
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Functional oligo- and polypeptide assemblies for photochemical, optical and electronic applications
Yohei
Yamamoto
 *a,
Wey Yih
Heah
a and
Kentaro
Tashiro
*b
*a,
Wey Yih
Heah
a and
Kentaro
Tashiro
*b
aInstitute of Pure and Applied Sciences, University of Tsukuba, 1-1-1, Tennodai, Tsukuba, Ibaraki 305-8573, Japan. E-mail: yamamoto@ims.tsukuba.ac.jp
bResearch Center for Macromolecules & Biomaterials, National Institute for Materials Science, 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan. E-mail: Tashiro.kentaro@nims.go.jp
First published on 24th June 2024
Abstract
The primary and secondary structures of peptides are useful as scaffolds to sequentially arrange functional groups of molecules. In this review, we review self-assembled functional peptides, whereby peptides with appropriate amino acid sequences can assemble using functional groups on their side chains. First, we apply our design strategies for the synthesis of peptide-based materials with sequenced side chains with polar moieties, organic dyes and metal complexes. The synthetic oligopeptides thus obtained exhibit inherent photoinduced charge separation and electrochemical redox activities, as well as responses to bio-sequences. Next, catalytic and photocatalytic oxidation reduction reactions and hydrogen evolution reactions are shown by utilizing the peptides with separated functionalities on both sides of β-sheets by hybridizing with electro- and photoactive graphene oxide and metal nanoparticles. Finally, the self-assembled natural proteins that form micrometre-scale spherical geometry and fibres are utilized for optical and electronic applications. The silk fibroin forms well-defined microspheres with smooth surface morphology, leading to properties suitable for use in optical resonators, which can sense external humidity because of the hygroscopic nature of silk fibroin. Dragline silk fibres can act as optical waveguides that can perform intermediate natural polymer-based optical logic operations. These functional peptides are utilizable for various applications in catalysis, optics and electronics.
Wider impactPeptides are one of the main components in our body and have an essential role in life activities. In this focused review, we show the possible application of peptide assemblies in not only living organisms and biomedical materials but also for other purposes, such as electrochemical, catalytic, optical, and electrical applications, by utilizing the potential self-organizing ability of peptides. For example, peptide β-sheets can arrange the functional units, such as positive/negative charges, electron donor/acceptor units, and metal nanoparticles, leading to efficient photoinduced charge separation and photocatalytic reactions. Furthermore, self-assembled peptide assemblies can be used as components of optical sensing and optical logic devices, as well as organic electronic devices. This review includes the fundamentals of peptide assembling structures and ways in which molecular functional groups assemble using peptide scaffolds. These insights will be valuable to find the potential of peptides and their assemblies for applications in various fields. |
1. Introduction
The ability to put components in their appropriate positions is crucial, not only to manufacturing machinery but also to engineering a wide range of materials.1 When these materials are supposed to be prepared through assembling molecules, an attractive option of compounds for this attempt would be peptides, as their intrinsic structural features allow control of functionalities over one-, two- and three-dimensional (1D, 2D and 3D) orientations.2,3 First, the primary structure of a single peptide chain is composed of a sequence of multiple monomer units, amino acids (AAs), whose side residue can be used as the scaffold to selectively construct a specific 1D sequence of multiple functionalities (Fig. 1). Second, each peptide can adopt a particular conformation, such as helix, strand and turn, which is dependent on its primary sequence, while single or multiple peptide chains form 2D sheets (β-sheet) that are composed of multiple strands aligned in parallel or anti-parallel fashion (Fig. 1). By taking advantage of this secondary structure-forming capability of peptides, specific 2D and 3D orientations of their side residues and functionalities are programmable through the judicious choice of the primary sequence of peptides (Fig. 1). When the length of a peptide chain becomes long enough to be regarded as a protein, an escalating number of possible 3D conformational outcomes allow its folding and self-assembly into non-natural microstructures, creating unprecedented materials. This review summarizes the attempts of researchers, including us, for more than a decade to design functional materials based on peptides. The reviewed studies mainly focus on two topics: (1) the development of novel synthetic strategies to reach unprecedented structures of peptide-based materials that are difficult to achieve by conventional methods4,5 and (2) exploration of the physical/chemical properties of these materials to find out their applications.6 | ||
| Fig. 1 Schematic representations of primary and secondary structures of peptides. AA: Amino acid, Rn: Side chain at the n-th AA. | ||
2. Strategies to design peptide-based materials
2.1. Covalent introduction of non-natural functionalities into peptide side residues
For the preparation of a peptide sequence with multiple non-natural functionalities that are covalently linked to side residues, two different synthetic approaches can be considered. The first approach is the sequentialization of amino acids, whose side residues have been modified in advance with one of the non-natural functionalities (Fig. 2a). This approach is theoretically useful to produce targeted sequences in a defect-free form as all required functionalities can be quantitatively attached at expected positions. It is also advantageous to introduce diverse functionalities into single peptide chains, where even a single type of amino acid side residue can be sufficiently used for site-specific covalent linkage of several different functionalities. This approach allowed us4,5,7,8 and other research groups9–12 to obtain several peptide sequences with multiple organic, as well as metal complex, moieties.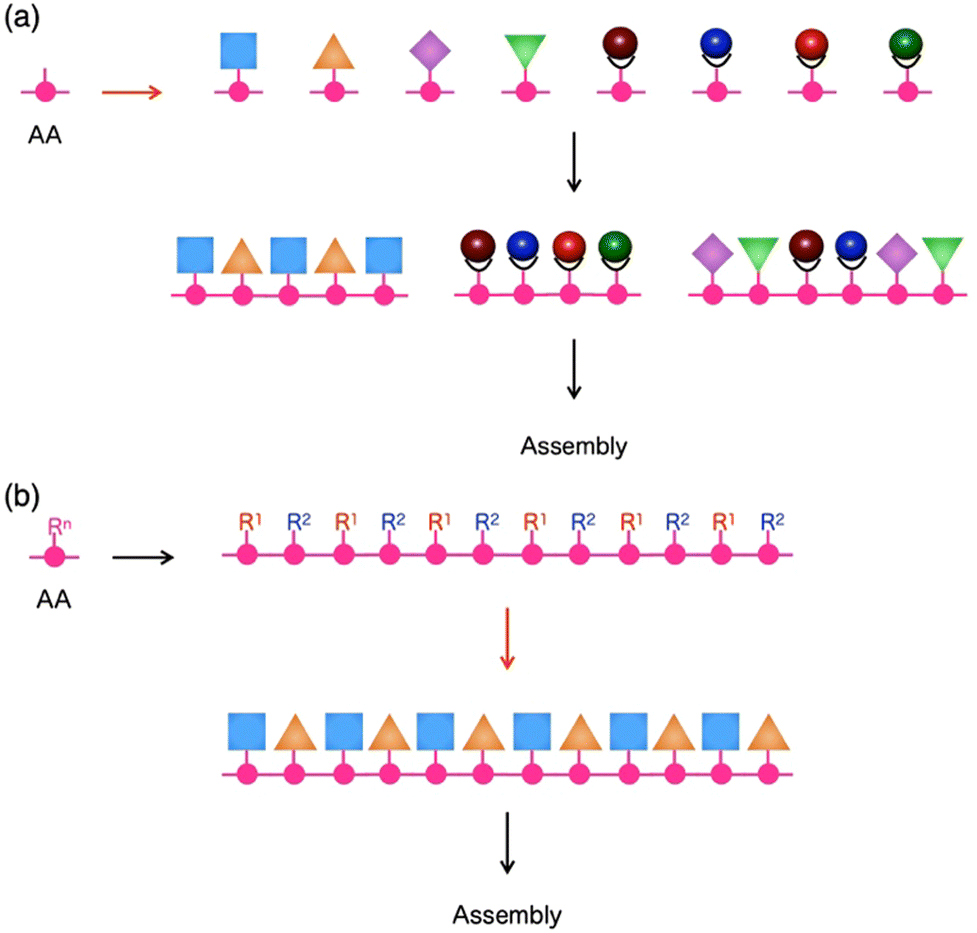 | ||
| Fig. 2 (a) Sequentialization of side-residue functionalized amino acids and (b) post-modification of the side residues of a natural peptide to prepare peptide sequences with multiple non-natural functionalities that are covalently linked to the side residues. Red arrows in schemes correspond to the steps of side-residue functionalization. | ||
As a representative example, a terpyridine-appended L-tyrosine derivative was designed for metalation with Pt(II), Rh(III), or Ru(II) ions to afford a series of metalated amino-acid monomers.4 These monomers were sequentially coupled using the solid-phase peptide synthesis technique13,14 to produce tyrosine oligomers with a specific heteronuclear multi-metallic sequence, such as Rh–Pt–Ru–Pt–Rh–Pt (Fig. 3a). As proved by our experimental results, this synthetic strategy effectively works for the preparation of shorter peptides with 10 or fewer functionalities in a single molecule. However, its straightforward application for the construction of longer sequences containing monomers with a good number of variations could be challenging due to the need to synthesise and coupling an increasing number of monomer structures.
 | ||
| Fig. 3 Molecular structures of side-residue functionalized peptides bearing (a) Rh–Pt–Ru–Pt–Rh–Pt and (b) Sm–Tb–Eu metal seaueces. | ||
The latter issue has been partly addressed by introducing a modular strategy, where shorter sequences prepared via solid-phase synthesis protocols were site-specifically linked in solution to produce longer linear or branched structures.5 To avoid the randomization of constructed metal sequences through undesired ligand–metal exchange or demetallation,15,16 it is crucial to choose the ligand structure judiciously. Because of the sufficiently low level of ligand–metal exchange activity, multidentate ligands, such as terpyridine,4 phenanthroline,17–19 and porphyrin,5 turned out to be useful choices. Later, another research group developed a cryptate-based ligand system that could bind and exchange labile lanthanoid ions firmly by taking advantage of its capability to encapsulate metal ions (Fig. 3b).12 The metal complex sequences, similar to underlying amino acid sequences, were found to strongly affect the self-assembly behaviour of these multi-metalated peptide species, where their gelation17 or amyloid-like fibril formation7 was promoted by the presence of particular sequences, while a heterotrimetallic peptide sequence exhibited an anti-Hofmeister trend in its anion-dependent aggregation.20
The second approach to introduce non-natural functionalities into the peptide side residues is the post-modification of the side residues in natural peptide sequences (Fig. 2b). This is currently regarded as a practical approach to obtaining side-residue functionalized long peptides. This approach is useful when research goals allow an imperfect conversion of the side residues into planned functionalities, as well as when it is difficult to perform a site-specific modification of the residues in a single peptide with several types of functionalities.21,22 While some of the side-residue functionalized peptides thus prepared have been used in the same way as their non-assembled forms,23 most were successively subjected to self-assembly to afford β-sheet-like secondary structures, where the functionalities attached can spontaneously adopt specific 2D and 3D orientations to serve particular roles.
2.2. Charge-separated peptide β-sheets with natural amino acids with charged side chains
Peptide β-sheet is an appropriate scaffold to arrange molecular functionality. Pioneering works on the self-assembly of the peptides with molecular functionality have been carried out by several groups, and excellent reviews on the fundamentals and applications of self-assembled peptide assemblies were published.24–31 In the present review, we introduce research results on arranging molecular functionality on the top and bottom sides of the β-sheet.Nakayama et al. prepared peptide β-sheets, where positively- and negatively charged side chains, were separated on the top and bottom sides of the β-sheets (Fig. 4a).8 The strategy for the formation of charge-separated peptide β-sheets is that, in the β-sheets, side chains of odd- and even-numbered amino acids are arranged on the opposite side of the β-sheet. According to the 9-fluorenyl methoxycarbonyl (Fmoc)-peptide synthetic protocol,13 18 Fmoc-pentapeptides, including valine (V, neutral), lysine (K, positive charge at the even position, R2 and/or R4), and glutamic acid (E, negative charge at the odd position, R1, R3, and/or R5), are synthesized, and their self-assembling behaviors in MeOH are investigated with the slow addition of Et2O vapour. By X-ray diffraction (XRD) and transmission electron microscopy (TEM) studies, Fmoc-pentapeptides with one E and K each were observed to form β-sheet structures in the solid state after evaporation of the solvent for the self-assembled structure. However, when the β-sheets were added to a glycine buffer (pH = 8.5), only selected sequences maintained the β-sheet structure, as revealed by the photoluminescence (PL) test using thioflavin (ThT) that selectively emits PL upon adsorption on the β-sheet. Especially, Fmoc-peptides with charged side chains at the central position (R3) keep the β-sheet structure with difficulty because of the charge repulsion of the charges aligned in the center of the antiparallel β-sheet (for example, Fmoc-VVEKV, Fig. 4b right). Accordingly, the necessary strategy for obtaining the charge-separated peptide β-sheet is to separate the charges in the neighboring β-strands to reduce the charge repulsion (for example, Fmoc-VVVKE, Fig. 4b left). In contrast, when two pairs of E and K are introduced in the Fmoc-pentapeptide, β-sheet structure is only formed from Fmoc-EKVKE and Fmoc-KEVEK are in the solid state, and all the other sequences sparsely form a β-sheet because of strong charge repulsion.
 | ||
| Fig. 4 (a) Schematic representations of parallel and antiparallel peptide β-sheets. (b) Schematic representations of charge-separated peptide β-sheets with stable (left) and unstable (right) charge distributions. Reproduced from ref. 8 with permission from Wiley-VCH, Copyright (2014). | ||
2.3. Photoinduced charge separation of peptide β-sheets from neighbouring electron donor–acceptor pairs
As proposed by Zhang et al., complementary charged peptides tend to form the β-sheets (Fig. 5a left).32 In Zhang's model, positively and negatively charged side chains (E and K) are arranged on one side of the β-sheet, while hydrophobic side chains (V) are assembled on the other side. The alternately arranged charges interact electrostatically with one another and compensate for the charges, which stabilizes the β-sheet structure. Inspired by this model, Nakayama et al. proposed β-sheet designs with photoinduced charge separation, where electron-donating tetraphenylporphyrin (TPP) and electron-accepting naphthalene diimide (NDI) were alternately anchored on one side of the β-sheet (Fig. 5a and b).33 In Nakayama's model, electron-donor and acceptor molecular moieties are arranged on one side of the peptide β-sheet, instead of positive and negative charges in the charge-complementary peptide β-sheet. | ||
| Fig. 5 (a) Schematic representations of complementarily charged peptides (left, Zhang's model) and electron donor–acceptor peptides (right, D–A model). (b) Molecular structures of pentapeptide, VENDIVKTPPVENDIV (left), electron-donating TPP derivative (right top) and electron-accepting NDI derivative (right bottom). (c) Schematic representations of the self-assembled structures of the heptapeptide by diffusion of IPE vapor into 30% TFE-CHCl3 (left) and 50% TFE-HFIP solutions (right). Reproduced from ref. 33 with permission from Chemical Society of Japan, Copyright (2017). | ||
For synthesis, amino acid dimers of TPP-anchored lysine and valine (Fmoc-KTTPV–OH) and NDI-anchored glutamic acid (Fmoc-ENDIV–OH) are synthesized, which are further connected with Fmoc-V to form a heptapeptide, VENDIVKTPPVENDIV. The heptapeptide self-assembles in aprotic 30% trifluoroethanol (TFE)-CHCl3 with slow addition of nonsolvent vapors of isopropyl ether (IPE). As confirmed by spectroscopy and X-ray diffraction, the heptapeptide forms a β-sheet structure. PL quenching studies indicate that photoinduced electron transfer occurs efficiently from TPP to NDI moieties in the pentapeptide. Further studies on the self-assembling structure reveal that the TPP moieties form a J-type aggregate in neutral conditions (30% TFE-CHCl3, Fig. 5c left). In contrast, under protic conditions (50% TFE-hexafluoroisopropanol (HFIP)), the heptapeptide sparsely forms β-sheet due to the electronic repulsion between protonated TPPs. The β-sheet formation begins rapidly when deprotonation of the TPP moieties is completed by the addition of IPE. In the β-sheet, the TPP and NDI moieties separately form H- and J-type aggregates, respectively (Fig. 5c right).
Segregated stacks of electron donor (D) and acceptor (A) molecules are achieved by utilizing the self-assembly behaviour of the peptides. Sanders et al. reported that A–D–A dyads of quarterthiophene (D) and oligopeptide with NDI (A) form 1D nanofibers in an aqueous media.34 Due to the separation of D and A in the fibrous assembly, photoinduced electron transfer occurs with a long-lived charge-separated state (Fig. 6). Examples of such precise control of segregated stacks of D and A in a nanostructure are reported for self-assembled nanotubes with coaxial structure, where an electron-donating core layer is laminated by an electron-accepting shell.35–37 Meanwhile, by utilizing the characteristics of a self-assembling peptide β-sheet, the segregated stack of electron donor and acceptor molecular moieties on the side chains can be achieved.
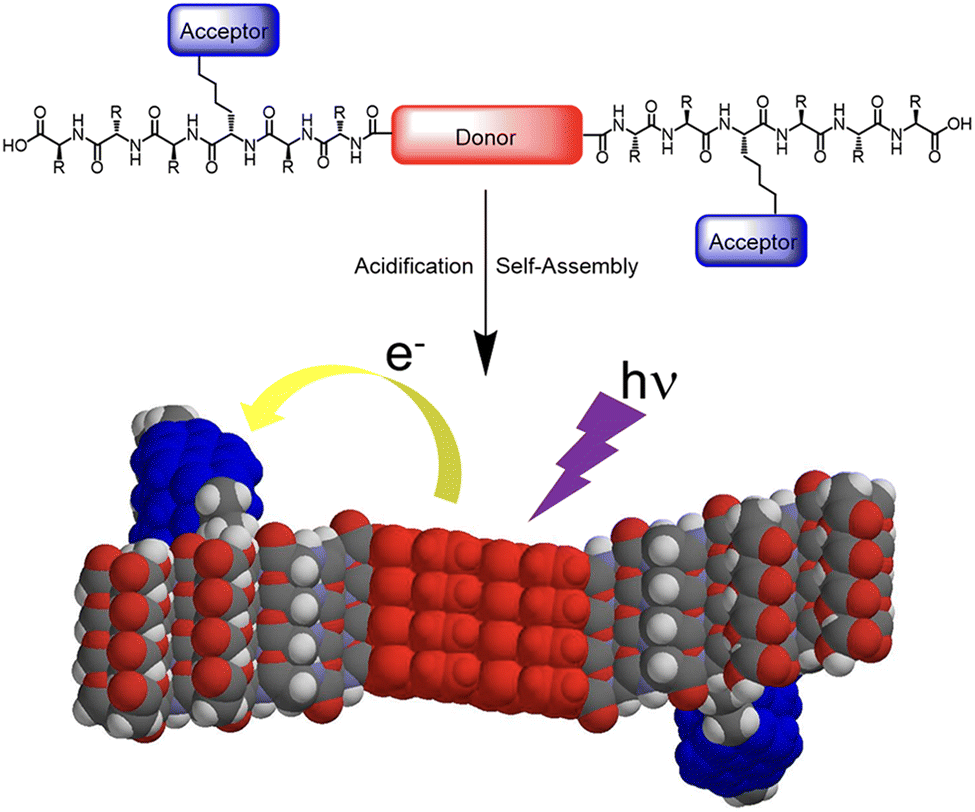 | ||
| Fig. 6 Illustration of self-assembly and electron transfer of a donor–acceptor π-peptide hybrid. Reproduced from ref. 34 with permission from American Chemical Society, Copyright (2016). | ||
2.4. Design strategy for helix-forming non-natural peptides
The helix is a representative secondary structure of peptides, which features a chiral 1D shape. As nature mostly uses the L-form of amino acid enantiomers as a result of long-term biotic/prebiotic evolution processes, helices found in nature possess single helicity. While individual helices are typically obtained through the intramolecular folding of a single peptide chain, they may sometimes consist of multiple peptides bound together. The ability of a peptide to form a helical structure is strongly governed by its primary structure, where several helix-forming amino acid sequences have been identified. Accordingly, the most straightforward design strategy for a helix is to adopt one such sequence as the main component of the targeted non-natural peptide chain.38 Intra-helix interactions between side residue functionalities also contribute to promoting or inhibiting helix formation.39Co-polymerization of γ-propargyl-L-glutamic acid N-carboxyanhydride and N-ε-2-[2-(2-methoxyethoxy)ethoxy]acetyl-L-lysine N-carboxyanhydride has been reported to create water-soluble polypeptides that adopt α-helical conformations.40 The acetylene terminal attached to the glutamate side residue was used for the click reaction to introduce another functionality into the polypeptides. When parent polypeptides were modified with a cell-binding short peptide, GRGDS (Gly–Arg–Gly–Asp–Ser), the resultant polypeptide could retain its helical content, promoting the adhesion of Chinese hamster ovary cells. Although, generally, helix-forming peptides must have a longer chain length than those for the β-sheets, Gly–Pro–Pro tripeptide bearing pyridyl moieties at its C- and N-terminals has been found to form a helical conformation upon coordination to the Ag(I) ion to afford a crystalline porous network.41 Owing to the helical conformation of this tripeptide, the crystal offers chiral nanochannels working for the chiroselective uptake of guest molecules.
3. Peptide β-sheets as crosslinkers and scaffolds for assembling catalytic metal nanoparticles (MNPs) and graphene oxide
In this section, applications for self-assembled peptide β-sheets with separated functionality on opposite sides are introduced. Function-separated peptide β-sheets are useful for assembling different components on the top and bottom sides of the β-sheet. The characteristics of the function-separated peptide β-sheets are utilized as dispersants of agglomerated metal nanoparticles (MNPs). They are further applicable for use as scaffolds in the assembly of MNPs and graphene oxide (GO) for photo- and electro-catalytic activity.3.1. Cysteine-containing peptide β-sheet as a redispersant of agglomerated MNPs
The strategy shown in Section 2.2 to prepare oligopeptide β-sheets with separated functionalities on the top and bottom sides of the β-sheet is useful for various applications. For example, a Fmoc-pentapeptide β-sheet with a positively charged amino group, such as lysine, on one side and a metal-binding thiol group, such as that of cysteine, on the other side of the β-sheet can redisperse agglomerated MNPs (M = Au, Cu, Pt and Pd, Fig. 7a).42 Surfactant-free Au nanoparticles (AuNPs) prepared by laser ablation technique are originally highly dispersed in water and organic solvents due to the negative charge on the surface of the AuNPs.43–47 However, the AuNPs gradually agglomerate after 6 months of preparation. By adding the β-sheet of Fmoc-VKVVC 1 in MeOH, the AuNPs are enwrapped by the β-sheet with the thiol group on one side, while the aminobutyl group of lysine on the other side of the β-sheet maintains the dispersion state of the β-sheets in water/MeOH mixed solvent. Finally, after 3 days of aging, agglomerated AuNPs are redispersed on the surface of the β-sheet with the colour of the solution turning from blue (agglomerated AuNPs) to original pink (dispersed AuNPs). A similar effect is observed for agglomerated CuNPs, PtNPs and PdNPs.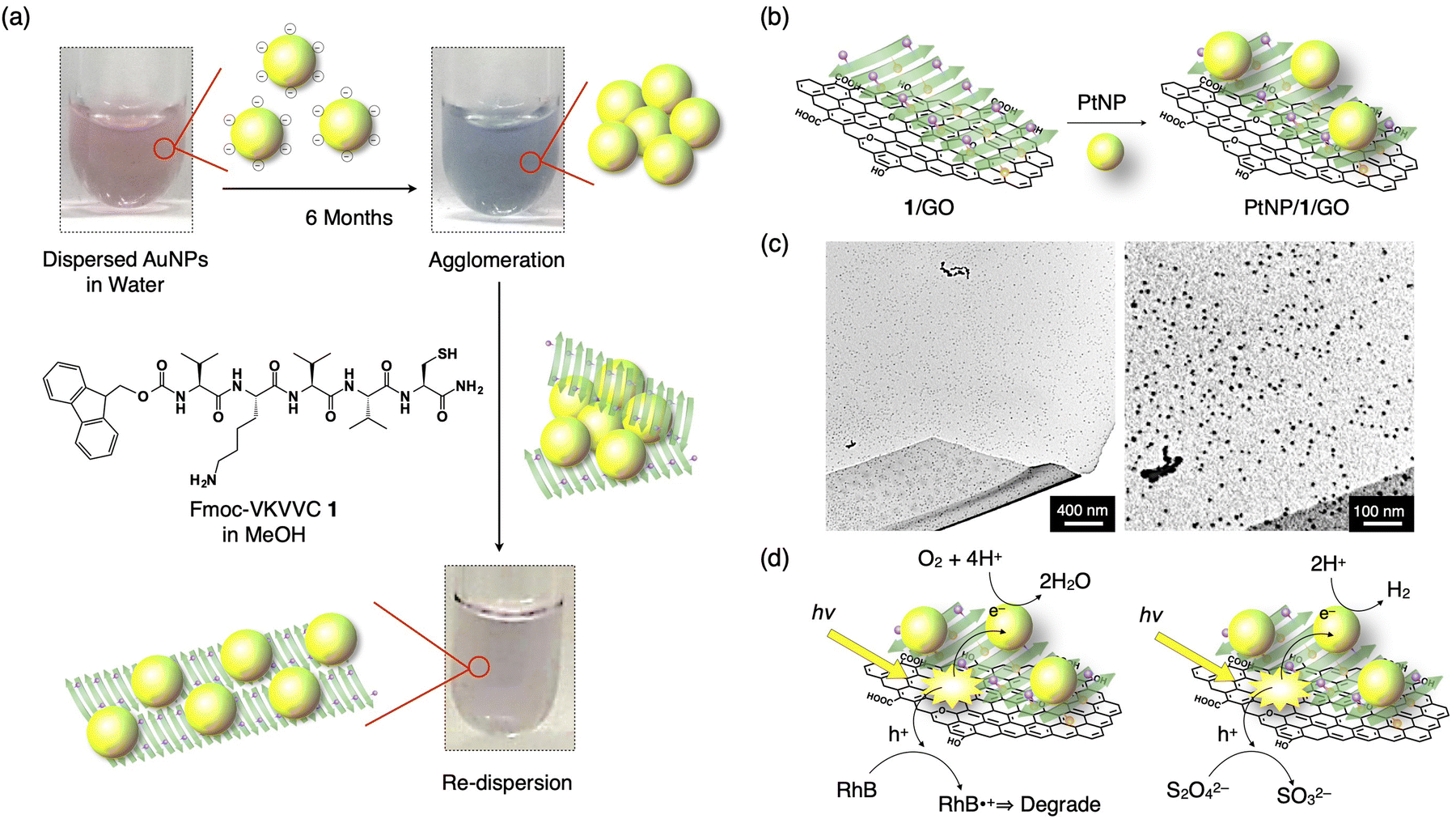 | ||
| Fig. 7 (a) Schematic representation of the redispersion of MNPs by an addition of Fmoc-VKVVC 1 (β-sheet) in MeOH. Photographs show as-prepared AuNP dispersion in water (left top), AuNP dispersion in water 6 months after preparation (right top) and that added by MeOH solution of the β-sheet of 1 aged for 3 days (right bottom). Yellow sphere, green arrow and purple sphere on the green arrow indicate AuNP, β-strand of 1 and thiol side chain, respectively. (b) Schematic representation of the hierarchical assembly of PtNPs, 1, and GO to form the PtNP/1/GO complex. (c) TEM micrographs of the PtNP/1/GO complex. (d) Schematic representations of the pathways of photogenerated hole and electron for dye degradation (left) and H2 evolution (left). Reproduced from ref. 42 (a) and 48 (b–d) with permission from Royal Society of Chemistry, Copyright (2015) and American Chemical Society, Copyright (2017). | ||
3.2. PtNPs/cysteine-containing peptide β-sheet/graphene oxide composite for oxygen reduction and hydrogen generation reactions
Using a function-separated β-sheet of 1, metal nanoparticles can be immobilized on a graphene oxide (GO) nanosheet. GO sheets have negatively charged carboxy and hydroxy groups, which readily interact with positively charged amino groups on one side of the β-sheet of 1. As a result, the β-sheet of 1 covers the surface of the GO sheet when mixed. By adding a solution of PtNPs to the β-sheet/GO composite, the PtNPs bind to the thiol group of the β-sheet to form a highly condensed PtNPs/1/GO ternary complex (Fig. 7b and c).48 Upon photoirradiation, the suspension of PtNPs/1/GO ternary composite displays remarkable photocatalytic activity: GO absorbs light, and thus, an excited electron transfers to PtNP, resulting in the oxidation–reduction reaction (ORR). The generated holes in GO are transferred to the dye added, Rhodamine B (RhB), which degraded it through the oxidation of RhB (Fig. 7d, left). When Na2S2O4 is added as a hole scavenger, the ternary complex displays a hydrogen evolution reaction (HER) upon white light irradiation (Fig. 7d, right).Another type of peptide/PtNP complex shows electrocatalytic ORR.49 The heptapeptide, AAKLVFF, forms a β-sheet which has the amino groups of the lysine side chain and a terminal of the peptide with a positive in water; therefore, the PtNPs with negative surface potential form a PtNP/β-sheet complex with it. The ORR activity of the PtNP/β-sheet complex is higher than the benchmark Pt/C electrocatalyst in terms of the onset potential and reaction kinetics. Furthermore, the complex displays one order of magnitude higher ORR mass activity than previously reported peptide-based ORR electrocatalysts, which are derived from well-dispersed PtNPs on the β-sheet, where 50% amine groups function as active sites for the catalytic reaction.
4. Self-assembled microstructures from natural proteins for optical and electronic applications
In this section, we introduce self-assembled natural proteins that can perform optical and electronic functions, such as optical sensors, optical resonators and lasers, optical waveguides, optical logic gates, and electronic transistors. By utilizing the characteristics of natural biodegradable polymers, bio- and environmentally-friendly devices can be developed.4.1. Self-assembled silk fibroins for optical sensing
Silk fibroin (SF) is a natural protein produced by the mulberry silkworm (Bombyx mori). It is a historically important merchandise, especially in medieval times, when it was considered a luxurious fabric. Due to its high mechanical properties, rather high refractive index (∼1.54) and hygroscopic properties, as well as high biocompatibility, SF is utilized for various applications as biomedical material,50 such as for optics and sensing. For example, Malinowski et al. prepared SF films with high transparency (>93%) and high haze (>65%), which enhanced the silicon photodiode efficiency by 14.9%.51 Furthermore, Li et al. utilized SF thin film with sub-micron thick as humidity sensing, where the increase in the film thickness by moisture adsorption created a large redshift in the reflectance peak with optical interference in the film.52Optical resonators are valuable for sensing applications with high sensitivity.53–56 Xu et al. fabricated a microtoroidal structure from SF with a diameter of 80 μm.57 The toroidal disk acted as a whispering gallery mode (WGM) optical resonator with a high Q-factor in the order of 105. Utilizing the high Q resonator, the authors demonstrated thermal sensing with a sensitivity of −0.72 nm K−1. When the disk was exposed to MeOH vapour and dried, the Silk I structure transformed to Silk II, thus leading to a much higher sensitivity of −1.17 nm K−1. This value is 8 times higher than previously reported values for silica and polymer-based WGM thermal sensors.58,59
Due to sharp PL peaks, WGM optical resonators are also utilized as highly sensitive humidity sensors (Fig. 8a).60 Using the water-in-oil mini-emulsion method with a natural surfactant, well-defined microspheres of SF are formed with an average diameter of 7.7 μm (Fig. 8b). The resultant SF microspheres, doped with a fluorescent dye, display clear WGM resonant peaks upon photoexcitation to a single microsphere (Fig. 8c). By increasing the relative humidity (RH) from 24 to 95% at 25 °C, the diameter of the SF microspheres increases with moisture absorption, accompanying the redshift of resonant PL peaks (Fig. 8d). The peak shift shows an almost linear relationship with the increase/decrease in the RH with a sensitivity of 187 pm/% RH (Fig. 8e). The SF resonator maintains its performance even at high humidity of 95% RH; good repeatability is seen upon switching the relative humidity between low (23% RH) and high RH (93%, Fig. 8f). The humidity sensing across a wide humidity range is suitable in a harsh environment.
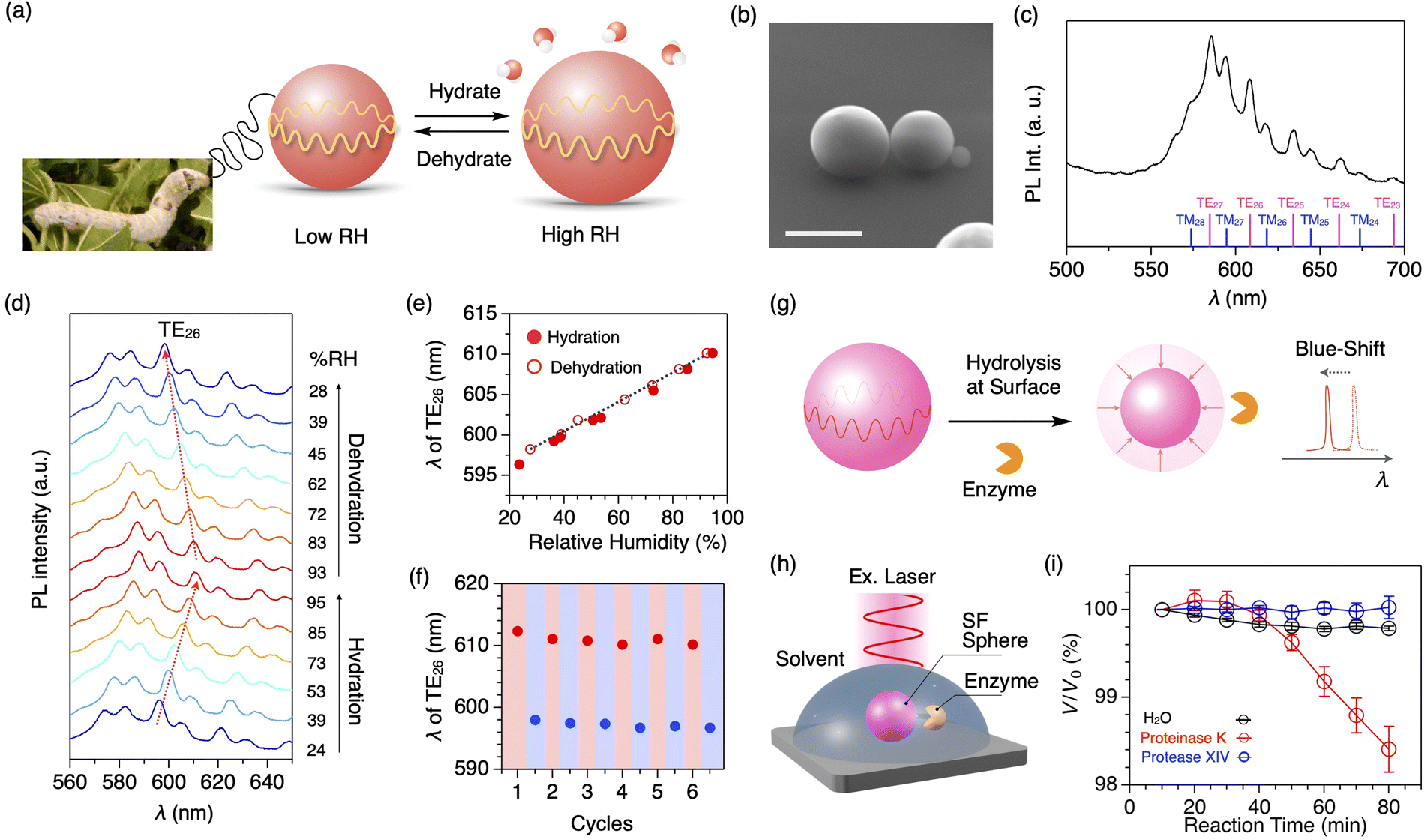 | ||
| Fig. 8 (a) Photograph of mulberry silkworm and a schematic representation of silk fibroin microsphere that expand or contract in response to surrounding humidity. (b) SEM micrograph of the SF microsphere. Scale bar: 5 μm. (c) PL spectra of a single SF microsphere upon excitation with a cw laser (λex = 450 nm). Each WGM peak is assigned as shown at the bottom. (d) Humidity-dependent PL spectra of a single SF microsphere upon excitation with a cw laser (λex = 450 nm). (e) Plot of the wavelength of the resonant peak of TE26 upon increasing (filled circle) and decreasing (open circle) the surrounding humidity. (f) Plot of the wavelength of the resonant peak of TE26 upon six cycles of hydration (red) and dehydration (blue) between 93 and 25% RH. (g) A schematic representation of the quantitative measurements of the hydrolysis reaction based on the spectral shift of the resonance peaks. (h) Schematic representation of optical monitoring of the degradation process of the SF microsphere by an enzyme. (i) Plots of V/V0 of the SF microsphere versus the incubation time under H2O (black) and aqueous solutions of proteinase K (blue) and protease XIV (red). Reproduced from ref. 60 (a–f) and 61 (g–i) with permission from the Royal Society of Chemistry, Copyright (2021) and Copyright (2023). | ||
The peptide bonds in SF are selectively cleaved by an enzyme called proteinase K. In general, the degradation process is monitored by the weight loss of proteins in solution through mixing with enzymes, centrifugation, extraction, drying, and weighing. However, it requires a gram-scale sample and takes time (several days to months). Additionally, it is difficult to investigate the beginning of the degradation process. Using the SF microsphere resonator, the degradation process of proteins can be monitored precisely (Fig. 8g).61 When a buffer solution containing proteinase K is added into a solvent containing the SF microspheres that are mounted on a quartz substrate (Fig. 8h), the peak shift of the WGM PL starts at 40 min after the addition of proteinase K (Fig. 8i). The peak shift is converted into the volume loss of the microsphere, indicating the cleavage of the SF protein by proteinase K.
Apart from proteins, several optical microresonators from natural polymers have been reported. For example, Wei et al. reported WGM microlasers made of microspheres of potato starch that had been doped with a fluorescent dye.62 The lasing peaks shift upon dehydration, where the structural transformation occurs from native B-type starch into A-type starch with double helix conformation. Recently, Suharman et al. reported thermally tolerant microsphere resonators from quasi-natural poly(lactic acid).63 Upon mixing poly(L-lactic acid) and poly(D-lactic acid), polymers form a stereocomplex, which enhances thermal stability. As a result, optical resonator properties are preserved even at 230 °C, which is 70 °C higher than the acceptable temperature for homochiral microspheres of poly(L-lactic acid) or poly(D-lactic acid).
4.2. Optical and electronic gate operations of natural proteins
Optical fibres require high transparency, high refractive index, and high mechanical and thermal stability, along with good processability. Polymer optical fibres have received increasing attention due to their high flexibility and processability. Synthetic polymer fibres are often utilized for transmitting optical signals,64 but natural fibres with high transparency are also utilizable. For example, Hendra et al. demonstrated the operation of an optical logic gate using fluorescent optical resonators that were interconnected with dragline spider silk microfibres.65 The optical loss coefficient of the natural dragline fibre was evaluated to be 0.03 dB μm−1, which is superior to that of polystyrene (PS) microfibres made by hand spinning (0.13 dB μm−1).64 By combining photoswitchable fluorescent microresonators, the switching gate and a NOT gate, their combined operations are demonstrated (Fig. 9a). | ||
| Fig. 9 (a) Matrix of optical gate operations and outputs, schematic representations, and corresponding optical and fluorescent microscopy images of the combination of switching and NOT gates. The microspheres are arranged on two dragline fibers. Upon photoexcitation of the microsphere on the left, blue-color PL is generated, which passes through the dragline fiber and reaches the microsphere at the center. The centre microsphere works as an optical gate, which controls any further transmission of light to output microspheres on the right. (b) Structure of OFET fabricated with albumen dielectrics with egg white; the schematic drawing of the denaturation of albumen protein and the crosslinking reaction under thermal treatments. Reproduced from ref. 65 (a) and 66 (b) with permission from Wiley-VCH, Copyright (2023) and Copyright (2011). | ||
For electronic gates, Chang et al. utilized chicken egg white, also called chicken albumen, as a dielectric layer of an organic field-effect transistor (OFET) (Fig. 9b).66 Using pentacene and C60 as hole- and electron-transporting layers, FET operations work with the hole and electron mobilities of 0.09 and 0.13 cm2 V−1 s−1, respectively. The dielectric properties of chicken albumen are more than double those of PS and polymethylmethacrylate (PMMA). The FET operation is observed using a flexible polyethylene naphthalene (PEN) substrate with high reproducibility after bending the substrate. Furthermore, complementary inverter operation was demonstrated. Wang et al. reported silk fibroin as a dielectric layer of flexible OFET with pentacene as an active layer and polyethylene terephthalate (PET) as the flexible substrate.67 The field-effect mobility was as high as 23.2 cm2 V−1 s−1 with a low operation voltage of −3 V.
5. Conclusions
This article comprehensively reviews recent progress in the design of peptide-based self-assembled materials. Given the ability of the peptides to control 1D, 2D and 3D orientations of functionalities attached, they are attractive scaffolds to design materials for various purposes, such as catalysts, sensing as well as biomedical treatments. Strategies, such as (1) side residue functionalization, (2) hybridization of the self-assembled form with other materials and (3) self-assembly into non-natural 3D structures, as described in this review, allowed the development of non-intrinsic properties and functions of the peptides. Since the structures and properties of the peptides have been improved through repetitive biotic and prebiotic selection processes, the fabrication of peptide-based materials can be regarded as a long-term nature–human collaboration, which will keep providing fruitful products as it has been already. State-of-the-art studies afford various applications of the peptides and their assemblies, including cyclic peptides, mainly for their drug, pharmaceutical and biomedical applications.68–73 However, by fully utilizing the inherent properties, such as precise self-assembly and self-organization, self-assembled peptides can be utilized as materials for practical applications for optics and electronics in biologically and environmentally friendly manners. The cost of materials, processing, durability, and useful properties are the next important issues for utilizing peptide assemblies as future materials and device applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
KT acknowledges the World Premier International Research Center (WPI) Initiative on Materials Nanoarchitectonics from MEXT, Japan and KAKENHI (JP23K04844) for funding. YY acknowledges the Japan Science and Technology Agency (JST) for the support by CREST (JPMJCR20T4) and KAKENHI (JP24H01693) for funding.References
- K. Tashiro, Synthetic Molecular Sequences in Materials Science, Springer, 2023 Search PubMed.
- J. Venkatraman, S. C. Shankaramma and P. Balaram, Chem. Rev., 2001, 101, 3131–3152 CrossRef CAS PubMed.
- I. W. Hamley, Angew. Chem., Int. Ed., 2007, 46, 8128–8147 CrossRef CAS PubMed.
- P. Vairaprakash, H. Ueki, K. Tashiro and O. M. Yaghi, J. Am. Chem. Soc., 2011, 133, 759–761 CrossRef CAS PubMed.
- K. V. Sajna, A. M. Fracaroli, O. M. Yaghi and K. Tashiro, Inorg. Chem., 2015, 54, 1197–1199 CrossRef CAS PubMed.
- Y. Yamamoto, H. Yamagishi, J.-S. Huang and A. Lorke, Acc. Chem. Res., 2023, 56, 1469–1481 CrossRef CAS PubMed.
- R. Rajmohan, G. Vrla, H. Ueki, K. Sajna, T. Takei, H. Ohtsu, M. Kawano, P. Vairaprakash and K. Tashiro, Chem. – Asian J., 2020, 15, 766–769 CrossRef CAS PubMed.
- T. Nakayama, T. Sakuraba, S. Tomita, A. Kaneko, E. Takai, K. Shiraki, K. Tashiro, N. Ishii, Y. Hasegawa, Y. Yamada, R. Kumai and Y. Yamamoto, Asian J. Org. Chem., 2014, 3, 1182–1188 CrossRef CAS.
- N. C. Yoder and K. Kumar, Chem. Soc. Rev., 2002, 31, 335–341 RSC.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and G. Sakellariou, Chem. Rev., 2009, 109, 5528–5578 CrossRef CAS PubMed.
- K. Isozaki, Y. Haga, K. Ogata, T. Naota and H. Takaya, Dalton Trans., 2013, 42, 15953–15966 RSC.
- E. Kreidt, W. Leis and M. Seitz, Nat. Commun., 2020, 11, 1346 CrossRef CAS PubMed.
- R. B. Merrifield, Solid phase peptide synthesis, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- P. Bose, P. K. Sukul, O. M. Yaghi, K. Tashiro and J. Visualized, J. Visualized Exp., 2016, 116, 54513 Search PubMed.
- K. Tanaka, G. H. Clever, Y. Takezawa, Y. Yamada, C. Kaul, M. Shionoya and T. Carell, Nat. Nanotechnol., 2006, 1, 190–194 CrossRef CAS PubMed.
- K. Takanashi, A. Fujii, R. Nakajima, H. Chiba, M. Higuchi, Y. Einaga and K. Yamamoto, Bull. Chem. Soc. Jpn., 2007, 80, 1563–1572 CrossRef CAS.
- A. M. Fracaroli, K. Tashiro and O. M. Yaghi, Inorg. Chem., 2012, 51, 6437–6439 CrossRef CAS PubMed.
- A. M. Fracaroli, G. Grover, H. Ohtsu, M. Kawano, F. Gándara, R. H. de Rossi, R. G. Weiss and K. Tashiro, Langmuir, 2023, 39, 7353–7360 CrossRef CAS PubMed.
- M. Criado-Gonzalez, N. Alegret, A. M. Fracaroli, D. Mantione, G. Guzmán-González, R. Del Olmo, K. Tashiro, L. C. Tomé, M. L. Picchio and D. Mecerreyes, Angew. Chem., Int. Ed., 2023, 62, e202301489 CrossRef CAS PubMed.
- P. K. Sukul, P. Bose, T. Takei, O. M. Yaghi, Y. He, M. Lee and K. Tashiro, Chem. Commun., 2016, 52, 1579–1581 RSC.
- T. J. Deming, Chem. Rev., 2016, 116, 786–808 CrossRef CAS PubMed.
- A. L. Wollenberg, T. M. O’Shea, J. H. Kim, A. Czechanski, L. G. Reinholdt, M. V. Sofroniew and T. J. Deming, Biomaterials, 2018, 178, 527–545 CrossRef CAS PubMed.
- P. Bose, T. Takei, X. Li, T. Minowa, R. Rajmohan, P. Vairaprakash and K. Tashiro, ChemBioChem, 2018, 19, 1706–1710 CrossRef CAS PubMed.
- L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2008, 41, 1674–1684 CrossRef CAS PubMed.
- H. A. M. Ardoña and J. D. Tovar, Bioconjugated Chem., 2015, 26, 2290–2302 CrossRef PubMed.
- Z. L. Yu, F. Tantakitti, T. Yu, L. C. Palmer, G. C. Schatz and S. I. Stupp, Science, 2016, 351, 497–502 CrossRef CAS PubMed.
- O. J. G. M. Goor, S. I. S. Hendrikse, P. Y. W. Dankers and E. W. Meijer, Chem. Soc. Rev., 2017, 46, 6621–6637 RSC.
- K. Tao, P. Makam, R. Aizen and E. Gazit, Science, 2017, 358, eaam9756 CrossRef PubMed.
- M. Coste, E. Suárez-Picado and S. Ulrich, Chem. Sci., 2022, 13, 909–933 RSC.
- T. Li, X.-M. Lu, M.-R. Zhang, K. Hu and Z. Li, Bioact. Mater., 2022, 11, 268–282 CAS.
- Y. Tian, J. Li, A. Wang, Q. Li, H. Jian and S. Bai, Macromol. Biosci., 2023, 23, 2300171 CrossRef CAS PubMed.
- S. Zhang, T. Holmes, C. Lockshin and A. Rich, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 3334–3338 CrossRef CAS PubMed.
- T. Nakayama, K. Tashiro, T. Takei and Y. Yamamoto, Chem. Lett., 2017, 46, 423–426 CrossRef CAS.
- A. M. Sanders, T. J. Magnanelli, A. E. Bragg and J. D. Tovar, J. Am. Chem. Soc., 2016, 138, 3362–3370 CrossRef CAS PubMed.
- Y. Yamamoto, T. Fukushima, Y. Suna, N. Ishii, A. Saeki, S. Seki, S. Tagawa, M. Taniguchi, T. Kawai and T. Aida, Science, 2006, 314, 1761–1764 CrossRef CAS PubMed.
- Y. Yamamoto, G. Zhang, W. Jin, T. Fukushima, N. Ishii, A. Saeki, S. Seki, S. Tagawa, T. Minari, K. Tsukagoshi and T. Aida, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21051–21056 CrossRef CAS PubMed.
- R. Charvet, Y. Yamamoto, T. Sasaki, J. Kim, K. Kato, M. Takata, A. Saeki, S. Seki and T. Aida, J. Am. Chem. Soc., 2012, 134, 2524–2527 CrossRef CAS PubMed.
- B. V. Popp and Z. T. Ball, J. Am. Chem. Soc., 2010, 132, 6660–6662 CrossRef CAS PubMed.
- R. P. Cheng, W.-R. Wang, P. Girinath, P.-A. Yang, R. Ahmad, J.-H. Li, P. Hart, B. Kokona, R. Fairman, C. Kilpatrick and A. Argiros, Biochemistry, 2012, 51, 7157–7172 CrossRef CAS PubMed.
- J. Cao, P. Hu, L. Lu, B. A. Chan, B.-H. Luo and D. Zhang, Polym. Chem., 2015, 6, 1226–1229 RSC.
- T. Sawada, A. Matsumoto and M. Fujita, Angew. Chem., Int. Ed., 2014, 53, 7228–7232 CrossRef CAS PubMed.
- T. Mizutaru, T. Sakuraba, T. Nakayama, G. Marzun, P. Wagener, C. Rehbock, S. Barcikowski, K. Murakami, J. Fujita, N. Ishii and Y. Yamamoto, J. Mater. Chem. A, 2015, 3, 17612–17619 RSC.
- S. Petersen, A. Barchanski, U. Taylor, S. Klein, D. Rath and S. Barcikowski, J. Phys. Chem. C, 2011, 115, 5152 CrossRef CAS.
- S. Ibrahimkutty, P. Wagener, A. Menzel, A. Plech and S. Barcikowski, Appl. Phys. Lett., 2012, 101, 103104 CrossRef.
- P. Nachev, D. D. van’t Zand, V. Coger, P. Wagener, K. Reimer, P. M. Vogt, S. Barcikowski and A. Pich, J. Laser Appl, 2012, 24, 042012 CrossRef.
- C. Rehbock, V. Merk, L. Gamrad, R. Streubel and S. Barcikowski, Phys. Chem. Chem. Phys., 2013, 15, 3057 RSC.
- M. Lau, I. Haxhiaj, P. Wagener, R. Intartaglia, F. Brandi, J. Nakamura and S. Barcikowski, Chem. Phys. Lett., 2014, 610–611, 256 CrossRef CAS.
- T. Mizutaru, G. Marzun, S. Kohsakowski, S. Barcikowski, D. Hong, H. Kotani, T. Kojima, T. Kondo, J. Nakamura and Y. Yamamoto, ACS Appl. Mater. Interfaces, 2017, 9, 9996–10002 CrossRef CAS PubMed.
- A. Jindal, K. Tashiro, H. Kotani, T. Takei, S. Reichenberger, G. Marzun, S. Barcikowski, T. Kojima, Y. Yamamoto and A. C. S. Appl, Energy Mater., 2019, 2, 6536–6541 CAS.
- T. Bernadette Aigner, E. DeSimone and T. Scheibel, Adv. Mater., 2018, 30, 1704636 CrossRef PubMed.
- C. Malinowski, F. He, Y. Zhao, I. Chang, D. W. Hatchett, S. Zhai and H. Zhao, RSC Adv., 2019, 9, 40792 RSC.
- Q. Li, N. Qi, Y. Peng, Y. Zhang, L. Shi, X. Zhang, Y. Lai, K. Wei, I. S. Kim and K.-Q. Zhang, RSC. Adv., 2017, 7, 17889 RSC.
- M. Loyez, M. Adolphson, J. Liao and L. Yang, ACS Sens., 2023, 8, 2440–2470 CrossRef CAS PubMed.
- N. Tanji, H. Yamagishi, K. Fujita and Y. Yamamoto, ACS Appl. Polym. Mater., 2022, 4, 1065–1070 CrossRef CAS.
- A. Qiagedeer, H. Yamagishi, M. Sakamoto, H. Hasebe, F. Ishiwari, T. Fukushima and Y. Yamamoto, Mater. Chem. Front., 2021, 5, 799–803 RSC.
- A. R. Anwar, M. Mur and M. Humar, ACS Photonics, 2023, 10, 1202–1224 CrossRef CAS PubMed.
- L. Xu, X. Jiang, G. Zhao, D. Ma, H. Tao, Z. Liu, F. G. Omenetto and L. Yang, Opt. Express, 2016, 24, 20825 CrossRef CAS PubMed.
- B.-B. Li, Q.-Y. Wang, Y.-F. Xiao, X.-F. Jiang, Y. Li, L. Xiao and Q. Gong, Appl. Phys. Lett., 2010, 96, 251109 CrossRef.
- Y.-Z. Yan, C.-L. Zou, S.-B. Yan, F.-W. Sun, Z. Ji, J. Liu, Y.-G. Zhang, L. Wang, C.-Y. Xue, W.-D. Zhang, Z. F. Han and J. J. Xiong, Opt. Express, 2011, 19, 5753 CrossRef CAS PubMed.
- W. Y. Heah, H. Yamagishi, K. Fujita, M. Sumitani, Y. Mikami, H. Yoshioka, Y. Oki and Y. Yamamoto, Mater. Chem. Front., 2021, 5, 5653–5657 RSC.
- A. Takeuchi, W. Y. Heah, Y. Yamamoto and H. Yamagishi, Chem. Commun., 2023, 59, 1477–1480 RSC.
- Y. Wei, X. Lin, C. Wei, W. Zhang, Y. Yan and Y. S. Zhao, ACS Nano, 2017, 11, 597–602 CrossRef CAS PubMed.
- Suharman, W. Y. Heah, H. Yamagishi and Y. Yamamoto, Nanoscale, 2023, 15, 19062–19068 RSC.
- Hendra, A. Takeuchi, H. Yamagishi, O. Oki, M. Morimoto, M. Irie and Y. Yamamoto, Adv. Funct. Mater., 2021, 2103685 CrossRef CAS.
- Hendra, H. Yamagishi, W. Y. Heah, A. D. Malay, K. Numata and Y. Yamamoto, Adv. Opt. Mater., 2023, 2202563 CrossRef CAS.
- J.-W. Chang, C.-G. Wang, C.-Y. Huang, T.-D. Tsai, T.-F. Guo and T.-C. Wen, Adv. Mater., 2011, 23, 4077–4081 CrossRef CAS PubMed.
- C.-H. Wang, C.-Y. Hsieh and J.-C. Hwang, Adv. Mater., 2011, 23, 1630 CrossRef CAS PubMed.
- Peptide Self-Assembly and Engineering, Fundamentals, Structures, and Applications, ed. Xuehai Yan, Wiley VCH, 2024 Search PubMed.
- D. Bhandari, S. Rafiq, Y. Gat, P. Gat, R. Waghmare and V. Kumar, Int. J. Pept. Res. Ther., 2020, 26, 139–150 CrossRef CAS.
- L. Wang, N. Wang, W. Zhang, X. Cheng, Z. Yan, G. Shao, X. Wang, R. Wang and C. Fu, Signal Transduction Targeted Ther., 2022, 7, 48 CrossRef CAS PubMed.
- M. Bejaoui, W. Y. Heah, A. K. O. Mizushima, M. Nakajima, H. Yamagishi, Y. Yamamoto and H. Isoda, ACS Appl. Bio Mater., 2024, 7, 1513–1525 CrossRef CAS PubMed.
- A. A. Vinogradov, Y. Yin and H. Suga, J. Am. Chem. Soc., 2019, 141, 4167–4181 CrossRef CAS PubMed.
- S. La Manna, C. Di Natale, V. Onesto and D. Marasco, Int. J. Mol. Sci., 2021, 22, 12662 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |