
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
First principles insights into stability of defected MXenes in water
Haohong
Song
 a and
De-en
Jiang
*ab
a and
De-en
Jiang
*ab
aInterdisciplinary Materials Science Program, Vanderbilt University, Nashville, TN 37235, USA
bDepartment of Chemical and Biomolecular Engineering, Vanderbilt University, Nashville, TN 37235, USA. E-mail: de-en.jiang@vanderbilt.edu
First published on 4th September 2023
Abstract
Two-dimensional transition metal carbides and nitrides, known as MXenes, have demonstrated remarkable performance in electrochemical energy storage and various other applications. Despite their potential, MXenes exhibit instability in aqueous solutions, and the role of defects in their aqueous stability is unclear. In this study, we report on the interfacial chemistry between water and defected Ti3C2O2 MXene at room temperature using first principles molecular dynamics simulations. We investigate how defects such as O vacancy, Ti vacancy, F terminal groups, and Ti–O vacancy pair influence the chemical interaction of water molecules with the basal plane of Ti3C2O2. Our results show that water molecules can repair the surface O vacancies, by dissociating to hydroxide and hydronium. On the other hand, F terminal groups cannot effectively block water chemisorption on the surface Ti, while Ti vacancies behave as a spectator species on the surface with respect to interaction with water. Ti3C2O2 with a Ti–O vacancy pair is found to behave like Ti3C2O2 with an O vacancy where a water molecule dissociates and refills the vacancy. These findings enrich our understanding of water interaction with defects on the MXene surfaces.
Introduction
Two-dimensional (2D) materials are an exciting area of research due to their unique properties and potential applications.1,2 Over the past decade, the MXenes as 2D carbides and nitrides have received increasing attention.3 These materials are characterized by the general formula Mn+1XnTx (n = 1, 2, 3, or 4), where M represents transition metal, X carbon and/or nitrogen, and T surface termination (–OH, –O, and/or –F).3 Due to their highly tunable compositions and electronic structure, as well as diverse surface chemistry,4 MXenes have shown great potential for a variety of applications, including supercapacitors,5,6 lithium-ion batteries,7–9 photoelectric devices,10 sensors,11 detectors,12 membranes,13,14 and catalysts.15–17Defects can impart new functionalities and augment MXenes' electrochemical behavior, which may be beneficial for applications such as electrocatalysis and energy storage.18,19 For instance, the introduction of ruthenium doping in Mo2CTX was found to significantly enhance the electrocatalytic activity for the nitrogen reduction reaction.20 Defect-engineered Ti2CT2 monolith enabled interfacial photothermal catalysis for high-yield solar hydrogen generation.21 Phosphorus doped Ti3C2Tx MXene electrodes can increase the capacitance.22 Ti defects and C defects in MXenes can affect the electronic transport behavior of MXene-based sensors.23 Moreover, MXenes with vacancy ordering have been prepared.17,24,25
On the other hand, the tendency of MXenes to convert to oxides over time has been a major concern,26 resulting in structural degradation and loss of performance.27–30 MXenes are susceptible to degradation in aqueous media and previous simulations31 suggested that water can attack the surface Ti atoms of a purely O-terminated Ti3C2O2 basal plane, leading to the hydrolysis of MXene. This finding from first principles molecular dynamics simulations corroborates the experimentally observed degradation trends of Ti3C2Tx and other MXenes in different solution/gas environments.27–30 Furthermore, in both battery and capacitor systems, the contact of MXene with water is often unavoidable when aqueous electrolytes are employed.19 Thus, it is desirable to know how defects on MXene surfaces would further impact their aqueous stability and one hopes that introduction of defects can modify their surface chemistry and electronic characteristics, leading to improved resistance to degradation.
In this study, our goal is to use Ti3C2O2 as a representative MXene material and perform first principles molecular dynamics simulations to investigate the chemical behavior of water at the interface with different types of defects. First principles methods have played an important role in understanding electrode–electrolyte interfaces32–34 and MXenes/anti-MXenes properties.35,36 Below we explain our methods and models.
Computational methods
Vienna ab initio simulation package (VASP)37,38 was used to perform first principles molecular dynamics (FPMD) simulations. The interaction between electrons and ions was described by the Projector Augmented Wave (PAW) method39 and electron exchange–correlation by the Perdew–Burke–Ernzerhof (PBE) variant of the Generalized Gradient Approximation (GGA).40 The Grimme's DFT+D3 method was used to account for van der Waals interactions.41 Plane-wave basis cutoff energy was set at 500 eV. We used an orthorhombic supercell (a = 10.45 Å; b = 9.04 Å) of the Ti3C2O2 monolayer (12 formula units) with two layers of water (corresponding to 15 water molecules per supercell) randomly distributed between MXene layers. The Brillouin zone was subjected to sampling through a Γ-centered 3 × 3 × 2 k-point mesh. Convergence criteria were 10−5 eV and 0.01 eV Å−1 for total energies and Hellmann–Feynman forces, respectively. After geometric optimization, non-spin-polarized FPMD simulations were performed within the NVT ensemble with a time step of 1 femtosecond (fs) and the Nose–Hoover thermostat42,43 for up to 45 ps at 298 K.Results and discussion
Common defects in MXenes include vacancies and minority termination groups, so we focus on the following major defect types: O vacancies; F functional groups; Ti vacancies; Ti–O vacancy pairs.O vacancies on Ti3C2O2
We first examined the impact of a surface oxygen vacancy on the Ti3C2O2 basal plane on the interaction with water. Fig. 1 shows the snapshots along the trajectory where the vacancy site is indicated by a red circle in the upper panel. One can see that at 0.2 ps a water molecule is orientating to prepare adsorption at the O vacancy (dashed black circle in Fig. 1b). Then the adsorbed H2O molecule dissociates to a surface OH group (dashed black circle and inset Fig. 1c) which fills or repairs the O vacancy site, while the proton from the water dissociation forms a hydronium ion (red dashed box in the upper panel of Fig. 1c) with another water molecule. At this stage, the MXene surface is partially hydroxylated and negatively charged, which prevents further chemisorption of water on the MXene surface. Furthermore, the proton on the surface (i.e., the hydroxyl proton) can reversibly shuttle between a surface O atom and a water O atom (Fig. 1c–e): surface-OH + H2O ⇌ surface-O + H3O+. Conversely, the generation of hydronium ions elevates water's acidity and increases negative charges on the MXene surface, which reorients the water molecules by pointing the O–H groups toward the surfaces (Fig. 1e), thereby distancing water O atoms from the surface Ti atoms and safeguarding the MXene from potential attack.31 In the meantime, the hydronium ion transfers its proton among water molecules in the water layer via the Grotthuss mechanism.44 So our FPMD simulations predict that O vacancies themselves are not stable at the interface with water, but after the reaction with water, further reaction of the MXene surface with water becomes less likely.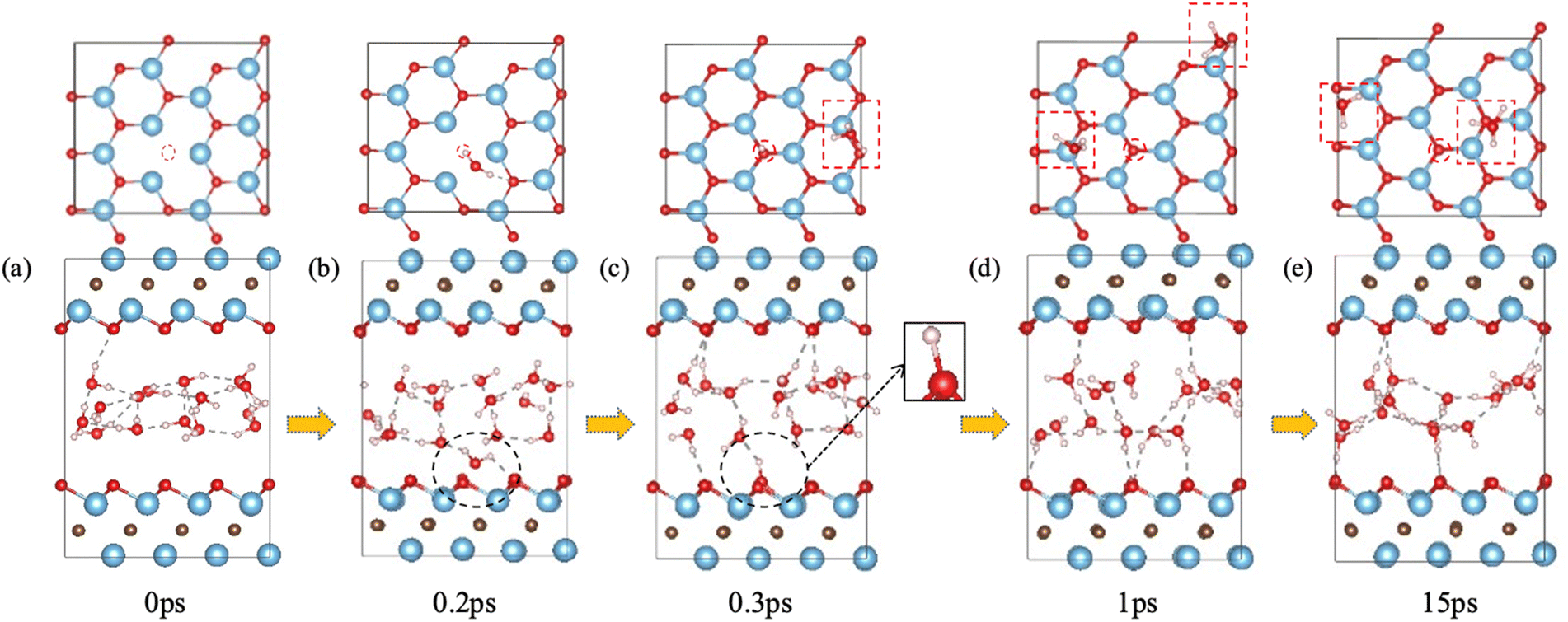 | ||
| Fig. 1 Interaction of O vacancy in Ti3C2O2 with water from FPMD trajectory: upper panel, top view (O vacancy indicated by a dashed red circle); lower panel, side view (the O vacancy is on the lower surface, within the dashed black circle). Ti, blue; O, red; C, gray; H, white. Red dashed boxes in (c)–(e) indicate hydronium ions. | ||
F-terminated MXenes
F groups often appear on the MXene surface after etching of the MAX phase by F-containing solutions.45 The stoichiometry of F as part of T in Ti3C2Tx can range from 0.2 to 0.8.46 So we have examined one to four F terminal groups on the both sides of the MXene flake, corresponding to a stoichiometry of 0.17 to 0.67 (Fig. 2a). Fig. 2b shows how the minimum distance between surface Ti atoms and O atoms in the water layer changes with time for different amount of F terminal groups. When there is no chemical interaction between water and the MXene surface, the distance should be above 3.0 Å. On the other hand, the distance would be below 2.0 Å when water chemisorbs on Ti to initiate a chemical attack. From Fig. 2b, one can see that the time of water chemisorption generally increases with the coverage of F terminal groups, even though the difference between 1F and 2F is not significant (2F happened ahead of 1F). Fig. 3 shows snapshots of water attacking a surface Ti in the case of Ti3C2O1.5F0.5 (where F replaces three O out of 12 on one surface of Ti3C3O2). One can see that water chemisorbs on a surface Ti first (dashed circle in Fig. 3b), followed by water dissociation and Ti pullout by –OH (dashed circles and insets in Fig. 3c and d). This behavior is similar to that of water on Ti3C3O2.31 In the case of the highest F coverage examined with four F groups on the surface, no water chemisorption was observed due to the limitation of our simulation timescale (∼30 ps). In other words, the F terminal group can passivate the MXene surface to a certain extent against water attack, but is unlikely to be very effective in preventing water in getting close to some surface Ti. Hence, our results suggest that oxidation by water are less likely at very high coverages of –F groups on the MXene surface.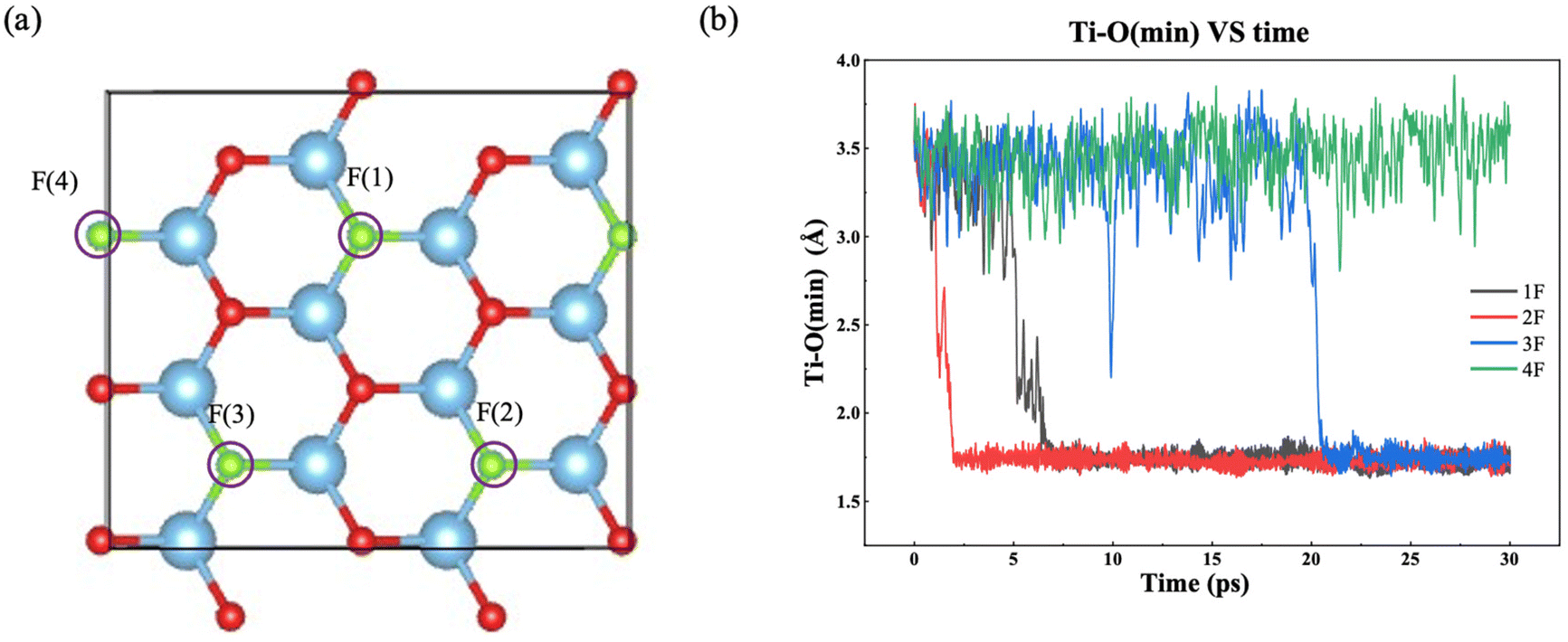 | ||
| Fig. 2 (a) Top view of Ti3C2O1.33F0.67 where F replaces four O atoms out of 12 on both surfaces of Ti3C2O2. (b) Closest contact between water molecules and surface Ti atoms, as measured the minimum Ti–O distance, over time for different amount of terminal F groups on the surface (1F to 4F mean that F replaces one to four O atoms out of 12 on both surfaces of Ti3C2O2, corresponding to chemical compositions of Ti3C2O1.83F0.17 to Ti3C2O1.33F0.67). | ||
 | ||
| Fig. 3 Interaction of Ti3C2O1.5F0.5 (where F replaces three O atoms out of 12 on both surfaces of Ti3C2O2) with water from FPMD trajectory: upper panel, top view; lower panel, side view. Black circles and insets indicate the site where water attacks surface Ti; red dashed boxes in (c)–(e) indicate hydronium ions. Ti, blue; O, red; C, gray; F, green; H, white. | ||
Ti vacancies on Ti3C2O2
It has been found that Ti vacancies are prevalent in the monolayer flakes and that their concentration can be controlled by the etchant concentration during preparation.47 We investigated water interaction with a Ti vacancy created on the surface of Ti3C2O2 (Fig. 4) and found that the water behavior is similar to that on defect-free Ti3C2O2: it starts with reversible water adsorption on Ti (Fig. 4a–e); then irreversible water adsorption on Ti and water dissociation take place (Fig. 4f). So Ti3C2O2 MXene with surface Ti vacancies is as susceptible as defect-free Ti3C2O2 toward water attack. In other words, with or without surface Ti vacancies, attack of the MXene surfaces by water is equally likely.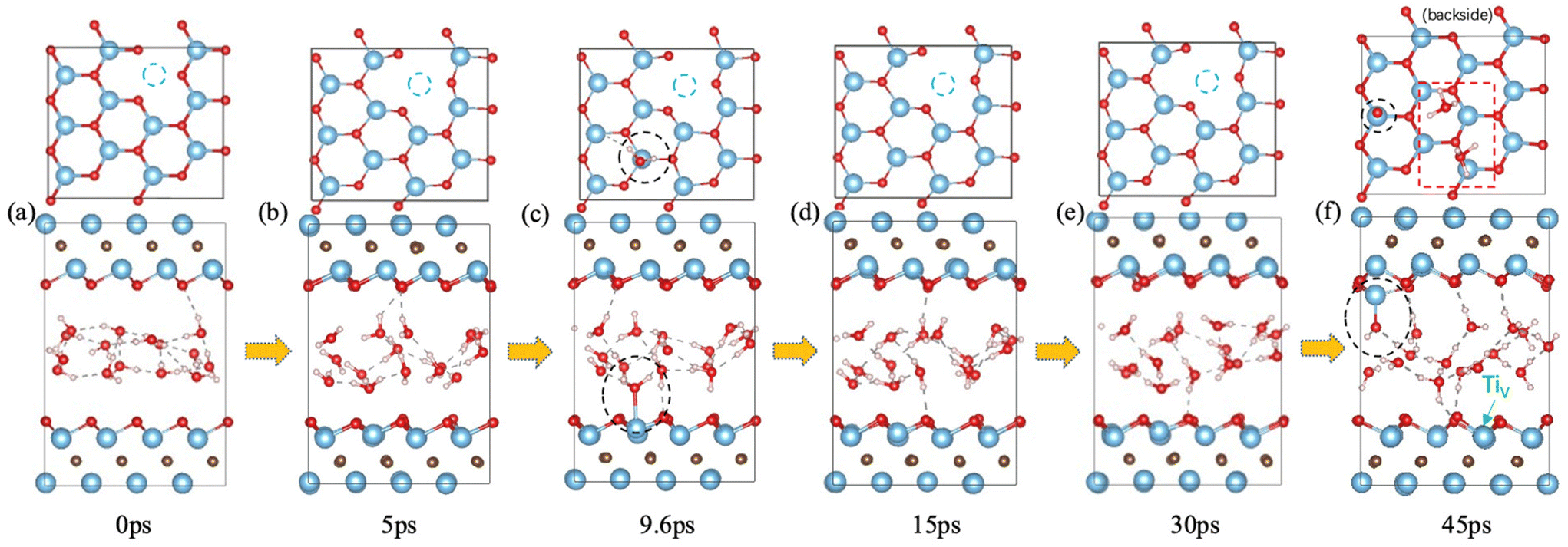 | ||
| Fig. 4 Interaction of Ti vacancy in Ti3C2O2 with water from FPMD trajectory: upper panel, top view (Ti vacancy indicated by a dashed blue circle); lower panel, side view (the Ti vacancy is on the lower surface, as indicated in f). Black circles and insets indicate the site where water attacks surface Ti; red dashed box in (f) indicates hydronium ions. | ||
Ti–O cluster vacancies on Ti3C2O2
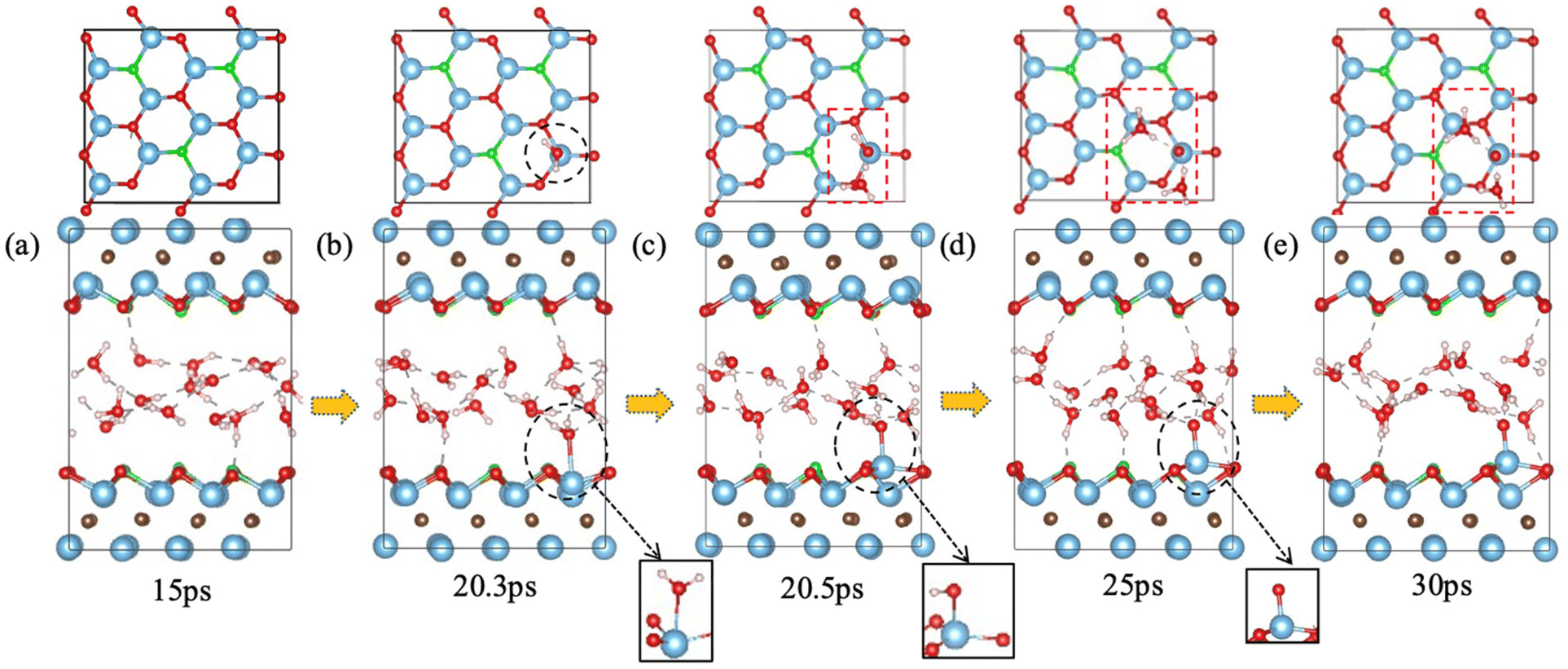
We also tested the scenario of Ti–O cluster vacancies on Ti3C2O2. The simplest case would be a Ti–O vacancy pair as shown in Fig. 5a upper panel. We found that the water behavior in this case is similar to that on Ti3C2O2 with just an O vacancy: a water molecule refills the O vacancy and dissociates to –OH and a proton that binds with another water to form a hydronium ion (Fig. 5b and c), and then the hydrogen-bond network reorganizes by pointing O–H groups toward the MXene surfaces to facilitate reversible proton transfer between the surface O atoms and the water layer (Fig. 5c–e). The newly formed –OH group on the original O vacancy is indicated by black dashed circles in Fig. 5c–e, while the hydronium ion (red dashed box in Fig. 5c upper panel) soon protonates a surface O atom to form a hydroxyl on the top surface (green circles in the lower panel of Fig. 5d and e). The Ti vacancy behaves as a spectator defect, not interacting directly with either O, –OH, or water.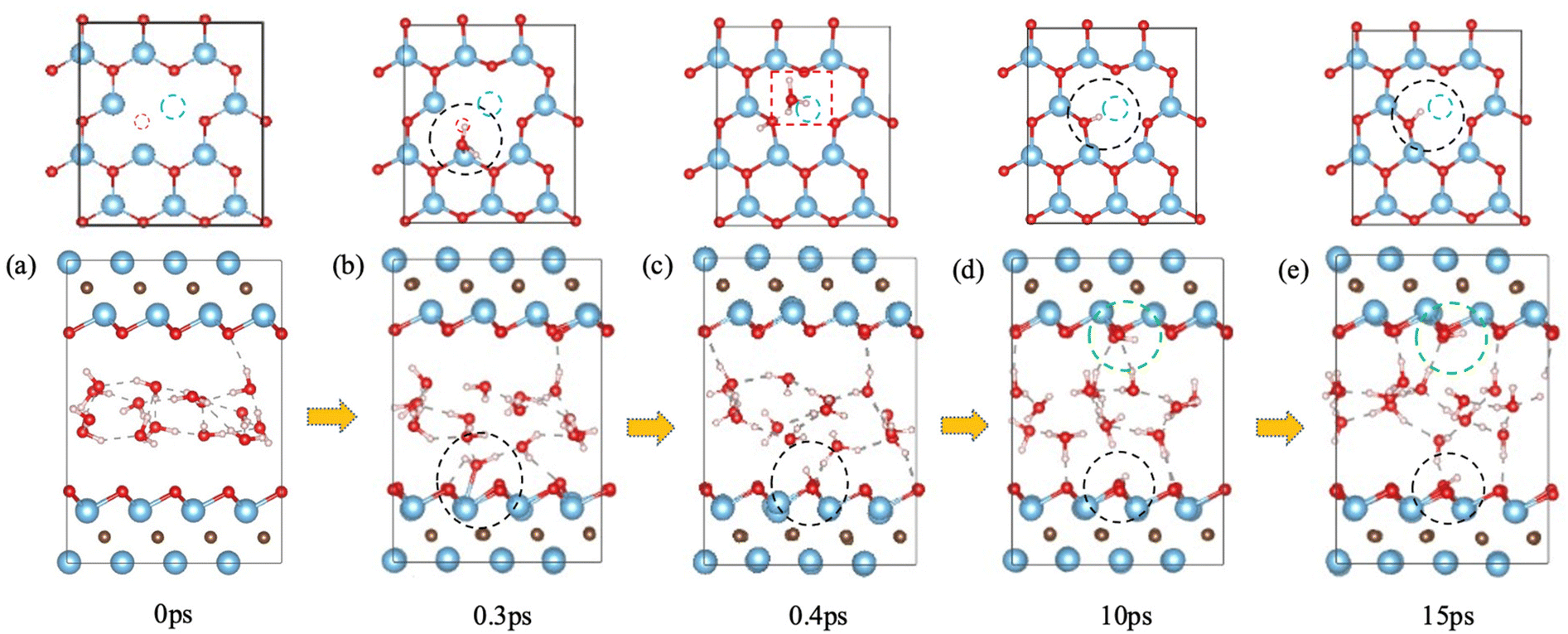 | ||
| Fig. 5 Interaction of Ti–O vacancy pair on Ti3C2O2 with water from FPMD trajectory: upper panel, top view (Ti and O vacancies indicated by dashed blue and red circles, respectively); lower panel, side view (the Ti–O vacancy pair is on the lower surface). Black circles indicate the site where water reacts with the surface O vacancy and leaves an –OH group on the surface; green circles in (d and e) indicate –OH formed by reversible proton transfer between surface O and a hydronium ion indicated by a red dashed box in (c). | ||
Implications on MXene stability in water
MXenes are susceptible to degradation; oxidation and stability of MXenes in different environments have been comprehensively reviewed recently.26 In the aqueous environment, it has been shown that hydrolysis of MXenes is an important channel of degradation, as evidenced by CH4 formation.27,28 In this work, our FPMD simulations captured a very short spatiotemporal window of the initial water adsorption and dissociation step on defected Ti3C2O2 MXenes, limited to a small system size (∼100 atoms) and time scale (∼45 ps). To examine the subsequent reactions in water with or without an oxidizer such as O2, advanced simulation techniques are needed, such as machine-learning force fields trained on FPMD data to extend the size and time scales of the simulations,48,49 so that more direct comparison with the experiment can be made regarding the MXene degradation process and mechanism under different environments.Conclusions
Using first principles molecular dynamics, we have simulated the interaction of defected-Ti3C2O2–MXene with confined water at room temperature. Defects examined include O vacancy, Ti vacancy, F terminal groups, and Ti–O vacancy pair. We found that water molecules refill the surface O vacancies, leading to –OH on the surface and hydronium in the water layer; the negatively charged surface blocks further water attack on Ti atoms, while proton can reversibly exchange between surface O atoms and water molecules. Although a slower rate of water attack on the surface Ti was observed for higher F coverages, F terminal groups could not effectively prevent water chemisorption on the surface Ti. The impact of surface Ti vacancies on the interaction with water was found to be minimal, so Ti3C2O2 with a Ti vacancy behaves like a defect-free Ti3C2O2 while Ti3C2O2 with a Ti–O vacancy pair behaves like Ti3C2O2 with an O vacancy. Overall, our FPMD simulations predict that O vacancies react with water first, which renders further reaction of the MXene surface with water less likely; very high coverages of –F groups on the MXene surface can prevent water oxidation; surface Ti vacancies do not influence oxidation of the MXene surfaces by water. These insights further our understanding of aqueous stability of real MXene materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is sponsored by the Fluid Interface Reactions, Structures, and Transport (FIRST) Center, an Energy Frontier Research Center funded by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences. This research used resources of the National Energy Research Scientific Computing Center, a DOE Office of Science User Facility supported by the Office of Science of the U.S. Department of Energy under contract no. DE-AC02-05CH11231.References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- A. H. Castro Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys., 2009, 81, 109–162 CrossRef CAS.
- A. VahidMohammadi, J. Rosen and Y. Gogotsi, Science, 2021, 372, eabf1581 CrossRef CAS PubMed.
- V. Kamysbayev, A. S. Filatov, H. Hu, X. Rui, F. Lagunas, D. Wang, R. F. Klie and D. V. Talapin, Science, 2020, 369, 979–983 CrossRef CAS PubMed.
- X. Wang, S. Kajiyama, H. Iinuma, E. Hosono, S. Oro, I. Moriguchi, M. Okubo and A. Yamada, Nat. Commun., 2015, 6, 6544 CrossRef CAS PubMed.
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2, 16098 CrossRef CAS.
- M. Naguib, J. Halim, J. Lu, K. M. Cook, L. Hultman, Y. Gogotsi and M. W. Barsoum, J. Am. Chem. Soc., 2013, 135, 15966–15969 CrossRef CAS PubMed.
- B. Ahmed, D. H. Anjum, M. N. Hedhili, Y. Gogotsi and H. N. Alshareef, Nanoscale, 2016, 8, 7580–7587 RSC.
- Q. Tang, Z. Zhou and P. Shen, J. Am. Chem. Soc., 2012, 134, 16909–16916 CrossRef CAS PubMed.
- E. Li, C. Gao, R. Yu, X. Wang, L. He, Y. Hu, H. Chen, H. Chen and T. Guo, Nat. Commun., 2022, 13, 2898 CrossRef CAS PubMed.
- Y. Pei, X. Zhang, Z. Hui, J. Zhou, X. Huang, G. Sun and W. Huang, ACS Nano, 2021, 15, 3996–4017 CrossRef CAS PubMed.
- T. Zhao, P. Xie, H. Wan, T. Ding, M. Liu, J. Xie, E. Li, X. Chen, T. Wang, Q. Zhang, Y. Wei, Y. Gong, Q. Wen, M. Hu, C.-W. Qiu and X. Xiao, Nat. Photonics, 2023, 17, 622–628 CrossRef CAS.
- L. Ding, Y. Wei, L. Li, T. Zhang, H. Wang, J. Xue, L. X. Ding, S. Wang, J. Caro and Y. Gogotsi, Nat. Commun., 2018, 9, 155 CrossRef PubMed.
- S. Hong, F. Al Marzooqi, J. K. El-Demellawi, N. Al Marzooqi, H. A. Arafat and H. N. Alshareef, ACS Mater. Lett., 2023, 5, 341–356 CrossRef CAS.
- L. M. Azofra, N. Li, D. R. MacFarlane and C. Sun, Energy Environ. Sci., 2016, 9, 2545–2549 RSC.
- Á. Morales-García, F. Calle-Vallejo and F. Illas, ACS Catal., 2020, 10, 13487–13503 CrossRef.
- C. Ling, L. Shi, Y. Ouyang and J. Wang, Chem. Mater., 2016, 28, 9026–9032 CrossRef CAS.
- Y. Tang, C. Yang, X. Xu, Y. Kang, J. Henzie, W. Que and Y. Yamauchi, Adv. Energy Mater., 2022, 12, 2103867 CrossRef CAS.
- X. Li, Z. Huang, C. E. Shuck, G. Liang, Y. Gogotsi and C. Zhi, Nat. Rev. Chem., 2022, 6, 389–404 CrossRef PubMed.
- W. Peng, M. Luo, X. Xu, K. Jiang, M. Peng, D. Chen, T. S. Chan and Y. Tan, Adv. Energy Mater., 2020, 10, 2001364 CrossRef CAS.
- Q. Zhang, L. Li, H. Zhang, N. He, B. Wang, D. Ying, X. Zhang, B. Jiang and D. Tang, Cell Rep. Phys. Sci., 2022, 3, 100877 CrossRef CAS.
- K. Liu, Q. Xia, L. Si, Y. Kong, N. Shinde, L. Wang, J. Wang, Q. Hu and A. Zhou, Electrochim. Acta, 2022, 435, 141372 CrossRef CAS.
- Y. Ma, Y. Cheng, J. Wang, S. Fu, M. Zhou, Y. Yang, B. Li, X. Zhang and C. W. Nan, InfoMat, 2022, 4, e12328 CrossRef CAS.
- Q. Tao, M. Dahlqvist, J. Lu, S. Kota, R. Meshkian, J. Halim, J. Palisaitis, L. Hultman, M. W. Barsoum, P. O. A. Persson and J. Rosen, Nat. Commun., 2017, 8, 14949 CrossRef PubMed.
- R. Meshkian, M. Dahlqvist, J. Lu, B. Wickman, J. Halim, J. Thornberg, Q. Tao, S. Li, S. Intikhab, J. Snyder, M. W. Barsoum, M. Yildizhan, J. Palisaitis, L. Hultman, P. O. A. Persson and J. Rosen, Adv. Mater., 2018, 30, e1706409 CrossRef PubMed.
- F. Cao, Y. Zhang, H. Wang, K. Khan, A. K. Tareen, W. Qian, H. Zhang and H. Agren, Adv. Mater., 2022, 34, e2107554 CrossRef PubMed.
- S. Huang and V. N. Mochalin, ACS Nano, 2020, 14, 10251–10257 CrossRef CAS PubMed.
- S. Huang and V. N. Mochalin, Inorg. Chem., 2019, 58, 1958–1966 CrossRef CAS PubMed.
- A. Lipatov, M. Alhabeb, M. R. Lukatskaya, A. Boson, Y. Gogotsi and A. Sinitskii, Adv. Electron. Mater., 2016, 2, 1600255 CrossRef.
- C. J. Zhang, S. Pinilla, N. McEvoy, C. P. Cullen, B. Anasori, E. Long, S.-H. Park, A. Seral-Ascaso, A. Shmeliov, D. Krishnan, C. Morant, X. Liu, G. S. Duesberg, Y. Gogotsi and V. Nicolosi, Chem. Mater., 2017, 29, 4848–4856 CrossRef CAS.
- T. Wu, P. R. C. Kent, Y. Gogotsi and D.-e. Jiang, Chem. Mater., 2022, 34, 4975–4982 CrossRef CAS.
- C. Zhan, W. Sun, Y. Xie, D. E. Jiang and P. R. C. Kent, ACS Appl. Mater. Interfaces, 2019, 11, 24885–24905 CrossRef CAS PubMed.
- T. A. Pham, K. E. Kweon, A. Samanta, M. T. Ong, V. Lordi and J. E. Pask, J. Phys. Chem. C, 2020, 124, 21985–21992 CrossRef CAS.
- B. C. Wood, T. Ogitsu, M. Otani and J. Biener, J. Phys. Chem. C, 2013, 118, 4–15 CrossRef.
- T. Wu and D.-e. Jiang, MRS Bull., 2023, 48, 253–260 CrossRef.
- J. Gu, Z. Zhao, J. Huang, B. G. Sumpter and Z. Chen, ACS Nano, 2021, 15, 6233–6242 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- W. G. Hoover, Phys. Rev. A, 1985, 31, 1695–1697 CrossRef PubMed.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- Y. Sun, C. Zhan, P. R. C. Kent, M. Naguib, Y. Gogotsi and D. E. Jiang, ACS Appl. Mater. Interfaces, 2020, 12, 763–770 CrossRef CAS PubMed.
- M. Naguib, O. Mashtalir, J. Carle, V. Presser, J. Lu, L. Hultman, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2012, 6, 1322–1331 CrossRef CAS PubMed.
- M. A. Hope, A. C. Forse, K. J. Griffith, M. R. Lukatskaya, M. Ghidiu, Y. Gogotsi and C. P. Grey, Phys. Chem. Chem. Phys., 2016, 18, 5099–5102 RSC.
- X. Sang, Y. Xie, M.-W. Lin, M. Alhabeb, K. L. Van Aken, Y. Gogotsi, P. R. C. Kent, K. Xiao and R. R. Unocic, ACS Nano, 2016, 10, 9193–9200 CrossRef CAS PubMed.
- S. Batzner, A. Musaelian, L. Sun, M. Geiger, J. P. Mailoa, M. Kornbluth, N. Molinari, T. E. Smidt and B. Kozinsky, Nat. Commun., 2022, 13, 2453 CrossRef CAS PubMed.
- L. Zhang, J. Han, H. Wang, R. Car and W. E, Phys. Rev. Lett., 2018, 120, 143001 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |