
Highly stretchable porous regenerated silk fibroin film for enhanced wound healing†
Fengchao
Sun
ab,
Dongdong
Xiao
cd,
Hui
Su
ab,
Zhiliang
Chen
ab,
Bijia
Wang
ab,
Xueling
Feng
 ab,
Zhiping
Mao
ab and
Xiaofeng
Sui
*ab
ab,
Zhiping
Mao
ab and
Xiaofeng
Sui
*ab
aKey Lab of Science and Technology of Eco-Textile, Ministry of Education, College of Chemistry, Chemical Engineering and Biotechnology, Donghua University, Shanghai, 201620, China
bShanghai Belt and Road Joint Laboratory of Textile Intelligent Manufacturing, Shanghai, 201620, China
cDepartment of Urology, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, 200001, China
dShanghai Key Laboratory of Tissue Engineering, Shanghai Ninth People's Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, 200011, China
First published on 9th January 2023
Abstract
Silk fibroin (SF) has received interest in tissue engineering owing to its biocompatibility, biodegradability, and favorable mechanical properties. However, the complex preparation, brittleness, and lack of pores in the structure of the silk fibroin film limit its application. Herein, we show that facile dissolution of SF in aqueous phosphoric acid followed by regeneration in aqueous ammonium sulfate ((NH4)2SO4) could afford highly stretchable films with nano-pores formed in the nonsolvent-induced phase separation process. The named phase separation, which determines the morphology and mechanical properties of the regeneration silk fibroin (RSF) films, is highly dependent on the (NH4)2SO4 concentration as well as the initial concentration of the SF solution. Therefore, the RSF films exhibit a tunable pore size ranging from 230 to 510 nm and excellent stretchability with tensile strain up to 143 ± 16%. Most interestingly, the RSF films were shown to support the proliferation of human skin fibroblasts in vitro as well as speed up full-thickness skin wound healing in a rat model. This work establishes an easy and feasible method to access porous RSF membranes that can be used for wound dressing in clinical settings.
Introduction
The skin is the biggest organ in the human body, covering the whole body's surface and performing a variety of physiological tasks.1 However, the vulnerability of skin to harm such as through burns, mechanical injuries, illness, or surgery can threaten skin health. Wound healing is a multi-stage process that includes hemostasis, inflammation, proliferation, and extracellular matrix remodeling.2 To speed up the healing process, it is critical to provide a favorable external environment by covering the wound with an appropriate dressing material. The optimal wound dressing should be biocompatible, dust bacteria resistant, breathable yet able to maintain an appropriate moist environment, and preferably biodegradable.3 Many biomaterials,4 such as cellulose, sodium alginate, chitosan, and silk fibroin (SF), have been exploited as wound dressings.SF is a structural protein obtained from degumming of silkworm cocoons. Due to its excellent mechanical properties, good biocompatibility, and programmable degradation,5,6 SF is widely used in medical applications,7 in the form of sutures, corneas, dressings, bone nails, blood vessels, and even wearable healthcare systems.8,9
The features of SF that make it ideal for skin wound healing are as follows: inducing a low inflammatory response;10,11 accelerating re-epithelialization and angiogenesis;12,13 promoting cell migration and proliferation through activating NF-κB, MEK and JNK signaling pathways;14,15 and accelerating blood coagulation by promoting platelet adhesion and aggregation.16
Typically, SF needs to be disassembled in specific dissolution systems and then reconstructed into various forms to satisfy various application requirements.17 Lithium bromide/water and calcium chloride/ethanol/water are the most extensively used systems for solubilizing SF.18 Both require long-term dialysis, desalting to create an aqueous solution of SF and additional induction of water insolubility of the film.19,20 Because the present preparation methods are time-consuming, a faster method of preparing regeneration silk fibroin (RSF) films is desirable.
Although various SF-based wound dressings having been developed for promoting skin regeneration, few works had systematically investigated the effect of pore structures in RSF films for this purpose. A given pore size in a dressing not only offers permeability but also promotes cell migration and adhesion.21 Existing strategies to introduce pores in the RSF films include direct addition of porogenic agents such as PEO22 and PEG23 or inducing the formation of nanoparticles by autoclaving treatment.24 These procedures are complicated, and leftover porogenic agents may negatively affect the mechanical properties and biocompatibility of the final film. As a result, producing porous RSF films in a quick and straightforward manner remains a challenge.
Herein, to address the challenges in the preparation of RSF films, we established a rapid method for the direct and controllable construction of porous RSF films enabled by nonsolvent-induced phase separation to meet the requirements of ideal wound dressings (Fig. 1). Phosphoric acid (H3PO4) has previously been used to dissolve SF, but further studies have been hindered by the fact that H3PO4 can cause SF degradation.25–27 Our recent study revealed that lowering of dissolution temperature could effectively prevent severe acid-catalyzed hydrolysis by H3PO4.28 Therefore, SF was dissolved in 85 wt% aqueous H3PO4 at −18 °C in the current work followed by regeneration in 20% (w/v) ammonium sulfate solution. The morphology and mechanical properties of RSF films prepared were tuned by controlling the regeneration process. The cell proliferation and in vivo wound healing effects of these RSF films as wound dressings were comprehensively evaluated. We believe that the named approach might be useful for producing high-performance silk-based films for wound-healing and other clinical applications in general.
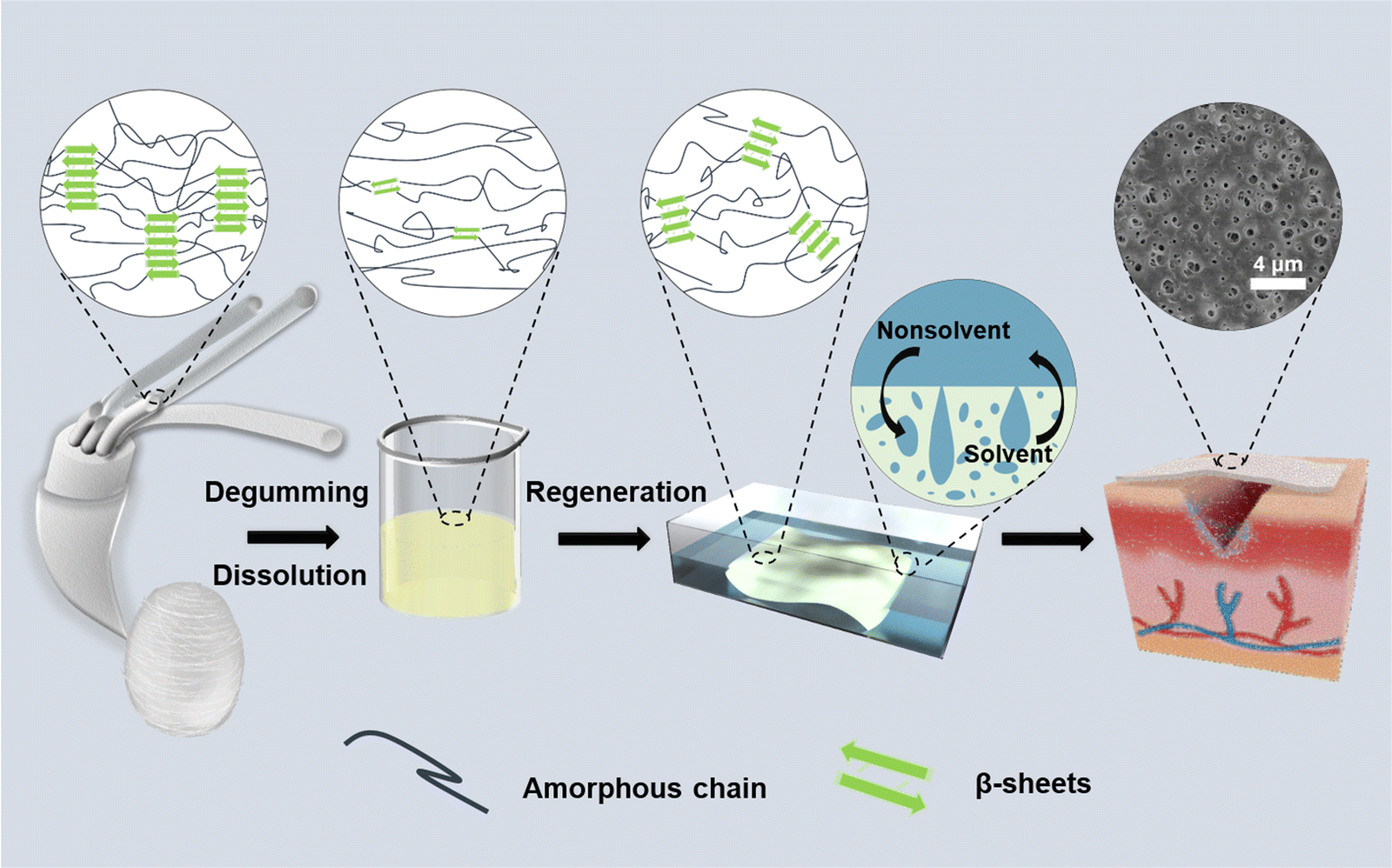 | ||
| Fig. 1 Schematic illustration of the preparation processes for the porous RSF films and corresponding crystal structure changes. | ||
Materials and methods
Materials
Silk fibers were purchased from Zhitai Silk Co., Ltd (Jiangxi, China) as raw materials. Phosphoric acid (H3PO4 85 wt%, PA) and ammonium sulfate ((NH4)2SO4) were purchased from Shanghai Titan Scientific Co., Ltd (Shanghai, China). Sodium carbonate (Na2CO3) and lithium bromide (LiBr) were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). All reagents were of analytical grade and used as received. For sample preparation, deionized water was used.Preparation of silk fibroin solution
Silk fibers were boiled for 30 minutes at 100 °C in 0.02 M Na2CO3 to remove sericin, rinsed completely with deionized water, and dried in an oven for 8 hours at 60 °C. The degummed silk fibers were then cut, and the 85 wt% phosphoric acid solution was pre-set at −18 °C. The fibers were stirred thoroughly into the phosphoric acid solution. The mixture was then placed at −18 °C for 1 h, with stirring every 15 min for 2 min. Optical microscopy was used to observe the dissolution process until the fibers disappeared completely. The obtained silk fibroin solution was de-bubbled by centrifugation at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min at 18 °C for subsequent use.
000 rpm for 10 min at 18 °C for subsequent use.
Fabrication of silk fibroin films
The silk fibroin solution was scraped on a glass plate at room temperature using a 1 mm-thick film scraper and subsequently submerged in a coagulation bath of (NH4)2SO4 solution at different concentrations at 15 °C for 50 min. The concentration of (NH4)2SO4 was varied from 15% (w/v) to 30% (w/v). The RSF films were completely washed with deionized water until neutral. The concentration of the silk fibroin solution was 10 wt%, unless otherwise mentioned.Preparation of lyophilized silk fibroin
To obtain lyophilized silk fibroin, the silk fibroin solution was dialyzed in distilled water for three days to remove phosphoric acid, then frozen at −80 °C for three hours and dried in a freeze-drier (Labconco FD5-3, USA) for 48 hours.Rheological behavior of silk fibroin solutions
A HAAKE MARS 60 rheometer (Thermo, USA) with a conical plate (35 mm, 1°) was used to test the rheological properties of the 10% silk fibroin solution.The oscillation frequency sweep test was performed at 25 °C to obtain the storage modulus (G′) and loss modulus (G′′) in the angular velocity range of 1 to 600 rad s−1. To ensure the test was performed in the linear viscoelastic region of the solution, a strain value of 3% and a frequency of 1 Hz were selected.
In order to characterize the gelation rate of the silk fibroin solution in different concentrations of (NH4)2SO4 (from 15% (w/v) to 30% (w/v)), the oscillation time sweep mode was selected with the strain and frequency set as 3% and 1 Hz, respectively. At 25 °C, 2 mL of varying concentrations of (NH4)2SO4 solution were added when the test was carried out for 60 s.
Intrinsic viscosity
The RSF films prepared from 25 °C and −18 °C phosphoric acid solutions were completely dissolved in a 9.8 M lithium bromide solution under 60 °C. The solutions were cooled and filtered into a capillary viscometer. The efflux times of diluted solutions of different concentrations were tested after the temperature stabilized at 25 °C. Each group was tested three times. The intrinsic viscosity of the silk fibroin solution was calculated and extrapolated according to the research.29Fourier transform infrared spectroscopy (FTIR)
FT-IR spectra of the RSF films were obtained from an FTIR Spectrophotometer (Spectrum Two, PERKI-NELMER, USA) over a wavelength range of 500–4000 cm−1. For each measure, 32 scans were recorded with a resolution of 4.0 cm−1. The secondary structures of the RSF films were evaluated by Fourier self-deconvolution of the amide I region (1580–1730) using peakfit 4.12. The following peaks were assigned to different secondary structures: 1620 cm−1 (β-sheet), 1650 cm−1 (random coil/helix) and 1700 cm−1 (β-turn).X-Ray diffraction (XRD) measurement
XRD patterns were collected from a DX-2700BH multifunctional X-ray diffractometer (Haoyuan, China), with Cu Kα radiation (λ = 1.5419 Å), operated at 40 kV and 40 mA with a step size of 0.03°. The profile of the RSF film was recorded at 2° min−1 in the 2θ scan angle range of 5°–50°.Morphology observation by SEM
SEM of the RSF film was conducted on a scanning electron microscope (TM-3030, Hitachi, Japan) with the voltage setup at 15 kV. The sample needs to be fixed on the conductive adhesive and sprayed with gold before SEM observation.ImageJ analysis
The pore size distribution and thickness of RSF films were calculated using ImageJ software based on the SEM surface and cross-sectional images, respectively. The percentage of the surface area covered by pores, which is defined as the surface porosity in this study, was also calculated using ImageJ.Mechanical measurement
The mechanical properties of RSF films in a wet state were tested using a UH6502 Universal Testing Machine (UH Measurement & Control, China). All samples were cut to 5 mm wide by 50 mm long and then stretched at a rate of 10 mm min−1 at 25 °C and 50% relative humidity.Water content measurement
The water content of the RSF film was calculated using the following equation:| WH2O = 100% × (Ww − Wd)/Ww |
Air permeability
The air permeability of the RSF films was measured under a differential pressure of 200 Pa using a capillary flow porometer (POROLUX 100FM, Belgium).Thermal analysis
The thermal degradation of silk fibroin materials was determined by a thermogravimetric analyzer (TG-209, NETZSCH) at 30–900 °C in a nitrogen atmosphere with a heating rate of 10 °C min−1.In vitro degradation experiment
The mass retention of the RSF film in PBS buffer (pH 7.4) and protease XIV solutions was used to evaluate degradation performance. Proteinase XIV was dissolved in PBS buffer to achieve a concentration of 1 U mL−1. The RSF10 was weighed and placed in 10 mL centrifuge tubes containing PBS or enzyme solution, respectively, with a sample-to-solution mass ratio of 1:100. The samples were incubated for 12, 24, 48, and 72 hours at 37 °C. They were removed at each time point, rinsed three times with deionized water, and finally oven dried at 60 °C for 4 h to achieve constant weight. The masses before (M0) and after (M1) incubation were recorded, and the residual mass (%) was obtained by dividing M1 by M0. Each group contained three replicated samples.
Cell culture and viability assay
The biocompatibility of RSF films was assessed using the Cell Counting Kit-8 (CCK-8) (Do-jindo Molecular Technologies, Inc., Kumamoto, Japan). Human skin fibroblasts (Shanghai Cell Library of the Chinese Academy of Science, Shanghai, China) were cultured in serum-free Dulbecco's Modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum. Cells were incubated in a humidified incubator at 37 °C with 5% CO2, and the medium was changed every 3 days. RSF films were sterilized with 75% ethanol for 30 min, then rinsed three times with PBS solution, cut, and placed in 96-well tissue culture plates (BD Falcon, Corning Inc., NY, USA). Subsequently, cells were inoculated onto the RSF films at a density of 1 × 105 per well. On days 1, 3, and 5, 500 μL of medium containing 50 μL of CCK-8 solution was added to each well. After 2 h of incubation at 37 °C in 5% humidified CO2, the cell proliferation effect was measured using CCK-8. The absorbance at 450 nm was then measured using a microplate reader (Varioskan, Thermo Fisher Scientific Inc., MA, USA). On day 5, the Live and Dead Cell Assay was used to evaluate the viability of human skin fibroblasts and assess cell morphology under microscopy.Wound healing assay
The Institutional Animal Care and Use Committee of Shanghai Ninth People's Hospital approved the study protocol (License number: HKDL [2016] 149). The wound healing-promoting properties of RSF films were investigated using a rat model of a full-thickness skin defect. Six-week-old male Sprague-Dawley rats (150–200 g) were used for the experiments. The rats were anesthetized by inhalation of isoflurane, and two circular defects with a diameter of 10 mm were created on their dorsum using a tissue punch and surgical scissors. Circular defects were covered by conventional wet gauze (control group) and RSF10. The RSF films were sterilized with ethanol and covered on the wound surface with an outer layer of gauze, finally fixed with adhesive 3 M. The control group was treated with gauze and adhesive. After the experiments, the SD rats were euthanised by inhalation of CO2. Wound regeneration was assessed by monitoring the wound on day 7 and day 21. Hematoxylin-eosin (HE) staining and Masson's trichrome (MT) staining were performed on the wound surface on day 21, and epidermal regeneration was assessed by histological analysis.Statistical analysis
At least three independent measurements were collected and expressed as a mean ± standard deviation for all experiments. Statistical comparisons were performed using ANOVA, and *p < 0.05 was recognized as statistically significant.Results and discussion
Morphology of the RSF films
The relative average molecular weight can be characterized by intrinsic viscosity. The intrinsic viscosity of SF solutions in aq. LiBr was used as an indirect indicator of the size (molecular weight) of the RSF samples. As shown in Fig. S1 (ESI†), the RSF prepared from the cold (−18 °C) aq. PA solution had significantly higher intrinsic viscosity (0.434 dL g−1) than the RSF prepared from the room temperature solution (0.373 dL g−1). These results suggest that degradation of SF in aq. PA was notably suppressed at −18 °C.30 So the dissolution temperature was chosen at −18 °C. Aqueous (NH4)2SO4 solution is commonly utilized in the fabrication of RSF materials because of its salting-out effect.31–33 SF molecules self-assemble by rearranging hydrophobic and hydrophilic regions to minimize the free energy of the system and thus adopting a more ordered conformation.34 In the nonsolvent-induced phase separation method, the constitution of the coagulation bath and the initial polymer concentration, both play a part in determining the surface and sub-surface morphology of the RSF films. The influence of coagulation bath concentrations was examined by scanning electron microscopy (Fig. 2). Phase separation occurs in the casting solution during the preparation process, generating a polymer-poor phase and a polymer-rich phase, which are then convert inverted to pores and the membrane matrix, respectively.35–37 As shown in Fig. 2, at a low (NH4)2SO4 concentration of 15% (w/v), the cross-section of the RSF film exhibits a tight porous structure while the film obtained from the 30% (w/v) (NH4)2SO4 bath exhibits finger-like macropores. The change in pore size at the surface and in the cross-section with varying (NH4)2SO4 concentration follows opposite trends (Fig. S2, ESI†). | ||
| Fig. 2 SEM images of the cross sections (a–h) and surface (i–l) of RSF films prepared from 15% (w/v) (NH4)2SO4 solution (a, e and i), from 30% (w/v) (NH4)2SO4 solution (b, f and j), and from 6 wt% SF concentrations (c, g and k) and 10 wt% SF concentrations (d, h and l) (20% (w/v) (NH4)2SO4. | ||
The outward diffusion of H3PO4 and the inward diffusion of water and (NH4)2SO4 (anti-solvent) during coagulation resulted in a phase separation process started from the interface. Higher (NH4)2SO4 concentration would lead to faster diffusion of the antisolvent, the former would speed up the coagulation of SF at the interface, resulting in a dense surface with less pores. The surface layer formed would in turn slow down the diffusive exchange of solvent and anti-solvent in the sublayer,38–40 therefore resulting in a different coagulant concentration gradient along the cross section.41 The steeper coagulant concentration gradient would result in transient liquid–liquid partitioning and the formation of asymmetric films with a relatively thin and dense top layer and finger-like macropores in between. Transient liquid–liquid partitioning gives rise to polymer-poor nuclei that would absorb the surrounding solvent and eventually develop into macropores.36 On the other hand, a coagulation bath of lower concentration afforded films with a sponge-like middle layer sandwiched by thick skin layers resulting from delayed liquid–liquid partitioning.
Furthermore, the influence of varying the SF concentration of the casting solution was investigated. The concentration of the coagulation bath was set to be 20% (w/v) and the regeneration was done at 15 °C. The cross sections of RSF films all contain macropores (Fig. 2 and Fig. S3a, ESI†). As the SF casting solution became more concentrated, the surface porosity of the RSF film significantly decreased from 26.2% to 2.4%, the average pore size also decreased from 0.51 ± 0.19 μm to 0.23 ± 0.09 μm (Fig. S3d, ESI†). In addition, the thickness of the RSF film gradually increased from 182 (RSF8) to 367 μm (RSF14) (Table S1, ESI†). Higher polymer concentration would yield a higher polymer-rich phase volume, resulting in decreased surface porosity.42 When the concentration of SF solution increased, the SF quickly aggregated on the surface, reducing the pore size of the RSF films.43 The porosity had a positive correlation to the water content of the film. For instance, RSF6 had an 86.63% water content (Table S1, ESI†). As the SF concentration increased, the water content in the RSF films decreased, and the film structure became more compact with lower porosity. As shown in Fig. S4 (ESI†), the air permeability of the RSF films increased from 48.8 to 61.2 mm s−1, demonstrating that the porous RSF films have an outstanding air permeability performance. Therefore, different regeneration bath concentrations can be used to prepare RSF films with varying microstructure architectures. Moreover, the thickness, surface porosity, and pore size distribution can be changed further by modifying the SF concentration.
Mechanical properties of RSF films
(NH4)2SO4 concentration and SF concentration greatly affected the mechanical properties of the RSF films. The effect of the coagulation process on the RSF films was investigated (Fig. 3a). Due to the dominance of the loss modulus (G′ < G′′), the resultant SF solution has a liquid-like behavior, which enables easy and rapid membrane scraping (Fig. S5a, ESI†). With the increase of (NH4)2SO4 concentration in the coagulating bath, the tensile strength (σb) and elongation at break (εb) of RSF films first increased from 0.30 ± 0.02 MPa and 88 ± 12% (15% (w/v)) to 0.39 ± 0.02 MPa and 143 ± 16% (20% (w/v)), then gradually decreased to 0.36 ± 0.03 MPa and 100 ± 11% (30% (w/v)), respectively (Fig. S6a, ESI†). The oscillatory time-sweep mode was used to describe the solution-gel transition32 behavior of the SF induced by the (NH4)2SO4 solution (Fig. S5b, ESI†). Briefly, 2 mL of (NH4)2SO4 solution of predetermined concentration (15–30% (w/v)) was gently added to the rheometer plate at approximately 60 s after the start of the test, and then the gelation point (G′ = G′′) was observed.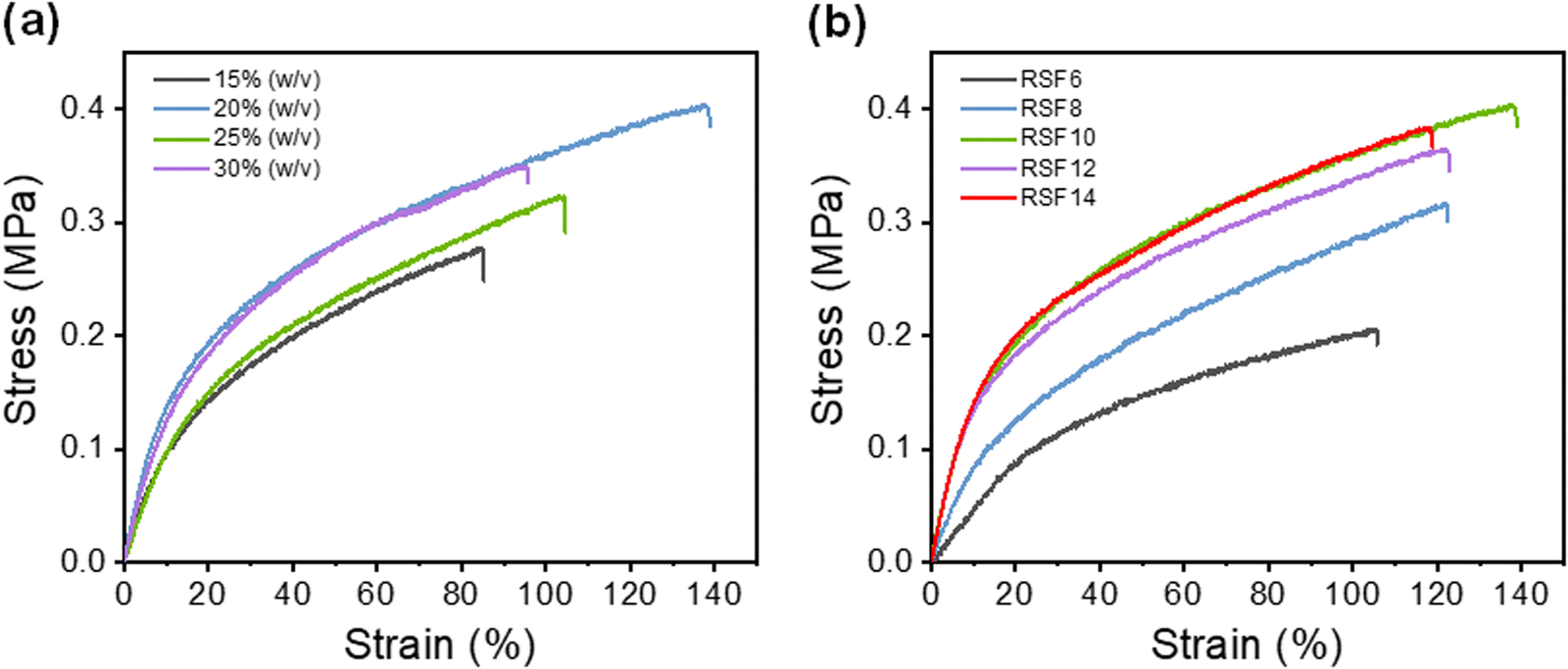 | ||
| Fig. 3 Typical stress–strain curves of wet RSF films prepared at (a) different (NH4)2SO4 concentrations ranging from 15% (w/v) to 30% (w/v) and (b) different SF concentrations ranging from 6 wt% to 14 wt% (20% (w/v) (NH4)2SO4 solution). | ||
The rate of gelation gradually increased with the increasing concentration of the regeneration bath, and gelation was accomplished in less than 5 minutes at 30% (w/v) (NH4)2SO4. A higher gelation rate leads to reduced strain for causing more defects within the RSF films,44 whereas slower gelation at lower coagulant concentrations would result in the formation of a more compact structure and inevitably lead to more severe hydrolysis due to prolonged exposure of SF to acidic conditions, which also worsen the mechanical properties of the film. As shown in the SEM, as the concentration of the regeneration bath grows, so does the number of finger-like holes in the film. The pores provide the film with a significant deformation capacity, but they also create stress concentration and lower its strength. As a result, the strain of RSF films will exhibit a tendency for an initial rise and subsequent decline as the concentration of the coagulating bath increases. A 20% (w/v) (NH4)2SO4 solution was chosen as the coagulating bath for further investigations.
The effect of the SF concentration on the mechanical properties of RSF films was also investigated (Fig. 3b). As the concentration of the SF solution increased from 6 wt% to 10 wt%, the tensile strength (σb) and elongation at break (εb) gradually increased from 0.22 ± 0.01 MPa to 0.39 ± 0.02 MPa and 111 ± 13% to 143 ± 16%, respectively. This is due to the fact that at higher SF concentrations, SF chains tend to aggregate, interact and entangle with each other more readily, leading to improved mechanical properties of the RSF films. However, further increase of SF concentrations led to a decrease in the elongation at break due to the presence of more macropores.
The extraordinarily high elongation of RSF may be attributed to the fact that water molecules plasticize the protein structure, weakening intermolecular interactions and making it more conducive to intermolecular chain slippage, and the porous structure of the film can undergo greater strain when being stretched. Furthermore, the lower β-sheet content also improves the elongation properties of RSF films. The maximum tensile strength of the RSF was 0.39 MPa with 143% strain. It is worth noting that the tensile elongation of our film is much higher than films made from FA/CaCl2 (70%),45 LiBr/H2O (50%),46 and CaCl2/EtOH/H2O solution (49.4%).47
Secondary structure change of silk fibroin
Fourier transform-infrared (FT-IR) spectroscopy and X-ray diffraction (XRD) were employed to investigate the change in SF's secondary structure during the dissolution-regeneration process. The amide I band, which reflects the secondary structure properties of the SF peptide chain48 is observed at 1600–1700 cm−1. Typically, degummed silk fiber shows a characteristic β-sheet peak at 1620 cm−1 while lyophilized SF exhibits a peak at 1645 cm−1 corresponding to random coils. As shown in Fig. 4a, the RSF films prepared herein display both peaks at 1620 and 1650 cm−1, suggesting that β-sheets and random coils co-exist in RSF. Subsequently, the IR curves within the amide I region were deconvoluted for quantitative analysis of the β-sheet and random coil contents (Fig. 4b). The degummed silk fiber, lyophilized SF, and RSF films were found to contain 50%, 16% and 23% of β-sheets, and 47%, 80%, and 73% of random coils, respectively (Fig. 4c), suggesting most of the β-sheets in natural SF were lost during solubilization and only part of them were recovered during regeneration.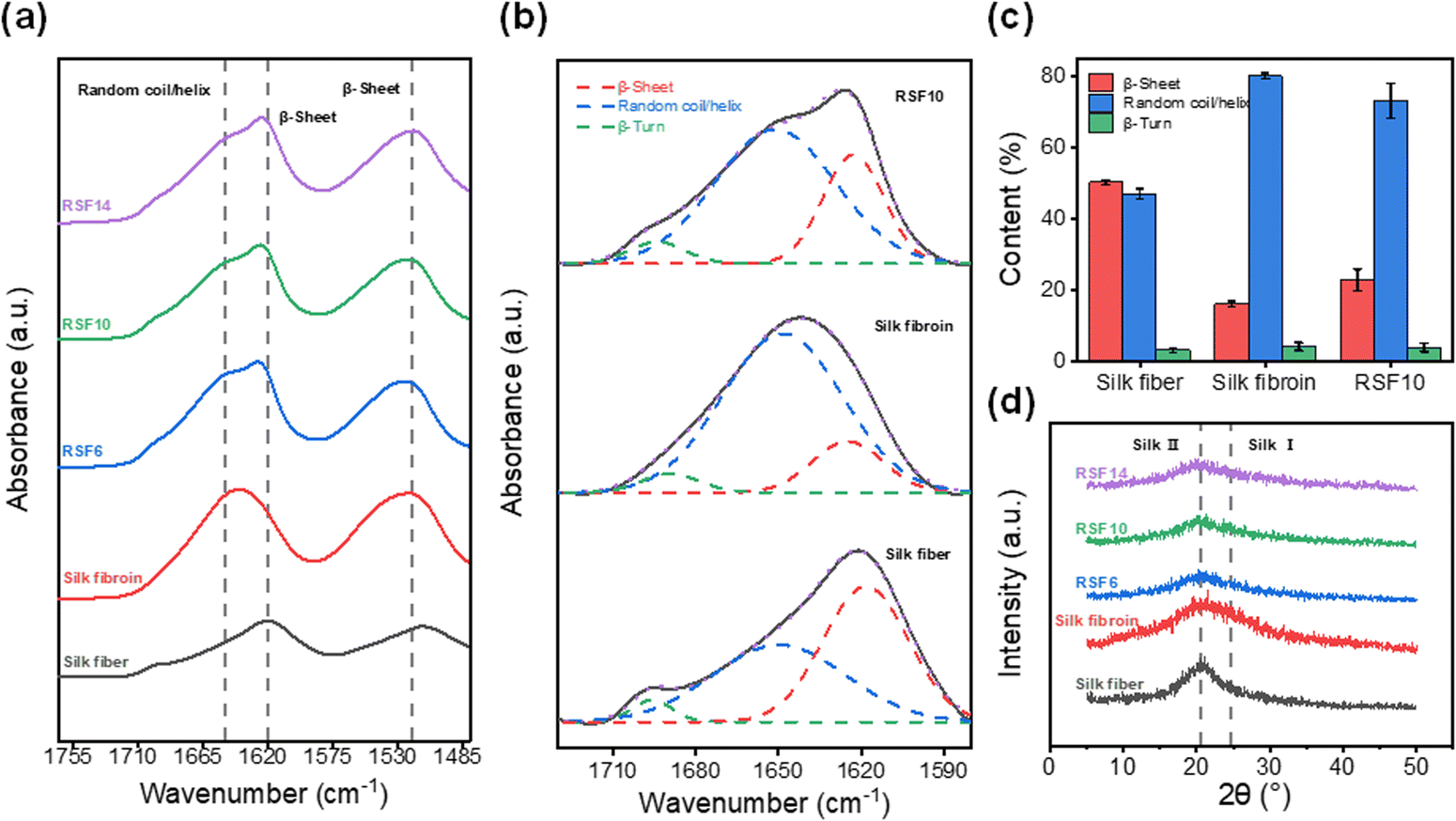 | ||
| Fig. 4 (a) FTIR spectra, (b) FTIR deconvolution, (c) secondary structure quantitative analysis, and (d) XRD profiles of degummed silk fiber, lyophilized silk fibroin, and RSF films. | ||
Similarly, it can be seen from XRD profiles that degummed silk fiber exhibits a strong characteristic β-sheets diffraction peak49 at about 20.5°; lyophilized SF shows the typical silk I diffraction peak at approximately 24.5°; and the RSF films display a weak β-sheet diffraction peak (Fig. 4d), revealing similar trend as the FT-IR results.
The thermal properties of RSF films in comparison to degummed silk and lyophilized SF were evaluated using a thermogravimetric analyzer (Fig. S7, ESI†). The lyophilized SF was found to have the largest bound water content (9.8%), whereas the degummed silk fiber has the lowest (3.8%). RSF films started to degrade at around 270 °C, which was 30 °C lower than that of the degummed silk. This was likely due to the partial loss of β-sheets, which contributes to weakened thermal stability50 in RSF films. Nevertheless, the RSF films are still sufficiently stable to heat for applications for biomedical applications.
Degradability and cytocompatibility
The RSF10 prepared using the non-solvent induced phase separation method has both sponge-like interpenetrating microporous structure and finger-like macropores, as well as the benefits of the pore structure of both lyophilized sponge and electrospun silk membrane, with good air permeability and water absorption properties, which may be helpful to promote wound healing. The porous substrate can improve cell viability when the average pore size of polymer film pores is reduced from the micron to the nanometer level.51 Therefore, RSF10 was selected for the following experiments due to its suitable pore size and mechanical properties.Degradability is an important property for biomaterials used in vitro and in vivo. The favorable degradability of SF has been well demonstrated, it was also shown that the rate of SF degradation could be efficiently controlled by altering the content of β-sheets.52,53 Phosphate-buffered saline (PBS) at neutral pH (pH = 7.4) and protease XIV were used to assess the in vitro degradation of RSF films. As seen in Fig. 5a, the degradation rate of RSF film became slower as the exposure period increased. The films degraded more quickly in the protease XIV solution than in PBS, as was expected. The remaining mass of RSF10 was about 83.1% and 57.2% after 72 h in PBS and protease XIV solutions, respectively, demonstrating appreciable in vivo biodegradability.
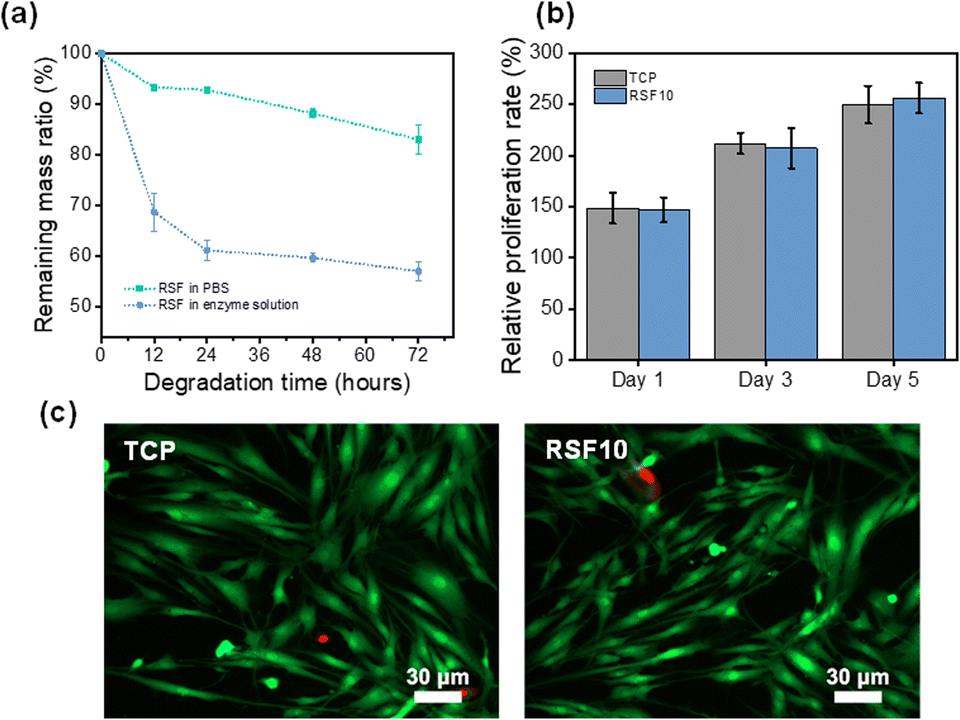 | ||
| Fig. 5 (a) Weight change of RSF10 in a PBS buffer (pH 7.4) and in protease XIV solution over time. (b) CCK-8 assay of cells' viability on the RSF10 and TCP for 5 day co-culture. Values are means ± SD for each group (n = 6). (c) Fluorescence microscopy images of cells cultured on the RSF10 for 5 days. | ||
One potential application of RSF films is as a wound dressing for preventing infection and encouraging skin tissue regeneration. To this end, it's critical to evaluate the RSF's biocompatibility for it will be in direct contact with the wound. Cell Counting Lit-8 (CCK-8) was used to examine the adhesion and proliferation of human skin fibroblasts on RSF films. Fig. 5b shows the proliferation of cells on RSF10 at 1, 3, and 5 days. The relative cell proliferation rate increased with increasing culture time and was comparable without a significant difference between RSF10 and tissue culture plate (TCP) on day 5. Further examination of the cell morphology on the fifth day revealed that the cells had a healthy spindle-shaped morphology and most cells were viable (Fig. 5c). The results demonstrated that RSF films are cyto-compatible with human skin fibroblasts.
In vivo wound healing assessment
A full-thickness skin defect rat model was established to test the wound-healing properties of RSF films. Digital pictures of rat wounds after various treatment materials and periods are shown in Fig. 6a. With increasing treatment time, the wound area was gradually decreased. Fig. 6b quantifies the percentage of residual wound area after various treatments. By day 7, all wound sites were crusted and the wound sizes were significantly reduced. In particular, the wound areas of the control group were statistically significantly larger than those of RSF10, 51.5 ± 3.3% and 31.9 ± 4.3%, respectively (p < 0.05). The favorable effect of nanopores on wound healing was also reported by Liu,24etc., where they found a greater reduction in wound area when using nano-porous SF films compared with non-porous controls. Notably, by day 21, wounds were almost completely healed, with a large amount of regenerated epidermis and hair covering the area, with a wound area of 6.8 ± 2.9% in the control group and a wound area of 4.2 ± 1.8% in RSF10. These findings show that the RSF film can clearly promote wound healing, probably because its porous structure can fulfill the wound's oxygen permeability throughout the healing process as well as the microenvironmental requirements for appropriate cell proliferation.24,54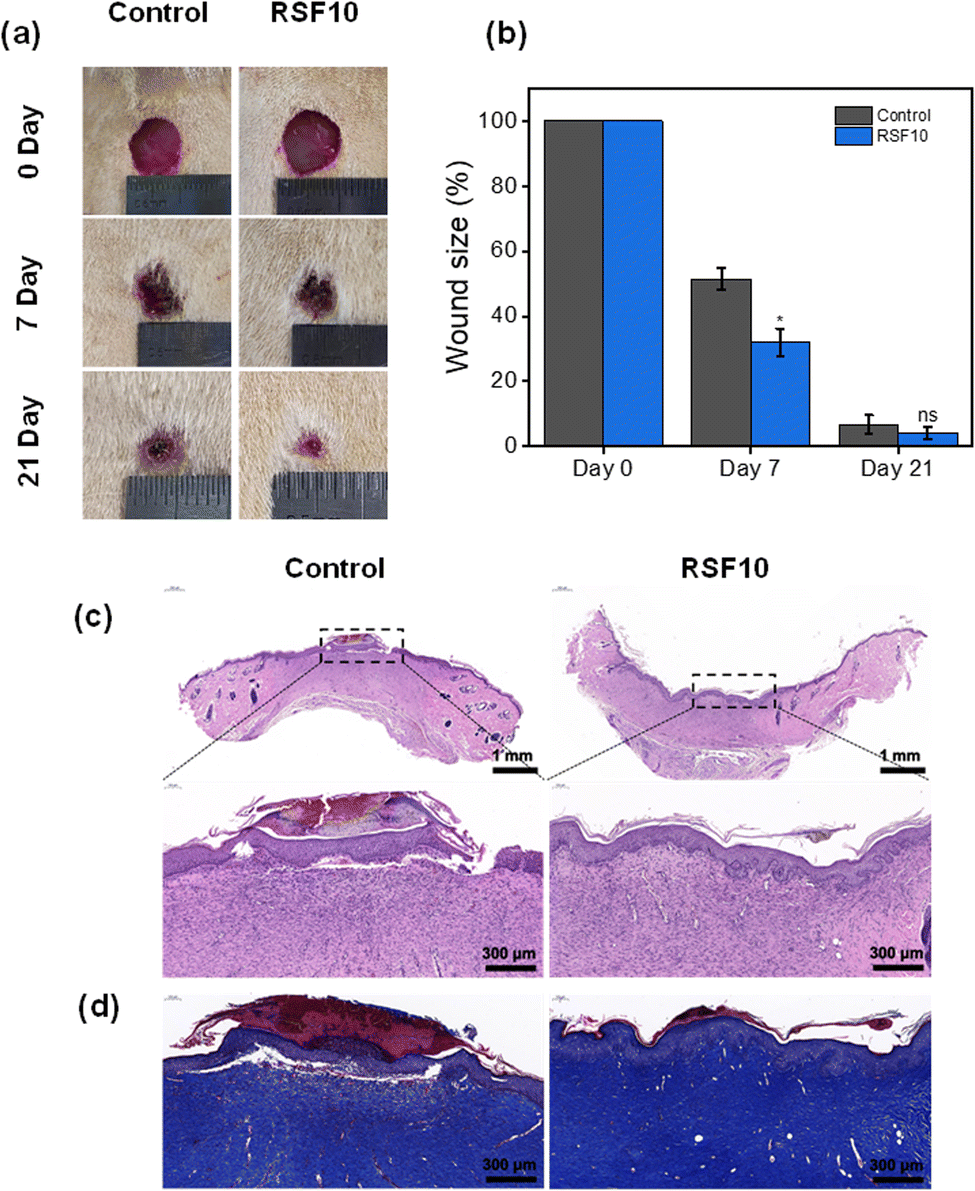 | ||
| Fig. 6 (a) Gross morphology of the healing progression of the full-thickness wounds treated with the different materials after 7 and 21 days. (b) Wound area measurements of the different materials after 7 and 21 days. Values are means ± SD for each group (n = 5, *P < 0.05). (c) Hematoxylin-eosin and (d) Masson's trichrome staining images of the wound sections. | ||
The wound healing effect of the RSF films at d21 was further evaluated using the hematoxylin-eosin (HE) stained histological images shown in Fig. 6c. Compared with the control group, which still had thick scabs in the defect region and a large number of inflammatory cells, the wounds treated with RSF films demonstrated greater epidermal remodeling and noticeably more neovascularization. Masson's trichrome (MT) staining was used to examine collagen deposition in the regenerated dermis (Fig. 6d). Compared to the control group, the RSF film group had a darker blue color with a tighter connection between the epidermis and the dermis, indicating better collagen growth and partial restoration of normal structure and function, further demonstrating its superior wound healing capabilities.
Conclusions
In summary, we have developed a facile method to prepare RSF films with tunable pore size, excellent stretchability, and outstanding biocompatibility by using aqueous phosphoric acid (85 wt%) as the dissolving solvent and aqueous ammonium sulfate (20% (w/v)) as the coagulating bath. The process is straightforward, efficient and environmentally friendly, and it does not require the use of complicated procedures such as dialysis. The tunable nano-porous structure resulted from the nonsolvent-induced phase separation process not only improved breathability, but also increased wound-healing speed. The RSF films exhibit outstanding stretchability, good biocompatibility, and excellent wound-healing effects. These results suggest that the RSF films are promising candidates for full-thickness skin defect repair in tissue engineering applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Fundamental Research Funds for the Central Universities (No. 2232022G-04) and International Cooperation Fund of Science and Technology Commission of Shanghai Municipality (No. 21130750100).References
- R. A. Clark, K. Ghosh and M. G. Tonnesen, J. Invest. Dermatol., 2007, 127, 1018–1029 CrossRef CAS PubMed.
- P. Martin, Science, 1997, 276, 75–81 CrossRef CAS PubMed.
- D. Chouhan, N. Dey, N. Bhardwaj and B. B. Mandal, Biomaterials, 2019, 216, 119267 CrossRef CAS PubMed.
- M. Naseri-Nosar and Z. M. Ziora, Carbohydr. Polym., 2018, 189, 379–398 CrossRef CAS PubMed.
- Y. Pei, L. Wang, K. Tang and D. L. Kaplan, Adv. Funct. Mater., 2021, 31, 2008552 CrossRef CAS.
- C. Vepari and D. L. Kaplan, Prog. Polym. Sci., 2007, 32, 991–1007 CrossRef CAS PubMed.
- L.-D. Koh, Y. Cheng, C.-P. Teng, Y.-W. Khin, X.-J. Loh, S.-Y. Tee, M. Low, E. Ye, H.-D. Yu, Y.-W. Zhang and M.-Y. Han, Prog. Polym. Sci., 2015, 46, 86–110 CrossRef CAS.
- C. Wang, K. Xia, Y. Zhang and D. L. Kaplan, Acc. Chem. Res., 2019, 52, 2916–2927 CrossRef CAS PubMed.
- Y. Zhang, H. Lu, X. Liang, M. Zhang, H. Liang and Y. Zhang, Acta Phys.-Chim. Sin., 2021, 0, 2103034 Search PubMed.
- L. Meinel, S. Hofmann, V. Karageorgiou, C. Kirker-Head, J. McCool, G. Gronowicz, L. Zichner, R. Langer, G. Vunjak-Novakovic and D. L. Kaplan, Biomaterials, 2005, 26, 147–155 CrossRef CAS PubMed.
- E. Bellas, T. J. Lo, E. P. Fournier, J. E. Brown, R. D. Abbott, E. S. Gil, K. G. Marra, J. P. Rubin, G. G. Leisk and D. L. Kaplan, Adv. Healthcare Mater., 2015, 4, 452–459 CrossRef CAS PubMed.
- E. S. Gil, B. Panilaitis, E. Bellas and D. L. Kaplan, Adv. Healthcare Mater., 2013, 2, 206–217 CrossRef CAS PubMed.
- D. Chouhan, T. U. Lohe, P. K. Samudrala and B. B. Mandal, Adv. Healthcare Mater., 2018, 7, e1801092 CrossRef PubMed.
- C. Martinez-Mora, A. Mrowiec, E. M. Garcia-Vizcaino, A. Alcaraz, J. L. Cenis and F. J. Nicolas, PLoS One, 2012, 7, e42271 CrossRef CAS PubMed.
- Y. R. Park, M. T. Sultan, H. J. Park, J. M. Lee, H. W. Ju, O. J. Lee, D. J. Lee, D. L. Kaplan and C. H. Park, Acta Biomater., 2018, 67, 183–195 CrossRef CAS PubMed.
- M. Santin, A. Motta, G. Freddi and M. Cannas, J. Biomed. Mater. Res., 1999, 46, 382–389 CrossRef CAS PubMed.
- B. L. Tardy, B. D. Mattos, C. G. Otoni, M. Beaumont, J. Majoinen, T. Kamarainen and O. J. Rojas, Chem. Rev., 2021, 121, 14088–14188 CrossRef CAS PubMed.
- H. Y. Wang, Y. Q. Zhang and Z. G. Wei, Crit. Rev. Biotechnol., 2021, 41, 406–424 CrossRef CAS PubMed.
- Y. Wang, B. J. Kim, B. Peng, W. Li, Y. Wang, M. Li and F. G. Omenetto, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 21361–21368 CrossRef CAS PubMed.
- B. D. Lawrence, F. Omenetto, K. Chui and D. L. Kaplan, J. Mater. Sci., 2008, 43, 6967–6985 CrossRef CAS.
- S. Werner, O. Huck, B. Frisch, D. Vautier, R. Elkaim, J. C. Voegel, G. Brunel and H. Tenenbaum, Biomaterials, 2009, 30, 2291–2301 CrossRef CAS PubMed.
- B. D. Lawrence, J. K. Marchant, M. A. Pindrus, F. G. Omenetto and D. L. Kaplan, Biomaterials, 2009, 30, 1299–1308 CrossRef CAS PubMed.
- M. Demura and T. Asakura, J. Membr. Sci., 1991, 59, 39–52 CrossRef CAS.
- J. Liu, R. Huang, G. Li, D. L. Kaplan, Z. Zheng and X. Wang, Biomacromolecules, 2021, 22, 546–556 CrossRef CAS PubMed.
- H. Ishizaka, Y. Watanabe, K. Ishida and O. Fukumoto, J. Jpn. Seric., 1989, 58, 87–95 CAS.
- J. Schurz, Nature, 1954, 173, 952–953 CrossRef CAS.
- C. S. Ki, K. H. Lee, D. H. Baek, M. Hattori, I. C. Um, D. W. Ihm and Y. H. Park, J. Appl. Polym. Sci., 2007, 105, 1605–1610 CrossRef CAS.
- H. Su, B. Wang, Z. Sun, S. Wang, X. Feng, Z. Mao and X. Sui, Carbohydr. Polym., 2022, 277, 118878 CrossRef CAS PubMed.
- A. I. Susanin, E. S. Sashina, N. P. Novoselov and M. Zaborkskii, Fibre Chem., 2017, 49, 88–96 CrossRef CAS.
- G. Freddi, G. Pessina and M. Tsukada, Int. J. Biol. Macromol., 1999, 24, 251–263 CrossRef CAS PubMed.
- W. Chen, F. Li, L. Chen, Y. Zhang, T. Zhang and T. Wang, Int. J. Biol. Macromol., 2020, 156, 633–639 CrossRef CAS PubMed.
- Y. Yao, B. J. Allardyce, R. Rajkhowa, C. Guo, X. Mu, D. Hegh, J. Zhang, P. Lynch, X. Wang, D. L. Kaplan and J. M. Razal, Biomacromolecules, 2021, 22, 788–799 CrossRef CAS PubMed.
- P. P. Madeira, M. G. Freire and J. A. P. Coutinho, J. Mol. Liq., 2020, 301, 112409 CrossRef CAS.
- R. A. Curtis, J. M. Prausnitz and H. W. Blanch, Biotechnol. Bioeng., 1998, 57, 11–21 CrossRef CAS PubMed.
- W. Ma, W. Ren, X. Bai, J. Pan, L. Huang, Q. Huang, Z. Guo and X. Wang, J. Water Process Eng., 2022, 45, 102504 CrossRef.
- D.-M. Wang, A. Venault and J.-Y. Lai, in Hollow Fiber Membranes, ed. T.-S. Chung and Y. Feng, Elsevier, 2021, pp. 13–56 DOI:10.1016/B978-0-12-821876-1.00009-3.
- M. I. Baig, E. N. Durmaz, J. D. Willott and W. M. de Vos, Adv. Funct. Mater., 2020, 30, 1907344 CrossRef CAS.
- G. R. Guillen, Y. Pan, M. Li and E. M. V. Hoek, Ind. Eng. Chem. Res., 2011, 50, 3798–3817 CrossRef CAS.
- M. Hopp-Hirschler and U. Nieken, J. Membr. Sci., 2018, 564, 820–831 CrossRef CAS.
- M. R. Cervellere, X. Qian, D. M. Ford, C. Carbrello, S. Giglia and P. C. Millett, J. Membr. Sci., 2021, 619, 118779 CrossRef CAS.
- H.-h Lin, Y.-h Tang, H. Matsuyama and X.-l Wang, J. Membr. Sci., 2018, 548, 288–297 CrossRef CAS.
- P. Tomietto, M. Carré, P. Loulergue, L. Paugam and J.-L. Audic, Polymer, 2020, 204, 122813 CrossRef CAS.
- H. H. Wang, J. T. Jung, J. F. Kim, S. Kim, E. Drioli and Y. M. Lee, J. Membr. Sci., 2019, 574, 44–54 CrossRef CAS.
- J. Yan, G. Zhou, D. P. Knight, Z. Shao and X. Chen, Biomacromolecules, 2010, 11, 1–5 CrossRef CAS PubMed.
- H. Fan, A. Huang, S. Zhou, Z. Hu, S. Yan and Q. Zhang, J. Phys.: Conf. Ser., 2021, 1790, 012067 CrossRef CAS.
- X. Hu, K. Shmelev, L. Sun, E.-S. Gil, S.-H. Park, P. Cebe and D. L. Kaplan, Biomacromolecules, 2011, 12, 1686–1696 CrossRef CAS PubMed.
- Q. Lü, C. Cao, H. Zhai and H. Zhu, Chin. Sci. Bull., 2004, 49, 1009–1011 CrossRef.
- Q. Wang, S. Ling, Q. Yao, Q. Li, D. Hu, Q. Dai, D. A. Weitz, D. L. Kaplan, M. J. Buehler and Y. Zhang, ACS Mater. Lett., 2020, 2, 153–160 CrossRef CAS.
- Z. Hu, S. K. Das, S. Yan, R. You, X. Li, Z. Luo, M. Li, Q. Zhang and D. L. Kaplan, Composites, Part B, 2020, 200, 108222 CrossRef CAS.
- C. Guo, C. Li, H. V. Vu, P. Hanna, A. Lechtig, Y. Qiu, X. Mu, S. Ling, A. Nazarian, S. J. Lin and D. L. Kaplan, Nat. Mater., 2020, 19, 102–108 CrossRef CAS PubMed.
- V. H. Fragal, T. S. Cellet, E. H. Fragal, G. M. Pereira, F. P. Garcia, C. V. Nakamura, T. Asefa, A. F. Rubira and R. Silva, J. Colloid Interface Sci., 2016, 466, 150–161 CrossRef CAS PubMed.
- Y. Cai, J. Guo, C. Chen, C. Yao, S. M. Chung, J. Yao, I. S. Lee and X. Kong, Mater. Sci. Eng., C, 2017, 70, 148–154 CrossRef CAS PubMed.
- V. P. Ribeiro, A. da Silva Morais, F. R. Maia, R. F. Canadas, J. B. Costa, A. L. Oliveira, J. M. Oliveira and R. L. Reis, Acta Biomater., 2018, 72, 167–181 CrossRef CAS PubMed.
- J. Ju, L. Hao, S. Yang, D. Wang, Y. Zhang, H. Yuan, X. Yin and Y. Tan, Adv. Funct. Mater., 2020, 30, 2006544 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tb01896a |
| This journal is © The Royal Society of Chemistry 2023 |