
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A polymer library enables the rapid identification of a highly scalable and efficient donor material for organic solar cells†
Martina
Rimmele
 a,
Zhuoran
Qiao
a,
Julianna
Panidi
a,
Francesco
Furlan
a,
Chulyeon
Lee
ab,
Wen Liang
Tan
c,
Christopher R.
McNeill
c,
Youngkyoo
Kim
b,
Nicola
Gasparini
*a and
Martin
Heeney
*ad
a,
Zhuoran
Qiao
a,
Julianna
Panidi
a,
Francesco
Furlan
a,
Chulyeon
Lee
ab,
Wen Liang
Tan
c,
Christopher R.
McNeill
c,
Youngkyoo
Kim
b,
Nicola
Gasparini
*a and
Martin
Heeney
*ad
aDepartment of Chemistry and Centre for Processable Electronics, Imperial College London, London, W12 0BZ, UK. E-mail: n.gasparini@imperial.ac.uk
bOrganic Nanoelectronics Laboratory and KNU Institute for Nanophotonics Applications (KINPA), Department of Chemical Engineering, School of Applied Chemical Engineering, Kyungpook National University, Daegu 41566, Republic of Korea
cDepartment of Materials Science and Engineering, Monash University, Wellington Road, Clayton, Victoria 3800, Australia
dKing Abdullah University of Science and Technology (KAUST), KAUST Solar Centre (KSC), Physical Sciences and Engineering Division (PSE), Thuwal, 23955–6900, Saudi Arabia. E-mail: martin.heeney@kaust.edu.sa
First published on 2nd August 2023
Abstract
The dramatic improvement of the PCE (power conversion efficiency) of organic photovoltaic devices in the past few years has been driven by the development of new polymer donor materials and non-fullerene acceptors (NFAs). In the design of such materials synthetic scalability is often not considered, and hence complicated synthetic protocols are typical for high-performing materials. Here we report an approach to readily introduce a variety of solubilizing groups into a benzo[c][1,2,5]thiadiazole acceptor comonomer. This allowed for the ready preparation of a library of eleven donor polymers of varying side chains and comonomers, which facilitated a rapid screening of properties and photovoltaic device performance. Donor FO6-T emerged as the optimal material, exhibiting good solubility in chlorinated and non-chlorinated solvents and achieving 15.4% PCE with L8BO as the acceptor (15.2% with Y6) and good device stability. FO6-T was readily prepared on the gram scale, and synthetic complexity (SC) analysis highlighted FO6-T as an attractive donor polymer for potential large scale applications.
 Martin Heeney | It is an honour to contribute this article to the special issue celebrating ten years of Materials Horizons. We contributed our first article back in 2013, when the journal was first starting under the leadership of Professor Seth Marder. It has been a pleasure as a reader, contributor and member of the advisory board to watch the journal flourish over this past decade, becoming one of the most important journals in the materials science community. Congratulations on the milestone, and I look forward to another decade of exciting breakthroughs. |
New conceptsThe nature of the side chain is vitally important to the performance of conjugated polymers in almost all of their applications, influencing their solubility, aggregation, self-assembly and phase segregation when blended. However side chain engineering is often laborious and time-consuming, since side chains are typically introduced early in the monomer synthesis. Here we develop a late-stage functionalization of an acceptor co-monomer, enabling the introduction of the side chain from a simple alcohol precursor in a single step. The resulting monomer is ready for polymerization, and we used this approach for the ready generation of a small library of polymers with five different side chains and three co-monomers. Such a library was used to rapidly identify a promising donor polymer, FO6-T, for use in organic photovoltaic devices. FO6-T exhibited a number of attractive properties, such as solubility in non-chlorinated solvents, good device stability and an efficiency over 15%. Furthermore the late-stage functionalization approach lends itself to upscaling, as highlighted by a synthetic complexity analysis. |
Introduction
Power conversion efficiencies of organic photovoltaic devices (OPVs) have continuously increased over the past 10 years, but a significant improvement in the efficiency of certified devices was achieved from 2018 to 2020.1 The step-change improvements in efficiency were largely driven by the development of new non-fullerene acceptors, especially Y6 and its derivatives.2–12 Careful optimisation of the NFA structure has led to further incremental improvements, but the focus is now returning to the donor polymer for record efficiencies.13–15 The design of wide band gap (WBG) polymers which are well matched with NFAs has led to impressive device efficiencies above 19%.16,17 This is related to increases in the device absorption bandwidth as well as higher open-circuit voltages (VOC) because of reduced energy losses.18–20However, some of the improvements in device efficiency have come at the cost of synthetic simplicity, with many of these materials requiring complex, multi-step synthesis. Numerous obstacles still need to be resolved in order for successful large-scale application.1,21,22 For example, scalability of the materials is crucial for commercialisation, but is often not considered in the design of new materials and hence well-performing materials are usually prepared with significant synthetic effort.23,24 The currently used high performing donor polymers such as PM6 and D18 are designed with a donor-linker-acceptor-linker architecture, which adds additional steps to the preparation.14,25,26 Many OPVs reaching record performance utilise PM6, which is prepared in at least a 10 step synthesis, giving a yield of 0.6% (Table S9, ESI†).26–29 Although there is undoubtedly room for optimisation in the synthetic route, the number of steps provides an indication of the overall structural complexity. Moreover, the number of steps and purification procedures also influence the energy requirements of the preparation, as well as the need for toxic/harmful reagents and solvents. Hence a condensed protocol can lead to more sustainable materials. Therefore, simplifying the structure of the materials is key to shortening synthetic protocols, thereby lowering the cost and increasing the sustainability.
Polymers such as poly(3-hexyl)thiophene (P3HT) and poly[(thiophene)-alt-(6,7-difluoro-2-(2-hexyldecyloxy)quinoxaline)] (PTQ10) are prepared using much less elaborate synthetic routes than PM6/D18, but suffer from other limitations. P3HT is limited by its relatively large band gap and its small ionisation potential, limiting the VOC and efficiency of solar cells.30 PTQ10 has been shown to possess excellent photovoltaic properties, as well as a short synthetic protocol.31 However, the synthesis is relatively costly compared to other donor polymers, mainly because of the starting materials, as highlighted by Rech and co-workers in a cost analysis of commonly used donor polymers.24 Therefore the challenges in the design of new donor polymers are not only limited to the electronic structure of the material and its match with the NFA, but also the synthetic complexity and cost of the material.
Conjugated donor polymers used in OPVs typically consist of three parts, the conjugated backbone, usually alternating donor (D) and acceptor (A) units, aliphatic side chains that render the polymer soluble, and functional groups to fine tune the band gap. The key considerations for the optimisation of electronic properties are both the conjugated framework and the functional groups, but the side chains also have a critical role in device performance.32–35 Whilst their primary role is to provide good solubility and processability to the polymer, they also influence polymer aggregation and self-assembly in both solution and the solid state, as well as phase segregation from the NFA in the blend.36,37
Given the importance of the side chains towards the overall device performance, and the difficulty in predicting which is the optimum side chain a priori, considerable synthetic effort is usually required to synthesise and screen different options. This can be especially time consuming given that the side chains are usually introduced at an early stage of the synthesis (in order to render monomer materials processable) and given the complexity of some donor systems it is not surprising that in many reported cases limited side chain examples are reported.38–43 Even for relatively simple systems such as PTQ10, there are no reported studies examining the influence of the side chain length to the best of our knowledge, although one report examines the effect of the branching point position.44 A methodology to readily create libraries of conjugated polymers with differing side chains is therefore attractive to help rapidly identify promising candidates and to facilitate understanding of the influence that small structural changes can have on film microstructure and device performance.
Herein, we report a series of donor polymers which are prepared in just two steps from a readily available precursor (which can also be prepared in just two steps from intermediates available at the kilogram scale). The side chain was introduced in the first synthetic step using a nucleophilic aromatic substitution (SNAr) reaction of a branched alcohol in good yield. Exploiting the ready availability of a range of branched alcohols, we prepared a library consisting of five different acceptor monomers containing branched ethers of systematically increasing lengths, with three different comonomers. The physical, optoelectronic and photovoltaic device properties of the resulting polymers were investigated, allowing for the rapid identification of the key structural factors. As a result, we were able to identify a donor polymer FO6-T that achieved an efficiency of 15.2% with Y6 as the acceptor in OPV devices and could be prepared via a simple synthetic protocol that was successfully scalable to the gram scale.
Design and synthesis
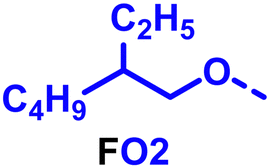
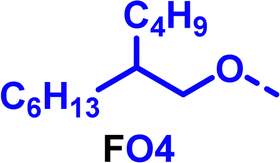
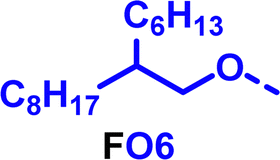
To create libraries of polymers with comparable structures to investigate the influence of small structural changes has traditionally been quite tedious.45–47 Generally, the synthesis of monomers is the most time-consuming process in preparing new materials and altering side chains often requires changes early in the (multi-step) synthetic route. Therefore, it is desirable to design monomers from easily accessible starting materials in which the side chain is introduced at the latest stage possible. Thus, libraries of polymers become accessible, and comparisons between structural attributes are possible. Considering the prevalence of donor–acceptor type polymers, in which an electron poor monomer is copolymerised with an electron rich comonomer, in OPV materials, we focused on routes to readily functionalise the acceptor comonomer. This was based on the fact that electron poor aromatics are generally amenable to nucleophilic aromatic substitution reactions, which have a well-documented history of scalability and commercialisation.48 A second factor relates to the fact that alcohols are good nucleophiles, and there is a large pool of commercially available branched alcohols (from the surfactant and additive industries). Hence, we chose the commonly deployed benzo[c][1,2,5]thiadiazole as the acceptor unit for the polymer and commercially available thiophene, 2,2′-bithiophene or thieno[3,2-b]thiophene as the donor units.Our starting point for these studies was 4,7-dibromo-5,6-difluorobenzo[c][1,2,5]thiadiazole (1), which is readily commercially available.49 We and others have previously reported that nucleophilic aromatic substitution (SNAr) reactions on the fluoro substituents of 5,6-difluorobenzo[c][1,2,5]thiadiazole derivatives are a useful method to introduce functionality, either on various comonomers or on the polymer itself.50–53 Gratifyingly, treatment of 1 with an alcohol in the presence of a base afforded the mono-substituted product in satisfying reaction yields between 63 and 80% after column chromatography. No competing bromide displacement was observed under our conditions, although small amounts of disubstituted products were formed. The facile nature of the reaction, coupled with the ready availability of a variety of primary alcohols with different side chain lengths, enabled the preparation of a library of five monomer materials with relatively small synthetic effort (Scheme 1). We focused on branched derivatives because of their higher solubilising power compared to their straight chain analogues, moving systematically from 2-ethylhexanol to 2-decyl-1-tetradecanol with four additional methylene groups added in each step.
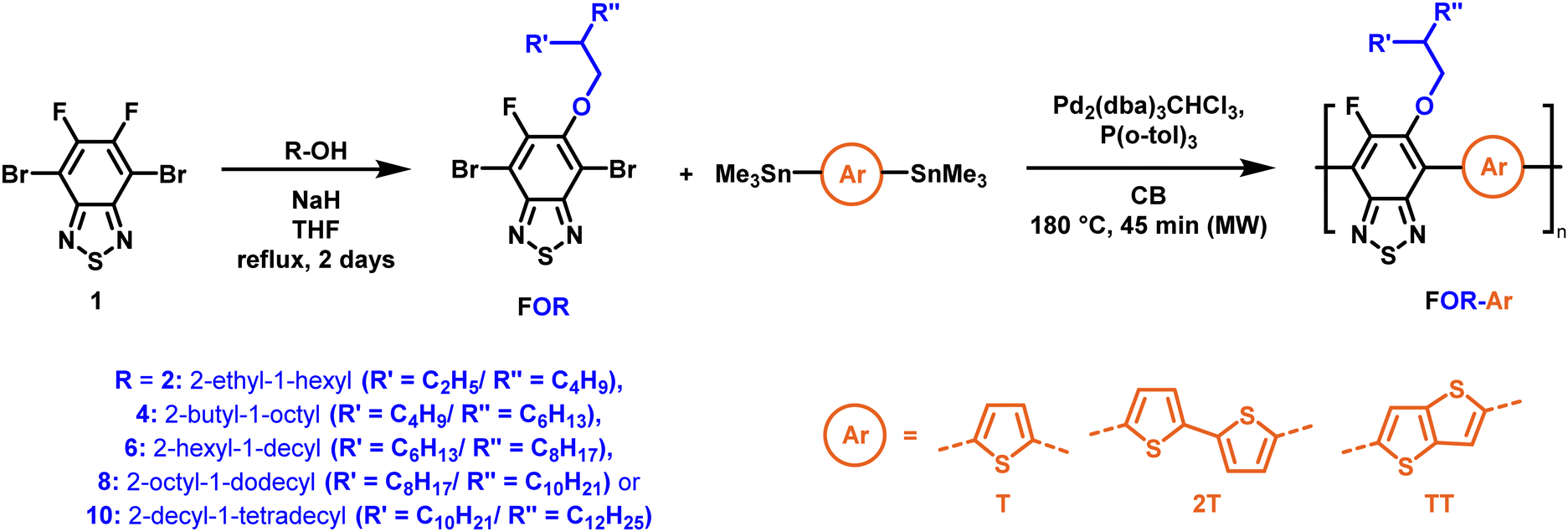 | ||
| Scheme 1 Two step polymer synthesis via nucleophilic aromatic substitution and subsequent polymerisation. | ||
With the monomers in hand, a library of conjugated polymers was rapidly produced by Stille polymerisation with the donor monomers (2,5-bis(trimethylstannyl)thiophene (T), 5,5′-bis(trimethylstannyl)-2,2′-bithiophene (2T) and 2,5-bis(trimethylstannyl)thieno[3,2-b]thiophene) (TT). All polymerisations were performed under identical microwave irradiation conditions in anhydrous chlorobenzene.54 Following work-up and Soxhlet washing, all polymers were isolated from their chloroform fractions. In general, yields of the isolated polymers were high, except for those with short side chains which exhibited very poor solubility in chloroform, where the majority of the product was insoluble (see Table 1). The structures of all polymers were confirmed by a combination of elemental analysis and NMR spectroscopy (see Table S1 and Fig. S1–S24, ESI†).
|
|
|
|
|
|
Yield | 17 mg, 9% | — | — |
|
196 mg, 93% | — | — | |
| 14.0 kDa, [33], 2.1 | ||||
| 14.5 mg mL −1 | ||||
|
M n, [n], Ð | 234 mg, 92% | 23 mg, 8% | 217 mg, 82% |
| 11.5 kDa, [24], 3.3 | 11.4 kDa, [20], 1.5 | 3.8 kDa, [7], 1.1 | ||
| 41.8 mg mL −1 | 2.9 mg mL −1 | 4.4 mg mL −1 | ||
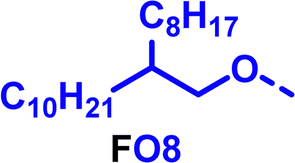
|
236 mg 91% | 270 mg, 88% | 205 mg, 94% | |
| 7.4 kDa [14], 2.2 | 15 kDa, [24], 1.6 | 4.8 kDa, [8], 1.4 | ||
| 8.0 mg mL −1 | 10.8 mg mL −1 | |||
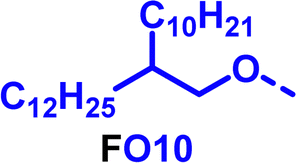
|
Solubility limit | 259 mg, 89% | 403 mg*, 86% | 440 mg*, 98% |
| 8.1 kDa, [14], 2.6 | 12.4 kDa, [18], 1.4 | 16.4 kDa, [25], 1.6 | ||
| 11.0 mg mL −1 | 39.8 mg mL −1 |
Our initial assessment focused on the polymer solubility in chloroform, which is a common device processing solvent. The solubility limit at room temperature of selected polymers was determined via a standard calibration curve method (Fig. S25 and S26, ESI†).33 As shown in Table 1, several trends were immediately evident. Focusing initially on the thiophene (T) copolymers, the polymer with the shortest 2-ethylhexyl group (FO2-T) exhibited poor solubility, with only oligomeric species extracted in chloroform in the Soxhlet extraction step. As the side chain length increased to the 2-butyloctyl group, the solubility increased to 14.5 mg mL−1 and by increasing the side chain with four additional methylene groups (FO6-T) a value of 41.8 mg mL−1 was reached, sufficiently high for ink formulation. Due to this high value, longer chains were not measured. Focusing now on the 2-hexyldecyl side chain (FO6), moving from thiophene (T) to bithiophene (2T) or thienothiophene (TT) results in drastically lower solubility with 4.4 mg mL−1 for TT and 2.8 mg mL−1 for 2T. Clearly increasing the length of the aromatic comonomer reduces the solubility significantly. Because of the poor solubility of FO6-2T and FO6-TT, the synthesis of shorter FO4 and FO2 chains was not attempted, especially in light of the processing of the materials for the preparation of OPV devices. Increasing the side chain for both 2T and TT polymers increased solubility in both cases, with the 2T polymer consistently lower than TT. We attribute this to the reduced percentage of the alkyl group to the repeat unit mass for 2T versus TT (or T).
Gel permeation chromatography (GPC) analysis in chlorobenzene (CB) was performed to determine the molecular weight distribution of the polymer fractions. The reduced solubility of FO2-T precluded the measurement in our set-up. Analysis of FO6-TT and FO8-TT showed very low molecular weight for both, which we attribute to a combination of low solubility (see solubility limits in Table 1) leading to early polymer precipitation in the reaction, and solution aggregates not passing through the GPC pre-filter. Since the molecular weight of the repeat units changes across the series, we include the average number of repeat units [n] in each polymer chain, based on the number average molecular weight in Table 1. This shows that most of the polymers fall within the range of 14–24 repeat units, allowing for a reasonable comparison of their properties.
Thermogravimetric analysis revealed the good stability of polymers FO10-T, FO10-2T and FO10-TT up to 336 °C, at which point a 5% weight loss was observed. No features could be observed by differential scanning calorimetry (DSC) measurements for any of the polymers (Fig. S27 and Fig. S28, ESI†).
Optoelectronic properties
The UV-vis absorption spectra of the polymers in chloroform solution and spun-cast thin films on glass are shown in Fig. 1 (respective photoluminescence (PL) spectra are shown in Fig. S29, ESI†). In solution, all polymers exhibit an absorption peak in the high energy region around 350–400 nm, as well as peaks in the lower energy region from 600–700 nm. The latter peaks exhibit a vibronic progression with pronounced 0–0 and 0–1 peaks, particularly for the 2T and TT polymer. Serial dilution measurements (Fig. S30, ESI†) demonstrate the spectral features and extinction coefficient did not change, suggesting that the vibronic character was not due to solution aggregation. This was further confirmed by the absence of any significant changes to the spectra upon heating (Fig. S31, ESI†). We therefore assign the vibronic character therefore to good planarity of the polymer backbones, possibly assisted by intermolecular non-covalent interactions between the oxygen of the ether and the fluorine with the adjacent thiophene or thienothiophene rings, which lead to longer conjugation lengths.55 All polymers exhibit small Stokes shifts, in further agreement with good backbone planarity. The Stokes shift is smallest for the TT polymers, which can be attributed to a more rigid and ordered polymer due to the fused comonomer.56 The TT polymer is also expected to exhibit a more linear backbone in comparison to the T or 2T polymers, due to the bond angles of the centrosymmetric TT.57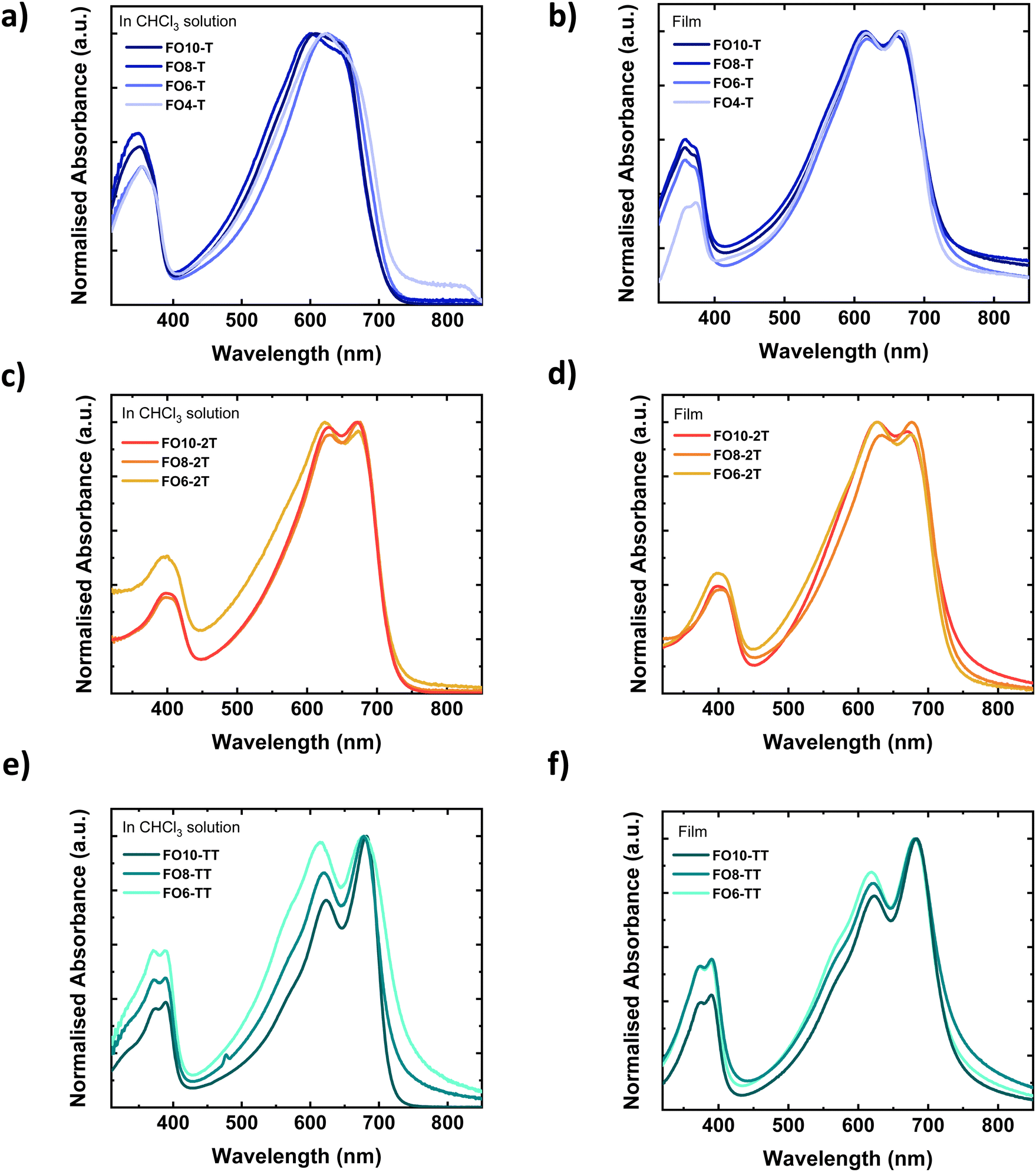 | ||
| Fig. 1 UV-vis absorption spectra of polymers in solution in CHCl3 (a) FOR-T polymers, (c) FOR-2T polymers, and (e) FOR-TT polymers, and in thin film (b) FOR-T polymers, (d) FOR-2T polymers and (f) FOR-TT polymers. The solutions were prepared at a 5 μM concentration and thin films were prepared from a 10 mg mL−1 solution and spin coated on glass slides. | ||
The choice of comonomer only has a minor effect on the absorption wavelength, with all materials exhibiting a similar optical band gap (Table 2). Moving from solution to the thin-film does not result in a significant red-shift, in further agreement with the polymers extended structure in solution. The ratio of the 0–0 and 0–1 peaks does change subtly, and the peaks become sharper for the T series upon solidification. The spectral shape does not vary significantly within a series of side chains, suggesting the effective conjugation length is reached. The exception is for the shorter alkyl chains of the TT series, which likely relates to their low molecular weight.
| Polymer | CVb | PESAc | |||||
|---|---|---|---|---|---|---|---|
| λ max,sol (nm) | λ max,film (nm) | E g,opt | PLmax, sol (nm) | HOMO (eV) | LUMO (eV) | I.P. (eV) | |
| a The optical bandgap (Eg,opt) was determined from the onset wavelength of the thin film absorption. b Energy levels were estimated from cyclic voltammetry onset potentials for oxidation and reductions peaks, using a Fc/Fc+ standard of −4.8 eV in the solid state as a thin film with an Ag/Ag+ reference electrode at a scan rate of 0.1 V s−1 with tetrabutylammonium hexafluorophosphate in acetonitrile (0.1 M) as the supporting electrolyte. c PESA measurements were performed in thin films spin coated on ITO. The error of both techniques is around ±0.1 eV. | |||||||
| FO4-T | 622, 661 | 616, 667 | 1.70 | 702 | −5.59 | −3.25 | −5.19 |
| FO6-T | 625, 650 | 619, 665 | 1.69 | 713 | −5.50 | −3.45 | −5.23 |
| FO8-T | 602, 645 | 616, 663 | 1.69 | 709 | −5.57 | −3.48 | −5.25 |
| FO10-T | 608, 645 | 617, 665 | 1.69 | 710 | −5.62 | −3.54 | −5.22 |
| FO6-2T | 625, 673 | 627, 676 | 1.69 | 722 | −5.34 | −3.47 | −5.00 |
| FO8-2T | 632, 678 | 634, 676 | 1.68 | 720 | −5.44 | −3.43 | −5.06 |
| FO10-2T | 631, 672 | 628, 670 | 1.68 | 725 | −5.45 | −3.38 | −5.05 |
| FO6-TT | 611, 678 | 619, 680 | 1.68 | 710 | −5.43 | −3.51 | −5.22 |
| FO8-TT | 621, 676 | 622, 682 | 1.66 | 704 | −5.52 | −3.55 | −5.20 |
| FO10-TT | 623, 682 | 625, 684 | 1.68 | 705 | −5.55 | −3.50 | −5.21 |
The energy levels of the three polymeric series were investigated as thin films by both cyclic voltammetry (CV) and photon electron spectroscopy in air (PESA). All polymers exhibited both oxidation and reduction peaks (Fig. S32, ESI†) by CV, which were converted to HOMO/LUMO energy levels relative to a ferrocene standard. PESA measured only the ionisation potential (I.P.). The error of both techniques is around ±0.1 eV. The absolute values obtained varied according to the technique, as often observed for conjugated polymers,58 but overall similar trends were found. The HOMO levels of both the T and TT polymers were clearly deeper than the more electron rich 2T comonomer by both techniques. The length of the side chain did not lead to significant differences in the HOMO in the PESA measurements.
We also examined the charge transport properties of the materials in a transistor device, in this case keeping a consistent side chain (O8) and varying the comonomer. Organic thin film transistors (OTFTs) were fabricated in bottom-contact top-gate architecture from FO8-T, FO8-2T and FO8-TT. Representative transfer and output characteristics of OTFTs are presented in Fig. S33 and S34 (ESI†) and a summary of the extracted parameters are shown in Table S3 (ESI†). Similar hole charge carrier transport is observed for all tested polymers on the order of 10−2 cm2 V−1 s−1 with a low threshold voltage. In particular FO8-T and FO8-TT showed decent hole mobility in the saturation regime of 4 × 10−2 cm2 V−1 s−1 with FO8-2T achieving 6 × 10−2 cm2 V−1 s−1, which is promising for such a structurally simple polymer. Moving to shorter side chains, in the case of FO6-T it did not lead to a significant difference compared to the longer FO8-T.
Photovoltaic properties
To explore the device performance of the polymer series, we fabricated bulk heterojunction organic solar cells. The devices were based on an inverted architecture of ITO/ZnO/active layer/MoOx/Ag (Fig. 2a), where the active layer consisted of a blend of the donor polymer and Y6 as the electron acceptor, keeping in all cases a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5 donor:acceptor ratio. Clear differences are observed dependent on comonomer and side chain length. Fig. 2b shows the current density–voltage characteristics of the solar cells under AM1.5G illumination, and the best photovoltaic parameters and their average values are collected in Table 3 and Fig. S35 (ESI†), respectively. We obtained a power conversion efficiency (PCE) of 14.1% for FO6-T:Y6 blends with a high short circuit current density (JSC) of 26.7 mA cm−2, open-circuit voltage (VOC) of 0.79 V and fill factor (FF) of 67% (Fig. 2b). Increasing or decreasing the side chain length decreased the performance for the T-series, resulting in decreasing FF and photocurrent. Keeping the same side chain and replacing the thiophene moiety with 2T (FO6-2T:Y6) and TT (FO6-TT:Y6) units also resulted in a drastic reduction in performance. Longer side chains are useful for increasing the solubility of polymers but can have a detrimental impact on the microstructure and therefore charge transport properties, whereas replacing T with 2T increases the HOMO energy level, resulting in lower VOC in the devices. For the TT series, we attribute the lower performance principally to microstructural changes, as discussed below. Interestingly, it is clear that the optimal side chain is dependent on the comonomer in each case, with 6 for T, 8 for 2T and 10 for TT, highlighting the complexity of finding the optimal side chain length for new materials.
:1.5 donor:acceptor ratio. Clear differences are observed dependent on comonomer and side chain length. Fig. 2b shows the current density–voltage characteristics of the solar cells under AM1.5G illumination, and the best photovoltaic parameters and their average values are collected in Table 3 and Fig. S35 (ESI†), respectively. We obtained a power conversion efficiency (PCE) of 14.1% for FO6-T:Y6 blends with a high short circuit current density (JSC) of 26.7 mA cm−2, open-circuit voltage (VOC) of 0.79 V and fill factor (FF) of 67% (Fig. 2b). Increasing or decreasing the side chain length decreased the performance for the T-series, resulting in decreasing FF and photocurrent. Keeping the same side chain and replacing the thiophene moiety with 2T (FO6-2T:Y6) and TT (FO6-TT:Y6) units also resulted in a drastic reduction in performance. Longer side chains are useful for increasing the solubility of polymers but can have a detrimental impact on the microstructure and therefore charge transport properties, whereas replacing T with 2T increases the HOMO energy level, resulting in lower VOC in the devices. For the TT series, we attribute the lower performance principally to microstructural changes, as discussed below. Interestingly, it is clear that the optimal side chain is dependent on the comonomer in each case, with 6 for T, 8 for 2T and 10 for TT, highlighting the complexity of finding the optimal side chain length for new materials.
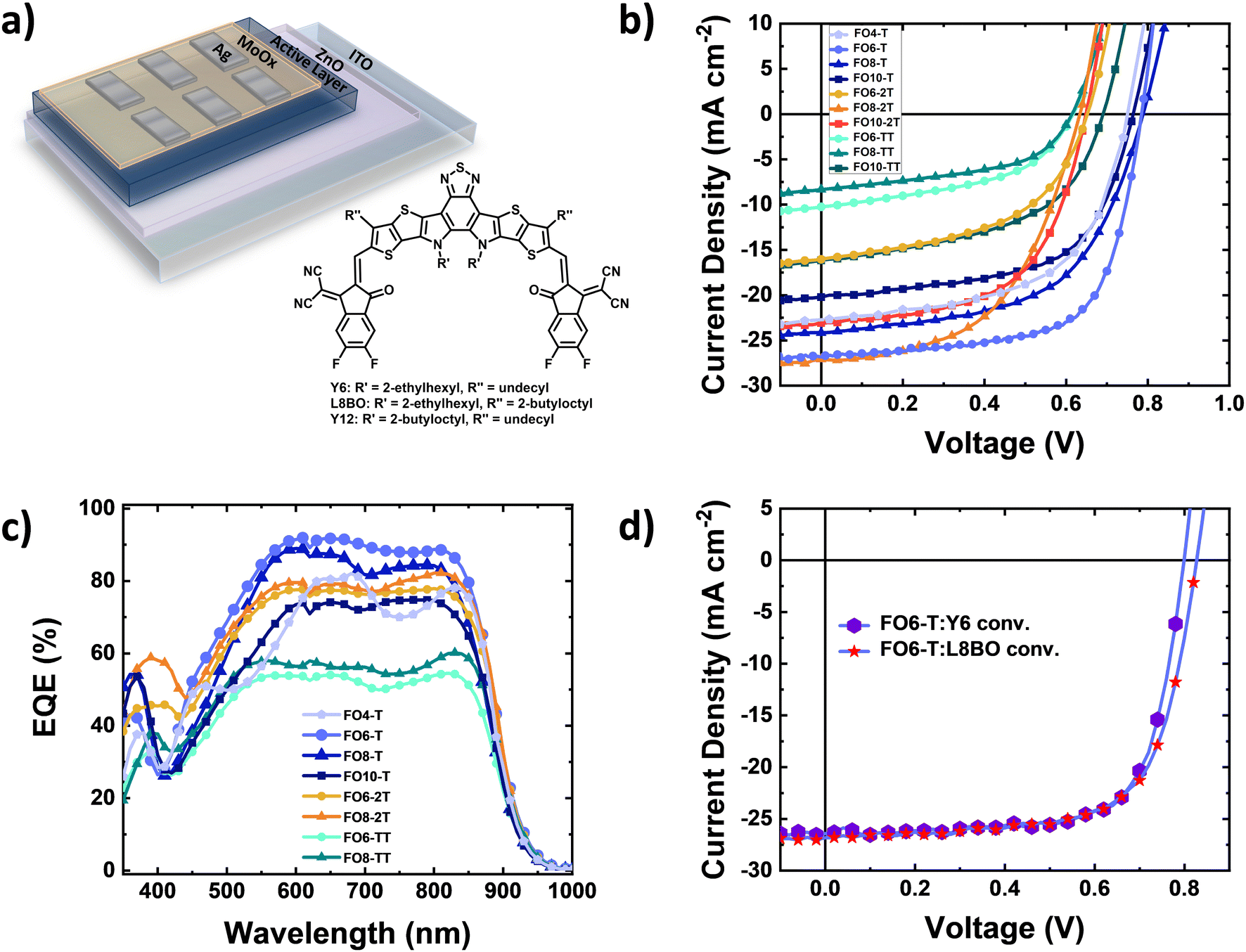 | ||
| Fig. 2 (a) Device architecture of the solar cells (inverted structure) and molecular structure of the NFAs, (b) J–V curves of devices under AM1.5G illumination, (c) EQE spectra of the OPV devices and (d) J–V curves for devices with conventional structure L8BO and Y6 as acceptors under AM1.5G illumination. | ||
| Blend | V OC (V) | J SC (mA cm2) | FF (%) | PCE (%) |
|---|---|---|---|---|
| a Conventional structure devices based on ITO/PEDOT/AL/PDINO/Ag. | ||||
| FO4-T:Y6 | 0.75 | 22.72 | 58 | 9.8 |
| FO6-T:Y6 | 0.79 | 26.70 | 67 | 14.1 |
| FO8-T:Y6 | 0.79 | 24.16 | 57 | 10.7 |
| FO10-T:Y6 | 0.76 | 20.20 | 60 | 9.3 |
| FO6-2T:Y6 | 0.65 | 16.04 | 50 | 5.3 |
| FO8-2T:Y6 | 0.63 | 27.07 | 53 | 9.0 |
| FO10-2T:Y6 | 0.65 | 23.06 | 58 | 8.7 |
| FO6-TT:Y6 | 0.61 | 10.24 | 48 | 3.0 |
| FO8-TT:Y6 | 0.61 | 8.32 | 51 | 2.6 |
| FO10-TT:Y6 | 0.69 | 16.14 | 51 | 5.7 |
| FO6-T:Y6 | 0.80 | 26.41 | 72 | 15.2 |
| FO6-T:L8BO | 0.83 | 26.82 | 69 | 15.4 |
To further elucidate the different performances of the devices, we investigated the optoelectronic properties of FO6-T and FO8-T-based devices. First, to confirm the high JSC in FO6-T:Y6 devices, we measured the external quantum efficiency (EQE). As depicted in Fig. 2c, FO6-T:Y6 solar cells show high EQE of around 90% and the integrated JSC is within the 5% margin compared to the one measured under the solar simulator light. We observed reduced light-to-current conversion for FO8-T:Y6 devices which can be associated with increased charge recombination in the devices.59 Longer side chains and replacing T with 2T and TT leads to a reduction of the EQE, in agreement with the JSC values extracted from J–V characteristics. To evaluate the charge recombination processes in FO6-T:Y6 and FO8-T:Y6 devices, we measured the current density–voltage characteristics at different light illumination.60,61 As depicted in Fig. S36a (ESI†), we calculated slopes of JSCvs. light intensity of 0.90 and 0.86 for FO6-T:Y6 and FO8-T:Y6, respectively. This indicates higher bimolecular recombination for FO8-T based OPV. On the other hand, we observed similar trap-assisted recombination (Fig. S36b, ESI†) for FO6-T:Y6 and FO8-T:Y6 devices, with values of 1 and 1.05 kT q−1, respectively.62–64
Based on the promising initial performance of FO6-T, we also evaluated the performance in conventional architecture based on ITO/PEDOT:PSS/Active layer/PDINO/Ag. The J–V characteristics under one sun illumination are depicted in Fig. 2d. We obtained higher PCE values of 15.2% due to the high FF of 72% (Table 3). Recently, a novel Y-family NFA, L8BO, has been reported for high performing OPV.6 To further test the potential of FO6-T, we blended it with L8BO and fabricated the devices in a conventional architecture. The J–V characteristics are depicted in Fig. 2d (and EQE in Fig. S37, ESI†). Notably, we obtained a PCE of 15.4%, together with a VOC of 0.83 V, FF of 69% and JSC of 26.82 mA cm−2, demonstrating the potential of our low synthetic complexity polymer.
To investigate the transport properties of the blends used, we measured the charge carrier mobility of FO6-T, FO8-T, and Y6 and their blends with the space-charge limited current (SCLC) method (Tables S4 and S5 and Fig S38 and S39, ESI†).65 We calculated the charge carried mobility using the Mott-Gurney equation in the trap-free regime, according to:
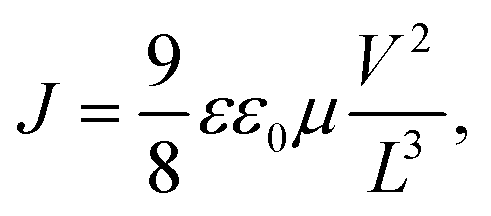
Finally, for real-world applications the halogenated solvents used to dissolve and coat the photoactive layer of a device should be replaced with non-toxic solvents. Due to the poor solubility of Y6 in non-chlorinated solvents,67 we replaced Y6 with BTP-4F-C12 (Y12) and fabricated inverted structure BHJ (bulk-heterojunction) solar cells with FO6-T:Y12 dissolved in o-xylene. We obtained a PCE exceeding 12.6% (see Table S6 and Fig. S40, ESI†), which further confirms the potential of FO6-T-based devices for upscaled production. Devices prepared with FO6-T:Y12 showed even better stability compared to Y6 blends, with a reduction in PCE of approximately 5% over 720 h of illumination (Fig. S42, ESI†).
Morphology
To correlate the device performance with the microstructure of the blends, we performed grazing-incidence wide-angle X-ray scattering (GIWAXS) measurements. Fig. 3 shows the 2D scattering patterns of the pristine polymers and their blends with Y6. In order to ensure a reasonable degree of solubility in all cases, we used the FO8 series to compare the effect of the comonomer. We also investigated FO6-T, as the best performing donor. All neat films show patterns characteristic of semicrystalline polymers, but interestingly, we see different preferred orientations for the polymers (see Fig. S43-46, ESI†). Thus, pristine FO6-T shows a mixture of face-on and edge-on orientations, as indicated by the out of plane lamellar (h00) and π–π (010) peaks, whereas FO8-T shows edge-on oriented features (out of plane lamellar (h00) peak and in plane π–π (010) peak). Changing the comonomer to 2T, FO8-2T scattering patterns are dominated by face-on orientation features, with minority edge-on orientation features. Contrarily, FO8-TT scattering patterns consist of only edge-on orientation features. By increasing the length of the side chain from FO6 to FO8, the lamellar stacking distance increases, as indicated by the shift of (h00) peaks to lower q-positions (Fig. S47, ESI†). In addition, a shorter π–π stacking difference is observed, which is reflected in a shift of the π–π (010) peak to a higher q-position. As expected, the distance of the backbone repeat unit does not show a change, indicated by the same (002) position. We conclude that the microstructure changes quite drastically between different side chains and comonomers, thus helping to explain the differences in FF and overall device performance. The orientation of the crystallites has a major influence on overall device performance, as face-on orientation facilitates charge transport in OPV devices.68 In line with this, for FO6-T, which shows mixed orientations of the crystallites, we observe better overall performance in the devices compared to the FO8-TT which shows only edge-on orientation of the crystallites. FO8-2T shows preferred face-on orientation, which facilitates charge transport, but for this series we observe an increase in the HOMO energy level, and hence we attribute the lower PCE principally to the drop in VOC.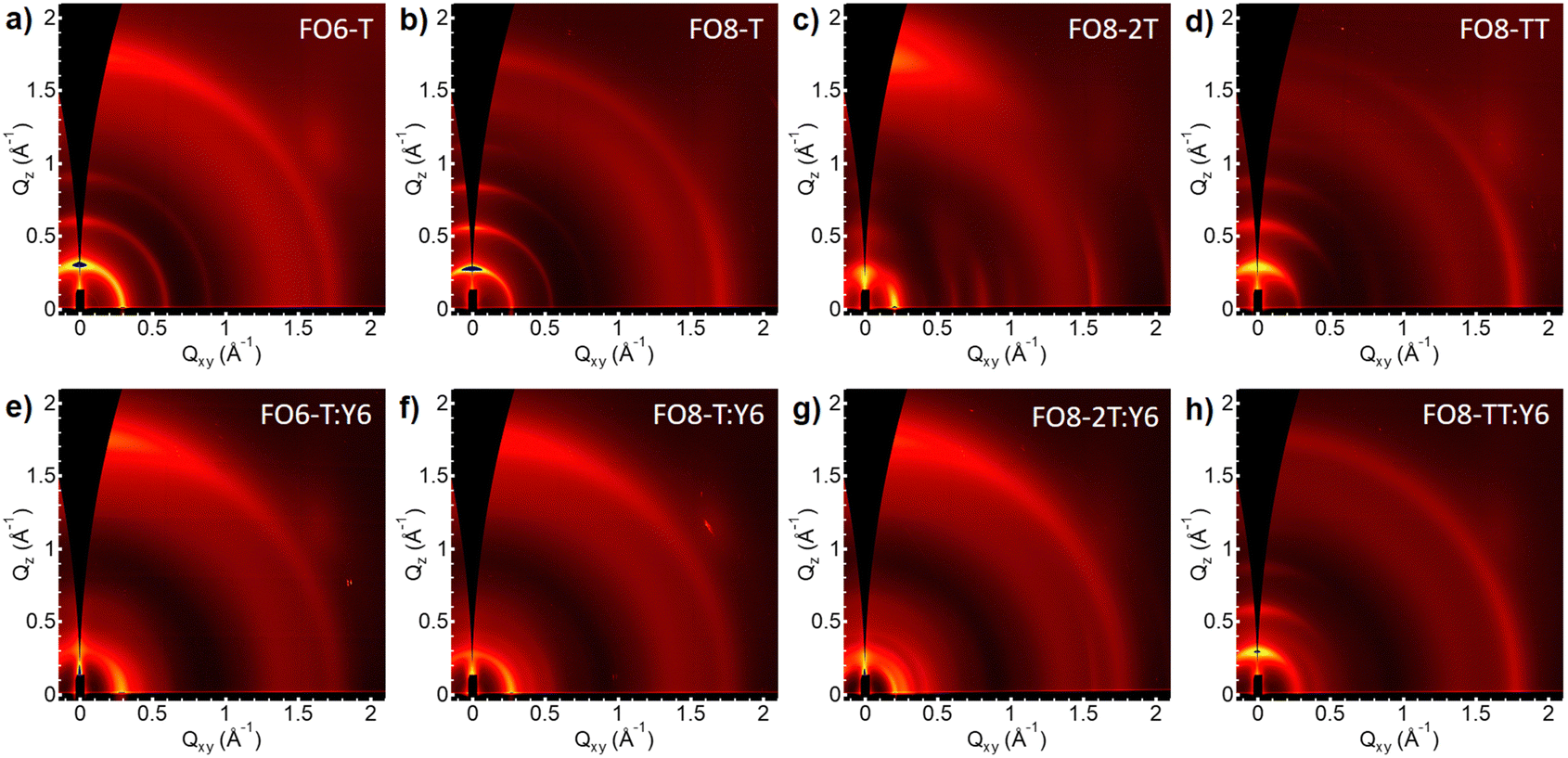 | ||
| Fig. 3 2D GIWAXS images of the polymers and their blends with Y6. | ||
When these polymers are blended with Y6 (Fig. 3e–h) we observed different microstructures. For FO6-T:Y6 the scattering pattern is dominated by the strong aggregation of Y6 with significant disruption to the FO6-T packing compared to the neat film, with only peaks attributable to (100) and (010) scattering observed (Fig. S43, ESI†). Nevertheless, the polymer maintains the beneficial mixture of edge-on and face-on orientations. In both FO8-T:Y6 and FO8-2T:Y6 the scattering features from the edge-on polymer are also strongly suppressed, with features mostly attributable to Y6 (Fig. S44-45, ESI†). However, the more linear polymer FO8-TT behaves differently in the blend (Fig. S46, ESI†), with a strong aggregation behaviour of the polymer maintained and the Y6 π–π staking disrupted, in agreement with the poor device performance.
To further analyse the microstructure of the blends, contact angle measurements were conducted and the respective surface energies and Flory–Huggins interaction parameters calculated.69,70 Polymer surface energy, and therefore the interaction parameter χ of the respective blends of polymer donors and Y6, changed quite significantly from FO6-T to FO8-T (0.23 and 0.78 respectively), as well as from FO4-T (0.53) (see Table S7, ESI†). A lower χ indicates a stronger interaction between the two materials and may help to promote a more optimal device microstructure. However no clear trend of surface energy with side chain was observed. We note that surface energy is also known to be molecular weight dependent, as a result of the reduced polymer free volume at the surface, with higher weight resulting in a larger surface energy,71 suggesting that molecular weight difference may also play a role.
Discussion and larger scale preparation of FO6-T
The results demonstrate that late-stage introduction of the side chain facilitates the synthesis of a series of polymers with systematic changes to the structure. Their ready synthesis allowed device screening to be performed in parallel, under similar conditions, leading to a rapid initial screening of device properties. Whilst device fabrication is unavoidably time consuming, such an approach still allowed screening of the polymer library. The rapid identification of the initial hit allowed for subsequent device optimisation by variation of processing conditions and acceptor. Our results highlight that there is no clear screening parameter which can be used to identify the most promising side chain without making the devices. Indeed, the reasons why FO6-T demonstrates the best performance are complex, and we feel that developing a full understanding is knowledge that is unlikely to transfer to other donor systems. Even within our own limited library, each different comonomer has a different optimal side chain. Therefore, we feel the power of the approach demonstrated here is that late stage side chain introduction allows for rapid identification of initially promising structures. A second advantage is that the structure identified will likely lend itself to larger scale preparation, important for practical applications, provided of course the acceptor building block can be readily prepared.To further explore the scalability of FO6-T in relation to other donor polymers, a synthetic complexity (SC) analysis was performed, following previously reported protocols.23,29 We note that the details on how the analysis is performed can vary widely, and thus it is essential to define the parameters beforehand and proceed as consistently as possible. We based the estimation on five parameters, the number of synthetic steps (NSS), the number of unit operations (NUO) meaning the number of work up and purification steps, the reciprocal overall yield (RY), the number of column chromatographies (NCC) and the number of hazardous chemicals (NHC), following the procedure of Riccardo Po and co-workers.29 Details on the implementation of the SC analysis can be found in the supporting information (Table S8 and S9, ESI†). We highlight that the numbers should not be over analysed and only give a broad indication of likely scalability. Clearly the synthetic procedures of most current polymers could be significantly improved, and procedures fit for small-scale preparation are unlikely to be used for larger scale. Nevertheless, the analysis does have some value in comparing materials, with significantly different values likely to highlight possible challenges in scalability.
The main result of the SC analysis (see Table S8, ESI†) is to highlight the significantly reduced complexity of FO6-T compared to the highest performing donors like PM6. Most of the reported polymers suffer from the high number of synthetic steps and consequentially low overall yields and large number of purification and work up steps. The most pertinent comparison is probably to PTQ10, another readily scalable material. Indeed PTQ10 and FO6-T share a common precursor, 3,6-dibromo-4,5-difluoro-1,2-benzenediamine, whose optimised synthesis has recently been reported.24FO6-T and PTQ10 can both be synthesised in three-steps from this intermediate, and both use the same tin comonomer. The major difference between the two relates to the introduction of the side chain. For PTQ10, this either requires alkylation with an alkyl bromide (which is prepared from the corresponding alcohol in an additional step), or Mitsunobu coupling directly with the alcohol. The use of the alcohol is preferred for cost reasons, but the Mitsunobu coupling has poor atom efficiency and requires relatively expensive DIAD/PPh3. For FO6-T we simply use the alcohol directly in the presence of a base, and for this reason the overall complexity is slightly lower. Here we note that in analogy to many other research scale polymers, we used Stille polymerisation, which is certainly undesirable from a scalability perspective due to the use of toxic organotin. However, many other options are possible for the second step of the polymerisation which do not use toxic organometallics. These include direct arylation polymerisation and/or Suzuki polymerisation and in our opinion are likely to further reduce SC. Generally, if materials are compared to each other, PCE is the parameter that is mainly considered. However, we suggest that comparing the SC of a new polymer to the state-of-the-art materials can help understand its relevance in the field, especially from an application point of view.
We conclude that high PCE is not the only factor to aim for when designing conjugated polymers for OPVs, especially with commercial application in mind. A complex and long-winded synthesis could diminish the overall significance of the material for commercialisation or application in large scale solar cells substantially.
Finally, we demonstrated the scalability of FO6-T moving from a 0.5 mmol to a 4 mmol scale. At this scale, the polymerisation was not feasible in the microwave reactor due to volume limitations. Hence the polymerisation was performed in a reaction flask heated in an oil bath at reflux (see the ESI†). The resulting solution was precipitated in MeOH and purified via Soxhlet extraction. After drying, a yield of 95% (1.8 g) was obtained. After preparing a second upscaled batch (1.5 g), the performances of the two batches were compared in OPV devices, and we found no significant difference between the two batches (see Fig. S48, ESI†). The successful upscaling reaction alongside the synthetic complexity analysis strongly indicate that this donor material FO6-T is a suitable candidate for application in large scale organic photovoltaics. The development of a tin-free polymerisation method would further assist this aim and will be the basis of future work.
Conclusion
In conclusion, we prepared a new series of polymers with a simple structure using a facile two-step synthetic protocol from a widely available precursor. This allowed the preparation of a library of eleven different polymers with differing alkyl chain lengths and comonomers. Increasing the alkyl chain length increased polymer solubility, whereas moving from thiophene to thienothiophene or bithiophene decreased it. The nature of the comonomer and the alkyl chain only had a minor influence on the optical properties, with all polymers exhibiting a similar optical band gap, and they all exhibited characteristics consistent with a highly planar backbone. This is in agreement with the good performance exhibited in field-effect transistor devices. The oxidation potential of the polymers was influenced by the nature of the comonomer. Preliminary device screening identified FO6-T as the most promising donor, and further optimisation lead to efficiencies of 15.4% with L8BO as the acceptor material in a conventional structure device (15.2% with Y6). Both devices exhibited promising stability under continual illumination at the maximum power point. A synthetic complexity analysis highlighted the potential of FO6-T for upscaling in comparison to a range of commonly used donor polymers. Finally, gram-scale synthesis was demonstrated, with the polymer exhibiting a similar performance to small scale batches.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We would like to thank the Engineering and Physics Science Research Council (EPSRC) (EP/V048686/1, EP/R513052/1 and EP/T028513/1), the Royal Society and Wolfson Foundation, and the King Abdullah University of Science and Technology (KAUST) Office of Sponsored Research (OSR) under Award No. OSR-2020-CRG8-4095 for the financial support. This work was performed in part on the SAXS/WAXS beamline72 at the Australian Synchrotron, part of ANSTO. YK and CL appreciate financial support from the National Research Foundation (NRF) of Korea (2021R1I1A3A04037494).References
- G. Bernardo, T. Lopes, D. G. Lidzey and A. Mendes, Adv. Energy Mater., 2021, 11, 2100342 CrossRef CAS.
- Y. Lin, J. Wang, Z.-G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- Q. He, P. Ufimkin, F. Aniés, X. Hu, P. Kafourou, M. Rimmele, C. L. Rapley and B. Ding, SusMat, 2022, 2, 591–606 CrossRef CAS.
- Y. Cui, H. Yao, J. Zhang, T. Zhang, Y. Wang, L. Hong, K. Xian, B. Xu, S. Zhang, J. Peng, Z. Wei, F. Gao and J. Hou, Nat. Commun., 2019, 10, 2515 CrossRef.
- C. Li, J. Zhou, J. Song, J. Xu, H. Zhang, X. Zhang, J. Guo, L. Zhu, D. Wei, G. Han, J. Min, Y. Zhang, Z. Xie, Y. Yi, H. Yan, F. Gao, F. Liu and Y. Sun, Nat. Energy, 2021, 6, 605–613 CrossRef CAS.
- K. Jiang, Q. Wei, J. Y. L. Lai, Z. Peng, H. K. Kim, J. Yuan, L. Ye, H. Ade, Y. Zou and H. Yan, Joule, 2019, 3, 3020–3033 CrossRef CAS.
- Y. Cui, H. Yao, J. Zhang, K. Xian, T. Zhang, L. Hong, Y. Wang, Y. Xu, K. Ma, C. An, C. He, Z. Wei, F. Gao and J. Hou, Adv. Mater., 2020, 32, 1908205 CrossRef CAS PubMed.
- J. Zhou, D. He, Y. Li, F. Huang, J. Zhang, C. Zhang, Y. Yuan, Y. Lin, C. Wang and F. Zhao, Adv. Mater., 2023, 35, 2207336 CrossRef CAS PubMed.
- S. Dai, J. Zhou, S. Chandrabose, Y. Shi, G. Han, K. Chen, J. Xin, K. Liu, Z. Chen, Z. Xie, W. Ma, Y. Yi, L. Jiang, J. M. Hodgkiss and X. Zhan, Adv. Mater., 2020, 32, 2000645 CrossRef CAS PubMed.
- Z. Luo, T. Liu, J. Oh, R. Ma, J. Miao, F. Ni, G. Zhang, R. Sun, C. E. Zhang, Z. Chen, Y. Zou, J. Min, C. Yang, H. Yan and C. Yang, Adv. Funct. Mater., 2022, 32, 2203200 CrossRef CAS.
- Y. Ma, R. Sun, Z. Chen, S. Zhang, D. Cai, S. Wan, W. Lin, S.-Q. Zhang, Q. Tu, W. Ma, J. Min, X. Hao and Q. Zheng, Nano Energy, 2023, 107, 108116 CrossRef CAS.
- T. Zhang, C. An, P. Bi, Q. Lv, J. Qin, L. Hong, Y. Cui, S. Zhang and J. Hou, Adv. Energy Mater., 2021, 11, 2101705 CrossRef CAS.
- T. Zhang, C. An, Y. Cui, J. Zhang, P. Bi, C. Yang, S. Zhang and J. Hou, Adv. Mater., 2022, 34, 2105803 CrossRef CAS.
- Z. a Li, C.-C. Chueh and A. K. Y. Jen, Prog. Polym. Sci., 2019, 99, 101175 CrossRef CAS.
- Y. Cui, Y. Xu, H. Yao, P. Bi, L. Hong, J. Zhang, Y. Zu, T. Zhang, J. Qin, J. Ren, Z. Chen, C. He, X. Hao, Z. Wei and J. Hou, Adv. Mater., 2021, 33, 2102420 CrossRef CAS.
- L. Zhu, M. Zhang, J. Xu, C. Li, J. Yan, G. Zhou, W. Zhong, T. Hao, J. Song, X. Xue, Z. Zhou, R. Zeng, H. Zhu, C.-C. Chen, R. C. I. MacKenzie, Y. Zou, J. Nelson, Y. Zhang, Y. Sun and F. Liu, Nat. Mater., 2022, 21, 656–663 CrossRef CAS PubMed.
- Z. Li, K. Jiang, G. Yang, J. Y. L. Lai, T. Ma, J. Zhao, W. Ma and H. Yan, Nat. Commun., 2016, 7, 13094 CrossRef CAS PubMed.
- Y. Cai, L. Huo and Y. Sun, Adv. Mater., 2017, 29, 1605437 CrossRef PubMed.
- C. An, Z. Zheng and J. Hou, Chem. Commun., 2020, 56, 4750–4760 RSC.
- M. Riede, D. Spoltore and K. Leo, Adv. Energy Mater., 2021, 11, 2002653 CrossRef CAS.
- L. W. T. Ng, S. W. Lee, D. W. Chang, J. M. Hodgkiss and D. Vak, Adv. Mater. Technol., 2022, 7, 2101556 CrossRef.
- M. Moser, A. Wadsworth, N. Gasparini and I. McCulloch, Adv. Energy Mater., 2021, 11, 2100056 CrossRef CAS.
- J. J. Rech, J. Neu, Y. Qin, S. Samson, J. Shanahan, R. F. Josey III, H. Ade and W. You, ChemSusChem, 2021, 14, 3561–3568 CrossRef CAS.
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiao, K. Sun, S. Yang, X. Zhang and L. Ding, Sci. Bull., 2020, 65, 272–275 CrossRef CAS.
- M. Zhang, X. Guo, W. Ma, H. Ade and J. Hou, Adv. Mater., 2015, 27, 4655–4660 CrossRef CAS PubMed.
- D. Qian, L. Ye, M. Zhang, Y. Liang, L. Li, Y. Huang, X. Guo, S. Zhang, Z. A. Tan and J. Hou, Macromolecules, 2012, 45, 9611–9617 CrossRef CAS.
- M. Zhang, X. Guo, S. Zhang and J. Hou, Adv. Mater., 2014, 26, 1118–1123 CrossRef CAS PubMed.
- R. Po, G. Bianchi, C. Carbonera and A. Pellegrino, Macromolecules, 2015, 48, 453–461 CrossRef CAS.
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS PubMed.
- C. Sun, F. Pan, H. Bin, J. Zhang, L. Xue, B. Qiu, Z. Wei, Z.-G. Zhang and Y. Li, Nat. Commun., 2018, 9, 743 CrossRef.
- J. Xu, H. Feng, Y. Liang, H. Tang, Y. Tang, Z. Du, Z. Hu, F. Huang and Y. Cao, J. Energy Chem., 2022, 66, 382–389 CrossRef CAS.
- L. Ye, W. Li, X. Guo, M. Zhang and H. Ade, Chem. Mater., 2019, 31, 6568–6577 CrossRef CAS.
- Q. Wang, M. Li, Y. Sui, Z. Wang, Z. Liang and Y. Geng, J. Mater. Chem. C, 2019, 7, 9581–9590 RSC.
- N. Leclerc, P. Chávez, O. A. Ibraikulov, T. Heiser and P. Lévêque, Polymers, 2016, 8, 11 CrossRef PubMed.
- A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS.
- C. Duan, F. Huang and Y. Cao, J. Mater. Chem., 2012, 22, 10416–10434 RSC.
- L. Biniek, S. Fall, C. L. Chochos, D. V. Anokhin, D. A. Ivanov, N. Leclerc, P. Lévêque and T. Heiser, Macromolecules, 2010, 43, 9779–9786 CrossRef CAS.
- C. L. Chochos and S. A. Choulis, Prog. Polym. Sci., 2011, 36, 1326–1414 CrossRef CAS.
- O. A. Ibraikulov, C. Ngov, P. Chávez, I. Bulut, B. Heinrich, O. Boyron, K. L. Gerasimov, D. A. Ivanov, S. Swaraj, S. Méry, N. Leclerc, P. Lévêque and T. Heiser, J. Mater. Chem. A, 2018, 6, 12038–12045 RSC.
- B. W. H. Saes, M. M. Wienk and R. A. J. Janssen, Chem. – Eur. J., 2020, 26, 14221–14228 CrossRef CAS PubMed.
- B. W. H. Saes, M. Lutz, M. M. Wienk, S. C. J. Meskers and R. A. J. Janssen, J. Phys. Chem. C, 2020, 124, 25229–25238 CrossRef CAS PubMed.
- J. Subbiah, C. J. Lee, V. D. Mitchell and D. J. Jones, ACS Appl. Mater. Interfaces, 2021, 13, 1086–1093 CrossRef CAS PubMed.
- D. Yuan, G. Qin, L. Zhang, F. Pan, R. Qiu, S. Lei, S. Deng and J. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 57654–57663 CrossRef CAS PubMed.
- A. Casey, S. D. Dimitrov, P. Shakya-Tuladhar, Z. Fei, M. Nguyen, Y. Han, T. D. Anthopoulos, J. R. Durrant and M. Heeney, Chem. Mater., 2016, 28, 5110–5120 CrossRef CAS.
- J. W. Jung, J. W. Jo, E. H. Jung and W. H. Jo, Org. Electron., 2016, 31, 149–170 CrossRef CAS.
- J. C. Gustafsson-Carlberg, O. Inganäs, M. R. Andersson, C. Booth, A. Azens and C. G. Granqvist, Electrochim. Acta, 1995, 40, 2233–2235 CrossRef CAS.
- D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed.
- -. d.-. The starting material 4, 6-difluorobenzo[c][1,2,5]thiadiazole, can be bought on a 100 g scale, is a commonly used monomer and can be purchased from over 60 suppliers (according to SciFinder). 100 g from Derthon was purchased at $2800 recently (Febuary 2022).
- A. Creamer, C. S. Wood, P. D. Howes, A. Casey, S. Cong, A. V. Marsh, R. Godin, J. Panidi, T. D. Anthopoulos, C. H. Burgess, T. Wu, Z. Fei, I. Hamilton, M. A. McLachlan, M. M. Stevens and M. Heeney, Nat. Commun., 2018, 9, 3237 CrossRef PubMed.
- A. Creamer, A. Casey, A. V. Marsh, M. Shahid, M. Gao and M. Heeney, Macromolecules, 2017, 50, 2736–2746 CrossRef CAS.
- P. Cong, Z. Wang, Y. Geng, Y. Meng, C. Meng, L. Chen, A. Tang and E. Zhou, Nano Energy, 2023, 105, 108017 CrossRef CAS.
- Z. Hu, Q. Huang, C. Liu, A. Song, L. Shao, Y. Bai, Z. Hu, K. Zhang, F. Huang and Y. Cao, Org. Mater., 2022, 4, 18–27 CrossRef CAS.
- S. Tierney, M. Heeney and I. McCulloch, Synth. Met., 2005, 148, 195–198 CrossRef CAS.
- T. Kharandiuk, E. J. Hussien, J. Cameron, R. Petrina, N. J. Findlay, R. Naumov, W. T. Klooster, S. J. Coles, Q. Ai, S. Goodlett, C. Risko and P. J. Skabara, Chem. Mater., 2019, 31, 7070–7079 CrossRef CAS.
- J.-L. Brédas, J. Cornil and A. J. Heeger, Adv. Mater., 1996, 8, 447–452 CrossRef.
- I. McCulloch, M. Heeney, M. L. Chabinyc, D. DeLongchamp, R. J. Kline, M. Cölle, W. Duffy, D. Fischer, D. Gundlach, B. Hamadani, R. Hamilton, L. Richter, A. Salleo, M. Shkunov, D. Sparrowe, S. Tierney and W. Zhang, Adv. Mater., 2009, 21, 1091–1109 CrossRef CAS.
- J. Bertrandie, J. Han, C. S. P. De Castro, E. Yengel, J. Gorenflot, T. Anthopoulos, F. Laquai, A. Sharma and D. Baran, Adv. Mater., 2022, 34, 2202575 CrossRef CAS PubMed.
- D. Baran, N. Gasparini, A. Wadsworth, C. H. Tan, N. Wehbe, X. Song, Z. Hamid, W. Zhang, M. Neophytou, T. Kirchartz, C. J. Brabec, J. R. Durrant and I. McCulloch, Nat. Commun., 2018, 9, 2059 CrossRef PubMed.
- L. J. A. Koster, V. D. Mihailetchi, H. Xie and P. W. M. Blom, Appl. Phys. Lett., 2005, 87, 203502 CrossRef.
- J. Vollbrecht, J. Lee, S.-J. Ko, V. V. Brus, A. Karki, W. Le, M. Seifrid, M. J. Ford, K. Cho, G. C. Bazan and T.-Q. Nguyen, J. Mater. Chem. C, 2020, 8, 15175–15182 RSC.
- C. M. Proctor, M. Kuik and T.-Q. Nguyen, Prog. Polym. Sci., 2013, 38, 1941–1960 CrossRef CAS.
- V. V. Brus, Org. Electron., 2016, 29, 1–6 CrossRef CAS.
- J. Vollbrecht, V. V. Brus, S.-J. Ko, J. Lee, A. Karki, D. X. Cao, K. Cho, G. C. Bazan and T.-Q. Nguyen, Adv. Energy Mater., 2019, 9, 1901438 CrossRef.
- G.-J. A. H. Wetzelaer, N. J. Van der Kaap, L. J. A. Koster and P. W. M. Blom, Adv. Energy Mater., 2013, 3, 1130–1134 CrossRef CAS.
- L. J. A. Koster, V. D. Mihailetchi and P. W. M. Blom, Appl. Phys. Lett., 2006, 88, 052104 CrossRef.
- S. Strohm, F. Machui, S. Langner, P. Kubis, N. Gasparini, M. Salvador, I. McCulloch, H. J. Egelhaaf and C. J. Brabec, Energy Environ. Sci., 2018, 11, 2225–2234 RSC.
- V. Vohra, K. Kawashima, T. Kakara, T. Koganezawa, I. Osaka, K. Takimiya and H. Murata, Nat. Photonics, 2015, 9, 403–408 CrossRef CAS.
- N. Gasparini, S. H. K. Paleti, J. Bertrandie, G. Cai, G. Zhang, A. Wadsworth, X. Lu, H.-L. Yip, I. McCulloch and D. Baran, ACS Energy Lett., 2020, 5, 1371–1379 CrossRef CAS.
- D. K. Owens and R. C. Wendt, J. Appl. Polym. Sci., 1969, 13, 1741–1747 CrossRef CAS.
- J.-H. Kim, A. Gadisa, C. Schaefer, H. Yao, B. R. Gautam, N. Balar, M. Ghasemi, I. Constantinou, F. So, B. T. O'Connor, K. Gundogdu, J. Hou and H. Ade, J. Mater. Chem. A, 2017, 5, 13176–13188 RSC.
- N. M. Kirby, S. T. Mudie, A. M. Hawley, D. J. Cookson, H. D. T. Mertens, N. Cowieson and V. Samardzic-Boban, J. Appl. Crystallogr., 2013, 46, 1670–1680 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3mh00787a |
| This journal is © The Royal Society of Chemistry 2023 |