
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA switchable route for selective transformation of ethylene glycol to hydrogen and glycolic acid using a bifunctional ruthenium catalyst†
Satabdee Tanaya
Sahoo
,
Aisa
Mohanty
,
Raju
Sharma
and
Prosenjit
Daw
 *
*
Department of Chemical Sciences, Indian Institute of Science Education and Research Berhampur, Transit Campus, (Govt. ITI Building), Engg. School Junction, Berhampur 760010, Odisha, India. E-mail: pdaw@iiserbpr.ac.in
First published on 27th June 2023
Abstract
The developed bifunctional NNN–Ru complex features a high catalytic efficiency for the selective production of hydrogen and glycolic acid from ethylene glycol under mild reaction conditions, where a TON of 6395 was achieved. Tuning the reaction conditions afforded further dehydrogenation of the organic substrate with higher hydrogen production, and a higher TON of 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 225 was attained. The scale-up reaction yielded 1230 mL of pure hydrogen gas under the optimized reaction conditions. The role of the bifunctional catalyst was studied and mechanistic investigations were performed.
225 was attained. The scale-up reaction yielded 1230 mL of pure hydrogen gas under the optimized reaction conditions. The role of the bifunctional catalyst was studied and mechanistic investigations were performed.
Sustainable hydrogen fuel production is encountered as a critical development to achieve a superior alternative to conventional fossil-based fuel owing to its high energy density and environmentally friendly properties.1,2 Over the past decades, rapid expansion in the utilization and upgrading of renewable biomass to sustainable fuel and commodity chemicals has gained significant interest.3 In this context, ethylene glycol (EG), owing to its properties, such as low cost, high potential for commercialization, a hydrogen storage capacity of 6.5 wt%, carbon neutral, and wide accessibility, makes it feasible to be utilized as a new substituent for energy material.4 Apart from the traditional methods for producing ethylene glycol from ethylene oxide and syngas, technologies from biomass-derived feedstocks and industrial waste (e.g. glycerol and cellulose) becoming increasingly important.5 Therefore, in the light of hydrogen economy, the utilization of this bio-derived feedstock for the extensive production of hydrogen and value-added chemicals by partial or complete oxidation via an acceptorless dehydrogenation pathway is eminently beneficial for a decarbonized sustainable future.6,7
Although the utilization of ethylene glycol for the synthesis of several value-added chemicals, such as acids,8–11 amides,12 polyethyleneimine,13 oligoester,14,15 and N-heteroarenes16 by dehydrogenative coupling has grown as a potential methodology for upgrading this feedstock nonetheless to maximize its utility, reforming ethylene glycol to produce hydrogen is of great concern.17,18
Various attempts have been devoted on heterogeneous catalysts for the transformation of ethylene glycol for hydrogen production, either through high-temperature steam reforming or via aqueous phase reforming (APR) over supported metal catalysts19–23 or metal-free graphene-based catalysts.24 However, many of these strategies operate at high reaction temperatures, requiring complex reaction setups, and a loss of selectivity is observed. Considering the harsh reaction conditions in heterogeneous systems, the current research interest has uncovered some homogeneous catalyst systems for the selective transformation of ethylene glycol to glycolic acid, where pure hydrogen gas was formed as the by-product via the acceptorless dehydrogenation process. Glycolic acid is a value-added product that has numerous industrial applications, including textiles, metal cleaning, adhesives, health-care products, leather processing, and especially biodegradable materials.25
Cole-Hamilton pioneered a Ru-based [RuH2(N2)(PPh3)3] molecular catalyst for the generation of hydrogen from alcoholic substrates, including ethylene glycol with a TOF of >1000 h−1.26 Notably, Zhang's group investigated catalysts based on Ir, and Rh for reforming ethylene glycol and achieved 81% glycolic acid at 100 °C after 10 h.27 Milstein's group reported glycolic acid synthesis from ethylene glycol by employing a Ru pincer system yielded 93% after 48 h.28 Recently, Maji and co-workers employed the Mn–MACHO complex for glycolic acid production at 140 °C with a TON of 2400 (Scheme 1a).29 Even though some notable progress has been made in the reforming of ethylene glycol concerning the upgradation of biomass to hydrogen and value-added products, the current scenario still requires efficient catalysts for achieving sustainable development.
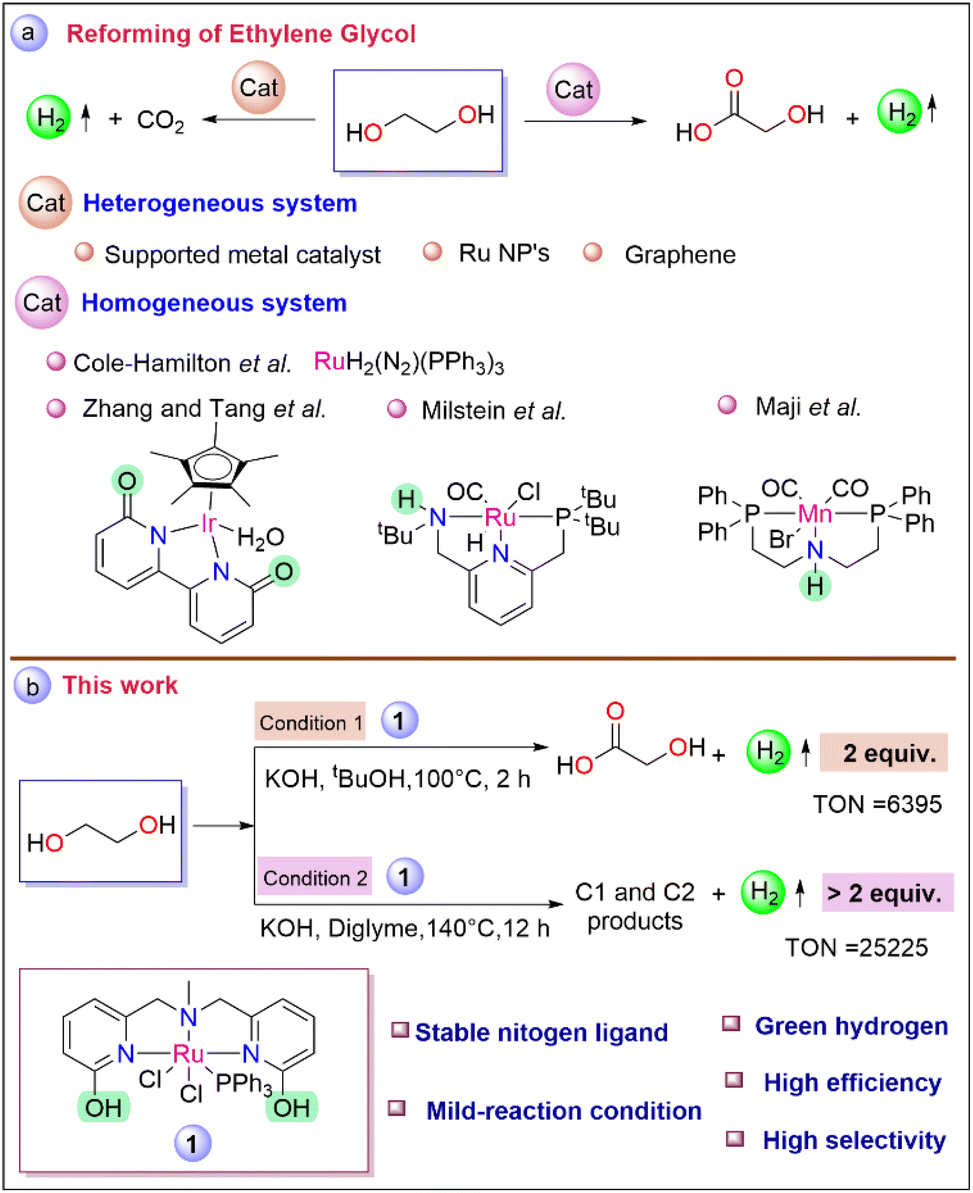 | ||
| Scheme 1 (a) Reforming of ethylene glycol by various heterogeneous and homogeneous catalysts. (b) This work. | ||
In our previous report, the catalytic performance of complex 1 (Scheme 1b) was explored for the generation of hydrogen from biomass-derived sorbitol, where the mechanistic studies revealed the fine-tuning of ligand architecture had a pivotal influence on the catalytic activity and facilitated the cooperative mechanism.30 To bring the development forward to the path of being put into effort here, we attempt to produce selectively green hydrogen from ethylene glycol under two different operating conditions. The first optimized strategy operates under relatively mild reaction conditions, leading to the selective production of glycolic acid with the evolution of pure hydrogen at a high initial rate. Meanwhile, under the second developed strategy, the selectivity for the production of more equivalent hydrogen gas from ethylene glycol was increased due to further dehydrogenation of glycolic acid, which has not been reported for the homogeneous catalyst system till now.
To develop the desired methodology for green hydrogen production from biomass-derived ethylene glycol, the initial studies include optimization of various reaction parameters such as a catalyst, base, solvent, temperature, and reaction time. To our delight, under the initial operating conditions (EG (2.503 mmol), 1 (1 mol%), KOH (6.25 mmol), in tBuOH (1 mL) at 100 °C for 2 h), 118 mL of the gas volume was obtained (Table 1, entry 1). Monitoring the progress of the reaction by measuring the gas volume generated during the catalysis revealed that the rate of the initial reaction was high, at an initial TOF of 469 h−1 (Fig. S2†). From the NMR analysis of the reaction mixture, the glycolic acid yield was found to be 94% with high selectivity after the complete conversion of ethylene glycol indicating efficient balancing of the total carbon atom (Fig. S19 and S20†). The purity of the evolved gas was evaluated by gas chromatography analysis (Fig. S16†) as well as by the chemical technique (utilizing the evolved gas for Pd/C catalyzed hydrogenation of styrene to ethylbenzene (section 10, Fig. S12†)). Further, the role of various solvents was screened under the mentioned conditions. However, the conversion efficiencies in protic solvents such as tBuOH, tAmOH, and mixtures of tBuOH and H2O were found to be impressive (entries 1, 2, 4 vs. 3) which indicates a protic solvent may assist the reaction pathway during the reforming of ethylene glycol.31,32 A similar reaction was also performed in the absence of catalyst 1, resulting in no formation of gas or glycolic acid, indicating the role of the catalyst (entry 5, Fig. S21 and S22†). Meanwhile, under optimized conditions (EG (2.503 mmol), KOH (6.25 mmol), in tBuOH at 100 °C for 2 h), the activity of the metal precursor (RuCl2(PPh3)3) was found to be insignificant with only 15% ethylene glycol conversion with 21 mL of pure hydrogen, indicating the role of ligand during the reaction (entry 6, Fig. S25, S26, and S16B-c†). Compared to the catalytic efficiencies of complex 1, complex 2 (see Fig. S1 in ESI†) was proven to have lower catalytic activity, highlighting the dual influence of the –OH group at the ortho position of the pyridyl arm compared to the only electronic influence of the –OMe group (entry 1 vs. 7, S16B-b†).33,34
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Conv.g (%) | GAg (%) | H2 in mL (equiv.h) |
| Reaction condition: ethylene glycol (2.503 mmol), KOH (6.25 mmol), catalyst (1 mol%), solvent (1 mL), at 100 °C for 2 h.a KOH (3.75 mmol).b KOH (1.25 mmol).c 90 °C.d 140 °C.e 12 h.f In the presence of Hg (100 mol%).g Conversion and yield were calculated by 1H NMR using 2,6-lutidine as an internal standard. In diglyme and in THF solvent, the conversion of ethylene glycol was not identifiable.h See Table S1† for equivalent calculation of hydrogen and carbon balance of reaction.i Experiments were repeated three times, and the average results were reported with an error limit of 5%. | |||||
| 1i | 1 | t BuOH | 100 | 94 | 118 (2.1) |
| 2i | 1 | t AmOH | 91 | 90 | 100 (1.78) |
| 3 | 1 | THF | — | 10 (0.18) | |
| 4i | 1 |
t
BuOH:H2O |
81 | 80 | 90 (1.6) |
| 5 | — | t BuOH | 0 | 0 | 0 |
| 6 | RuCl2(PPh3)3 | t BuOH | 15 | 5 | 21 (0.37) |
| 7i | 2 | t BuOH | 40 | 29 | 50 (0.89) |
| 8a | 1 | t BuOH | 59 | 49 | 75 (1.33) |
| 9b | 1 | t BuOH | 28 | 3 | 35 (0.62) |
| 10c,i | 1 | t BuOH | 65 | 59 | 78 (1.39) |
| 11d,i | 1 | t BuOH | 100 | 70 | 130 (2.31) |
| 12d | 1 | Diglyme | — | 52.7 | 146 (2.59) |
| 13d,e,i | 1 | Diglyme | — | 0 | 180 (3.2) |
| 14d,e,i | 1 | Diglyme:H2O |
— | 25 | 152 (2.7) |
| 15f,i | 1 | t BuOH | 82 | 81 | 96 (1.71) |
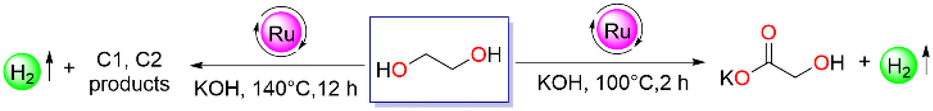
Furthermore, the crucial role of the substrate-to-base ratio was depicted, where a reduced amount of glycolic acid (49% and 3%) and 75 ml and 35 ml H2 gas were obtained by lowering the base loading to 1.5 and 0.5 equivalents, respectively (entries 8 and 9). Also, on decreasing the temperature to 90 °C under optimized conditions, the conversion of ethylene glycol and yield of glycolic acid reduced to 65% and 59%, respectively with an evolution of 78 mL of hydrogen gas volume (entry 10). Surprisingly, under similar conditions at 140 °C, 130 mL of gas volume was evolved, with decreased glycolic acid selectivity, indicating a higher temperature is beneficial to get a higher gas volume (entry 11). Furthermore, when the reaction was investigated in high boiling diglyme solvent at 140 °C for 2 h, 52.7% of glycolic acid was obtained with 146 mL (more than 2 equivalents) of gas volume after passing through a double alkali solution (entry 12). The purity of the gas was also analyzed by chemical technique (Fig. S14†). Indeed, monitoring the reaction time after 12 h, 180 mL of hydrogen gas was obtained with the complete transformation of glycolic acid (entry 13, Fig. S16c, S23 and S24†), whereas in the diglyme:H2O (9:1) mixture under optimised conditions (EG (2.503 mmol), KOH (6.25 mmol) at 140 °C for 12 h) 152 mL of gas volume resulted with 25% of glycolic acid remaining in the reaction (entry 14). The lower yield of glycolic acid is owing to its subsequent dehydrogenation to H2 and other C1, and C2 products (oxalate, formate, and CO2) in the presence of catalyst 1, and thus resulted in a significant drop in a carbon depiction.22 Additionally, when catalyst 1 was subjected to the mercury drop experiment under optimized conditions for glycolic acid production, no significant decrease in catalytic efficiency of 1 was observed, indicating the homogeneity of the reaction medium (entry 15, section 9, Fig. S11†).
Further, the effect of catalyst loading was demonstrated under both optimized conditions. A reduced yield of glycolic acid was obtained by lowering the substrate-to-catalyst ratio under the initial optimized conditions (EG (2.503 mmol), KOH (6.25 mmol) in tBuOH at 100 °C, 2 h). Particularly, with 0.5 mol% and 0.1 mol% of catalyst 1 loading, 57 mL and 41 mL of hydrogen gas were collected (Table 2, entries 2 and 3), respectively. Pleasingly, with 0.01 mol% catalyst loading, a TON of 6395 was achieved in tBuOH after 2 h with a gas volume of 36 mL and with only 33% EG conversion (entry 4). Meanwhile, under the second optimized conditions in diglyme at 140 °C after 12 h an even higher TON of 25225 was obtained with 142 mL of gas evolution on decreasing catalyst loading to 0.01 mol% (entry 5). Encouraged by the catalytic efficiency of complex 1, the reaction was scaled up to 25.03 mmol of EG with 0.05 mol% of catalyst loading in diglyme at 140 °C and 1.23 L of pure hydrogen gas was collected after a period of 48 h with a glycolic acid yield of 80% (entry 6, section 5, Fig. S33, S34, and S35 in ESI†). For commercialization, solvent-free reforming of ethylene glycol is highly desirable, and accordingly, the catalytic activity of 1 (0.05 mol%) was also evaluated in neat condition with 25.03 mmol of substrate loading at 140 °C (Table 2, entry 7). Pure 1.4 L of H2 gas was collected (Fig. S3†) (analyzed by GC-TCD, Fig. S16B-a†) over a period of 96 h with a glycolic acid yield of 82% (Fig. S36 and S37†).
| Entry | Cat. loading | Conv.% | GA% | H2 in mL | TONe |
|---|---|---|---|---|---|
| Reaction conditions: ethylene glycol (2.503 mmol), KOH (2.5 equivalent w.r.t. ethylene glycol), at 100 °C.a In diglyme at 140 °C.b 25.03 mmol in diglyme for 48 h at 140 °C.c 25.03 mmol in neat condition at 140 °C over 96 h.d After acid workup free glycolic acid.e TON and TOF were calculated on the basis of the mmol of hydrogen gas produced during the reaction. See the ESI† for the TON calculation.f Experiments were repeated three times, and the average results were reported with an error limit of 5%. | |||||
| 1f | 1 | 100 | 94 | 118 | 210 |
| 2 | 0.5 | 51 | 23 | 57 | 203 |
| 3f | 0.1 | 35 | 12 | 41 | 728 |
| 4 | 0.01 | 33.5 | 9 | 36 | 6395 |
| 5a,f | 0.01 | — | 62 | 142 | 25225 |
| 6b | 0.05 | — | 86.6 (80)d | 1230 | 4370 |
| 7c | 0.05 | 94 | 82 | 1400 | 4974 |
To understand the possible involvement of various intermediates in the catalytic process, a couple of control experiments were performed. As indicated in the mechanistic pathway (Scheme 2A), glyoxal can be a potential intermediate during the dehydrogenation of ethylene glycol, which may undergo either base or metal-catalyzed rearrangement to produce glycolic acid. On the treatment of glyoxal under optimised conditions (glyoxal (2.503 mmol), KOH (6.25 mmol), in diglyme at 140 °C), without any catalyst resulted in 85% of glycolic acid although no gaseous product was observed (Fig. S27†), whereas, in presence of 1, 35% of glycolic acid was obtained with the evolution of 32 mL gas volume (Fig. S28†), which depicted that the formed glycolic acid in the presence of catalyst 1 may decompose further to other C1, C2 products (Scheme 2B-a). Therefore, glycolic acid was subjected to degradation in diglyme at 140 °C for 12 h (Scheme 2B-b). In the absence of any catalyst, only 2% of glycolic acid was converted, whereas 20% of glycolic acid decomposition was observed with 20 mL of gas volume in the presence of 1 (Fig. S29 and S30†). From the gas chromatography analysis of the produced gas, the presence of a mixture of H2 and CO2 was detected (Fig. S17b†). The lower conversion of glycolic acid depicts that the decomposition of glycolic acid is a slow process and may also be assisted by the protonic source. Accordingly, a 1:1 mixture of ethylene glycol and glycolic acid was subjected to dehydrogenation. Under standard conditions, a gas volume of 100 mL and 50% conversion of glycolic acid resulted, indicating a solvent/substrate-assisted proton shuttle pathway may follow for the further dehydrogenation of glycolic acid (Fig. S31†). Following the possible reaction pathway, dehydrogenation of formic acid was also performed under the standard optimized conditions, which resulted in 50 mL of gas volume with 95% consumption of the formic acid (Fig. S32 and S17c†).
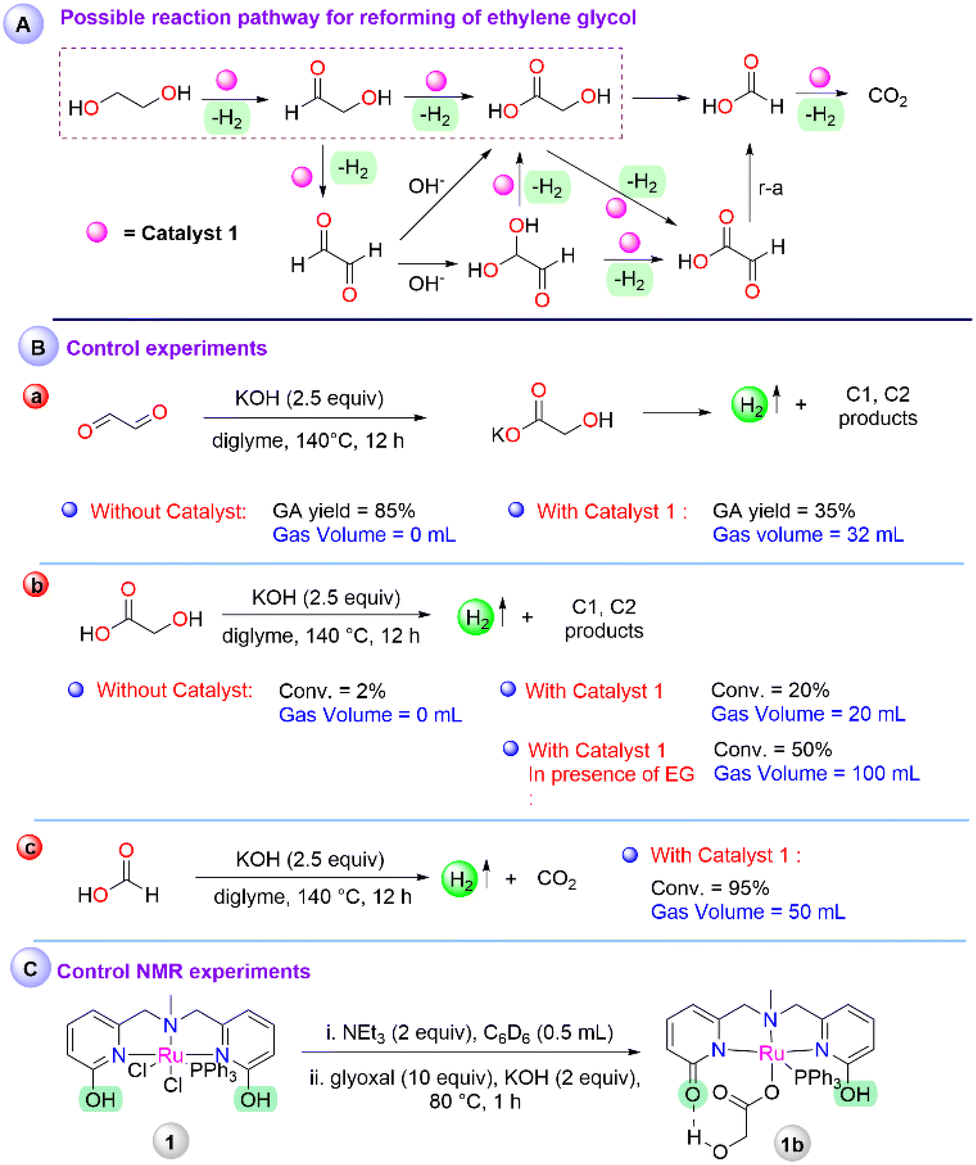 | ||
| Scheme 2 (A) Possible reaction pathway for reforming of ethylene glycol, (B) control experiments and (C) elucidation of the active intermediate by control NMR experiments. | ||
Further, to obtain an insight into the active intermediate involved at the metal center, catalyst 1 was treated with 2 equiv. of NEt3, and the formation of 1a was observed as reported earlier (Scheme S1†).30 In the same reaction, the addition of glyoxal (10 equiv.) resulted in a peak at 42.03 ppm in 31P NMR, probably due to the interaction of glyoxal, followed by the addition of KOH (2.5 equiv. w.r.t. glyoxal) and heating at 80 °C for 1 h, resulting in a peak at 50.69 ppm in 31P NMR, indicating glycolate coordinated intermediate 1b (section 8a in ESI†). The treatment of 1 with NEt3, followed by glycolic acid addition showed the same peak in 31P NMR, which was further characterized by HRMS and depicted a peak at m/z 684.12 (Scheme 2C, Fig. S4, S5, and S38†). Despite several attempts, the structural characterization of 1b was unsuccessful, although the details of NMR studies and secondary coordination sphere interaction with the metal complex with glycolate were highlighted.30 Additionally, from the mass analysis of the reaction mixture, peaks at m/z = 649.12 and 608.10 were attributed to the deprotonated complex (1a), indicated as an active intermediate (Fig. S39†). This result ruled out catalyst deactivation by glycolate coordination during the reforming of ethylene glycol. Further, to elucidate the identity of Ru-hydride, which is a key intermediate after β-hydride elimination, an NMR scale ethylene glycol reaction was performed. Treatment of 1 with NEt3 (forming 1ain situ) and the addition of 10 equiv. of ethylene glycol afforded a new peak at 47.15 ppm in 31P NMR, which upon heating for 2 h at 80 °C disappeared (Fig. S6†). At this stage, the 1a peak was the major in 31P NMR and no hydride peak was observed in the negative region in 1H NMR for the corresponding Ru–H species. However, performing a similar experiment in an H2 atmosphere instead of N2, a doublet peak at −26.23 ppm attributed to the Ru–H intermediate was observed (Scheme S1†).35 Along with that, the existence of a hydrogen-bonded protic arm due to the appearance of peaks at 10.08 ppm and 13.79 ppm was also observed (Fig. S7 and S8†). On the other hand, in the absence of ethylene glycol, no Ru–H species were observed under the H2 atmosphere (Fig. S9 and S10†). It is noteworthy to mention that the hydride intermediate involved during catalysis is only stable under the hydrogen atmosphere. Therefore, a fast H2 evolution may take place when performing the experiments in an N2 atmosphere, depicting the role of protic arm functionality in the secondary coordination sphere for the ligand assisted proton shuttle and effectiveness of catalyst 1 for the high TOF value.36
Importantly, the synergetic effect arising by the metal–ligand cooperative via lactam–lactim tautomerism by the introduction of the pyridonate moiety within the metal complex has emerged as a powerful tool for enhancement of the catalytic activity.37 On the basis of our previous observations and current experimental studies, a possible reaction pathway for the dehydrogenation of ethylene glycol is predicted in Scheme 3. Initial treatment of the base could afford active deprotonated intermediate A, which could undergo metal–ligand cooperation to activate ethylene glycol, resulting in alkoxy complex B, where the additional hydrogen bond helps the substrate recognition via secondary coordination sphere interaction. The β-hydride elimination of B could eliminate the aldehyde product with the hydride intermediate C (as observed in Scheme S1†), which can produce dihydrogen via the proton shuttle hydrogen elimination with the regeneration of active catalyst A. The resulting carbonyl compound could undergo further dehydrogenation by active complex A to produce more hydrogen and other C1, and C2 products (Scheme 2A).
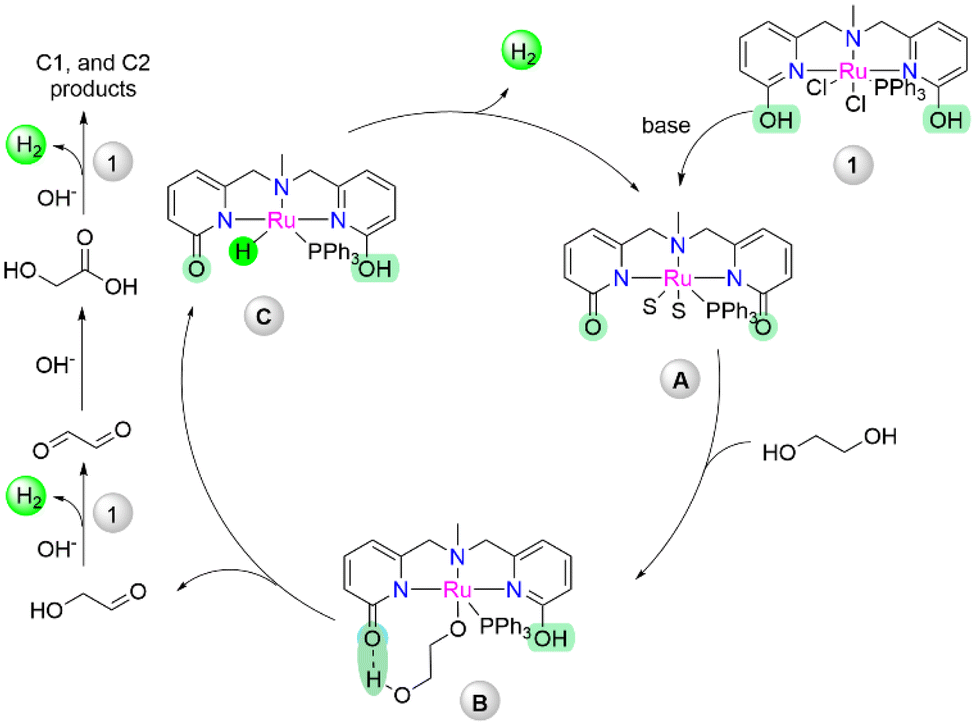 | ||
| Scheme 3 Possible reaction mechanism of reforming of ethylene glycol catalyzed by 1. | ||
Conclusions
In conclusion, the developed NNN–Ru bifunctional catalyst is a highly efficient catalyst for reforming of ethylene glycol under two switchable operating conditions. A high TON of 6395 and an even higher TON of up to 25225 were achieved at 100 °C and 140 °C, respectively. To the best of our knowledge, this is the first molecular catalyst system that can produce more than two equivalents of hydrogen during the reforming of ethylene glycol ever reported under mild operating conditions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
For financial support, P. D. gratefully acknowledged IISER Berhampur, and SERB, DST, India for the startup research grand (SRG/2020/000424). S. T. S. thanks CSIR, New Delhi, for the fellowship. A. M. and R. S. are thankful to IISER Berhampur for their fellowship. We are thankful for the Central Advance Instrument Facility (CAIF) at IISER Berhampur.References
- D. Wei, X. Shi, R. Qu, K. Junge, H. Junge and M. Beller, ACS Energy Lett., 2022, 7, 3734–3752 CrossRef CAS.
- R. H. Crabtree, Chem. Rev., 2017, 117, 9228–9246 CrossRef CAS PubMed.
- A. Kumar, P. Daw and D. Milstein, Chem. Rev., 2022, 122, 385–441 CrossRef CAS PubMed.
- V. Yadav, G. Sivakumar, V. Gupta and E. Balaraman, ACS Catal., 2021, 11, 14712–14726 CrossRef CAS.
- H. Yue, Y. Zhao, X. Ma and J. Gong, Chem. Soc. Rev., 2012, 41, 4218–4244 RSC.
- Y. X. Chen, A. Lavacchi, H. A. Miller, M. Bevilacqua, J. Filippi, M. Innocenti, A. Marchionni, W. Oberhauser, L. Wang and F. Vizza, Nat. Commun., 2014, 5, 1–6 Search PubMed.
- J. Rana, S. T. Sahoo and P. Daw, Tetrahedron, 2021, 99, 132473 CrossRef CAS.
- M. Lee, H. Byeon and H.-Y. Jang, J. Org. Chem., 2022, 87, 4631–4639 CrossRef CAS PubMed.
- M. Trincado, K. Kühlein and H. Grützmacher, Chem. – Eur. J., 2011, 17, 11905–11913 CrossRef CAS PubMed.
- J. Wu, L. Shen, Z. N. Chen, Q. Zheng, X. Xu and T. Tu, Angew. Chem., Int. Ed., 2020, 59, 10421–10425 CrossRef CAS PubMed.
- L. Shen, Z. N. Chen, Q. Zheng, J. Wu, X. Xu and T. Tu, ACS Catal., 2021, 11, 12833–12839 CrossRef CAS.
- Y. Q. Zou, Q. Q. Zhou, Y. Diskin-Posner, Y. Ben-David and D. Milstein, Chem. Sci., 2020, 11, 7188–7193 RSC.
- A. C. N. Brodie, A. E. Owen, J. S. Kolb, M. Bühl and A. Kumar, Angew. Chem., Int. Ed., 2023, e202306655 Search PubMed.
- Y. Q. Zou, N. von Wolff, A. Anaby, Y. Xie and D. Milstein, Nat. Catal., 2019, 2, 415–422 CrossRef CAS PubMed.
- Q. Q. Zhou, Y. Q. Zou, Y. Ben-David and D. Milstein, Chem. – Eur. J., 2020, 26, 15487–15490 CrossRef CAS PubMed.
- P. Daw, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2018, 140, 11931–11934 CrossRef CAS PubMed.
- M. T. Azizan, A. Aqsha, M. Ameen, A. Syuhada, H. Klaus, S. Z. Abidin and F. Sher, Biomass Convers. Biorefin., 2023, 13, 8441–8464 CrossRef PubMed.
- P. D. Vaidya and J. A. Lopez-Sanchez, ChemistrySelect, 2017, 2, 6563–6576 CrossRef CAS.
- D. Mei, V. L. Dagle, R. Xing, K. O. Albrecht and R. A. Dagle, ACS Catal., 2016, 6, 315–325 CrossRef CAS.
- R. R. Davda, J. W. Shabaker, G. W. Huber, R. D. Cortright and J. A. Dumesic, Appl. Catal., B, 2003, 43, 13–26 CrossRef CAS.
- G. Pipitone, G. Zoppi, R. Pirone and S. Bensaid, Int. J. Hydrogen Energy, 2021, 47, 151–180 CrossRef.
- T. Van Haasterecht, T. W. Van Deelen, K. P. De Jong and J. H. Bitter, Catal. Sci. Technol., 2014, 4, 2353–2366 RSC.
- A. Kumar, B. Priya and S. K. Singh, ACS Sustainable Chem. Eng., 2023, 11, 3999–4008 CrossRef CAS.
- I. Esteve-Adell, N. Bakker, A. Primo, E. J. M. Hensen and H. García, ChemistrySelect, 2017, 2, 6338–6343 CrossRef CAS.
- D. Si, B. Xiong, L. Chen and J. Shi, Chem Catal., 2021, 1, 941–955 CrossRef CAS.
- D. Morton and D. J. Cole-Hamilton, J. Chem. Soc., Chem. Commun., 1988, 2, 1154–1156 RSC.
- Y. Zhan, W. Hou, G. Li, Y. Shen, Y. Zhang and Y. Tang, ACS Sustainable Chem. Eng., 2019, 7, 17559–17564 CrossRef CAS.
- Y. Q. Zou, N. von Wolff, M. Rauch, M. Feller, Q. Q. Zhou, A. Anaby, Y. Diskin-Posner, L. J. W. Shimon, L. Avram, Y. Ben-David and D. Milstein, Chem. – Eur. J., 2021, 27, 4715–4722 CrossRef CAS PubMed.
- S. Waiba, M. Maiti and B. Maji, ACS Catal., 2022, 12, 3995–4001 CrossRef CAS.
- S. T. Sahoo, A. Mohanty, R. Sharma, S. R. Rout, R. Dandela and P. Daw, Organometallics, 2023, 42, 745–751 CrossRef CAS.
- F. J. De Zwart, V. Sinha, M. Trincado, H. Grützmacher and B. De Bruin, Dalton Trans., 2022, 51, 3019–3026 RSC.
- V. Sinha, N. Govindarajan, B. De Bruin and E. J. Meijer, ACS Catal., 2018, 8, 6908–6913 CrossRef CAS PubMed.
- W. H. Wang, J. T. Muckerman, E. Fujita and Y. Himeda, ACS Catal., 2013, 3, 856–860 CrossRef CAS.
- W. H. Wang, S. Xu, Y. Manaka, Y. Suna, H. Kambayashi, J. T. Muckerman, E. Fujita and Y. Himeda, ChemSusChem, 2014, 7, 1976–1983 CrossRef CAS PubMed.
- L. N. Dawe, M. Karimzadeh-Younjali, Z. Dai, E. Khaskin and D. G. Gusev, J. Am. Chem. Soc., 2020, 142, 19510–19522 CrossRef CAS PubMed.
- M. Iguchi, H. Zhong, Y. Himeda and H. Kawanami, Chem. – Eur. J., 2017, 23, 17788–17793 CrossRef CAS PubMed.
- T. Shimbayashi and K. I. Fujita, Catalysts, 2020, 10, 635 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dt01671d |
| This journal is © The Royal Society of Chemistry 2023 |