
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-healable dynamic poly(urea-urethane) gel electrolyte for lithium batteries†
Fermin
Elizalde
a,
Julia
Amici
 b,
Sabrina
Trano
b,
Giulia
Vozzolo
a,
Robert
Aguirresarobe
a,
Daniele
Versaci
b,
Silvia
Bodoardo
b,
David
Mecerreyes
a,
Haritz
Sardon
*a and
Federico
Bella
*b
b,
Sabrina
Trano
b,
Giulia
Vozzolo
a,
Robert
Aguirresarobe
a,
Daniele
Versaci
b,
Silvia
Bodoardo
b,
David
Mecerreyes
a,
Haritz
Sardon
*a and
Federico
Bella
*b
aPOLYMAT, University of the Basque Country UPV/EHU, Joxe Mari Korta Center Avda. Tolosa 72, 20018 Donostia-San Sebastian, Spain. E-mail: haritz.sardon@ehu.es
bDepartment of Applied Science and Technology, Politecnico di Torino, Corso Duca degli Abruzzi 24, 10129 Turin, Italy. E-mail: federico.bella@polito.it
First published on 31st May 2022
Abstract
Self-healing materials are actively studied in order to extend the lifetime and performance of batteries. Dynamic covalent networks have recently emerged as one of the best self-healable materials which allow thermosets to be reprocessed and recycled. Among all the different exchangeable bonds studied over the last few years, hindered urea bonds appear to be one of the most feasible options to create self-healable materials due to their exchange activation at low temperatures. Although this chemistry is very popular in composite and coating applications, it has not been considered for designing self-healable materials for batteries. In this work we synthesize a membrane containing dynamic hindered urea crosslinking points by reacting tris[2-(isopropylamino)ethyl]amine with hexamethylene diisocyanate, followed by the addition of polyethylene glycol. It is proved that this newly designed material possesses self-healable properties and higher ionic conductivity than the commercially available liquid electrolyte embedded in a porous Celgard® 2500 separator. The polyurethane gel electrolyte shows very homogeneous Li plating and stripping in Li symmetrical cells and is also compatible with Li-mediated electrochemical ammonia synthesis approaches. Most importantly, after severely mechanically damaging the gel membrane, the polymer electrolyte shows great recovery of the electrochemical properties, experiencing more than 100 charge/discharge cycles (after cutting) at C/5 rate.
1. Introduction
Over the past few decades, Li-ion batteries (LIBs) have enabled the development of portable electronic devices (mobile phones, laptops, digital cameras, etc.) and are also the best choice for electric vehicles.1–3 This technology is based on the rocking chair concept, where Li ions shuttle between electrodes during charge and discharge cycles.4–6 Unfortunately, this technology is now reaching its theoretical energy density value and therefore will probably not be able to meet the foreseen future energy needs.7 One strategy to enhance the energy density of these energy storage devices is to directly use Li-metal as an anode material, thanks to its high theoretical capacity and low reduction potential.8,9 Nevertheless, the practicality of Li-metal batteries is limited to the inevitable damage provoked by Li dendrite growth,10,11 which can either propagate through the separator after repetitive cycling processes, leading to the final short-circuiting of the cell, or, for the thinner ones, break from the roots, forming the so-called “dead lithium”.12,13 The aforementioned Li dendrite growth is mainly caused by the spontaneous formation of an unstable solid electrolyte interphase, which turns out to be fragile and heterogeneous with variable spatial resistance, thus inducing uneven Li ion flow and random Li deposition underneath.14 This mechanical instability and heterogeneity of the layer has become the main issue for the commercialization of batteries based on Li-metal anodes.15 In this scenario, the scientific community is fully aware that Li-metal batteries need continuous development to reach further and more demanding applications. Furthermore, the scenario of Li-mediated ammonia electrosynthesis through Li–N2 cells has recently emerged16,17 and stable Li+ conducting polymer membranes represent a strategic component.Taking into account the growing awareness of global sustainability and the increasing demand for energy storage and conversion devices, one of the greatest upcoming challenges nowadays is to develop effective, safe and recyclable electrochemical devices.18 With the aim of overcoming undesired failures, solid polymer electrolytes (SPEs) have emerged as mechanically robust materials with low flammability properties, improved safety and good thermal stability.19–23 However, when unwanted scratches form in the material, these advantages disappear, resulting in catastrophic failure.24
Self-healing materials based on covalent adaptable networks (CANs) have been studied for several applications,25 but their exploitation is still a promising target for Li-metal batteries. Up to now, most published studies for this application have reported mainly self-healing mechanisms based on the formation of hydrogen bonds,26 and the research activity in much more related to the development of self-healing binders than SPEs.27 Few examples can be found in the literature, such as aliphatic disulfides24 and boronic esters.28 As previously reported, Li-metal batteries can lose efficiency and present safety problems after the electrolyte/separator component is damaged.1,29 Indeed, as can be seen in Fig. 1A, when the SPE is not able to self-repair, a simple scratch (that can occur during battery operation under real conditions) can create an easy pathway for faster dendrite growth and eventual short-circuiting of the whole cell.30 In contrast, self-healable polymer electrolytes can rapidly eliminate scratches and avoid the final shorting of the device, allowing for a longer and safer cycle life (Fig. 1B).
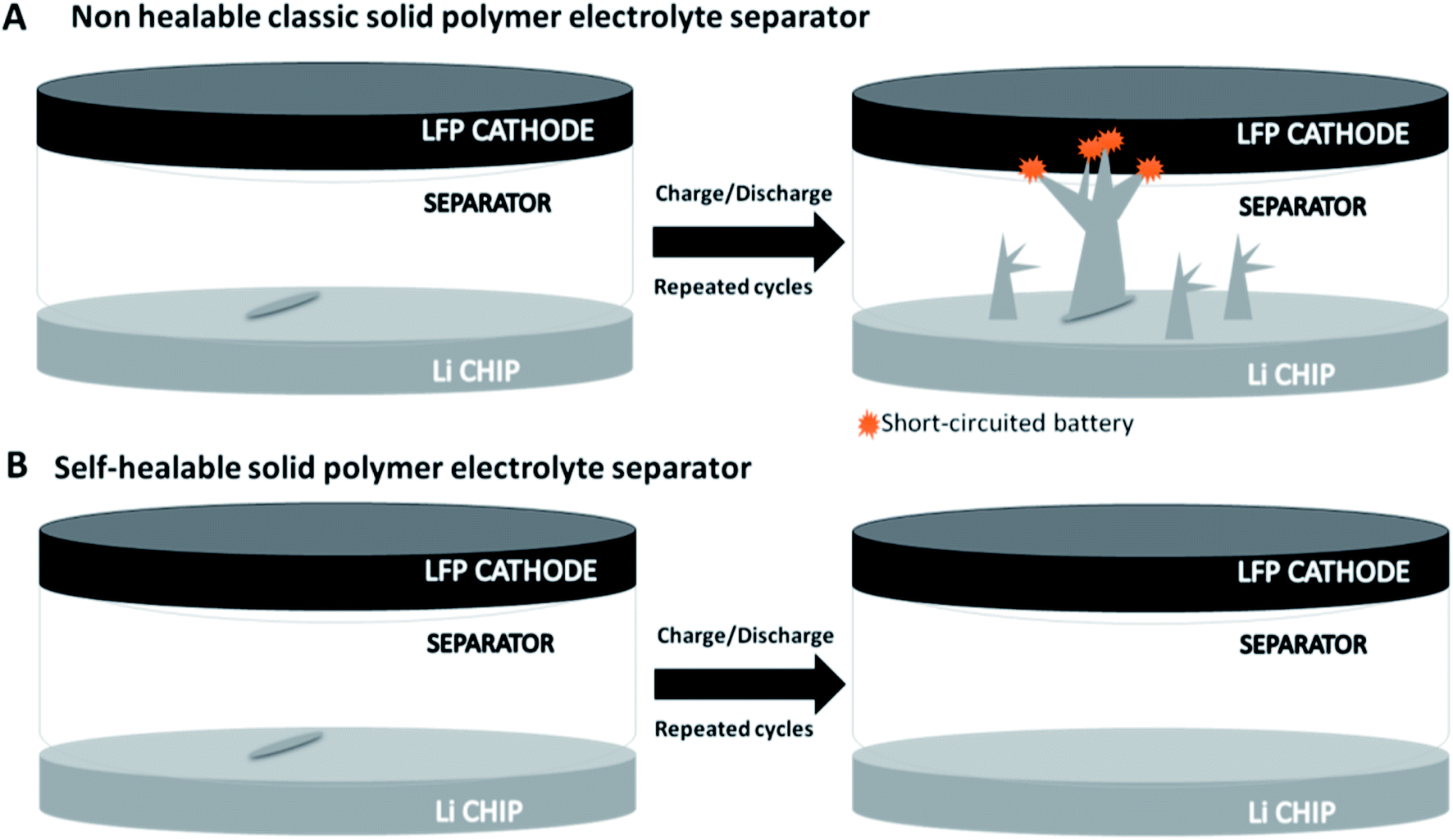 | ||
| Fig. 1 (A) Scratched classic SPE separator before and after repeated charge and discharge cycles. (B) Scratched self-healable SPE separator to avoid dendrite growth after repeated charge/discharge cycles. | ||
Polyurethanes are one of the most versatile families of polymers and are commercially available as coatings, elastomers, textile fibers, adhesives and rigid or flexible foams.29,31 Their adaptable synthesis allows ad hoc material properties tailored to their final application.32,33 Besides, most of the starting reactants (common polyols, isocyanates and small molecular weight diols/diamines) are well known and have been employed industrially for several decades; polyurethanes still offer an open door for the introduction of more sophisticated compounds that could play a key role in advanced energy storage applications. As an example, single-ion conducting polyurethane electrolytes were studied by Porcarelli et al.,34 while a review article on polyurethane-based polymer electrolytes for LIBs has recently summarized the main advantages.35
Recently, Cheng et al. reported the first dynamic polyurea thermosets based on hindered urea bonds (HUBs).36 They concluded that the introduction of bulky substituents on the nitrogen atom weakens the planarity of the amide bond, reducing the stability of the urea bonds and resulting in the dissociation of isocyanate and amine groups under mild conditions. In comparison with other exchangeable bonds, HUBs significantly weakened the bonding energy of the amide bond and reduced the reverse reaction conditions. They showed intrinsically dynamic reaction without catalyst at room temperature, thus allowing the self-healing of HUB-based thermosets under ambient conditions. Since this discovery, these dynamic bonds have gained incredible attention due to the excellent self-healing, recycling and shape-memory properties that they exhibit.37 However, to the best of our knowledge this successful chemistry has not yet been considered by the scientific community operating in the electrochemical energy storage field.
In this work, we have selectively synthesized dynamic crosslinking points based on HUBs to subsequently create a self-healable crosslinked poly(urea-urethane) network by the addition of polyethylene glycol 2000 (PEG2000); this matrix will be referred to, from now on, as the HUB-PU network. After checking the effective self-healing behavior of the newly proposed membrane, complete electrochemical characterization was carried out to assess its potential application as gel-polymer electrolyte in Li-metal batteries, and the results were compared to those obtained for a Celgard® 2500 separator embedded with liquid electrolyte. Galvanostatic cycling demonstrated—just after cell assembly—similar performances between the newly proposed SPE and Celgard® 2500. Our final goal is to investigate the self-healing ability of the HUB-PU membrane and its performance in batteries after cutting and healing cycles as compared to the commonly used porous separators.
2. Results and discussion
2.1 Synthesis of a self-healable crosslinked poly(urea-urethane) network: the HUB-PU network
Fig. 2A describes the synthetic route to cross-linked poly(urea-urethane) networks in two steps by reacting first a trifunctional bulky secondary amine with a diisocyanate in order to prepare a functional urethane polyurea precursor. In the second step, the HUB-PU network is formed by reacting with PEG2000. Every step of the reported procedure was followed by FTIR spectroscopy, as shown in Fig. 2B. As can be seen in the spectra, during the synthesis of the HUB-PU network, the isocyanate stretching band at 2256 cm−1 completely disappeared and a new band, corresponding to the formation of the urea group, appeared at 1618 cm−1, followed by the corresponding band of urethane group at 1717 cm−1. A representative picture of the crosslinked HUB-PU film is also shown in Fig. 2C.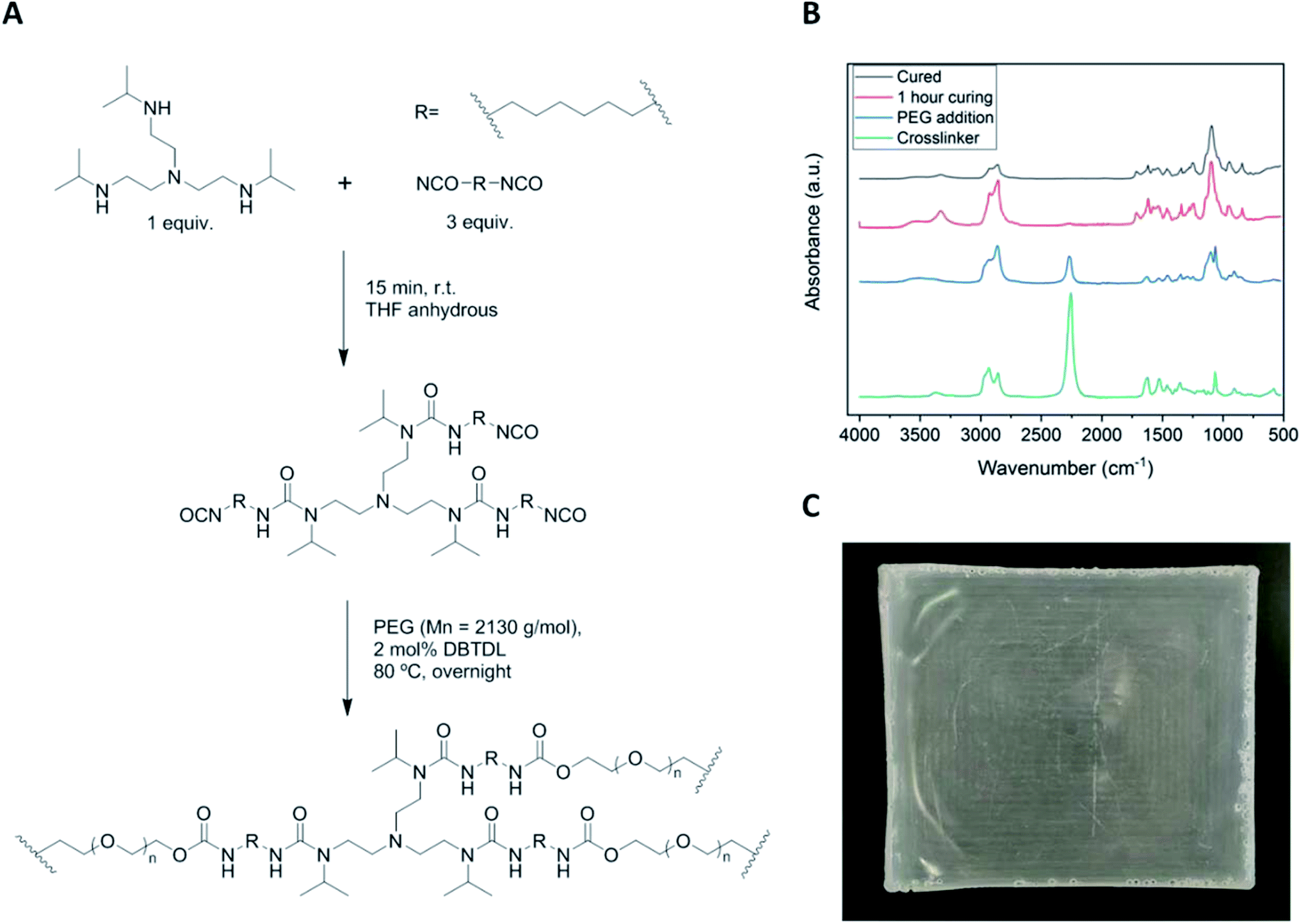 | ||
| Fig. 2 (A) Synthetic procedure for obtaining cross-linked aliphatic polyurethanes with dynamic crosslinking points based on HUBs. (B) Representative FTIR spectra of the reaction between tris[2-(isopropylamino)ethyl]amine and hexamethylene diisocyanate (green spectrum). The spectrum obtained after addition of PEG2000 to the mixture (blue spectrum) and the spectrum obtained after 1 h of curing (red spectrum) are also shown. Finally, the black spectrum corresponds to the final poly(urea-urethane) film crosslinked for 12 h at 80 °C. (C) Representative image of one of the synthesized HUB-PU networks. | ||
2.2 Self-healing ability of the HUB-PU network
The self-healing ability of the synthesized dynamic network was characterized by optical microscopy. Firstly, the self-healing behavior of the HUB-PU network (before being activated with Li salt) was analyzed under pressure and heating up to 80 °C. This temperature was applied in order to be above the melting temperature of PEG2000, thus increasing chain mobility within the membrane. We noticed that, depending on the pressure applied, the self-healing time could vary between 4 h (with a squeezer clamp, Fig. S1†) and 12 h (sample placed between two glass pieces and closed with paper clips). A representative image is given in Fig. 3A.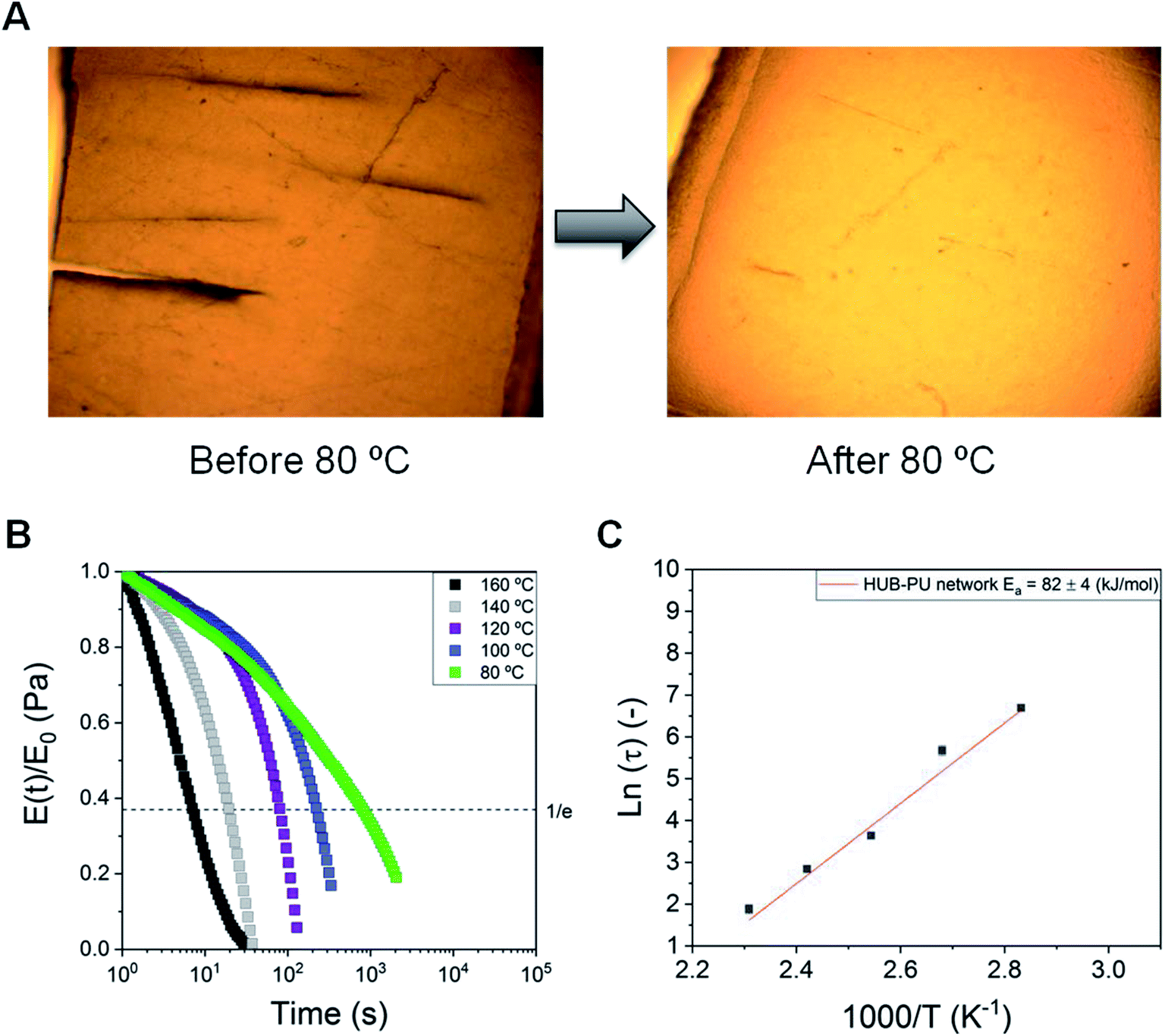 | ||
| Fig. 3 (A) Scratch disappearance in a raw dynamic poly(urea-urethane) network membrane after being kept overnight at 80 °C with pressure (applied by closing two glass pieces with paper clips). (B) Stress–relaxation analysis for the HUB-PU network, performed between 80 and 160 °C. (C) Arrhenius plot of characteristic relaxation times of the HUB-PU network. | ||
As regards the rheological study of the dynamic behavior of the HUB-PU network, stress-relaxation measurements were carried out at 80, 100, 120, 140 and 160 °C (Fig. 3B). As can be seen, samples showed a decay in the relaxation moduli with significant temperature dependence. The relaxation curves showed fast relaxation under mild conditions and the material did not show any sign of degradation even at 160 °C. The characteristic Arrhenius plot and activation energy for the HUB-PU network are shown in Fig. 3C. This plot was obtained from stress–relaxation measurements carried out between 80 °C and 160 °C. It can be concluded that the obtained characteristic low activation energy value is common for dissociative CANs.
In order to properly understand the dynamic behavior of the sample when used under real conditions, the swollen membrane (i.e., the HUB-PU network activated with a typical Li-metal battery electrolyte) was also analyzed, under pressure, to properly see the scratch disappearance (Fig. S2A†). Clearly, the swelling of the membrane with liquid electrolyte decreased its crystallinity (i.e., the membrane became translucent), thus enhancing chain mobility and allowing the self-healing of the scratch even at room temperature. Such an achievement demonstrated the very promising features of the newly designed SPE for application in Li-metal batteries. Stress–relaxation measurements of the swollen membrane also confirmed the dynamicity of the material at room temperature (Fig. S2B†).
2.3 Electrochemical characterization of gel-polymer electrolyte based on a swollen HUB-PU network
A thorough electrochemical characterization was carried out in order to understand and analyze the most important electrochemical characteristics of the HUB-PU network, also comparing them to those featuring the commercial separator Celgard® 2500.A mandatory step to activate the proposed polymer electrolyte is to soak the membrane in a standard liquid electrolyte, i.e. 1.0 M LiPF6 in a mixture of ethylene carbonate and diethyl carbonate (EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DEC 1:1, v/v), for at least 30 min; such a step permits the uptake of the solvated Li ions and the liquid phase, thus ensuring a good ionic mobility within the polymeric network.
:DEC 1:1, v/v), for at least 30 min; such a step permits the uptake of the solvated Li ions and the liquid phase, thus ensuring a good ionic mobility within the polymeric network.
As a result of this process, a self-standing polymer electrolyte membrane was obtained, that was subsequently cut and used in lab-scale Li-metal cells (ECC-Std geometry). The electrolyte uptake was measured with time (Fig. S3†) and, overall, it was observed that the membrane was able to swell up to 590% of its initial weight.
Ionic conductivity represents the first key parameter to evaluate electrolyte performance. The temperature dependence of ionic conductivity obtained in a temperature range from 20 to 60 °C for both the HUB-PU network and Celgard® 2500 is shown in Fig. 4A. Noteworthily, the HUB-PU network presented a higher ionic conductivity than the commercial separator over the whole temperature range. A possible explanation could be that the activation of the newly proposed membrane with the liquid electrolyte resulted in multidirectional swelling of the polymer molecular chains, forming more amorphous regions and therefore providing effective channels for Li ion migration.38
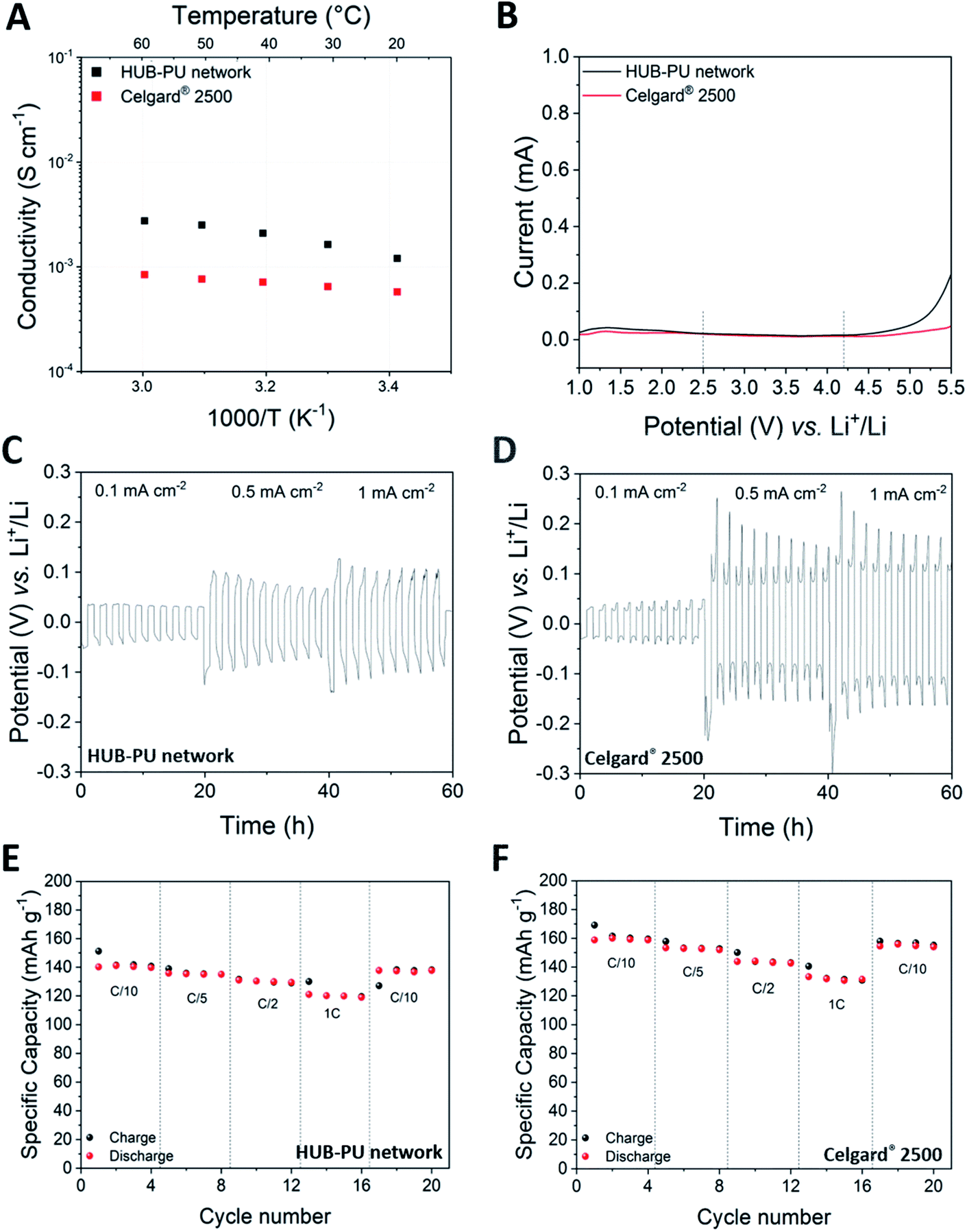 | ||
| Fig. 4 (A) Ionic conductivity values at different temperatures for the HUB-PU network and Celgard® 2500 electrolytes. (B) ESW of the HUB-PU network and Celgard® 2500. (C and D) Potential vs. test time of Li stripping and plating for a symmetrical Li/Li cell at various current densities and at room temperature, for the HUB-PU network and Celgard® 2500 samples, respectively. (E and F) Charge/discharge performances of Li/electrolyte/LiFePO4 cells at various C-rates in the presence of the HUB-PU network and Celgard® 2500 electrolytes, respectively. | ||
The electrochemical stability window (ESW) of the electrolyte, i.e. the figure of merit that defines the voltage range in which the electrolyte can work safely, was the second feature we investigated for the HUB-PU membrane. This assessment is very important for a new polymeric matrix proposed for Li-metal batteries, since if the electrochemical potential of the anode is above the electrolyte reduction potential, it could lead to the reduction of the electrolyte. Similarly, if the electrochemical potential of the cathode is below the electrolyte oxidation potential, it could lead to the oxidation of the electrolyte.39 Taking into account that the charge and discharge cycles of cells based on the Li/membrane/LiFePO4 cathode architecture are usually performed between 2.5 and 4.2 V, the ESW of our HUB-PU electrolyte must be wider than this potential range. According to Fig. 4B, the HUB-PU network showed a stability window that fully satisfies the operation conditions of a Li-metal cell. As regards Celgard® 2500, it showed an even wider ESW; overall, both materials were demonstrated to be perfectly stable in the range of interest and were subsequently subjected to further electrochemical characterization.
Galvanostatic cycling measurements were initially carried out on symmetric Li/Li cells to study the plating and stripping behavior of Li, which may determine Li dendrite nucleation and growth.40 Potential profiles of Li stripping and plating at various current densities (i.e., 0.1, 0.5 and 1 mA cm−2) at 25 °C were reported for the HUB-PU network and Celgard® 2500 samples in Fig. 4C and D, respectively. When the current densities were increased from 0.1 to 0.5 and 1 mA cm−2, a great change of overpotential was noticed in both materials, this increase being much more noticeable for Celgard® 2500. These electrochemical tests confirmed that both materials were quite efficient against dendrite growth at low current density (0.1 mA cm−2). At higher values, visible changes appeared in the voltage profiles. In particular, the cell with Celgard® 2500 showed a peaking shape, which has already been reported in the literature as the result of different (and unfavored) kinetic pathways for reactions at the electrode/electrolyte interface.41 Such shape change was not detected in the cell assembled with the HUB-PU network. Overall, these observations demonstrated a much easier plating/stripping process and stabilized interface for the HUB-PU network, indicating restrained Li dendrite growth and highly stable Li plating/stripping reversibility.42 The latter has also been proved by the Li plating and stripping behaviour (see Fig. S4†) of the Li/HUB-PU membrane/Cu cell, the Coulombic efficiency values of which, even for higher current densities, are encouraging and worthy of further analysis.
To further study and compare the performance of the HUB-PU network and Celgard® 2500, Li-metal cells with a LiFePO4 (LFP) cathode were assembled and tested at room temperature. Fig. 4E and F present the cell performances at different charge and discharge rates (from C/10 to 1C) in the presence of the HUB-PU network or Celgard® 2500, respectively. The newly proposed polymer electrolyte showed high capacity values at every C-rate, even if slightly lower than those shown by Celgard® 2500 (121 mA h g−1 and 131 mA h g−1 at 1C, respectively). Despite this, the cell capacities obtained remain rather impressive for a polymer electrolyte obtained by a new synthetic process, never explored before by the electrochemistry community, and it is able to work at room temperature (especially at 1C). Nevertheless, it is also noteworthy to highlight the more stable specific capacities provided by the HUB-PU network, when compared to the Celgard® 2500 counterpart, after 200 cycles at constant C/10 current (Fig. S5†) with 93.35% of the 10th cycle capacity retained for the HUB-PU network, against 89.78% for Celgard® 2500.
2.4 Performance of Li-metal cells after mechanical damage and healing of the HUB-PU gel polymer electrolyte
The charge/discharge capacity comparison at C/5 rate was performed between the HUB-PU network and Celgard® 2500 samples in order to analyze how each membrane was able to recover after being cut, i.e. mechanical damage. To properly carry out this characterization, first each membrane was cycled at the abovementioned rate for 10 cycles. Right after this, both cells were put into the glovebox and were disassembled. Carefully, the LFP cathode was removed and, with a surgeon's knife, each membrane got a similar cut (see Fig. 5A and B). Successively, the electrochemical cells were reassembled and the cycling test was resumed at the same C/5 rate. The obtained results were very different. As shown in Fig. 5C and D, the self-healable membrane was able to charge and discharge again, giving capacity values very close to the ones obtained during the first 10 cycles. In particular, the capacity just before the cut was around 161 mA h g−1, while just after the cut 159 mA h g−1 was recovered. Indeed, after the cut, the HUB-PU network showed really good performance for another 50 cycles, with capacity retention of 94.78% at the 50th cycle with respect to the 10th cycle. Later, this capacity decreases slowly to lower values (as typical for non-optimized lab-scale cells), but it still works even after 100 cycles (Fig. S6†), retaining 79.68% the capacity at the 10th cycle. Of note, from our literature research (see Table S1†), we conclude that no previous studies were carried out with polymer electrolytes directly cut in the electrochemical cell and cycled while still damaged.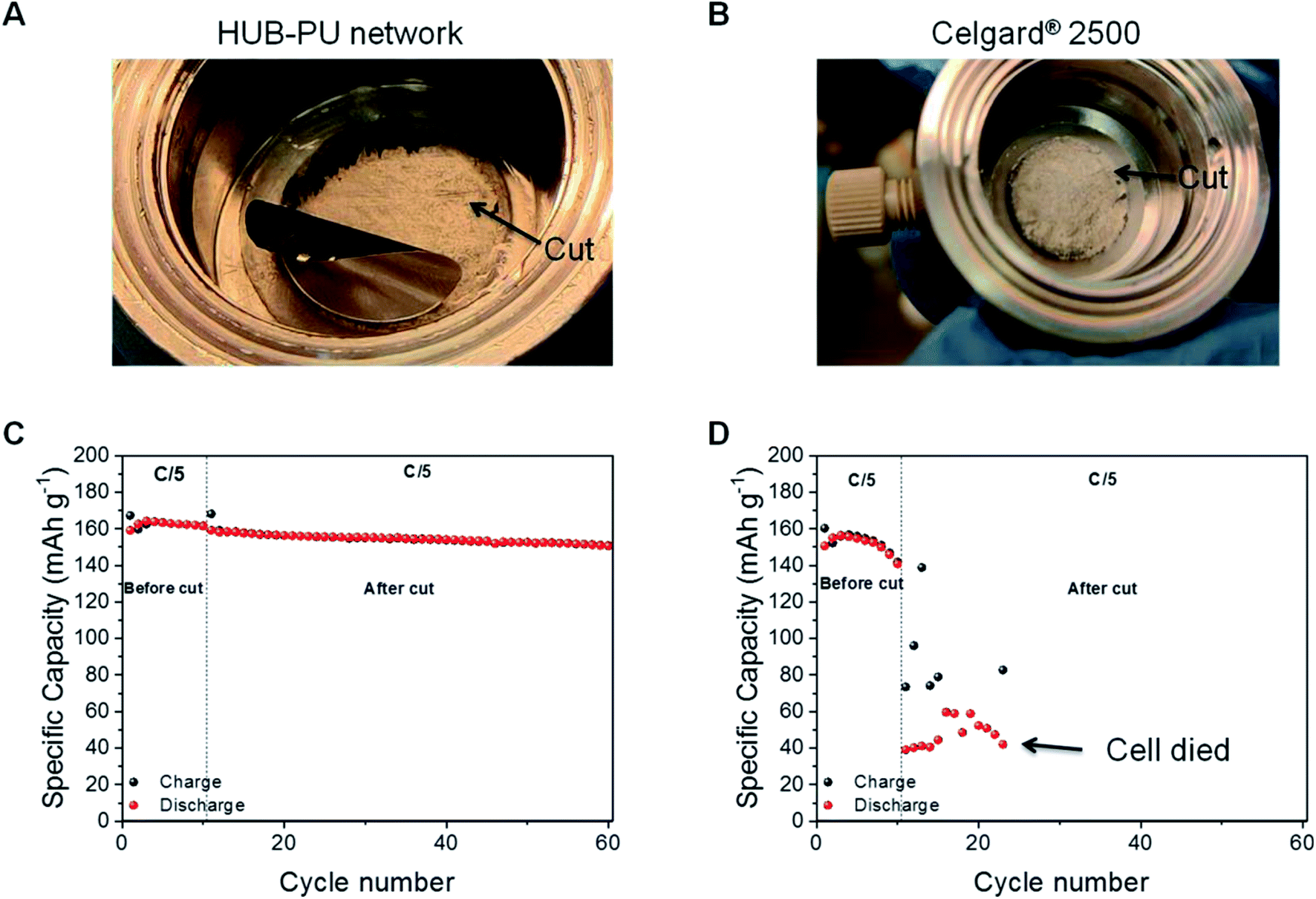 | ||
| Fig. 5 (A and B) Optical proof of the cut carried out inside the glove box on both the self-healable HUB-PU network and Celgard® 2500 membrane after 10 cycles at C/5 rate. (C and D) Specific capacity (mA h g−1) vs. cycle number at C/5 rate for the HUB-PU network and Celgard® 2500 membrane, respectively. The first 10 cycles are carried out before the cut, while the subsequent cycles refer to the period after the cut. | ||
On the other hand, in the case of the commercial Celgard® 2500 membrane, Fig. 5D clearly shows its incapability to recover from the cut (further confirmed by its potential profiles in Fig. S7†). After the scratch, the cell worked with lower capacity values compared to the first ten cycles. Indeed, the charge capacity of the first cycle after the cut is 27% of that before the cut and the corresponding discharge capacity is only 39 mA h g−1. Moreover, the subsequent charge capacity values seemed quite random, clearly showing the ongoing failure of the cell.
3. Conclusion
In summary, in this paper a polyurethane network containing dynamic crosslinking points based on HUBs has been synthesized and effectively used for the first time as a self-healable polymer electrolyte in Li-metal batteries. Noteworthily, this novel membrane has shown superior ionic conductivity to the typical liquid electrolyte embedded in commercial porous Celgard® 2500 separators. Furthermore, to the best of our knowledge, self-healing electrolytes have never been electrochemically cycled while still damaged, only when already healed. In this paper, the self-healing efficiency of HUB-PU membranes has been compared after cutting the materials and cycling them in lab-scale prototypes at C/5 rate. In the case of the reference commercial membrane, after the scratch the capacity value dropped to half of the previous values and the electrochemical performances rapidly deteriorated. In contrast, the novel HUB-PU membrane showed good recovery after mechanical damage and the capacity values remained constant and similar to the initial ones with 161 mA h g−1 before the cut and 159 mA h g−1 just after. The HUB-PU polymer electrolyte with the self-healing feature has been able to cycle for more than 100 cycles after damage/healing. Overall, we believe that the findings of this work contribute to optimizing and enhancing the implementation of covalent adaptable linkages (more specifically, HUBs) in the growing technology transfer of safe, solid-state and self-repairing materials for electrochemical energy storage, as well as Li–N2 cells prepared for sustainable ammonia electrosynthesis.4. Experimental section
4.1 Materials
Tris[2-(isopropylamino)ethyl]amine (90%), dibutyltin dilaurate (DBTDL, 95%), hexamethylene diisocyanate (HDI, ≥98.0%) and anhydrous tetrahydrofuran (THF, ≥99.9%) were purchased from Merck and used as received. PEG2000 was purchased from Merck and dried in an oven (70 °C) overnight prior to use. 1.0 M LiPF6 solution in EC:DEC (1:1, v/v) was purchased from Solvionic and used in the glove box. Poly(vinylidenedifluoride) (PVDF-HSV900:ADX160 90:10 wt:wt, Arkema; 10 wt% in N-methyl-2-pyrrolidinone (NMP) solution), Li disk (Chemetall Foote Corporation, Ø 16 mm), polypropylene polymeric membrane (Celgard® 2500, 25 μm-thick, Ø 19 mm), LFP (Aleees) and carbon black powder (C65, C-NERGY Super C65, Timcal) were used as received.
4.2 Synthesis of the HUB-PU network
In order to synthesize the self-healable crosslinked poly(urea-urethane) network (Fig. 2A), HDI (1.5 mmol, 252 mg) was dissolved, under quite diluted conditions, using anhydrous THF (13 mL) as a solvent. The solution was kept stirring vigorously at room temperature. Subsequently, tris[2-(isopropylamino)ethyl]amine (0.5 mmol, 136 mg) was added dropwise in order to form free isocyanate group end capped hindered urea trimmers. The mixture was stirred for 15 min and, successively, previously dried PEG2000 (0.75 mmol, 1.6 g) was added to the solution. Once PEG2000 was properly dissolved, DBTDL (2 mol% with respect to NCO content, 9.5 mg) was added and, after 5 min stirring, the mixture was deposited in a rectangular Teflon mold. Before the curing step, the mixture was degassed in a vacuum oven and then heated up to 80 °C overnight.4.3 Cathode preparation
To obtain the LFP electrode as a cathode for the cell testing, a slurry consisting of LFP, C65 and PVDF, at a weight ratio of 70:20:10, was mixed in NMP solvent. Then, the slurry was coated on an Al foil and dried overnight at room temperature. The as-obtained electrode was cut into 15 mm-diameter discs and dried under vacuum at 120 °C for 4 h.
4.4 Membrane activation
The HUB-PU network membrane was cut into 12 mm discs and dried under vacuum at 50 °C for 12 h before being transferred to a glovebox and activated by swelling in 1.0 M LiPF6 in EC:DEC (1:1, v/v) for 30 min.
4.5 FTIR spectroscopy
FTIR spectra were obtained using an FTIR spectrophotometer (Nicolet is20, Thermo Scientific Inc.) equipped with the ATR feature with a diamond crystal. Spectra were recorded between 4000 and 600 cm−1 with a spectrum resolution of 4 cm−1. All spectra were averaged over 16 scans.4.6 Stress–relaxation measurements
Stress–relaxation experiments to obtain the relaxation modulus G(t) were carried out using an ARES rheometer (Rheometrics) under the conditions indicated in the manuscript, using a film tension fixture and 5% strain. The employed samples had a width between 1.30 and 1.70 mm, and a thickness between 0.15 and 0.35 mm. The temperature dependence of relaxation time is described by the Arrhenius equation: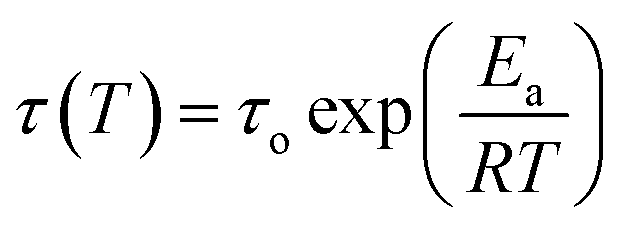 | (1) |
4.7 Electrochemical characterization
The electrochemical testing was carried out with the aim of comparing the newly designed HUB-PU membrane with a commercial separator (Celgard® 2500) impregnated with 300 μL of the above-mentioned liquid electrolyte.The ionic conductivity was determined by electrochemical impedance spectroscopy (EIS) in the frequency range between 100 kHz and 1 Hz at open circuit potential, using a VSP-3e potentiostat (BioLogic Science Instruments). The activated membrane was sandwiched between two stainless steel blocking electrodes (ECC-Std test cells, EL-CELL GmbH). The assembled cells were kept in a climatic chamber (model MK53 E2.1 by BINDER GmbH) and tested between 20 and 60 °C. The resistance of the electrolyte was given by the high-frequency intercept of the Nyquist plot. The ionic conductivity was calculated at each temperature using eqn (2):
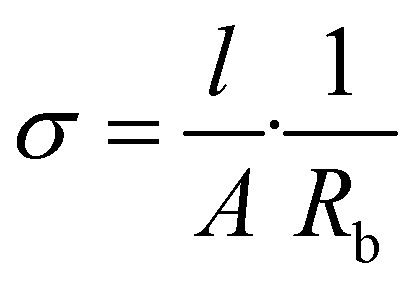 | (2) |
The ESW was evaluated by linear sweep voltammetry carried out on a stainless steel/HUB-PU membrane/Li cell using a potentiostat (CH Instruments, Inc.), at a scan rate of 0.5 mV s−1 from 0.5 to 6 V vs. Li+/Li at room temperature.
The effect of the membrane on Li plating and stripping was studied using a Li/Li symmetrical cell configuration, with the HUB-PU membrane sandwiched in between and using the VSP-3e potentiostat. The current density and the related discharge capacity were 0.1 mA cm−2 and 0.1 mA h cm−2, 0.5 mA cm−2 and 0.5 mA h cm−2, 1 mA cm−2 and 1 mA h cm−2, respectively. EIS measurement was performed, between 100 kHz and 1 Hz at open circuit potential, on fresh cells and after 10 cycles at each current density.
The reversibility of Li plating and stripping in a Li/HUB-PU membrane/Cu cell has been tested by galvanostatic cycling. Li was plated for 5 cycles on Cu for 1 h at 0.1 mA cm−2 and then stripped at the same current density up to a voltage cut-off of 0.5 V. Subsequently, increasing current densities of 0.5 and 1 mA cm−2 were applied, with related higher limiting voltages of 0.5 and 1.5 V, respectively, for 5 cycles under each condition. The reversibility of Li deposition is described by the Coulombic efficiency expressed as the ratio between the areal capacity (mA h cm−2) of the stripping and the Li plating areal capacity, times 100.
For testing of lab-scale prototypes, cells with an architecture based on LFP/HUB-PU membrane/Li were assembled. Galvanostatic cycling was performed to assess the lifetime and rate performances on an Arbin BT-2000 battery tester. The tests were carried out at room temperature, in three replicas.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No. 948769, project title: SuN2rise).References
- J. Xie and Y. C. Lu, Nat. Commun., 2020, 11, 9 CrossRef PubMed.
- P. K. D. Pramanik, N. Sinhababu, B. Mukherjee, S. Padmanaban, A. Maity, B. K. Upadhyaya, J. B. Holm-Nielsen and P. Choudhury, IEEE Access, 2019, 7, 8930492 Search PubMed.
- S. Li, S. Q. Zhang, L. Shen, Q. Liu, J. B. Ma, W. Lv, Y. B. He and Q. H. Yang, Adv. Sci., 2020, 7, 1903088 CrossRef CAS PubMed.
- P. Hundekar, S. Basu, J. Pan, S. F. Bartolucci, S. Narayanan, Z. Yang and N. Koratkar, Energy Storage Mater., 2019, 20, 291 CrossRef.
- M. A. A. M. Abdah, M. Mokhtar, L. T. Khoon, K. Sopian, N. A. Dzulkurnain, A. Ahmad, Y. Sulaiman, F. Bella and M. S. Su'ait, Energy Rep., 2021, 7, 8677 CrossRef.
- Y. Zhu, G. Chen, J. Sun, J. Wang, T. Wu, W. Dai and L. Lu, Mater. Technol., 2020, 35, 572 CrossRef CAS.
- Y. Liu, R. Zhang, J. Wang and Y. Wang, iScience, 2021, 24, 102332 CrossRef CAS PubMed.
- N. Wu, Y. R. Shi, S. Y. Lang, J. M. Zhou, J. Y. Liang, W. Wang, S. J. Tan, Y. X. Yin, R. Wen and Y. G. Guo, Angew. Chem., Int. Ed., 2019, 58, 18146 CrossRef CAS PubMed.
- Y. Yu, Y. Bin Yin, J. L. Ma, Z. W. Chang, T. Sun, Y. H. Zhu, X. Y. Yang, T. Liu and X. B. Zhang, Energy Storage Mater., 2019, 18, 382 CrossRef.
- M. Alidoost, A. Mangini, F. Caldera, A. Anceschi, J. Amici, D. Versaci, L. Fagiolari, F. Trotta, C. Francia, F. Bella and S. Bodoardo, Chem.–Eur. J., 2022, 28, e202104201 CrossRef CAS PubMed.
- J. Amici, C. Torchio, D. Versaci, D. Dessantis, A. Marchisio, F. Caldera, F. Bella, C. Francia and S. Bodoardo, Polymers, 2021, 13, 1625 CrossRef CAS PubMed.
- K. H. Chen, K. N. Wood, E. Kazyak, W. S. Lepage, A. L. Davis, A. J. Sanchez and N. P. Dasgupta, J. Mater. Chem. A, 2017, 5, 11671 RSC.
- X. Q. Zhang, X. B. Cheng and Q. Zhang, Adv. Mater. Interfaces, 2018, 5, 1 Search PubMed.
- K. Chen, D. Y. Yang, G. Huang and X. B. Zhang, Acc. Chem. Res., 2021, 54, 632 CrossRef CAS PubMed.
- P. Ding, Z. Lin, X. Guo, L. Wu, Y. Wang, H. Guo, L. Li and H. Yu, Mater. Today, 2021, 51, 449 CrossRef CAS.
- Y. Yang, N. Zhang, Z. Zou, X. Yi and J. Liu, Chem. Eng. J., 2022, 435, 135148 CrossRef CAS.
- K. Kim, Y. Chen, J. I. Han, H. C. Yoon and W. Li, Green Chem., 2019, 21, 3839 RSC.
- N. Verdier, G. Foran, D. Lepage, A. Prébé, D. Aymé-Perrot and M. Dollé, Polymers, 2021, 13, 1 CrossRef PubMed.
- Y. Chen, G. Xu, X. Liu, Q. Pan, Y. Zhang, D. Zeng, Y. Sun, H. Ke and H. Cheng, RSC Adv., 2018, 8, 39967 RSC.
- N. N. M. Radzir, S. A. Hanifah, A. Ahmad, N. H. Hassan and F. Bella, J. Solid State Electrochem., 2015, 19, 3079 CrossRef CAS.
- H. B. Youcef, B. Orayech, J. M. L. Del Amo, F. Bonilla, D. Shanmukaraj and M. Armand, Solid State Ionics, 2020, 345, 115168 CrossRef CAS.
- K. Deng, Q. Zeng, D. Wang, Z. Liu, Z. Qiu, Y. Zhang, M. Xiao and Y. Meng, J. Mater. Chem. A, 2020, 8, 1557 RSC.
- D. M. Shin, J. E. Bachman, M. K. Taylor, J. Kamcev, J. G. Park, M. E. Ziebel, E. Velasquez, N. N. Jarenwattananon, G. K. Sethi, Y. Cui and J. R. Long, Adv. Mater., 2020, 32, 1905771 CrossRef CAS PubMed.
- Y. H. Jo, S. Li, C. Zuo, Y. Zhang, H. Gan, S. Li, L. Yu, D. He, X. Xie and Z. Xue, Macromolecules, 2020, 53, 1024 CrossRef CAS.
- K. M. Lindenmeyer, R. D. Johnson and K. M. Miller, Polym. Chem., 2020, 11, 5321 RSC.
- Y. Cheng, X. Xiao, K. Pan and H. Pang, Chem. Eng. J., 2020, 380, 122565 CrossRef CAS.
- T. Munaoka, X. Yan, J. Lopez, J. W. F. To, J. Park, J. B. H. Tok, Y. Cui and Z. Bao, Adv. Energy Mater., 2018, 8, 1703138 CrossRef.
- B. B. Jing and C. M. Evans, J. Am. Chem. Soc., 2019, 141, 18932 CrossRef CAS PubMed.
- C. Chang, Y. Yao, R. Li, Z. H. Guo, L. Li, C. Pan, W. Hu and X. Pu, Nano Energy, 2022, 93, 106871 CrossRef CAS.
- P. Jaumaux, Q. Liu, D. Zhou, X. Xu, T. Wang, Y. Wang, F. Kang, B. Li and G. Wang, Angew. Chem., Int. Ed., 2020, 59, 9134 CrossRef CAS PubMed.
- F. Elizalde, R. H. Aguirresarobe, A. Gonzalez and H. Sardon, Polym. Chem., 2020, 11, 5386 RSC.
- X. Lopez De Pariza, T. Erdmann, P. L. Arrechea, L. Perez, C. Dausse, N. H. Park, J. L. Hedrick and H. Sardon, Chem. Mater., 2021, 33, 7986 CrossRef CAS.
- R. H. Aguirresarobe, S. Nevejans, B. Reck, L. Irusta, H. Sardon, J. M. Asua and N. Ballard, Prog. Polym. Sci., 2021, 114, 101362 CrossRef CAS.
- L. Porcarelli, K. Manojkumar, H. Sardon, O. Llorente, A. S. Shaplov, K. Vijayakrishna, C. Gerbaldi and D. Mecerreyes, Electrochim. Acta, 2017, 241, 526 CrossRef CAS.
- Z. Lv, Y. Tang, S. Dong, Q. Zhou and G. Cui, Chem. Eng. J., 2022, 430, 132659 CrossRef CAS.
- H. Ying, Y. Zhang and J. Cheng, Nat. Commun., 2014, 5, 3218 CrossRef PubMed.
- Q. Zhang, S. Wang, B. Rao, X. Chen, L. Ma, C. Cui, Q. Zhong, Z. Li, Y. Cheng and Y. Zhang, React. Funct. Polym., 2021, 158, 104807 CrossRef.
- Q. Guo, Y. Han, H. Wang, S. Xiong, Y. Li, S. Liu and K. Xie, ACS Appl. Mater. Interfaces, 2017, 9, 41837 CrossRef CAS PubMed.
- C. F. N. Marchiori, R. P. Carvalho, M. Ebadi, D. Brandell and C. M. Araujo, Chem. Mater., 2020, 32, 7237 CrossRef CAS.
- L. Porcarelli, C. Gerbaldi, F. Bella and J. R. Nair, Sci. Rep., 2016, 6, 19892 CrossRef CAS PubMed.
- K. N. Wood, E. Kazyak, A. F. Chadwick, K. H. Chen, J. G. Zhang, K. Thornton and N. P. Dasgupta, ACS Cent. Sci., 2016, 2, 790 CrossRef CAS PubMed.
- C. A. Calderón, A. Vizintin, J. Bobnar, D. E. Barraco, E. P. M. Leiva, A. Visintin, S. Fantini, F. Fischer and R. Dominko, ACS Appl. Energy Mater., 2020, 3, 2020 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ta02239g |
| This journal is © The Royal Society of Chemistry 2022 |