treatment options, such as the standard chemotherapy drug gemcitabine, fall short for extreme cases, with 5-year expected survival in 5% of patients.4 This is primarily due to the fact that PDAC exhibits substantial stromal components, comprising more than half of the tumor mass, along with complex tumor blood vessel structures.5 These factors collectively exert significant barriers for the effective treatment and delivery of drugs.6 Addressing these challenges of pancreatic cancer chemotherapy involves effective targeting of metabolic pathways, the acidic tumor microenvironment, and neighboring cancer stem cells.7 These obstacles must be overcome to improve the outcomes for individuals battling pancreatic cancer. Currently, the available treatments include radiation therapy, surgical resection, and chemotherapy.8 Delivering antitumor agents specifically to tumors in pancreatic cancer is crucial to enhance treatment effectiveness while minimizing damage to healthy tissues.
The intestinal lymphatic system (ILS) is an attractive biological pathway for providing intestinal absorption for medications administered through the oral route.9 However, the presence of the ILS in the small intestine presents challenges in efficiently delivering drug vehicles bypassing the stomach environment. Enterocytes and microfold (M) cells play a crucial role in the transport of drugs through the ILS. Reports have claimed that M cells exhibit phagocytic and transcytosis activities for particulate constructs from the gut lumen compared to enterocytes.10
In this study, we developed an effervescent-based nano-gas (NG) system for site-specific drug delivery for the treatment of pancreatic cancer. We incorporated luteolin (LUT), which has significant anticancer potential, but its use is currently limited due to associated side effects with respect to its nonspecific reach and hydrophobic nature.11–13 We addressed the major issues associated with LUT and transformed it into an efficient and effective treatment for pancreatic cancer. We observed that the NPs (nanomicelles) had a particle size of 450 ± 100 nm with a −27.8 mV surface potential. The NPs showed a 2-fold increase in luteolin solubility with a sustained drug-release profile.13 Here, we explored the distribution of formulation by IVIS imaging in a rat model and observed that after disassembly in intestinal pH due to the eudragit coating, the drug was absorbed by the intestinal cells and could then significantly accumulate in the pancreas via the ILS through M-cell mediated internalization. The potential antitumor effects of luteolin were enhanced by promoting its solubility, permeability, active targeting, and its potential to suppress survival signaling pathways, which enabled it to promote apoptosis and further block the tumor cell proliferation in pancreatic cancer. In vivo assessment on a xenograft pancreatic cancer model showed that the LUT-NPs treatment was effective in terms of tumor regression, as evidenced by several molecular studies. We observed that after successful accumulation in pancreas, LUT acted by downregulating the expression of the cell proliferation markers and metastatic markers VEGF-A, FAK, Ki-67, and TNF-α, as well as by upregulating caspase-3 expression at the malignant site, ultimately resulting in tumor cell death and the inhibition of metastasis.
To the best of our knowledge, this effervescent-based nanocarrier system is pH-sensitive and convenient, and can be specifically designed to facilitate targeted drug delivery to both the lymphatic system and tumor sites following oral administration.
Materials and methods
Investigation of intestinal permeability and internalization through a Caco-2 cells monolayer
Caco-2 cells monolayers were allowed to grow on a polycarbonate Costar transwell (0.4 mm) plate featuring six wells per plate, supplied by Corning Costar Corp, New York, USA. Cells were maintained until their transepithelial electrical resistance (TEER) was observed to be around 350 ± 50 Ω cm2, indicating a cell monolayer growth.14 Further this monolayer was treated with LUT-NPs (LUT-loaded nanoparticles) for 1 h. Drug permeability was measured through apical junctional complex (AJC) using immunofluorescence staining. After washing three times with cold PBS, the cells were fixed with 4% paraformaldehyde (PFA) for 10 min, followed by blocking with 2% BSA to avoid nonspecific binding for 2 h at room temperature. Further the cells were incubated with primary antibody (mouse anti-E-cadherin, Abcam, Cambridge, MA, USA) overnight at 4 °C. After washing twice with phosphate-buffered saline with Tween 20 (PBST), the cells were incubated with secondary antibody (Anti-Mouse TRITC secondary antibody, Abcam, MA, USA) for 2 h at 37 °C. Further, DAPI antifade was used for counterstaining the cell nucleus, and the cells were then observed using confocal laser scanning microscopy (CLSM; Zeiss LSM780, Carl Zeiss, Jena GmbH, Germany).Next, to determine the cellular internalization of the nanoparticles against the Caco-2 monolayer, the cells were treated with different samples (NPs, LUT, and LUT-NPs). The uptake was assessed based on the fluorescence intensity uptake of the nanomicelles. The uptake was quantified by confocal laser scanning microscopy.
Animal study
In vivo safety study
Assessment of the in vivo safety of the formulation, specifically for acute toxicity, was carried out on Wistar rats. The animals were distributed into two groups (control group, NG-treated group), with six animals in each group (n = 6). The NG group was treated with blank formulation (containing citric acid:SDS:SBC, 100 mg kg−1) for 14 days through oral administration, and the untreated group was used as a control. Further the body weight and any sign of toxicity were closely monitored throughout the treatment schedule. The animals were sacrificed after 14 day treatment and all the vital organs were isolated, and subsequently immersed in phosphate-buffered formalin. Further, the organs were molded in paraffin wax and cut into 5 μm thin sections for histological analysis. We also assessed the biochemical parameter to evaluate organ function.
Biodistribution study of the NPs
To investigate the distribution of nanoparticles in the biological system, we synthesized effervescent enteric-coated granules (ICG w NG: citric acid/SDS/SBC/ICG) and enteric-coated capsules of ICG (indocyanine green). To mimic an enteric coating, we loaded free ICG into an enteric-coated capsule (size 9, Torpac Ltd, USA). Here, ICG worked as a fluorescent absorbed drug molecule, and was utilized to locate the distribution of the drug. The study was conducted on Wistar rats (three groups, with each group n = 3). All the animals were fasted the day before treatment and after that they were received the oral treatment of free ICG, ICG w/o NG, and ICG w NG formulation. The animals were euthanized at predetermined time points and the gastro intestinal tract (GIT) as well as all vital organs (brain, heart, kidney, liver, lungs, and pancreas) were collected. The organs were imaged for ex vivo analysis and the fluorescence intensity of tissue was determined using an IVIS® Lumina Series system (PerkinElmer, USA). Regions of interest (ROIs) from the images obtained were identified and quantified as average radiance using Living ImageJ software 4.0.Determination of the route of drug transportation
In this study, we evaluated the intestinal lymphatic pathway of drug transportation, which plays the main role in the delivery of the drug-loaded NPs from the intestine to the pancreas, utilizing two methods: in situ loop formation study and Peyer's patches ligation assay.15–17First, we performed the in situ loop formation study, in which the Wistar rats were used. The animals (n = 3) were fasted for 12 h, and after that the rats were anesthetized using the EZ-7000 classic system. Further, we opened the abdominal cavity of the animals by cutting a 1 cm area in the midline part of the body to expose the small intestinal and allow ligating the ascending part of the small intestine; further, we washed the segment with PBS (pH 7.4). The loop formed by ligation of the descending part of small intestine was injected with the ICG-NPs formulation. Then we placed the loop part in the abdominal region and closed the part by suturing. After 3 h of treatment, the animals were sacrificed, and the small intestinal segment and intestinal lymphatic system were harvested. The harvested organs were visualized under the IVIS system and the fluorescence intensity of NPs was imaged and quantified. The nanoparticles were analyzed for colocalization in intestinal cells. The immunofluorescence staining of cytokeratin 8 expressed on the enterocytes of the small intestine (anti-cytokeratin 8 primary antibody, Invitrogen, USA; Alexa Fluor® 647 secondary antibody, Abcam) was performed on a cryosection of the isolated intestinal loop.18 Furthermore, consecutively we performed Peyer's patches ligation assay to confirm the NPs internalization through M cells, through applying Peyer's patches (PPs) to Wistar rats that had been ligated. In brief, the Wistar rats (n = 3) were first anesthetized with the EZ classical system, and after that a small cut was made in the middle region of the abdomen. We selected the ileum part, which is abundant with PP. We ligated both ends of the PP with silk suture without rupturing the blood vessel, and we further incorporated 50 μl of ICG-labeled NPs suspension into a Peyer's patch segment, and then closed the abdominal cavity by suturing. After 3 h of treatment, the rats were sacrificed and we removed the ligated patch loop (minimum three Peyer's patches were isolated). The excised PPs were cleansed in PBS (pH 7.4) four times on ice to remove the mucosal fluid and unentrapped particles.15 The colocalization of NPs with M cells was observed by confocal laser scanning microscopy (CLSM). The loops were immersed in 4% paraformaldehyde (PFA), and these samples were further treated with OsO4.19
Therapeutic efficacy assessment
On the basis of the results of our in vitro experiments, we performed the therapeutic efficacy assessment of the nanoparticles with a tumor xenograft mouse model. The mice were injected with pancreatic cancer (PANC-1) cells. The animals were distributed to compare the anticancer effect of free LUT and its nanoformulation. We developed the pancreatic cancer xenograft model with PANC-1 cells. The human pancreatic adenocarcinoma cell line (PANC-1) was procured from NCCS, Pune, India. The cell viabilities were assessed by the trypan blue exclusion method. The cells suspension was injected at a concentration of 10 × 106 cells per mice in the right lower flank of the mice.Results and discussion
Formulation development and characterization. In previous work, we successfully developed an effervescent-based nanocarrier (NG) system,13 which was basically made up with bicarbonate and acidic agents capable of producing gas carriers for the drug LUT and could ultimately transform into LUT-loaded nanomicelles (LUT-NPs).Assessment of epithelial permeability and internalization of the NPs
In this study sodium bicarbonate (SBC), citric acid, SDS, and LUT were properly mixed and then we prepared the granules in polyvinyl–pyrrolidone (PVP). Further these formed granules were coated with Eudragit® S-100 (pH > 7 sensitive polymer). After reaching the intestine, the outer coating of eudragit dissolves and the effervescent granules ultimately release their contents. The citric acid has 3 dissociable protons that undergo ionization and lose 3 protons to form citrate ion, making it acidic. The SBC instantly reacts in the acidic environment to release CO2 bubbles, which are stabilized by SDS with the formation of a layer.33 The surfactant layer plays a key role in transforming the micelles-type nanomicelles due to the presence of the lipophilic part in the SDS. The nanomicelles were 450 ± 100 nm in size with a negative surface (−27.8 mV) potential (Fig. 1A–D).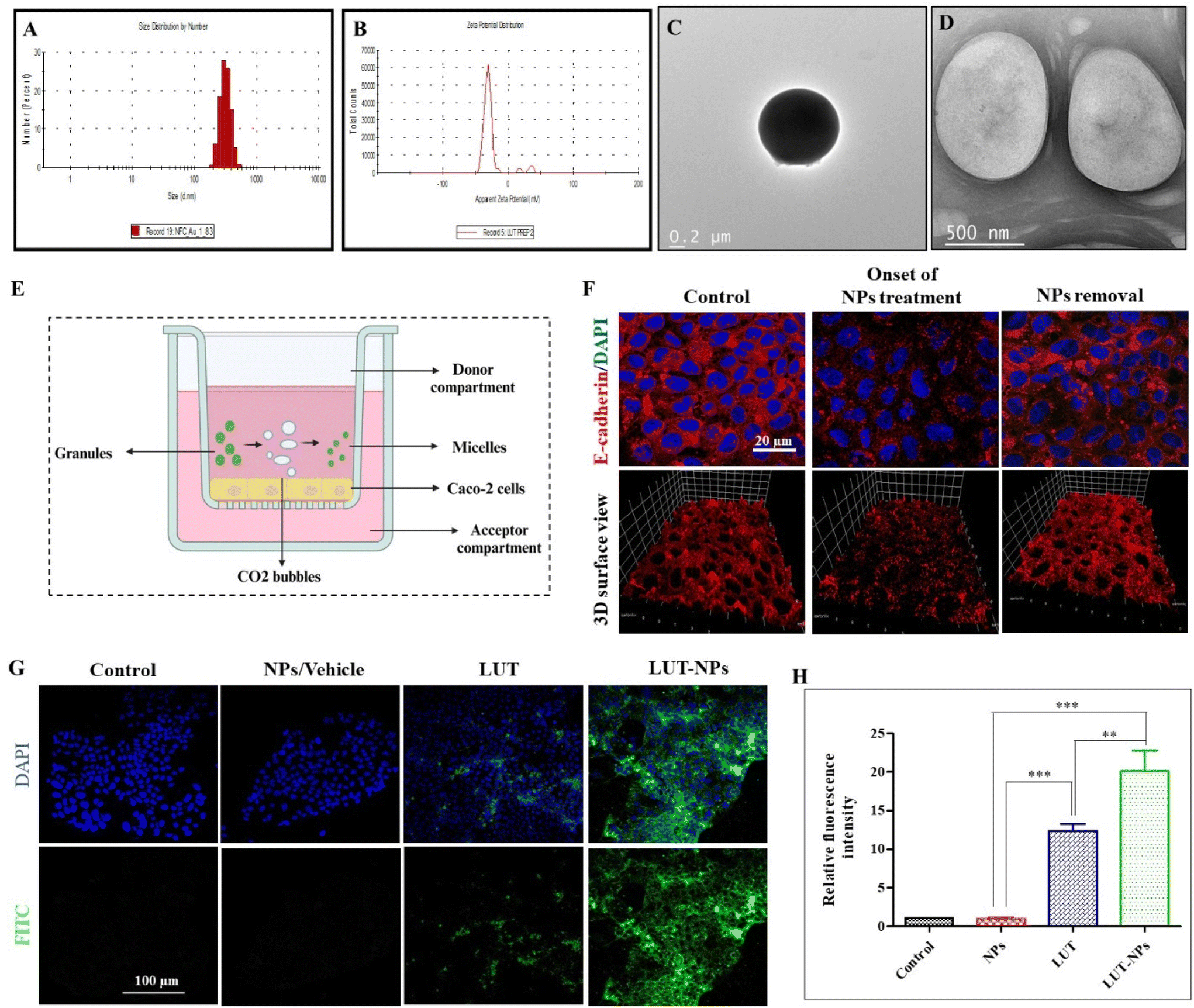 | ||
| Fig. 1 Characterization of the nanomicelles. (A) Particle-size distribution. (B) Surface charge. (C and D) Representative TEM images of the NPs (figure is reused from our pervious paper,13 copyright reserved to Elsevier, Lic. no. 5752501392891). In vitro intestinal permeability assay. (E) Schematic illustration of the experimental design for epithelial permeability assessment in the Caco-2 monolayer. (F) Change in the Caco-2 monolayer ultrastructure after the onset and after 2 h of formulation treatment (NPs). (G) Confocal fluorescent images of the internalized NPs after treatment with NPs, LUT, and LUT-NPs of Caco-2 cells. (H) Qualitative fluorescence analysis of the internalized nanoparticles using CLSM: with significant difference defined as ***P < 0.001 and **P < 0.01. | ||
The intracellular junction consists of two major proteins: adherens junctions (AJs) proteins and tight junctions (TJs) proteins. AJs not only induce the adhesion of plasma membranes but also promote the assembly of TJs. Consequently, any changes to AJs can modulate the structure of the TJs, ultimately affecting epithelial permeability.20 E-cadherin is a calcium (Ca2+)-dependent adhesion molecule that works as the major transmembrane protein in AJs. Consequently, the presence of extracellular Ca2+ is important for the development of the apical junctional complex (AJC) and intercellular connections.21
In our previous study, we observed the potential of the effervescent-based nanocarrier system, which enhanced the epithelial permeability due to the calcium chelation properties of SDS and citric acid.22 For that reason, we further elucidated the mechanism of the intestinal permeability in the presence of NPs. The Caco-2 monolayer cell model was basically developed from human colorectal adenocarcinoma cells that are frequently used for the prediction of in vitro permeability.23
To assess epithelial permeability, we performed immunofluorescence staining for E-cadherin (AJs protein) expressed on the Caco-2 monolayer at onset and after treatment with NPs. Confocal analysis indicated that the initial treatment induction caused changes in the Caco-2 cells monolayer by internalization of the E-cadherin protein (red color) due to depletion of the extracellular calcium level. In previous research work, we reported that extracellular calcium was reduced to Ca2+ due to the chelation properties of the formulation components,13 which can further affect the cell–cell interactions and enhance the intestinal permeability of drug molecules.24 For confirmation of the reversible alteration after NPs removal, again immune fluorescence staining was performed on the Caco-2 cells monolayer 2 h after stopping treatment, with Fig. 1F showing the reformation of the apical junction protein.
The internalization of the drug-loaded nanomicelles by the Caco-2 cell monolayers, cultivated on the membrane surface of a transwell chamber, as depicted in Fig. 1E, was analyzed via confocal laser scanning microscopy (CLSM). In order to mimic the internalization of drug-loaded NPs (LUT-NPs) though the intestinal epithelial layer, again the monolayer was treated with LUT-NPs, NPs, and free LUT. Fig. 1G shows the LUT-NPs-treated group had the maximum internalization of micellar particles. Here, the drug-loaded micelles internalization was proportional to the fluoresce intensity. The intensity of the fluorescence signal was notably higher when compared to the group treated with free ICG (**P < 0.01) (Fig. 1H), possibly due to the reduction of drug aggregation and the improvement of drug absorption.
In vivo acute toxicity study
To evaluate the in vivo safety profile of the effervescent-based nanocarrier (NG), an acute oral toxicity study was conducted on Wistar rats. In course of 14-day in vivo toxicity assessment, the rats were administered a daily dose of effervescent granules (NG) orally, while the untreated rats served as the control group (Fig. 2A). Assessment of the body weight is essential to know the health and direct parameters to assess in an in vivo toxicity study.25 Initially, we assessed the body weight of the animals, and observed that there was a non-significant reduction in the body weight in the treated group compared to the untreated group, as explained in Fig. 2B (ns, P > 0.05). Further we performed organ toxicity assessment based on biomarkers for the liver and kidney. The serums were collected at the end of the study and the biochemical parameters were assessed, such as alanine transferase (ALAT), serum glutamic-oxaloacetic transaminase (SGOT), serum glutamic pyruvic transaminase (SGPT), and aspartate aminotransferase (ASAT). The results of the biochemical assessment (including liver and kidney function tests) indicated their alterations were not-significantly different (ns, P > 0.05) after 14-day oral dosing compared to the untreated group (Fig. 2C). Further we performed a study to assess histological changes in the vital organs, such as liver, kidney, brain, and spleen. Hematoxylin and eosin (H&E) staining was performed on vital organ, and the results (Fig. 2D) indicated that no histological changes could be observed in any vital organ. Hence, no significant changes were observed in the histological and biochemical parameters or body weight after NG treatment, suggesting our formulation is non-toxic and safe for biological systems.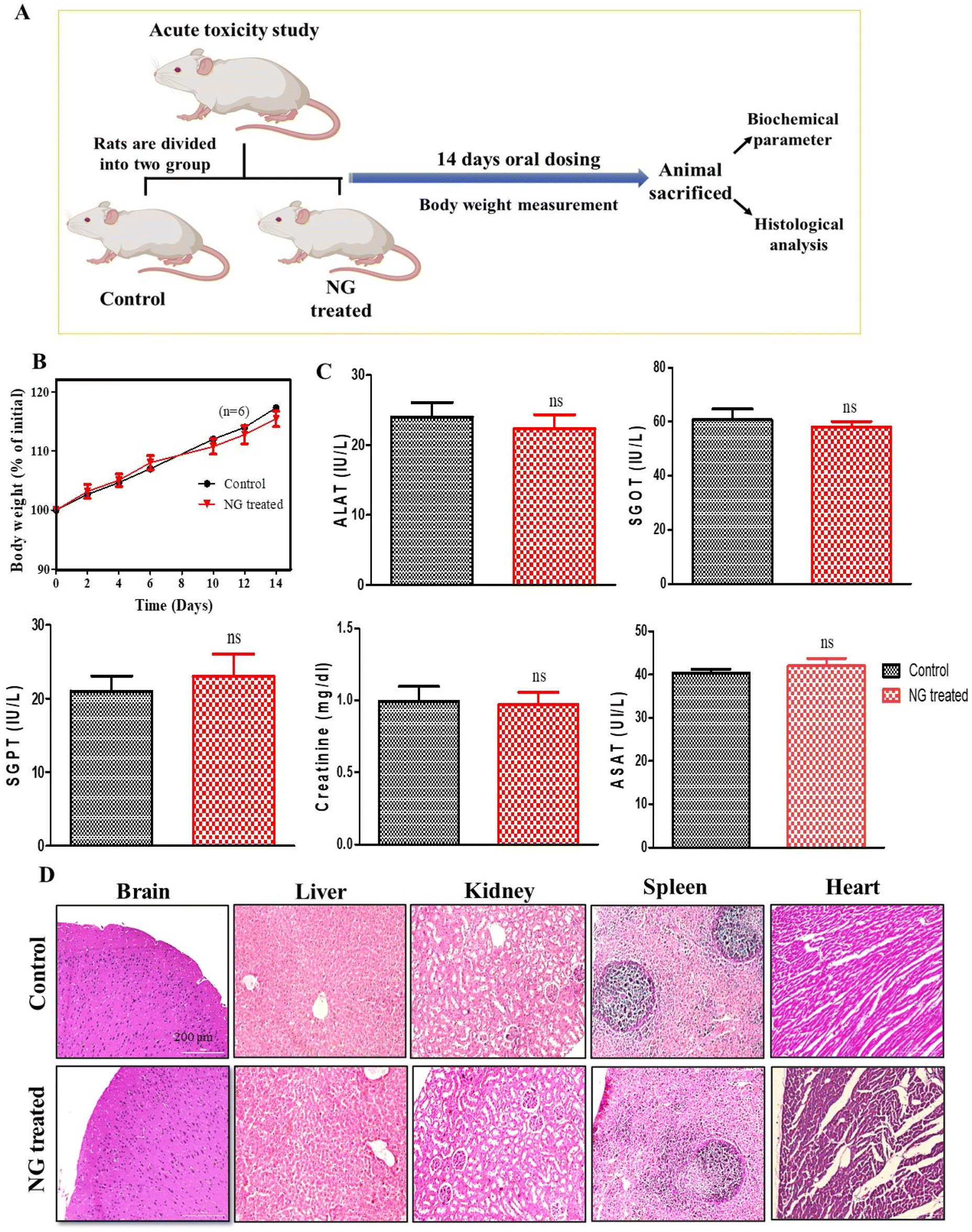 | ||
| Fig. 2 In vivo safety study. (A) Schematic depiction of the treatment schedule employed for the assessment of toxicity. (B) Representation of % body weight change from day 0 to day 14. (C) Graphical representation of the biochemical parameters for assessing liver toxicity (ALAT, SGOT, and SGPT) and kidney toxicity (creatinine and ASAT), data represented as mean ± SD, n = 6, ns = non-significant. (D) H&E staining microscopic photographs of all the vital organs, scale bar 200 μm. (*H&E – hematoxylin and eosin, ALAT – alanine transferase, SGOT – serum glutamic–oxaloacetic transaminase, SGPT – serum glutamic pyruvic transaminase, ASAT – aspartate aminotransferase). | ||
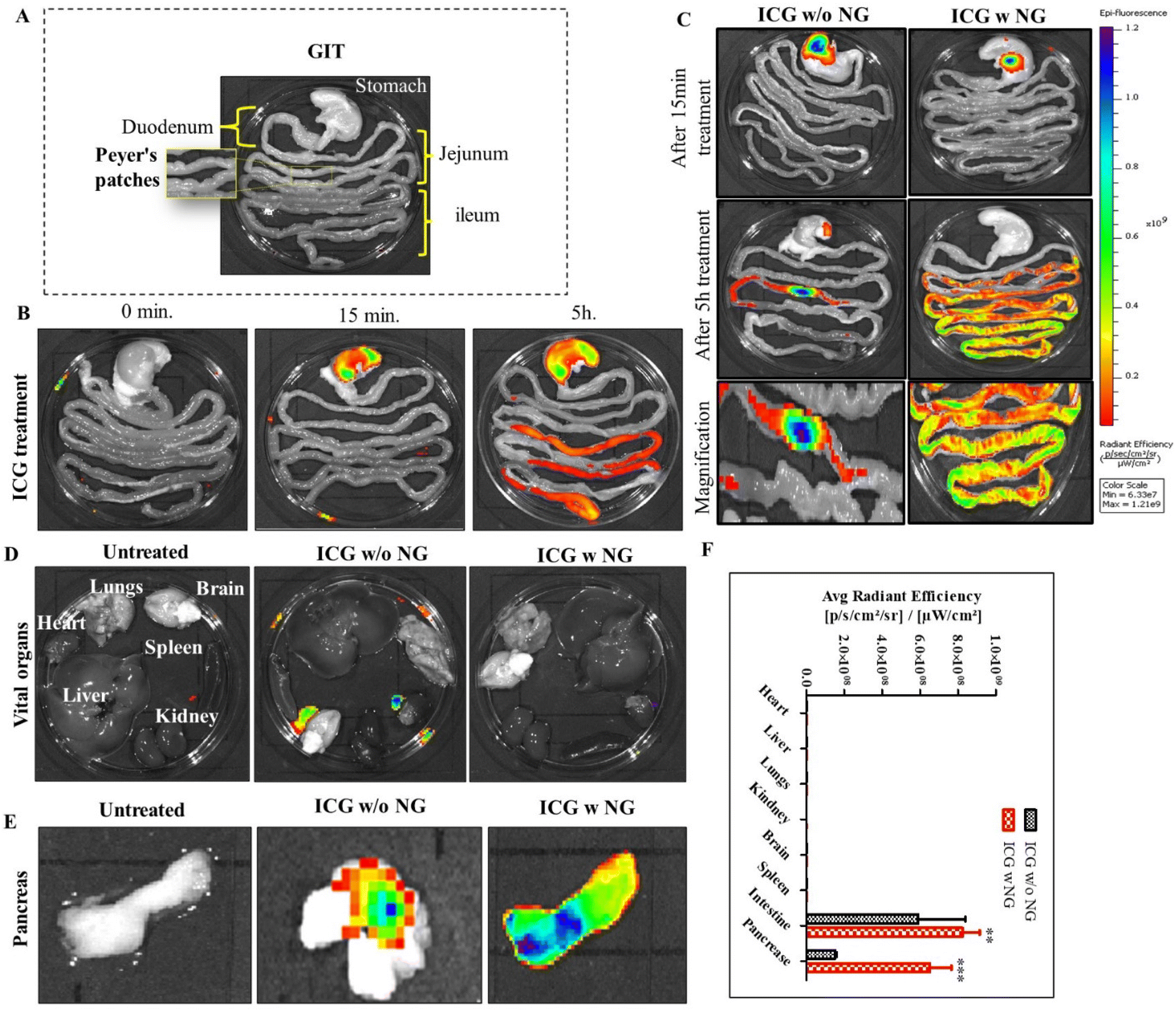 | ||
| Fig. 3 Representative images of the IVIS imaging. (A) Ex vivo IVIS imaging of the gastrointestinal tract of untreated and control rats. (B) IVIS images of free ICG distribution in the GIT. (C) IVIS fluorescence analysis of ICG w/o NG (without NG) and ICG w NG (with NG). (D and E) Ex vivo imaging of all vital organs (heart, kidney, liver, lung, spleen, brain, and pancreas). (F) Fluorescence intensities of the organs. ROI analysis of the fluorescence intensity is expressed as photons per second per square centimeter per steradian (p per s per cm2 per sr). In each experiment, we included n = 3 animals in every group. Level of significance: **P < 0.01 and ***P < 0.001. | ||
Route of drug delivery
The ILS represents a unique biological route for oral drug delivery, with the potential for selective targeting of the pancreas by mesenteric transportation.26 The ILS involves a basic biological absorption mechanism for microorganisms, macromolecules, and particles. To understand the intestinal absorption mechanism of the drug-loaded nanomicelles, a schematic illustration is presented in Fig. 4A, where NPs absorption is carried out by two types of cells: enterocytes or M cells. To understand whether the nanoparticles are uptake by the enterocytes or M cells we performed an in situ loop formation study and a Peyer's patches ligation assay. The in situ loop formation study was performed to understand the uptake of NP through the enterocytes, and Peyer's patch ligation assay was performed to understand the NPs uptake by the M cells. Fig. 4B presents a schematic depiction of the intestinal villi. First, we performed the intestinal loop method, which is a classical method to study the intestinal absorption of oral nanoformulations.27 We performed immunofluorescence staining to the enterocytes using the cytokeratin-8 antibody (Fig. 4C). We observed that some ICG-tagged NPs (NP) were internalized by the enterocytes of the villi of the small intestine and the green fluorescent NPs were colocalized in the enterocytes, stained with cytokeratin-8 (Cyto-8) for showing a red colored fluorescence (Fig. 4C). Furthermore, we revealed the internalization or absorption of the NPs in the mesenteric lymphatic system by IVIS imaging, where Fig. 4D shows an illustration for mesenteric lymph node (part of the ILS), and Fig. 4E shows the IVIS image of the NP uptake with fluorescence scattered through the mesenteric lymph capillaries of the intestine. The study revealed that the NPs followed the mesenteric lymphatic pathway for absorption through the intestine.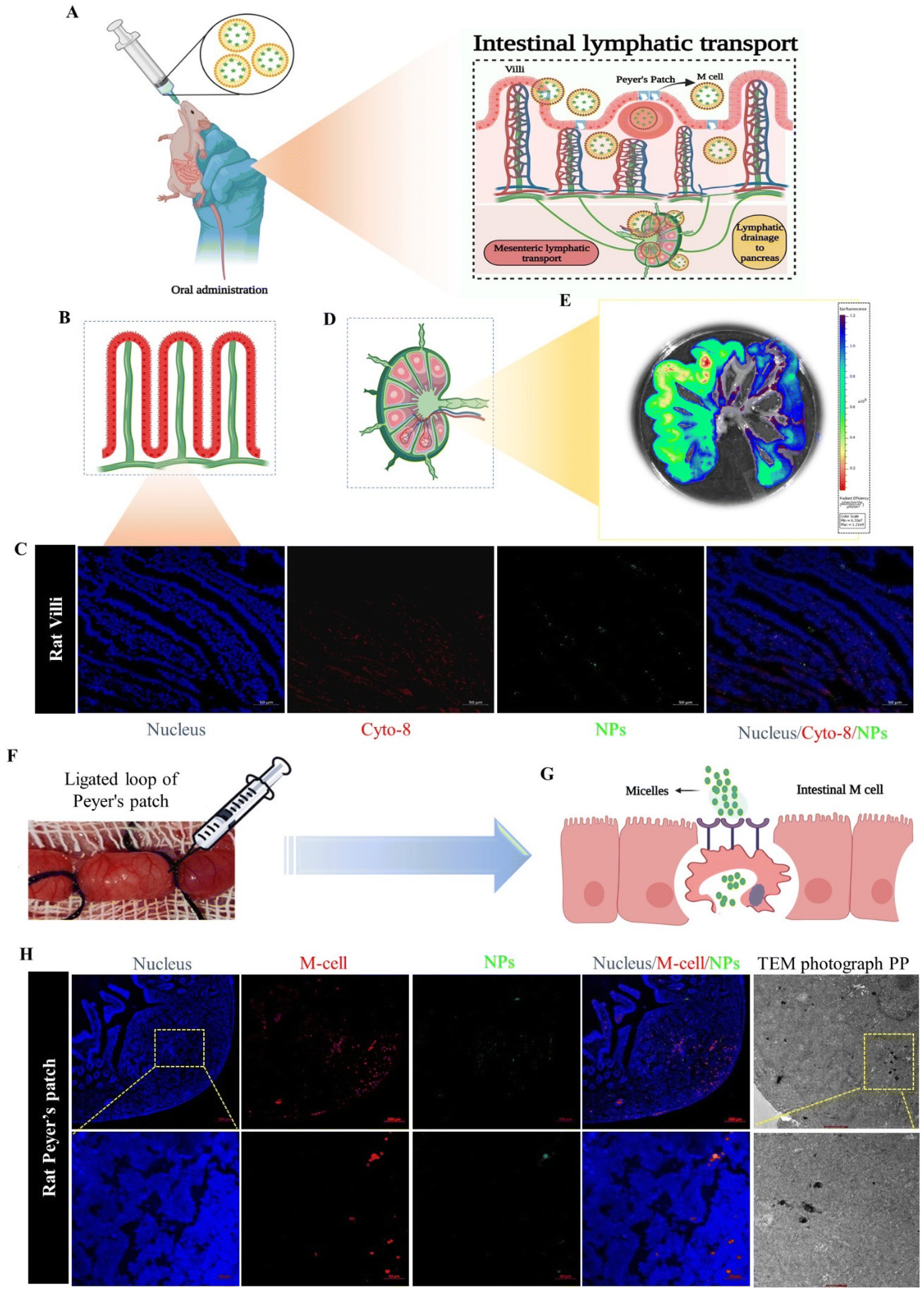 | ||
| Fig. 4 (A) Illustration of the drug-delivery route by the intestinal lymphatic pathway after oral drug administration. In situ intestinal loop formation study. (B) Schematic depiction of the intestinal villi. (C) Immunofluorescence staining of cytokeratin-8 (cyt-8) performed on a villi segment, scale bar: 50 μm. (D) Mesenteric lymphatic node (part of the intestinal lymphatic system). (E) Ex vivo IVIS fluorescence images of the mesenteric lymphatic system (MLS). (F) Representation of the intestinal Peyer's patch ligation assay. ICG-loaded NPs were introduced into a ligated Peyer's patch loop using a syringe. (G) Immunofluorescence staining and TEM analysis of M cells, indicating the co-localization of the NPs (ICG-loaded nanomicelles) in Peyer's patches 2 h after treatment. Scale bar: 200 and 50 μm. In each experiment, we included n = 3 animals in every group. | ||
In the Peyer's patch ligation assay, the in vivo localization of nanoparticles in M cells was revealed.28,29Fig. 4F and G present the intestinal Peyer's patch ligation assay results, where ICG-loaded NPs (NPs) were introduced into a ligated Peyer's patch loop using a syringe. M cells in the follicle-associated epithelium covering the Peyer's patch were detected by whole-mount immunostaining with anti-Gp2 antibody. We observed that the NPs were efficiently colocalized in M cells. The NPs undergo transcytosis mediated by specialized epithelial M cells (Fig. 4H). Further internalization of the NPs into PPs was confirmed by TEM imaging of the internalized nanomicelles within the ligated Peyer's patch (Fig. 4H).
Both assays revealed that the NPs were internalized through the M cells as well as enterocytes of the intestine; however, most of the NPs were uptaken by M cells due to the internalization through M cells being governed by the size and surface charge of the particles. M cells have the capability to capture negatively charged particles efficiently, due to the minimal electric repulsion for the particles in M cells compared to enterocytes.30,31 Additionally, the cellular internalization of nanoparticles was contingent upon the size of particles: small-sized particles (less than 100 nm) could be uptaken by enterocytes, whereas larger particles were more suited to being internalized by M cells.32
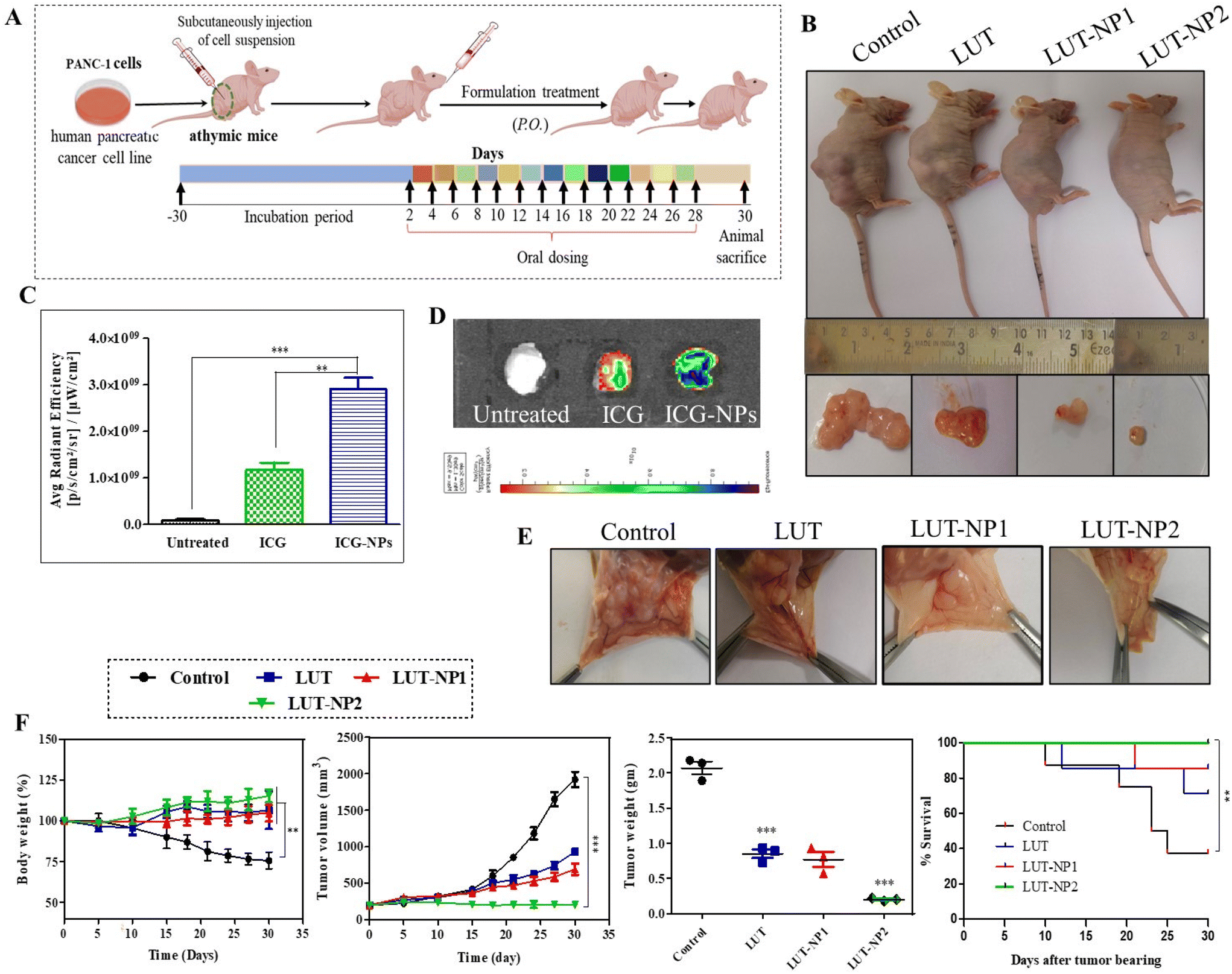 | ||
| Fig. 5 LUT-NP-mediated antitumor efficacy in a subcutaneous PANC-1 xenograft model. (A) Treatment schedule and protocol for the study. Disease animals (tumor-bearing mice) received oral treatment of LUT at a dose of 100 mg kg−1 p.o.−1 or oral administration of the nanoformulation LUT-NP1/LUT-NP2 at an equivalent dose of LUT = 100 mg kg−1 and 50 mg kg−1, respectively. (B) Photographs of the tumor-bearing mice and the removed tumor at the end of the study following various treatments: LUT, LUT-NP1, and LUT-NP2. (C) Ex vivo IVIS imaging was conducted to assess intratumor NP accumulation after one-day oral treatment. The fluorescence intensity was quantified in the regions of interest by IVIS imaging software. (D) Ex vivo IVIS images of solid tumors. (E) Observation of in vivo intratumor angiogenesis (blood vessels formation) inhibition after the end of treatment. (F) Body weight, tumor growth, and survival curve monitored till 30 days of treatment course, and the tumor weight of isolated solid tumors was noted. Each group contained six mice (n = 6), except the disease (control) group, which had eight (n = 8) due to mortality. All data represented as the mean ± SD, *P < 0.05; **P < 0.01; ***P < 0.001. ns, not significant. | ||
Angiogenesis plays a key role in tumor growth and metastasis by supplying oxygen with the formation of new blood vessels, thus providing essential nutrients for the growth of the tumor.32 Preventing the development of blood vessels in the tumor vasculature can impede tumor progress.35 Consequently, we further investigated the effect of LUT and LUT-NPs on tumor angiogenesis (Fig. 5E). We observed there were less blood vessels formed in LUT-NP1 due to the anti-angiogenic effect of the compound, whereas the control group tumors showed more blood vessels were developed and distributed into the whole tumor mass. The LUT-NP2-treated group tumor showed a notable anti-angiogenic activity compared to the LUT-treated group. These results revealed that LUT was able to reduce the angiogenesis of the tumor, but potential anti-angiogenic activity was found in the LUT-NP2-treated group. Further, this nanoformulation could target various anti-angiogenic pathways, offering an alternative to traditional therapy and proving beneficial for reducing both tumor growth and metastasis.
The efficacy of the formulation was confirmed by monitoring the body weight, tumor growth and survival rate of the mice throughout the treatment schedule (Fig. 5F). In the disease group (control), the body weight was slightly increased up to 1 week, but after development of the tumor the body weight continuously reduced. The body weight of the control group was significantly reduced compared to the treatment group (***p < 0.001) due to disease-associated cachexia.36 Furthermore, there was no significant alteration in the mouse weight observed in the LUT, LUT-NP1, and LUT-NP2-treated groups during the treatment schedule, which reflected the antitumor effect of the treatment. Along with the body weight, the tumor volume of all the groups was measured during a complete course of treatment. The tumor volume graph shows that the tumor volume of the untreated group (control) was exceptionally increased, and at the end of the study, the tumor volume was increased up to 1919.33 mm3 due to aggressive tumor growth. The anticancer activity of LUT was reflected in the LUT-treated group, where the tumor volume (933.22 mm3) was significantly (***p < 0.001) decreased compared to the control group. Similar effects were observed in the low-dose formulation (LUT-NP1)-treated group, where the tumor volume was 700 mm3 at the end of the experiment. When comparing the experimental LUT-NP2-treated group with the other treatment groups, it was evident that its antitumor efficacy was significantly higher than for LUT-NP1 and LUT (with significant difference, P < 0.005). The tumor volume differentiation remained constant to about 203.33 mm3. We measured the weight of the excised tumors at the study completion, wherein the LUT-NP2-treated group exhibited a significant higher reduction of tumor weight than the LUT- and LUT-NP1-treated groups (*P < 0.05). LUT (at a dose of 100 mg kg−1) showed an anticancer effect in terms of tumor volume and tumor weight reduction against PANC-1 cells tumor, but the efficacy of treatment was more potentially observed in the LUT-NP2-treated group, however at a low dose of LUT in the nanocarrier, the tumor-shrinking behavior was similar to that of the LUT-treated group due to NG formulation. The potential antitumor effect was evident from the changes in survival rates during the treatment as well as the reduced tumor volumes and weights at the end of the study.
In our previous work, we determined the anticancer mechanism of LUT against the PANC-1 cell line; on this basis, we explored the in vivo anticancer mechanism of LUT. The tumor mass was isolated at the end of study and histological assessments were carried out through H&E staining, Masson's trichrome staining, and immunohistochemical staining (IHC). H&E staining of the tumor sections clearly reflected the typical tumor morphology, such as higher tumor cells number, active mitosis, high nuclear to cytoplasmic ratio, and large hyperchromatic nuclei in the control group (depicted in Fig. 6A); whereas on the other hand, the tumor samples of the treated mice, namely LUT, LUT-NP1, and LUT-NP2 groups, showed marked differences in tumor cell morphology, like cell necrosis pyknosis (karyopyknosis), cell morphology destruction, and tumor cells reduction (Fig. 6A). Further, the collagen expression (indicated with red arrow) in the tumor mass was identified by Masson's trichrome staining (Fig. 6A), revealing that the distribution of collagen was effectively reduced in the LUT-NP2-treated group compared to the LUT-treated group. This observation was further confirmed by the quantification of the tumor collagen content using a Sircol assay (as shown in Fig. 6C). This revealed a notable decrease (*P < 0.05) in collagen content in the tumor samples of the LUT- and LUT-NP1-, and LUT-NP2-treated groups compared to the control group. The reduction in collagen in LUT-NP2 was higher compared to the control group (**P < 0.01). Further, we assessed the expression levels of VEGF-A (vascular endothelium growth factor A), caspase-3, FAK (Focal Adhesion Kinase), and TNF-α (tumor necrotic factor) through IHC (Fig. 6B) of the tumor histological sections. Our results revealed that the tumor sections of the disease group (control) showed upregulated expressions levels of VEGF-A, FAK, and TNF-α, as evidenced by immunopositive brown color cells (indicated with red arrow). The expression levels of these markers were significantly reduced in all treatment groups (LUT, LUT-NP1, and LUT-NP2). While assessing the expressions of caspase-3, it was observed that caspase-3 was downregulated in the disease condition but upregulated in the treatment groups compared to the disease group. LUT-NP2 showed a significant inhibition of VEGF-A, FAK, and TNF-α and also upregulated the expression of caspase-3. We also quantified the number of immunopositive cells for specific markers with the help of Image J software, and calculated the number of immunopositive cell and compared these with the results from IHC (shown in Fig. 6D–G), with the results presented in Fig. 6B. The results indicated that LUT-NPs had the ability to effectively downregulate the expressions of VEGF-A, FAK, and TNF-α as well as upregulate caspase-3 expression at malignant sites, consequently augmenting the antitumor efficacy of LUT in pancreatic cancer in a dose-dependent manner.
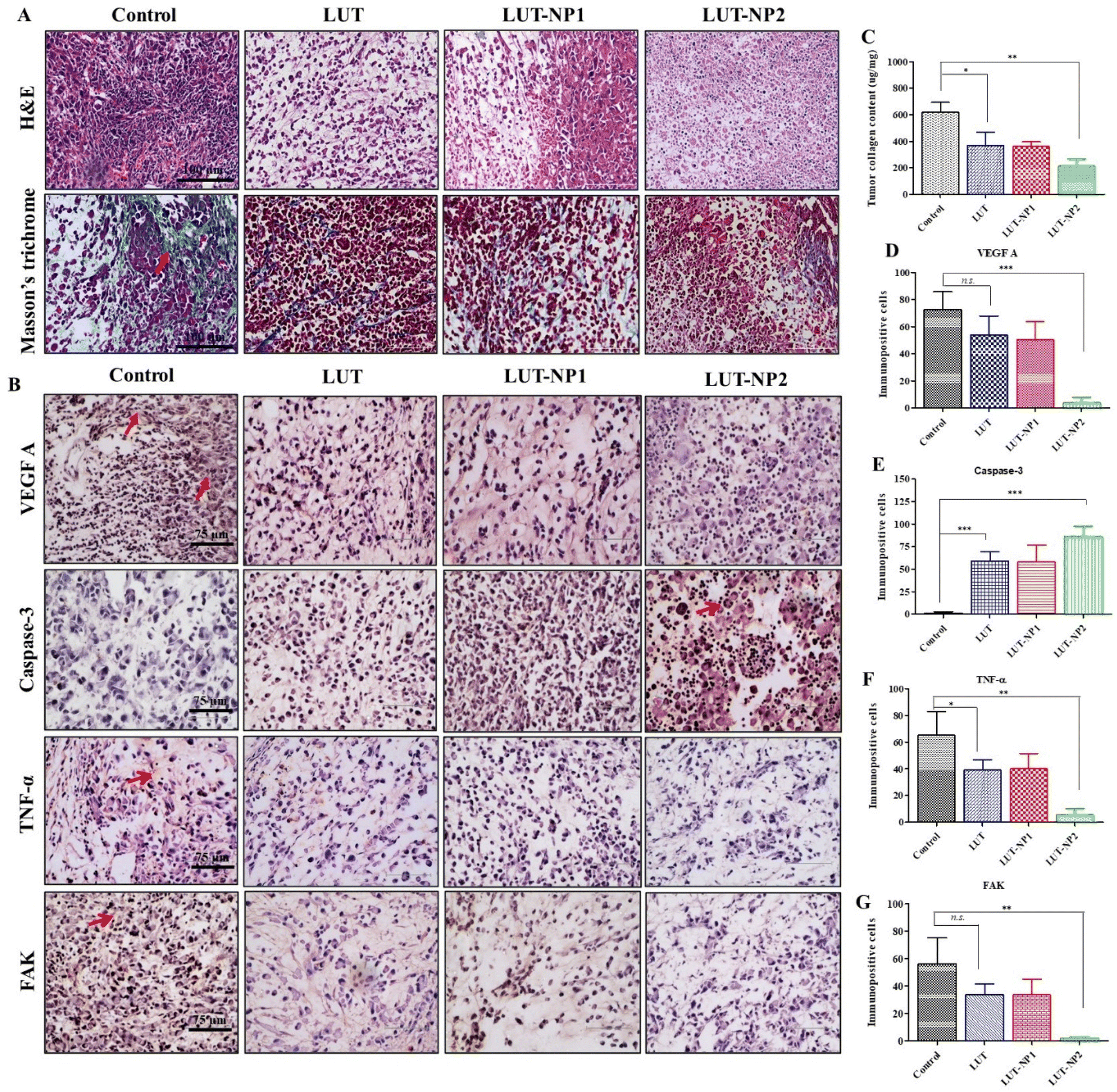 | ||
| Fig. 6 Pictorial and graphical representations of histological staining. (A) Histological analysis with H&E staining and Masson's trichrome staining; red color arrow indicates collagen staining on tumor sections, scale bar = 100 μm. (B) Immunohistochemical staining for assessing the expressions of VEFG A, caspase-3, FAK, and TNF-α, scale bar = 75 μm; red color arrow represents protein expression. (C) Overall collagen content within the tumor tissue was quantified with the Sircol Soluble Collagen Assay Kit, supplied by ThermoFisher Scientific. Quantification of immunopositive cells: (D) VEFG A, (E) caspase-3, (F) TNF-α, and (G) FAK. All data points are represented as the mean ± SD. (n = 3). p* < 0.05, **p < 0.01, ***p < 0.001, ns = not significant compared with the disease control. | ||
Based on the IHC results, gene and protein expression analyses were carried out to provide insights into the molecular mechanisms of LUT in pancreatic cancer. Hence, we performed RT-PCR analysis of VEGF-A, caspase-3, TNF-α, and PTK2/FAK (Fig. 7A). We also determined the gene expression of Ki-67 due to its strong correlation with tumor cell proliferation and tumor metastasis in cancer.37 We observed that LUT treatment significantly (*P < 0.05) increased caspase-3 expression in pancreatic tumors. We also observed a fourfold upregulation in caspase-3 gene expression in the LUT-NP2-treated group compared to the disease control group. We observed that the caspase-3 gene expression in the LUT-NP2-treatment group was significantly (***P < 0.001) upregulated compared to the LUT- and LUT-NP1-treatment groups.
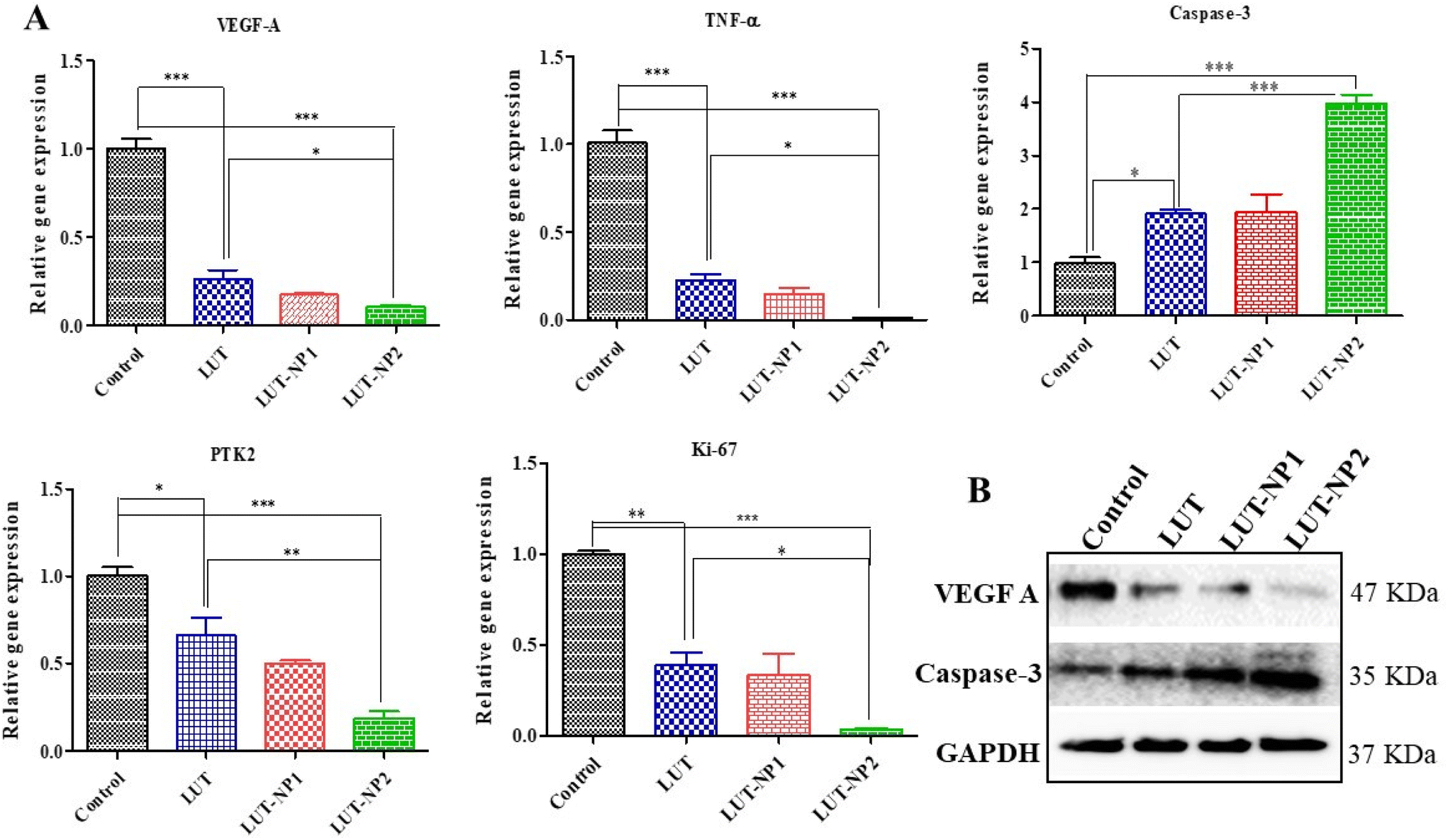 | ||
| Fig. 7 Anticancer mechanism and efficacy of LUT/LUT-NPs against pancreatic cancer. (A) mRNA expression analysis via RT-PCR. (B) Western blot analysis, which was conducted to assess alterations in the expressions of VEGF-A and caspase-3 after treatment. *p < 0.05, **p < 0.01, ***p < 0.001. | ||
The gene expression levels of VEGF-A, TNF-α, PTK2 (Protein Tyrosine Kinase), and Ki-67 were downregulated in the LUT-NP2-treated group compared to the untreated and disease groups, by approximately three-fold. Comparison of the expressions between the LUT-treated group and LUT-NP2-treated group showed significant reductions in the levels of VEGF-A, TNF-α, PTK2, and Ki-67 genes.
Furthermore, we performed western blot analysis to confirm the protein expressions of caspase-3 and VEGF-A after the treatment (as shown in Fig. 7B). We observed that the LUT-treated group showed a substantially increased expression of caspase-3 and decreased expression of VEGF-A compared to the control or disease group. In the comparison between the LUT and LUT-NP2 potential, we observed that LUT-NP2 enhanced the expression of apoptotic protein and reduced the expression of proliferative protein.
All the results suggest that free LUT has the ability to downregulate the expression of cell proliferative markers and upregulate the apoptotic protein in pancreatic cancer, but when incorporating it in the nanocarrier system, the anticancer potency of LUT-NP2 is more profound and potent compared to free LUT after oral administration due to its solubility, stability, and permeability enhancement effect along with the targeted drug delivery.
Conclusions
In summary, the aforementioned results collectively illustrate that the effervescent-based nano-gas carrier could efficiently transport the poorly water-soluble drug LUT to the target site after oral administration. Biocompatible LUT-NPs can overcome the intestinal epithelial barrier in a noninvasive manner, while the surface charge and particle size of the nanomicelles help in M-cell-medicated lymphatic drug transport, and from there the drug can accumulate in the pancreatic tumor mass and show anticancer efficacy, which improved the survival rate of tumor-bearing mice.We investigated the anticancer mechanism of LUT against pancreatic cancer. LUT showed significant therapeutic potency by activating the caspase-3 mediated apoptosis pathway and blocked tumor angiogenesis and metastasis by downregulating VEGF A, FAK, TNF-α, and Ki-67 against PANC-1 tumors when delivered in the form of nanomicelles. We expect that our drug-delivery approach through the lymphatic transport system (ILS) has significant translational potential for the treatment of pancreatic diseases.
Author contributions
Archana Karole: Conceptualization, methodology, investigation, writing – original draft, writing – review and editing. Yirivinti Hayagreeva Dinakar: Methodology and investigation. Poonam Sagar: Investigation. Shyam Lal Mudavath: Conceptualization, supervision, writing – review and editing, funding acquisition.Conflicts of interest
Authors declare no conflict of interests with regards to the submitted work.Acknowledgements
All schematic illustrations were made using BioRender software. Archana Karole thanks Institute of Nano Science and Technology for providing the doctoral fellowship. This work is supported by the Government of India for funding under DST-SERB (EEQ/2020/000563).References
- J. Ushio, A. Kanno, E. Ikeda, K. Ando, H. Nagai, T. Miwata, Y. Kawasaki, Y. Tada, K. Yokoyama, N. Numao, K. Tamada, A. K. Lefor and H. Yamamoto, Diagnostics, 2021, 11, 1–13 CrossRef PubMed.
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- T. J. Grant, K. Hua and A. Singh, Prog. Mol. Biol. Transl. Sci., 2016, 144, 241–275 CrossRef CAS PubMed.
- M. Amrutkar and I. P. Gladhaug, Cancers, 2017, 9, 1–23 CrossRef PubMed.
- A. Neesse, P. Michl, K. K. Frese, C. Feig, N. Cook, M. A. Jacobetz, M. P. Lolkema, M. Buchholz, K. P. Olive, T. M. Gress and D. A. Tuveson, Gut, 2011, 60, 861–868 CrossRef PubMed.
- N. Kaushik, S. B. Borkar, S. K. Nandanwar, P. K. Panda, E. H. Choi and N. K. Kaushik, J. Nanobiotechnol., 2022, 20, 1–23 CrossRef PubMed.
- W. Chen, J. Dong, J. Haiech, M. C. Kilhoffer and M. Zeniou, Stem Cells Int., 2016, 16, 1–38 Search PubMed.
- S. Md, N. A. Alhakamy, H. M. Aldawsari, J. Ahmad, W. S. Alharbi and H. Z. Asfour, J. Drug Delivery Sci. Technol., 2021, 64, 102555 CrossRef CAS.
- Y. Mao, X. Wang, C. Chen, Q. Zhao, Y. Liu, J. Zhang and S. Wang, Acta Pharm. Sin. B, 2022, 12, 4501–4518 CrossRef CAS PubMed.
- A. Dillon and D. D. Lo, Acta Pharm. Sin. B, 2019, 10, 1–13 Search PubMed.
- S. Song, Z. Su, H. Xu, M. Niu, X. Chen, H. Min, B. Zhang, G. Sun, S. Xie, H. Wang and Q. Gao, Cell Death Dis., 2017, 8, 1–9 CrossRef CAS PubMed.
- D. Majumdar, K.-H. Jung, H. Zhang, S. Nannapaneni, X. Wang, A. R. M. R. Amin, Z. Chen, Z. G. Chen and D. M. Shin, Cancer Prev. Res., 2014, 7, 65–73 CrossRef CAS PubMed.
- A. Karole, S. Parvez, R. S. Thakur and S. L. Mudavath, J. Drug Delivery Sci. Technol., 2022, 69, 103167 CrossRef CAS.
- S. Parvez, A. Karole and S. L. Mudavath, Biochim. Biophys. Acta, Gen. Subj., 2022, 1866, 130157 CrossRef CAS PubMed.
- S. Fukuda, K. Hase and H. Ohno, J. Visualized Exp., 2011, 1–5 Search PubMed.
- M. Garinot, V. Fiévez, V. Pourcelle, F. Stoffelbach, A. des Rieux, L. Plapied, I. Theate, H. Freichels, C. Jérôme, J. Marchand-Brynaert, Y. J. Schneider and V. Préat, J. Controlled Release, 2007, 120, 195–204 CrossRef CAS PubMed.
- T. Sugiyama, A. Yamamoto, Y. Kawabe, T. Uchiyama, Y. S. Quan and S. Muranishi, Biol. Pharm. Bull., 1997, 20, 812–814 CrossRef CAS PubMed.
- R. Moll, W. W. Franke, D. L. Schiller, B. Geiger and R. Krepler, Cell, 1982, 31, 11–24 CrossRef CAS PubMed.
- E. Y. Chuang, K. J. Lin, T. Y. Huang, H. L. Chen, Y. B. Miao, P. Y. Lin, C. T. Chen, J. H. Juang and H. W. Sung, ACS Nano, 2018, 12, 6389–6397 CrossRef CAS PubMed.
- Y. B. A. Kang, S. Rawat, J. Cirillo, M. Bouchard and H. M. Noh, Biofabrication, 2013, 5, 45008 CrossRef CAS PubMed.
- L.-W. Hsu, Y.-C. Ho, E.-Y. Chuang, C.-T. Chen, J.-H. Juang, F.-Y. Su, S.-M. Hwang and H.-W. Sung, Biomaterials, 2013, 34, 784–793 CrossRef CAS PubMed.
- E. Y. Chuang, K. J. Lin, P. Y. Lin, H. L. Chen, S. P. Wey, F. L. Mi, H. C. Hsiao, C. T. Chen and H. W. Sung, Biomaterials, 2015, 64, 115–124 CrossRef CAS PubMed.
- F. Araújo, P. Fonte, H. A. Santos and B. Sarmento, J. Diabetes Sci. Technol., 2012, 6, 1486–1497 CrossRef PubMed.
- B. T. Helfand, M. G. Mendez, J. Pugh, C. Delsert and R. D. Goldman, Mol. Biol. Cell, 2003, 14, 5069–5081 CrossRef CAS PubMed.
- E.-Y. Chuang, K.-J. Lin, F.-Y. Su, F.-L. Mi, B. Maiti, C.-T. Chen, S.-P. Wey, T.-C. Yen, J.-H. Juang and H.-W. Sung, J. Controlled Release, 2013, 172, 513–522 CrossRef CAS PubMed.
- S. Hirono, M. Tani, M. Kawai, K.-I. Okada, M. Miyazawa, A. Shimizu, K. Uchiyama and H. Yamaue, Dig. Surg., 2012, 29, 132–139 CrossRef PubMed.
- W. Liu, H. Pan, C. Zhang, L. Zhao, R. Zhao, Y. Zhu and W. Pan, Int. J. Mol. Sci., 2016, 17(7), 1171 CrossRef PubMed.
- S. Hapfelmeier, M. A. E. Lawson, E. Slack, J. K. Kirundi, M. Stoel, M. Heikenwalder, J. Cahenzli, Y. Velykoredko, M. L. Balmer, K. Endt, M. B. Geuking, R. Curtiss 3rd, K. D. McCoy and A. J. Macpherson, Science, 2010, 328, 1705–1709 CrossRef CAS PubMed.
- R. L. Owen and A. L. Jones, Gastroenterology, 1974, 66, 189–203 CrossRef CAS.
- O. Borges, A. Cordeiro-da-Silva, S. G. Romeijn, M. Amidi, A. de Sousa, G. Borchard and H. E. Junginger, J. Controlled Release, 2006, 114, 348–358 CrossRef CAS PubMed.
- K. M. Bennett, S. L. Walker and D. D. Lo, Infect. Immun., 2014, 82, 2860–2871 CrossRef PubMed.
- G. M. Tozer, C. Kanthou and B. C. Baguley, Nat. Rev. Cancer, 2005, 5, 423–435 CrossRef CAS PubMed.
- B. R. Smith, E. E. B. Ghosn, H. Rallapalli, J. A. Prescher, T. Larson, L. A. Herzenberg and S. S. Gambhir, Nat. Nanotechnol., 2014, 9, 481–487 CrossRef CAS PubMed.
- Y. B. Miao, K. H. Chen, C. T. Chen, F. L. Mi, Y. J. Lin, Y. Chang, C. S. Chiang, J. T. Wang, K. J. Lin and H. W. Sung, Adv. Mater., 2021, 33, 2100701 CrossRef CAS PubMed.
- M. Kohandel, M. Kardar, M. Milosevic and S. Sivaloganathan, Phys. Med. Biol., 2007, 52, 3665 CrossRef CAS PubMed.
- M. Hegde, U. D. Daimary, S. Girisa, A. Kumar and A. B. Kunnumakkara, Exp. Biol. Med., 2022, 247, 713–733 CrossRef CAS PubMed.
- N. A. Hamilton, T.-C. Liu, A. Cavatiao, K. Mawad, L. Chen, S. S. Strasberg, D. C. Linehan, D. Cao and W. G. Hawkins, Surgery, 2012, 152, 107–113 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr06638j |
| This journal is © The Royal Society of Chemistry 2024 |