
Platinum-complexed phosphorous-doped carbon nitride for electrocatalytic hydrogen evolution†
Forrest
Nichols
 a,
Qiming
Liu
a,
Jasleen
Sandhu
a,
Zahra
Azhar
a,
Rafael
Cazares
a,
Rene
Mercado
a,
Frank
Bridges
b and
Shaowei
Chen
*a
a,
Qiming
Liu
a,
Jasleen
Sandhu
a,
Zahra
Azhar
a,
Rafael
Cazares
a,
Rene
Mercado
a,
Frank
Bridges
b and
Shaowei
Chen
*a
aDepartment of Chemistry and Biochemistry, University of California, 1156 High Street, Santa Cruz, California 95064, USA. E-mail: shaowei@ucsc.edu
bDepartment of Physics, University of California, 1156 High Street, Santa Cruz, California 95064, USA
First published on 21st September 2021
Abstract
Sustainable hydrogen gas production is critical for future fuel infrastructure. Here, a series of phosphorous-doped carbon nitride materials were synthesized by thermal annealing of urea and ammonium hexafluorophosphate, and platinum was atomically dispersed within the structural scaffold by thermal refluxing with Zeise's salt forming Pt–N/P/Cl coordination interactions, as manifested in X-ray photoelectron and absorption spectroscopic measurements. The resulting materials were found to exhibit markedly enhanced electrocatalytic activity towards the hydrogen evolution reaction (HER) in acidic media, as compared to the P-free counterpart. This was accounted for by P doping that led to a significantly improved charge carrier density within C3N4, and the sample with the optimal P content showed an overpotential of only −22 mV to reach the current density of 10 mA cm−2, lower than that of commercial Pt/C (−26 mV), and a mass activity (7.1 mA μg−1Pt at −70 mV vs. reversible hydrogen electrode) nearly triple that of the latter. Results from the present study highlight the significance of P doping in the manipulation of the electronic structures of metal/carbon nitride nanocomposites for high-performance HER electrocatalysis.
Introduction
Globally, renewable electricity is projected to reach 33% of the total installed power capacity by 2025, surpassing electricity produced by coal and becoming the largest segment of electricity generation around the world.1 With renewable electricity on the rise, electrochemical water splitting for hydrogen generation provides a promising technology for sustainable energy storage and conversion.2,3 Currently, more than 95% of the world's hydrogen gas is produced through steam methane reforming, which produces a significant amount of carbon dioxide byproduct.4 Acceptance of electrochemical water splitting as a viable means for hydrogen gas generation will require improvements in electrocatalyst design and usage of platinum within the material to lower device costs and hydrogen gas conversion efficiencies, as platinum remains the catalyst of choice towards the hydrogen evolution reaction (HER), in particular, in acidic media.5–7 Numerous routes have been taken to achieve this goal including the use of platinum oxide particles,8,9 manipulation of nanoparticle morphologies,10,11 and decreased particle size down to the single platinum atom regime.12,13Significant improvement in Pt mass efficiency can be achieved through the utilization of Pt single atom sites because of maximal surface exposure and utilization of Pt within the catalyst materials.13–22 For instance, Cheng et al.12 recently deposited Pt single atoms on nitrogen-doped carbon nanosheets by utilizing an atomic layer deposition (ALD) approach, and the resulting Pt single atom catalysts (SAC) showed a mass activity of 10.1 mA μg−1Pt in a 0.5 M H2SO4 electrolyte, 37 times that of Pt/C (0.27 mA μg−1Pt), at the applied potential of −50 mV vs. reversible hydrogen electrode (RHE). Fang and associates recently deposited Pt single atoms onto a nitrogen-doped carbon framework of approximately 300 nm in diameter also through an ALD process, and the catalyst exhibited a low overpotential (η10) of −19 mV to reach the current density of 10 mA cm−2 in a 0.5 M H2SO4 electrolyte. Operando synchrotron X-ray absorption spectroscopy studies revealed partial release of the Pt single atoms resulting in undercoordination. Further analysis by extended X-ray absorption fine structure (EXAFS) measurements and theoretical simulations suggested that a high 5d orbital occupancy, close to zero valency of Pt, was responsible for the high HER activity.
However, recent reports have shown Pt in a lower orbital occupancy, such as Pt oxide, can facilitate HER efficiently as well. For example, Yu and colleagues recently reported on Pt–O as a superior active site to Pt0 for HER with the use of a platinum tungsten oxide (PtW6O24) spherical particles of ca. 20 nm in diameter.23 The material was prepared from a mixture of Na2Pt(OH)6 and Na2WO4 followed by collection of precipitates and washing. Electrochemical measurements revealed a mass activity of 20.175 mA μg−1Pt for HER at the overpotential of −70 mV in 0.5 M H2SO4, significantly better than commercial Pt/C (20 wt%, 0.398 mA μg−1Pt). Cao and co-workers10 employed a one-pot synthesis route using Pt(acac)2 and Ni(acac)2 metal precursors and produced Pt–Ni branched nanocages of approximately 200 nm in diameter with a thinner branching morphology spreading out from the center (50 nm), which exhibited a mass activity nearly 2.5-fold higher than commercial Pt/C. These findings confirm that Pt does not need to possess high orbital occupancy to efficiently catalyse HER. To this end, an array of interesting Pt-based catalysts can be devised for the efficient evolution of hydrogen through unique synthetic methods beyond the ALD process.
Note that ALD, used in these previous studies for the synthesis of Pt-based SAC, requires the use of sophisticated and expensive instrumentation. It would be of great significance to develop low-cost, effective procedures for the preparation of Pt SAC. A range of synthetic approaches toward Pt SAC have been studied and well documented in recent reviews.24,25 These synthetic approaches include wet-chemistry, pyrolysis of metal–organic frameworks, electrodeposition, high-temperature atom trapping from bulk particles, and vacancy/defect immobilization methods. For example, Wang and colleagues demonstrated an electro-filtration strategy to synthesize Pt single atom sites using a Pt foil and graphite carbon paper. In this electroplating strategy Pt single atoms were created through oxidation of the Pt foil counter electrode followed by Pt single atom deposition onto the carbon paper working electrode. A graphene oxide membrane was used as a filter between the counter and working electrodes to control the Pt single atom diffusion rate and prevent Pt particle formation. Scalability of this process is of somewhat concern since production of the Pt SAC is limited by the size of the electrochemical cell and working electrode.
In recent studies,16,26 it was observed that ion complexation by simple thermal refluxing can be exploited to effectively embed isolated metal centers within the structural skeleton of graphitic carbon nitride (C3N4) by taking advantage of the pyridine-like nitrogen moieties, where the incorporation of metal centers was found to lead to charge redistribution and enhanced electrical conductivity of the semiconducting C3N4 matrix. For example, Zeng and colleagues reported on the synthesis of single-atom Pt confined by interlayer C3N4 for efficient photocatalytic hydrogen evolution.27 The synthesis process involved mixing of potassium-intercalated C3N4 with Pt(NH3)4Cl2 in deionized water under vigorous stirring at room temperature over a 24 h period. Incorporation of Pt single atoms into the C3N4 framework significantly improved the photocatalytic hydrogen evolution efficiency to 22.65 mmol (H2) g−1 h−1 with an 8.7 wt% Pt loading. The semiconductor properties of C3N4 are ideal for such photocatalytic processes; however, this leads to poor electron carrier capabilities when applied to electrocatalysis. Fortunately, deliberate modifications to the carbon nitride framework can be made to improve the electrocatalytic efficiency of C3N4. One such modification involves heteroatom doping within the C3N4 framework to improve electrical conductivity of the support material.28–30 For example, Zhang and co-workers developed a phosphorous-doped C3N4 solid which demonstrated enhanced electrical conductivity.29 Experimentally, 1-butyl-3-methylimidazolium hexafluorophosphate was added as a phosphorous source during the polycondensation of dicyandiamide (DCDA) forming P-doped C3N4, which was confirmed in spectroscopic and electrochemical measurements. Such P-doped C3N4 materials have been implemented for electrochemical applications. Qiu and co-workers reported on a three-dimensional P-doped C3N4 for enhanced metal-free oxygen reduction reaction (ORR) applications, which was prepared by polycondensation of DCDA and nitrilotris(methylene)-triphosphoric acid as a phosphorous source at 600 °C. P-doping was seen to substantially improve the ORR half-wave potential and limiting current, likely due to improved electrical conductivity as found in electrochemical impedance measurements. Notably, heteroatom doping of semiconductor materials is well studied as a tool for tuning the band gap structure,31–33 where doping of higher periodic elements typically leads to a narrower band gap and lower highest occupied molecular orbital (HOMO) levels.34 Thus, the larger atomic size of phosphorous, versus carbon and nitrogen, is anticipated to lead to improved conformation and conductivity of C3N4. This is the primary motivation of the present study.
Herein, P-doped C3N4 was first prepared via a facile thermal annealing procedure using urea and ammonium hexafluorophosphate as the precursors, into which were then embedded Pt metal centers by thermal refluxing with Zeise's salt forming Pt–N/P/Cl coordination bonds, as attested in spectroscopic measurements. Mott–Schottky analysis showed that P doping led to a marked improvement of the charge carrier density and electrical conductivity. Electrochemical tests demonstrated that the resulting PtCNP nanocomposites outperformed the P-free counterparts, and the sample with the optimal P content showed an η10 of only −22 mV, lower than that of commercial Pt/C (−26 mV), and a mass activity (7.1 mA μg−1Pt at −70 mV vs. RHE) nearly triple that of the latter. The results suggest that P-doped C3N4 may be used as a unique structural scaffold to optimize the use of Pt for HER electrocatalysis.
Experimental section
Chemicals
Urea (CO(NH2)2, certified ACS, Fisher Chemicals), potassium trichloro(ethylene)platinate(II) hydrate (Zeise's salt, KPtCl3(C2H4), Sigma-Aldrich), ammonium hexafluorophosphate (NH4PF6, Acros Organics), potassium hydroxide (KOH, Fisher Chemicals), platinum on carbon (Pt/C, nominally 20 wt%, Alfa Aesar), carbon black (Vulcan XC 72R), and Nafion 117 (Sigma-Aldrich) were used as received. All solvents were obtained through typical commercial sources and used as received, too. Water was supplied with a Barnstead Nanopure water system (18.3 MΩ cm).Synthesis of (phosphorous-doped) polymeric carbon nitride
P-doped C3N4 was synthesized by a modified method from previous reports,29 as illustrated in Scheme 1. Briefly, urea (10 g) was ground to a fine powder with a designated amount of NH4PF6 (i.e., 0.5, 1.0, 2.5, or 5 g), and the mixture was placed in a crucible, covered, heated in air to 300 °C at a rate of 2 °C min−1 and held at 300 °C for 3 h. The resulting white solid was ground to a fine powder, heated again in air to 520 °C at a rate of 2 °C min−1 and held at this temperature for 4 h. The produced yellow solid was collected, washed with Nanopure water, and dried in a vacuum oven overnight, affording P-doped C3N4 that was referred to as CNP1, CNP2, CNP3, and CNP4, respectively. Undoped C3N4 was produced in the same manner but without the addition of NH4PF6.9,35 | ||
| Scheme 1 Schematic of the synthetic procedure of PtCNP. | ||
Synthesis of platinum–carbon nitride nanocomposites
Platinum was incorporated into the carbon nitride scaffold obtained above by following a procedure used for the synthesis of platinum bipyridine complexes (Scheme 1).36 Briefly, the P-doped C3N4 prepared above (50 mg) was dispersed in a mixture of acetone (4 mL) and isopropanol (1 mL) by sonication for 1 h. In a separate vial, Zeise's salt (50 mg) was dissolved in acetone (5 mL) under magnetic stirring in an ice bath. KOH (20 mg) was dissolved in a minimum amount of water and added to the Zeise's salt solution under stirring for 40 min, where KCl salt precipitates could be seen accumulated on the walls of the glass vial. The carbon nitride dispersion was immersed into an ice bath under vigorous stirring for 10 min, into which was added the Zeise's salt solution. After magnetic stirring for 40 min, the resulting mixture was centrifuged at 6000 rpm for 10 min, and the precipitate was collected and washed 5 times with acetone to remove any unreacted platinum salts and contaminants. It should be noted that the colour of the supernatant changed from light yellow to clear after the subsequent washes, indicating a saturated loading of platinum into carbon nitride and the subsequent removal of unreacted metal salt upon washing. The material was dried in a vacuum oven overnight, affording platinum containing carbon nitrides, which were denoted as PtCNP1, PtCNP2, PtCNP3, PtCNP4, respectively. The P-free sample (PtCN) was prepared in the same manner except that undoped C3N4 was used instead of P-doped C3N4.Characterization
Scanning electron microscopic (SEM) images were collected on a Thermo Scientific Apreo scanning electron microscope with high/low-vacuum compatibility and a 400 nA beam current equipped with an Oxford instruments energy dispersive X-ray spectroscopy (EDS) attachment. Transmission electron microscopy (TEM) and scanning transmission electron microscopy (STEM) measurements were carried out on a JOEL JEM 2100F microscope. X-ray diffraction (XRD) patterns were acquired with a Rigaku Smartlab Diffractometer, where the samples were deposited on an Al plate and the measurements were conducted at a step of 0.02° and a scan rate of 3° min−1. X-ray photoelectron spectroscopy (XPS) measurements were performed with a Phi 5400/XPS instrument equipped with an Al Kα source operated at 350 W and 10−9 torr. X-ray absorption spectroscopy (XAS) measurements were carried out at 10 K on beamline 4–1 at the Stanford Synchrotron Radiation Lightsource using an Oxford liquid helium cryostat. Inductively coupled plasma-optical emission spectrometry (ICP-OES) measurements were conducted with a SPECTROBLUE SOP instrument.Electrochemistry
Electrochemical measurements were performed with a CHI 710 workstation, and electrochemical impedance measurements were carried out with a Gamry Reference 600 instrument. A glassy carbon electrode (5.60 mm in diameter, 0.246 cm2) was used as the working electrode, while a Ag/AgCl (1.0 M KCl) electrode and a graphite rod were used as the reference and counter electrodes, respectively. The Ag/AgCl electrode was calibrated versus a RHE and all potentials in the present study were referenced to this RHE, unless specified otherwise. To prepare the catalyst ink, 2 mg of the nanocomposites obtained above was mixed with carbon black (3 mg) and sonicated for 10 min in isopropanol (1 mL), followed by the addition of Nafion (40 μL) and sonication for an additional 20 min. 30 μL of the prepared ink was dropcast onto the surface of the glassy carbon electrode and dried at room temperature, corresponding to a catalyst mass loading of 0.244 mg cm−2. Finally, 6 μL of Nafion (20 wt%) in isopropanol was added to the prepared catalyst surface and allowed to dry, before the electrode was immersed into an electrolyte solution for data collection.Results and discussion
The material structures were first characterized by electron microscopic measurements. From the SEM images in Fig. 1a and b, one can see that PtCNP2 exhibits a flaky morphology that closely resembles that of typical polymeric carbon nitride,29,35 and the wrinkled, porous nature of the material likely arose from oxygen-containing gases generated during the polymerization of the urea precursor resulting in punctures and holes in the material matrix. The corresponding EDS elemental maps are depicted directly below panel (a), where one can readily see an even distribution of all elements characterized including C, N, O, P, Pt, and Cl. The porous, layered structure of the composite is also evident in the TEM images shown in Fig. 1c and d. Notably, no particulates can be resolved in the TEM images of the sample, suggesting successful atomic dispersion of Pt within the C3N4 skeletons. Similar structural characteristics can also be seen with other samples in the series, as manifested in both SEM (Fig. S1†) and TEM measurements (Fig. S2†). | ||
| Fig. 1 (a) SEM image of the PtCNP2 sample, with the EDS elemental maps shown at the bottom for C, N, P, Pt, Cl, and O. (b) High-magnification SEM image for the red box area in panel (a). (c and d) TEM images of PtCNP2. Scale bars are (a) 5 μm, (b) 1 μm, (c) 50 nm and (d) 10 nm. | ||
The sample structures were then examined by XRD measurements. From Fig. S3,† one can see that in addition to the diffraction patterns of the Al plate (substrate), the PtCN, PtPCN1 and PtCNP2 composites all exhibited a small peak at 2θ = 27°, consistent with results reported previously for orthorhombic polymeric carbon nitride materials.35 Notably, this peak shifted to a somewhat smaller angle of ca. 26° in PtCNP3 and became significantly broadened in PtCNP4, likely due to the doping of larger-sized P into the C3N4 framework which resulted in deterioration of the material crystallinity at high P concentration. Furthermore, no diffraction features of Pt, PtO, or PtO2 can be resolved in any of the samples, in agreement with the results from SEM and TEM measurements which suggested the absence of metal (oxide) nanoparticles.
The elemental composition and valence state was then investigated by XPS measurements. From the survey spectra in Fig. S4a,† the PtCNP samples can be seen to contain a series of well-defined peaks, at ca. 75 eV for Pt 4f electrons, 133 eV for P 2p, 200 eV for Cl 2p, 285 eV for C 1s, 400 eV for N 1S, and 530 eV for O 1s. PtCN exhibited a similar profile except for the P 2p peak. Based on the integrated peak areas, the elemental compositions of the samples were then estimated and listed in Table S1.† One can see that the C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N atomic ratios of the samples all deviated appreciably from the theoretical value of 0.75 for pristine C3N4, likely the result of a combined contribution of structural defects and heteroatom doping.37,38 Notably, with increasing P doping, the C:N ratio increases accordingly in the order of PtCN (0.86) < PtCNP1 ≈ PtCNP2 (0.96) < PtCNP3 (1.04) < PtCNP4 (2.75), likely due to the incorporation of oxygen and phosphorous into carbon nitride replacing nitrogen. In fact, one can see that the oxygen content increased from 3.71% for PtCN to 6.38%, 4.73%, 13.19%, and 55.53% for PtCNP1, PtCNP2, PtCNP3, and PtCNP4, respectively; and the corresponding phosphorous content was found to increase from 0% to 1.31%, 1.11%, 6.32%, and 11.03%. The corresponding Pt contents were estimated to be 0.73, 1.69, 1.69, 1.58 and 0.32 at% (or 9.72, 19.52, 19.59, 17.45, and 3.38 wt%), which were substantially higher than those obtained in ICP-OES measurements (1.8, 1.8, 5.1, 0.6, and 0.8 wt%). As XPS is a surface probing technique, this suggests enrichment of Pt on the carbon nitride surface. This follows reasonably from the catalyst preparation methods, where the Pt salt could only complex at the surface upon contact with the previously prepared C3N4. Note that preventing the incorporation of Pt toward the inside of the C3N4 support is beneficial for electrocatalysis as only the surface Pt will be involved in electrochemical processes, thereby improving mass activity of the electrocatalysts (vide infra). Furthermore, high doping levels of P appeared to hinder Pt incorporation, likely due to the highly defective scaffold, and CNP2 represented the optimal structure for Pt complexation. Notably, the Cl:Pt ratios of all but PtCNP4 were found to be close to 1:1, at 0.79 for PtCN, 0.95 for PtCNP1, 1.02 for PtCNP2, and 0.74 for PtCNP3, suggesting that the incorporation of Pt into the nanocomposites was due to partial replacement of the original Cl ligands of the Zeise's salt precursor (which features a Cl:Pt ratio of 3) (Scheme 1).
:N atomic ratios of the samples all deviated appreciably from the theoretical value of 0.75 for pristine C3N4, likely the result of a combined contribution of structural defects and heteroatom doping.37,38 Notably, with increasing P doping, the C:N ratio increases accordingly in the order of PtCN (0.86) < PtCNP1 ≈ PtCNP2 (0.96) < PtCNP3 (1.04) < PtCNP4 (2.75), likely due to the incorporation of oxygen and phosphorous into carbon nitride replacing nitrogen. In fact, one can see that the oxygen content increased from 3.71% for PtCN to 6.38%, 4.73%, 13.19%, and 55.53% for PtCNP1, PtCNP2, PtCNP3, and PtCNP4, respectively; and the corresponding phosphorous content was found to increase from 0% to 1.31%, 1.11%, 6.32%, and 11.03%. The corresponding Pt contents were estimated to be 0.73, 1.69, 1.69, 1.58 and 0.32 at% (or 9.72, 19.52, 19.59, 17.45, and 3.38 wt%), which were substantially higher than those obtained in ICP-OES measurements (1.8, 1.8, 5.1, 0.6, and 0.8 wt%). As XPS is a surface probing technique, this suggests enrichment of Pt on the carbon nitride surface. This follows reasonably from the catalyst preparation methods, where the Pt salt could only complex at the surface upon contact with the previously prepared C3N4. Note that preventing the incorporation of Pt toward the inside of the C3N4 support is beneficial for electrocatalysis as only the surface Pt will be involved in electrochemical processes, thereby improving mass activity of the electrocatalysts (vide infra). Furthermore, high doping levels of P appeared to hinder Pt incorporation, likely due to the highly defective scaffold, and CNP2 represented the optimal structure for Pt complexation. Notably, the Cl:Pt ratios of all but PtCNP4 were found to be close to 1:1, at 0.79 for PtCN, 0.95 for PtCNP1, 1.02 for PtCNP2, and 0.74 for PtCNP3, suggesting that the incorporation of Pt into the nanocomposites was due to partial replacement of the original Cl ligands of the Zeise's salt precursor (which features a Cl:Pt ratio of 3) (Scheme 1).
Further structural insights were obtained in high-resolution XPS analysis. Fig. 2a depicts the C 1s spectra of the sample series, where two major components can be deconvoluted for all samples at ca. 288.0 and 284.5 eV, due to the sp2 hybridized carbon within the carbon nitride matrix (C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) and defected carbon (Csp3), respectively.39–41 Interestingly, the ratio between the areas of these two peaks decreased from 4.14 for PtCN to 1.99 for PtCNP1, 2.07 for PtCNP2, 1.16 for PtCNP3, and 0.3 for PtCNP4, again, suggesting the formation of an increasingly defective C3N4 skeleton with increasing phosphorous doping. Additionally, for the PtCNP samples, a third peak can be resolved at ca. 286 eV, which can be ascribed to C–P bond, signifying successful P doping of C3N4.28,42
C) and defected carbon (Csp3), respectively.39–41 Interestingly, the ratio between the areas of these two peaks decreased from 4.14 for PtCN to 1.99 for PtCNP1, 2.07 for PtCNP2, 1.16 for PtCNP3, and 0.3 for PtCNP4, again, suggesting the formation of an increasingly defective C3N4 skeleton with increasing phosphorous doping. Additionally, for the PtCNP samples, a third peak can be resolved at ca. 286 eV, which can be ascribed to C–P bond, signifying successful P doping of C3N4.28,42
 | ||
| Fig. 2 High resolution XPS spectra of the (a) C 1s, (b) N 1s, (c) Pt 4f, and (d) P 2p electrons of the PtCN, PtCNP1, PtCNP2, PtCNP3, and PtCNP4 samples. | ||
The corresponding N 1s spectra are shown in Fig. 2b, where two major components can be resolved, C–NC at ca. 398.4 eV and tertiary N–(C)3 at ca. 399 eV.39–41 The exact binding energy of C–NC varied slightly, 398.49 eV for PtCN, 398.21 eV for PtCNP1, 398.36 eV for PtCNP2, 398.29 eV for PtCNP3, and 398.47 eV for PtCNP4. For tertiary N–(C)3, the binding energy was very close among the P-free PtCN (399.05 eV), and low-P PtCNP1 (398.86 eV) and PtCNP2 (398.99 eV), whereas for PtCNP3 and PtCNP4 that contained a high P dopant concentration, the binding energy was substantially higher at 399.62 and 399.78 eV, respectively. This suggests that at low P doping, the C3N4 skeleton structure remained largely intact, whereas at high doping, the matrix became increasingly defective, as manifested in XRD measurements (Fig. S3†).
The high-resolution scans of the Pt 4f region are depicted in Fig. 2c. In all samples, deconvolution yields two doublets, where the major one at ca. 72.5/75.8 eV can be ascribed to the 4f5/2/4f7/2 electrons of Pt(II), whereas the minor one at 74.3/77.6 eV to those of Pt(IV) (Table S2†).9 One can see that the 4f7/2 binding energy of both Pt(II) and Pt(IV) in the PtCNP series diminished somewhat, as compared to those of P-free PtCN, implying that P doping led to electron enrichment of the Pt centres, and there appears a volcano variation of the binding energies with P dopant concentration, with PtCNP2 at the maxima (72.54 and 74.34 eV). A reverse trend is observed in the binding energy of the P 2p electrons (Fig. 2d), where PtCNP2 exhibited the lowest binding energy (132.88 eV) of the 2p3/2 electrons (Table S3†). This suggests charge transfer between Pt and P within the P-doped samples, and such an interaction (charge transfer) was the strongest in PtCNP2 among the series. Note that the binding energy of 132.88 eV is consistent with that observed for P–C bond, again, confirming the successful doping of P into the C3N4 structure.43
Fig. S4b† depicts the corresponding high-resolution Cl 2p spectra, where the Cl 2p3/2 binding energy can be identified at 198.00, 197.74, 197.79, 197.83, and 198.15 eV for PtCN, PtCNP1, PtCNP2, PtCNP3, and PtCNP4 (Table S3†), respectively. These binding energies are consistent with those observed for Pt–Cl containing complexes reported previously in the literature.44,45
Further insights into the Pt electronic state and coordination structures were obtained through XAS measurements in the range of 11.5 to 12.0 keV corresponding to the Pt L3 edge. Fig. 3a depicts the X-ray absorption near edge structures (XANES) for the sample series, the Zeise's salt precursor (orange curve), and a Pt foil (cyan curve). The obtained data were reduced and analysed using the RSXAP package.46 The Pt foil (cyan curve) can be seen to exhibit a strong absorption peak at 11.566 keV, which is referred to as the white line and attributed to the Pt 2p → 5d transition (L3 edge).47,48 The white line peak position for the Zeise's salt occurs at 11.567 keV, and the remaining samples exhibit a peak position of 11.568 keV. The white line amplitude in this region typically increases with decreasing 5d orbital occupancy (i.e., increasing average valence state). One can see that the white line amplitude increases in the order of Pt foil < Zeise's salt < PtCNP1 ≈ PtCNP2 ≈ PtCNP3 ≈ PtCNP4 < PtCN (inset to Fig. 3a). This suggests (i) a non-metallic valence state of Pt in the PtCN and PtCNP samples, and (ii) a somewhat lower Pt valence state in PtCNP than in PtCN.48–50 These findings are consistent with results from XPS measurements (Table S2†). At higher energies, one can see that the PtCN and PtCNP samples exhibited a very similar extended X-ray absorption fine structure (EXAFS), which was analogous to that of the Zeise's salt but markedly different from that of the Pt foil. This can also be manifested in the corresponding R-space profiles in Fig. 3b, where the same Fourier transform range (3.5–12 Å−1) was used for each sample. A plot of the k space for the sample series can be seen in Fig. S5.† The R-space plots depict a clear change in the Pt environment in PtCN and PtCNP, with a smaller amplitude (increasing disorder), as compared to the two reference samples of Zeise's salt (orange curve) and Pt foil (cyan curve). Note that the peak structure near 2.4 Å, corresponding to the Pt–Pt pairs within the Pt foil (cyan curve), is not found in any of the other samples, whereas the primary peak near 1.9 Å in Zeise's salt (orange curve) corresponding to Pt–Cl pairs is shifted to a lower R position for the PtCN and PtCNP samples and becomes broadened. Of note, the PtCN and PtCNP samples exhibit very similar R-space spectra with a broad peak near 1.7 Å, suggesting a consistent coordination environment around the Pt centres.
 | ||
| Fig. 3 (a) Normalized Pt L3 edge XANES for the sample series in comparison to Pt Zeise's salt precursor (orange) and Pt foil (cyan). All edge steps are normalized to an edge height of 1. The energy range selected to normalize the edge is 11.7–12.0 keV. Measurements were performed at a temperature of 10 K to reduce thermal disorder within samples. Inset is the zoom in of the white line region. (b) Pt L3 edge R-space EXAFS data for the sample series and two reference samples, Pt Zeise's salt (orange) and Pt foil (cyan). The FT window is from 3.5 to 12.0 Å−1, rounded using a Gaussian function of width, 0.2 Å−1. The fast-oscillating function is the real part R of the FT, and the amplitude is (R2 + I2)1/2, whereas I is the imaginary part of the FT. Samples have been shifted vertically for better comparison. | ||
The EXAFS spectra were then fitted to quantify the bond lengths and coordination numbers using the rsFit function within the RSXAP software. The FEFF7 software was used to calculate the theoretical functions for each Pt pair (Pt–N/C, Pt–Cl/P).52 The samples were fitted using four Pt–N/C paths and one Pt–Cl/P path created from the atomic positions in the structures depicted in Fig. 4a and b with distances from Pt of 2.0 Å (Pt–N), 2.5 Å (Pt–N/C), 3.5 Å (Pt–N/C), 4.1 Å (Pt–N/C), and 2.3 Å (Pt–Cl/P), respectively. The structure for fittings was created using a heptazine-based carbon nitride matrix. Pt was introduced into the gap between three tris-s-tiazine structures with the inclusion of one remaining Cl, and P was incorporated by replacement of a nearby N and bonded to C within the carbon nitride framework, as is consistent with the XPS findings (Fig. 2). The structure was relaxed using density functional theory to minimize the geometric strain. The coordinates of each atom within the relaxed structure were used as input to FEFF7 to create the 5 Pt paths used for EXAFS fittings. The first path arises from the 4 nearest N neighbors to Pt, shown by direct bonds in Fig. 4a and b. The second path arises from the next nearest neighbors to Pt, one Cl for structure 4a and one Cl and one P in structure 4b. As PtCN contained no P, P is absent in the structure used for PtCN fittings (Fig. 4a). Paths 3, 4, and 5 arise from C and N atoms farther away from the central Pt atom, between 2.5 and 4.1 Å. These atoms are not directly bonded to Pt, but nonetheless are responsible for some structure in the raw spectra for each sample. At farther distances from Pt there will be more possible neighbors and therefore we see an increase in the coordination number at these distances. The number of neighbors in the first Pt–N/C path of both fittings was fixed at 4 to be consistent with the structures in Fig. 4a and b. The number of neighbors in all other paths were allowed to vary. The Debye–Waller parameter (σ2) for all paths remained low between 0.003 and 0.04. Fittings were performed in the R range of 1.3 to 3.7 Å for both samples, and the results are shown graphically in Fig. 4c and d, respectively (also listed in Table 1). Due to the proximity of the atomic number of C (6), N (7), P (15), and Cl (17), the Pt–N and Pt–C paths are indistinguishable from one another through EXAFS fitting. Similarly, Pt–Cl and Pt–P paths are also indistinguishable. Therefore, the coordination number estimated from the fitting for a given path represents the sum of either Pt–C/Pt–N or Pt–Cl/Pt–P coordination. From the fitting results, the Pt–N/C bond length (r) for the first shell atoms was estimated to be 2.01 Å for PtCN and 2.03 Å for PtCNP2, both with a coordination number of ca. 4, suggesting a slightly stronger bonding interaction in the former, in agreement with the somewhat higher binding energy of the Pt 4f electrons in XPS measurements (Table S2†). This bond length is consistent with the proposed structure and the expected bond length of a Pt–N coordination complex. Meanwhile, the Pt–Cl/P bond length showed identical bond lengths of 2.31 Å, with the respective coordination number of 0.75 and 1.65. As mentioned earlier in XPS measurements, the atomic ratio of Cl:Pt was ca. 0.79 for PtCN (Table S1†), very consistent with the coordination number of 0.75, suggesting a single Pt–Cl bond remained in PtCN. In XPS measurements, PtCNP2 showed a Cl:Pt ratio of 1.02, also indicating a single Pt–Cl coordination. Thus, the Pt–Cl/P coordination number of 1.65 in PtCNP2 suggests that additional coordination most likely arose from a Pt–P environment.
 | ||
| Fig. 4 Ball and stick model representations of the fitting structures for (a) PtCN and (b) PtCNP2. Coordination to the centre Pt atom indicates first and second shell atoms used for fitting. Atoms are colour coded with carbon (brown), nitrogen (gray), platinum (cyan), chlorine (green), and phosphorous (light red). EXAFS fitting results shown in R-space for (c) PtCN and (d) PtCNP2. Raw R-space data is shown in solid with fits shown in gray circles. Fittings are performed in the R range of 1.3 to 2.5 Å. | ||
| Sample | Pt–N/C | Pt–Cl/P | Pt–N/C | Pt–N/C | Pt–N/C | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| r (Å) | CN | σ 2 | r (Å) | CN | σ 2 | r (Å) | CN | σ 2 | r (Å) | CN | σ 2 | r (Å) | CN | σ 2 | |
| PtCN | 2.01 | 4.0 | 0.0066 | 2.31 | 0.75 | 0.0026 | 2.53 | 4.0 | 0.0059 | 3.54 | 19.5 | 0.040 | 4.14 | 12.70 | 0.024 |
| PtCNP2 | 2.03 | 4.0 | 0.0092 | 2.31 | 1.65 | 0.014 | 2.52 | 4.0 | 0.0060 | 3.51 | 14.1 | 0.035 | 4.15 | 9.02 | 0.014 |
Note that EXAFS fittings in Fig. 4c and d were performed on the first primary peak beginning at 1.3 Å and extended to the structure near 3.7 Å using three additional Pt–N/C paths in 9–19 atom clusters, consistent with the structures depicted in Fig. 4a and b. The results show no interaction between Pt–Pt neighbours and hence the absence of metallic Pt or PtOx, which is consistent with atomic dispersion of Pt within the sample, as suggested in TEM measurements (Fig. 1 and S2†). Thus, based on the XPS and EXAFS fitting results, two structural models were constructed for PtCN and PtCNP2, as depicted in Fig. 4a and b, respectively, involving PtN4Cl and PtN4PCl nearest neighbours. Note there are several equivalent ways to place P on one of the nearby N/C sites.
The electrocatalytic activity towards HER was then analysed and compared in acidic media. Note that cyclic voltametric measurements of PtCNP2 within the potential range of 0 to +1.2 V in a N2-saturated H2SO4 solution reveals no noticeable hydrogen adsorption–desorption feature (Fig. S6†), further confirming the absence of Pt and PtOx nanoparticles within the sample (vide ante). Fig. 5a depicts the HER polarization curves in N2-saturated 0.5 M H2SO4 of the various catalysts loaded onto a glassy carbon electrode. Notably, the metal-free C3N4 and CNP2 exhibit only a minimal HER activity, and the activity was markedly improved with the incorporation of Pt forming PtCN and PtCNP. In fact, one can see that PtCNP2 even outperformed commercial Pt/C catalyst. This can be readily seen in Fig. 5b, where the η10 was estimated to be −383 mV for PtCN, −116 mV for PtCNP1, −22 mV for PtCNP2, −217 mV for PtCNP3, −304 mV for PtCNP4, and −26 mV for Pt/C. That is, P-doping significantly enhanced the HER activity of PtCN, and PtCNP2 stood out as the best HER catalyst among the series, with a performance even better than that of Pt/C. Further contrast is observed in Fig. 5c depicting the mass activity (MA, by normalizing the HER current to the Pt mass determined by ICP-OES measurements) obtained at −0.070 V vs. RHE, which is 0.028 mA μg−1Pt for PtCN, 0.771 mA μg−1Pt for PtCNP1, 7.1 mA μg−1Pt for PtCNP2, 0.226 mA μg−1Pt for PtCNP3, 0.05 mA μg−1Pt for PtCNP4, and 2.387 mA μg−1Pt for Pt/C. Again, PtCNP2 possessed the best performance, with the MA nearly triple that of Pt/C.
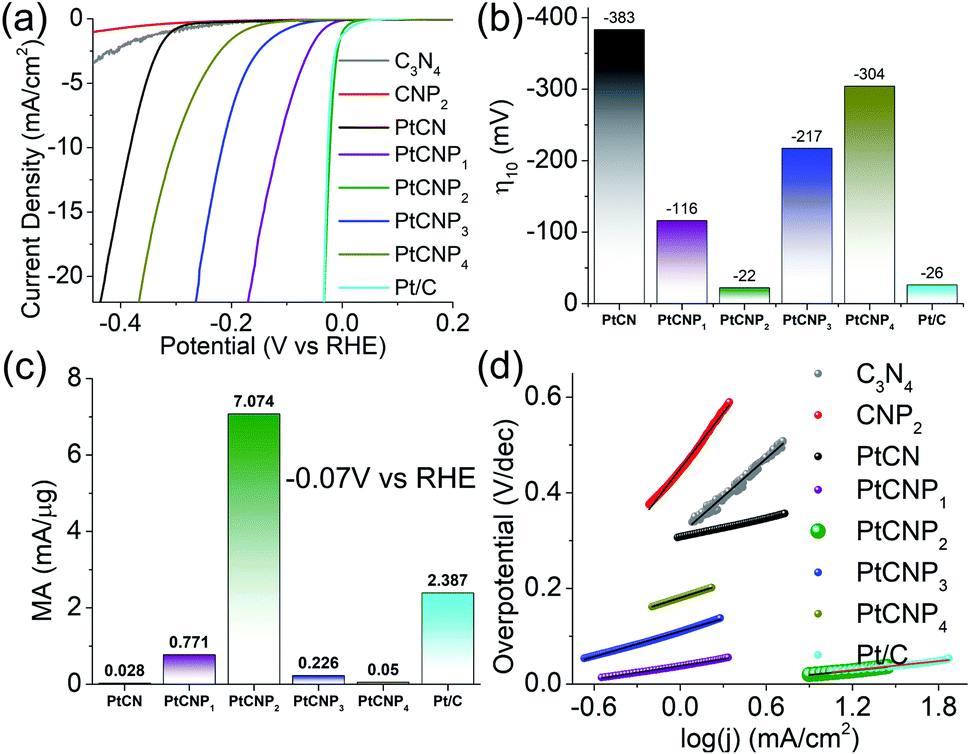 | ||
| Fig. 5 (a) HER polarization curves on various electrocatalysts in 0.5 M H2SO4 at the rotation rate of 1600 rpm, potential sweep rate of 10 mV s−1 and 85% iR compensation. (b) Corresponding η10 from the HER polarization curves in panel (a). (c) Mass activity at −0.07 V vs. RHE per μg of Pt calculated from ICP-OES measurements. (d) Tafel plots derived from panel (a). Note the overlapping data points between the Pt/C and PtCNP2 samples. | ||
Fig. 5d depicts the Tafel plots for the various samples. Based on linear regression, the Pt-free samples can be seen to display a high Tafel slope of 265.4 mV dec−1 for C3N4 (grey) and 390.8 mV dec−1 for CNP2 (red), which became substantially reduced for the PtCN and PtCNP samples, 65.7 mV dec−1 for PtCN, 47.3 mV dec−1 for PtCNP1, 31.2 mV dec−1 for PtCNP2, 88.2 mV dec−1 for PtCNP3, 95.7 mV dec−1 for PtCNP4, and 31.5 mV dec−1 for Pt/C. That is, PtCNP2 exhibited the lowest Tafel slope, suggesting most favorable electron-transfer kinetics. This also suggests that the Tafel or recombination reaction is the rate determining step,51 as observed with commercial Pt/C, in which two adsorbed hydrogen join at the surface of the electrocatalyst and desorb to form molecular hydrogen.
Electrochemical impedance measurements were then carried out to evaluate the HER charge-transfer resistance (Rct). Fig. 6a depicts the Nyquist plots produced at the overpotential of −10 mV in 0.5 M H2SO4, which were fitted by using EC-Lab's software V11.10 with the equivalent circuit shown in the figure inset (R1 is the solution resistance, Rct is the charge-transfer resistance, C is the double-layer capacitance, and Wd is the Warburg diffusion term). At the selected overpotential, Rct was found to vary significantly among the samples, 2926 Ω for PtCN, 102.6 Ω for PtCNP1, 19.3 Ω for PtCNP2, 5862 Ω for PtCNP3, and 6851 Ω for PtCNP4, consistent with the variation of their HER activity as manifested in the HER polarization curves (Fig. 5a).
 | ||
| Fig. 6 (a) Nyquist plots for the sample series produced from electrochemical impedance measurements at −10 mV overpotential. Inset is the Randle's equivalent circuit where R1 is the solution resistance, Rct is the charge-transfer resistance, C is the double-layer capacitance, and Wd is the Warburg diffusion term. (b) Mott–Schottky measurements for the sample series performed in 0.1 M Na2SO4. Potentials are vs. an Ag/AgCl reference electrode. Regression taken within the linear portion of each sample is shown as a black solid line. Y-axis on the left correspond to samples PtCN, PtCNP1, and PtCNP4 while y-axis on the right corresponds to PtCNP2 and PtCNP3. | ||
To gain further insight into the charge-transfer capability of the samples, Mott–Schottky analysis was performed in 0.1 M Na2SO4. From Fig. 6b, all samples can be seen to possess a positive slope along the linear region of each curve, suggesting n-type semiconductor behavior.26 The flat band potential (Efb) for PtCN was calculated to be −0.92 V vs. Ag/AgCl and an anodic shift was observed upon phosphorous doping to −0.61 V for PtCNP1 and −0.60 V for PtCNP2, whereas a further increase of the phosphorous dopant concentration was found to result in a slight cathodic shift of Efb to −0.65 V for PtCNP3 and −0.68 V for PtCNP4. Note that the formal potential for hydrogen evolution (H+/H2) is −0.59 V vs. Ag/AgCl in 0.1 M Na2SO4.26 A positive shift toward −0.59 V found in the PtCNP1 and PtCNP2 samples suggests a narrowing of the energy barrier, while the slight negative shift in PtNP3 and PtCNP4 suggests a broadening of the energy barrier. These findings are consistent with the HER polarization curves in Fig. 5a and the electrocatalytic activity trend observed. Furthermore, the charge carrier density (Nd) of the catalyst can be calculated by  where e is the elementary charge of an electron (1.602 × 10−19 C), ε is the dielectric constant (10),52ε0 is the permittivity in a vacuum (8.85 × 10−12 Fm−1), and (m) is the slope of the Mott–Schottky linear regression. Nd was calculated to be 8.66 × 1016 for PtCN, 4.30 × 1016 for PtCNP1, 6.60 × 1017 for PtCNP2, 6.55 × 1017 for PtCNP3, and 6.32 × 1016 for PtCNP4. That is, PtCNP2 possessed the highest charge carrier density (with PtCNP3 being the close second) among the sample series, which is nearly 10 times that of PtCN. These results suggest phosphorous doping within the carbon nitride framework reaches an optimum level for charge carrier density for PtCNP2, in coincidence with the best HER activity.
where e is the elementary charge of an electron (1.602 × 10−19 C), ε is the dielectric constant (10),52ε0 is the permittivity in a vacuum (8.85 × 10−12 Fm−1), and (m) is the slope of the Mott–Schottky linear regression. Nd was calculated to be 8.66 × 1016 for PtCN, 4.30 × 1016 for PtCNP1, 6.60 × 1017 for PtCNP2, 6.55 × 1017 for PtCNP3, and 6.32 × 1016 for PtCNP4. That is, PtCNP2 possessed the highest charge carrier density (with PtCNP3 being the close second) among the sample series, which is nearly 10 times that of PtCN. These results suggest phosphorous doping within the carbon nitride framework reaches an optimum level for charge carrier density for PtCNP2, in coincidence with the best HER activity.
Conclusions
In summary, a series of phosphorous-doped carbon nitride materials were synthesized by thermal annealing of urea and NH4PF6. A facile chelation procedure was exploited for the complexation of isolated Pt centers into the carbon nitrile matrix using of an activated Pt(II) Zeise's salt as the precursor. XPS and XAS measurements confirmed the atomic dispersion of Pt into the structural scaffold with Pt–N/P/Cl coordination. The resulting materials were found to exhibit remarkable electrocatalytic activity towards HER in acidic media, and the results suggest that P doping was critical in enhancing the electrical conductivity and HER electron-transfer kinetics of the composite catalysts. Among the series, the PtCNP2 sample was found to possess the best HER electrocatalytic performance, with a mass activity almost three-times that of commercial Pt/C benchmark. This was coincident with the highest charge carrier density and lowest charge-transfer resistance. Results from the present study highlight the unique advantage of P doping in manipulating the electronic properties and enhancing the electrocatalytic activity of metal–carbon nanocomposites for electrocatalysis in important electrochemical energy technologies.Author contributions
S. C. conceived the study. F. N. carried out the majority of the work. J. S., Z. A., and R. C. assisted in data acquisition. Q. M. L. assisted in result analysis and theoretical simulations. R. M. performed the XPS work. F. N. collected and analyzed the XAS data under the supervision of F. B. All authors contributed to result discussion and writing of the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported, in part, by the National Science Foundation (CHE-2003685 and CHE-1900235). TEM and XPS studies were carried out at the National Center for Electron Microscopy and Molecular Foundry, Lawrence Berkeley National Laboratory, which is supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. XAS experiments were performed at the Stanford Synchrotron Radiation Lightsource (SSRL), which is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract DE-AC02-76SF00515. We would like to thank Brandon Cheney for the assistance collecting SEM and EDS mapping results. We also thank Jeremy Barnett for the assistance with sample preparation and XRD data collection using the Rigaku Smartlab Diffractometer that was funded by NSF MRI grant number 1126845.References
- https://www.iea.org/reports/renewables-2020 .
- J. Greeley, T. F. Jaramillo, J. Bonde, I. B. Chorkendorff and J. K. Nørskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed.
- D. Voiry, H. Yamaguchi, J. Li, R. Silva, D. C. B. Alves, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy and G. Eda, Nat. Mater., 2013, 12, 850–855 CrossRef CAS PubMed.
- K. Liu, C. Song and V. Subramani, Hydrogen and Syngas Production and Purification Technologies, John Wiley & Sons, 2010 Search PubMed.
- A. Eftekhari, Int. J. Hydrogen Energy, 2017, 42, 11053–11077 CrossRef CAS.
- B. D. Adams and A. C. Chen, Mater. Today, 2011, 14, 282–289 CrossRef CAS.
- Z. Zhao, H. Liu, W. Gao, W. Xue, Z. Liu, J. Huang, X. Pan and Y. Huang, J. Am. Chem. Soc., 2018, 140, 9046–9050 CrossRef CAS PubMed.
- J. Liu, Y. Li, X. Zhou, H. Jiang, H. G. Yang and C. Li, J. Mater. Chem. A, 2020, 8, 17–26 RSC.
- F. Nichols, J. E. Lu, R. Mercado, R. Dudschus, F. Bridges and S. W. Chen, Chem.–Eur. J., 2020, 26, 4136–4142 CAS.
- Z. M. Cao, H. Q. Li, C. Y. Zhan, J. W. Zhang, W. Wang, B. B. Xu, F. Lu, Y. Q. Jiang, Z. X. Xie and L. S. Zheng, Nanoscale, 2018, 10, 5072–5077 RSC.
- X. Tian, X. Zhao, Y. Su, L. Wang, H. Wang, D. Dang, B. Chi, H. Liu, E. J. Hensen and X. W. D. Lou, Science, 2019, 366, 850–856 CrossRef CAS PubMed.
- N. Cheng, S. Stambula, D. Wang, M. N. Banis, J. Liu, A. Riese, B. W. Xiao, R. Y. Li, T.-K. Sham and L.-M. Liu, Nat. Commun., 2016, 7, 13638 CrossRef CAS PubMed.
- X. P. Yin, H. J. Wang, S. F. Tang, X. L. Lu, M. Shu, R. Si and T. B. Lu, Angew. Chem., Int. Ed., 2018, 57, 9382–9386 CrossRef CAS PubMed.
- B. Z. Lu, L. Guo, F. Wu, Y. Peng, J. E. Lu, T. J. Smart, N. Wang, Y. Z. Finfrock, D. Morris, P. Zhang, N. Li, P. Gao, Y. Ping and S. W. Chen, Nat. Commun., 2019, 10, 631 CrossRef CAS.
- Q. M. Liu, Q. X. Li and S. W. Chen, Curr. Opin. Electrochem., 2020, 21, 46–54 CrossRef CAS.
- Y. Peng, B. Z. Lu, L. M. Chen, N. Wang, J. E. Lu, Y. Ping and S. W. Chen, J. Mater. Chem. A, 2017, 5, 19499 RSC.
- Y. Wang, H. Su, Y. He, L. Li, S. Zhu, H. Shen, P. Xie, X. Fu, G. Zhou, C. Feng, D. Zhao, F. Xiao, X. Zhu, Y. Zeng, M. Shao, S. W. Chen, G. Wu, J. Zeng and C. Wang, Chem. Rev., 2020, 120, 12217–12314 CrossRef CAS PubMed.
- B. Lu, Q. Liu, F. Nichols, R. Mercado, D. Morris, N. Li, P. Zhang, P. Gao, Y. Ping and S. W. Chen, Research, 2020, 2020, 9167829 CAS.
- W. Y. Zhu and S. W. Chen, Electroanalysis, 2020, 32, 2591–2602 CrossRef CAS.
- J. Peng, Y. Chen, K. Wang, Z. Tang and S. W. Chen, Int. J. Hydrogen Energy, 2020, 45, 18840–18849 CrossRef CAS.
- B. Lu, Q. Liu and S. W. Chen, ACS Catal., 2020, 10, 7584–7618 CrossRef CAS.
- Q. M. Liu, Y. Peng, Q. X. Li, T. He, D. Morris, F. Nichols, R. Mercado, P. Zhang and S. W. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 17641–17650 CrossRef CAS.
- F.-Y. Yu, Z.-L. Lang, L.-Y. Yin, K. Feng, Y.-J. Xia, H.-Q. Tan, H.-T. Zhu, J. Zhong, Z.-H. Kang and Y.-G. Li, Nat. Commun., 2020, 11, 490 CrossRef PubMed.
- Z. H. Pu, I. S. Amiinu, R. L. Cheng, P. Y. Wang, C. T. Zhang, S. C. Mu, W. Y. Zhao, F. M. Su, G. X. Zhang, S. J. Liao and S. H. Sun, Nano-Micro Lett., 2020, 12, 21 CrossRef CAS.
- J. Kim, H. E. Kim and H. Lee, ChemSusChem, 2018, 11, 104–113 CrossRef CAS.
- Y. Peng, W. Z. Pan, N. Wang, J. E. Lu and S. W. Chen, ChemSusChem, 2018, 11, 130–136 CrossRef CAS.
- Z. Zeng, Y. Su, X. Quan, W. Choi, G. Zhang, N. Liu, B. Kim, S. Chen, H. Yu and S. Zhang, Nano Energy, 2020, 69, 104409 CrossRef CAS.
- H. Yan, C. Tian, L. Wang, A. Wu, M. Meng, L. Zhao and H. Fu, Angew. Chem., Int. Ed., 2015, 127, 6423–6427 CrossRef.
- Y. J. Zhang, T. Mori, J. H. Ye and M. Antonietti, J. Am. Chem. Soc., 2010, 132, 6294–6295 CrossRef CAS PubMed.
- S. Guo, Z. Deng, M. Li, B. Jiang, C. Tian, Q. Pan and H. Fu, Angew. Chem., Int. Ed., 2016, 128, 1862–1866 CrossRef.
- J. P. Paraknowitsch and A. Thomas, Energy Environ. Sci., 2013, 6, 2839–2855 RSC.
- X. W. Wang, G. Z. Sun, P. Routh, D. H. Kim, W. Huang and P. Chen, Chem. Soc. Rev., 2014, 43, 7067–7098 RSC.
- S. B. Yang, L. J. Zhi, K. Tang, X. L. Feng, J. Maier and K. Mullen, Adv. Funct. Mater., 2012, 22, 3634–3640 CrossRef CAS.
- Z. F. Hu, Z. R. Shen and J. C. Yu, Green Chem., 2017, 19, 588–613 RSC.
- A. Lazauskas, J. Baltrusaitis, L. Puodziukynas, M. Andrulevicius, G. Bagdziunas, D. Volyniuk, S. Meskinis, G. Niaura, T. Tamulevicius and V. Jankauskaite, Carbon, 2016, 107, 415–425 CrossRef CAS.
- M. Benedetti, C. R. Girelli, D. Antonucci, S. A. De Pascali and F. P. Fanizzi, Inorg. Chim. Acta, 2014, 413, 109–114 CrossRef CAS.
- H.-X. Han and B. J. Feldman, Solid State Commun., 1988, 65, 921–923 CrossRef CAS.
- K. Ogata, J. F. D. Chubaci and F. Fujimoto, J. Appl. Phys., 1994, 76, 3791–3796 CrossRef CAS.
- V. N. Khabashesku, J. L. Zimmerman and J. L. Margrave, Chem. Mater., 2000, 12, 3264–3270 CrossRef CAS.
- Q. J. Xiang, J. G. Yu and M. Jaroniec, J. Phys. Chem. C, 2011, 115, 7355–7363 CrossRef CAS.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2010, 26, 3894–3901 CrossRef CAS PubMed.
- C. Lv, Q. Yang, Q. Huang, Z. Huang, H. Xia and C. Zhang, J. Mater. Chem. A, 2016, 4, 13336–13343 RSC.
- H. Wu, Z. Chen, Y. Wang, E. Cao, F. Xiao, S. Chen, S. Du, Y. Wu and Z. Ren, Energy Environ. Sci., 2019, 12, 2697–2705 RSC.
- C. Andersson and R. Larsson, Chem. Scr., 1977, 11, 140–144 CAS.
- D. T. Clark, D. Briggs and D. B. Adams, J. Chem. Soc., Dalton Trans., 1973, 169–172 RSC.
- C. H. Booth, see: http://lise.lbl.gov/RSSAXP/, 2010.
- J. A. Horsley, J. Chem. Phys., 1982, 76, 1451–1458 CrossRef CAS.
- A. N. Mansour, J. W. Cook Jr and D. E. Sayers, J. Phys. Chem., 1984, 88, 2330–2334 CrossRef CAS.
- D. Friebel, D. J. Miller, C. P. O'Grady, T. Anniyev, J. Bargar, U. Bergmann, H. Ogasawara, K. T. Wikfeldt, L. G. M. Pettersson and A. Nilsson, Phys. Chem. Chem. Phys., 2011, 13, 262–266 RSC.
- H. Yoshida, S. Nonoyama, Y. Yazawa and T. Hattori, Phys. Scr., 2005, 2005, 813 CrossRef.
- M. Zeng and Y. Li, J. Mater. Chem. A, 2015, 3, 14942–14962 RSC.
- F. H. Abd El-kader, M. A. Moharram, M. G. Khafagia and F. Mamdouh, Spectrochim. Acta, Part A, 2012, 97, 1115–1119 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental data. See DOI: 10.1039/d1ta06240a |
| This journal is © The Royal Society of Chemistry 2022 |