
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSalt-mediated, plasmonic field-field/field-lattice coupling-enhanced NIR-II photodynamic therapy using core-gap-shell gold nanopeanuts†
Naresh
Kuthala
a,
Munusamy
Shanmugam
a,
Xiangyi
Kong
b,
Chi-Shiun
Chiang
 c and
Kuo Chu
Hwang
*a
c and
Kuo Chu
Hwang
*a
aDepartment of Chemistry, National Tsing Hua University, Hsinchu 30013, Taiwan, Republic of China. E-mail: kchwang@mx.nthu.edu.tw
bDepartment of Breast Surgical Oncology, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100021, China
cDepartment of Biomedical Engineering and Environmental Sciences, National Tsing Hua University, Hsinchu 30013, Taiwan, Republic of China
First published on 9th May 2022
Abstract
Plasmonic field-field coupling-induced enhancement of the optical properties of dye molecules in the nanogaps among metal nanoparticle clusters and thin films has attracted significant attention especially in disease-related theranostic applications. However, it is very challenging to synthesize plasmonic core-gap-shell nanostructures with a well-controlled nanogap, uniform shape, and distances to maximize the plasmonic field-field coupling between the core and the shell. Herein, we synthesized Au@gap@AuAg nanopeanut-shaped core-gap-shell nanostructures (Au NPN) and tuned their optical absorption from near-infrared region-I (NIR-I) to near-infrared region-II (NIR-II) by filling their nanogap with a high dielectric NaCl(aq) aqueous solution, which led to a dramatic redshift in the plasmonic absorption band by 320 nm from 660 to 980 nm and a 12.6-fold increase (at 1064 nm) in the extinction coefficient in the NIR region (1000–1300 nm). Upon filling the nanogap with NaCl(aq) aqueous solution, the Au NPN6.5(NaCl) (i.e., ∼6.5 nm nanogap)-mediated NIR-II photodynamic therapy effect was dramatically enhanced, resulting in a much longer average lifespan of >55 days for the mice bearing a murine colon tumor and treated with Au NPN6.5(NaCl) plus 1064 nm light irradiation compared to the mice treated with Au NPN6.5 + 1064 nm light irradiation (without nanogap filled with dielectric NaCl(aq), 40 d) and the doxorubicin-treated group (23 d). This study demonstrates a simple but effective method to tune and maximize the plasmonic field-field coupling between the metal shell and metal core of core-gap-shell nanostructures, the plasmonic field-lattice interactions, and biomedical applications for the treatment of tumors. Overall, our work presents a new way to enhance/maximize the plasmonic field-field and field-lattice coupling, and thus the performance/sensitivities in nanogap-based bioimaging, sensing, and theranostic nanomaterials and devices.
New conceptsPlasmonic field-field coupling interactions between metal nanoparticles are well known to spectroscopic scientists and have been utilized in the surface-enhanced Raman sensing of biological species and probes. Although the magnitude of plasmonic field-field coupling is known to be influenced by many factors, it has never been reported that the plasmonic field-field coupling can be tuned via the addition of salt solution, and thus has never been applied in the therapeutic treatment of cancers. Herein, we report a simple but very effective method for tuning and maximizing the plasmonic field-field and field-lattice interactions between the metal core and metal shell in an Au@gap@AuAg nanopeanut-like core-gap-shell (Au NPN) nanostructure. We demonstrate for the first time that salt-filling in the nanogaps of plasmonic core-gap-shell nanostructures can also enhance the plasmonic field-lattice interactions, leading to enhanced photothermal conversion efficiencies, X-ray scattering cross sections in XRD measurements, a 2-fold increase in the singlet oxygen sensitization quantum yield, >10-fold increase in the SERS intensities of surface-absorbed dye molecules, enhanced photodynamic therapy effects in destroying solid tumors and prolonged average lifespan of diseased mice. Our study sheds new light on enhancing the detection sensitivities and limits of biosensing devices based on plasmonic field-field coupling. |
Introduction
Surface plasmon resonance (SPR) is a unique property of metallic nanostructures.1,2 The plasmonic field generated from a single, isolated metal nanoparticle or rough metal surface is known to enable a large enhancement in the Raman scattering intensity of dye molecules absorbed on the metal surface via the plasmonic field-enhanced Raman scattering cross section of the dye molecules with incident photons.3 This phenomenon is well-known as surface-enhanced Raman scattering (SERS). The enhancement factor for the SERS phenomenon from a single, isolated metal nanoparticle is in the range of 1 × 105 to 1 × 106.3 Besides SERS, the plasmonic field is also known to enhance the fluorescence emission4,5 and infrared absorption6,7 of dye molecules nearby or absorbed on the surface of metal nanoparticles. Simple-structured metallic nanoparticles, such as nanospheres,8 nanodisks,9 nanorods,10 nanotriangles,11 nanocubes,12 nanorings,13 nanoshells,14 nanostars,15 nanocrescents,16 nanocrosses,17 and nanocages,18 were reported to generate plasmonic field upon photo irradiation. The SPR-enhanced opto-properties of metal nanostructures have attracted significant attention and have been widely applied in different research areas, such as bio-sensing/imaging,19–22 photodetectors,23–25 plasmon-enhanced solar cells,26,27 surface-enhanced Raman scattering (SERS),28,29 cancer therapy and drug delivery.30–34 Recently, the plasmonic field generated from isolated metal nanoparticles was utilized to mediate singlet oxygen generation in the absence of any organic photosensitizers for cancer treatment in xenograft mice models.35–40When simple-structured nanoparticles gather to form complicate-structured aggregates, such as metal nanoparticle clusters and self-assembled structures, metal nanoparticle on metal thin film (i.e., so called nanoparticle-on-mirror, NPoM)41–44 and core-gap-shell structures, some new opto-properties may appear, which uniquely belong to these more complicated nanostructures and cannot be found in simple-structured nanomaterials. For example, plasmonic field-field coupling interactions through the nanogap space between two adjacent plasmonic fields can occur in the above-mentioned complicated metal nanostructures, but not in single, isolated nanoparticles. Plasmonic field-field coupling through the nanogaps between two adjacent metal nanoparticles, among metal nanoparticle clusters,45–54 in core-gap-shell metal nanostructures,53–56 and in NPoM39–42 was found to result in an enormously large plasmonic field enhancement factor (EF) in the range of 1.0 × 108 to >1010 due to the Raman scattering of dye molecules located within the nanogap,55–58 which is 3–4 orders larger than that generated from a single, isolated metal nanoparticle (EF = 1 × 105–1.0 × 106).3 The huge plasmonic field-field coupling in nanogaps with an EF larger than 1.0 × 108 is believed to be large enough to allow the observation of the Raman scattering of a single molecule.41–44,57 The observation of single molecule Raman scattering is not possible from single, isolated metal nanoparticle systems due to their insufficient enhancement factors. The huge plasmonic field enhancement in metal nanoparticle arrays and metal nanoparticles on metal thin films is frequently utilized in sensing devices to enhance the fluorescence emission or Raman scattering of dye molecules siting in the nanogap cavities.4,5,41,43 When dye molecules are encapsulated in the nanogap region of core-gap-shell nanostructures or nanoparticle arrays, the Raman scattering intensities can be dramatically enhanced by the coupled plasmonic fields between the metal core and the metal shell or between the nanoparticles in nanoparticle aggregates/arrays. This is the so-called gap-enhanced Raman tag (GERT), which is commonly used to trace/image the location and distribution of core-gap-shell nanomaterials and tumor sites in biological systems.55–58 The huge nanogap-enhanced plasmonic field-field coupling phenomenon was utilized to detect cancer biomarkers with an ultralow detection limit of 10 fM.57
Besides enhancing the fluorescence and Raman scattering intensities, the plasmonic field-field coupling in nanogaps was also found to cause a redshift in the localized surface plasmon resonance (LSPR) bands of aggregated metal nanoparticles/thin films by 35–250 nm.45–47 Rod-shaped gold nanorattles were shown to have a redshift in their LSPR band by ∼75 nm compared to isolated Au NRs.54 Au opening-nanoshell ordered arrays were also shown to exhibit a redshift in their LSPR from the visible to NIR region upon a decrease in the nanogap distance from 16 to 2 nm between adjacent Au nanoshells.53 The most well-known example is the aggregation of gold nanoparticles (Au NPs), leading to a color change from red for single isolated Au NPs (LSPR absorption band in the short UV region) to blue color for aggregated Au NPs (i.e., LSPR band shifted to long visible region).48–50 This aggregation-induced red-to-blue color transition of Au NPs was utilized in colorimetric sensing devices to detect various types of biological species.51,52 Gold nanorod-in-shell was reported to act as an effective photothermal therapeutic (PTT) reagent in the NIR-II biological window using a 1064 nm high laser power of 3 W cm−2 for excitation, which exceeds the skin threshold value of 420 mW cm−2 at 1064 nm.59,60 Most of the therapeutic models were studied in the NIR-I BW (650–950 nm).61,62 However, recently, isolated metal nanomaterials were used for NIR PDT therapeutic treatment of cancers in the NIR-II biological window.33,34,63,64
In the literature, many theoretical calculations and experimental observations have confirmed and illustrated that the plasmonic coupling between two adjacent plasmonic fields leads to a redshift in the LSPR bands of nanostructures. For example, theoretical calculation using a discrete dipole approximation (DDA) method showed that the magnitude of coupling between two adjacent plasmonic fields and the extent of the redshift of LSPR bands will increase upon decreasing the nanogap distance between two metal nanoparticles,65,66 increasing the number of coupling plasmonic fields in a metal nanoparticle chain,67 and increasing the medium dielectric constant in the nanogap between two plasmonic nanoparticles.68–70 The magnitude of plasmonic field-field coupling was also found to be strongly dependent on the relative orientation of two gold nanorods.71,72 In the case of hollow Au nanoframes, the magnitude of plasmonic field-field coupling between the outer and the inner LSPR fields and the extent of the redshift in coupled LSPR absorption bands increased as the solvent dielectric constant increased.70 It is also well established in the literature that the extent of the redshift in the LSPR absorption band is linearly correlated with the strength of the plasmonic field-field coupling between two adjacent plasmonic fields. Theoretical simulation also predicted that the magnitude of plasmonic field-field coupling is independent of the aspect ratio in the case of core-gap-shell nanostructures. The above-mentioned experimental observations, including redshift in the LSPR absorption bands of assembled nanoparticles,45–47 gold nanorattles,54 and aggregation-induced red-to-blue color transition of Au NPs48–52 can be well predicted and explained by theoretical calculations based on the plasmonic field-field coupling between two adjacent plasmonic fields. Experimentally, it was also observed that the extent of the redshift in the LSPR bands of an Ag nanoparticle array continuously increased upon increasing the refractive index in the nanogap from 1.0 (air) to 1.49 (filling the nanogaps by PMMA).73 In another study, it was observed that coating a dielectric layer (i.e., TiO2) on Au nanodisks dramatically led to a redshift in the LSPR absorption bands of a 2D Au nanodisk array.74 Overall, it is well established and confirmed by both theoretical calculations and experimental observations that the magnitude of the plasmonic field-field coupling between two adjacent plasmonic metals and the extent of the redshift in the LSPR absorption bands are strongly influenced by many factors, including the nanogap distance, the dielectric constant of the nanogap medium, the number of coupling plasmonic fields, and relative orientation of two plasmonic fields.
Currently, the plasmonic field enhancement in nanostructures with nanogaps has been mostly applied to bioimaging and sensing but very rarely applied to enhance in vivo therapeutic efficacies for cancer treatments.75,76 One of the reasons for this is that it is difficult to maintain metal nanoparticles in a cluster form with a constant nanogap distance in biological systems, and meanwhile still be able to pass through various biological barriers to reach tumor/disease sites. In the case of core-gap-shell nanostructures, it is difficult and challenging to synthesize nanostructures with a uniform nanogap and a suitable nanogap distance to achieve the maximum plasmonic field-field coupling between the metal core and the metal shell. Core-gap-shell nanostructures with irregular nanogap shapes/distances are always obtained, making it very difficult to conduct theoretical simulation and experimental optimization of the plasmonic field-field coupling between the core and the shell. When the nanogap in a core-gap-shell nanostructure is too large, the plasmonic field-field coupling among the core and the shell becomes very weak, which behaves like a single isolated metal nanoparticle. Therefore, it is crucial to develop alternative/simple ways to tune the plasmonic coupling strengths in core-gap-shell nanostructures and to maximize the enhancement factors of their fluorescence/Raman scattering intensities and other opto-properties, allowing their use in more applications, in particular, in the field of cancer diagnosis and therapeutic treatments.
Herein, we demonstrate that the plasmonic field-field interactions of core-gap-shell metal nanostructures can be tuned by adjusting the medium dielectric constant by filling high dielectric NaCl(aq) aqueous solution in the nanogaps. We demonstrate that filling NaCl(aq) aqueous solution into the nanogap of Au@gap@AuAg core-gap-shell nanopeanut-shaped nanostructures (Au NPNs) resulted in an amazingly large LSPR band redshift of 320 nm from 660 nm (NIR-I) to 980 nm (NIR-II) and a 12.6-fold increase (at 1064 nm) in the extinction coefficients in the NIR region. We also demonstrate the biomedical application of the Au@gap@AuAg core-gap-shell nanostructures to treat tumors with salt-enhanced NIR-II PDT efficacy for the first time. We observed that the average lifespan of mice bearing CT-26 colon tumor and treated with Au NPN6.5(NaCl) + 1064 nm light irradiation was prolonged to >55 days, which was much longer than that of the mice treated with Au NPN6.5 + 1064 nm (without filling NaCl in the nanogap, 40 d) and doxorubicin-treated mice group (23 d).
Results and discussion
Characterization of Au NPNs with 2.0 and 6.5 nm nanogaps
In this study, we synthesized gold nanopeanuts (Au NPNs) with nanogaps of ∼2.0 nm and ∼6.5 nm (average) between the Au NR core and the AuAg shell according to literature procedures with slight modification.59,60 In brief, the Au NPNs were prepared via a three-step process. All nanostructures were well characterized. Firstly, gold nanorods (Au NRs) were prepared with an aspect ratio of 4.0, which exhibited the LSPR maximum at 780 nm. Further, the Au NRs were coated with an Ag shell of different thicknesses (7–8 nm and 9–10 nm), which was controlled by tuning the volume of AgNO3 (1 mM) added to the Au NR-containing solution. The coating of a thin layer of Ag shell resulted in a blue shift in the LSPR band (at 780 nm) of the Au NRs. The Au@Ag core–shell NRs possessed two LSPR bands located at 390 and 670 nm (for 7–8 nm Ag shell) and 402 and 604 nm (for 9–10 nm Ag shell), respectively (Fig. S1(a) and (b), ESI†). The final step of the synthesis involved galvanic replacement of the Ag shell by Au3+ ions via the addition of 1 mM HAuCl4·3H2O to a solution containing the Au@Ag core–shell nanorods. The Ag shell served as a sacrificial reducing agent to reduce the Au3+ ions to form a hollow AuAg shell (vide infra) with a nanogap to the Au NR core.59,60 This galvanic replacement process is similar to that reported in the literature using Ag nanoparticles as a sacrificial electron donor for the preparation of Au nanocages or Au nanoframes.77–82 Depending on the thickness of the Ag shell layer and the amount of Au ions, the as-produced AuAg shell was intact or had small nanopores (like Au nanocage) or large nanopores (like Au nanoframe) on the outer shell. Au NPNs with different nanogaps were formed by controlling the amount of 1 mM HAuCl4·3H2O (volume V = 700 μL for ∼2.0 nm average nanogap and V = 400 μL for 6.5 nm average nanogap) added to the solution. The nanogaps were generally irregular with different nanogap distances at different locations. The values of 2.0 and 6.5 nm are the average nanogap distances. The nanogaps of the Au NPNs of 2.0 and 6.5 nm were filled with 1.67 M NaCl aqueous solution by soaking the Au NPNs in 1.67 M NaCl aqueous solution overnight, which were designated as Au NPN2.0(NaCl) and Au NPN6.5(NaCl), respectively. The NaCl(aq) aqueous solution filled the nanogap region through the nanopores on the Au shell via capillary action. The LSPR bands for Au NPN2.0 appeared at 530 and 700 nm, whereas that of Au NPN6.5 nm was located at 525 and 660 nm. The morphological evaluation of the Au NPNs was performed via TEM, as shown in Fig. 1(a)–(g). The as-prepared Au NRs had an average longitudinal length of ∼52 nm and average transverse diameter of ∼13 nm with an aspect ratio of ∼4.0. The TEM images clearly showed that no morphological collapse or aggregation occurred after the NaCl(aq) aqueous solution filled the nanogaps (see Fig. 1(f) and (g)). Fig. S2 (ESI†) shows the TEM-EDX analysis of Au NPN6.5, where the presence of both Ag and Au elements can be clearly seen in the shell. The ratio of Ag:Au is 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :64 in atomic percentage in the Au NPN6.5 sample, as evidenced by point spectral analysis. Interestingly, upon filling NaCl(aq) aqueous solution in the nanogaps between the core and the shell, both Au NPN2.0(NaCl) and Au NPN6.5(NaCl) showed enhanced broad LSPR absorption in the NIR region, extending from 500 nm to 1350 nm, which is attributed to the NaCl(aq)-mediated plasmonic field-field coupling between the core and the shell (see Fig. 2(a)). When the metal nanoparticles are brought together by salt-induced aggregation, two adjacent plasmonic fields can couple, leading to a redshift in their LSPR bands,45–47 and possibly an enhancement in their LSPR band absorption (or extinction coefficients) (note that this point has never been reported in the literature). This process is highly disfavored by the dilution of the nanoparticle concentration in solution, given that a low concentration of metal nanoparticles prevents aggregation. Alternatively, the redshift and enhancement in the LSPR bands of the Au NPNs could be due to the salt-in-nanogap-mediated plasmonic field-field coupling between the metal core and the metal shell, as proposed in this study. This latter process is an intrinsic property of a core-gap-shell nanoparticle and is independent of the nanoparticle concentration in solution. To differentiate whether our observed redshift and enhancement in NIR absorption originated from the aggregation-induced plasmonic field-field coupling or from salt-in-nanogap-mediated plasmonic field-field coupling, we compared the UV-visible-NIR absorption spectral profiles of high and low Au NPN6.5(NaCl) concentrations in solution by diluting the nanoparticle concentration by 10 times. The blue-green spectrum (top 1 curve) shown in Fig. 2a was obtained from an Au NPN6.5(NaCl)-1.67 M NaCl aqueous solution with an OD of ∼1.0 at 1064 nm. Upon 10 times dilution (still in a 1.67 M NaCl aqueous solution), the OD value of Au NPN6.5(NaCl) became nearly 10-fold smaller (see the bottom pink curve in Fig. 2a). The UV-visible-NIR spectrum of the low-concentration Au NPN6.5(NaCl) sample was magnified by 9.5× (see the dashed green-brown curve in Fig. 2a) and compared with that obtained from the high-concentration sample. As shown in Fig. 2a, the profile of the UV-visible-NIR spectrum from the low-nanoparticle concentration sample in the NIR region is nearly the same as that from the high nanoparticle concentration sample (the green-blue curve) but is distinctly different from that of Au NPN6.5 (without salt in the nanogaps, blue curve). The redshift and enhancements in the LSPR bands in the NIR region were still observed for the low concentration Au NPN6.5(NaCl) sample. This result clearly demonstrates that the redshift and enhancements in the LSPR bands in the NIR region for the Au NPN6.5(NaCl) core-gap-shell nanostructure originate from the salt-in-nanogap-mediated plasmonic field-field coupling, but not (or very minor) from aggregation-induced plasmonic field-field coupling.
:64 in atomic percentage in the Au NPN6.5 sample, as evidenced by point spectral analysis. Interestingly, upon filling NaCl(aq) aqueous solution in the nanogaps between the core and the shell, both Au NPN2.0(NaCl) and Au NPN6.5(NaCl) showed enhanced broad LSPR absorption in the NIR region, extending from 500 nm to 1350 nm, which is attributed to the NaCl(aq)-mediated plasmonic field-field coupling between the core and the shell (see Fig. 2(a)). When the metal nanoparticles are brought together by salt-induced aggregation, two adjacent plasmonic fields can couple, leading to a redshift in their LSPR bands,45–47 and possibly an enhancement in their LSPR band absorption (or extinction coefficients) (note that this point has never been reported in the literature). This process is highly disfavored by the dilution of the nanoparticle concentration in solution, given that a low concentration of metal nanoparticles prevents aggregation. Alternatively, the redshift and enhancement in the LSPR bands of the Au NPNs could be due to the salt-in-nanogap-mediated plasmonic field-field coupling between the metal core and the metal shell, as proposed in this study. This latter process is an intrinsic property of a core-gap-shell nanoparticle and is independent of the nanoparticle concentration in solution. To differentiate whether our observed redshift and enhancement in NIR absorption originated from the aggregation-induced plasmonic field-field coupling or from salt-in-nanogap-mediated plasmonic field-field coupling, we compared the UV-visible-NIR absorption spectral profiles of high and low Au NPN6.5(NaCl) concentrations in solution by diluting the nanoparticle concentration by 10 times. The blue-green spectrum (top 1 curve) shown in Fig. 2a was obtained from an Au NPN6.5(NaCl)-1.67 M NaCl aqueous solution with an OD of ∼1.0 at 1064 nm. Upon 10 times dilution (still in a 1.67 M NaCl aqueous solution), the OD value of Au NPN6.5(NaCl) became nearly 10-fold smaller (see the bottom pink curve in Fig. 2a). The UV-visible-NIR spectrum of the low-concentration Au NPN6.5(NaCl) sample was magnified by 9.5× (see the dashed green-brown curve in Fig. 2a) and compared with that obtained from the high-concentration sample. As shown in Fig. 2a, the profile of the UV-visible-NIR spectrum from the low-nanoparticle concentration sample in the NIR region is nearly the same as that from the high nanoparticle concentration sample (the green-blue curve) but is distinctly different from that of Au NPN6.5 (without salt in the nanogaps, blue curve). The redshift and enhancements in the LSPR bands in the NIR region were still observed for the low concentration Au NPN6.5(NaCl) sample. This result clearly demonstrates that the redshift and enhancements in the LSPR bands in the NIR region for the Au NPN6.5(NaCl) core-gap-shell nanostructure originate from the salt-in-nanogap-mediated plasmonic field-field coupling, but not (or very minor) from aggregation-induced plasmonic field-field coupling.
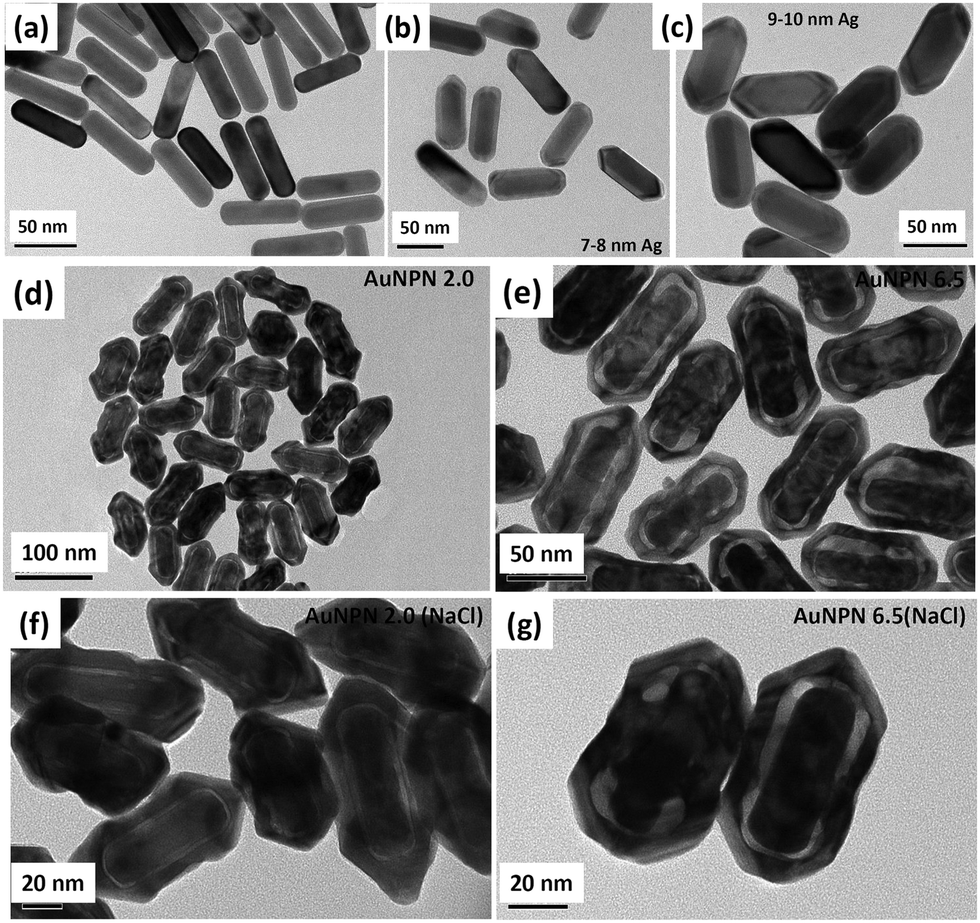 | ||
| Fig. 1 TEM images of as-synthesized gold and gold–silver nanostructures. TEM images of (a) Au NRs with an aspect ratio of 4.0. (b) and (c) Au@Ag core–shell NRs with different Ag thicknesses of 7–8 nm and 9–10 nm, respectively. (d) and (e) Au NPNs with two different average nanogaps of 2.0 nm and 6.5 nm, respectively. (f) and (g) Au NPNs with an average nanogap of 2.0 nm, and 6.5 nm, respectively, with NaCl(aq) filled in the nanogaps. | ||
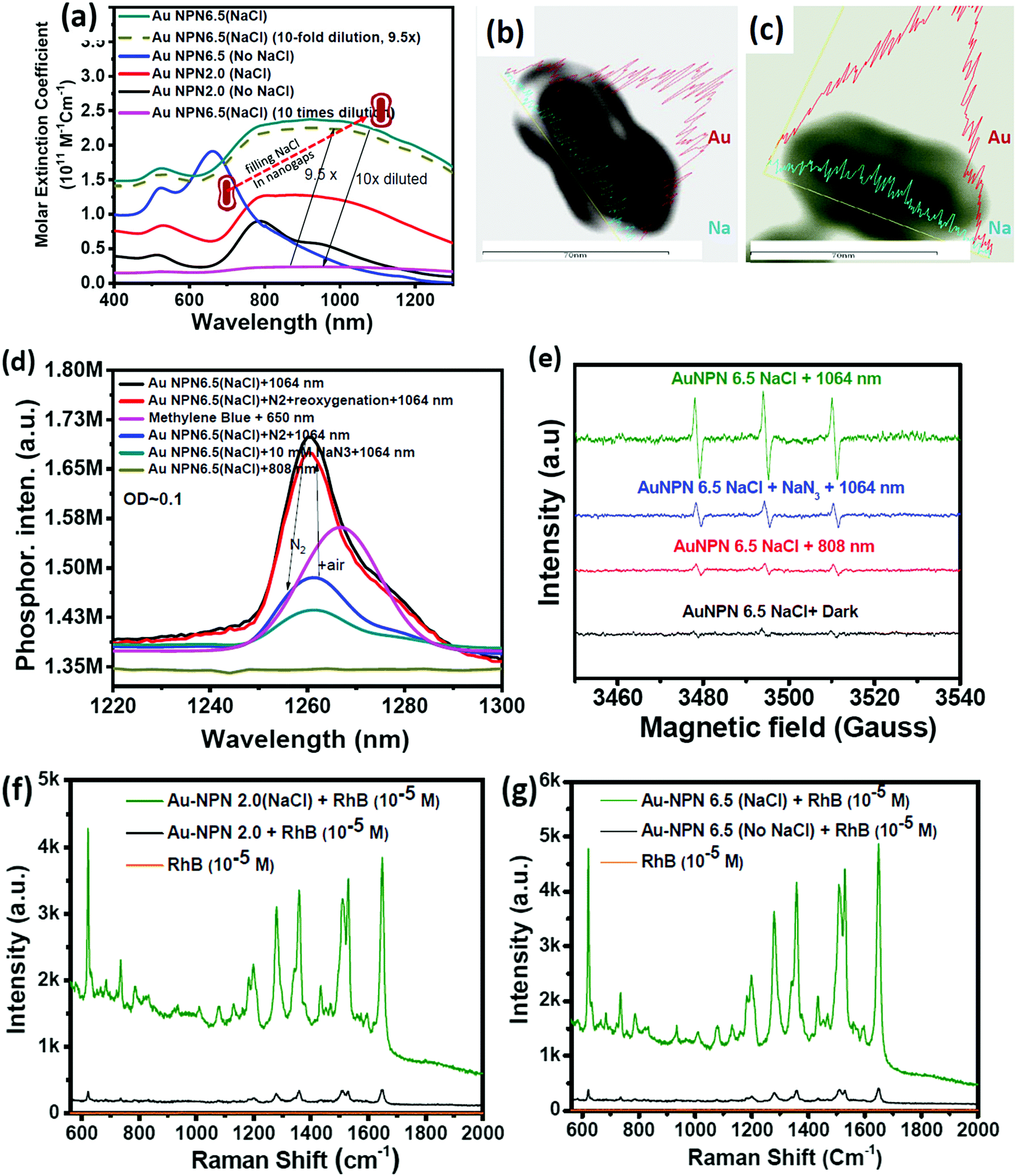 | ||
| Fig. 2 Characteristic properties of Au NPNs upon filling NaCl(aq) in their nanogaps. (a) Molar extinction coefficients of the as-synthesized Au NPNs with different nanogaps of 2.0 nm and 6.5 nm and that for Au NPN2.0(NaCl) and Au NPN6.5(NaCl). TEM-EDX line scan mapping intensities of the Au and Na elements in (b) Au NPN6.5 and (c) Au NPNs6.5(NaCl) nanostructures. The intensities of Au and Na elements are plotted in the figures. The scale bar is 70 nm. (d) Singlet 1O2 phosphorescence emission spectra generated from photo-excitation of methylene blue (λex = 650 nm) and Au NPN6.5(NaCl) in 1.67 M NaCl aqueous solution (excitation wavelength λex = 808 or 1064 nm) in the presence of air (black curve), N2 atmosphere (blue curve)-reoxygenation (red curve) and 10 mM NaN3 (blue-green curve). (e) EPR spectra of singlet oxygen generated from anti-EGFR-Au NPN6.5(NaCl) nanostructures upon NIR light (808 or 1064 nm) photo-excitation. Addition of 10 mM sodium azide (blue color line) led to partial suppression of the singlet O2 EPR signals. (f) and (g) Surface-enhanced Raman spectra of surface-bound Rhodamine B molecules on Au NPN2.0 and Au NPN6.5 before and after filling NaCl(aq) aqueous solution in their nanogaps, respectively. | ||
In the literature, it has been reported that the oscillation frequency of surface plasmon resonance is in the THz domain.83 Increasing the salt (NaCl) concentration will lead to an increase in the dielectric constant of the aqueous solution.84 Therefore, the redshift from NIR-I (650–950 nm) to NIR-II (1000–1350 nm) in the current gold core-gap-shell nanopeanut system can be well explained by the increase in the surrounding medium dielectric properties, and thus the enhanced plasmonic field-field couplings between the outer shell and the inner shell, and the metal core and the metal shell upon filling NaCl(aq) solution in the nanogap through the nanopores on the outer shells of the Au NPNs (Fig. S3, ESI†). The nanopore regions have a broken or no Au outer shell, and thus appear brighter than the other nanogap regions in the transmission electron microscopy images (Fig. S3, ESI†). Some have clear edges and some have irregular edges. This method for the identification of nanopores is the same as that reported in the literature for gold nanocages/nanoframes.77–82 Dynamic light scattering (DLS) measurements using low concentrations of Au NPN samples with OD= 0.1 at 1064 nm showed that the particle size distribution of the Au NPNs remained nearly the same after NaCl(aq) aqueous solution filled their nanogaps, indicating that the large redshift in the LSPR absorption bands of the Au NPNs is not due to aggregation (Fig. S4, ESI†). As stated in the Introduction, it is well reported in the literature that the LSPR band of plasmonic metal nanoparticles is very sensitive to the dielectric properties of the local environment.68 Thus, the introduction of a high dielectric salts, such as NaCl(aq), in the nanogap region is expected to enhance the plasmonic field-field coupling between the Au core and AuAg shell. This enhancement was more pronounced in the case of Au NPN6.5(NaCl) as compared to Au NPN2.0(NaCl). Further, the line scan TEM-EDX line scan mapping of Au NPN6.5(NaCl)(1.67 M) clearly showed the presence of the Au and Na elements (Fig. 2(b) and (c)). The Na element was observed in the nanogap region of the Au NPN6.5(NaCl) nanostructures but not in the Au NPN6.5 samples. The data clearly showed that after soaking in NaCl aqueous solution overnight, the percentage of Na element in the nanogap region increased dramatically, indicating the entry of NaCl(aq)via capillary action through the nanopores on the outer surface of nanostructures into the nanogap of the Au NPN6.5 core-gap-shell nanostructure. Fig. 2(b) and (c) provide direct evidence for the diffusion/filling of NaCl(aq) aqueous solution in the nanogap region of the Au@gap@AgAu core-gap-shell nanostructures. The overall procedure for the preparation of Au NPN6.5(NaCl) is schematically presented in Scheme 1.
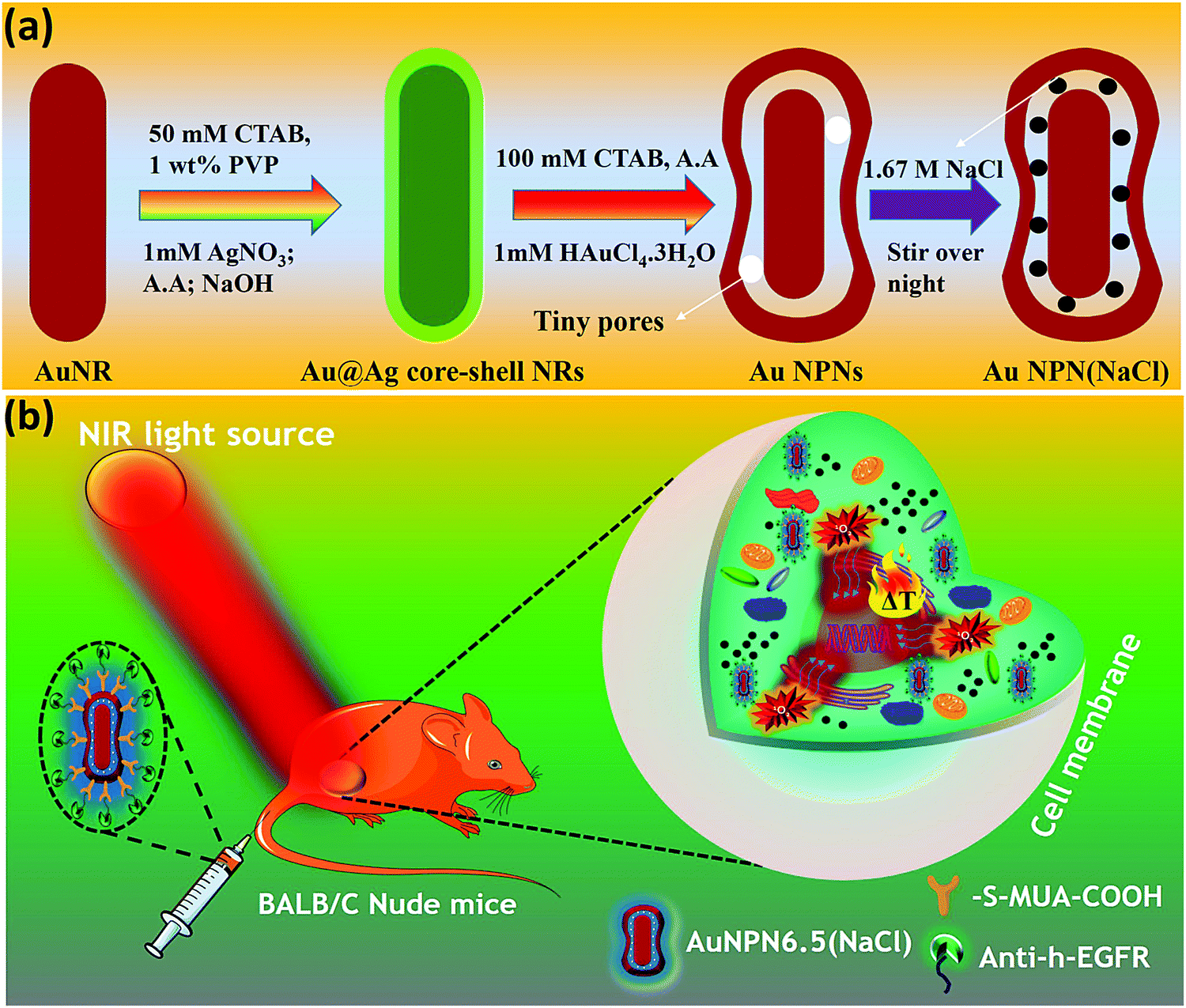 | ||
| Scheme 1 Synthesis and in vivo therapeutic scheme for targeted Au NPN6.5(NaCl) in BALB/C nude mice. | ||
The molar extinction coefficient for Au NPN2.0(NaCl) is 0.96 × 1011 M−1 cm−1 at 1064 nm, whereas Au NPN6.5(NaCl) has a larger extinction coefficient of 1.95 × 1011 M−1 cm−1 at 1064 nm (Fig. 2(a)), which is 6–8 orders higher than that of conventional organic dyes and 2–3 orders higher than that of gold nanoparticles. Besides the large molar extinction coefficients, we also found that the Au NPNs could sensitize the formation of singlet 1O2 upon photo excitation by 1064 nm (but not by 808 nm) light, as evidenced by the characteristic singlet oxygen phosphorescence at ∼1260 nm (Fig. 2(d) and Fig. S5b, ESI†). In the presence of 10 mM NaN3, the singlet 1O2 phosphorescence emission was partially quenched with a lower intensity (blue color line). Upon bubbling the solution with N2 gas for 10 min, the 1260 nm emission intensity was dramatically suppressed, which could be restored by bubbling the solution with air for 10 min. The non-zero 1260 nm emission intensity under an N2 atmosphere is probably due to some residual molecular O2 inside the nanogap region, which could not be easily removed by bubbling N2 gas for 10 min in the solution. In the literature, it was reported that the emission maximum wavelength of singlet O2 phosphorescence distributes in a wide range from 1263 to 1279 nm,85–88 depending on the local environment, solvent polarity and solvent polarizability.85–88 The emission maximum wavelength represents the LUMO–HOMO energy gap of singlet oxygen. The singlet O2 emission wavelength at 1260 nm for the current Au NPN6.5(NaCl) in a 1.67 M aqueous solution is slightly higher than that reported in the literature, indicating that the LUMO–HOMO energy gap of singlet O2 in the current system is slightly higher than that commonly reported in the literature. Possibly, the highly dielectric polar aqueous environment stabilizes the slightly polar HOMO energy level of the triplet ground state O2 or de-stabilizes the non-polar LUMO energy level of the singlet excited O2, leading to a slightly larger LUMO-HOMO energy gap, and thus slight blue shift in the singlet oxygen phosphorescence emission maximum wavelength.
The singlet 1O2 quantum yields were determined to be 0.20 and 0.42 at 1064 nm for Au NPN6.5 (without filling NaCl(aq) aqueous solution in the nanogaps) and Au NPN6.5(NaCl), respectively, using low concentrations of Au NPNs in solution with OD = 0.1 at 1064 nm to avoid the incident light scattering problem and the inner filter effect, i.e., absorption of singlet O2 phosphorescence intensity by Au NPNs in the region of 1200–1300 nm.89 (Please see ESI,† for details regarding the measurements of singlet oxygen formation quantum yields). It appears that filling NaCl(aq) aqueous solution in the nanogaps could enhance the plasmonic field-lattice interaction, and thus the singlet oxygen sensitization quantum yield in the Au NPN6.5(NaCl) nanostructures. In the literature, it was reported that the singlet oxygen phosphorescence quantum yield from a surface-bound, photoexcited photosensitizer could be enhanced by a plasmonic field generated from simple, isolated metal nanoparticles;90,91 however, it has never been reported to be enhanced by simply adding a salt-containing aqueous solution to the nanogaps of core-gap-shell nanostructures. The singlet oxygen phosphorescence lifetime measurements were carried out in D2O (Fig. S6(a), ESI†). For Au NPN6.5(NaCl), the singlet oxygen phosphorescence emission lifetime decay curve is composed of two components with a short-lived component (34.7%) having a lifetime of 0.9 μs and a long-lived component (65.3%) having a lifetime of 9.0 μs. The results indicate that the singlet O2 generated from anti-EGFR-AuNPN 6.5(NaCl) in D2O upon 1064 nm photo-excitation has two distinctly different environments, most probably one from the singlet O2 inside the nanogap regions of the anti-EGFR-Au NPN 6.5(NaCl) and another one outside the core-gap-shell nanostructure and free in the D2O solution. It is known that many factors including physical energy transfer/vibrational collision processes and chemical reaction quenching processes can lead to a shorter singlet oxygen phosphorescence lifetime.92 The long-lived component with a lifetime of 9.0 μs is slightly shorter than that reported in the literature, i.e., 18 μs93 to 68 μs.94–96 The detailed mechanism of the singlet oxygen relaxation/lifetimes in the current heterogeneous system is complicated and different from those in homogeneous systems, which is not the main focus of this study and deserves a separate study in the future.
Besides the detection of the singlet oxygen phosphorescence emission spectrum, we also detected the formation of hydroxyl radicals (Fig. S6(b), ESI†) and singlet oxygen via EPR measurements using 2,2,6,6-tetramethylpiperidine (TEMP) as a singlet oxygen-trapping reagent upon 1064 nm NIR light irradiation of the Au NPN6.5(NaCl) nanostructures (Fig. 2(e)). The TEMP-singlet oxygen adduct EPR signal has a characteristic 1:1:1 triplet pattern, which can be dramatically suppressed by the presence of a singlet oxygen-specific quencher, i.e., sodium azide, further confirming the identity of the singlet oxygen. In contrast, 808 nm light irradiation of Au NPN6.5(NaCl) generated a negligible amount of singlet oxygen. The hydroxyl radical has a characteristic 1:2:2:1 quartet pattern, which can be dramatically suppressed by the presence of a hydroxyl radical-specific quencher, i.e., mannitol, further confirming the identity of the hydroxyl radials. In contrast, 808 nm light irradiation of Au NPN6.5(NaCl) generated a negligible amount of hydroxyl radicals. In the literature, it is well known that hydroxyl radicals can be derived from singlet oxygen via a multistep process of superoxide formation (via electron transfer to singlet oxygen), hydrogen peroxide formation, and dark Fenton reaction of hydrogen peroxides with reducing metal ions/reagents.97 Therefore, it is not surprising to observe the formation of hydroxyl radicals, given that singlet oxygen was formed upon 1064 nm NIR light irradiation of the Au NPN6.5(NaCl) nanostructure.
Au NPN6.5(NaCl) exhibited a larger extinction coefficient as compared to Au NPN2.0(NaCl) under the same conditions, implying its promising bio-application for cancer treatment. To introduce tumor targeting ability, Au NPN6.5(NaCl) was surface-modified with anti-h-EGFR antibody for subsequent in vivo applications. Briefly, the surface of Au NPN6.5(NaCl) was initially conjugated with 11-mercapto undecanoic acid by following a literature procedure with slight alterations, and then coupled to anti-h-EGFR antibody using EDC coupling agent. The successful functionalization was confirmed by the appearance of the characteristic 1632 cm−1 (amide bond C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching) and 2900 cm−1 (amide bond N–H stretching) bands in its FT-IR spectrum (Fig. S7, ESI†).
O stretching) and 2900 cm−1 (amide bond N–H stretching) bands in its FT-IR spectrum (Fig. S7, ESI†).
XRD and SERS enhancement
Upon filling 1.67 M NaCl(aq) aqueous solution in the nanogap, Au NPNs were found to exert slightly enhanced XRD intensities compared to that of the Au NPNs (Fig. S8, ESI†). It was clearly observed that the major (111) XRD peaks of Au NPNs 2.0 and the (200) peak for Au NPN6.5 were enhanced after filling the nanogap with NaCl(aq) aqueous solution. The mechanism responsible for the enhanced XRD signals is not clear at this stage, which may be due to the plasmonic field-enhanced scattering interactions between the incident X-ray beams and metal crystalline lattices, similar to the working mechanism for plasmonic field-enhanced Raman scattering between incident photons with molecular vibrations observed in SERS. Besides an enhancement in the XRD intensity, we also found that the primary SERS signals of the surface-bound organic molecules on the Au NPNs could also be further enhanced upon filling NaCl(aq) aqueous solution in the nanogaps. Upon filling the gaps with 1.67 M NaCl(aq), it could clearly be seen that the SERS signal at 1650 cm−1 for the surface-bound Rhodamine B molecules was further enhanced by 11.7- and 14.8-fold for both Au NPN2.0(NaCl) and Au NPN6.5(NaCl) (Fig. 2(f) and (g)), respectively. Consistent with the results in the NIR plasmonic absorption, the enhancement in the SERS signals was more pronounced in Au NPN6.5(NaCl) compared to that from Au NPN2.0(NaCl). To the best of our knowledge, this is the first literature report showing salt-mediated enhancement effects on all the NIR plasmonic absorption, XRD and SERS characteristics of plasmonic core-gap-shell metal nanostructures.Mechanism of filling NaCl(aq) aqueous solution in the nanogaps
In the synthesis of the gold core-gap-shell nanopeanut structure,59,60 the Au NR@Ag core–shell nanorod was synthesized first. Then, the silver shell nanorod was used as the sacrificial electron donor to reduce Au ions to generate the AuAg outer shell layer. This galvanic replacement process is similar to that was reported in the literature using Ag nanoparticles as a sacrificial electron donor for the preparation of Au nanocages or Au nanoframes.77 Depending on the thickness of the Ag shell layer and the amount of Au ions, the as-produced Au outer shell may be intact or have small holes (like Au nanocage) or large holes (like Au nanoframe). In the case of the intact Au core-gap-shell, the external NaCl(aq) aqueous solution will not be able to enter the inner nanogap region. However, in the case of the slightly broken Au shell, the external NaCl(aq) will be able to slowly diffuse and fill the nanogap region via capillary action through the nanopores on the outer shell. The diffusion/filling and the diffusing-out rates of NaCl(aq) will be strongly dependent on the size of the nanopores on the AuAg outer shell. For the Au core-gap-shell nanopeanuts with small nanopores on the AuAg outer shell, it takes a relatively longer time for the external NaCl(aq) aqueous solution to diffuse into the nanogaps, and likewise it will be difficult for the trapped NaCl(aq) in the nanogap (or takes a longer time) to diffuse out. Note that the physiological NaCl concentration is 0.125 M. The NaCl aqueous solution inside the nanogap will stop diffusing out when the internal NaCl(aq) becomes nearly equal to the outside physiological NaCl concentration, i.e., 0.125 M. In addition, the surface adsorption of proteins, such as bovine serum albumin (BSA) on Au NPN nanostructures, may partially block NaCl(aq) from diffusing out of the nanogaps. For all the experiments, we soaked the Au@gap@AuAg core-gap-shell nanoparticles in NaCl aqueous solution overnight to allow the diffusion of the NaCl aqueous solution into the nanogap. For the Au@gap@AuAg having an intact AuAg outer shell, the NaCl aqueous solution could not diffuse into the nanogap, and thus no Na element was expected to be in the nanogap region after overnight soaking in NaCl-containing aqueous solution. In addition, the XPS analysis (see Fig. S9(a) and (b), ESI†) of both Au NPN6.5(NaCl) and Au NPN6.5 showed that the Na 1s (at ∼ 1072 eV) and Na KLL (at ∼498 eV) XPS peaks could be clearly observed from the Au NPN6.5(NaCl)(1.67 M) sample, but not in the Au NPN6.5 sample, respectively. The atomic percentage of Na element obtained from the XPS measurements is ∼15% in the Au NPN6.5(NaCl)(1.67 M) sample and nearly zero in the Au NPN6.5 sample. The large percentage of Na element in Au NPN6.5(NaCl)(1.67 M) is consistent with the filling of NaCl(aq) aqueous solution in the nanogap region and cannot be from the surface adhesion of residual NaCl(aq) on the outer surface of Au NPN6.5(NaCl)(1.67 M). Both the TEM-EDX line scan data and XPS data strongly support the presence of NaCl(aq) aqueous solution in the nanogap, which cannot be explained by residual, if any, NaCl(aq) on the surface of the Au NPN6.5(NaCl) nanostructure. The NaCl detected in the TEM-EDX line scan mapping and XPS measurements cannot originate from the surface adsorbed layer on the Au@gap@AuAg core-gap-shell nanopeanuts, given that NaCl is highly soluble in water and has no special affinity with the surface of Au. According to general chemistry textbooks, all sodium salts are highly soluble in water due to the very large exothermic hydration energy (∼−97 kcal mol−1) of the Na+ cation with water molecules and the large entropy gain upon their dissolution in water.Further, to obtain the optimal NaCl concentration for maximum plasmonic field-field coupling, the absorption spectrum of Au NPN6.5(NaCl) was plotted as a function of NaCl concentration (Fig. S10, ESI†), which shows that an NaCl concentration of ∼1.67 M can lead to the maximum redshift and highest LSPR absorption at 1064 nm, and thus the maximum plasmonic field-field coupling effect. Besides NaCl, filling other high dielectric salts, such as NaBr, NaI, MgSO4, MgCl2, and ((NH4)2SO4), in the nanogaps all increased the medium dielectric properties in the nanogap region, and thus led to enhanced plasmonic field-field coupling, redshift in the LSPR bands, and enhanced NIR absorption of Au NPN6.5. However, the optimal salt concentrations for the maximum plasmonic field-field coupling were not the same for different salts due to the differences in their dielectric constants (see experimental results shown in Fig. S11, ESI†). Overall, the biocompatibility of NaCl makes it suitable to be used and filled in Au NPN6.5(NaCl)(1.67 M) for therapeutic applications. To measure the amount of NaCl(aq) inside the nanogap region, we washed the Au NPN6.5(NaCl)(1.67 M) sample with DI water 6 times, and then conducted ICP-MS measurements to determine the Na element concentration. Our ICP-MS measurements (data not shown) indicated that the amount of NaCl present in the gaps of Au NPN6.5(NaCl)(1.67 M) is ∼3.019 ppm.
Stability and salt diffusing-out rate from Au NPN6.5(NaCl)
To gain more information regarding the stability of the NaCl(aq) aqueous solution in the nanogaps, we carried out salt filling-in and diffusing-out experiments (Fig. 3). As shown in Fig. 3(a), Au NPN6.5 (the black curve) has a very weak absorption in the NIR (800–1300 nm) region. Upon dispersion in a 1.67 M NaCl aqueous solution for 5 min, the NIR absorption of Au NPN6.5 immediately increased to a very large extent (see the red curve in Fig. 3(a)), followed by a further slow increase and redshift in the broad LSPR absorption band in the next 12 h. It has been reported in the literature that the LSPR absorption band of plasmonic metal nanoparticles will redshift upon increasing the dielectric constant of the surrounding medium,73 where increasing the dielectric constant of the external solvent will enhance the coupling between the outer and the inner plasmonic fields of hollow plasmonic metal nanostructures, leading to a redshift in the LSPR absorption band.70 The increase in the LSPR absorption for the first 5 min of immersion of Au NPN6.5 in 1.67 M NaCl aqueous solution is most probably due to the sudden increase in the dielectric property of the surrounding solvent, which enhanced the plasmonic field-field coupling between the outer and inner plasmonic field of the AgAu shell alone (without involving the Au NR core),70 rather than enhanced the plasmonic field-field coupling between the AgAu shell and the Au NR core. This is because it is unlikely that the NaCl(aq) aqueous solution diffused and filled the nanogap completely within 5 min of immersion. Upon slow diffusion and filling of NaCl(aq) in the nanogap region within the next 12 h, the high dielectric NaCl aqueous solution could further mediate and promote the plasmonic field-field coupling among the outer shell field, inner shell field and core plasmonic field, leading to a further NIR redshift and increase in the LSPR absorption band of Au NPN6.5(NaCl). Specifically, the overall plasmonic field-field coupling is composed of two parts, as follows: (a) the plasmonic field-field coupling between the outer shell and the inner shell alone (without or weakly involving the metal core) and (b) the plasmonic field-field coupling between the AuAg shell and the Au NR core. The former is strongly and rapidly influenced by sudden changes in the external surrounding solvent dielectric constant, whereas the latter is strongly influenced by the medium dielectric constant in the nanogap. The LSRP absorption band rise profile shown in Fig. 3(a) indicates that the filling of NaCl(aq)via capillary action in the nanogap region is a slow process.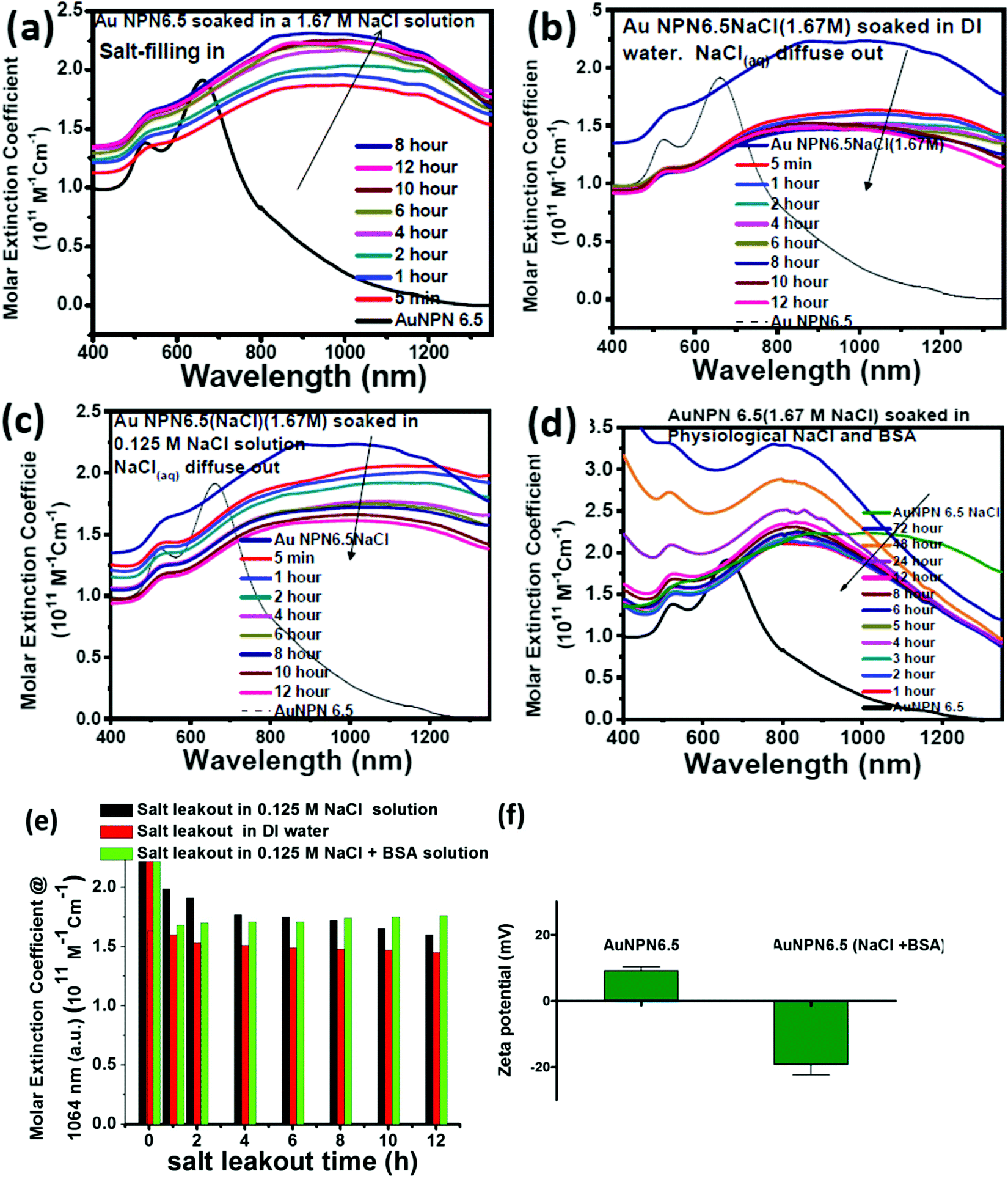 | ||
| Fig. 3 (a) UV-visible-NIR absorption spectrum of Au NPN6.5 in DI water (black curve) and spectra obtained upon dispersing Au NPN6.5 in 1.67 M NaCl aqueous solution with different soaking times. (b) and (c) UV-visible-NIR absorption spectrum of Au NPN6.5(NaCl) (1.67 M) in 1.67 M NaCl aqueous solution (navy blue curve), and spectra obtained upon dispersing Au NPN6.5(NaCl)(1.67 M) in (b) DI water and (c) 0.125 M NaCl aqueous solution with different soaking times. (d) UV-visible-NIR absorption spectrum of Au NPN6.5(NaCl) (1.67 M) in 1.67 M NaCl aqueous solution (navy blue curve) and the spectra obtained upon dispersing Au NPN6.5(NaCl) (1.67 M) in 0.125 M NaCl + BSA (∼30 mg mL−1) aqueous solution with different soaking times. (e) LSPR band intensity at 1064 nm for the salt diffusing-out experiments in DI water (red bars), 0.125 M NaCl aqueous solution (black bars) and 0.125 M NaCl + BSA aqueous solution (green bars). (f) Zeta potential measured for AuNPN6.5 and AuNPN6.5(NaCl) after salt diffusion out experiments in 0.125 M NaCl + BSA aqueous solution. | ||
To investigate the diffusing-out process of NaCl(aq) from the nanogaps of Au NPN6.5(NaCl)(1.67 M) to the external aqueous phase, we carried out salt diffusing-out experiments by dispersing Au NPN6.5(NaCl)(1.67 M) in DI water (Fig. 3(b)), 0.125 M NaCl aqueous solution (Fig. 3(c)), and 0.125 M NaCl + BSA aqueous solution (Fig. 3(d)). The NaCl concentration in the physiological condition is 0.125 M. As shown in Fig. 3(b), upon dispersion in DI water, the LSPR absorption band of Au NPN6.5(NaCl)(1.67 M) immediately decreased to a certain extent, followed by a slow decrease process. The rapid decrease in the LSPR absorption intensity is due to the sudden decrease in the surrounding solvent dielectric constant from 1.67 M NaCl to DI water, which caused a rapid decrease in the plasmonic field-field coupling between the outer AuAg shell surface and the inner AuAg shell surface. The slow decrease in the LSPR absorption intensity is consistent with the slow diffusion of the Na+(aq)/Cl−(aq) hydrated ions from the nanogaps to the external DI water phase, and thus the decrease in the medium dielectric constant in the nanogap region, leading to a decrease in the plasmonic field-field coupling strength between the outer AgAu shell and the inner Au NR core. When the outside solvent contained high dielectric species, such as 0.125 M NaCl or (0.125 M NaCl + BSA protein), the rapid LSPR absorption decrease shown in Fig. 3(d) was absent. In fact, the surface absorption (vide infra) of negatively charged BSA proteins on the surface of Au NPN6.5(NaCl) could enhance the overall LSPR absorption and slow down the salt diffusing-out rate, as seen in Fig. 3(d). The salt diffusing-out profiles of Au NPN6.5(NaCl)(1.67 M) in the above-mentioned three conditions are shown in Fig. 3(e), where the decrease in the LSPR absorption intensity at 1064 nm is plotted as a function of the soaking time. When Au NPN6.5(NaCl)(1.67 M) was dispersed in 0.125 M NaCl (physiological NaCl concentration) aqueous solution (Fig. 3(c)), the LSPR band intensity decay profile (see black bars in Fig. 3(e)) is similar to that (red bars in Fig. 3(e)) in DI water, but to a lesser extent due to the smaller decrease in the external surrounding solvent dielectric constant. After 12 h soaking in 0.125 M NaCl solution, the LSPR absorption at 1064 nm (with an extinction coefficient of 1.61 × 1011 M−1 cm−1) for Au NPN6.5(NaCl) dropped by 27% but still 8.7-fold higher than that (0.18 × 1011 M−1 cm−1) of Au NPN6.5 without NaCl(aq) in its nanogap. It was observed that the decay in the LSPR absorption band intensity became much slower with a longer soaking time. In addition, besides NaCl, many other salts are present in the physiological condition, including KCl, CaCl2, and MgCl2. All these high dielectric salts can also contribute to enhance the plasmonic field-field couplings between the outer plasmonic field and the inner plasmonic field of the AgAu shell, as well as the plasmonic field-field coupling between the AgAu shell and the Au NR core. Therefore, the stability or the extent of the decrease in the salt-mediated plasmonic field-field coupling (and thus the enhanced LSPR absorption in the NIR region) in Au NPN6.5(NaCl) is still within an acceptable range. The salt diffusing-out rate can be further tuned or slowed down by designing Au NPN nanostructures with smaller nanopores on the outer AgAu shell. The zeta potential for Au NPN6.5 was determined to be +9 mV, whereas that for Au NPN6.5(NaCl) was −19 mV after the salt diffusing out experiments in 0.125 M NaCl + BSA aqueous solution. This result indicates that the negatively charged BSA binds to the surface of the Au NPN6.5(NaCl) nanostructures, preventing the salt from diffusing out and stabilizing the salt-filled Au NPN6.5(NaCl) nanostructures.
The photothermal conversion efficiency (PCE, η) for both AuNPN6.5 and AuNPN6.5(NaCl) was measured to be: η = 6.4% (at 808 nm) and 5% (at 1064 nm) for AuNPN6.5, as well as η = 28.1% (at 808 nm) and 15. 1% (at 1064 nm) for AuNPN6.5(NaCl) (see ESI,† for details on the measurements and experimental data shown in Fig. S12). This enhanced photothermal conversion efficiency phenomenon upon filling NaCl(aq) aqueous solution in the nanogaps was also observed in the singlet oxygen sensitization quantum yields, i.e., 0.20 for AuNPN6.5 and 0.42 for AuNPN6.5(NaCl), upon 1064 nm laser excitation. It appears that filling NaCl(aq) in the nanogaps not only enhanced the plasmonic field-field coupling between the AuAg shell and the Au NR core, but also enhanced the plasmonic field-lattice interactions, which promoted nonradiative decay of plasmonic excited states and further led to enhanced photothermal conversion efficiencies and other optical properties.
The mechanism of ROS generation in Au NPN6.5(NaCl)
In 2011, our group discovered and reported for the first time that in the absence of any organic photosensitizer, photo excitation of metal nanoparticles can lead to the generation of singlet oxygen,35,37,98 which was later confirmed by many other groups.99–101 Experimentally, it was observed that 1064 nm NIR light irradiation of Au NPNs could lead to the generation of singlet O2, whereas 808 nm light irradiation did not lead to the formation of singlet O2, as evidenced by the absence of the characteristic singlet O2 phosphorescence emission at ∼1265 nm. The excitation spectrum of Au NPNs for singlet O2 generation is shown in the ESI,† Fig. S5a. Not all excitation wavelengths can lead to the generation of singlet O2 upon photo irradiation of Au NPNs. To generate singlet O2, the wavefunctions of the Au NPN plasmonic excited states should have good similarity and overlap with the singlet excited state of the triplet ground state molecular oxygen to allow effective plasmonic energy transfer from the localized surface plasmon resonance (LSPR) excited states of Au NPNs to the ground state molecular O2. Gold nanoparticles are known to have many LSPR states, which are localized and have different excited state wavefunctions, and thus different extents of wavefunction similarity/overlap with the singlet excited state wavefunction of molecular O2. Different photo excitation wavelengths, such as 808 and 1064 nm, will lead to different LSPR excited states, which have different LSPR excited-state wavefunctions, different extents of wavefunction similarity/overlap with the singlet excited state wavefunction of molecular O2, and thus different abilities to induce the generation of singlet oxygen. This may be the reason why 1064 nm excitation light can lead to the formation of singlet O2, whereas 808 nm light cannot. This possible mechanism was previously discussed in a literature review article.102In vitro PDT/PTT cellular experiments
To investigate the possibility of using the Au NPNs as NIR-I and –II dual-modal photodynamic and photothermal therapy reagents, in vitro cellular experiments were carried using murine CT-26 colon carcinoma cancer cells. Given that Au NPN6.5(NaCl) has a much larger extinction coefficient in the NIR region than Au NPN2.0(NaCl), it was chosen as the nanomaterial for in vivo cancer treatment applications. After incubating the murine CT-26 colon carcinoma cancer cells with different amounts of Au NPN6.5 (or Au NPN6.5(NaCl)), the cancer cells were irradiated with an 808 (200 mW cm−2, 10 min) or 1064 nm (200 mW cm−2, 11 min) laser. The laser irradiation time was adjusted according to the extinction coefficients at 808 and 1064 nm, allowing the amounts of photons being absorbed by Au NPN6.5(NaCl) to be the same for both 808 and 1064 nm. For example, the extinction coefficient at 1064 nm is 0.909-fold that at 808 nm. Therefore, the laser irradiation time for the 1064 nm wavelength was set to be 1.1-fold longer (10 min × 1.1 = 11 min) than that set for 808 nm, causing the photon fluxes for both wavelengths to be the same. Then, the corresponding photothermal therapy and photodynamic therapy effects could be fairly compared. The MTT cell experiments were carried out at 37 °C and 4 °C, respectively.Fig. 4(a) and (b) show that Au NPN6.5(NaCl) had negligible to low dark cellular cytotoxicity toward the murine CT-26 colon carcinoma cancer cells in a dose-dependent manner. However, upon 1064 nm laser irradiation, ∼71.5% cellular death, together with 13% dark cellular death, was observed due to the photo-induced cytotoxicity at a dose of 50 μg mL−1 of Au NPN6.5(NaCl) at 37 °C. At a dose of 50 μg/mL, the Au NPN6.5(NaCl) + 1064 nm laser condition resulted in 1.73-fold higher cellular death than Au NPN6.5 + 1064 (without filling NaCl(aq) aqueous solution in the nanogaps), 1.48-fold higher than that for Au NPN6.5(NaCl) + 808, and 2.53-fold higher than that for Au NPN6.5 + 808 nm at 37 °C. The cellular death followed the order of Au NPN6.5(NaCl) + 1064 > Au NPN6.5(NaCl) + 808 > Au NPN6.5 + 1064 > Au NPN6.5 + 808. The results clearly showed that filling NaCl(aq) in the nanogap of Au NPN6.5 led to greater photo-induced cellular death for both 1064 and 808 nm, which was due to the salt filling-enhanced light absorption (or larger extinction coefficients). When irradiated with 1064 nm, more cell death was observed compared to that for 808 nm light irradiation, which is attributed to combined NIR-II PDT and NIR-II PTT effects for 1064 nm compared to the pure NIR-PTT effect for 808 nm light irradiation (vide infra). To estimate the relative contributions/fractions of PDT/PTT-induced cellular death, the cell viability experiments were carried out at a low temperature of 4 °C. The percentage PDT vs. PTT was calculated according to the literature method (please see ESI,† for detailed procedure).36–38 As shown in Fig. 4, it can clearly be seen that 1064 nm NIR light irradiation of the murine CT-26 colon carcinoma cancer cells incubated with Au NPN6.5(NaCl) (at a dose of 50 μg mL−1) led to 71.5% (=57% PDT + 14.5% PTT) cellular death at 37 °C and 67% (=55% PDT + 12% PTT) cellular death at 4 °C, which are much higher than that obtained from Au NPN6.5 without filling NaCl(aq) aqueous solution in the nanogap. It should be noted that in the absence of Au NPN6.5 and Au NPN6.5(NaCl), laser light irradiation alone did not cause any noticeable cell death under a laser power of 200 mW cm−2 for both 808 nm (10 min irradiation) and 1064 nm (11 min irradiation) (see Fig. S13, ESI†). These results unambiguously show that filling NaCl(aq) aqueous solution in the nanogap of Au NPN6.5 led to much more effective photothermal (808 nm irradiation) and photodynamic (1064 nm irradiation) therapy on inducing cancer cell death under the same NIR photo irradiation conditions. As shown in Fig. 4(a) and (b), it can also be seen that the cancer cell killing effect was more pronounced for 1064 nm NIR light irradiation compared to 808 nm light irradiation. The wavelength effect on killing cancer cells is due to the fact that ROS-based photodynamic therapy (from 1064 nm light irradiation) is more effective in killing cancer cells compared to the hyperthermia-based photothermal therapy effect (via infra). The cancer cell killing mechanism of photothermal therapy is based on the hyperthermia-induced generation of heat-shock proteins (HSP), and subsequently HSP-mediated cell apoptosis. When the local temperature is slightly higher than the normal physiological temperature of 37 °C, cells will produce HSPs to help stabilize other cellular proteins from being denatured and malfunctioning, thus protecting the cells from thermal damage. Specifically, the major function of HSPs is to protect cells from malfunctions and deaths. However, when the local cell temperature is higher than 42–45 °C for a period of 15–30 min or 50 °C for 4–6 min, the over-expressed HSPs will then trigger the cell apoptosis process to destroy cells. Specifically, a low amount of HSPs can protect cellular proteins from denature and prolong the life of cells.90,91 In contrast, excess HSPs will trigger the cellular death/apoptosis process.103 Upon photo irradiation, the temperature rising profile in cancer tissues is not even within tumor tissues due to the uneven uptake/distribution of nanoparticles in tumor tissues. The threshold hyperthermia conditions may not be reached for some areas in tumor tissues, where a smaller number of nanomaterials accumulates and/or low intensity of excitation light is provided. The harmful hyperthermia effect of PTT will be partially cancelled by the protection effects of HSPs. It also requires accumulation of many photon energies to reach or surpass the harmful hyperthermia condition. In addition, PTT is strongly dependent on the outside cellular temperature, and will be significantly suppressed upon lowering the temperature surrounding cancer tissues. The uneven uptake of nanomaterials by cancer cells due to the poorly and unevenly developed blood vessels in cancer tissues and low light intensity will also lead to in-efficient PTT effects.
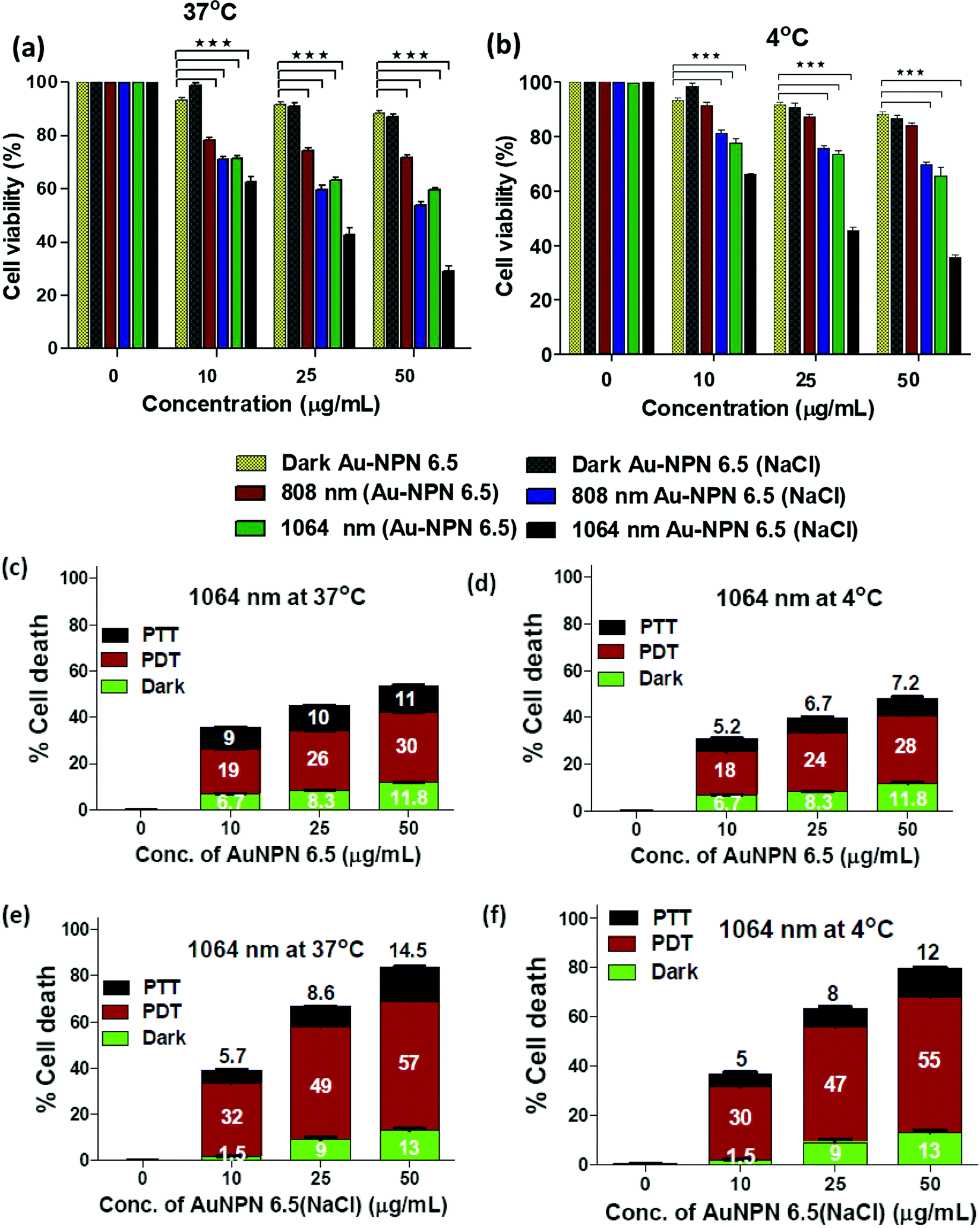 | ||
| Fig. 4 Cell viability of Au NPN6.5 upon 1064 nm NIR light irradiation at 37 °C and 4 °C. Cell viability assay for Au NPNs at different conditions in murine CT-26 colon carcinoma cancer cells: (a) Au-NPN6.5 and Au NPN6.5(NaCl) at 37 °C and (b) Au-NPN6.5 and Au-NPN6.5(NaCl) at 4 °C. Relative contributions of PDT and PTT to cellular death under 1064 nm photo irradiation of (c) Au-NPN6.5 at 37 °C, (d) Au NPN6.5 at 4 °C, (e) Au NPN6.5(NaCl) at 37 °C, and (f) Au NPN6.5(NaCl) at 4 °C. | ||
In contrast, ROS-based photodynamic therapy relies on chemically highly reactive oxygen species, such as 1O2, O2−˙, and ˙OH radicals and H2O2, to kill cancer cells. Due to their high chemical reactivities and powerful oxidation abilities, ROS lead to cellular damage, whereas in the case of PTT, a single photon energy is not enough to create a harmful hyperthermia condition. The chemical reactivity of ROS is nearly independent of the local temperature in cancer tissues and will not be suppressed by low temperatures in cancer cells. Unlike HSPs, ROS do not have a cellular protection effect. All ROS contribute to cancer cell damages and deaths. This is why ROS-based PDT is more effective in destroying cancer cells as compared to hyperthermia-based PTT effects under the same conditions. Experimentally, we also observed that under the condition of equal amounts of photons being absorbed by the same nanomaterial, NIR PDT always led to greater cellular death and the corresponding NIR PTT.36–38,63,104
Further, cellular uptake experiments were carried out using confocal laser scanning microscopy (CLSM) to determine the uptake efficiency of Au NPNs by cancer cells. The observation of bright fluorescence in the Cy5.5 channel confirmed the efficient uptake of anti-h-EGFR-Au NPN6.5(NaCl)-Cy5.5 by the murine CT-26 colon carcinoma cancer cells (Fig. 5(a) and (b)). Further, the targeted anti-h-EGFR-Au NPN6.5(NaCl)-treated CT-26 cells showed a significantly higher allophycocyanin (APC) fluorescence intensity than the Au NPN6.5(NaCl) (without surface-coated anti-h-EGFR)-treated CT-26 cells (Fig. S14, ESI†), indicating the important role of the anti-h-EGFR moieties in endowing the Au NPNs cancer cell-targeting ability. The extent of cellular death was determined using the Calcein AM and PI staining assays (see Fig. 5(c) and (d)), respectively. A higher extent of cellular death was observed in the case of anti-h-EGFR-Au NPN6.5(NaCl) + 1064 nm compared to the cases of (i) anti-h-EGFR-Au NPN6.5 + 1064 nm and (ii) anti-h-EGFR-Au-NPN6.5(NaCl) + 808 nm light irradiation. The data is consistent with that obtained from the MTT assay. This is mainly due to the enhanced NIR absorption of Au NPNs upon filling NaCl(aq) aqueous solution in their nanogap.
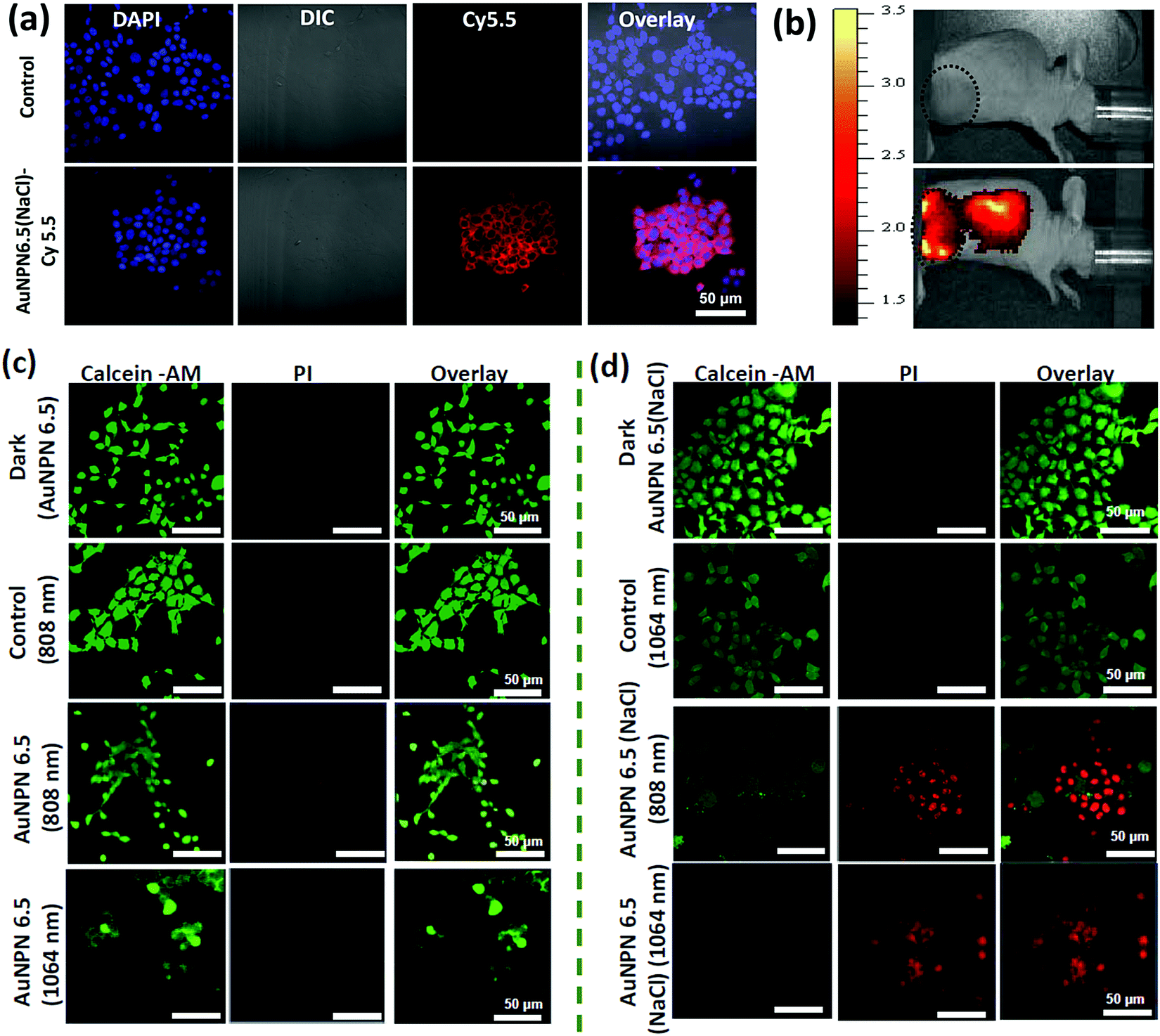 | ||
| Fig. 5 In vitro cellular uptake of Au NPNs. IVIS imaging for in vivo uptake and cell death analysis. (a) Murine CT-26 colon carcinoma cancer cells fed with anti-h-EGFR-Au-NPN6.5(NaCl)-Cy5.5. (b) IVIS imaging for in vivo location/distribution of anti-h-EGFR-Au-NPN6.5(NaCl)-Cy5.5 (via i.v. injection, bottom image), whereas a PBS-injected mouse was used as the control for comparison (top image). (c) and (d) Calcein-AM/PI staining of CT-26 cancer cells treated with Au-NPN6.5 or Au-NPN6.5(NaCl) under different conditions as labeled on the side of each figure and under laser irradiation of 808 and 1064 nm, respectively. Cells not incubated with anti-h-EGFR-Au-NPN6.5(NaCl) were used as the control. | ||
To investigate the origin (hyperthermia vs. ROS) of the cellular death, the CT-26 colon cancer cells were incubated with Au NPN6.5 or Au NPN6.5(NaCl) + DCFH-DA, and then irradiated with 808 and 1064 nm lasers. The flow cytometry results showed that Au NPN6.5(NaCl) + 1064 at 37 °C generated a 31-fold higher level of ROS relative to that measured in the dark control group. Further, in the in vitro ROS experiments (Fig. S15(a), ESI†), the addition of 50 mM NaN3 (a known singlet oxygen quencher) significantly suppressed the amount of ROS, suggesting that the major component of ROS is singlet oxygen. When Au NPN6.5 (without filling NaCl(aq) in the nanogap) was irradiated with 808 and 1064 nm lasers, a negligible amount of ROS was detected, which is attributed to the poor absorption (or small extinction coefficients) of Au NPN6.5 at these two wavelengths. According to the intra cellular ROS experiments, it is clear that Au NPN6.5(NaCl) + 1064 nm could lead to a higher extent of cellular death due to its higher molar extinction coefficient compared to that of its counterpart without filling NaCl(aq) in the nanogap. It is known that thermal stress can also induce the generation of ROS in cells.105 As shown in Fig. S15a (ESI†), the amount of thermal stress-induced ROS generated by 808 nm light irradiation for the Au NPNs was negligible or much lower than that from PDT-induced ROS (i.e., generated by 1064 nm light irradiation of Au NPNs). Further, the generation of ROS was confirmed by confocal imaging. As shown in Fig. S16 (ESI†), the Au NPN6.5(NaCl) + 1064 nm system generated a higher amount of ROS, as evidenced by the bright green fluorescence from the DCFH-DA probe compared to the other systems without NaCl(aq) aqueous solution filled in the nanogap or irradiation by 808 nm light (see Fig. S16, ESI†). The ROS data are consistent with the cellular death data, showing that the Au NPN6.5(NaCl) + 1064 nm system has the highest amount of ROS and cellular death among the samples. When a singlet oxygen (1O2) specific sensor, i.e., singlet oxygen sensor green (SOSG), was used to detect the ROS, bright green fluorescence was observed, further confirming that the major ROS generated from the Au NPN6.5(NaCl) + 1064 nm system is singlet oxygen (Fig. S17, ESI†). To examine whether the photothermal effect was also involved in the cellular death, the amount of heat shock protein (HSP70) was determined for different irradiation conditions. As shown in Fig. S18(a) and (b) (ESI†), the amounts of HSP70 for Au NPN6.5(NaCl) + 808 nm light irradiation at 37 °C and Au NPN6.5(NaCl) + 1064 at 37 °C are 8.5-fold and 4-fold, respectively, relative to that for the dark control group. The results confirm that 808 nm light irradiation of Au NPN6.5(NaCl) mostly results in a photothermal therapeutic effect, whereas 1064 nm light irradiation of Au NPN6.5(NaCl) results in the combination of photothermal (minor) and photodynamic (major) therapy effects for killing cancer cells.
In vivo mice experiments
Inspired by the in vitro results, in vivo mice experiments were carried out using BALB/C nude mice. CT-26 colon cancer cells were implanted on the right leg muscle area of the mice. When the tumor grew and reached a size of 3–4 mm in diameter (or a volume of 14–33 mm3), the nanomaterial-photo irradiation treatments were started. In all the in vivo therapeutic models, anti-h-EGFR-Au NPN6.5 and anti-h-EGFR-Au NPN6.5(NaCl) were injected intravenously with a dose of 25 mg kg−1. The Au NPN6.5(NaCl) nanopeanuts were surface-modified with Cy5.5 and anti EGFR via EDC coupling, and then injected into the mice through tail vein injection. At 24 h after i.v. injection, the mice were euthanized and their major organs were collected to analyze the biodistribution of the nanomaterials. Their organs were digested according to standard protocols, and the amount of nanomaterials in all the organs was determined via inductively coupled plasma mass spectrometry (ICP-MS). The amount of anti-h-EGFR-Au NPN6.5(NaCl) at the tumor site was determined to be 46 μg Au per g tumor tissue, which accounts for ∼13.6% of the total amount of anti-h-EGFR-Au NPN6.5(NaCl) injected (Fig. S15(b), ESI†). The targeting and accumulation of the gold core-gap-shell nanostructures at the tumor site were investigated via in vivo IVIS imaging of mice iv injected with anti-h-EGFR-Au NPN6.5(NaCl)-Cy5.5, where the anti-h-EGFR-Au NPN6.5(NaCl) nanoparticles were further modified with a red fluorescent probe, Cy5.5. According to Fig. 5(b), the red fluorescence (Cy5.5 channel) at the tumor site clearly indicates the effective accumulation of anti-h-EGFR-Au NPN6.5(NaCl)-Cy5.5 at the tumor site. The red fluorescence at the Cy5.5 channel is consistent with the ICP-MS results for the accumulation of gold nanopeanuts at the tumor sites. After confirming the successful targeting by anti-h-EGFR-Au NPN6.5(NaCl)-Cy5.5, the mice were separated randomly into eight groups. Subsequently, in vivo temperature elevation experiments were carried out to monitor the temperature changes at the tumor site upon NIR light irradiation (Fig. 6(a)–(f)). In the case of Au NPN6.5(NaCl) + 808 nm light irradiation, a temperature elevation of ΔT = 19.5 °C was observed, which is significantly higher than that for Au NPN6.5 + 808 (ΔT = 9.7 °C), Au NPN6.5(NaCl) + 1064 (ΔT = 5.7 °C) and Au NPN6.5 + 1064 (ΔT = 2.8 °C). The temperature elevation profiles (see Fig. 6(g)) together with the HSP70 and ROS data confirm that the Au NPN6.5(NaCl) + 808-treated mouse group underwent tumor ablation purely through the PTT effect, whereas the Au NPN6.5(NaCl) + 1064-treated mouse group received combined PDT (major) and PTT (minor) effects. In addition, the results also clearly demonstrate that filling NaCl(aq) aqueous solution in the nanogap could significantly enhance the PTT effect (via enhanced NIR light absorption). Further, for nanomaterials to be used in the clinical treatment of tumors, many factors have to be considered, including their biocompatibility, metabolic tolerance, immune response, and long-term cytotoxicity. In the current study, we investigated the short and mid-term cytotoxicities of anti-h-EGFR-Au NPN6.5(NaCl). BALB/C nude mice were treated with anti-h-EGFR-Au NPN6.5(NaCl) at a dose of 50 mg kg−1 and the hepatic and nephritic functions of the mice were examined at the time point of 45 days post-i.v. injection of anti-h-EGFR-Au NPN6.5(NaCl) (see Fig. S19, ESI†). No noticeable differences were observed between the anti-h-EGFR-Au NPN6.5(NaCl)-treated and the PBS-treated control groups. The results clearly showed that the anti-h-EGFR-Au NPN6.5(NaCl) is biocompatible and safe without any noticeable short and mid-term cytotoxicities to the liver and kidney. In addition, the mouse group treated with anti-h-EGFR-Au NPN6.5(NaCl) + 1064 nm showed an average half-life span of >55 days (see the data below in Fig. 6(i)), which also indicates the biocompatibility and low short- and midterm-cytotoxicities of anti-h-EGFR-Au NPN6.5(NaCl). The investigation of the long-term (>6 months) cytotoxicity and metabolism pathways of anti-h-EGFR-Au NPN6.5(NaCl) deserve a separate study in the future. It is generally accepted and believed that gold nanomaterials have good biocompatibility and low cytotoxicities.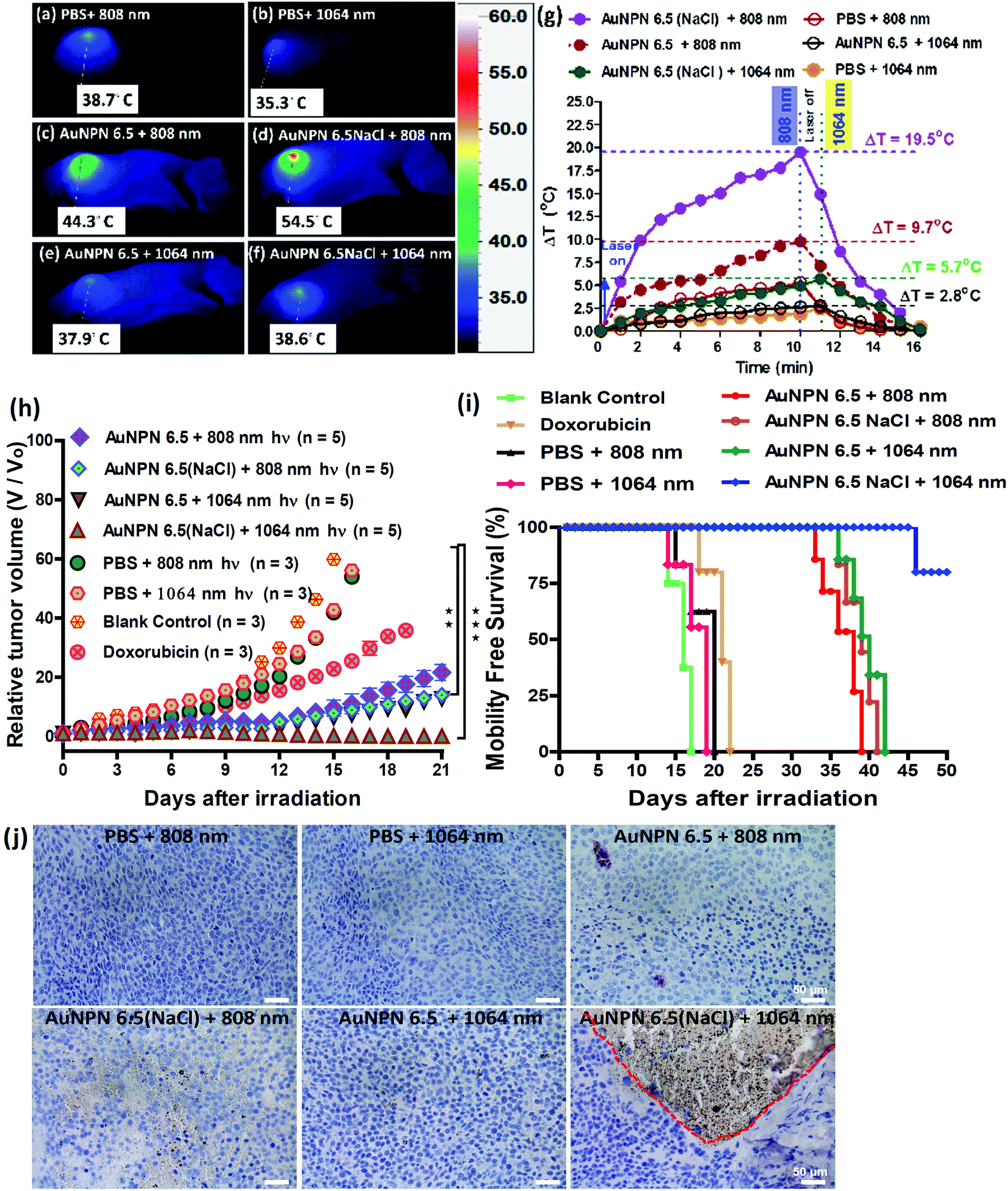 | ||
| Fig. 6 In vivo therapeutic xenograft model. (a)–(f) Thermal imaging of tumor sites with CT-26 colon cancer. No necrotic cell death was observed in the major organs, such as the liver and spleen. 1064 nm light irradiation led to more necrotic cell death than 808 nm light irradiation. Similar results were observed in the immunohistochemistry (IHC) staining by caspase-3 antibody for apoptotic cellular death (see (j) below) at the tumor region. (g) Temperature elevation (ΔT) vs. time of irradiation profiles for various groups of therapy. (h) Tumor growth curves for different treatment groups. (i) Survival rate profiles for the mouse groups during therapy. (j) Caspase-3 antibody (ab2302) staining of tumor tissues under different conditions, as labeled in the figure. | ||
To investigate the photo-therapeutic effects of anti-h-EGFR-Au NPN6.5(NaCl) on the in vivo destruction of solid colon tumor, mouse-bearing tumors were irradiated with either 808 nm (200 mW cm−2, 10 min) or 1064 nm (200 mW cm−2, 11 min) NIR laser light at 24 h after iv injection of nanomaterials with a dose of 25 mg kg−1. The tumor growths were monitored up to 55 days of therapy (Fig. 6(h)). Among them, the tumor growth in the anti-h-EGFR-AuNPN6.5(NaCl) + 1064 nm-treated group was nearly completely inhibited. The therapy images at day 0, 10, and 21 are shown in Fig. S20(a) (ESI†). The median survival rate for anti-h-EGFR-Au NPN6.5(NaCl) + 1064 nm was longer than 55 days, which far exceeds that of the anti-h-EGFR-Au NPN6.5 + 1064 (no NaCl(aq) in the nanogap, 40 d)-treated, doxorubicin (23 days)-treated, and blank control (16 days) groups (Fig. 6i). As shown in Fig. 6j, the amount of apoptotic cells (see the brown-colored cells) followed the sequence of AuNPN6.5(NaCl) + 1064 nm ≫ AuNPN6.5(NaCl) + 808 nm > AuNPN6.5 + 1064 nm ≈ AuNPN6.5 + 808 nm, indicating that (a) the presence of NaCl(aq) aqueous solution in the nanogap dramatically enhances the plasmonic field-field coupling between the Au NR core and the AuAg shell, further leading to an increase in light absorption in both NIR regions, enhancing the NIR-I PTT (808 nm) and NIR-II PDT (1064 nm) effects on inducing the cancer cell apoptosis process and (b) in the presence of NaCl(aq) aqueous solution in the nanogap, NIR-II PDT (1064 nm) has a much higher effect than NIR-I PTT (808 nm) on inducing apoptotic cancer cell death. These results are consistent with both the tumor growth curves (Fig. 6h) and the average lifespan data (Fig. 6i). During the therapy period, no abnormalities were observed in the body weights of the mice (Fig. S20(b), ESI†). Further, after 10 days of therapy, the liver, spleen, and tumor were collected for H&E staining analysis to determine the extent of necrotic cellular death (Fig. S21, ESI†). Clear necrotic damage was observed in the tumor tissues, but not in the other major organs. Previously, Au NPN2.0 was used to generate hyperthermia for killing LLC/LL2 lung cancer cell-based tumors using very strong 1064 nm (3 W cm−2, 5 min) laser light.59,60 However, the observed cell death was mistakenly assumed to be due to pure photothermal therapy effects without measuring the amounts of heat shock proteins and ROS. The salt-enhanced plasmonic field-field coupling between the metal core and the metal shell, as well as the effects on enhancing the PTT/PDT therapeutic effects were not realized in the prior work.59,60
According to the above-mentioned data, we demonstrated that filling NaCl(aq) aqueous solution into the nanogap in Au core-gap-shell NPNs could enhance both the NIR-I PTT and NIR-II PDT effects for destroying a solid colon tumor. The core-gap-shell plasmonic metal nanostructures have a very large interfacial area between the core and the shell compared to the few contact points in nanoparticle cluster/array systems, providing a very good system to allow plasmonic field-field coupling to occur. As described in the Introduction, the LSPR of metal nanoparticles is very sensitive to the dielectric function of their local environment.68–70 Thus, filling the high dielectric NaCl(aq) aqueous solution in the nanogap of Au NPN6.5 provided a means to tune the medium dielectric property in the nanogap, and thus maximize the plasmonic field-field coupling between the metal shell and the metal core, which further led to enhanced opto-properties, including a 3-fold enhancement in SERS, large LSPR broad redshift by 320 nm, and 12.6-fold increase (at 1064 nm) in the extinction coefficient in the NIR regime. The larger the plasmonic field-field coupling, the larger the redshift in the LSPR absorption bands. The increase in the NIR extinction coefficients dramatically enhanced the ability of Au NPN6.5(NaCl) to absorb NIR light and exert NIR-I PTT and NIR-II PDT for the treatment of tumors with much better cancer cell killing efficacies. The anti-h-EGFR-chelated Au NPN6.5(NaCl) system could target and destroy the solid tumor very effectively under an ultra-low laser power density (1064 nm, 200 mW cm−2) due to the plasmonic field-enhanced absorption (or extinction coefficients) in the far NIR regime.
Conclusion
In summary, we reported a simple but effective method to tune and maximize the plasmonic field-field coupling between the shell and the core of core-gap-shell nanostructures and the plasmonic field-lattice interactions, and also demonstrated its biomedical application for the treatment of tumors. By filling 1.67 M of high dielectric NaCl(aq) aqueous solution into the nanogap, the LSPR absorption band of Au NPN6.5(NaCl) was red-shifted by 320 nm from 660 to 980 nm accompanied with an increase in the LSPR absorption in the NIR region, or ∼12.6-fold increase in the extinction coefficient at 1064 nm. The large extent of the redshift and increase in the NIR extinction coefficient in the LSPR absorption band were due to the combined effects of the high dielectric salt-enhanced plasmonic field-field coupling between the outer shell surface and the inner shell surface of the AuAg shell and the plasmonic field-field coupling between the AuAg shell and the Au NR core. Besides enhancing the plasmonic field-field coupling in the metal core-gap-shell nanostructures, salt-filling in the nanogaps of the plasmonic core-gap-shell nanostructures could also enhance the plasmonic field-lattice interactions, leading to enhanced photothermal conversion efficiencies, X-ray scattering cross sections in the XRD measurements, a 2-fold increase in the singlet oxygen sensitization quantum yield, and >10-fold increase in the SERS intensities of surface-absorbed dye molecules. We also demonstrated the biomedical applications of the salt-enhanced NIR-I PTT and NIR-II PDT effects in destroying a solid tumor in mice, leading to a prolonged average lifespan (>55 d) of the mice bearing murine colon tumors and treated with anti-h-EGFR-Au NPN6.5(NaCl) plus 1064 nm NIR light irradiation, which was much better than that of the mice treated with anti-h-EGFR-Au NPN6.5 + 1064 group (without filling salt, 40 d), the doxorubicin-treated group (23 d), and blank control (16 days) groups. Simple salt-filling in the nanogaps of the metal core-gap-shell nanostructures had a very dramatic enhancement effect on prolonging the average lifespan of the NIR-II PDT-treated mice group. To the best of our knowledge, this is the first literature report on salt-mediated, plasmonic field-field/field-lattice coupling-enhanced optical properties and their biomedical application in enhancing the NIR-II PDT effects for destroying solid tumors using an ultralow dose of laser light irradiation (1064 nm, 200 mW cm−2, 11 min). Overall, our work presents a new way to enhance/maximize the plasmonic field-field/lattice coupling, and thus the performance/sensitivities in nanogap-based bioimaging, sensing, and theranostic nanomaterials and devices.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We are grateful to the financial support from the Ministry of Science & Technology, Taiwan; as well as the Frontier Research Center on Fundamental and Applied Sciences of Matters, National Tsing Hua University, Taiwan.References
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS.
- S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 8410–8426 CrossRef CAS.
- J. Langer, D. Jimenez de Aberasturi, J. Aizpurua, R. A. Alvarez-Puebla, B. Auguié, J. J. Baumberg, G. C. Bazan, S. E. J. Bell, A. Boisen, A. G. Brolo, J. Choo, D. Cialla-May, V. Deckert, L. Fabris, K. Faulds, F. J. García de Abajo, R. Goodacre, D. Graham, A. J. Haes, C. L. Haynes, C. Huck, T. Itoh, M. Käll, J. Kneipp, N. A. Kotov, H. Kuang, E. C. Le Ru, H. K. Lee, J.-F. Li, X. Y. Ling, S. A. Maier, T. Mayerhöfer, M. Moskovits, K. Murakoshi, J.-M. Nam, S. Nie, Y. Ozaki, I. Pastoriza-Santos, J. Perez-Juste, J. Popp, A. Pucci, S. Reich, B. Ren, G. C. Schatz, T. Shegai, S. Schlücker, L.-L. Tay, K. G. Thomas, Z.-Q. Tian, R. P. Van Duyne, T. Vo-Dinh, Y. Wang, K. A. Willets, C. Xu, H. Xu, Y. Xu, Y. S. Yamamoto, B. Zhao and L. M. Liz-Marzán, ACS Nano, 2020, 14, 28–117 CrossRef CAS PubMed.
- A. Kinkhabwala, Z. Yu, S. Fan, Y. Avlasevich, K. Müllen and W. E. Moerner, Nat. Photonics, 2009, 3, 654–657 CrossRef CAS.
- K. J. Russell, T.-L. Liu, S. Cui and E. L. Hu, Nat. Photonics, 2012, 6, 459–462 CrossRef CAS.
- M. Osawa, K.-I. Ataka, K. Yoshii and Y. Nishikawa, Appl. Spectrosc., 1993, 47, 1497–1502 CrossRef CAS.
- F. Neubrech, C. Huck, K. Weber, A. Pucci and H. Giessen, Chem. Rev., 2017, 117, 5110–5145 CrossRef CAS PubMed.
- J.-H. Lee, H.-Y. Cho, H. K. Choi, J.-Y. Lee and J.-W. Choi, Int. J. Mol. Sci., 2018, 19, 2021 CrossRef CAS PubMed.
- V. K. S. Hsiao, Y. B. Zheng, B. K. Juluri and T. J. Huang, Adv. Mater., 2008, 20, 3528–3532 CrossRef CAS.
- H. Chen, L. Shao, Q. Li and J. Wang, Chem. Soc. Rev., 2013, 42, 2679–2724 RSC.
- C. Wang, B. Liu and X. Dou, Sens. Actuators, B, 2016, 231, 357–364 CrossRef CAS.
- B. Gao, G. Arya and A. R. Tao, Nat. Nanotechnol., 2012, 7, 433–437 CrossRef CAS PubMed.
- E. M. Larsson, J. Alegret, M. Käll and D. S. Sutherland, Nano Lett., 2007, 7, 1256–1263 CrossRef CAS PubMed.
- C. Radloff and N. J. Halas, Nano Lett., 2004, 4, 1323–1327 CrossRef CAS.
- F. Hao, C. L. Nehl, J. H. Hafner and P. Nordlander, Nano Lett., 2007, 7, 729–732 CrossRef CAS PubMed.
- R. Bukasov, T. A. Ali, P. Nordlander and J. S. Shumaker-Parry, ACS Nano, 2010, 4, 6639–6650 CrossRef CAS PubMed.
- N. Verellen, P. Van Dorpe, C. Huang, K. Lodewijks, G. A. E. Vandenbosch, L. Lagae and V. V. Moshchalkov, Nano Lett., 2011, 11, 391–397 CrossRef CAS PubMed.
- Y. Xia, W. Li, C. M. Cobley, J. Chen, X. Xia, Q. Zhang, M. Yang, E. C. Cho and P. K. Brown, Acc. Chem. Res., 2011, 44, 914–924 CrossRef CAS PubMed.
- A. J. Haes and R. P. Van Duyne, J. Am. Chem. Soc., 2002, 124, 10596–10604 CrossRef CAS PubMed.
- A. V. Kabashin, P. Evans, S. Pastkovsky, W. Hendren, G. A. Wurtz, R. Atkinson, R. Pollard, V. A. Podolskiy and A. V. Zayats, Nat. Mater., 2009, 8, 867–871 CrossRef CAS PubMed.
- J. R. Mejía-Salazar and O. N. Oliveira, Chem. Rev., 2018, 118, 10617–10625 CrossRef PubMed.
- J. N. Anker, W. P. Hall, O. Lyandres, N. C. Shah, J. Zhao and R. P. Van Duyne, Nat. Mater., 2008, 7, 442–453 CrossRef CAS PubMed.
- M. Dai, H. Chen, R. Feng, W. Feng, Y. Hu, H. Yang, G. Liu, X. Chen, J. Zhang, C.-Y. Xu and P. Hu, ACS Nano, 2018, 12, 8739–8747 CrossRef CAS PubMed.
- Y. Lu, S. Feng, Z. Wu, Y. Gao, J. Yang, Y. Zhang, Z. Hao, J. Li, E. Li, H. Chen and S. Lin, Nano Energy, 2018, 47, 140–149 CrossRef CAS.
- M. Gong, R. Sakidja, Q. Liu, R. Goul, D. Ewing, M. Casper, A. Stramel, A. Elliot and J. Z. Wu, Adv. Opt. Mater., 2018, 6, 1701241 CrossRef.
- P. Shen, G. Wang, B. Kang, W. Guo and L. Shen, ACS Appl. Mater. Interfaces, 2018, 10, 6513–6520 CrossRef CAS PubMed.
- G. Tagliabue, A. S. Jermyn, R. Sundararaman, A. J. Welch, J. S. DuChene, R. Pala, A. R. Davoyan, P. Narang and H. A. Atwater, Nat. Commun., 2018, 9, 3394 CrossRef PubMed.
- X. Wang, G. Ma, A. Li, J. Yu, Z. Yang, J. Lin, A. Li, X. Han and L. Guo, Chem. Sci., 2018, 9, 4009–4015 RSC.
- K.-L. Lee, C.-Y. Hung, M.-Y. Pan, T.-Y. Wu, S.-Y. Yang and P.-K. Wei, Adv. Mater. Interfaces, 2018, 5, 1801064 CrossRef.
- W. Liu, X. Li, W. Li, Q. Zhang, H. Bai, J. Li and G. Xi, Biomaterials, 2018, 163, 43–54 CrossRef CAS PubMed.
- T. Bao, W. Yin, X. Zheng, X. Zhang, J. Yu, X. Dong, Y. Yong, F. Gao, L. Yan, Z. Gu and Y. Zhao, Biomaterials, 2016, 76, 11–24 CrossRef CAS PubMed.
- X. Cai, W. Gao, L. Zhang, M. Ma, T. Liu, W. Du, Y. Zheng, H. Chen and J. Shi, ACS Nano, 2016, 10, 11115–11126 CrossRef CAS PubMed.
- S. Dong, Y. Dong, T. Jia, S. Liu, J. Liu, D. Yang, F. He, S. Gai, P. Yang and J. Lin, Adv. Mater., 2020, 32, 2002439 CrossRef CAS PubMed.
- J. Xu, R. Shi, G. Chen, S. Dong, P. Yang, Z. Zhang, N. Niu, S. Gai, F. He, Y. Fu and J. Lin, ACS Nano, 2020, 14, 9613–9625 CrossRef CAS PubMed.
- R. Vankayala, A. Sagadevan, P. Vijayaraghavan, C.-L. Kuo and K. C. Hwang, Angew. Chem., Int. Ed., 2011, 50, 10640–10644 CrossRef CAS PubMed.
- P. Kalluru, R. Vankayala, C.-S. Chiang and K. C. Hwang, Angew. Chem., Int. Ed., 2013, 52, 12332–12336 CrossRef CAS PubMed.
- R. Vankayala, Y.-K. Huang, P. Kalluru, C.-S. Chiang and K. C. Hwang, Small, 2014, 10, 1612–1622 CrossRef CAS PubMed.
- P. Vijayaraghavan, C.-H. Liu, R. Vankayala, C.-S. Chiang and K. C. Hwang, Adv. Mater., 2014, 26, 6689–6695 CrossRef CAS PubMed.
- R. Vankayala, C. L. Kuo, K. Nuthalapati, C.-S. Chiang and K. C. Hwang, Adv. Funct. Mater., 2015, 25, 5934–5945 CrossRef CAS.
- P. Kalluru, R. Vankayala, C.-S. Chiang and K. C. Hwang, Adv. Funct. Mater., 2016, 26, 7908–7920 CrossRef CAS.
- D. F. Cruz, C. M. Fontes, D. Semeniak, J. Huang, A. Hucknall, A. Chilkoti and M. H. Mikkelsen, Nano Lett., 2020, 20, 4330–4336 CrossRef CAS PubMed.
- R. Chikkaraddy, B. de Nijs, F. Benz, S. J. Barrow, O. A. Scherman, E. Rosta, A. Demetriadou, P. Fox, O. Hess and J. J. Baumberg, Nature, 2016, 535, 127–130 CrossRef CAS PubMed.
- J. J. Baumberg, J. Aizpurua, M. H. Mikkelsen and D. R. Smith, Nat. Mater., 2019, 18, 668–678 CrossRef CAS PubMed.
- B. de Nijs, F. Benz, S. J. Barrow, D. O. Sigle, R. Chikkaraddy, A. Palma, C. Carnegie, M. Kamp, R. Sundararaman, P. Narang, O. A. Scherman and J. J. Baumberg, Nat. Commun., 2017, 8, 994 CrossRef PubMed.
- T. J. Norman, C. D. Grant, D. Magana, J. Z. Zhang, J. Liu, D. Cao, F. Bridges and A. Van Buuren, J. Phys. Chem. B, 2002, 106, 7005–7012 CrossRef CAS.
- T. Ung, L. M. Liz-Marzán and P. Mulvaney, Colloids Surf., A, 2002, 202, 119–126 CrossRef CAS.
- R. Bardhan, S. Mukherjee, N. A. Mirin, S. D. Levit, P. Nordlander and N. J. Halas, J. Phys. Chem. C, 2010, 114, 7378–7383 CrossRef CAS.
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607–609 CrossRef CAS PubMed.
- J. Liu and Y. Lu, J. Am. Chem. Soc., 2004, 126, 12298–12305 CrossRef CAS PubMed.
- S. K. Ghosh and T. Pal, Chem. Rev., 2007, 107, 4797–4862 CrossRef CAS PubMed.
- K. Sato, K. Hosokawa and M. Maeda, J. Am. Chem. Soc., 2003, 125, 8102–8103 CrossRef CAS PubMed.
- H. Aldewachi, T. Chalati, M. N. Woodroofe, N. Bricklebank, B. Sharrack and P. Gardiner, Nanoscale, 2018, 10, 18–33 RSC.
- G. Liu, Y. Li, G. Duan, J. Wang, C. Liang and W. Cai, ACS Appl. Mater. Interfaces, 2012, 4, 1–5 CrossRef CAS PubMed.
- Y. Khalavka, J. Becker and C. Sönnichsen, J. Am. Chem. Soc., 2009, 131, 1871–1875 CrossRef CAS PubMed.
- L. Lin, M. Zapata, M. Xiong, Z. Liu, S. Wang, H. Xu, A. G. Borisov, H. Gu, P. Nordlander, J. Aizpurua and J. Ye, Nano Lett., 2015, 15, 6419–6428 CrossRef CAS PubMed.
- N. G. Khlebtsov, L. Lin, B. N. Khlebtsov and J. Ye, Theranostics, 2020, 10, 2067–2094 CrossRef CAS PubMed.
- D.-K. Lim, K.-S. Jeon, J.-H. Hwang, H. Kim, S. Kwon, Y. D. Suh and J.-M. Nam, Nat. Nanotechnol., 2011, 6, 452–460 CrossRef CAS PubMed.
- J.-M. Nam, J.-W. Oh, H. Lee and Y. D. Suh, Acc. Chem. Res., 2016, 49, 2746–2755 CrossRef CAS PubMed.
- M.-F. Tsai, S.-H. G. Chang, F.-Y. Cheng, V. Shanmugam, Y.-S. Cheng, C.-H. Su and C.-S. Yeh, ACS Nano, 2013, 7, 5330–5342 CrossRef CAS PubMed.
- K.-W. Hu, T.-M. Liu, K.-Y. Chung, K.-S. Huang, C.-T. Hsieh, C.-K. Sun and C.-S. Yeh, J. Am. Chem. Soc., 2009, 131, 14186–14187 CrossRef CAS PubMed.
- B. Jang, J.-Y. Park, C.-H. Tung, I.-H. Kim and Y. Choi, ACS Nano, 2011, 5, 1086–1094 CrossRef CAS PubMed.
- H. P. Tham, H. Chen, Y. H. Tan, Q. Qu, S. Sreejith, L. Zhao, S. S. Venkatraman and Y. Zhao, Chem. Commun., 2016, 52, 8854–8857 RSC.
- M. Shanmugam, N. Kuthala, R. Vankayala, C.-S. Chiang, X. Kong and K. C. Hwang, ACS Nano, 2021, 15, 14404–14418 CrossRef CAS PubMed.
- D. Wei, Y. Chen, Y. Huang, P. Li, Y. Zhao, X. Zhang, J. Wan, X. Yin, T. Liu, J. Yin, Z. Liu, Q. Zhang, J. Wang and H. Xiao, Nano Today, 2021, 41, 101288 CrossRef CAS.
- C.-h. Zhang, J. Zhu, J.-j. Li and J.-w. Zhao, J. Appl. Phys., 2015, 117, 063102 CrossRef.
- M. Dhiman, A. Maity, A. Das, R. Belgamwar, B. Chalke, Y. Lee, K. Sim, J.-M. Nam and V. Polshettiwar, Chem. Sci., 2019, 10, 6594–6603 RSC.
- S. J. Barrow, A. M. Funston, D. E. Gómez, T. J. Davis and P. Mulvaney, Nano Lett., 2011, 11, 4180–4187 CrossRef CAS PubMed.
- M. M. Miller and A. A. Lazarides, J. Phys. Chem. B, 2005, 109, 21556–21565 CrossRef CAS PubMed.
- P. K. Jain and M. A. El-Sayed, Nano Lett., 2008, 8, 4347–4352 CrossRef CAS PubMed.
- M. A. Mahmoud and M. A. El-Sayed, J. Am. Chem. Soc., 2010, 132, 12704–12710 CrossRef CAS PubMed.
- C. Tabor, D. V. Haute and M. A. El-Sayed, ACS Nano, 2009, 3, 3670–3678 CrossRef CAS PubMed.
- A. K. Sahu and S. Raj, Gold Bull., 2022, 55, 19–29 CrossRef CAS.
- M. K. Kinnan, S. Kachan, C. K. Simmons and G. Chumanov, J. Phys. Chem. C, 2009, 113, 7079–7084 CrossRef CAS.
- L. Deng, Y. Zhai, Y. Chen, N. Wang and Y. Huang, J. Phys. D: Appl. Phys., 2020, 53, 155103 CrossRef CAS.
- C. Ayala-Orozco, C. Urban, M. W. Knight, A. S. Urban, O. Neumann, S. W. Bishnoi, S. Mukherjee, A. M. Goodman, H. Charron, T. Mitchell, M. Shea, R. Roy, S. Nanda, R. Schiff, N. J. Halas and A. Joshi, ACS Nano, 2014, 8, 6372–6381 CrossRef CAS PubMed.
- M. Sun, F. Liu, Y. Zhu, W. Wang, J. Hu, J. Liu, Z. Dai, K. Wang, Y. Wei, J. Bai and W. Gao, Nanoscale, 2016, 8, 4452–4457 RSC.
- J. Xiao and L. Qi, Nanoscale, 2011, 3, 1383–1396 RSC.
- K.-K. Liu, S. Tadepalli, L. Tian and S. Singamaneni, Chem. Mater., 2015, 27, 5261–5270 CrossRef CAS.
- P. Singh, S. Roy and A. Jaiswal, J. Phys. Chem. C, 2017, 121, 22914–22925 CrossRef CAS.
- P. Singh, T. A. F. König and A. Jaiswal, ACS Appl. Mater. Interfaces, 2018, 10, 39380–39390 CrossRef CAS PubMed.
- N. Gandra, C. Portz and S. Singamaneni, Adv. Mater., 2014, 26, 424–429 CrossRef CAS PubMed.
- A. Jaiswal, L. Tian, S. Tadepalli, K.-K. Liu, M. Fei, M. E. Farrell, P. M. Pellegrino and S. Singamaneni, Small, 2014, 10, 4287–4292 CAS.
- X. Zhang, J. He, Y. Wang and F. Liu, Sci. Rep., 2016, 6, 18902 CrossRef CAS PubMed.
- W. Qiao, K. Yang, A. Thoma and T. Dekorsy, J. Infrared, Millimeter, Terahertz Waves, 2012, 33, 1029–1038 CrossRef CAS.
- A. Brombergt and C. S. Foote, J. Phys. Chem., 1989, 93, 3968 CrossRef.
- Q. Li, F. Chen, W. Zhao, M. Xu, B. Fang, Y. Zhang, L. Duo, Y. Jin and F. Sang, Bull. Korean Chem. Soc., 2007, 28, 1656 CrossRef CAS.
- M. Sakamoto, T. Tachikawa, M. Fujitsuka and T. Majima, Langmuir, 2009, 25, 13888 CrossRef CAS PubMed.
- J. M. Wessels and M. A. J. Rodgers, J. Phys. Chem., 1995, 99, 17586 CrossRef CAS.
- M. Bregnhoj, S. Rodal-Cedeira, I. Pastoriza-Santos and P. R. Ogilby, J. Phys. Chem. C, 2018, 122(27), 15625–15634 CrossRef.
- Y. Zhang, K. Aslan, M. J. R. Previte and C. D. Geddes, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1798–1802 CrossRef CAS PubMed.
- O. Planas, N. Macia, M. Agut, S. Nonell and B. Heyne, J. Am. Chem. Soc., 2016, 138, 2762–2768 CrossRef CAS PubMed.
- A. Sagadevan, K. C. Hwang and M. D. Su, Nat. Commun., 2017, 8, 1812 CrossRef PubMed.
- R. Dědič, A. Svoboda, J. Pšenčík, L. Lupínková, J. Komenda and J. Hála, J. Luminescence, 2003, 102–103, 313–317 CrossRef.
- C. Schweitzer and R. Schmidt, Chem. Rev., 2003, 103(5), 1685–1758 CrossRef CAS PubMed , p1704.
- P. R. Ogilby and C. S. Foote, J. Am. Chem. Soc., 1983, 105, 3423–3430 CrossRef CAS.
- B. A. Lindig, M. A. J. Rodgers and A. P. Schaap, J. Am. Chem. Soc., 1980, 102, 5590–5593 CrossRef CAS.
- J. B. Feix and B. Kalyanaraman, Arch. Biochem. Biophys., 1991, 291, 43–51 CrossRef CAS PubMed.
- R. Vankayala, C.-L. Kuo, A. Sagadevan, P.-H. Chen, C.-S. Chiang and K. C. Hwang, J. Mater. Chem. B, 2013, 1, 4379–4387 RSC.
- G. Pasparakis, Small, 2013, 9, 4130–4134 CrossRef CAS PubMed.
- S. J. Chadwick, D. Salah, P. M. Livesey, M. Brust and M. Volk, J. Phys. Chem. C, 2016, 120, 10647–10657 CrossRef CAS PubMed.
- M. R. Younis, G. He, J. Qu, J. Lin, P. Huang and X.-H. Xia, Adv. Sci., 2021, 2102587 CrossRef CAS PubMed.
- R. Vankayala and K. C. Hwang, Adv. Mater., 2018, 30, 1706320 CrossRef PubMed.
- F. R. Sharp, S. M. Massa and R. A. Swanson, Trends Neurosci., 1999, 22, 97–99 CrossRef CAS PubMed.
- N. Kuthala, R. Vankayala, C.-S. Chiang and K. C. Hwang, Adv. Funct. Mater., 2020, 30, 2002940 CrossRef CAS.
- A. V. Fedyaeva, A. V. Stepanov, I. V. Lyubushkina, T. P. Pobezhimova and E. G. Rikhvanov, Biochemistry, 2014, 79, 1202–1210 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d1nh00631b |
| This journal is © The Royal Society of Chemistry 2022 |