
Benchtop nickel-catalyzed reductive coupling of aldehydes with alkynes and ynamides†
Aankhi
Khamrai
and
Venkataraman
Ganesh
 *
*
Department of Chemistry, Indian Institute of Technology Kharagpur, Kharagpur, West Bengal 721302, India. E-mail: ganesh.v@chem.iitkgp.ac.in
First published on 24th August 2023
Abstract
We demonstrate the potential of Ni(COD)(DQ), a bench-stable Ni0 complex, as a catalyst for the reductive coupling of aldehydes with alkynes and ynamides, providing silylated allyl alcohols with excellent yields and regioselectivities. Mass spectrometric identification of the intermediates and DFT studies supported the proposed mechanism.
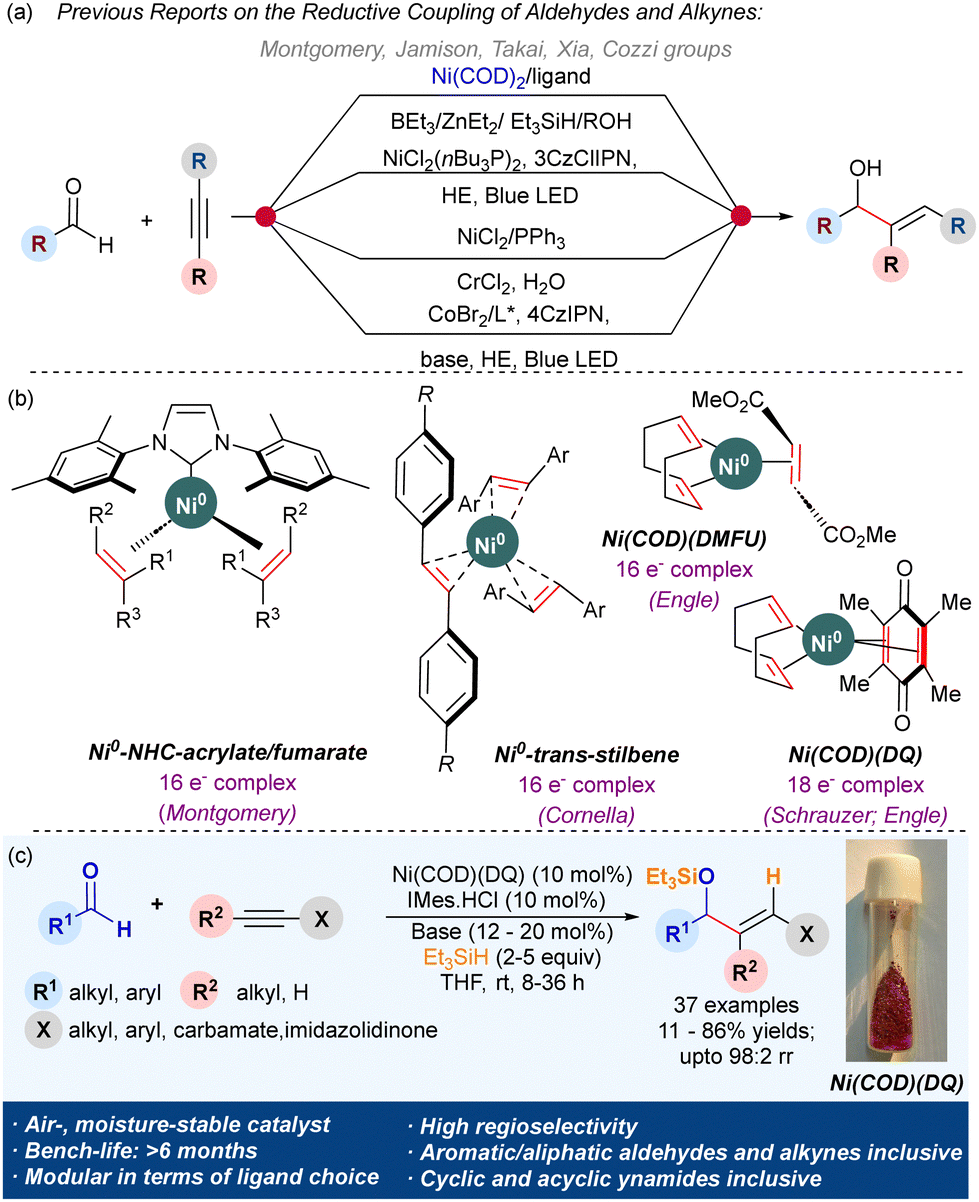
The reductive coupling of alkynes and aldehydes provides a ubiquitous stereoselective route to allyl alcohols (Scheme 1a).1 Due to the wide applications of allyl alcohols as a potent synthetic intermediate and a prominent fragment in natural products, drugs, perfumes, etc.,2 much focus has been devoted to their stereoselective synthesis. Specifically, the nickel-catalyzed reductive coupling approach has gained popularity due to the high regio-/stereocontrol observed in the products and the use of inexpensive metal.1d–f,3 Ni-complexes also exhibit a lower barrier for oxidative addition and reductive elimination compared to the platinum group metal complexes.4 However, this exceptional reactivity of Ni0 complexes makes them highly air and moisture sensitive. The major commercial Ni0 source, Ni(COD)2, degrades within a few hours on the benchtop. The catalytic activity of other stable commercial sources like Ni(CO)4, Ni(PPh3)4, Ni(COD)(DQ), etc., are under-explored.5
 | ||
| Scheme 1 (a) Reductive coupling of alkynes and aldehydes; (b) bench-stable Ni0 precatalysts; (c) present work. | ||
Various attempts to improve the benchtop stability and leverage the unique reactivity of Ni0 complexes have been reported.6 Montgomery uncovered the exceptional stability of Ni0–NHC 16e− complexes ligated with electron-deficient olefins (EDOs) like fumarates and acrylates (Scheme 1b).7 Fumarate complexes demonstrated superior air stability than acrylates and exhibited remarkable catalytic activity in the alkyne/aldehyde reductive coupling and amination reaction of aryl chlorides. Then, Engle and coworkers reported a wide range of Ni0(COD)(EDO) complexes like Ni(COD)(DMFU) (16e− complex),8 Ni(COD)(DQ) (18e− complex), etc.9 that are exceptionally stable over the benchtop and catalyzed C–C and C–N bond forming reactions efficiently.9,10 They used Ni(COD)(DQ)/SIPr.HCl system for the catalytic amination of aryl chlorides.10a The Cornella group utilized para-substituted stilbenes as ligands to achieve Ni(stb)3 (16e− complexes) with excellent air stability and catalytic activity.11
Motivated by the efficiency of Montgomery's Ni(NHC)(EDO)2 complexes in the reductive coupling of alkynes, we envisaged the access to Ni(NHC)(EDO)2 complexes in a modular fashion by reacting NHC salts with Ni(COD)(DQ) to catalyze alkyne/aldehyde reductive coupling (Scheme 1c). The in situ generated Ni(NHC)(DQ) complexes could eliminate the requirement of a glovebox for this transformation.
We began our investigation by subjecting Ni(COD)(DQ) (10 mol%) with various monophosphines, bis-phosphines, and free-NHCs (10 mol%) for the reductive coupling of benzaldehyde 1a and 1-phenyl-1-propyne 2a in THF at 45 °C, with triethyl silane as the reducing agent. However, with phosphine ligands, the reaction failed to provide the desired coupled product 3aa. With free-IPr as a ligand, the reaction provided the silylated allyl alcohol 3aa in 20% yield with poor regioselectivity. With IMes carbene, at 45 °C, the resulting active catalyst provided the desired silylated allyl alcohol 3aa in a 73% isolated yield and 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 rr. Other imidazolium salts like SIPr.HCl, ICy.HBF4 and IBn.HBF4 failed to provide 3aa. Cs2CO3 was the suitable base for forming the active catalyst via deprotonation of the NHC salts. With Ni(COD)(DQ) (10 mol%), IMes.HCl (10 mol%), Cs2CO3 (20 mol%) in THF at rt using triethyl silane, the reaction provided the best yield of 3aa (82% NMR yield) entirely on the benchtop without relying on a glovebox (see ESI†). With 2 mol% loading, a diminished yield of 3aa (13%) was obtained.
:3 rr. Other imidazolium salts like SIPr.HCl, ICy.HBF4 and IBn.HBF4 failed to provide 3aa. Cs2CO3 was the suitable base for forming the active catalyst via deprotonation of the NHC salts. With Ni(COD)(DQ) (10 mol%), IMes.HCl (10 mol%), Cs2CO3 (20 mol%) in THF at rt using triethyl silane, the reaction provided the best yield of 3aa (82% NMR yield) entirely on the benchtop without relying on a glovebox (see ESI†). With 2 mol% loading, a diminished yield of 3aa (13%) was obtained.
With the optimized conditions, the substrate scope of the aldehydes was initially studied with 1-phenyl-1-propyne 2a. With benzaldehyde 1a, the reaction provided the corresponding silylated allyl alcohol 3aa in a 79% isolated yield with 97:3 regioselectivity (Scheme 2). Substrates bearing electronically diverse substituents like –F, –Cl, electron-withdrawing –CF3, –CO2Et, and –OMe were compatible under the reaction conditions giving the desired products 3ba–3fa with good yields (71–79%) and regioselectivity (97:3). Whereas, substrates bearing electron-donating groups in conjugation with the aldehyde functionality as in the case of 1g and 1h showed diminished reactivity. At 45 °C, 1g and 1h were successfully transformed to the desired allylic alcohols 3ga and 3ha in moderate yields (54 and 21%, respectively). Similarly, α-branched aliphatic aldehydes 1i–1k smoothly reacted under the standard conditions to provide the corresponding silylated allyl alcohols 3ia–3ka in good to excellent yields (68–80%) and high regioselectivities. Contrarily, linear chain aliphatic aldehydes (1l, m) provided diminished yields of 3l, m (20 and 43%, respectively) under standard conditions. To investigate if the competing base-mediated enolization of linear aliphatic aldehydes was responsible for the diminished yields, IMes carbene was employed directly instead of generating it in situ from the imidazolium salt using a base (Cs2CO3). To our delight, under base-free conditions, 1m provided the desired product 3ma in an enhanced yield (55%, Scheme 2).
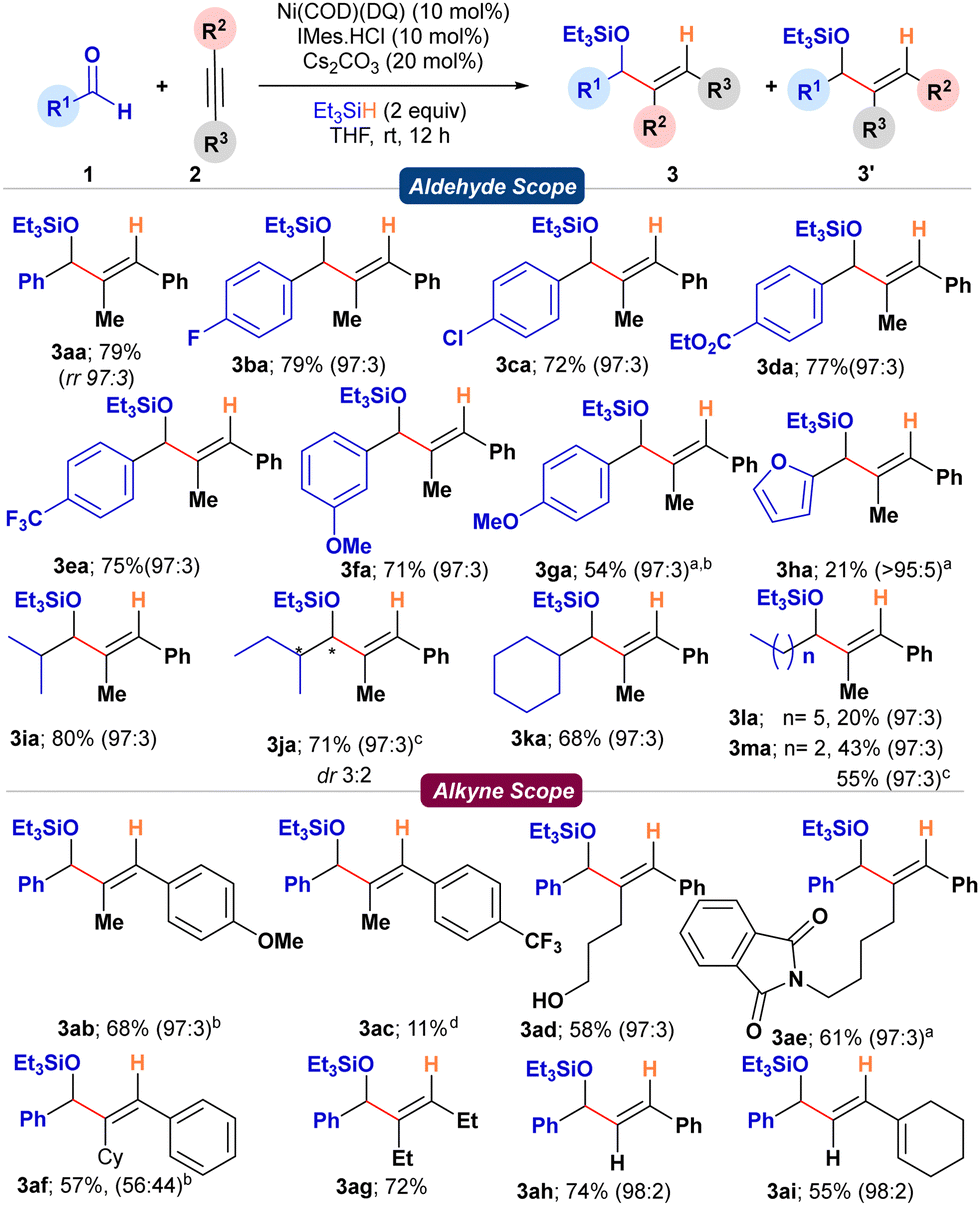 | ||
| Scheme 2 Substrate scope for the reductive coupling of aldehydes and alkynes; aperformed at 45 °C for 24 h; bisolated as free alcohol; cwith free IMes carbene; dstirred for 36 h. | ||
Then, we studied the scope of alkynes with benzaldehyde 1a as the standard aldehyde partner. With 1-(p-anisyl)-1-propyne 1b, the reaction provided the desired silylated allylic alcohol 3ab in 68% yield and 97:3 dr (Scheme 2). However, with an electron-withdrawing –CF3 group at the para-position, the reaction was sluggish to provide the silylated allyl alcohol 3ac in poor yield (11%) even after 36 h. This contrast in reactivity could be attributed to the poor initial coordination of the electron-deficient alkyne with the metal center. The reaction tolerated substrates bearing coordinating functionalities like –OH (2d) and phthalimides (2e), providing the desired functionalized allyl alcohol derivatives 3ad and 3ae in 58 and 61% yields, respectively, without compromising the regioselectivity. However, increasing the steric bulk on the aliphatic chain by introducing cyclohexyl moiety (2f) significantly affected the regioselectivity, providing the product 3af with 57% yield and 1:1 regioselectivity. Dialiphatic substituted (2g) and terminal alkynes (2h, i) smoothly delivered the corresponding products (3ag–3ai) in good yields (55–74%) and excellent regioselectivities. Terminal alkyne 2i was an enyne, and under standard conditions, the formation of the silylated diene-allylic alcohol 3ai is noteworthy (Scheme 2).
We then focused on studying the reductive coupling of ynamides using the bench-stable Ni(COD)(DQ) precatalyst (Scheme 3). The previous report by Sato and coworkers on the reductive coupling restricted to N-alkynyl oxazolidinones with aldehydes using Ni(COD)2/NHC catalyst is noteworthy.12 A quick optimization of the reaction conditions for the reductive coupling of 1a and ynamide 4b with Ni(COD)(DQ) revealed KOtBu (12 mol%) as the suitable base (see ESI†). Applying the above conditions to simple N-Boc-protected ynamide 4a with 1a as partner provided 5aa in good yield (52%; rr >95:5). Due to the inherent electronic bias in ynamides and the coordinating ability of the Boc-group, the reductive coupling proceeded with excellent regiocontrol (>95:5). Under Sato's conditions, using Ni(COD)2/NHC catalyst, 4a provided the coupled product in comparable yield (61% NMR yield; rr > 95:5). The reaction tolerated –F, –Cl, and –CO2Et functionalities on the aldehyde's aromatic ring, providing the corresponding enamide allyl alcohol derivatives 5ab–5ad in good yields (52–59%; rr > 95:5). Similarly, N-alkynyl oxazolidinone 4b smoothly reacted with a range of aryl, heteroaryl, and alkyl aldehydes to provide the corresponding silylated allyl alcohols (5ba–5bk) in good to excellent yields (29–86%). At a 1.8 mmol scale, 1a and 4b efficiently converted to product 5ba in 78% yield under standard conditions. Chiral N-alkynyl oxazolidinone (4c) derived from L-alanine with 1a, under the standard conditions, provided 5ca in 64% yield and 85:15 dr. The scope of N-alkynyl substrates was further expanded to N-alkynyl imidazolidinone. To our delight, N-alkynyl imidazolidinone 4d reacted smoothly with 1a to provide the corresponding product 5da in 51% yield (rr > 95:5). Intrigued by this result, we attempted the double-reductive coupling of N,N′-dialkynyl imidazolidinone 4e with aldehyde 1a. Pleasingly, the reaction provided the double reductive coupling product 5ea in 33% yield and 1:1 dr. Other N-alkynyl substrates bearing an amide or Ts-group failed to provide the coupling products (Scheme 3).
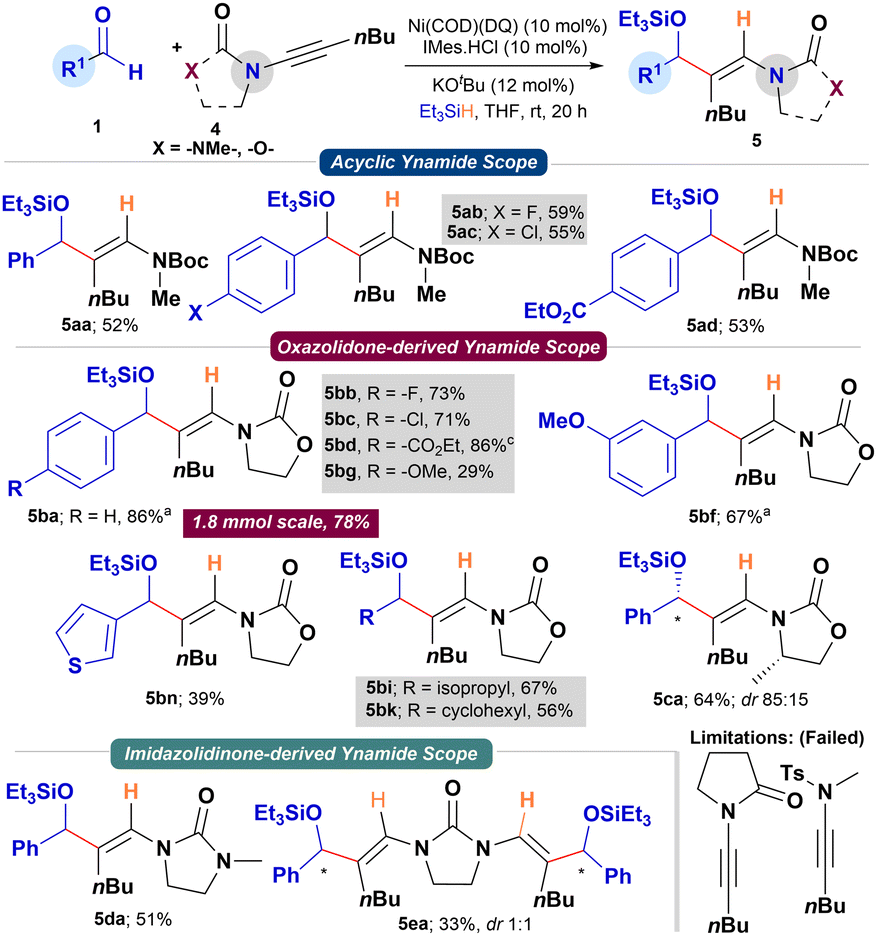 | ||
| Scheme 3 Scope for the reductive coupling of aldehydes and ynamides. a8 h reaction time. | ||
Further, we performed control experiments to determine the precatalyst's benchtop stability and understand the mechanism of the reductive coupling. Engle and coworkers demonstrated the exceptional thermal stability (up to 200 °C) of Ni(COD)(DQ) and other related complexes.9 Here, we studied Ni(COD)(DQ) precatalyst's potential to catalyze the reductive coupling upon prolonged storage over the benchtop (>6 months). The reaction of 1a and 2a under standard reaction conditions, with freshly prepared precatalyst, provided 3aa in 81% yield and 97:3 rr (Scheme 2). The same batch of the precatalyst was stored on the benchtop under ambient conditions without any special precautions. The catalytic activity was tested at regular intervals for up to 200 days, and the yields of 3aa were recorded (see ESI†). Even after 200 days, only minor changes in the catalytic activity (79% yield of 3aa) were observed. Intrigued by the stability of the precatalyst, we further explored if the coupling reaction was tolerant to ambient conditions in the presence of air and moisture. However, the starting materials 1a and 2a failed to react under ambient conditions. A similar outcome was observed when we performed the reaction using degassed moist THF as the solvent, excluding oxygen. These experiments suggested that the precatalyst under the reaction conditions first converted to a more reactive Ni0 species, which drives the catalytic cycle. The EDO ligand (DQ) that stabilized the Ni0 precatalyst could be decomplexing under the reaction conditions followed by subsequent coordination of the starting materials. TLC analysis of the reaction mixture indeed showed a spot corresponding to DQ immediately after adding the starting materials, and the spot remained until the end of the reaction (see ESI†). GCMS trace confirmed the presence of DQ at the end of the reaction. After the completion of the reaction, this spot was separated by column chromatography and characterized by 1H NMR to be DQ (recovery: 83%).
We then attempted the characterization of intermediates by mass spectrometry (MS) analysis by drawing aliquots for analysis at specific stages of the experiment. Mass spectrometry analysis of the mixture prior to the addition of substrates 1a and 2a revealed the formation of [Ni0(NHC)(DQ)] (B, Scheme 4). The mixture was again analyzed by MS after adding the substrates 1a and 2a, followed by stirring at rt for 1 h. To our delight, the signal corresponding to the five-membered oxanickelacycle intermediate was observed, and notably, the DQ ligand was dislodged, supporting our previous observations. DFT supported the preferential decomplexation of COD over DQ from Ni(COD)(DQ), followed by the addition of IMes. The formation of B was thermodynamically more favorable compared to [Ni(IMes)(COD)] by 26 kcal mol−1 (Scheme 4). Enhanced back-bonding into DQ in complex B was evident from the natural charges obtained from the NBO analysis.
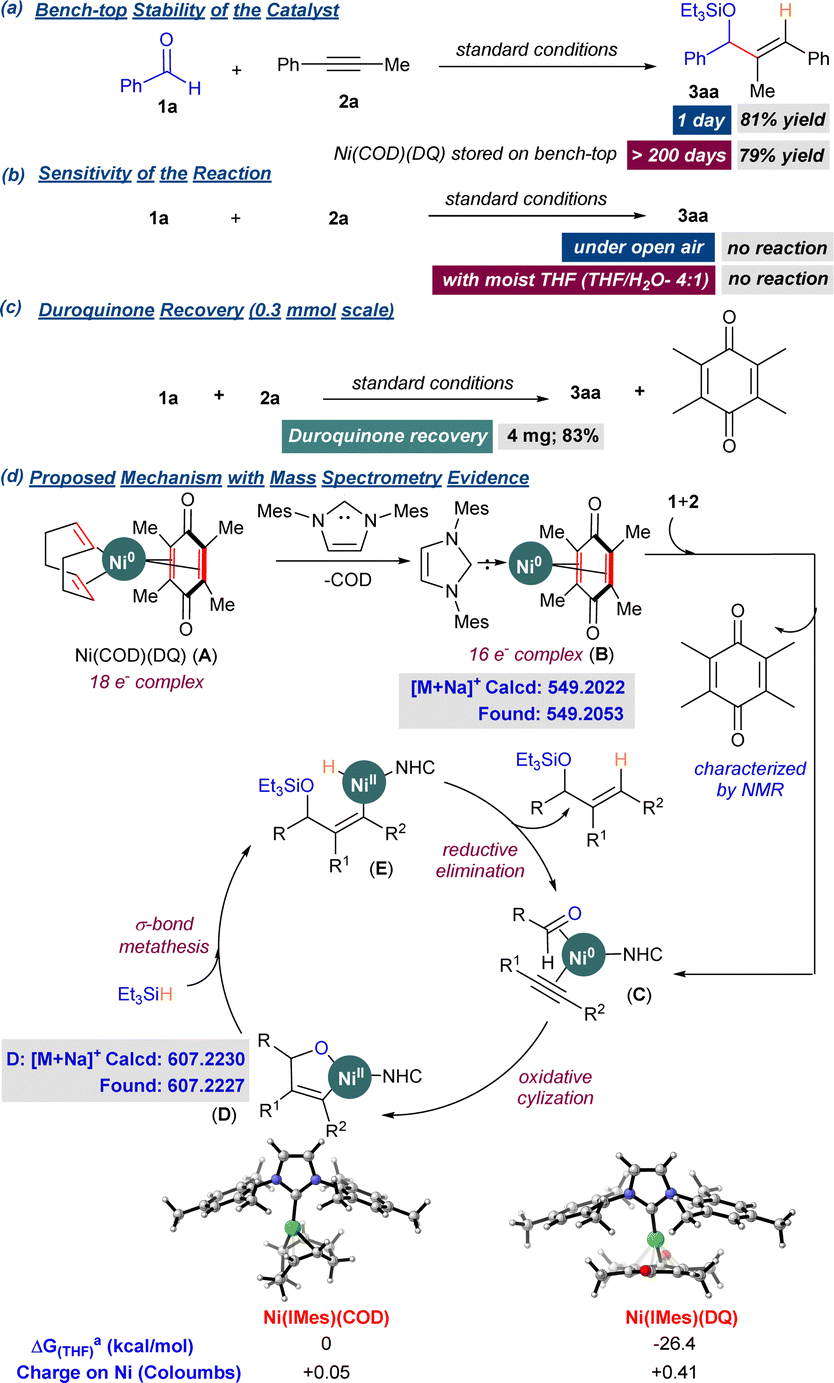 | ||
| Scheme 4 Control experiments and plausible mechanism. aGibbs free energies were calculated at M062X(SMD)/def2-TZVP (THF). Charges (NBO) at M062X/def2-TZVP. | ||
Based on the above observations and the literature on the reductive coupling of aldehydes and alkynes, a plausible mechanism has been presented for the Ni(COD)(DQ)-mediated reductive coupling. Initially, the IMes.HCl salt in the presence of Cs2CO3 enables the formation of IMes carbene. The carbene on complexation with Ni(COD)(DQ) precatalyst provides B (16e− complex). Our attempts to isolate complex B rendered unsuccessful due to extreme air sensitivity. Adding 1 and 2 triggers the decomplexation of DQ, enabling the substrate coordination resulting in complex C. C then undergoes oxidative cyclization to provide the oxanickelacycle D. The intermediate D further undergoes a σ-bond metathesis with Et3SiH to provide the Ni–H intermediate E, which on reductive elimination provides the coupled product 3 and active catalyst is turned over (Scheme 4).
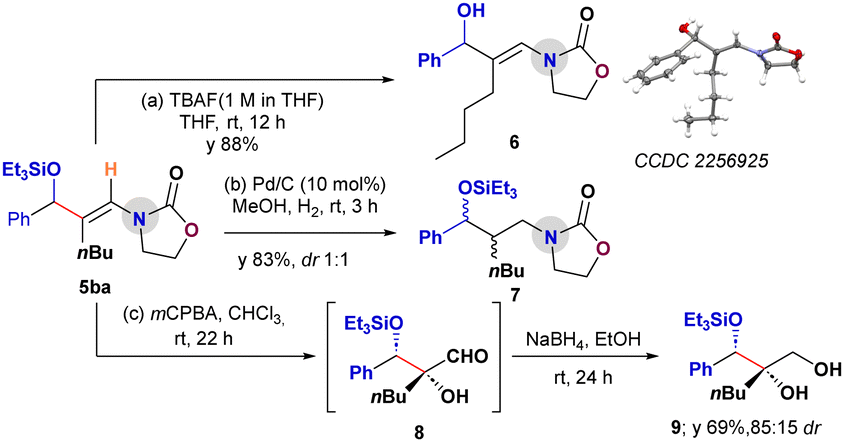
A series of downstream functionalizations of the aldehyde–ynamide coupled product 5ba was planned to demonstrate the synthetic utility of the products (Scheme 5).13 First, the deprotection of the TES-group was successfully carried out on 5ba with TBAF to provide the enamidoalcohol 6 in 88% yield without disturbing the enamide group.13a The structure of 6 was confirmed by X-ray crystallography. Further, the reduction of 5ba with 10% Pd/C and H2 provided the saturated oxazolidinone functionalized alcohol derivative 7 in 83% yield (dr 1:1).13b Finally, treating 5ba with mCPBA under standard epoxidation conditions13c resulted in a cascade epoxidation/oxazolidinone-assisted ring-opening followed by hydrolysis to provide the aldehyde 8. In situ reduction of 8 with NaBH4 delivered the silylated triol 9 in 69% yield (dr 85:15; Scheme 5).
 | ||
| Scheme 5 Synthetic utility of product 5ba. | ||
In conclusion, the study showed that the bench-stable Ni(COD)(DQ) can achieve reductive coupling with comparable efficiency to Ni(COD)2, reducing storage and handling costs. The precatalyst successfully coupled various aromatic/aliphatic aldehydes with alkyne and ynamide partners, providing the products with high yields and regioselectivities. The methodology demonstrated a modular approach for in situ Ni(NHC)(EDO) complex generation, enabling faster ligand screening for enantioselective catalysis. The exceptional stability and catalytic activity of Ni(COD)(DQ) on the bench for over six months offer the potential for user-friendly Ni0 chemistry.
We thank PMRF (F. No. 2400618), SERB (CRG/2022/003322; RJN/085/2018) for the financial support; NSM by C-DAC, MeitY, for computing; & DST (SR/FST/CSII-026/2013) for NMR facility.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) K. Takai, S. Sakamoto, T. Isshiki and T. Kokumai, Tetrahedron, 2006, 62, 7534–7539 CrossRef CAS; (b) Y.-L. Li, S.-Q. Zhang, J. Chen and J.-B. Xia, J. Am. Chem. Soc., 2021, 143, 7306–7313 CrossRef CAS PubMed; (c) F. Calogero, G. Magagnano, S. Potenti, A. Gualandi, A. Fermi, P. Ceroni and P. Giorgio Cozzi, Adv. Synth. Catal., 2022, 364, 3410–3419 CrossRef CAS; (d) K. M. Miller, W.-S. Huang and T. F. Jamison, J. Am. Chem. Soc., 2003, 125, 3442–3443 CrossRef CAS PubMed; (e) G. M. Mahandru, G. Liu and J. Montgomery, J. Am. Chem. Soc., 2004, 126, 3698–3699 CrossRef CAS PubMed; (f) Y.-L. Zheng and M. Ye, Chin. J. Chem., 2020, 38, 489–493 CrossRef CAS.
- (a) D. McTavish and E. M. Sorkin, Drugs, 1991, 42, 65–89 CrossRef CAS PubMed; (b) R. M. Moslin and T. F. Jamison, J. Org. Chem., 2007, 72, 9736–9745 CrossRef CAS PubMed; (c) K. S. Woodin and T. F. Jamison, J. Org. Chem., 2007, 72, 7451–7454 CrossRef CAS PubMed.
- (a) E. Oblinger and J. Montgomery, J. Am. Chem. Soc., 1997, 119, 9065–9066 CrossRef CAS; (b) W.-S. Huang, J. Chan and T. F. Jamison, Org. Lett., 2000, 2, 4221–4223 CrossRef CAS PubMed; (c) H. A. Malik, G. J. Sormunen and J. Montgomery, J. Am. Chem. Soc., 2010, 132, 6304–6305 CrossRef CAS PubMed; (d) E. P. Jackson and J. Montgomery, J. Am. Chem. Soc., 2015, 137, 958–963 CrossRef CAS PubMed; (e) H. Wang, G. Lu, G. J. Sormunen, H. A. Malik, P. Liu and J. Montgomery, J. Am. Chem. Soc., 2017, 139, 9317–9324 CrossRef CAS PubMed.
- V. P. Ananikov, ACS Catal., 2015, 5, 1964–1971 CrossRef CAS.
- (a) P. W. Jolly and G. Wilke, in The Organic Chemistry of Nickel, ed. P. W. Jolly and G. Wilke, Academic Press, 1974, pp. 244–328 Search PubMed; (b) G. N. Schrauzer and H. Thyret, Z. Naturforsch. B, 1962, 17, 73–76 CrossRef.
- (a) J. E. Dander, N. A. Weires and N. K. Garg, Org. Lett., 2016, 18, 3934–3936 CrossRef CAS PubMed; (b) N. D. Clement, K. J. Cavell and L.-L. Ooi, Organometallics, 2006, 25, 4155–4165 CrossRef CAS.
- (a) A. J. Nett, S. Cañellas, Y. Higuchi, M. T. Robo, J. M. Kochkodan, M. T. Haynes, J. W. Kampf and J. Montgomery, ACS Catal., 2018, 8, 6606–6611 CrossRef CAS PubMed; (b) M. T. Robo, A. R. Frank, E. Butler, A. J. Nett, S. Cañellas, P. M. Zimmerman and J. Montgomery, Organometallics, 2022, 41, 3293–3300 CrossRef CAS PubMed.
- N. Kim, V. T. Tran, O. Apolinar, S. R. Wisniewski, M. D. Eastgate and K. M. Engle, Synlett, 2021, 1570–1574 CAS.
- V. T. Tran, N. Kim, C. Z. Rubel, X. Wu, T. Kang, T. C. Jankins, Z.-Q. Li, M. V. Joannou, S. Ayers, M. Gembicky, J. Bailey, E. J. Sturgell, B. B. Sanchez, J. S. Chen, S. Lin, M. D. Eastgate, S. R. Wisniewski and K. M. Engle, Angew. Chem., Int. Ed., 2023, 62, e202211794 CrossRef CAS PubMed.
- (a) V. T. Tran, Z.-Q. Li, O. Apolinar, J. Derosa, M. V. Joannou, S. R. Wisniewski, M. D. Eastgate and K. M. Engle, Angew. Chem., Int. Ed., 2020, 59, 7409–7413 CrossRef CAS PubMed; (b) T. You and J. Li, Org. Lett., 2022, 24, 6642–6646 CrossRef CAS PubMed.
- (a) L. Nattmann and J. Cornella, Organometallics, 2020, 39, 3295–3300 CrossRef CAS; (b) L. Nattmann, R. Saeb, N. Nöthling and J. Cornella, Nat. Catal., 2020, 3, 6–13 CrossRef CAS.
- (a) N. Saito, T. Katayama and Y. Sato, Org. Lett., 2008, 10, 3829–3832 CrossRef CAS PubMed; (b) N. Saito, T. Katayama and Y. Sato, Heterocycles, 2011, 82, 1181–1187 CrossRef CAS PubMed.
- (a) D. A. Evans, T. B. Dunn, L. Kværnø, A. Beauchemin, B. Raymer, E. J. Olhava, J. A. Mulder, M. Juhl, K. Kagechika and D. A. Favor, Angew. Chem., Int. Ed., 2007, 46, 4698–4703 CrossRef PubMed; (b) Q. Jiang, T. Yang, Q. Li, G.-M. Liang, Y. Liu, C.-Y. He, W.-D. Chu and Q.-Z. Liu, Org. Lett., 2023, 25, 3184–3189 CrossRef CAS PubMed; (c) H. Xiong, R. P. Hsung, L. Shen and J. M. Hahn, Tetrahedron Lett., 2002, 43, 4449–4453 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2256925 (6). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc03322h |
| This journal is © The Royal Society of Chemistry 2023 |