
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective cyanation of propargylic C–H bonds via cooperative photoredox and copper catalysis†
Yunshun
Deng‡
a,
Ronghua
Lu‡
b,
Pinhong
Chen
 *b and
Guosheng
Liu
*ab
*b and
Guosheng
Liu
*ab
aDepartment of Chemistry, University of Science and Technology of China, Hefei, 230026, China. E-mail: gliu@mail.sioc.ac.cn
bState Key Laboratory of Organometallic Chemistry, and Shanghai Hongkong Joint Laboratory in Chemical Synthesis, Center for Excellence in Molecular Synthesis, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Shanghai 200032, China. E-mail: pinhongchen@mail.sioc.ac.cn
First published on 21st March 2023
Abstract
Herein, we report an enantioselective cyanation of propargylic C–H bonds by combining photoredox catalysis with a copper-catalyzed radical relay in which the propargylic radical was generated by an intramolecular 1,5-HAT process. This reaction provides easy access to optically pure propargyl nitrile compounds under mild conditions.
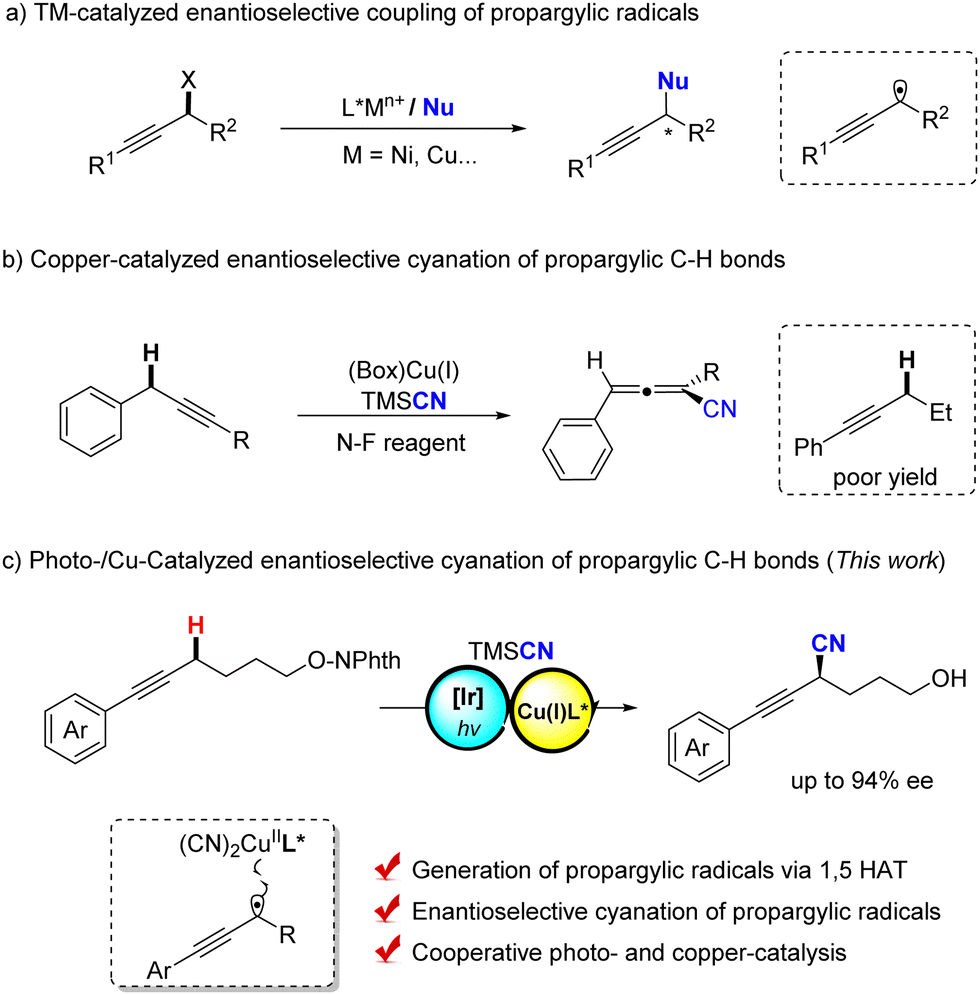
Optically pure alkynes are important unsaturated compounds that are frequently found in natural products, bioactive compounds and polymer materials1 as well as important and useful synthons in organic synthesis.2 Therefore, the synthesis of these chiral alkynes has received much attention and exploring efficient methods is of great significance. The transition metal-catalyzed propargylic functionalization reaction serves as a powerful and attractive tool.3 For instance, transition metal-catalyzed enantioselective propargylic substitutions of propargyl esters, chlorides and alcohols by different nucleophiles have been extensively reported.4 In contrast, owing to the easy availability of simple alkynes, functionalization of propargylic C–H bonds presents the most efficient streamline for their synthesis;5 however, asymmetric reactions still remain a formidably challenging task.6
Recently, transition metal-catalyzed asymmetric radical transformations (ARTs) have been rapidly developed as a powerful tool to construct enantiomeric organic compounds.7 For instance, Fu and coworkers reported an elegant work on the nickel-catalyzed enantioselective arylation of propargylic radicals, which derived from propargylic bromides (Scheme 1a).7d In addition, Xiao and Lu disclosed a dual photo- and copper-catalyzed asymmetric cyanation of propargylic acetates, where the propargylic radical intermediate was generated and trapped by chiral Cu(II) cyanide species in an enantioselective manner.8 However, for the Kharasch-type propargylic C–H oxidation9 that also involves the propargylic radical intermediates, the enantioselective control is extremely difficult and the asymmetric version is far from success.9c
 | ||
| Scheme 1 Catalytic asymmetric functionalization of propargylic radicals. | ||
Since 2016, our group has demonstrated copper-catalyzed radical relays as a powerful strategy for asymmetric radical reactions,10 such as cyanation of benzylic and allylic C–H bonds,11 in which a carbon-centred radical could be enantioselectively trapped by a chiral (Box)CuII(CN)2 species.12 Moreover, the asymmetric arylation and alkynylation of benzylic C–H bonds were also achieved via this stragety.13–15 Very recently, we have disclosed an enantioselective cyanation of propargylic C–H bonds for the construction of structurally diverse chiral allenyl nitriles.16 However, we found that the substrate of phenyl-substituted alkynes exhibited extremely poor reactivity toward the asymmetric C–H cyanation, where the BDE of propargylic C–H bonds is significantly higher than those of previous substrates, resulting in the insufficient HAT process under the reaction conditions. In comparison, the intermolecular 1,5-hydrogen atom transfer pathway has been widely used for the generation of sp3 carbon radicals, thereby achieving the functionalization of remote sp3 C–H bonds.17 Therefore, we reasoned that, if a 1,5-HAT process can be developed to cooperate with copper catalysis, the enantioselective cyanation of propargylic C–H bonds would be realized. Herein, we communicate a novel enantioselective cyanation of propargylic C–H bonds via copper-catalyzed radical relay, which allows for the straightforward and efficient synthesis of optically pure propargylic cyanoalcohols from readily accessible hydroxyl-tethered alkynes (Scheme 1c).
With our previous study,18 the reaction of 1a was started under cooperative photoredox and copper catalysis. As shown in Table 1, the reaction of 1a and TMSCN under irradiation of 2 × 3 W blue LEDs, with fac-Ir(ppy)3 (2 mol %) and Cu(CH3CN)4BF4 (10 mol %)/L1 (15 mol %) in DCM at room temperature can afford the desired cyanation product in a 40% yield with 84% ee (entry 1). It should be noted that the generated alcohol product 2a′ will react with TMSCN to form TMS protected product 2a, without the detection of allenyl nitrile. The radical coupling reaction with chiral Cu(II) cyanides is significantly affected by the steric hindrance of radicals. In this reaction, the result might be attributed to the less steric propargylic radical than allenyl radical. Other photocatalysts with higher oxidation potentials, such as Ir(ppy)2(dtbbpy)PF6 ([Ir]-1) and Ru(bpy)3Cl2 ([Ru]) did not affect the enantioselectivity (84% ee), but afforded the product mixture in lower yields (entries 2, 3). The organic photocatalyst Eosin Y was not suitable for this asymmetric cyanation reaction (entry 4). Further optimization of solvents indicated that the reaction in chlorobenzene afforded product 2a and 2a′ in a comparable yield with the same ee value, and the mass balance was better than that of the reaction in DCM (entries 5–7). Replacement of the cyclopropane unit in the box ligand with an acyclic one slightly increased the yield, but without the loss of enantioselectivity and L2 was the best (entries 8–11). Since the active hydroxyl group in 2a′ will react with TMSCN to generate 2a, the yield can be improved to 80% with 4.0 equivalents of TMSCN, lower catalyst loading and prolonged reaction time making the reaction complete (entries 12 and 13). Finally, lowering the reaction temperature can increase the enantioselectivity of the reaction to 88% (entry 14). Control experiments indicated that both photocatalyst and visible light are essential for this reaction (entry 15).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Photo cat. | Ligand | Solvent | conversion | 2a+2a′ Yieldb (ee) |
| a The reactions were conducted on a 0.1 mmol scale with photocatalyst (2 mol%) and Cu(CH3CN)4BF4 (10 mol%) in solvent (1 mL) at room temperature, irradiated with 2 × 3 W blue LEDs. b Crude 1H NMR yield with CH2Br2 as an internal standard; an enantiomeric excess (ee) value of 2a′ was determined by HPLC on a chiral stationary phase. c With photocatalyst (1 mol%), Cu(CH3CN)4BF4 (5 mol%). d With 4.0 equiv. TMSCN and 60 h. e At 10 °C for 72 h. f Without Ir(ppy)3 or in the dark. [Ir]-1 = Ir(ppy)2(dtbbpy)PF6. [Ru] = Ru(bpy)3Cl2. | |||||
| 1 | Ir(ppy)3 | L1 | DCM | 77% | 40% (84)% |
| 2 | [Ir]-1 | L1 | DCM | 12% | 11% (84%) |
| 3 | [Ru] | L1 | DCM | 15% | Trace (n. d.) |
| 4 | EosinY | L1 | DCM | 0 | 0% |
| 5 | Ir(ppy)3 | L1 | DMF | 13% | 0% |
| 6 | Ir(ppy)3 | L1 | PhCl | 37% | 37% (84%) |
| 7 | Ir(ppy)3 | L1 | CH3CN | 66% | 36% (71%) |
| 8 | Ir(ppy)3 | L2 | DCM | 100% | 57% (84%) |
| 9 | Ir(ppy)3 | L3 | DCM | 95% | 51% (84%) |
| 10 | Ir(ppy)3 | L2 | PhCl | 59% | 55% (84%) |
| 11 | Ir(ppy)3 | L3 | PhCl | 71% | 51% (84%) |
| 12c | Ir(ppy)3 | L2 | PhCl | 100% | 62% (84%) |
| 13cd | Ir(ppy)3 | L2 | PhCl | 100% | 80% (84%) |
| 14cde | Ir(ppy)3 | L2 | PhCl | 100% | 78% (88%) |
| 15f | — | L2 | DCM | 0 | 0 |
|
|||||
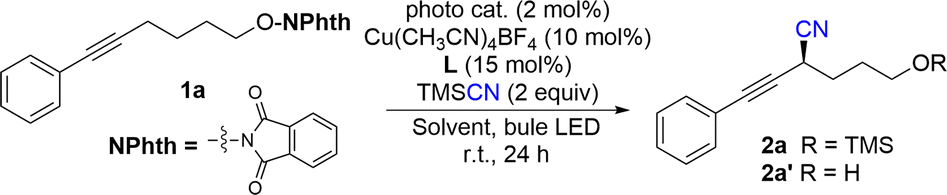
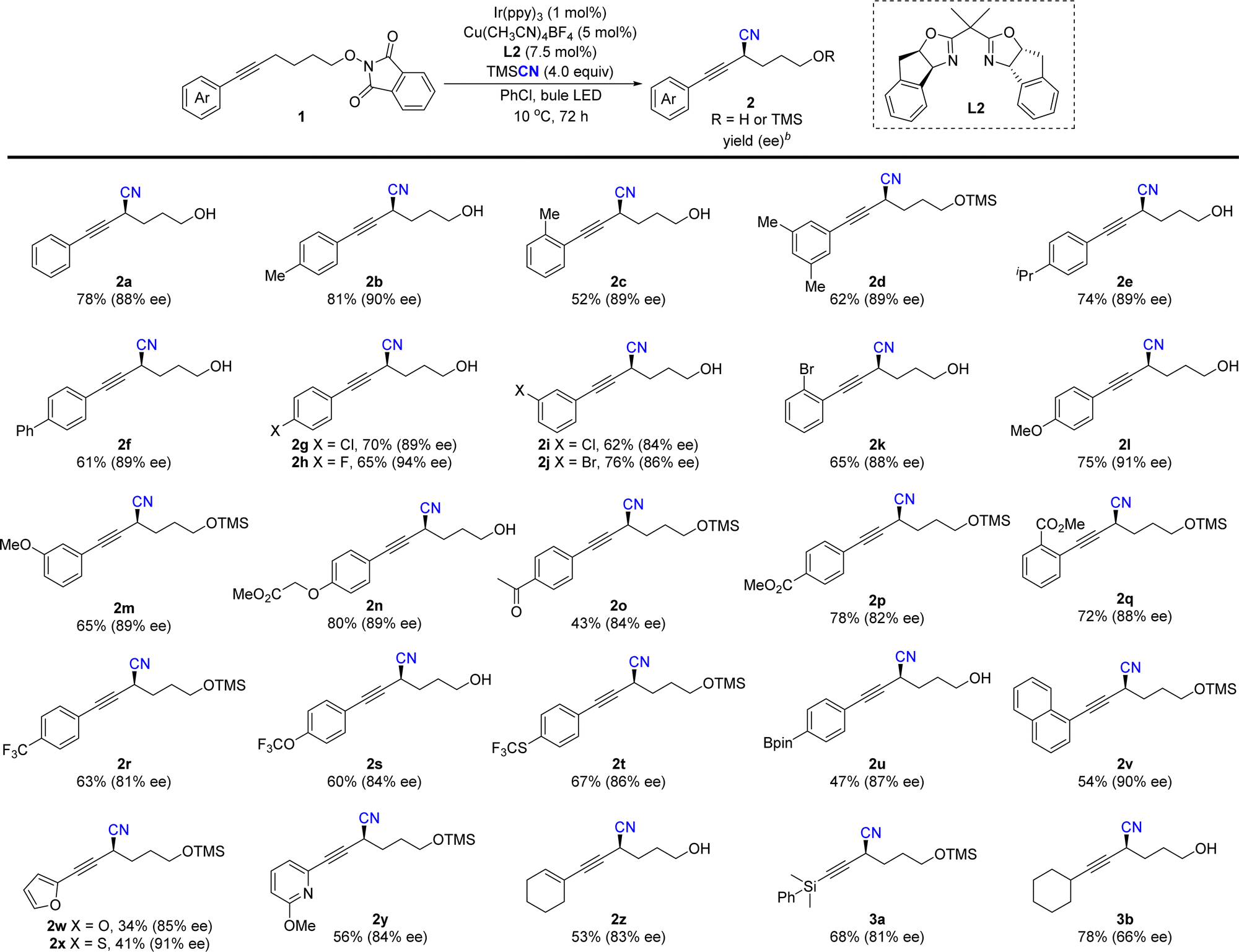
With the optimized reaction conditions in hand, the substrate scope and functional group tolerance were further examined. As shown in Table 2, various phenylethynyl-tethered NHP esters derived from simple alcohols were compatible with the reaction conditions to provide the corresponding products 2a–2q in good yields (43–81%) and excellent enantioselectivities (84–94% ee). A series of functional groups, such as halides, ethers, ketones and esters, were very well tolerated. Fluorine-containing groups such as trifluoromethyl, trifluoromethoxyl and thiotrifluoromethyl groups were tolerated in this reaction (2r–2t). Moreover, aryl boronic ester (Bpin) was also tolerated in the reaction to provide product 2u with excellent enantioselectivity (87% ee), which allows for further transformation. Substrates with meta- and ortho-substituent aryl groups did not affect the reaction. The reaction of 1-alkynyl naphthalene proceeded well to give 2v in 54% yield with 90% ee. Notably, heterocycles including furyl and thianaphthenyl were also suitable for the enantioselective propargylic C–H cyanation to give products 2w and 2x in slightly decreased yields and good enantioselectivities. The reason might be due to the conversion of the corresponding free alcohol products to racemic allenyl nitriles in the presence of fluorides. A 2-methoxyl pyridyl group was also tolerated to deliver 2y in a 56% yield with an 84% ee. More importantly, in addition to aryl-substituted alkynes, enyne 1z was also compatible, giving the target product 2z in 53% yield and 83% ee and the silyl-substituted allkynyl substituents were suitable to give 3a in 68% yield with slightly decreased enantioselectivity. However, the reaction of the alkyl substituted alkyne afforded 3b only in moderate enatioselectivity.
To get more insights into the reaction mechanism, we first measured the reduction potential of 1a to be E0/−1p = −1.51 V vs. SCE in MeCN (see ESI†). The reduction potential of the excited-state photosensitizer Ir(ppy)3* is (E1/2 (IrIV/III = −1.73 V (vs. SCE)),19 which indicates that the excited-state photosensitizer can be quenched by substrate 1a. At the same time, Stern–Volmer fluorescence quenching experiments demonstrated the emission intensity of IrIII* in the presence of substrate 1a (see ESI†). Finally, the light turn on–off experiments suggested a possible nonchain radical process in this reaction (See ESI†).
Based on our mechanistic experiments and previous studies,16,18 we proposed a mechanism as depicted in Scheme 2. Firstly, the photocatalyst Ir(ppy)3 was excited to generate Ir(ppy)3* under the irradiation of a blue LED. The photoexcited Ir(ppy)3* could undergo single electron transfer (SET) reduction with substrate 1 to form radical anion int.I. Subsequently, the radical anion could quickly generate oxygen radical int.II through a cleavage of the N–O bond. Then, the oxygen radical int.II was converted into propargylic radical int.IIIvia a 1,5 -HAT process. At the same time, the oxidized Ir(ppy)3+ was then reduced by L*CuICN to regenerate the ground-state Ir(ppy)3 and formed L*CuIICN rapidly, which could further react with TMSCN to form L*CuII(CN)2. Finally the combination of L*CuII(CN)2 with the propargylic radical gives the target product with a reported stereoinduction model.8
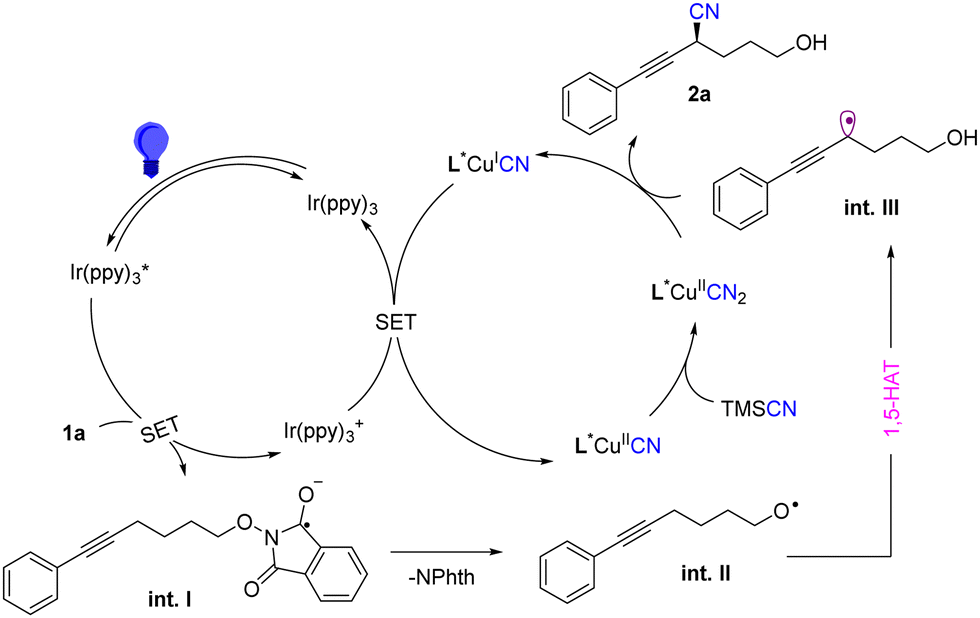 | ||
| Scheme 2 Plausible mechanism. | ||
In conclusion, we have developed an asymmetric cyanation of intramolecular propargylic C–H bonds via copper-catalyzed radical relay, in which a propargylic radical was formed via a photoredox-catalyzed intramolecular 1,5-HAT process. This reaction provides easy access to optically pure propargyl nitrile compounds under very mild conditions. Fluorescence quenching experiments indicated that the reaction was initiated by the oxidative quenching of excited-state photosensitizers.
We are grateful for financial support from the National Key R&D Program of China (2021YFA1500100), the National Natural Science Foundation of China (91956202, 92256301, 21790330, 21821002, and 22171279), the Science and Technology Commission of Shanghai Municipality (20JC1417000, 21520780100, and 19590750400), the Youth Innovation Promotion Association of Chinese Academy of Sciences (No. Y2022074), and the International Partnership Program of the Chinese Academy of Sciences (121731KYSB20190016).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) A. Boulton, B. Davis, D. Durden, L. Dyck, A. Juorio, X. Li, I. Paterson and P. Yu, Drug Dev. Res., 1997, 42, 150 CrossRef CAS; (b) B. M. Trost, and C.-J. Li, Modern Alkyne Chemistry: Catalytic and Atom-Economic Transformations, Wiley-VCH, New York, 2014 Search PubMed; (c) D. Huang, Y. Liu, A.-J. Qin and B.-Z. Tang, Polym. Chem., 2018, 9, 2853 RSC.
- (a) K. Nicholas, Acc. Chem. Res., 1987, 20, 207 CrossRef CAS; (b) C. Li, Acc. Chem. Res., 2010, 43, 581 CrossRef CAS PubMed; (c) R. Gleiter and D. B. Werz, Chem. Rev., 2010, 110, 4447 CrossRef CAS PubMed; (d) V. K. Tiwari, B. B. Mishra, K. B. Mishra, N. Mishra, A. S. Singh and X. Chen, Chem. Rev., 2016, 116, 3086 CrossRef CAS PubMed.
- (a) Y. Miyake, S. Uemura and Y. Nishibayashi, ChemCatChem, 2009, 1, 342 CrossRef CAS; (b) C.-H. Ding and X.-L. Hou, Chem. Rev., 2011, 111, 1914 CrossRef CAS PubMed; (c) D.-Y. Zhang and X.-P. Hu, Tetrahedron Lett., 2015, 56, 283 CrossRef CAS.
- (a) K. Nakajima, M. Shibata and Y. Nishibayashi, J. Am. Chem. Soc., 2015, 137, 2472 CrossRef CAS PubMed; (b) R.-Z. Li, H. Tang, K. Yang, L.-Q. Wan, X. Zhang, J. Liu, Z. Fu and D. Niu, Angew. Chem., Int. Ed., 2017, 56, 7213 CrossRef CAS PubMed; (c) W. Shao, Y. Wang, Z.-P. Yang, X. Zhang and S.-L. You, Chem. – Asian J., 2018, 13, 1103 CrossRef CAS PubMed; (d) X. Gao, R. Cheng, Y.-L. Xiao, X.-L. Wan and X. Zhang, Chemistry, 2019, 5, 2987 CrossRef CAS.
- (a) D. Cheng and W. Bao, J. Org. Chem., 2008, 73, 6881 CrossRef CAS PubMed; (b) R. D. Grigg, J. W. Rigoli, S. D. Pearce and J. M. Schomaker, Org. Lett., 2012, 14(280), 6 Search PubMed; (c) T. Wang, W. Zhou, H. Yin, J.-A. Ma and N. Jiao, Angew. Chem., Int. Ed., 2012, 51, 10823 CrossRef CAS PubMed; (d) H. Lu, C. Li, H. Jiang, C. L. Lizardi and X. P. Zhang, Angew. Chem., Int. Ed., 2014, 53, 7028 CrossRef CAS PubMed; (e) Y. Wang, J. Zhu, A. C. Durham, H. Lindberg and Y.-M. Wang, J. Am. Chem. Soc., 2019, 141, 19594 CrossRef CAS PubMed.
- (a) M. Ju, E. E. Zerull, J. M. Roberts, M. Huang and J. M. Schomaker, J. Am. Chem. Soc., 2020, 142, 12930 CrossRef CAS PubMed; (b) T. Li, X. Cheng, P. Qian and L. Zhang, Nat. Catal., 2021, 4, 164 CrossRef CAS PubMed.
- (a) G. C. Fu, ACS Cent. Sci., 2017, 3, 692 CrossRef CAS PubMed; (b) J. Choi and G. C. Fu, Science, 2017, 356, eaaf7230 CrossRef PubMed; (c) Q. Lu and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 49 CrossRef CAS PubMed; (d) S. W. Smith and G. C. Fu, J. Am. Chem. Soc., 2008, 130, 12645 CrossRef CAS PubMed; (e) A. J. Oelke, J. Sun and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 2966 CrossRef CAS PubMed; (f) N. D. Schley and G. C. Fu, J. Am. Chem. Soc., 2014, 136, 16588 CrossRef CAS PubMed; (g) F.-D. Lu, X. Jiang, L.-Q. Lu and W.-J. Xiao, Acta Chim. Sin., 2019, 77, 803 CrossRef CAS; (h) L. An, F.-F. Tong, S. Zhang and X. Zhang, J. Am. Chem. Soc., 2020, 142, 11884 CrossRef CAS PubMed.
- F.-D. Lu, D. Liu, L. Zhu, L.-Q. Lu, Q. Yang, Q.-Q. Zhou, Y. Wei, Y. Lan and W.-J. Xiao, J. Am. Chem. Soc., 2019, 141, 6167 CrossRef CAS PubMed.
- (a) H. Kropf, R. Schröer and R. Fösing, Synthesis, 1977, 894 CrossRef CAS; (b) L. X. Alvarez, M. L. Christ and A. B. Sorokin, Appl. Catal., A, 2007, 325, 303 CrossRef CAS; (c) J. S. Clark, K. F. Tolhurst, M. Taylor and S. Swallow, Tetrahedron Lett., 1998, 39, 4913 CrossRef CAS.
- For reviews, see: (a) F. Wang, P. Chen and G. Liu, Acc. Chem. Res., 2018, 51, 2036 CrossRef CAS PubMed; (b) Z. Zhang, P. Chen and G. Liu, Chem. Soc. Rev., 2022, 51, 1640 RSC; (c) F. Wang, P. Chen and G. Liu, Nat. Synth., 2022, 1, 107 CrossRef.
- For some examples, see: (a) W. Zhang, F. Wang, S. D. McCann, D. Wang, P. Chen, S. S. Stahl and G. Liu, Science, 2016, 353, 1014 CrossRef CAS PubMed; (b) J. Li, Z. Zhang, L. Wu, W. Zhang, P. Chen, Z. Lin and G. Liu, Nature, 2019, 574, 516 CrossRef CAS PubMed.
- For selected asymmetric radical cyanations, see: (a) F. Wang, D. Wang, X. Wan, L. Wu, P. Chen and G. Liu, J. Am. Chem. Soc., 2016, 138, 15547 CrossRef CAS PubMed; (b) D. Wang, F. Wang, P. Chen, Z. Lin and G. Liu, Angew. Chem., Int. Ed., 2017, 56, 2054 CrossRef CAS PubMed; (c) D. Wang, N. Zhu, P. Chen, Z. Lin and G. Liu, J. Am. Chem. Soc., 2017, 139, 15632 CrossRef CAS PubMed; (d) G. Zhang, L. Fu, P. Chen, J. Zou and G. Liu, Org. Lett., 2019, 21, 5015 CrossRef CAS PubMed; (e) W. Zhuang, P. Chen and G. Liu, Chin. J. Chem., 2021, 39, 50 CrossRef CAS; (f) S. Zhou, G. Zhang, L. Fu, P. Chen, Y. Li and G. Liu, Org. Lett., 2020, 22, 6299 CrossRef CAS PubMed; (g) L. Wu, L. Wang, P. Chen, Y.-L. Guo and G. Liu, Adv. Synth. Catal., 2020, 362, 2189 CrossRef.
- (a) W. Zhang, L. Wu, P. Chen and G. Liu, Angew. Chem., Int. Ed., 2019, 58, 6425 CrossRef CAS PubMed; (b) L. Fu, Z. Zhang, P. Chen, Z. Lin and G. Liu, J. Am. Chem. Soc., 2020, 142, 12493 CrossRef CAS PubMed.
- For selected asymmetric arylations, see: (a) L. Wu, F. Wang, X. Wan, D. Wang, P. Chen and G. Liu, J. Am. Chem. Soc., 2017, 139, 2904 CrossRef CAS PubMed; (b) D. Wang, L. Wu, F. Wang, X. Wan, P. Chen, Z. Lin and G. Liu, J. Am. Chem. Soc., 2017, 139, 6811 CrossRef CAS PubMed; (c) L. Wu, F. Wang, P. Chen and G. Liu, J. Am. Chem. Soc., 2019, 141, 1887 CrossRef CAS PubMed.
- For selected asymmetric alkynylation, see: (a) L. Fu, S. Zhou, X. Wan, P. Chen and G. Liu, J. Am. Chem. Soc., 2018, 140, 10965 CrossRef CAS PubMed; (b) Z. Hu, L. Fu, P. Chen and G. Liu, Org. Lett., 2020, 23, 129 CrossRef PubMed.
- R. Lu, T. Yang, X. Chen, W. Fan, P. Chen, Z. Lin and G. Liu, J. Am. Chem. Soc., 2021, 143, 14451 CrossRef CAS PubMed.
- (a) M. E. Wolff, Chem. Rev., 1963, 63, 55 CrossRef CAS; (b) L. M. Stateman, K. M. Nakafuku and D. A. Nagib, Synthesis, 2018, 1569 CAS.
- Z. Cheng, P. Chen and G. Liu, Acta. Chim. Sin., 2019, 77, 856 CrossRef CAS.
- K. C. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and compound characterization data. See DOI: https://doi.org/10.1039/d3cc00410d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |