
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dyes as efficient and reusable organocatalysts for the synthesis of cyclic carbonates from epoxides and CO2†
Jing
Chen
 a,
Giulia
Chiarioni
a,
Gert-Jan W.
Euverink
b and
Paolo P.
Pescarmona
*a
a,
Giulia
Chiarioni
a,
Gert-Jan W.
Euverink
b and
Paolo P.
Pescarmona
*a
aChemical Engineering Group, Engineering and Technology Institute Groningen (ENTEG), University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: p.p.pescarmona@rug.nl
bBiotechnology and Applied Microbiology Group, Engineering and Technology Institute Groningen (ENTEG), University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands
First published on 10th October 2023
Abstract
Inexpensive dyes available at the industrial scale, namely, rhodamine B (RhB), rhodamine 6G (Rh6G) and methylene blue (MB), were investigated as organocatalysts for the cycloaddition of CO2 to styrene oxide to yield styrene carbonate under solvent-free conditions (80 °C, 10 bar CO2, 24 h). Each of these dyes consists of a bulky cation, and a chloride anion that can act as a nucleophilic catalytic species in the target reaction. In order to prepare additional catalysts, the Cl− containing dyes were ion-exchanged with KX (X = Br, I) to afford their counterparts with Br− or I− as the anion. Among this set of nine organocatalysts (three dyes, each with three types of halide), the highest yield of styrene carbonate was obtained with Rh6G-I, and trends were identified based on the nature of the organic cation and halide, with the latter having a much larger impact on the activity (I− > Br− > Cl−). Additionally, we explored the effect of adding H2O as a green, inexpensive hydrogen bond donor acting as a co-catalyst, further optimising the styrene carbonate yield (96% with RhB-I and Rh6G-I in the presence of 50 mg H2O). However, the activity of these organocatalysts was only modest if the reaction temperature was decreased to 45 °C. To tackle this limitation, we designed a tailored yet straightforward modification of RhB-I to synthesise a bifunctional organocatalyst bearing a hydrogen bond donor in proximity of the iodide anion (RhB-EtOH-I). This strategy proved successful and the RhB-EtOH-I catalyst achieved a major increase in styrene carbonate yield (29% after 18 h at 45 °C, 10 bar CO2) compared to the RhB-I/H2O catalytic system (7%). The RhB-EtOH-I catalyst was also versatile and promoted the conversion of a broad scope of epoxides with good to high cyclic carbonate yields under relatively mild reaction conditions (60 °C, 10 bar, 24 h). Although these dye organocatalysts were homogeneous, RhB-EtOH-I could be easily recovered by precipitation with diethyl ether and reused without any loss of catalytic activity. Additionally, we demonstrated that nanofiltration was an effective technique for removing the dye organocatalysts from the cyclic carbonate, affording a high purity product (≤0.1 ppm of RhB in propylene carbonate). The metal-free nature of the optimum organocatalyst (RhB-EtOH-I), its facile preparation and the low cost and commercial availability of its precursors, its promising activity under mild reaction conditions and its reusability are all assets in the context of green chemistry and for potential large-scale applicability.
Introduction
The utilisation of CO2 to synthesise valuable chemicals, polymers, and fuels has been a growing research area in recent years.1 One of the drivers behind these research efforts is the public concern about climate change arising from the current high atmospheric CO2 concentration caused by anthropogenic CO2 emissions (ca. 35 Gton CO2 emissions in 2021).1,2 Another reason for investigating the conversion of CO2 into useful products is its renewable nature, abundancy and low toxicity, which makes it a green, inexpensive C1 feedstock that can partially substitute fossil-based resources.3 However, CO2 is a rather stable and inert molecule as the carbon atom is present in its highest oxidised state.4 The thermodynamic limitation can be overcome by reacting CO2 with high-energy substrates, such as epoxides, hydrogen or amines, while the kinetics of CO2 conversion can be improved by utilising appropriate catalysts.5,6 Particularly, the reaction of CO2 and epoxides to yield cyclic carbonates is a promising route in the context of green chemistry as it has a theoretical 100% atom efficiency, it can be generally carried out under solvent-free conditions at relatively mild temperature and CO2 pressure, and the obtained cyclic carbonates typically have low toxicity, high boiling point and high polarity, leading to a wide range of applications as green solvents, components for electrolyte solutions for lithium-based batteries, monomers for the preparation of polymers, and reactants for organic synthesis.7,8The simplest and most affordable catalysts for the cycloaddition of CO2 to epoxides are halide salts, among which the ones that show the best catalytic performance are organic halides such as imidazolium salts, quaternary ammonium salts and phosphonium salts.9–11 These compounds possess a halide nucleophilic species, which catalyses the reaction by causing the initial ring opening of the epoxide, followed by nucleophilic attack of the obtained alkoxide on CO2 and finally by ring closure with formation of the cyclic carbonate product (Scheme S1†).10,12 Among the organic halides, phosphonium salts such as PPNCl (bis(triphenylphosphine) iminium chloride) display high catalytic activity due to the delocalisation of the positive charge and the bulky nature of the cation, which make the halide more readily available for acting as a nucleophile.13–15 However, phosphonium salts are not ideal from the green chemistry point of view because their synthesis involves toxic organophosphines.16,17 More generally, these organocatalysts are significantly less active than catalytic systems that also contain metal centres acting as Lewis acids, which accelerate the reaction by activating the epoxide towards the nucleophilic attack by the halide.12,18,19 State-of-the-art metal-based homogeneous and heterogeneous catalysts enable the reaction of CO2 with epoxides under milder conditions (i.e. T ≤ 45 °C and pCO2 ≤ 10 bar) compared to the organic halides alone.12,18,20 However, the costly and complex synthesis of these metal-based catalysts, and the toxicity of some of the employed metals, are concerns for large-scale applications.21,22 Hydrogen bond donors (HBDs) are a green alternative to metal centres as Lewis acid species that has been increasingly investigated in recent years. HBDs can activate the epoxide by forming a hydrogen bond with its oxygen atom.10,23,24 Several types of HBD compounds have been explored, ranging from organic acids25,26 and alcohols27,28 to phenolic derivatives,29,30 silanediols,31 and H2O.15,27 Notably, bio-based compounds are often rich in HBD groups and several of them (e.g. amino acids, chitosan, cellulose, lignin) have been used for this application.32 HBDs are generally utilised as part of a binary catalytic system in combination with an organic halide, and most commonly with Bu4NX or PPNX (X = Cl, Br, I).15,28,30 Compared to organic HBDs, H2O is preferable in the context of green chemistry as it is non-toxic, inexpensive, abundant, renewable and, in general, easier to separate.15 An attractive alternative is to design a single-component bifunctional catalyst in which the nucleophile and an HBD group (e.g. –OH, –COOH, or –NH–) are present in the same compound, and preferably in close proximity to each other to enhance their cooperation.33–35 This type of metal-free bifunctional catalyst design has been developed both for homogeneous and heterogeneous catalysts.10,23,36,37 The homogeneous ones generally display higher activity but suffer from complex and costly catalyst separation and recycling.7,22 This limitation is intrinsically overcome with heterogeneous catalysts, though this typically comes at the cost of lower catalytic activity due to worse accessibility for the active sites.37–42 Therefore, designing metal-free bifunctional catalysts that combine the advantages of homogeneous and heterogeneous catalysts while avoiding their limitations is a desirable yet challenging target.
In this work, we aimed at tackling this challenge by studying and optimising the use of rhodamine 6G (Rh6G) and rhodamine B (RhB) as organocatalysts for the cycloaddition of CO2 to epoxides yielding cyclic carbonates. Both compounds (Fig. 1) are well known for their application as dyes.43 We choose to investigate these compounds as they have several attractive features for the target application: (i) they contain a chloride anion that can act as a nucleophile and can be easily exchanged for other halides (bromide, iodide), thus allowing the catalytic behaviour to be tuned; (ii) they contain a bulky cation with the positive charge being delocalised over the xanthene-based core;44 (iii) they contain HBD groups (–COOH in RhB and –NH– in Rh6G); (iv) they have a considerable molecular mass (479 g mol−1), which is significantly higher than that of cyclic carbonates (e.g. 102 g mol−1 for propylene carbonate) and which is expected to enable their separation from the reaction mixture by nanofiltration;45 and (v) they are inexpensive and commercially available on a large scale. Additionally, in the case of RhB, tuning of the functional groups can be achieved through reaction of the carboxyl group through a simple, accessible protocol (Scheme 1). Designing and developing a catalyst that has all these features, and thus combining highly accessible active sites with easy separation, would represent a clear green advance in the conversion of CO2 into cyclic carbonates.
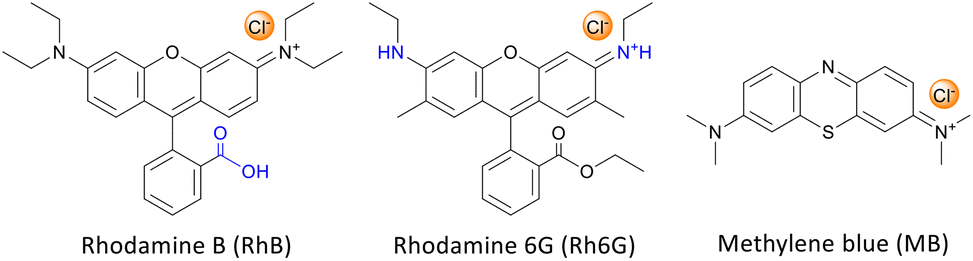 | ||
| Fig. 1 Dyes used in this work as organocatalysts for the cycloaddition of CO2 to epoxides. | ||
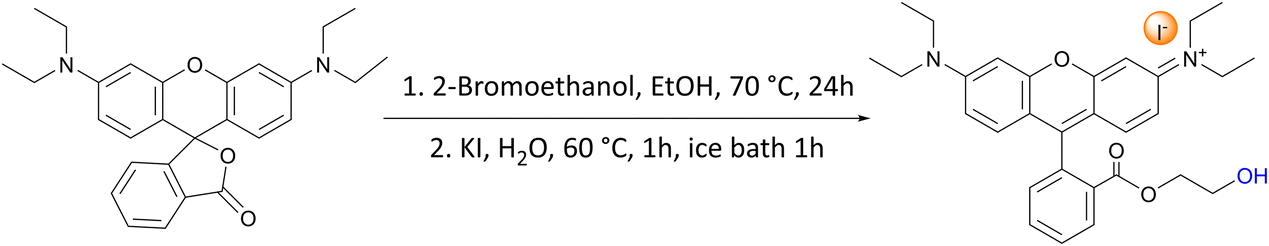 | ||
| Scheme 1 Synthesis of RhB-EtOH-I. | ||
Results and discussion
Evaluation of dyes as organocatalysts for the cycloaddition of CO2 to styrene oxide
In this study, rhodamine B (RhB) and rhodamine 6G (Rh6G) were investigated as metal-free bifunctional homogeneous catalysts for the cycloaddition of CO2 to epoxides leading to cyclic carbonates. Previous studies showed that these organocatalysts in their original chloride form showed only moderate activity in the conversion of CO2 into cyclic carbonates, and needed to be combined with organic bases or organic halide salts to achieve acceptable catalytic performance.46,47 Here, we demonstrate that the catalytic performance of RhB and Rh6G can be boosted dramatically through a series of tailored and yet straightforward modifications. The first, notable enhancement of the catalytic activity of RhB and Rh6G was achieved by ion exchange of the dye with either KBr or KI. This set of organocatalysts (RhB-X and Rh6G-X, with X = Cl, Br, I) was tested for the conversion of CO2 and styrene oxide into styrene carbonate under relatively mild conditions (80 °C, 10 bar CO2, 24 h). This was selected as the test reaction because styrene oxide is a rather challenging substrate for the cycloaddition reaction (compared for example to the often reported propylene oxide or epichlorohydrin).48 The performance of these organocatalysts was compared to that of methylene blue (MB) and its ion-exchanged derivatives (MB-X, with X = Cl, Br, I), as this dye is characterised by a smaller cation and does not contain HBD groups (Fig. 1). As shown in Table 1, the organocatalysts containing Cl− as an anion were rather inefficient towards the conversion of CO2 and styrene oxide into styrene carbonate (Table 1, entries 1–3). On the other hand, RhB-Br and MB-Br gave intermediate yields of cyclic carbonate product (entries 4 and 5) and all the organocatalysts with iodide as an anion achieved good catalytic activity (entries 7–9); the highest styrene carbonate yield (81%) was obtained with Rh6G-I. The selectivity towards the cyclic carbonate product was virtually complete in all these tests. The observed trend of increasing activity in the order of Cl− < Br− < I− was observed before for other catalysts active in the cycloaddition reaction and can be correlated to the leaving ability of the three halides, which followed the same order.15,23,49 This implies that with these catalysts the ring closure with the formation of the cyclic carbonate, during which the halide leaves the reaction intermediate (see Scheme S1†), is the rate-determining step of the reaction. However, the leaving ability of the halide acting as a nucleophile is not sufficient to explain the much lower activity of Rh6G-Br compared to RhB-Br and MB-Br (entries 4–6) while its counterpart in iodide form, Rh6G-I, shows higher activity than RhB-I and MB-I (entries 7–9). To understand this complex behaviour, it is necessary to take the solubility of the different (ion-exchanged) dyes in the reaction mixture into account (Table S1†). Incomplete solubility of the organocatalyst in the reaction mixture is undesirable as the fraction that remains undissolved would be much less accessible for the epoxide and CO2 and would thus contribute much less to the catalytic activity. Visual assessment of the solubility of each catalyst before the reaction, either at room temperature or at the reaction temperature (80 °C), and again at room temperature at the end of the catalytic test showed some general trends: (i) the solubility of all dyes increases in the presence of styrene carbonate and it is higher at the end of the reaction at room temperature than before the reaction, unless the carbonate yield is extremely low; (ii) the dyes in iodide form are the most soluble in the reaction mixture, followed by those in chloride and bromide forms; (iii) for each halide, RhB is the most soluble dye, followed by MB and Rh6G. These trends enable us to explain the observed low catalytic performance of Rh6G-Br, and also suggest that large differences in activity as a function of the nature of the halide are not only due to the trend in leaving ability discussed above but are also related to the solubility of the catalyst in the reaction mixture.|
|
|||
|---|---|---|---|
| Entry | Organocatalyst | Cyclic carbonate yielda [%] | Cyclic carbonate selectivitya [%] |
| Reaction conditions: styrene oxide (20 mmol), organocatalyst (1 mol% relative to the epoxide), o-xylene (1.5 mmol) as an internal standard, solvent (if used): propylene carbonate (PC), HBD (if used): H2O, 80 °C, 10 bar CO2, 24 h.a The yield and selectivity were determined by 1H NMR using o-xylene as internal standard. | |||
| 1 | MB-Cl | 5 | >99 |
| 2 | RhB-Cl | 4 | >99 |
| 3 | Rh6G-Cl | 6 | >99 |
| 4 | MB-Br | 33 | >99 |
| 5 | RhB-Br | 34 | >99 |
| 6 | Rh6G-Br | 4 | >99 |
| 7 | MB-I | 66 | >99 |
| 8 | RhB-I | 71 | >99 |
| 9 | Rh6G-I | 81 | >99 |

Another factor that is expected to influence the catalytic activity of organic halides is the nature of the cation, which in the case of the dyes studied in this work can have two effects: (i) the size and delocalisation of the positive charge over the cation affect the electrostatic interaction with the halide, thus determining its availability to act as a nucleophile;7,15,48 and (ii) the presence of functional groups that can contribute to the catalytic performance. Besides the –COOH group in RhB, which can act as an HBD to activate the epoxide towards nucleophilic attack by the halide,23 there are three types of N-containing groups in the dyes: (1) R2HN(+) groups (in Rh6G); (2) R3N(+) groups (in RhB and MB); and (3) N-heterocyclic group (in MB). Both type (1) and type (2) sites have partial iminium and partial amine character because the positive charge is delocalised over the organic cation (conventionally, one of the two groups is drawn as an amino group and the other as an iminium group – see Fig. 1).50 The type (1) groups in Rh6G can in principle act as hydrogen bond donors. However, the delocalisation of the positive charge implies an HBD behaviour that is intermediate between that of a secondary amine and that of an iminium group, with the latter being expected to be more effective as an HBD.51,52 The type (2) groups do not contain H atoms that can act as HBDs. On the other hand, this group could in principle activate CO2 as a base.53 However, the delocalisation of the positive charge implies that the basicity of these groups is weak.54 The type (3) group in MB is part of the thiazine ring located between the two aromatic rings. The resonance effect of the aromatic rings is expected to lead to a decrease in electron density in the heterocyclic ring and, as a consequence, to weak basicity for this group.52,54 The anticipated overall weak basicity of MB is demonstrated by the relatively high acid dissociation constant of the conjugate acid of MB (pKa = 3.8)55 compared to the much lower acidity of the conjugate acid of the bases that are known to activate CO2, such as DBU (conjugate acid pKa = 24.3) and DMAP (conjugate acid pKa = 18.0).56,57 Based on these considerations, type (2) and (3) groups in RhB and MB are not expected to behave as active sites for the CO2 cycloaddition reaction, whereas type (1) groups may act as mild HBDs. In order to evaluate the effect of the cation, the discussion can be focussed on the dyes in iodide form, for which the solubility in the reaction mixture is not a limiting factor (Table S1†). Among these organocatalysts, those containing a larger cation with an HBD group (Rh6G-I and RhB-I) displayed the expected higher activity compared to MB-I (Table 1, entries 7–9). However, the significantly higher styrene carbonate yield obtained with Rh6G-I compared to RhB-I suggests that the HBD group of the latter (–COOH) is relatively inefficient at promoting the cycloaddition reaction. This can be attributed to the distance of –COOH from the nitrogen atoms in RhB-I, close to which the iodide is more likely to be found. It has been shown that minimising the distance between the HBD group and the positively charged atom is vital for improving the catalytic activity of organocatalysts in the cycloaddition of CO2 to epoxides.34,58 In the RhB structure, the carboxyphenyl group is preferably oriented perpendicularly to the plane of the xanthene-based core,59 imposing a long distance between the –COOH group and the nitrogen atoms, and thus limiting its contribution as an HBD.
Optimisation of the catalytic activity of the dyes by adding a solvent
In order to overcome the poor solubility in the initial reaction medium of MB-X and Rh6G-X when X = Cl or Br (Table S1†), we utilised a previously reported strategy based on using a cyclic carbonate as a solvent.15 The choice of using a cyclic carbonate to increase the solubility of the dye in the reaction mixture is not only motivated by the high polarity, high boiling point and low or no toxicity of these compounds,8 but also by the fact that no separation is needed if the carbonate used is the same as the one produced during the reaction. Indeed, with several of the dyes tested, we observed that the solubility in the reaction mixture at room temperature was significantly higher at the end of the catalytic test (Table S1†), as a consequence of the formation of styrene carbonate. For practical reasons, we studied the effect of adding a cyclic carbonate as a solvent by using propylene carbonate (PC), due to its commercial availability. The effect of PC on the solubility and catalytic activity of MB-X and Rh6G-X was evaluated by gradually increasing its amount in the initial reaction solution (Fig. 2). In all cases, the addition of PC had a beneficial effect on the solubility of the dye, though the amount required differed (Table S1†). It was observed that, for each type of halide X (Cl or Br), MB-X was more soluble in the reaction mixture than Rh6G-X. Additionally, the dyes with X = Cl show higher solubility than their counterparts with X = Br (Table S1†). The enhanced solubility of the MB-X and Rh6G-X organocatalysts upon adding PC as a solvent resulted in the anticipated increase in activity (Fig. 2), with the improvement being most remarkable for MB-Cl, for which the yield of styrene carbonate increased from 5% without any PC to 61% when the reaction was carried out with 1.4 mL of PC as a solvent (Fig. 2). The optimum amount of PC used as a solvent differed between MB-Cl, MB-Br, Rh6G-Cl and Rh6G-Br, but in all cases the catalytic performance ultimately decreased when a larger amount of PC was used, most likely due to reduced contact between the catalyst and substrate caused by dilution of the reaction mixture.15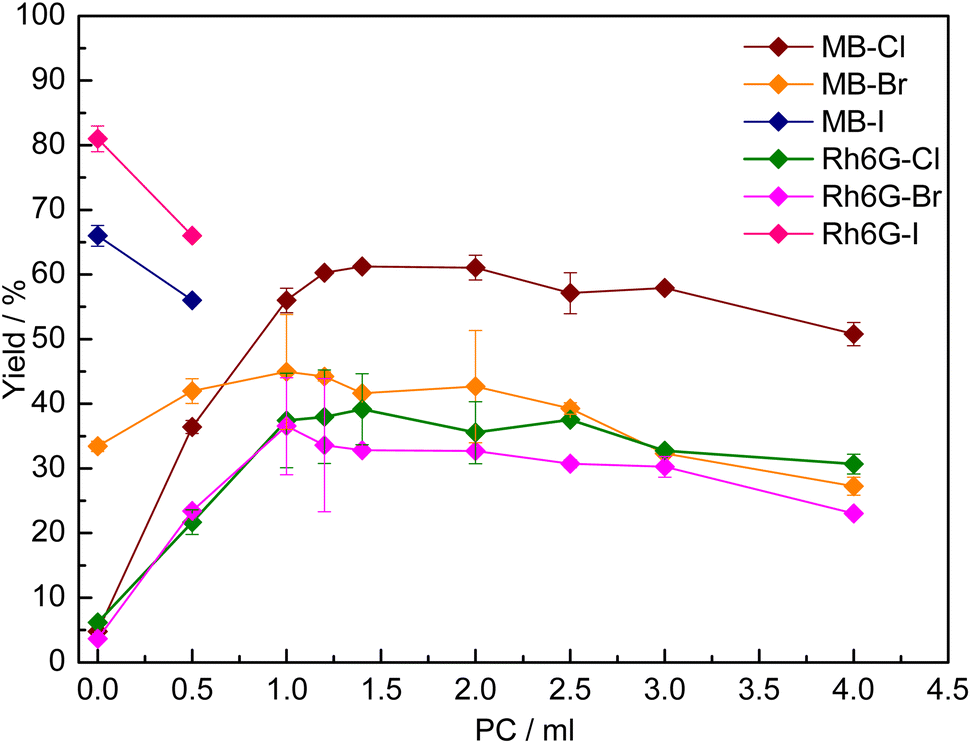 | ||
| Fig. 2 Effect of the use of propylene carbonate (PC) as a solvent on the catalytic activity of MB-X and Rh6G-X (X = Cl, Br, I) in the reaction of CO2 with styrene oxide. Reaction conditions: styrene oxide (20 mmol), organocatalyst (1 mol% relative to the epoxide), o-xylene (1.5 mmol) as internal standard, 80 °C, 10 bar CO2, 24 h. | ||
On the other hand, in the case of MB-I and Rh6G-I, the addition of even a small amount of PC as a solvent (0.5 mL) had a detrimental effect on the catalytic activity (Fig. 2). This can be rationalised by considering that these dyes were already rather soluble in the initial reaction mixture (Table S1†). Therefore, in this case, the addition of a solvent was not beneficial and caused only an undesired dilution of the reaction mixture.
Optimisation of the catalytic activity of the dyes by adding H2O as an HBD
With the purpose of further improving the catalytic activity of MB-X, RhB-X and Rh6G-X (X = Cl, Br, I) in the CO2 cycloaddition reaction, we investigated the effect of adding H2O as a green, inexpensive HBD. In the case of MB-X and Rh6G-X with X = Cl or Br, we also used 1 mL of PC as a solvent to improve the solubility of the dye in the reaction mixture (vide supra). When employing 50 mg H2O as an HBD, the styrene carbonate yield was significantly improved with all the dye organocatalysts with the exception of MB-Cl (Fig. 3). Remarkably, the use of H2O as an HBD allowed almost complete conversion of styrene oxide to be achieved with the dyes having iodide as the anion (92–96% yield for MB-I, RhB-I, Rh6G-I). The selectivity towards styrene carbonate was slightly decreased by the presence of H2O due to the hydrolysis of styrene oxide into the corresponding styrene diol (1-phenyl-1,2-ethanol).15 However, the formation of this side product was minimal (96% yield of styrene carbonate with 97% selectivity when using the optimum catalyst, Rh6G-I). The only dye for which the addition of H2O as an HBD was detrimental to the catalytic activity was MB-Cl. This drop in activity is attributed to the polar protic nature of H2O, which implies that the partially positive charge on its H atoms lead to ion–dipole interactions with the halides, thus causing a shielding effect that deceases the nucleophilicity of the halide.15,60 The magnitude of this effect diminishes by decreasing the strength of the base (Cl− > Br− > I−) and by increasing the size of the anion (Cl− < Br− < I−).15,61 Therefore, the addition of water is beneficial if its effect as an HBD is more substantial than the shielding effect, and this should happen in the order of I− > Br− > Cl−.15,23,34,49 In this work, this is the case for dyes that contain iodide and bromide anions. For the dyes with chloride, the shielding effect becomes the dominant factor for MB-Cl but not for RhB-Cl and Rh6G-Cl. The difference in behaviour between the three dyes with chloride anions is attributed to the presence of HBD groups in the structures of RhB and Rh6G, which can also interact with the partially negative charge on the O atoms of H2O through hydrogen bonding62 and thus mitigate the shielding effect.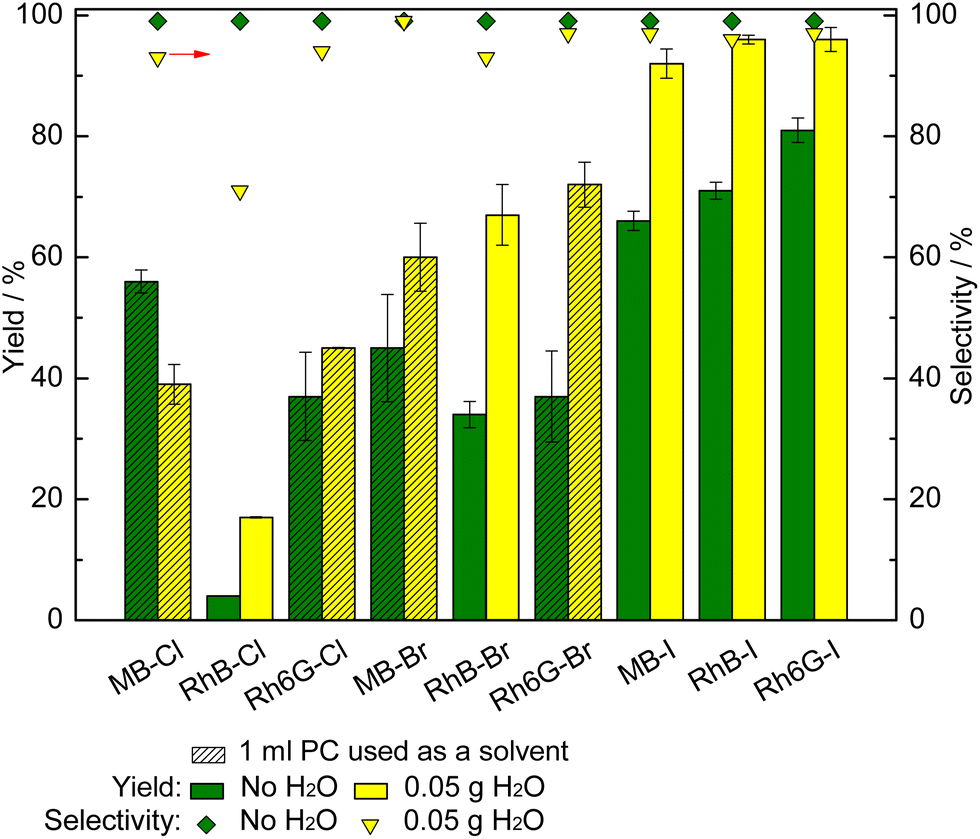 | ||
| Fig. 3 Effect of using H2O as an HBD on the catalytic activity of (ion-exchanged) dyes as organocatalysts for the conversion of styrene oxide and CO2 into styrene carbonate. Reaction conditions: styrene oxide (20 mmol), organic catalyst (1 mol% relative to the epoxide), o-xylene (1.5 mmol) as internal standard, 80 °C, 10 bar CO2, 24 h. | ||
Ion–dipole interactions are also expected to take place between the HBD groups of RhB-X and Rh6G-X and the halide, with their magnitude being a function of the strength of the base (Cl− > Br− > I−).63 Such intramolecular interactions can decrease the nucleophilicity of the halide and thus negatively affect the catalytic activity.64 This can explain why MB-X, which does not contain HBD groups, is more active than RhB-X and Rh6G-X in the experiments without the addition of H2O when X = Cl and, to a lesser extent, when X = Br, while the trend is inverted when X = I (Fig. 3, green bars).
With the dyes that proved to be the most active organocatalysts, i.e. those carrying iodide anions (MB-I, RhB-I, Rh6G-I), we studied the effect of decreasing the catalyst loading to 0.5 mol% (relative to the styrene oxide) in the presence of 50 mg H2O in the cycloaddition of CO2 to styrene oxide (Fig. 4). The results showed that in the presence of H2O as an HBD, similar or slightly higher catalytic activity was obtained compared with the same dyes employed with a double loading (1 mol%) but without H2O. This demonstrates that the amount of organocatalyst can be largely reduced by the use of H2O as an HBD co-catalyst.
 | ||
| Fig. 4 Effect of using H2O as an HBD on the catalytic activity of dye-I (dye = MB, RhB, Rh6G) combined with a lower catalyst loading relative to the epoxide (0.5 mol% in tests with H2O, 1 mol% in tests without H2O). Reaction conditions: styrene oxide (20 mmol), o-xylene (1.5 mmol) as internal standard, 80 °C, 10 bar CO2, 24 h. | ||
Enhancement of the catalytic activity by functionalisation of RhB-I
Once the reaction conditions had been optimised and the most promising dye organocatalysts had been identified (MB-I, RhB-I and Rh6G-I), we continued our study by investigating their performance in the cycloaddition of CO2 to styrene oxide at a milder reaction temperature and slightly shorter reaction time (45 °C, 10 bar CO2, 18 h). Under these conditions, the catalytic activity was markedly lower compared to the tests performed at 80 °C and only low yields of the styrene carbonate product were achieved, with a maximum of 11% with Rh6G-I (Table 2, entries 1–3). Additionally, in all these reactions, a nearly constant yield of the styrene diol by-product was obtained (2–3%), which suggests that the hydrolysis of styrene oxide depends on the presence of H2O and is catalysed by the carbonic acid generated by the dissolution of CO2 in the reaction mixture but is largely independent of the dye. As a consequence of the formation of the diol, the selectivity towards styrene carbonate decreased with its yield (Table 2, entries 1–3). In line with what was reported above, adding propylene carbonate as a solvent in the reaction catalysed by MB-I or Rh6G-I did not improve the yield of styrene carbonate (Table 2, entries 4 and 5). However, it did suppress to a large extent the formation of the diol by-product and thus led to much higher selectivity towards styrene carbonate, probably as a consequence of dilution of the carbonic acid that catalysed the hydrolysis reaction.|
|
|||||
|---|---|---|---|---|---|
| Entry | Organocatalyst | Yielda [%] | Selectivitya [%] | PC as solvent [mL] | H2O [mg] |
| Reaction conditions: styrene oxide (20 mmol), organocatalyst (1 mol% relative to the epoxide), o-xylene (1.5 mmol) as an internal standard, solvent (if used): propylene carbonate (PC), HBD (if used): H2O, 45 °C, 10 bar CO2, 18 h.a The yield and selectivity were determined by 1H NMR using o-xylene as internal standard.b Taken from ref. 15.c Reaction conditions: styrene oxide (60 mmol), organocatalyst (1 mol% relative to the epoxide), o-xylene (4.7 mmol) as internal standard, 45 °C, 10 bar CO2, 18 h.d The value in brackets is the isolated yield of styrene carbonate obtained by column separation.e 60 °C, CO2 balloon, 24 h, avoiding exposure to light.f 60 °C, CO2 balloon, 24 h, under visible light irradiation (wavelength range: 400–700 nm). | |||||
| 1 | MB-I | 1 | 18 | 0 | 0.05 |
| 2 | RhB-I | 7 | 67 | 0 | 0.05 |
| 3 | Rh6G-I | 11 | 77 | 0 | 0.05 |
| 4 | MB-I | 7 | >99 | 3.9 | 0.05 |
| 5 | Rh6G-I | 10 | >99 | 2.0 | 0.05 |
| 6 | RhB-EtOH-I | 29 | >99 | 0 | 0 |
| 7 | RhB-EtOH-I | 32 | 93 | 0 | 0.05 |
| 8 | RhB-EtOH-I | 34 | 88 | 0 | 0.10 |
| 9b | Bu4NI | 13 | ≥99 | 0 | 0 |
| 10b | PPNI | 20 | ≥99 | 0 | 0 |
| 11c | RhB-EtOH-I | 34 (29)d | >99 | 0 | 0 |
| 12e | RhB-EtOH-I | 14 | >99 | 0.5 | 0.05 |
| 13f | RhB-EtOH-I | 11 | >99 | 0.5 | 0.05 |
These results prompted us to envisage a strategy to further improve the catalytic activity of these dye organocatalysts. For this purpose, we introduced a tailored functionalisation of RhB-I. The –COOH group present in RhB proved to be inefficient as an HBD for activating styrene oxide, most likely due to the large distance between this group and the nitrogen atoms close to which the iodide nucleophile was expected to be found (vide supra). Based on this hypothesis, we decided to modify RhB-I by introducing a terminal ethanol moiety by the facile reaction of the carboxyl group of RhB-I with 2-bromoethanol (Scheme 1). In this way, we aimed at minimising the distance between the HBD group (–OH) and the nucleophile.34,58 Our strategy proved successful and the modified dye (RhB-EtOH-I) achieved a major improvement in the yield of styrene carbonate (from 7 to 29%, compare entries 2 and 6 in Table 2), with virtually complete selectivity (>99%). This result is significantly superior to the activity of benchmark commercial organocatalysts, namely, Bu4NI or PPNI (Table 2, entries 9 and 10).15 An additional advantage of having an HBD as a functional group within the dye compared to the use of H2O as an HBD is that the formation of the diol side product is prevented, leading to the observed full selectivity towards styrene carbonate (Table 2). Moreover, since RhB-EtOH-I already contains an effective HBD, the addition of H2O only led to a minor increase in the catalytic activity but resulted in a lower selectivity towards styrene carbonate due to the formation of the diol (Table 2, entries 7 and 8). It is worth noting that RhB-EtOH-I was fully soluble in the styrene oxide reaction medium even at room temperature (Table S2†), and this probably contributed to its high activity in the cycloaddition reaction with CO2. Next, we demonstrated that the synthesis of styrene carbonate using the RhB-EtOH-I catalyst could be achieved with a similar yet slightly higher yield in a three-fold scale-up (Table 2, entry 11). The formed styrene carbonate product was purified by column separation with n-hexane/ethyl acetate (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as the eluent, achieving an isolated yield of 29% (which was only slightly lower than the 34% yield calculated based on analysis of the crude reaction mixture). 1H NMR and 13C NMR spectra (see Fig. S21 and S22†) demonstrated the high purity of the collected solid styrene carbonate.
:1) as the eluent, achieving an isolated yield of 29% (which was only slightly lower than the 34% yield calculated based on analysis of the crude reaction mixture). 1H NMR and 13C NMR spectra (see Fig. S21 and S22†) demonstrated the high purity of the collected solid styrene carbonate.
The performance of our optimum catalyst (RhB-EtOH-I) was compared to state-of-the-art bifunctional homogeneous organocatalysts, including phosphonium,35,58,65 imidazolium64,66–68 and ammonium salts,33,34,69–73 and to selected halide-free catalysts,74–78 of which some are (partially) bio-based compounds (see Table S4†). Although a fully meaningful comparison is hindered by the different conditions under which these organocatalysts have been tested in the literature, it can be concluded that our RhB-EtOH-I is fully competitive with the state-of-the-art catalysts, and is able to achieve good cyclic carbonate yields with a relatively low catalyst loading (1 mol% relative to the epoxide) at significantly milder temperatures compared to those employed for most of the organocatalysts in the literature. When compared to the catalysts that were tested at similar temperatures (45 and 60 °C), RhB-EtOH-I displayed a similar performance with only a slightly lower TON and/or TOF compared to the most active catalysts, which however have the drawback of being based on phosphonium salts and, therefore, are less desirable in terms of greenness as their synthesis involves toxic organophosphines.16,17
Investigation of the effect of light on the catalytic activity of the dyes
The cycloaddition of CO2 to styrene oxide has also been reported to occur using photocatalysts under visible light irradiation.79 Multiple studies claimed that photocatalytic materials, such as porphyrin-based MOFs, can promote the conversion of CO2 and epoxides into cyclic carbonates at room temperature when irradiated with a visible light source (e.g. a Xe or LED lamp).80–82 However, the temperature increase in the photoreactor caused by the irradiation was often not investigated, leaving doubts about whether the CO2 cycloaddition was thermally or phototriggered.83 A few studies critically evaluated the temperature of the photocatalytic system and performed blank CO2 cycloaddition reaction tests under the system temperature without light irradiation,84 or stabilised the system temperature around room temperature by conducting the photocatalytic tests in a water bath.85 In this way, for specific photocatalytic systems, the influence of temperature was accounted for and the promoting effect of visible light irradiation on the CO2 cycloaddition reaction was demonstrated. Since dyes absorb light in the visible range and can act as sensitisers,86 we investigated a possible photocatalytic contribution to their activity by carrying out our test reactions in a photoreactor (Fig. S3†) under irradiation with visible light using our optimum catalyst, RhB-EtOH-I. The parent RhB has visible light absorption peaking at 554 nm, and by irradiation of a RhB solution with visible light (wavelengths longer than 400 nm), the photoactivated RhB can produce singlet and triplet excited states.87 RhB-EtOH-I has a similar visible light absorption spectrum to that of RhB (Fig. S4†), and yet displays significantly higher activity than the latter in the CO2 cycloaddition reaction (Table 2, entry 2 vs. entry 6), which suggests that the observed activity is not of photocatalytic origin. Indeed, the yield of styrene carbonate obtained with RhB-EtOH-I under visible light irradiation did not show any improvement compared to that obtained under the same conditions (including constant T = 60 °C) but without exposure to visible light (Table 2, entries 12 and 13). Therefore, we can conclude that there is no photochemical contribution to the observed activity of RhB-EtOH-I in the reaction of CO2 with styrene oxide yielding styrene carbonate.Versatility of dye organocatalysts with different epoxide substrates
The dye organocatalysts with the most promising activity in the cycloaddition of CO2 to styrene oxide (RhB-I, Rh6G-I and RhB-EtOH-I) were investigated further by screening their performance with a broad scope of epoxides. This study of the versatility of the organocatalysts was carried out with the assistance of 50 mg H2O as an HBD (Table 3). In the cases in which the dye organocatalyst was not fully soluble in the reaction mixture at room temperature (Table S3†), a minimum amount of propylene carbonate (Table 3) was used as a solvent to optimise the solubility. However, this was not beneficial for the tests with propylene oxide, and in such cases the results without additional solvent were reported. The reaction temperature was 60 °C and the initial CO2 pressure was 10 bar in most of the tests, apart from those with the more challenging epoxides (1,2-epoxyhexane, cyclohexene oxide and limonene oxide), for which harsher conditions were employed (Table 3). In general, RhB-EtOH-I was the most active of the three dye organocatalysts, achieving good to excellent cyclic carbonate yields (from 50 to 92%, Table 3). The only substrate for which poor results were achieved is limonene oxide, a sterically hindered internal epoxide that is well-known for being recalcitrant to conversion into the corresponding cyclic carbonate;88,89 negligible conversion of this compound was observed in all tests despite the harsher conditions that were employed (entries 19–21). Notably, RhB-EtOH-I achieved very high propylene carbonate yield (92%, see entry 3) although it was not fully soluble in the reaction mixture even at the end of the test (Table S3†). This implies that the activity might still be improved by enhancing the solubility of the catalyst. Some general trends were observed based on the inherent steric and electronic characteristics of the epoxides.48 Low steric hindrance around the epoxide ring facilitates its nucleophilic attack by the halide anion and the consequent ring-opening step that initiates the cycloaddition reaction (Scheme S1†),28,64 thus explaining the high conversion and cyclic carbonate yield achieved with propylene oxide, especially with Rh6G-I and RhB-EtOH-I (entries 2 and 3). Electron-withdrawing species, such as the chlorine in epichlorohydrin or the oxygen atom in allyl glycidyl ether, also promote nucleophilic attack on the epoxide ring, thus explaining the high conversion and cyclic carbonate yield obtained with these substrates (entries 4–9). Cyclohexene oxide is an internal epoxide substrate for which the conversion into the cyclic carbonate is further hindered by the geometric strain created by the two connected rings in cyclohexene carbonate.15,90 Therefore, harsh reaction conditions (120 °C, 10 bar CO2, 18 h) were necessary to achieve good yields of this cyclic carbonate (entries 16–18). Notably, with cyclohexene oxide the highest carbonate yield (65%) was obtained with Rh6G-I, possibly as a consequence of the higher temperature used in these tests, which increased the otherwise rather low solubility of this dye in the reaction mixture (Table S3†). With all the epoxides tested, the undesired hydrolysis side reaction leading to the conversion of the epoxide into the corresponding diol was limited to less than 1% yield, implying that the selectivity towards the target cyclic carbonate product was ≥98% in all the tests in which the conversion was >30%.| Entry | Catalyst | Epoxide | Cyclic carbonate product | T [°C] | PC as solventa [mL] | Yieldb [%] | Selectivityb [%] |
|---|---|---|---|---|---|---|---|
| Reaction conditions: epoxide (20 mmol), organocatalyst (1 mol% relative to the epoxide), o-xylene (1.5 mmol) as an internal standard, 50 mg H2O as a HBD, solvent (if used): propylene carbonate (PC), 60 °C, 10 bar CO2, 24 h.a The minimum amount of PC required to dissolve the catalyst was used in each test.b The yield and selectivity were determined by 1H NMR using o-xylene as internal standard.c 80 °C, 10 bar CO2, 24 h.d 120 °C, 30 bar CO2, 18 h. | |||||||
| 1 | RhB-I |
|
|
60 | 0 | 59 | 98 |
| 2 | Rh6G-I | 60 | 0 | 87 | ≥99 | ||
| 3 | RhB-EtOH-I | 60 | 0 | 92 | ≥99 | ||
| 4 | RhB-I |
|
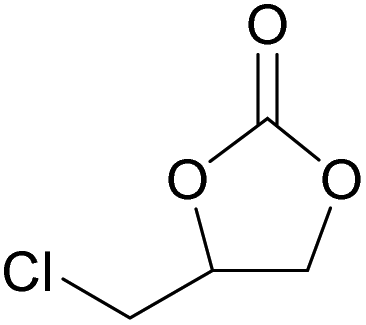
|
60 | 0 | 86 | ≥99 |
| 5 | Rh6G-I | 60 | 1.0 | 86 | ≥99 | ||
| 6 | RhB-EtOH-I | 60 | 0 | 86 | ≥99 | ||
| 7 | RhB-I |
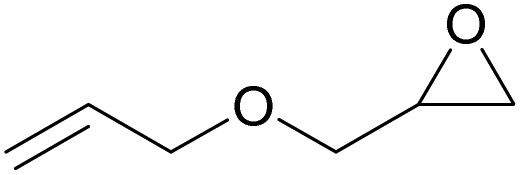
|
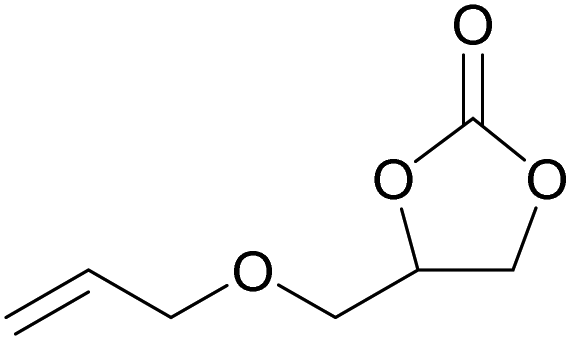
|
60 | 0 | 30 | ≥99 |
| 8 | Rh6G-I | 60 | 0.5 | 74 | ≥99 | ||
| 9 | RhB-EtOH-I | 60 | 0 | 87 | ≥99 | ||
| 10 | RhB-I |
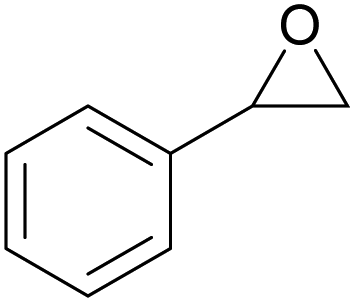
|
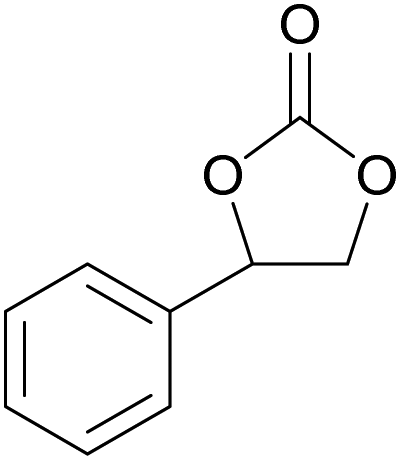
|
60 | 0 | 28 | 94 |
| 11 | Rh6G-I | 60 | 0.5 | 48 | ≥99 | ||
| 12 | RhB EtOH-I | 60 | 0.5 | 54 | ≥99 | ||
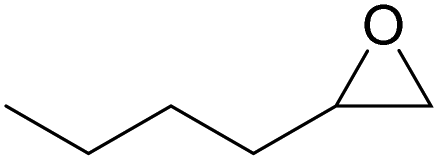
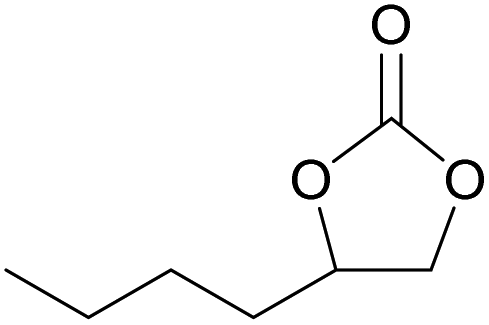
| 13c | RhB-I |
|
|
80 | 1.0 | 47 | ≥99 |
| 14c | Rh6G-I | 80 | 0.7 | 45 | ≥99 | ||
| 15 | RhB-EtOH-I | 60 | 0.5 | 52 | ≥99 | ||
| 16d | RhB-I |
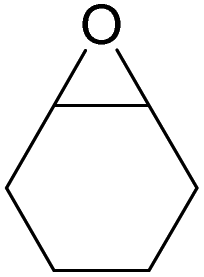
|
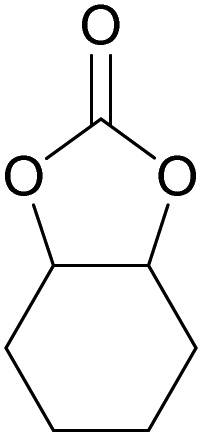
|
120 | 0.5 | 28 | ≥99 |
| 17d | Rh6G-I | 120 | 0.5 | 65 | ≥99 | ||
| 18d | RhB-EtOH-I | 120 | 0.5 | 50 | ≥99 | ||
| 19d | RhB-I |
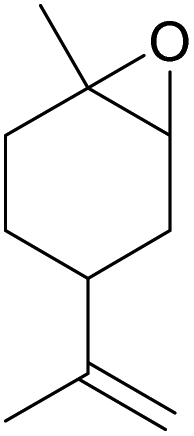
|
|
120 | 0.5 | <1 | ≥99 |
| 20d | Rh6G-I | 120 | 0.5 | <1 | ≥99 | ||
| 21d | RhB-EtOH-I | 120 | 0.5 | <1 | ≥99 | ||
Recovery and reuse of the dye organocatalyst
To assess the stability and reusability of the RhB-EtOH-I organocatalyst, we conducted a test using propylene oxide as a substrate under our standard reaction conditions (60 °C, 10 bar CO2, 24 h), achieving a propylene carbonate yield of 59% (see Fig. S1†). The dye organocatalyst could then be readily recovered from the reaction solution by precipitation using diethyl ether as an antisolvent. The latter is a volatile compound (b.p. 35 °C) that can be easily removed from the high boiling cyclic carbonates by distillation and reused. With this approach, RhB-EtOH-I was recovered in a yield of 64%, and the integrity of the compound was demonstrated by 1H and 13C NMR analysis (see Fig. S23–S26†). The separated organocatalyst solid was nearly pure, with a practically negligible fraction of propylene carbonate (0.8 wt% relative to RhB-EtOH-I). The recycled RhB-EtOH-I achieved the same yield (59%) and selectivity (>99%) towards propylene carbonate as the fresh catalyst (see Fig. S1†), thus demonstrating that its performance was fully retained upon recycling. The cumulative TON over the two catalytic runs was 119 (see Fig. S1†). Although this approach proved to be efficient at recovering and reusing the RhB-EtOH-I organocatalyst, the propylene carbonate obtained after removing diethyl ether still contained some RhB-EtOH-I, as indicated by its light purple colour. This might represent a limitation for the practical application of the cyclic carbonate product. To overcome this issue, we chose to explore the use of nanofiltration91 as a sustainable separation technique to purify cyclic carbonates. The choice of this strategy stems from the fact that nanofiltration techniques have been reported to be efficient at removing dyes during water treatment,92,93 and from the availability of a commercial nanofiltration membrane with a suitable cut-off size (400 Dalton). This is expected to enable the target separation by taking advantage of the large size of the dyes we used as catalysts (MM = 615 g mol−1 for RhB-EtOH-I, MM = 479 g mol−1 for RhB) compared to that of the cyclic carbonate products (MM = 102 g mol−1 for propylene carbonate, MM = 164 g mol−1 for styrene carbonate). The nanofiltration experiments were conducted with an aqueous solution containing RhB and propylene carbonate to simulate the reaction mixture at the end of our catalytic experiments. RhB was used in these tests as this dye is commercially available, is soluble in the aqueous medium in which the nanofiltration membrane operates (while RhB-EtOH-I has poor solubility in water), and has a lower molecular mass than our optimum catalyst, implying that if the separation is efficient with RhB it will work at least equally well with RhB-EtOH-I. In the nanofiltration setup (Fig. 5), a hollow fibre membrane with a cut-off size of 400 Da was installed, which allowed propylene carbonate to permeate through the membrane and RhB to be rejected and thus remain in the concentrated feedstock. This approach proved very successful: 99% of RhB remained in the concentrated feedstock (Table 4), with only a very minor fraction permeating through the nanofiltration membrane, resulting in a light pink aqueous solution in the permeate with less than 5 ppm RhB (in contrast to the dark purple colour of the feedstock, see Fig. S2†). The concentration of propylene carbonate in the permeate was nearly the same as in the feedstock (≥95% permeation, see Table 4), demonstrating that the majority of propylene carbonate could be recovered. In order to achieve an even higher degree of removal of the dye from the permeate containing propylene carbonate, we conducted a second nanofiltration test with an aqueous solution containing 5 ppm RhB, i.e. mimicking the concentration of the permeate obtained in the first nanofiltration test. This allowed us to achieve virtually complete removal of RhB from the permeate (≤0.1 ppm, see Table 5), which ensured a colourless permeate (Fig. S2†). This demonstrates that by combining two consecutive nanofiltration steps, a very high efficiency in the separation of the dye organocatalyst can be achieved. Notably, the permeate had a high flux of 11.62 L m−2 h−1, indicating the potential for achieving high output in the purification of the cyclic carbonate by nanofiltration. Finally, we investigated if the dye remained stable throughout the separation process by nanofiltration. For this purpose, we analysed the samples before and after the nanofiltration experiment by UV-vis spectroscopy. The position of the characteristic band of RhB in the visible range (λmax = 555 nm, Fig. S5†) was the same in all cases, indicating that RhB remained intact during the nanofiltration process.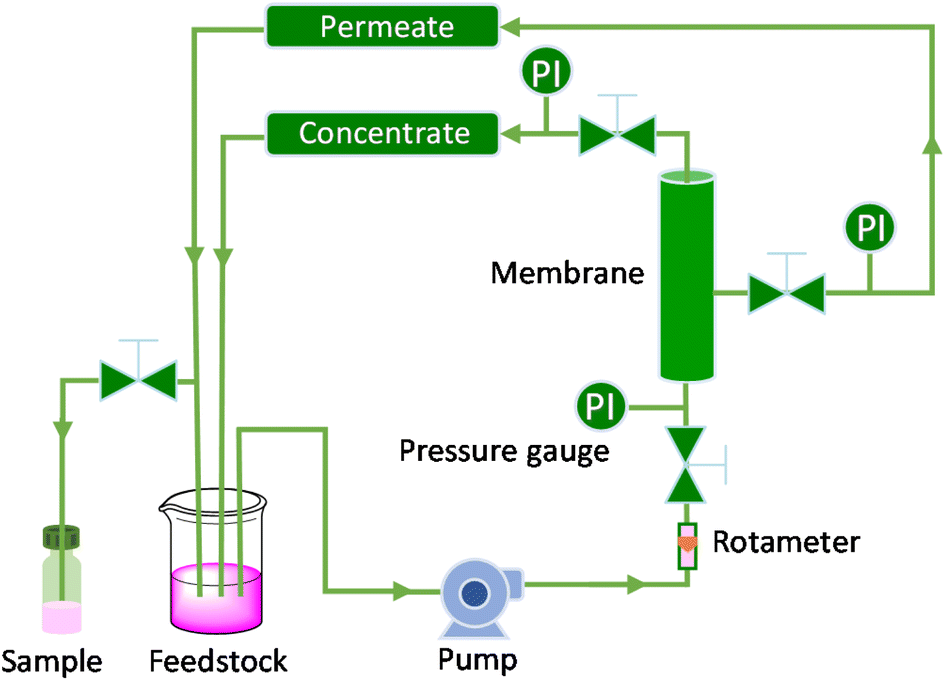 | ||
| Fig. 5 Scheme of the nanofiltration setup employed for the separation of the dye catalyst (PI = pressure indicator). | ||
| Time [min] | Feedstock samples | Permeate samples | Rejection [%] | Permeation [%] | ||
|---|---|---|---|---|---|---|
| [RhB] [ppm] | [PC] [mmol L−1] | [RhB] [ppm] | [PC] [mmol L−1] | |||
| Conditions: rhodamine B 0.51 g, propylene carbonate 10.01 g, H2O 1 L, pressure 1 barg, feed flow rate 50 L h−1. | ||||||
| 0 | 419 | 76 | — | — | — | — |
| 60 | 425 | 77 | 3 | 73 | 99 | 95 |
| 90 | 419 | 73 | 3 | 70 | 99 | 96 |
| 120 | 413 | 74 | 4 | 72 | 99 | 97 |
| 150 | 404 | 74 | 4 | 72 | 99 | 97 |
| Time [min] | [RhB]feed [ppm] | [RhB]permeate [ppm] | Rejection [%] |
|---|---|---|---|
| Conditions: rhodamine B 0.005 g, propylene carbonate 0.14 g, H2O 1 L, pressure 1 barg, feed flow rate 50 L h−1. | |||
| 0 | 5.1 | 5.1 | - |
| 60 | 6.1 | 0.1 | 98 |
| 90 | 6.4 | 0.1 | 98 |
| 120 | 6.7 | 0.1 | 98 |
| 150 | 7.0 | 0.1 | 99 |
By combining the results obtained by precipitation and nanofiltration, we demonstrated that the RhB-based organocatalysts can be recovered and reused without loss of activity and that cyclic carbonate with a very high degree of purity can be isolated. However, the degree of recovery of the RhB-EtOH-I catalyst is not yet complete. Further work can aim at improving this step either by tuning the solvent used for the precipitation or by optimising the nanofiltration approach to enable operating directly with the reaction mixture (the membrane used in this work requires an aqueous environment). The last option would require an investigation of the compatibility of the nanofiltration membrane with cyclic carbonates and, if this proved not to be feasible, exploring alternative nanofiltration membranes that could operate with our reaction mixture. Furthermore, it will be necessary to optimise the nanofiltration conditions, for example by recirculating the solution containing the catalyst and by increasing the pressure to decrease the volume and thus increase the concentration of the catalyst in the solution that is not permeated. The target would be to obtain a supersaturated solution of the catalyst, from which the compound would precipitate spontaneously, allowing its recovery. If the above-mentioned points are effectively tackled, the recovery of the catalyst and the purification of the cyclic carbonate product would become possible using exclusively nanofiltration technology.
Conclusions
In this work, we showed that tailored yet straightforward modifications of commercial dyes enabled us to dramatically boost their performance as organocatalysts for the cycloaddition of CO2 to epoxides to produce cyclic carbonates. First, we increased the activity of inexpensive and commercially available dyes (RhB, Rh6G, MB, all containing chloride anions) in the cycloaddition of CO2 to styrene oxide (80 °C, 10 bar CO2, 24 h) by means of ion exchange with KX (X = Br, I). The dye organocatalysts containing I− as the anion showed the highest catalytic activity as a consequence of the better leaving ability of iodide compared to bromide and chloride and of the worse solubility of the organocatalysts containing Cl− and Br−. The latter issue was tackled by adding a cyclic carbonate as a green solvent, which led to a significantly higher styrene carbonate yield. A further improvement of the catalytic performance was achieved by using H2O as a green and inexpensive hydrogen bond donor (HBD) to activate the epoxide towards nucleophilic attack by the iodide anion. The combination of all these strategies enabled excellent styrene carbonate yields to be reached in the catalytic tests at 80 °C (96% with RhB-I and Rh6G-I in the presence of 50 mg H2O). However, the performance of these organocatalysts at a lower temperature (45 °C) was less satisfactory, with only rather low styrene carbonate yields (maximum of 11% with Rh6G-I). In order to achieve a good performance under these mild conditions, we functionalised RhB-I by the straightforward reaction of its carboxyl group with 2-bromoethanol. The obtained bifunctional RhB-EtOH-I catalyst was characterised by a terminal ethanol moiety that was estimated to be located in close proximity to the iodide nucleophile, and this feature was anticipated to enhance its effectiveness as an HBD for promoting the catalytic reaction. The approach proved very successful and RhB-EtOH-I displayed a much higher styrene carbonate yield at 45 °C (29%) compared to the other dye organocatalysts, without requiring the addition of water as an HBD. The obtained yield is also significantly higher compared to that obtained with benchmark organocatalysts such as Bu4NI (13%) and PPNI (20%) under the same reaction conditions. Moreover, the synthesised RhB-EtOH-I organocatalyst showed good to excellent cyclic carbonate yields with a broad scope of epoxide substrates including the challenging cyclohexene oxide. Finally, we demonstrated that the RhB-EtOH-I catalyst could be easily recovered by precipitation with diethyl ether and reused without any loss of catalytic activity. Purification of the cyclic carbonate product from the dye organocatalyst was achieved by nanofiltration. By means of two consecutive nanofiltration steps, it was possible to attain virtually complete removal of the dye (>99.98%) with excellent permeation of the cyclic carbonate (≥95%). In summary, we introduced novel dye organocatalysts with highly promising activity and selectivity in the cycloaddition of CO2 to different epoxides yielding the corresponding cyclic carbonates under mild reaction conditions. Besides the relevance of the high activity and selectivity in an important reaction in the context of green chemistry, namely, the conversion of CO2 into cyclic carbonates, these catalysts display several additional attractive green features: they are metal free; they can be easily prepared from inexpensive, commercially available dyes; they are homogeneous catalysts and yet can be efficiently separated from the cyclic carbonate products and reused.Experimental section
Materials
Rhodamine 6G (Rh6G, 95% purity), rhodamine B (RhB, 95% purity), rhodamine B base (RhB base, 97% purity), methylene blue (MB), styrene oxide (97%), allyl glycidyl ether (99.9%), epichlorohydrin (≥99%), 1,2-epoxyhexane (97%), cyclohexene oxide (98%), (+)-limonene oxide (mixture of cis and trans, 97%), propylene carbonate (99.7% purity), magnesium sulphate (MgSO4, ≥99.5%), potassium iodide (KI, ≥99%), imidazole (≥99%), and deuterated solvents (CDCl3, CD3OD, and D2O used as solvents for NMR spectroscopy) were purchased from Sigma-Aldrich. Diethyl ether (AR grade), ethanol (AR grade) and ethyl acetate (AR grade) were obtained from Biosolve chemicals. Dichloromethane (AR grade) and n-hexane (99%) were purchased from Macron fine chemicals. 2-Bromoethanol (96%), propylene oxide (99.5%), and potassium bromide (KBr, ≥99%) were obtained from Fisher Scientific. Acetonitrile (99.9%) and o-xylene (≥99%) were purchased from Honeywell. Sodium bromide (NaBr, ≥99%) was obtained from ThermoFisher Scientific. All the chemicals were used without further purification.Characterisation of the modified dyes
Analysis of the modified dyes (see below) by NMR spectroscopy was carried out on a Varian Mercury Plus 400 MHz or Agilent MR 400 MHz apparatus. The NMR spectra were referenced using residual solvent peaks. Several techniques were employed: 1H NMR, 13C NMR, 1H-13C-HSQC (heteronuclear single quantum coherence), 1H-13C-HMBC (heteronuclear multiple bond correlation). Elemental analysis for H, C, N and S was carried out on a Vario Micro Cube elemental analyser.Ion exchange of the dyes
For a typical ion-exchange experiment, 2.01 g dye (4.18 mmol of RhB and Rh6G, 6.25 mmol of MB) was dissolved in either 130 mL (RhB) or 150 mL of deionised water (Rh6G and MB) and then mixed with a three-fold molar excess of KBr or KI pre-dissolved in 3–5 mL deionised water under stirring. A precipitate of the ion-exchanged dye started to form while adding the potassium halide solution. The mixture was stirred at 60 °C for 1 h and then placed in an ice bath for 1 h. The solid was separated from the supernatant by Büchner filtration, after which the solid product was washed with ca. 1 L deionised cold water. The obtained product was dissolved in the minimum amount of acetonitrile (150 to 600 mL) and dried with magnesium sulphate. The solid drying agent was removed by centrifugation, and the solvent was removed by reduced-pressure distillation to recover the product. The product (RhB-X, Rh6G-X, MB-X, with X = Br or I) was dried in a vacuum oven at 80 °C overnight. RhB-Br: 1.02 g, 47% yield. Rh6G-Br: 0.77 g, 35% yield, MB-Br: 0.62 g, 26% yield, RhB-I: 1.20 g, 50% yield, Rh6G-I; 1.73 g, 71% yield, MB-I: 0.90 g, 34% yield. The elemental composition of the exchanged dyes was determined by elemental analysis. Theor. for MB-Br: C 52.75 wt%, H 4.98 wt%, N 11.53 wt%, S 8.80 wt%; found: C 51.04 wt%, H 4.89 wt%, N 11.46 wt%, S 9.56 wt%. Theor. for MB-I: C 46.72 wt%, H 4.41 wt%, N 10.22 wt%, S 7.79 wt%; found: C 45.84 wt%, H 4.61 wt%, N 10.19 wt%, S 7.32 wt%. Theor. for RhB-Br: C 64.25 wt%, H 5.97 wt%, N 5.35 wt%; found: C 65.30 wt%, H 6.08 wt%, N 5.62 wt%. Theor. for RhB-I: C 58.95 wt%, H 5.48 wt%, N 4.91 wt%; found: C 58.68 wt%, H 5.57 wt%, N 4.93 wt%. Theor. for Rh6G-Br: C 64.25 wt%, H 6.97 wt%, N 5.35 wt%; found: C 65.05 wt%, H 6.02 wt%, N 5.53 wt%. Theor. for Rh6G-I: C 58.95 wt%, H 5.48 wt%, N 4.91 wt%; found: C 59.49 wt%, H 5.54 wt%, N 5.01 wt%.Synthesis of RhB-EtOH-Br
RhB-EtOH-Br was synthesised through the reaction of RhB base with 2-bromoethanol (Scheme 1). In a typical synthesis, RhB base (2.03 g, 4.52 mmol) and a three-fold excess of 2-bromoethanol (1.70 g, 13.57 mmol) were dissolved in 30 mL ethanol. The mixture was stirred at 70 °C for 24 h. After the reaction, the solvent was removed under reduced pressure. The obtained crude product was washed with ethyl acetate (30 mL × 9 times). The solid was further purified by dissolving it in 30 mL dichloromethane, followed by extraction with a saturated aqueous solution of NaBr (30 mL × 6 times) to remove RhB that might be present as a side product. MgSO4 was added to dry the organic solution and then removed by centrifugation. The purified RhB-EtOH-Br product was obtained by removing the solvent under reduced pressure, followed by drying at 80 °C overnight in a vacuum oven (brown powder, 2.15 g, 83% yield).
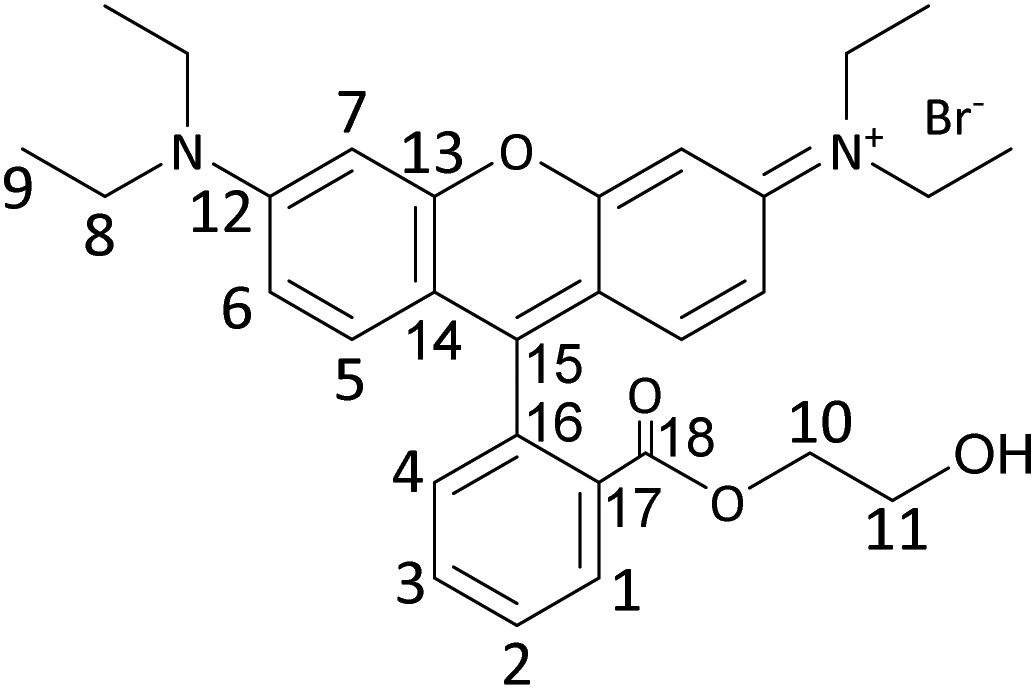
Synthesis of RhB-EtOH-I
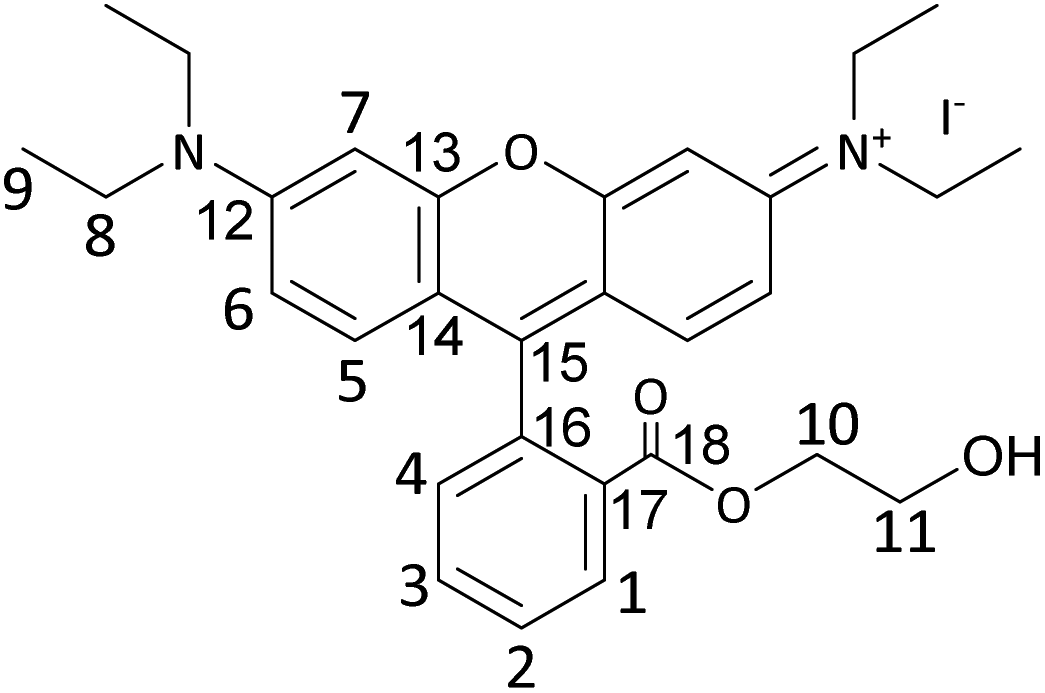
RhB-EtOH-Br (1.96 g, 3.52 mmol) was dissolved in 30 mL deionised water while stirring at 60 °C. Then, an aqueous solution of KI (1.76 g, 10.56 mmol in ca. 2 mL H2O) was added to the above solution upon stirring, leading to the precipitation of RhB-EtOH-I. The solution was stirred in an oil bath at 60 °C for 1 h and then placed in an ice bath and stirred for 1 h. The precipitate was recovered by centrifugation and washed with ca. 200 mL deionised H2O. Next, the obtained solid was dissolved in 30 mL dichloromethane and then recrystallised by adding 180 mL diethyl ether to the solution. Afterwards, the recrystallised solid was dissolved in 30 mL dichloromethane and dried with MgSO4. The drying agent was removed by centrifugation, and the solvent was removed under reduced pressure to obtain the solid product. The product was dried in a vacuum oven overnight at 80 °C (brown–gold powder, 1.81 g, 85% yield).
Catalytic tests
The catalytic tests for the cycloaddition of CO2 to produce cyclic carbonates were carried out in a high-throughput unit manufactured by Integrated Lab SC Solution (ILS), allowing to carry out the reaction with (supercritical) CO2 up to 200 bar and 200 °C. The reactor unit consists of two modules that can be operated independently: (1) a single batch reactor with a borosilicate window that allows observing the phase behaviour; (2) a block with 10 batch reactors that enables 10 catalytic tests to be performed simultaneously under the same reaction conditions. Each batch reactor is stirred individually with a magnetic stirring bar. Pressurisation of the reactors with CO2 is achieved using a ISCO pump. Each batch reactor is equipped with a burst disk to prevent over-pressurisation. In the 10-reactor block, each reactor chamber is equipped with a shutoff valve to avoid cross-contamination between reactors. Both modules were employed for the catalytic tests in this work. In a typical test, the epoxide (20 mmol), organocatalyst (1 mol% loading relative to epoxide), deionised water (50 mg) if added, and o-xylene (1.5 mmol) as NMR internal standard were placed in the glass vial (46 mL volume, 30 mm external diameter) with a magnetic stirring bar. The prepared sample vial was closed with a screw cap with a silicon/polytetrafluoroethylene (PTFE) septum. The septum was pierced with two needles for gas exchange. Then, each glass vial was placed in a batch reactor. After closing the reactor lid, the reactor module was first flushed with 5 bar N2 and then with 5 bar CO2 to remove air and then depressurised. Next, the reactor module was pressurised with CO2, warmed up to the target temperature, and further pressurised with CO2 to the desired pressure (if necessary). The reactor was operated under these conditions for 18 or 24 h while stirring at 950 rpm. Then, the stirring plate was turned off, and the reactor was cooled down by cooling water and then depressurised to atmospheric pressure. Finally, the reactor module was opened, and the glass vials were taken out. NMR samples were prepared by adding 10 mg of reaction solution to 0.7 mL of CDCl3. The conversion of the epoxide, and the yield and selectivity of the cyclic carbonate were calculated based on the 1H NMR spectra measured with Varian Mercury Plus 400 MHz and Agilent MR 400 MHz spectrometers (see Fig. S6–S12† for representative spectra), using the following equations: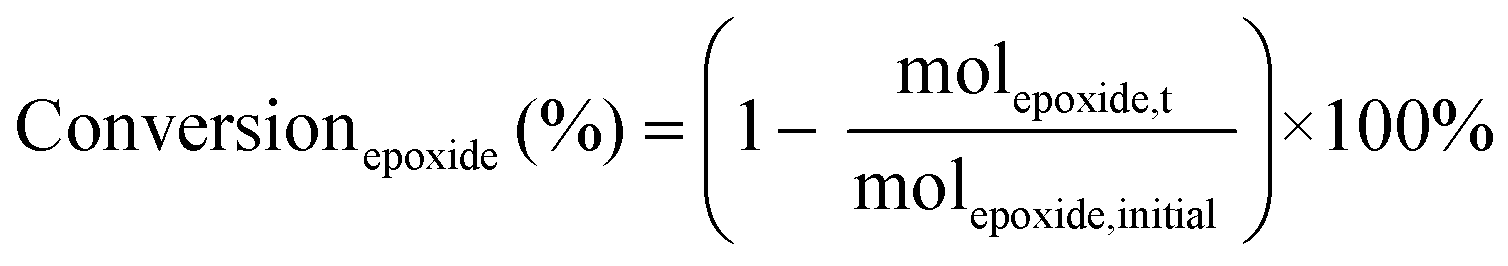
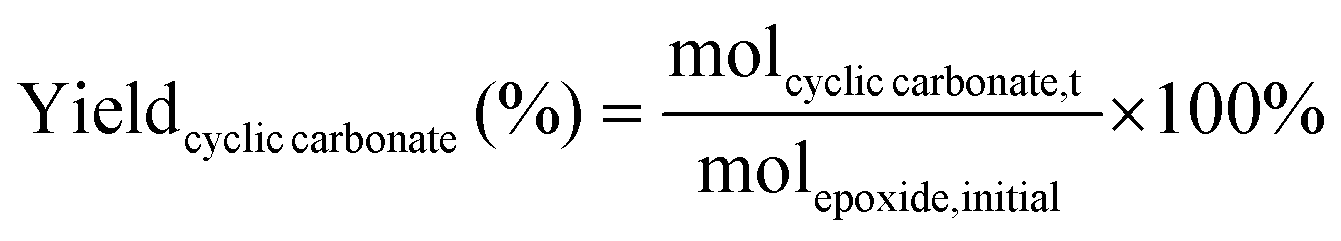

The molepoxide,t and the molcyclic carbonate,t were determined based on the integration of the 1H NMR signals of the epoxide and carbonate rings, respectively, followed by normalisation based on the integration of the signals of the internal standard.
The formation of the diol side product was investigated by gas chromatography–mass spectrometry analysis (GC-MS), which was carried out on an Agilent Hewlett-Packard-HP 6890 instrument (Rxi®-5 Sil MS column, 30 m, 0.25 mm) equipped with an Agilent Hewlett-Packard 5973 MSD mass spectrometer. No additional side products were observed by GC-MS or 1H NMR.
Selected catalytic tests were performed in duplicate or, in the few cases in which the reproducibility degree was not satisfactory, multiple times. For these experiments the average conversion and average yield are reported, while the standard deviation is provided in the form of error bars.
Three-fold scaled-up synthesis and purification of styrene carbonate
The synthesis of styrene carbonate through the reaction of CO2 cycloaddition to styrene oxide (reaction conditions: 45 °C, 10 bar CO2, 18 h) was repeated on a three-fold larger scale compared to the standard catalytic tests in this work, i.e. with 7.37 g (60.15 mmol) of styrene oxide, 0.37 g (0.60 mmol) of RhB-EtOH-I, 0.50 g (4.71 mmol) of o-xylene as internal standard. For the rest, the procedure was identical to the one described for our standard catalytic tests. At the end of the experiment, a small aliquot of the reaction mixture (ca. 10 mg) was dissolved in 0.7 mL CDCl3 and analysed by 1H NMR to determine the conversion of styrene oxide, and the yield and selectivity of styrene carbonate. Then, the styrene carbonate product was purified by silica-gel column separation (dcolumn = 4 cm, lcolumn = 31 cm, loaded with ca. 80 g of silica) using n-hexane/ethyl acetate (3:1) as the eluent (800–1400 mL). To avoid column overload with the reaction mixture sample, which would reduce the column separation performance, the column separation was conducted with separate batches, each with a portion of the reaction mixture (ca. 2.0 g of the total 8.90 g of reaction mixture). Additional column separation was conducted if the collected styrene carbonate fractions still contained impurities. The purified styrene carbonate was obtained as a solid after removing the eluent by rotary evaporation and was dried at 80 °C overnight in a vacuum oven. The separated batches were finally merged (2.89 g, isolated yield 29%) and analysed by 1H and 13C NMR on an Agilent MR 400 MHz spectrometer (for this analysis, 20 mg of the sample were dissolved in 0.7 mL CDCl3). The obtained spectra are presented in Fig. S21 and S22†.
Recovery and reuse of the RhB-EtOH-I catalyst
The reusability test of the RhB-EtOH-I catalyst was carried out using propylene oxide as the substrate under our standard reaction conditions (60 °C, 10 bar CO2, 24 h), but with all amounts scaled-up by a factor of 2, i.e. propylene oxide: 2.36 g (40.60 mmol), fresh RhB-EtOH-I catalyst: 0.25 g (0.40 mmol), and o-xylene as internal standard: 0.32 g (3.05 mmol). For the rest, the procedure was identical to the one described for our standard catalytic tests. At the end of the experiment, a small aliquot of the reaction mixture (ca. 10 mg) was dissolved in 0.7 mL CDCl3 and analysed by 1H NMR to determine the conversion of propylene epoxide, and the yield and selectivity of propylene carbonate. Then, the reaction mixture in the glass vial (without the cap) was warmed up to 80 °C for 1 h in a well-closed fume hood to remove the residual propylene oxide. The RhB-EtOH-I catalyst in the reaction mixture was recovered by precipitation using diethyl ether as an antisolvent. First, the reaction mixture was divided into two identical portions and transferred into two centrifugation tubes (50 mL volume for each tube), followed by slow addition of 40 mL diethyl ether to each tube through a dropping funnel (10–15 min in total), leading to the precipitation of the solid RhB-EtOH-I catalyst. Then, the precipitate was settled by centrifugation (4000 rpm, 5 min) and the supernatant was easily removed by pouring it out from the centrifugation tube. Then, 40 mL diethyl ether was added to each tube, and the suspension was sonicated for 10 min. The solid was again settled by centrifugation, the supernatant was discarded and the solid catalyst was dried at 80 °C overnight in a vacuum oven (brown–gold solid, 0.16 g, 0.26 mmol, corresponding to 64% recovery). The fresh and recovered RhB-EtOH-I catalysts were analysed by 1H and 13C NMR on a Bruker Avance NEO 600 MHz spectrometer (for this analysis, 20 mg of the sample were dissolved in 0.7 mL CDCl3). The obtained spectra are presented in Fig. S23–S26.†The recovered RhB-EtOH-I catalyst was reused in a second catalytic run under the same reaction conditions as in the initial catalytic activity test, maintaining the same catalyst-to-substrate ratio and adjusting the quantities based on the recovered amount of catalyst, i.e. propylene oxide: 1.48 g (25.54 mmol), recovered RhB-EtOH-I catalyst: 0.16 g (0.26 mmol), and o-xylene as internal standard: 0.21 g (1.9 mmol). At the end of the experiment, the reaction mixture was analysed by 1H NMR as after the first catalytic run.
Purification of the cyclic carbonate by nanofiltration
The nanofiltration experiments were conducted in the setup depicted in Fig. 5. The aqueous feedstock containing RhB and propylene carbonate was placed in a 2 L beaker and fed into the system by a pump (rotary vane pump, model: PO211). A rotameter was connected to the pump to measure the feedstock flow rate. The pressure exerted on the feedstock, concentrate and permeate was controlled by three needle valves and measured by the pressure gauges (PI, pressure indicator) on each line. A hollow fibre membrane (code MP025 dNF 40, purchased from NX-filtration) with an effective area of 0.05 m2 and a cut-off size of 400 Da was installed in this setup, which allowed small compounds (<400 Da) to permeate through the membrane and the large ones (>400 Da) to be rejected and thus remain in the concentrate. During nanofiltration, the output flows of both concentrate and permeate were placed in the feedstock beaker to keep the feedstock at a constant concentration. Small volume samples (10 mL) were taken at regular time intervals from the permeate (5 samplings).The nanofiltration experiment was performed with an aqueous feedstock (1 L) containing RhB (0.51 g) and propylene carbonate (10.01 g) at 1 barg, 25 °C for 2.5 h. Before starting the nanofiltration test, the aqueous solution was circulated in the system for 15 min without applying pressure to condition the pump and the concentrate line with the aqueous feedstock. Then, a 10 mL sample was taken to measure the initial concentration of RhB and propylene carbonate in the system. Next, the pressure exerted on the concentrate was adjusted to 1 barg and the feedstock flow rate was set at 50 L h−1 to start the nanofiltration test. After the system ran for 1 h to reach steady state, a 10 mL sample was taken from both the permeate and the (recirculating) feedstock every 30 min, for evaluation of the membrane separation efficiency. The system was then washed out with water until the outlet aqueous solution was colourless.
The efficiency in the separation of RhB from propylene carbonate (PC) was estimated based on the following equations:
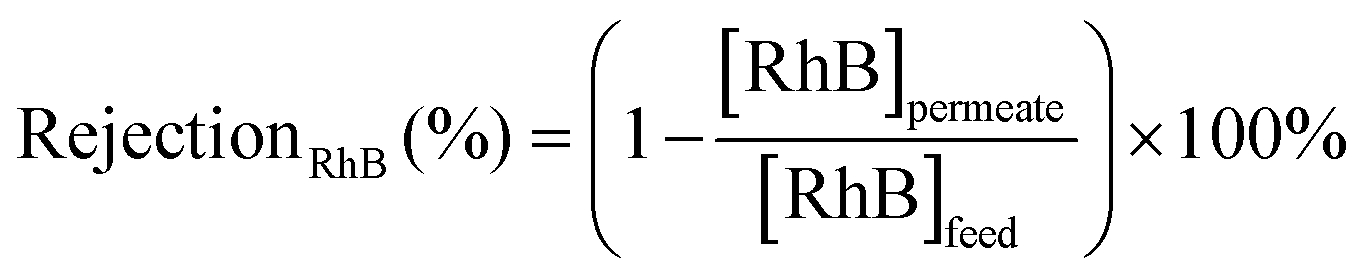
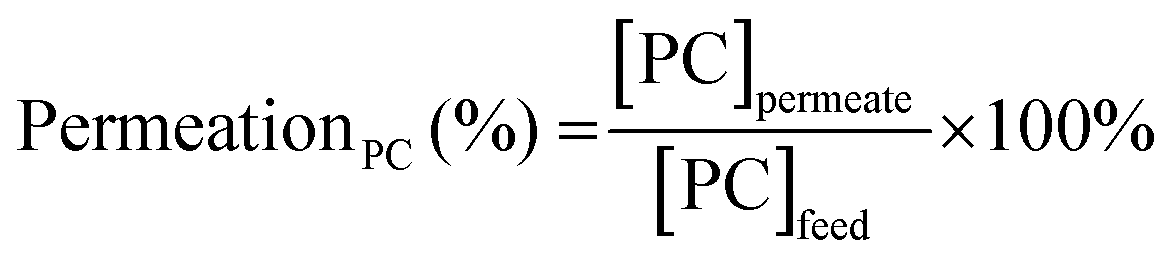
The concentration of RhB was quantified by UV-vis analysis (Agilent Cary 60 UV-vis). Before measuring the samples, 5 standard aqueous RhB samples with concentrations ranging from 1 to 12 ppm were prepared and measured by UV-vis (wavelength range: 200–600 nm) to obtain a linear plot of absorbance vs. concentration. The concentration of RhB in the feedstock samples (recirculating) was significantly higher than that of the permeate, above the concentration ranges of the calibration curve. Therefore, before measurement these samples were diluted by adding a 0.2 g sample from the recirculating feedstock to a 20 mL glass vial and mixing it with 10.0 g deionised water. The permeate samples had low RhB concentrations and were measured directly by UV-vis spectroscopy without dilution. The absorbance was obtained from the UV-vis spectra (at a wavelength of 554 nm) and then used to calculate the RhB concentration by means of the calibration curve (using Lambert–Beer law). The moles of propylene carbonate were calculated based on the 1H NMR spectra measured on a Varian Mercury Plus 400 MHz or Agilent MR 400 MHz spectrometer. The NMR samples were prepared by mixing a 2.5 g sample from the nanofiltration experiment and 0.03 g imidazole (NMR internal standard) in a 20 mL glass vial. Then, 10 mg of this solution was added to 0.7 mL of D2O.
To measure the permeate flux, water was fed into the system at the flow rate of 50 L h−1 with the pressure set at 1 barg. A certain amount of permeate was collected (V = 100 mL) and the time (t) needed to reach such a permeate volume was noted. Then, the permeate flux (Jperm) was calculated using the following equation:
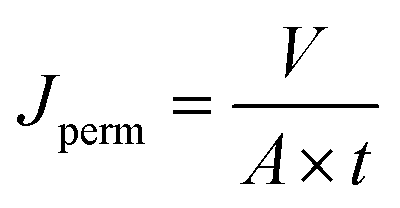
Photochemical tests
The effect of visible light irradiation on the catalytic activity of RhB-EtOH-I was determined in a photoreactor operated under the conditions specified in Fig. S3.† For this test, RhB-EtOH-I (0.12 g, 0.2 mmol), styrene oxide (0.48 g, 20 mmol), propylene carbonate (0.61 g), and H2O (0.05 g) were placed into the same type of glass vial used for the catalytic test described above. The glass vial was then closed with a screw cap with a silicon/polytetrafluoroethylene (PTFE) septum. Then, the septum was pierced with two needles for gas exchange. One of the needles was connected to the gas cylinder by a plastic tube, and the other one was open to the air, allowing the release of gas from the glass vial. The glass vial was flushed with N2 through the needle connected to the N2 cylinder and then flushed with CO2 by switching the connection to the CO2 cylinder. Afterwards, the glass vial was connected to a balloon by a plastic tube through one of the needles in the glass vial, while the other one was still attached to the CO2 cylinder. In this way, the glass vial was filled with CO2 until the balloon was inflated. The needle was disconnected from the CO2 cylinder and immediately attached to one syringe, which prevented CO2 loss from the reactor while also preventing pressure build up in the reactor. Finally, the sample was placed in an oil bath in the photoreactor, which had two LED lights on each side (Fig. S3†). As a reference, a second sample with the same amounts of RhB-EtOH-I, styrene oxide, propylene carbonate and H2O but with the glass vial and cap completely covered with aluminium film to avoid exposure to light was tested together with the first sample. Both samples were magnetically stirred at 60 °C for 24 h. At the end of the test, an aliquot of each sample was taken and analysed by 1H NMR to determine the yield and selectivity as described above in the section on the catalytic tests.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We are grateful for financial support from the China Scholarship Council for the Ph.D. grant of Jing Chen. We acknowledge technical support from Marcel de Vries, Henk van de Bovenkamp and Rick van der Reijd, and analytical support from Léon Rohrbach and Gert-Jan Boer. We are also thankful for elemental analysis support from Hans van der Velde.References
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS PubMed
.
- Z. Liu, Z. Deng, S. J. Davis, C. Giron and P. Ciais, Nat. Rev. Earth Environ., 2022, 3, 217–219 CrossRef PubMed
- R. Chauvy and G. De Weireld, Energy Technol., 2020, 8, 2000627 CrossRef
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed
- C. G. Okoye-Chine, K. Otun, N. Shiba, C. Rashama, S. N. Ugwu, H. Onyeaka and C. T. Okeke, J. CO2 Util., 2022, 62, 102099 CrossRef CAS
- A. J. Kamphuis, F. Picchioni and P. P. Pescarmona, Green Chem., 2019, 21, 406–448 RSC
- P. P. Pescarmona, Curr. Opin. Green Sustainable Chem., 2021, 29, 100457 CrossRef CAS
- H. Buettner, L. Longwitz, J. Steinbauer, C. Wulf and T. Werner, Top. Curr. Chem., 2017, 375, 1–56 CrossRef CAS PubMed
- F. D. Bobbink and P. J. Dyson, J. Catal., 2016, 343, 52–61 CrossRef CAS
- M. Alves, B. Grignard, R. Mereau, C. Jerome, T. Tassaing and C. Detrembleur, Catal. Sci. Technol., 2017, 7, 2651–2684 RSC
- R. R. Shaikh, S. Pornpraprom and V. D'Elia, ACS Catal., 2018, 8, 419–450 CrossRef CAS
- J. Xie, F. Chen, M. Li and N. Liu, J. CO2 Util., 2022, 62, 102100 CrossRef CAS
- Y. Qin, H. Guo, X. Sheng, X. Wang and F. Wang, Green Chem., 2015, 17, 2853–2858 RSC
- Y. A. Alassmy and P. P. Pescarmona, ChemSusChem, 2019, 12, 3856–3863 CrossRef CAS PubMed
- N. Aoyagi, Y. Furusho and T. Endo, Tetrahedron Lett., 2013, 54, 7031–7034 CrossRef CAS
- B. Chatelet, L. Joucla, J. P. Dutasta, A. Martinez and V. Dufaud, Chem. – Eur. J., 2014, 20, 8571–8574 CrossRef CAS PubMed
- T. K. Pal, D. De and P. K. Bharadwaj, Coord. Chem. Rev., 2020, 408, 213173 CrossRef CAS
- Y. Jiang, D. Li, Y. Zhao and J. Sun, J. Colloid Interface Sci., 2022, 618, 22–33 CrossRef CAS PubMed
- F. Zhou, S.-L. Xie, X.-T. Gao, R. Zhang, C.-H. Wang, G.-Q. Yin and J. Zhou, Green Chem., 2017, 19, 3908–3915 RSC
- G. V. Motuzova, T. M. Minkina, E. A. Karpova, N. U. Barsova and S. S. Mandzhieva, J. Geochem. Explor., 2014, 144, 241–246 CrossRef CAS
- G. Laugel, C. Carvalho Rocha, P. Massiani, T. Onfroy and F. Launay, Adv. Chem. Lett., 2013, 1, 195–214 CrossRef CAS
- M. Liu, X. Wang, Y. Jiang, J. Sun and M. Arai, Catal. Rev., 2019, 61, 214–269 CrossRef CAS
- S. Shirakawa, K. Okuno, R. Nishiyori and M. Hiraki, Heterocycles, 2021, 103, 94–109 CrossRef PubMed
- J. Wang and Y. Zhang, ACS Catal., 2016, 6, 4871–4876 CrossRef CAS
- S. Arayachukiat, C. Kongtes, A. Barthel, S. V. C. Vummaleti, A. Poater, S. Wannakao, L. Cavallo and V. D'Elia, ACS Sustainable Chem. Eng., 2017, 5, 6392–6397 CrossRef CAS
- M. Liu, K. Gao, L. Liang, F. Wang, L. Shi, L. Sheng and J. Sun, Phys. Chem. Chem. Phys., 2015, 17, 5959–5965 RSC
- L. Wang, G. Zhang, K. Kodama and T. Hirose, Green Chem., 2016, 18, 1229–1233 RSC
- M. Alves, B. Grignard, S. Gennen, R. Mereau, C. Detrembleur, C. Jerome and T. Tassaing, Catal. Sci. Technol., 2015, 5, 4636–4643 RSC
- C. J. Whiteoak, A. Nova, F. Maseras and A. W. Kleij, ChemSusChem, 2012, 5, 2032–2038 CrossRef CAS PubMed
- A. M. Hardman-Baldwin and A. E. Mattson, ChemSusChem, 2014, 7, 3275–3278 CrossRef CAS PubMed
- C. Claver, M. B. Yeamin, M. Reguero and A. M. Masdeu-Bulto, Green Chem., 2020, 22, 7665–7706 RSC
- M. Liu, X. Li, L. Liang and J. Sun, J. CO2 Util., 2016, 16, 384–390 CrossRef CAS
- H. Büttner, K. Lau, A. Spannenberg and T. Werner, ChemCatChem, 2015, 7, 459–467 CrossRef
- S. Liu, N. Suematsu, K. Maruoka and S. Shirakawa, Green Chem., 2016, 18, 4611–4615 RSC
- L. Guo, K. J. Lamb and M. North, Green Chem., 2021, 23, 77–118 RSC
-
F. Milocco, G. Chiarioni and P. P. Pescarmona, in Catalysis for Enabling Carbon Dioxide Utilization, Academic Press, 2022, pp. 151–187 Search PubMed
- T. Biswas and V. Mahalingam, Sustainable Energy Fuels, 2019, 3, 935–941 RSC
- W.-L. Dai, S.-L. Luo, S.-F. Yin and C.-T. Au, Appl. Catal., A, 2009, 366, 2–12 CrossRef CAS
- Y. Xie, Z. Zhang, T. Jiang, J. He, B. Han, T. Wu and K. Ding, Angew. Chem., Int. Ed., 2007, 46, 7255–7258 CrossRef CAS PubMed
- C. Calabrese, F. Giacalone and C. Aprile, Catalysts, 2019, 9, 325 CrossRef
- N. A. Raju, D. Prasad, P. M. Srinivasappa, A. V. Biradar, S. S. Gholap, A. K. Samal, B. M. Nagaraja and A. H. Jadhav, Sustainable Energy Fuels, 2022, 6, 1198–1248 RSC
- M. Beija, C. A. Afonso and J. M. Martinho, Chem. Soc. Rev., 2009, 38, 2410–2433 RSC
- K. Hiraoka, D. Asakawa and R. Takaishi, Surf. Interface Anal., 2013, 45, 968–972 CrossRef CAS
- S. Cheng, D. L. Oatley, P. M. Williams and C. J. Wright, Water Res., 2012, 46, 33–42 CrossRef CAS PubMed
- Q. Gong, H. Luo, D. Cao, H. Zhang, W. Wang and X. Zhou, Bull. Korean Chem. Soc., 2012, 33, 1945–1948 CrossRef CAS
- F.-T. Wu, L. Wu and C.-N. Cui, Tetrahedron, 2021, 83, 131965 CrossRef CAS
- P. P. Pescarmona and M. Taherimehr, Catal. Sci. Technol., 2012, 2, 2169–2187 RSC
- X. Liu, S. Zhang, Q.-W. Song, X.-F. Liu, R. Ma and L.-N. He, Green Chem., 2016, 18, 2871–2876 RSC
- T. Vosgrone and A. J. Meixner, ChemPhysChem, 2005, 6, 154–163 CrossRef CAS PubMed
- Z.-Z. Yang, L.-N. He, C.-X. Miao and S. Chanfreau, Adv. Synth. Catal., 2010, 352, 2233–2240 CrossRef CAS
- Y. Valadbeigi, Comput. Theor. Chem., 2020, 1188, 112947 CrossRef CAS
- J. Sun, W. Cheng, Z. Yang, J. Wang, T. Xu, J. Xin and S. Zhang, Green Chem., 2014, 16, 3071–3078 RSC
- M. Lõkov, S. Tshepelevitsh, A. Heering, P. G. Plieger, R. Vianello and I. Leito, Eur. J. Org. Chem., 2017, 2017, 4475–4489 CrossRef
- F. Chen, E. Zhao, T. Kim, J. Wang, G. Hableel, P. J. T. Reardon, S. J. Ananthakrishna, T. Wang, S. Arconada-Alvarez, J. C. Knowles and J. V. Jokerst, ACS Appl. Mater. Interfaces, 2017, 9, 15566–15576 CrossRef CAS PubMed
- L. Wang, K. Kodama and T. Hirose, Catal. Sci. Technol., 2016, 6, 3872–3877 RSC
- I. Kaljurand, A. Kutt, L. Soovali, T. Rodima, V. Mäemets, I. Leito and I. A. Koppel, J. Org. Chem., 2005, 70, 1019–1028 CrossRef CAS PubMed
- H. Büttner, J. Steinbauer and T. Werner, ChemSusChem, 2015, 8, 2655–2669 CrossRef PubMed
- J. C. Delgado and R. G. Selsby, Photochem. Photobiol., 2013, 89, 51–60 CrossRef CAS PubMed
- A. J. Parker, Q. Rev., Chem. Soc., 1962, 16, 163–187 RSC
- A. K. Soper and K. Weckstrom, Biophys. Chem., 2006, 124, 180–191 CrossRef CAS PubMed
- T. I. Morrow and E. J. Maginn, Fluid Phase Equilib., 2004, 217, 97–104 CrossRef CAS
- S. Foltran, J. Alsarraf, F. Robert, Y. Landais, E. Cloutet, H. Cramail and T. Tassaing, Catal. Sci. Technol., 2013, 3, 1046–1055 RSC
- J. Sun, L. Han, W. Cheng, J. Wang, X. Zhang and S. Zhang, ChemSusChem, 2011, 4, 502–507 CrossRef CAS PubMed
- D. Wei-Li, J. Bi, L. Sheng-Lian, L. Xu-Biao, T. Xin-Man and A. Chak-Tong, Appl. Catal., A, 2014, 470, 183–188 CrossRef
- M. H. Anthofer, M. E. Wilhelm, M. Cokoja, M. Drees, W. A. Herrmann and F. E. Kühn, ChemCatChem, 2015, 7, 94–98 CrossRef CAS
- Y. Li, B. Dominelli, R. M. Reich, B. Liu and F. E. Kühn, Catal. Commun., 2019, 124, 118–122 CrossRef CAS
- C. Yue, D. Su, X. Zhang, W. Wu and L. Xiao, Catal. Lett., 2014, 144, 1313–1321 CrossRef CAS
- K. R. Roshan, T. Jose, D. Kim, K. A. Cherian and D. W. Park, Catal. Sci. Technol., 2014, 4, 963–970 RSC
- H.-F. Jiang, B.-Z. Yuan and C.-R. Qi, Chin. J. Chem., 2008, 26, 1305–1308 CrossRef CAS
- C. Carvalho Rocha, T. Onfroy, J. Pilmé, A. Denicourt-Nowicki, A. Roucoux and F. Launay, J. Catal., 2016, 333, 29–39 CrossRef CAS
- J. Tharun, G. Mathai, R. Roshan, A. C. Kathalikkattil, K. Bomi and D. W. Park, Phys. Chem. Chem. Phys., 2013, 15, 9029–9033 RSC
- Y. Zhou, S. Hu, X. Ma, S. Liang, T. Jiang and B. Han, J. Mol. Catal. A: Chem., 2008, 284, 52–57 CrossRef CAS
- Z. Yue, M. Pudukudy, S. Chen, Y. Liu, W. Zhao, J. Wang, S. Shan and Q. Jia, Appl. Catal., A, 2020, 601, 117646 CrossRef CAS
- J. Tharun, K. R. Roshan, A. C. Kathalikkattil, D.-H. Kang, H.-M. Ryu and D.-W. Park, RSC Adv., 2014, 4, 41266–41270 RSC
- Y. Hao, D. Yuan and Y. Yao, ChemCatChem, 2020, 12, 4346–4351 CrossRef CAS
- X. Wu, C. Chen, Z. Guo, M. North and A. C. Whitwood, ACS Catal., 2019, 9, 1895–1906 CrossRef CAS
- H. Tong, Y. Qu, Z. Li, J. He, X. Zou, Y. Zhou, T. Duan, B. Liu, J. Sun and K. Guo, J. Chem. Eng., 2022, 444, 135478 CrossRef CAS
- P. D. Harvey, J. Mater. Chem. C, 2021, 9, 16885–16910 RSC
- N. Sharma, S. S. Dhankhar and C. M. Nagaraja, Microporous Mesoporous Mater., 2019, 280, 372–378 CrossRef CAS
- Z. W. Huang, K. Q. Hu, L. Mei, C. Z. Wang, Y. M. Chen, W. S. Wu, Z. F. Chai and W. Q. Shi, Inorg. Chem., 2021, 60, 651–659 CrossRef CAS PubMed
- Z. W. Huang, K. Q. Hu, L. Mei, X. H. Kong, J. P. Yu, K. Liu, L. W. Zeng, Z. F. Chai and W. Q. Shi, Dalton Trans., 2020, 49, 983–987 RSC
- L. G. Ding, B. J. Yao, W. X. Wu, Z. G. Yu, X. Y. Wang, J. L. Kan and Y. B. Dong, Inorg. Chem., 2021, 60, 12591–12601 CrossRef CAS PubMed
- Q. Yang, H. Peng, Q. Zhang, X. Qian, X. Chen, X. Tang, S. Dai, J. Zhao, K. Jiang, Q. Yang, J. Sun, L. Zhang, N. Zhang, H. Gao, Z. Lu and L. Chen, Adv. Mater., 2021, 33, 2103186 CrossRef CAS PubMed
- C. Liu, H. Niu, D. Wang, C. Gao, A. Said, Y. Liu, G. Wang, C.-H. Tung and Y. Wang, ACS Catal., 2022, 12, 8202–8213 CrossRef CAS
- C. Chen, W. Zhao, P. Lei, J. Zhao and N. Serpone, Chem. – Eur. J., 2004, 10, 1956–1965 CrossRef CAS PubMed
- SatoshiI. Horikoshi, A. Saitou and H. Hidaka, Environ. Sci. Technol., 2003, 37, 5813–5822 CrossRef CAS PubMed
- A. J. Kamphuis, M. Tran, F. Picchioni and P. P. Pescarmona, Green Chem. Eng., 2022, 3, 171–179 CrossRef
- A. J. Kamphuis, F. Milocco, L. Koiter, P. P. Pescarmona and E. Otten, ChemSusChem, 2019, 12, 3635–3641 CrossRef CAS PubMed
- V. Aomchad, S. Del Gobbo, P. Yingcharoen, A. Poater and V. D'Elia, Catal. Today, 2021, 375, 324–334 CrossRef CAS
- Y. Xu, H. Huang, G. Ying, J. Zhang, Y. Wu, S. Wu and Y. Yang, J. Mater. Res. Technol., 2020, 9, 11675–11686 CrossRef CAS
- A. Dobrak-Van Berlo, I. F. J. Vankelecom and B. Van der Bruggen, J. Membr. Sci., 2011, 374, 138–149 CrossRef CAS
- J. Lin, W. Ye, H. Zeng, H. Yang, J. Shen, S. Darvishmanesh, P. Luis, A. Sotto and B. Van der Bruggen, J. Membr. Sci., 2015, 477, 183–193 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: scheme showing the reaction mechanism, results of solubility tests, results of reusability tests, pictures of the used setups, and NMR and UV-vis spectra. See DOI: https://doi.org/10.1039/d3gc02161k |
| This journal is © The Royal Society of Chemistry 2023 |