
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrification of a Milstein-type catalyst for alcohol reformation†
Damien
Tocqueville‡
 a,
Francesco
Crisanti‡
a,
Julian
Guerrero
a,
Esther
Nubret
a,
Marc
Robert
ab,
David
Milstein
c and
Niklas
von Wolff
*a
a,
Francesco
Crisanti‡
a,
Julian
Guerrero
a,
Esther
Nubret
a,
Marc
Robert
ab,
David
Milstein
c and
Niklas
von Wolff
*a
aLaboratoire d’Electrochimie MoléculaireUniversité Paris Cité, CNRS, Paris F-75006, France. E-mail: niklas.von-wolff@u-paris.fr
bInstitut Universitaire de France (IUF), Paris F-75005, France
cDepartment of Molecular Chemistry and Materials Science, The Weizmann Institute of Science, Rehovot 7610001, Israel
First published on 18th October 2022
Abstract
Novel energy and atom efficiency processes will be keys to develop the sustainable chemical industry of the future. Electrification could play an important role, by allowing to fine-tune energy input and using the ideal redox agent: the electron. Here we demonstrate that a commercially available Milstein ruthenium catalyst (1) can be used to promote the electrochemical oxidation of ethanol to ethyl acetate and acetate, thus demonstrating the four electron oxidation under preparative conditions. Cyclic voltammetry and DFT-calculations are used to devise a possible catalytic cycle based on a thermal chemical step generating the key hydride intermediate. Successful electrification of Milstein-type catalysts opens a pathway to use alcohols as a renewable feedstock for the generation of esters and other key building blocks in organic chemistry, thus contributing to increase energy efficiency in organic redox chemistry.
In order to achieve the goals of the Sustainable Development Scenario (SDS) of the International Energy Agency, the chemical industry's emission should decline by around 10% before 2030.1,2 This could be achieved by increasing energy efficiency and the usage of renewable feedstocks. In this respect, molecular electrocatalytic alcohol oxidation could be powerful tool by potentially providing energy and atom efficiency for organic synthesis and energy applications.2–7 Besides the use of aminoxyl-derivatives,8–13 especially the seminal work of Vizza, Bianchini and Grützmacher demonstrated that (transfer)-hydrogenation (TH) catalysts could be activated electrochemically and used in a so-called “organometallic fuel cell”.14 Other TH systems are however mostly limited to two electron oxidations of secondary or benzylic alcohols (Scheme 1A).15–21
 | ||
| Scheme 1 (A) Advantages/limitation of electrochemical homogeneous alcohol oxidation using well-defined catalysts. (B) Current efforts to electrify acceptor-less alcohol dehydrogenation (AAD) systems due to their large range of application in thermal catalysis. | ||
As an exception, Waymouth et al. recently reported an example of the intramolecular coupling of vicinal benzylic alcohols to the corresponding esters.19,22 In order to extend the range of possible catalysts candidates, the Waymouth group recently also explored the possibility to use an iron-based acceptor-less alcohol dehydrogenation (AAD) catalysts23 for electrocatalytic alcohol oxidation (Scheme 1B).24 The stability under electrochemical conditions in this case is limited to <2 turnovers, but it opens the door to explore a wide range of AAD reactions under electrochemical conditions. Here, we demonstrate that a commercially available Milstein-type AAD catalyst (1)25 is competent for the electrocatalytic alcohol oxidation of ethanol to ethyl acetate and acetate (Scheme 1B).
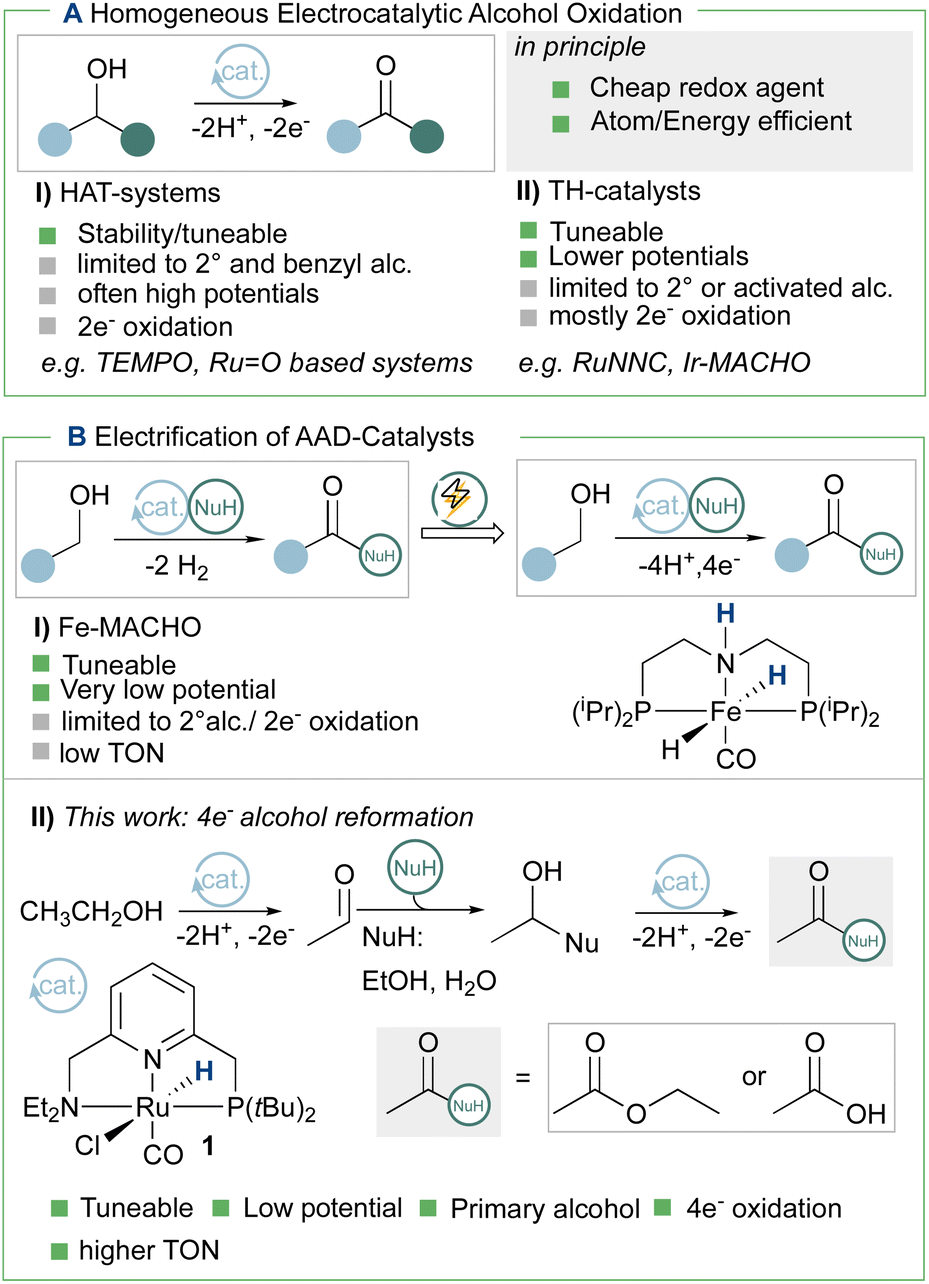
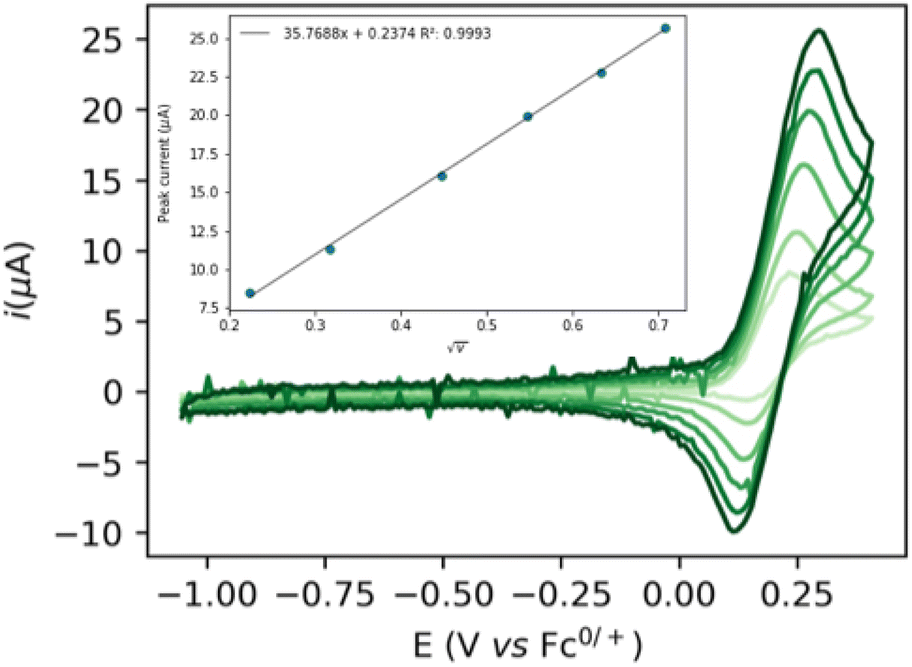
The cyclic voltammogram (CV) of complex 1 (Fig. 1) shows a quasi-reversible diffusive one electron oxidation wave at 0.2 V (all potentials are referenced vs. Fc+/Fc0) in 0.2 M NaPF6 THF/DFB (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (DFB = 1,2 difluoro benzene) assigned to the Ru(II)–Ru(III) couple (see ESI, section 2.2†). The addition of 1 to a 10 mM sodium ethoxide (NaOEt) solution in 200 mM ethanol (EtOH) in 0.1 M NaPF6 (2:1 THF/DFB) gives rise to several waves at ca. −0.5, 0.0 and 0.2 V with currents significantly higher than in the absence of catalysts or substrate, indicative of possible catalytic turnover (Fig. 2). Gradual increase of the EtOH concentration from 200 mM to 1 M is accompanied by the disappearance of the first wave at −0.5 V, while a new oxidation wave appears at ca. −0.25 V (Fig. 2, light to dark green traces).
:1) (DFB = 1,2 difluoro benzene) assigned to the Ru(II)–Ru(III) couple (see ESI, section 2.2†). The addition of 1 to a 10 mM sodium ethoxide (NaOEt) solution in 200 mM ethanol (EtOH) in 0.1 M NaPF6 (2:1 THF/DFB) gives rise to several waves at ca. −0.5, 0.0 and 0.2 V with currents significantly higher than in the absence of catalysts or substrate, indicative of possible catalytic turnover (Fig. 2). Gradual increase of the EtOH concentration from 200 mM to 1 M is accompanied by the disappearance of the first wave at −0.5 V, while a new oxidation wave appears at ca. −0.25 V (Fig. 2, light to dark green traces).
 | ||
| Fig. 1 Scan rate dependence of a 1 mM solution of 1 in in 2:1 THF/DFB + 0.2 M NaPF6 (from light to dark green: 0.05, 0.1, 0.2, 0.3, 0.4 and 0.5 V s−1, 3 mm GC electrode). Inset: evolution of the peak current as a function of the square root of the scan rate. | ||
 | ||
| Fig. 2 CVs of 10 mM NaOEt (grey) and of 5 mM 1 + 5 mM NaOEt with increasing concentrations of EtOH (from light to dark green: 200, 400, 600, 800 and 1000 mM) in 2:1 THF/DFB + 0.2 M NaPF6. Scan rate 0.1 V s−1, electrode: 3 mm diameter GC electrode. | ||
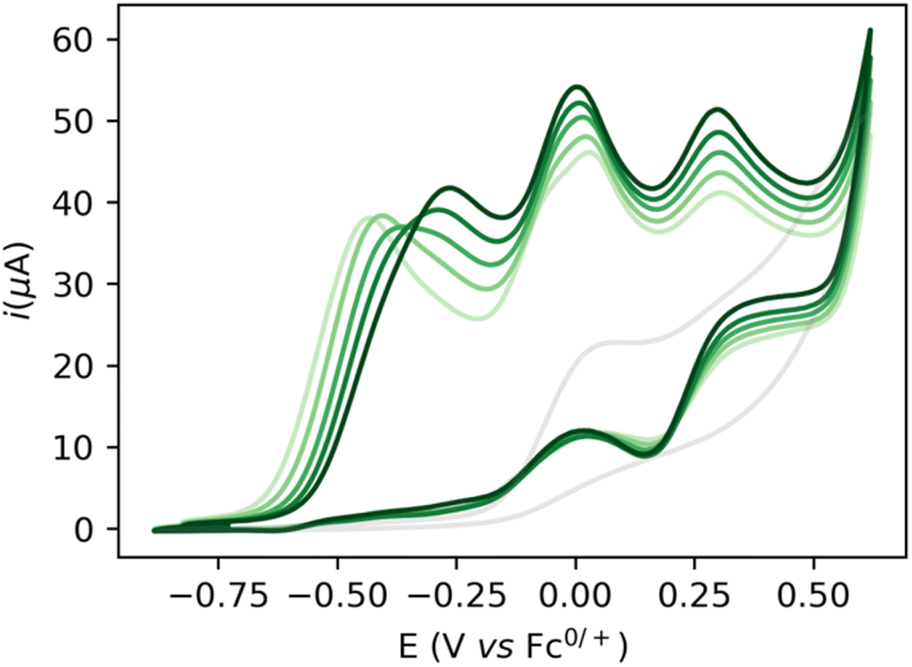
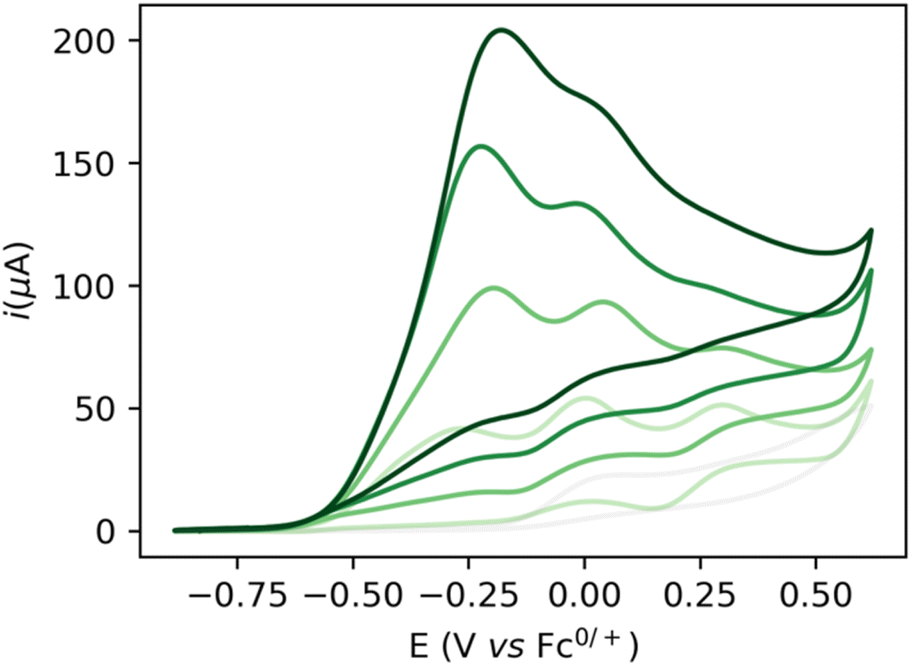
Increasing the base loading gradually from 5 to 20 mM yields a stark increase of current at this new wave at ca. −0.25 V (Fig. 3). Using (TBA)PF6 instead of NaPF6 (used to avoid Hofmann-elimination26) gave similar results (see ESI, section 2.2–2.5 and section 4†). In order to assess catalytic turnover under preparative conditions, controlled potential electrolysis (CPE) was performed. CPE experiments were run in pure ethanol (to reduce cell resistance) in the presence of 0.1 M electrolyte of well soluble bases (e.g. NaOEt, LiOH, see ESI section 4†). CPE in 0.1 M LiOH with 1 mM 1 at E = 0 V vs. Fc0/+ delivered ca. 15 mM of acetate and 6 mM of ethyl acetate, corresponding to 21 turnovers (per 4 electrons, or 42 turnovers per two electrons) and a faradaic efficiency (FE) of ca. 62% (see ESI section 4.3†). In the absence of applied potential (OCP, open circuit potential), no ethyl acetate was formed (see ESI, section 4.4†). Likewise, in the absence of catalyst, the passed charge was significantly lower (7C vs. 40C) with no detected formation of ethyl acetate. The low FE could be due to catalyst degradation, as Ru-nanoparticle formation is observed on the electrode post CPE (confirmed by SEM/Elemental mapping, see ESI section 5†). Noteworthy, rinse-test CPE and a CPE using a simple Ru-precursor, RuCl3, did not show any ethyl acetate formation and gave similar results to blank experiments, indicating that Ru-nanoparticles are probably not the active catalyst species and that catalyst instability could be responsible for low FE. Further studies are underway to fully understand catalyst speciation under preparative conditions (see ESI section 4.7†) the observed catalytic activity of 1 compares well in terms of TON and product selectivity with other molecular homogeneous TH systems, with most systems being limited to the two-electron oxidation of secondary or benzylic alcohols. The Waymouth group reported a NNC ruthenium pincer for the oxidation of isopropanol to acetone with a TON of 4.18 The same group reported on the usage of phenoxy mediators with an iridium pincer complex, reaching a TON of 8 for the same reaction.22 Bonitatibus and co-workers demonstrated the activity of an iridium-based systems with a TON of 32 for the formation of p-benzaldehyde.17 Appel and co-workers reported on a nickel (TON = 3.1)15 and a cobalt triphos systems (TON = 19.9)16 for benzaldehyde formation from benzyl alcohol. To the best of our knowledge, there is only one acceptor-less alcohol dehydrogenation (AAD) catalyst that has been activated electrochemically so-far,24 generating acetone with a TON <2. Only a handful of molecular systems are known to catalyze the electrochemical four electron alcohol reformation to esters, however at significantly higher potentials (1.15 V vs. Fc+/Fc0).2,27,28 Thus, although not designed for electrochemical applications, 1 shows high activity for the challenging 4 electron oxidation of aliphatic substrates.
 | ||
| Fig. 3 CV of 5 mM NaOEt (grey), 5 mM of 1 + 1 M EtOH with varying concentrations of base (5, 10, 15, and 20 mM NaOEt, light to dark green) in 2:1 THF/DFB + 0.2 M NaPF6. Scan rate 0.1 V s−1, electrode: 3 mm diameter GC electrode. | ||
To achieve the transposition from thermal to electrochemical TH, both Grützmacher et al. and Waymouth took advantage of a fast equilibrium between the alcohol substrate and a metal hydride intermediate that could be readily oxidized. The chemistry of ruthenium pincer AAD systems is well studied (Scheme 2)25,29–33 and allows for a putative assignment of the observed CV-behavior. In the presence of excess base and alcohol (Fig. 2 and 3), 1 is expected to yield dearomatized complex 2,25 as well as the alkoxide species 3.25,32 We might therefore assign the first wave at −0.5 V to the oxidation of dearomatized complex 2 and the wave around 0 V to the oxidation of the alkoxide complex 3. Indeed, independently synthesized samples of 2 and 3 (in the presence of excess ethanol) give rise to oxidation half-waves at −0.45 V and −0.1 V respectively (see ESI, section 3 and 5.2†). This is also in agreement with the observed behavior upon increasing the alcohol concentration with the expected consumption of dearomatized species 2 and concomitant disappearance of the first oxidation wave at −0.5 V. The equilibrium between 2, 3 and 4 has been reported32 and addition of excess ethanol to 2 is thus not only generating 3, but also is expected to deliver 4 (Scheme 2). The appearance of a new anodic wave at ca. −0.25 V (Fig. 2) is thus attributed to the increasing formation of 4 upon addition of larger amounts of EtOH. Complex 4 is relatively unstable in solution,25,32,33 and decomposes in the presence of electrolyte (see ESI section 3.1†). DFT calculations were thus used to predict its oxidation potential (see ESI, section 6†), which was in reasonable agreement with the observed wave (−0.19 V). The DFT calculations also confirmed the assignment of the other waves related to the dearomatized complex 2 (−0.33 V) and the ethoxide species 3 (−0.1 V). A more detailed mechanistic analysis remains currently hampered by the chemical instability of 4 under the employed reaction conditions, as well as difficulties to isolate 3 in the solid state (limiting kinetic measurements). DFT calculations were thus used to get a better view on possible reaction pathways (Schemes 2, 3 and ESI section 6.3†). The oxidation of 4 at −0.19 V (DFT) yields the radical cation 5, with a calculated pKa in THF of 8.2. In the presence of NaOEt, 5 should thus deprotonate readily to give radical 6, which has an extremely negative oxidation potential of −2.1 V. At the potential it is generated, 6 should thus directly be oxidized to cationic complex 7. This cationic species 7 has a calculated pKa of 22.7 in THF, which is in good agreement with experimental data from the Saouma group on a similar system.26 The high pKa of 7 in THF also validates the need for a strong base (e.g. NaOEt) to reform dearomatized 2. Both Grützmacher and co-workers,14 as well as Waymouth24 have noted that the accelerating effect during electrocatalysis stems from the oxidation of a metal hydride intermediate that is generated by fast chemical steps. In order to verify this hypothesis and to exclude an electrochemical activation of this hydride formation step, transition state barriers were computed (Scheme 3). Taking the dearomatized complex 2 as a reference point, a first step will form the alkoxide species 3 (TS0 = 21.2 kcal mol−1). Oxidizing 2 to 8 slows down the formation of the alkoxide species (TS0ox = 27.5 kcal mol−1), most-likely due to decreased basicity of the ligand. From the alkoxide species 3 dihydride 4 is formed via a linear, charge-separated transition state TS1 (15.7 kcal mol−1). The role of such linear transition states was highlighted recently in the case of ruthenium pincer catalysis for alcohol oxidation.34–37 In principle, it might be envisioned that the oxidation of the metal center could be an additional driving force for this hydride abstraction step. However, after oxidation, the energy span38,39 rises by about 11 kcal mol−1 (TS1ox = 24.7 kcal mol−1). Likewise, a beta-hydride elimination via side-arm opening is not accelerated either by oxidation (TS2ox = 37.5 kcal mol−1, see ESI section 6.4†). It thus seems that the generation of 4 is not accelerated by electron transfer steps and relies on a thermally activated chemical step. Importantly, alkoxide solutions were shown to be excellent hydride donors electrochemically, further corroborating that under the employed basic conditions, generation of 4 from 3 should be fast.40 Oxidation of 4 to 5 also doesn't accelerate thermal intramolecular release of H2 (TS3Box = 37.5 kcal mol−1), which is significantly higher than neutral thermal H2-releasing states (TS3A and TS3B). The experimentally observed acceleration via electron-transfer is thus proposed to follow a classical ECEC mechanism initiated by the oxidation of 4 to 5 (at roughly −0.19 V (DFT)), followed by deprotonation and re-oxidation as described above, finally delivering 2 at the electrode surface. Importantly, at the electrode surface 2 and 3 should be oxidized at the employed potentials, but based on DFT-calculations, these pathways are thought to be non-productive (Scheme 3) and could explain the low catalyst life-time and degradation under electrochemical conditions.
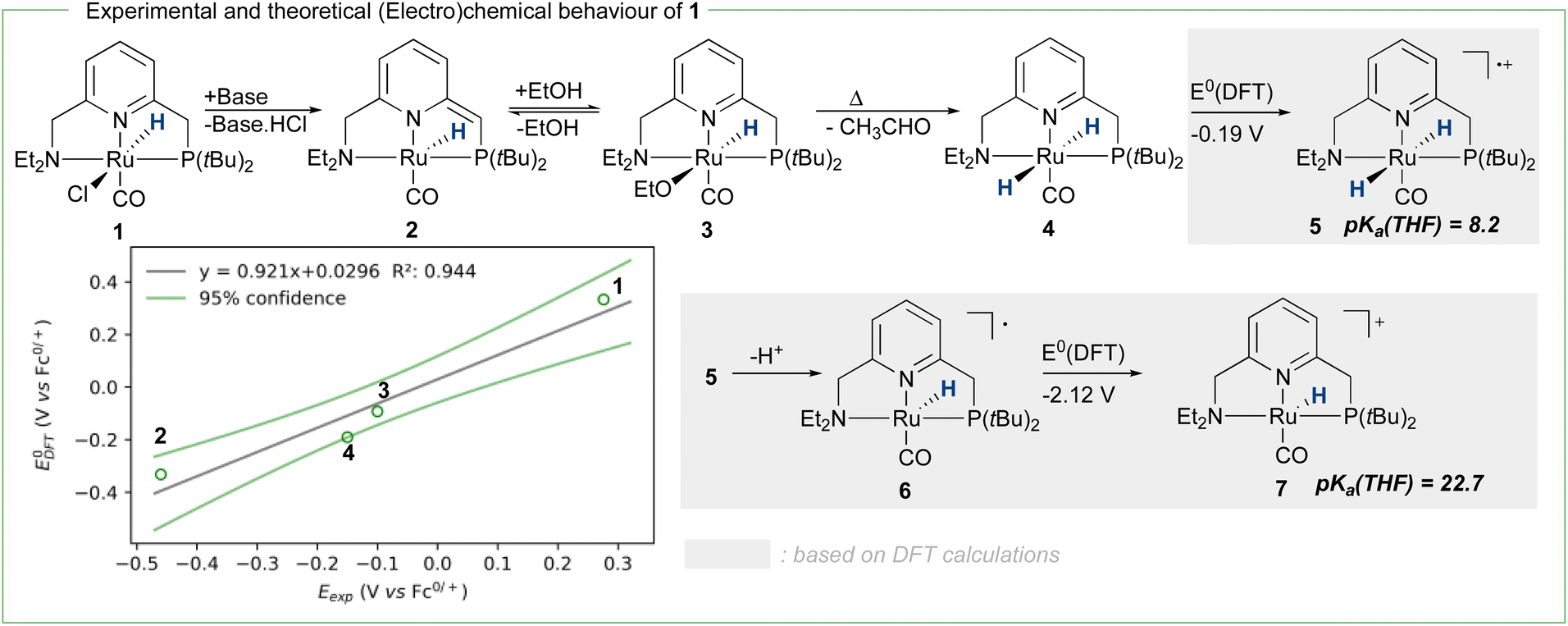 | ||
| Scheme 2 Reactivity of pyridine-based ruthenium complexes via dearomatization/aromatization, as well as DFT-based. | ||
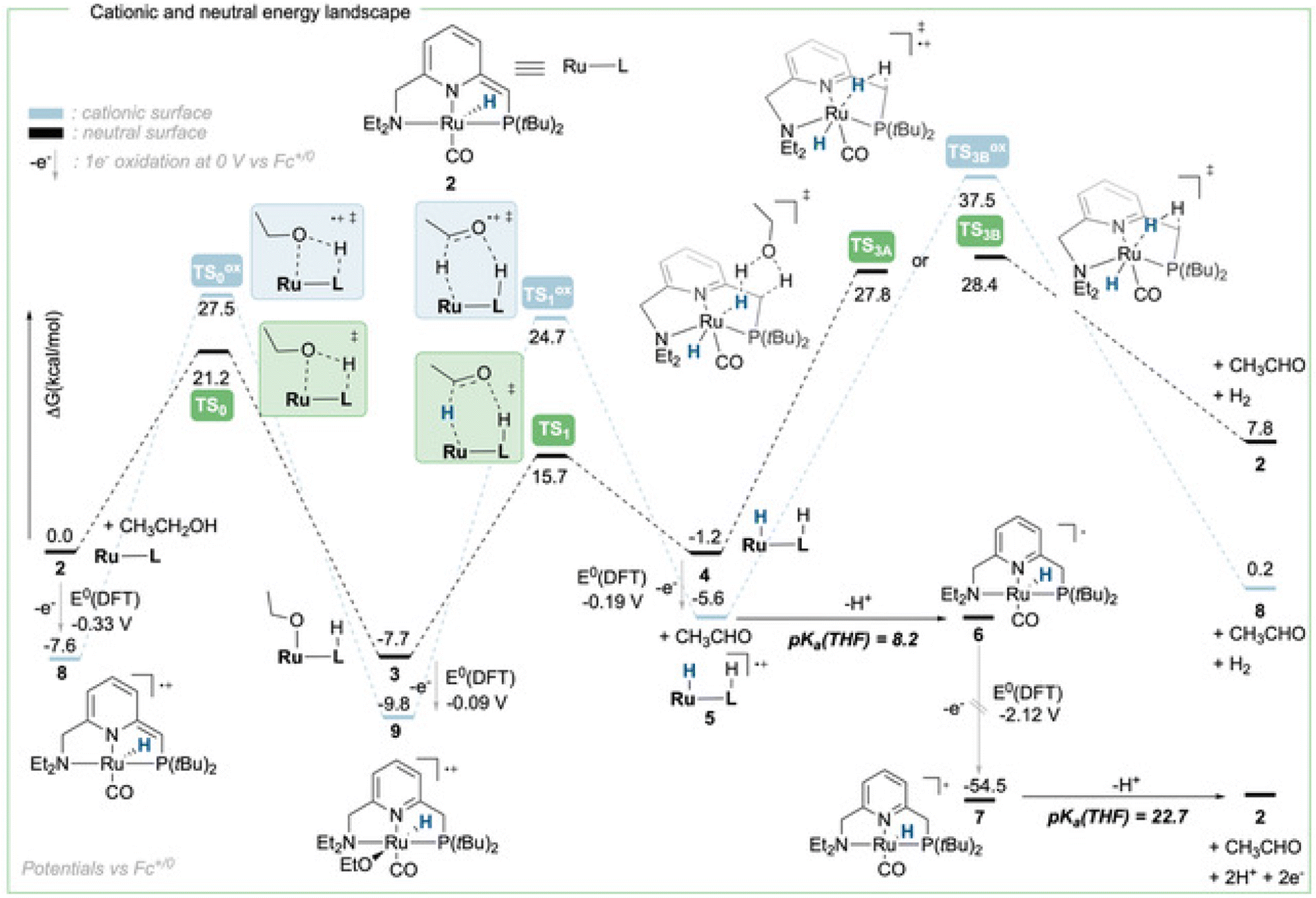 | ||
| Scheme 3 DFT-calculated energy landscape for the neutral (black dotted lines and bars) and cationic surface (blue dotted lines and bars) of ethanol dehydrogenation starting from 2 or its cationic analogue 8. | ||
Conclusions
In conclusion, a Milstein-type catalyst (1) was activated electrochemically for the dehydrogenation of a primary alcohol to the corresponding ester. CV and DFT studies were used to propose a putative catalytic cycle, based on the chemical generation of dihydride intermediate 4 and its electrochemical conversion. It was shown that 1 can be used under preparative conditions yielding the four-electron oxidation products ethyl acetate and acetate, thus confirming successful electrification. We are currently exploring the electrochemical activation to other AAD systems and substrates, as well as to investigate the mechanism in more details. This will hopefully allow to address adverse deactivation pathways, as well as to design more active catalysts.Data availability
All computed structures, including energies and thermochemistry is available free of charge via the ioChem-BD online repository under the following link: https://doi.org/10.19061/iochem-bd-6-117.Author contributions
Conceptualization: N. v. W. data curation: N. v. W., D. T., F. C. Electrochemistry: N. v. W., D. T., F. C. chemical synthesis: N. v. W., D. T. product analysis. N. v. W., D. T., F. C., E. N. Tests for nanoparticles: F. C. DFT calculations: N. v. W. SEM/Elemental Mapping: J. G. funding acquisition: N. v. W. writing original draft; N. v. W. reviewing: N. v. W., M. R., D. M.Conflicts of interest
There are no conflicts to declare.Acknowledgements
NvW is thankful for funding from the IdEx Université de Paris 2019, ANR-18-IDEX-0001. Computations were performed using HPC resources from GENCI-CINES (Grant 2019AP010811227 and AD010812061R1).Notes and references
- The Future of Petrochemicals – Analysis, https://www.iea.org/reports/the-future-of-petrochemicals, accessed October 26, 2021 Search PubMed.
- N. von Wolff, O. Rivada-Wheelaghan and D. Tocqueville, ChemElectroChem, 2021, 8, 4019–4027 CrossRef CAS.
- M. Bellini, M. Bevilacqua, A. Marchionni, H. A. Miller, J. Filippi, H. Grützmacher and F. Vizza, Eur. J. Inorg. Chem., 2018, 2018, 4393–4412 CrossRef CAS.
- A. W. Cook and K. M. Waldie, ACS Appl. Energy Mater., 2020, 3, 38–46 CrossRef CAS.
- Y. Holade, K. Servat, S. Tingry, T. W. Napporn, H. Remita, D. Cornu and K. B. Kokoh, ChemPhysChem, 2017, 18, 2573–2605 CrossRef CAS PubMed.
- J. Chen, S. Lv and S. Tian, ChemSusChem, 2019, 12, 115–132 CrossRef CAS.
- F. Wang and S. S. Stahl, Acc. Chem. Res., 2020, 53, 561–574 CrossRef CAS.
- A. Badalyan and S. S. Stahl, Nature, 2016, 535, 406 CrossRef CAS PubMed.
- J. E. Nutting, M. Rafiee and S. S. Stahl, Chem. Rev., 2018, 118, 4834–4885 CrossRef CAS PubMed.
- M. Rafiee, M. Alherech, S. D. Karlen and S. S. Stahl, J. Am. Chem. Soc., 2019, 141, 15266–15276 CrossRef CAS PubMed.
- M. Rafiee, Z. M. Konz, M. D. Graaf, H. F. Koolman and S. S. Stahl, ACS Catal., 2018, 8, 6738–6744 CrossRef CAS.
- A. C. Cardiel, B. J. Taitt and K.-S. Choi, ACS Sustainable Chem. Eng., 2019, 7, 11138–11149 CrossRef CAS.
- B. J. Taitt, M. T. Bender and K.-S. Choi, ACS Catal., 2020, 10, 265–275 CrossRef CAS.
- S. P. Annen, V. Bambagioni, M. Bevilacqua, J. Filippi, A. Marchionni, W. Oberhauser, H. Schönberg, F. Vizza, C. Bianchini and H. Grützmacher, Angew. Chem., Int. Ed., 2010, 49, 7229–7233 CrossRef CAS PubMed.
- C. J. Weiss, E. S. Wiedner, J. A. S. Roberts and A. M. Appel, Chem. Commun., 2015, 51, 6172–6174 RSC.
- S. P. Heins, P. E. Schneider, A. L. Speelman, S. Hammes-Schiffer and A. M. Appel, ACS Catal., 2021, 11, 6384–6389 CrossRef CAS.
- P. J. Bonitatibus, M. P. Rainka, A. J. Peters, D. L. Simone and M. D. Doherty, Chem. Commun., 2013, 49, 10581–10583 RSC.
- E. A. McLoughlin, K. C. Armstrong and R. M. Waymouth, ACS Catal., 2020, 10, 11654–11662 CrossRef CAS.
- K. R. Brownell, C. C. L. McCrory, C. E. D. Chidsey, R. H. Perry, R. N. Zare and R. M. Waymouth, J. Am. Chem. Soc., 2013, 135, 14299–14305 CrossRef CAS PubMed.
- M. Buonaiuto, A. G. De Crisci, T. F. Jaramillo and R. M. Waymouth, ACS Catal., 2015, 5, 7343–7349 CrossRef CAS.
- K. M. Waldie, K. R. Flajslik, E. McLoughlin, C. E. D. Chidsey and R. M. Waymouth, J. Am. Chem. Soc., 2017, 139, 738–748 CrossRef CAS.
- C. M. Galvin and R. M. Waymouth, J. Am. Chem. Soc., 2020, 142, 19368–19378 CrossRef CAS PubMed.
- M. Trincado, J. Bösken and H. Grützmacher, Coord. Chem. Rev., 2021, 443, 213967 CrossRef CAS.
- E. A. McLoughlin, B. D. Matson, R. Sarangi and R. M. Waymouth, Inorg. Chem., 2020, 59, 1453–1460 CrossRef CAS PubMed.
- J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2005, 127, 10840–10841 CrossRef CAS PubMed.
- C. L. Mathis, J. Geary, Y. Ardon, M. S. Reese, M. A. Philliber, R. T. VanderLinden and C. T. Saouma, J. Am. Chem. Soc., 2019, 141, 14317–14328 CrossRef CAS PubMed.
- G. J. Matare, M. E. Tess, Y. Yang, K. A. Abboud and L. McElwee-White, Organometallics, 2002, 21, 711–716 CrossRef CAS.
- M. E. Tess, P. L. Hill, K. E. Torraca, M. E. Kerr, K. A. Abboud and L. McElwee-White, Inorg. Chem., 2000, 39, 3942–3944 CrossRef CAS PubMed.
- C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790 CrossRef CAS PubMed.
- J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed.
- T. He, J. C. Buttner, E. F. Reynolds, J. Pham, J. C. Malek, J. M. Keith and A. R. Chianese, J. Am. Chem. Soc., 2019, 141, 17404–17413 CrossRef CAS PubMed.
- D. G. Gusev, Organometallics, 2020, 39, 258–270 CrossRef CAS.
- J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2006, 45, 1113–1115 CrossRef CAS PubMed.
- N. Govindarajan, V. Sinha, M. Trincado, H. Grützmacher, E. J. Meijer and B. de Bruin, ChemCatChem, 2020, 12, 2610–2621 CrossRef CAS.
- V. Sinha, N. Govindarajan, B. de Bruin and E. J. Meijer, ACS Catal., 2018, 8, 6908–6913 CrossRef CAS PubMed.
- Y.-Q. Zou, N. von Wolff, M. Rauch, M. Feller, Q.-Q. Zhou, A. Anaby, Y. Diskin-Posner, L. J. W. Shimon, L. Avram, Y. Ben-David and D. Milstein, Chem.–Eur. J., 2021, 27, 4715–4722 CrossRef CAS PubMed.
- P. A. Dub and J. C. Gordon, ACS Catal., 2017, 7, 6635–6655 CrossRef CAS.
- S. Kozuch, C. Amatore, A. Jutand and S. Shaik, Organometallics, 2005, 24, 2319–2330 CrossRef CAS.
- C. Amatore, A. Jutand, F. Khalil, M. A. M'Barki and L. Mottier, Organometallics, 1993, 12, 3168–3178 CrossRef CAS.
- C. Amatore, J. Badoz-Lambling, C. Bonnel-Huyghes, J. Pinson, J. M. Saveant and A. Thiebault, J. Am. Chem. Soc., 1982, 104, 1979–1986 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Including experimental procedures, CV-characterization of key intermediates, CPE results, DFT-calculated redox potentials and BDFEs. See DOI: https://doi.org/10.1039/d2sc04533h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |