
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optimisation of PET glycolysis by applying recyclable heterogeneous organocatalysts†
Zsuzsanna
Fehér
 a,
Johanna
Kiss
a,
Péter
Kisszékelyi
a,
János
Molnár
b,
Péter
Huszthy
a,
Levente
Kárpáti
b and
József
Kupai
*a
a,
Johanna
Kiss
a,
Péter
Kisszékelyi
a,
János
Molnár
b,
Péter
Huszthy
a,
Levente
Kárpáti
b and
József
Kupai
*a
aDepartment of Organic Chemistry and Technology, Faculty of Chemical Technology and Biotechnology, Budapest University of Technology and Economics, Műegyetem rkp. 3., H-1111 Budapest, Hungary. E-mail: kupai.jozsef@vbk.bme.hu
bLaboratory of Plastics and Rubber Technology, Faculty of Chemical Technology and Biotechnology, Budapest University of Technology and Economics, Műegyetem rkp. 3., H-1111 Budapest, Hungary
First published on 12th October 2022
Abstract
Chemical depolymerisation, or solvolysis, can be a sustainable plastic recycling method, as a circular economy can be achieved by recovering the pure monomers. Polyethylene terephthalate (PET) is a ubiquitous plastic material with short-life application and slow biodegradation, so its waste management needs to be continuously improved. In this study, we tested three commercially available organocatalyst-modified silica gels in the glycolysis of PET and developed another, functionalized with triazabicyclodecene (TBD), which was also tested. Organocatalysts are efficient in PET glycolysis, but their recyclability needs to be improved for industrial application. The applied heterogeneous modified silica gels can be recovered easily by filtration. Si-TEA catalyst was chosen for reaction optimisation because it has the highest thermal stability and good catalytic activity. The PET glycolysis process was optimised by fractional factorial experimental design and response surface methodology. Under optimal reaction conditions (PET (384 mg, 2 mmol), ethylene glycol (1.41 mL, 25.2 mmol), Si-TEA (15.5 mol%), 190 °C, 1.7 h), 88.5% non-isolated bis(2-hydroxyethyl) terephthalate (BHET) monomer yield was obtained. Si-TEA and Si-TBD catalysts were recycled in five reaction cycles, and with both catalysts, high cumulative BHET yields (89 and 88%, respectively) were achieved. Additionally, environmental energy impacts were calculated for the two catalysts and were compared favourably with other organocatalysts in the literature. A process scale-up was also implemented. Finally, it has been verified that modified silica gels have much higher catalytic activities than native silica gel, as solvolytic reactions using the former catalysts took a significantly shorter time.
1 Introduction
As global plastic production increases exponentially,1 there is a growing demand for environmental awareness and sustainable plastic recycling methods. Amongst the most prevalent plastic materials, polyethylene terephthalate (PET) is the one that becomes waste in the highest proportion of its produced quantity (97% of 33 million tons in 20151). This is because PET is primarily used as a short-life packaging material.2 Because of its slow biodegradation and inadequate waste management, adaptive and well-designed recycling strategies are needed to prevent plastic's harmful effect on the environment and improve the sustainability of plastic use.3Plastic recycling methods fall into four categories: primary (industrial scrap recycling), secondary (mechanical), tertiary (chemical), and quaternary (incineration) recycling.4 Mechanical recycling is still considered the most favourable due to its lower energy requirement than chemical recycling. In the case of PET bottles, the mechanical recycling technology is indispensable, particularly when there is a deposit refund system that can provide clean, homogeneous material for bottle-to-bottle recycling.5 However, if there is no such possibility, chemical recycling can be adapted in the case of contaminated, heterogeneous waste streams. Chemical recycling can be split into three categories: solvent purification, chemical depolymerisation (solvolysis or chemolysis), and thermal depolymerisation (thermal cracking or thermolysis). Among these technologies, solvolysis, which means the depolymerisation of plastic waste into its monomers and oligomers by a nucleophile reagent acting also as the solvent,6 has the most promise to complement mechanical recycling. This is because it can live up to the demands made for chemical recycling that mechanical recycling sometimes struggles to achieve: infinite virgin-grade recycling is technically feasible, food-grade products can be produced, and the removal of contaminants is possible.5 Sustainability requires the development of new methods to recover material of the same quality as the original one in an economical and environmentally friendly way, for example, using recyclable catalysts in PET solvolysis.
Based on the applied nucleophile in solvolysis, PET is most often depolymerised by hydrolysis, methanolysis, glycolysis, and aminolysis. Glycolysis is the simplest and earliest applied method of PET depolymerisation.7 This transesterification reaction proceeds with glycol excess at high temperatures (170–200 °C). It stands out with the advantage that its product, bis(2-hydroxyethyl) terephthalate (BHET), can be used to produce PET in only one polycondensation step.8,9 Moreover, on average, it has the lowest environmental energy impact among the solvolytic methods applied to rebuild PET (hydrolysis, methanolysis, and glycolysis),9 and it is the most advanced in demonstrating commercial viability on a larger scale.5
The glycolysis of PET proceeds through at least three stages: oligomers, dimer, and monomer, as previously reported in the literature.7 First, ethylene glycol diffuses into the polymer, causing the polymer to swell up, thus increasing the diffusion rate. It was shown earlier that the depolymerisation of PET with high polymerisation degree into PET with low polymerisation degree, preferentially leading to the dimer, would proceed in a relatively short time interval. So, the conversion of the dimer into BHET would be the rate-determining step.10 The transformation of dimer into BHET monomer is a reversible process, and an equilibrium exists between the BHET monomer and the remaining amount of the dimer.
Catalysts are usually applied in the solvolysis of PET due to its high chemical stability and low solubility.11 The most common catalysts in PET glycolysis are metal catalysts (metal salts,12–15 metal oxides,16–19 and others20–22), basic organocatalysts,12,23–25 ionic liquids,26–29 and deep eutectic solvents.30–33 Metal catalysts usually have high activity and thermal stability if somewhat lower selectivity in some cases, but recovery from the reaction mixture is challenging.9 Ionic liquids (ILs) offer environmentally friendly alternatives to metal catalysts. However, their manufacturing is complex and costly.34 The purification processes are cumbersome if the catalysts contain metal, thus causing the procedures to be expensive,23 and sometimes, only low monomer yields can be obtained with them.29 Deep eutectic solvents emerge as new alternatives with similar characteristics to ILs. However, as a novel method, only a few applications are described in the literature, in which sometimes high catalyst loading and reagent excess are applied.31 Organocatalysts are efficient in PET glycolysis with usually good monomer yield; however, this area also needs improvements for implementation in the industry: as the majority of them are relatively expensive, and organocatalyst loadings are often much higher than metal-based catalysts,35 their recyclability needs to be improved, for example, by grafting them to a solid support. These catalysts are prone to degradation; thus, their thermal stability must be evaluated.23
There are numerous parameters that influence the solvolysis of PET, which can be examined systematically by experimental design. The advantage of experimental design and its analysis is that a process can be described explicitly at the least amount of time and expense. Factorial experimental design is an efficient and straightforward method, and it is commonly used to determine the effects of independent variables that significantly influence the outcome of a process. With the help of experimental design, the numerous methods and catalysts used in PET glycolysis can be compared and evaluated more efficiently. Chen and co-workers applied a 23 factorial experimental design to study the two- and three-factor interaction effects of reaction temperature, catalyst![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PET ratio, and reaction time on the glycolysis of PET. They used cobalt acetate or manganese acetate as catalysts.36,37 Aguado and co-workers also studied a fourth independent variable, the reagent:PET ratio using Taguchi's parameter design while applying zinc acetate as a catalyst.38 A similar design was applied by Zawadzki and co-workers with sodium metasilicate catalyst to optimise the synthesis of PET polyol from which they prepared polyurethane adhesives.39 Van-Pham and co-workers applied a Box–Behnken design using sodium bicarbonate catalyst.40 There are multiple papers about applying design of experiments in PET glycolysis for zinc acetate catalyst.41–43 Seeing the advantages of experimental design in these studies, e.g., finding the economically optimal reaction conditions to obtain high yield and determining the effects of factors influencing the process, it is worth improving a catalytic recycling process by this method.
:PET ratio, and reaction time on the glycolysis of PET. They used cobalt acetate or manganese acetate as catalysts.36,37 Aguado and co-workers also studied a fourth independent variable, the reagent:PET ratio using Taguchi's parameter design while applying zinc acetate as a catalyst.38 A similar design was applied by Zawadzki and co-workers with sodium metasilicate catalyst to optimise the synthesis of PET polyol from which they prepared polyurethane adhesives.39 Van-Pham and co-workers applied a Box–Behnken design using sodium bicarbonate catalyst.40 There are multiple papers about applying design of experiments in PET glycolysis for zinc acetate catalyst.41–43 Seeing the advantages of experimental design in these studies, e.g., finding the economically optimal reaction conditions to obtain high yield and determining the effects of factors influencing the process, it is worth improving a catalytic recycling process by this method.
In this work, multiple commercially available functionalized silica gels (modified with trialkylguanidine (Si-GUA), trialkylamine (Si-TEA), or dialkylthiourea (Si-THU)), and a newly synthesised modified silica gel (modified with triazabicyclodecene (Si-TBD)) catalyst were tested in PET solvolysis with ethylene glycol (EG). These heterogeneous catalysts are easily recoverable, which is necessary for being green and efficient catalysts. To identify the effect of organocatalysts grafted to silica support, the catalytic activities of the modified silica gels were compared to unmodified silica gel (Si). To characterise the catalysts, different methods were used. The prepared Si-TBD catalyst was analysed by scanning electron microscopy with energy dispersive X-ray analysis (SEM-EDX). The thermal stability of all four modified silica gels was determined by thermogravimetric analysis combined with differential scanning calorimetry (TG-DSC). The reaction conditions, such as reaction temperature, catalyst:PET ratio, EG:PET ratio, and reaction time, were optimised using fractional factorial experimental design and response surface methodology. During the optimisation study, the yield of BHET was determined by high-performance liquid chromatography (HPLC), which was compared with the isolated yield in some cases. The recyclability of the two most efficient catalysts was investigated in multiple cycles.
2 Experimental
2.1 Materials
PET flakes from HUKE Ltd (Sárvár, Hungary) were used for glycolytic reactions. Post-consumer PET bottles were washed and ground into small flakes (thickness 0.41 ± 0.08 mm; (9.9 ± 2.9 mm) × (6.8 ± 2.2 mm)). Modified silica gels (Si-TEA, Si-GUA, Si-THU, silica gel functionalized with glycidoxy groups (Si-GLY)) were purchased from SiliCycle Inc, the unmodified silica gel (Si) and toluene from Merck. Ethylene glycol and TBD were purchased from Sigma-Aldrich.2.2 Preparation of TBD-modified silica gel (Si-TBD)
Si-GLY (5 g, 5.55 mmol glycidyl group), toluene (45 mL), and TBD (927 mg, 6.66 mmol) were added to a two-necked round-bottomed flask. The reaction mixture was stirred under argon atmosphere and heated under reflux for 10 hours. The solid product was filtered on a sintered glass filter and washed with a mixture of dichloromethane/methanol 5:1 (15 mL), followed by a mixture of dichloromethane/methanol/triethylamine 5:1:0.05 (2 × 15 mL), then again with dichloromethane/methanol 5:1 (2 × 15 mL). The product was dried in a drying oven at 80 °C for 2 hours to give a mass of 5.36 g—the molecular loading of Si-TBD was 0.58 ± 0.09 mmol g−1, determined by SEM-EDX elemental analysis.
2.3 General procedure for PET glycolysis
The catalyst, PET flakes (384 mg, 2 mmol), and ethylene glycol were added to a 5 mL sealable vial. The reagent and catalyst ratios were calculated in relation to the amount of PET repeating unit (MW = 192.2 g mol−1). The reactions were carried out under argon atmosphere, and magnetic stirring was used. A sand bath was used for heating, and the internal temperature (170–195 °C) was monitored by a thermometer placed in a separate 5 mL sealed vial filled with ethylene glycol. After the appropriate reaction time (1–2 h), the reaction mixture was allowed to cool to room temperature, and then HPLC samples were prepared to determine the non-isolated BHET yield. A MeCN:H2O = 5:1 mixture (3 × 2 mL) was added to the reaction mixture and filtered on a sintered glass filter to separate the catalyst and any remaining solid oligomers from the desired product. The solid residue was washed with MeCN:H2O = 5:1 solvent mixture (3 × 3 mL). The filtrate was then transferred to a 25 mL volumetric flask and filled with the above-mentioned solvent mixture to 25 mL. For the HPLC sample, 50 μL of this solution was taken, and 950 μL of MeCN:H2O = 5:1 solvent mixture was added.
The BHET was isolated by analogy with the method of Zhang and co-workers.44 For determining the isolated BHET yield, the acetonitrile was removed from the filtrate (MeCN:H2O = 5:1 mixture containing BHET, EG, and BHET dimer/trimer by-products) under reduced pressure, and the volume of the remaining aqueous solution was extended by water to 70 mL. Then, the water-insoluble oligomers (dimer and, in some cases, trimer based on HPLC-MS) were removed by filtration using a filter paper, and the filtrate was evaporated to approx. 4 mL under reduced pressure. The resulting solution was stored for 12 hours in a refrigerator (4 °C) to give BHET as white crystals. These crystals were filtered on a sintered glass filter and washed with the mother liquor, then with water.
The main product was identified by 1H, 13C nuclear magnetic resonance spectroscopy (NMR), high-resolution mass spectrometry (HRMS), and Fourier-transform infrared spectroscopy (FTIR), and the BHET dimer side-product was identified by 1H, 13C NMR, and HRMS.
BHET yield (non-isolated) was calculated based on HPLC calibration method using an external standard, by fitting a line to five data points (see Fig. S11 in the ESI†). The non-isolated yield was also compared with the isolated yield in some cases. The isolated yield was calculated as
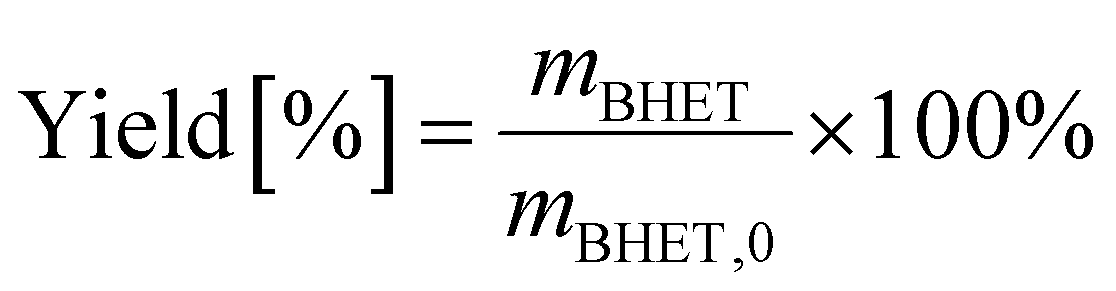 | (1) |
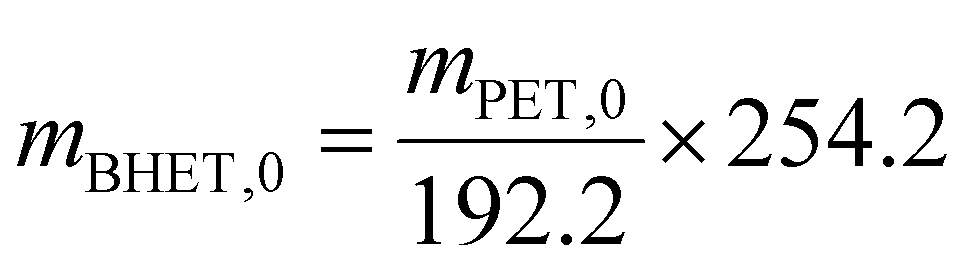 | (2) |
PET conversion was calculated as
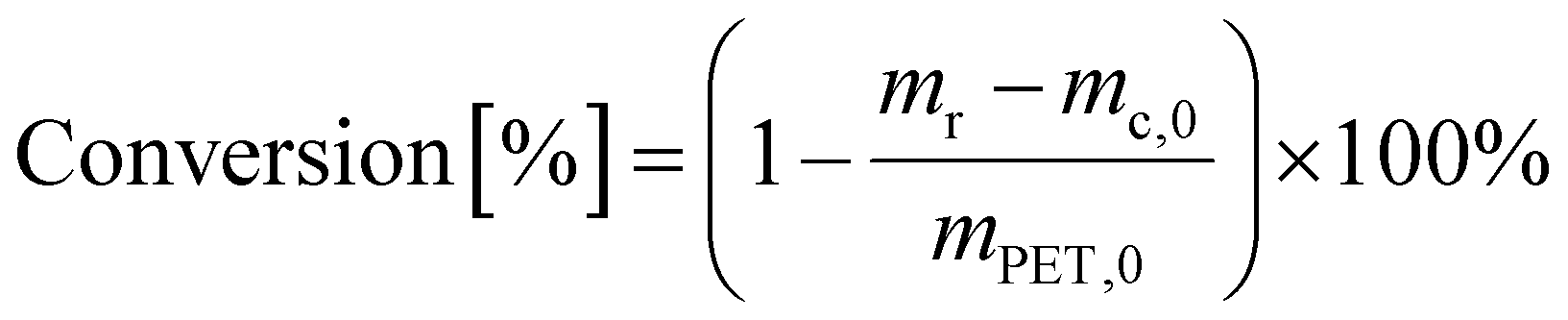 | (3) |
2.4 Catalyst recycling
For the catalyst recycling, 5 reaction cycles were carried out with Si-TEA and Si-TBD catalysts under optimal conditions. Each cycle was repeated once. To maintain the optimal ratio of the parameters, the mass of PET and the amount of ethylene glycol were reduced in proportion to the mass of the recycled catalyst. If the reaction did not proceed with complete conversion, the solid residue remaining after filtration was transferred to the next reaction cycle.2.5 Scale-up of PET glycolysis process
:H2O = 5:1 mixture (15 mL) was added to the reaction mixture and filtered on a sintered glass filter to separate the catalyst and any remaining solid oligomers from the desired product. The solid residue was washed with the above-mentioned solvent mixture (3 × 5 mL). The acetonitrile was removed from the filtrate under reduced pressure, and the volume of the remaining aqueous solution was extended by water to approx. 420 mL. The water-insoluble oligomers (dimer and trimer) were removed by filtration using a filter paper, and the filtrate was evaporated to approx. 25 mL under reduced pressure. The resulting solution was stored for 12 hours in a refrigerator (4 °C) to give BHET as white crystals. These were filtered on a sintered glass filter and washed with the mother liquor, then with water.
:H2O = 5:1 mixture (20 mL) was added to the reaction mixture and filtered on a sintered glass filter to separate the catalyst and any remaining solid oligomers from the desired product. The solid residue was washed with the above-mentioned solvent mixture (6 × 20 mL). The filtrate was then transferred to a 500 mL volumetric flask and filled with the above-mentioned solvent mixture to 500 mL. For the HPLC sample, 60 μL of this solution was taken, and 940 μL of MeCN:H2O = 5:1 solvent mixture was added.
For determining the isolated BHET yield, the acetonitrile was removed from the filtrate (MeCN:H2O = 5:1 mixture containing BHET, EG, and by-products) under reduced pressure, and the volume of the remaining aqueous solution was extended by water to 1.3 L. Then, the water-insoluble oligomers (dimer and trimer) and smaller-sized catalyst fraction (see Section 3.5) were removed by filtration using a filter paper (the solid residue on the filter paper was washed with a methanol:ethyl acetate = 1:1 mixture to recover the smaller-sized catalyst fraction by dissolving the oligomers), and the filtrate was evaporated to approx. 53 mL under reduced pressure. The resulting solution was stored for 12 hours in a refrigerator (4 °C) to give BHET as white crystals. These crystals were filtered on a sintered glass filter and washed with the mother liquor, then with water.
HPLC was performed using a Shimadzu LCMS-2020 device equipped with a HALO C18 (2.7 μm; 4.6 × 150 mm) column. The samples were eluted with gradient elution, using eluent A (0.1% HCOOH in H2O) and eluent B (MeCN). The flow rate was set to 0.8 mL min−1. The initial condition was 5% eluent B, followed by a linear gradient to 100% eluent B by 8 min; from 8 to 13 min, 100% eluent B was retained; and from 13 to 14 min, it went back by a linear gradient to 5% eluent B, which was retained from 14 to 15 min. The column temperature was kept at 40 °C, and the injection volume was 1 μL.
2.6 Experimental design
The optimisation of PET glycolysis was conducted by response surface methodology with gradient method. Statistica software (TIBCO Software Inc.) was applied for data analysis at 5% significance level (to calculate the regression model and perform analysis of variance (ANOVA)). 24-1 fractional factorial design was applied with two centre point experiments, and all experiments were repeated once. The effects of four independent variables (i.e., reaction temperature, catalyst:PET ratio in mol%, EG:PET molar ratio, and reaction time) were investigated. Two dependent variables were chosen as responses, BHET yield (Y) and PET conversion.
2.7 Characterisation
Infrared spectra were recorded on a Bruker Alpha-T FTIR spectrometer. Silica gel 60 F254 (Merck) plates were used for thin-layer chromatography (TLC). Ratios of solvents are given in volumes (mL mL−1). Melting points were measured in a Boetius micro-melting point apparatus, and were uncorrected.NMR spectra were recorded on a Bruker Avance 500 MHz spectrometer (500.13 MHz for 1H, 125.76 MHz for 13C) in DMSO-d6 and were referenced to residual solvent proton signals (δH = 2.50) and solvent carbon signals (δC = 39.51). All chemical shifts are reported in parts per million (ppm). Coupling constants (J) are given in Hz.
HPLC-MS was performed using a Shimadzu LCMS-2020 device, equipped with a Reprospher 100 C18 (5 μm; 100 × 3 mm) column and a positive/negative double ion source (DUIS±) with a quadrupole MS analyser in a range of 50–1000 m/z. The samples were eluted with gradient elution, using eluent A (0.1% HCOOH in H2O) and eluent B (0.1% HCOOH in MeCN). The flow rate was set to 1.5 mL min−1. The initial condition was 5% eluent B, followed by a linear gradient to 100% eluent B by 1.5 min; from 1.5 to 4.0 min, 100% eluent B was retained; and from 4 to 4.5 min, it went back by a linear gradient to 5% eluent B, which was retained from 4.5 to 5 min. The column temperature was kept at 30 °C, and the injection volume was 1 μL. The purity of the compounds was assessed by HPLC with UV detection at 215 and 254 nm. High-resolution mass spectrometry was measured on a Q-TOF Premier mass spectrometer for BHET, and on a Bruker MicroTOF II instrument for the BHET dimer. The ionisation method was electrospray ionisation (ESI) operated in positive ion mode.
The stability of the catalysts was investigated by thermogravimetric analysis and differential scanning calorimetry (TG-DSC). A PerkinElmer STA 6000 instrument was used. During the measurement, the samples of about 30 mg were heated from 25 °C to 190 °C at a heating rate of 10 °C min−1 and then held at the latter temperature for 360 min.
Si-TBD was analysed using a JEOL JSM-5500LV scanning electron microscope (SEM) at high vacuum with 10–20 kV accelerating voltage. The elemental analysis of Si-TBD was carried out with energy dispersive X-ray analysis (EDX with Si(Li) detector) applying 10 kV accelerating voltage and sampling time of 40 s.
The physical properties of the commercially available functionalized silica gels (particle size – measured by laser diffraction, specific surface area – measured by nitrogen adsorption/desorption method at 77 K, molecular loading – measured by elemental analysis, or titration in the case of Si-GLY), which were given by the manufacturer, SiliCycle Inc, can be found in Table S1 in the ESI.†
HS-GC-MS was performed using a Shimadzu GCMS-QP2010 device equipped with an Rtx-5 (30 m × 0.32 mm × 0.25 μm) column and an AOC-5000 auto-injector. The oven temperature was initially held at 40 °C for 2 minutes, increased to 320 °C at 10 °C min−1 with the final temperature held for 5 minutes. The sample holder was thermostated for 45 minutes at 190 °C before the measurement. The carrier gas was argon, the linear flow rate on the column was set to 50 cm s−1, and the split ratio was 1:10. The injector was held at 300 °C. The interface and the ion source temperature were set at 250 °C, the syringe temperature was 150 °C, and the detector voltage was 1.3 kV. Taking the molecular weights of the expected molecules into consideration, the mass scale was adjusted between mass to charge ratio (m/z) of 29–600.
3 Results and discussion
3.1 Synthesis and characterisation of catalysts
The Si-TBD was prepared from Si-GLY by reacting with TBD in toluene (Scheme 1). The Si-GLY used as starting material has a particle size of 40–63 μm, a specific surface area of 494 m2 g−1, and an active loading (the molecular loading measured by titration by the manufacturer, SiliCycle Inc) of 1.11 mmol g−1. Then, leaching of TBD from the catalyst was investigated: the product was stirred in a solvent mixture (dichloromethane/methanol 5:1) in which TBD was highly soluble, filtered, and washed. Analysis of the filtrate by HPLC-MS showed that less than 5% of the TBD on the catalyst surface (based on molecular loading) leached off from the catalyst.
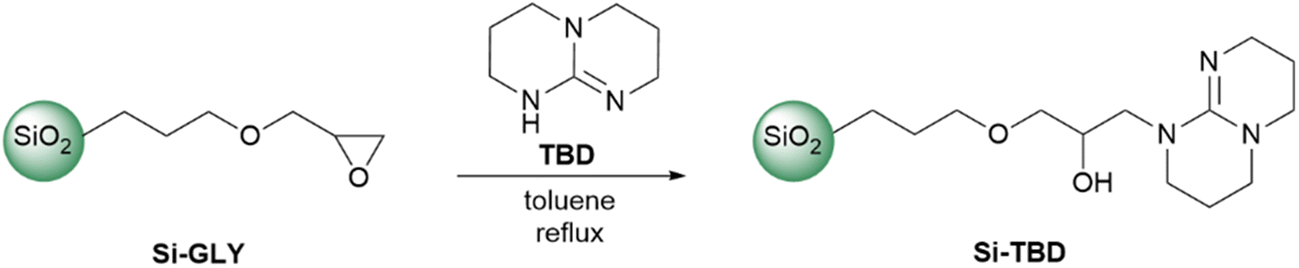 | ||
| Scheme 1 Preparation of TBD-functionalized silica gel. | ||
In addition to the synthesised Si-TBD, three commercially available functionalized silica gels, Si-GUA, Si-THU, or Si-TEA, were investigated (Fig. 1). The physical properties (particle size, specific surface area, molecular loading) of the commercially available functionalized silica gels, which were given by the manufacturer, SiliCycle Inc, can be found in Table S1 in the ESI.† To find the catalyst with the highest catalytic activity and recyclability while preserving the catalytic activity among the modified silica gels tested, we investigated their thermal stability by thermogravimetric analysis and differential scanning calorimetry (TG-DSC), being held at 190 °C (the temperature which is most commonly used in PET glycolysis) for 360 min. From the TG curves, which are similar for each tested silica gel (see Fig. S7–S10 in the ESI†), the mass loss of each sample was determined. For each of the modified silica gels, a theoretical mass loss was also calculated based on the molecular loading, assuming that all the organic units are eliminated from the modified silica gel. The theoretical and measured mass losses of the catalysts are summarised in Table 1. Since Si-TEA showed the lowest mass loss in the TG-DSC measurement, it is assumed to have the highest thermal stability. Si-TBD also showed a much lower mass loss than the theoretical maximum.
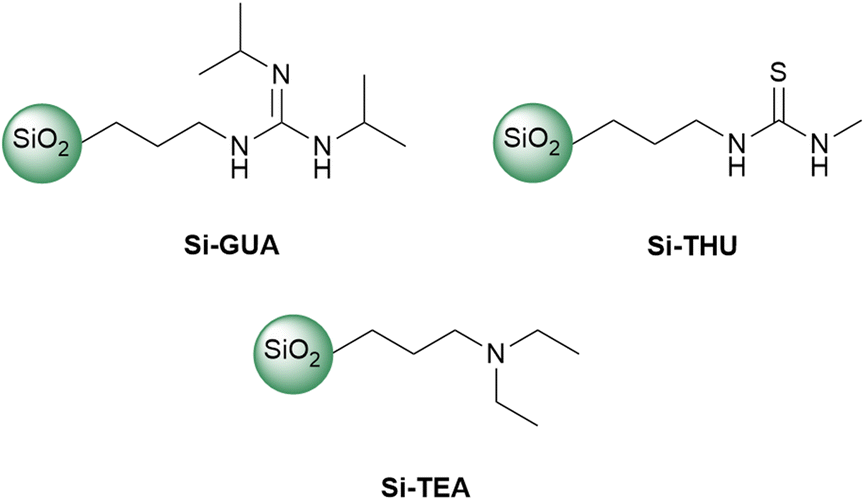 | ||
| Fig. 1 The applied, commercially available functionalized silica gels. | ||
| Modified silica gels | Mass loss (%) | Measured/theoretical maximum ratio (%) | |
|---|---|---|---|
| Theoretical maximum | Measured | ||
| Si-GUA | 16 | 12 | 75 |
| Si-TEA | 10 | 2 | 25 |
| Si-THU | 11 | 8 | 73 |
| Si-TBD | 15 | 8 | 53 |
During our experiments, the reactions are conducted in inert atmosphere. Otherwise, the tertiary amines in the catalysts could react with oxygen to give N-oxides, and then the Cope elimination of N-oxides would produce alkenes. There was no significant degradation of the catalysts observed. The thermal stability of silica gel-supported organocatalysts was reported earlier.45
The morphology of the Si-TBD catalyst was investigated by SEM. Comparing its SEM image (Fig. 2(a)) with the starting material, Si-GLY (Fig. 2(b)), both modified silica gels have irregular particle shapes with similar particle sizes. Therefore, it can be assumed that the particle size distribution did not change during the modification of Si-GLY (40–63 μm, measured by laser diffraction by SiliCycle Inc for Si-GLY). The molecular loading of Si-TBD was determined by SEM-EDX analysis based on nitrogen content. Si-GLY was used as background for the calculation.
 | ||
| Fig. 2 SEM images of Si-TBD (a) and Si-GLY (b). | ||
3.2 Catalyst screening
TBD is a bifunctional catalyst that can activate both an ester and alcohol through hydrogen bonding.46 Numerous nitrogen-based organocatalysts were screened for the depolymerisation of PET earlier in the literature.25,47 The catalytic activity was found to correlate with the basicity, but in the presence of short-chain diols such as ethylene glycol, which serve as a cocatalyst for activating the ester carbonyl groups via hydrogen bonding, the bifunctionality of TBD is less important, especially in the presence of excess ethylene glycol.25 The synthesised catalyst, Si-TBD loses the bifunctionality because of the grafting onto silica support (and thus, its basicity decreases – based on the reported pKa of the conjugated acid of methyl-TBD48), but as we use excess ethylene glycol, this probably does not hinder the catalytic activity greatly because of the latter findings. Thus, we wanted to screen other silica gel-grafted nitrogen-based organocatalysts also, that can be easily accessed commercially, so we chose based on literature analogies: Si-TEA based on triethylamine,49 Si-THU based on urea44 and another guanidine-based catalyst presumably similar to Si-TBD, Si-GUA.The catalytic activities of the commercially available modified silica gels (Si-GUA, Si-TEA, Si-THU, with a particle size of 40–63 μm) and the newly synthesised Si-TBD were investigated in PET glycolysis (Scheme 2), and the results are shown in Fig. 3. In addition to the modified silica gels, unmodified silica gel (Si, also with a particle size of 40–63 μm) was also tested, and reactions without catalyst were also performed. The depolymerisation rate was low without catalyst (9% conversion), while it was slightly higher (34% conversion, 26% BHET yield) with Si. Thus, even the silica support has catalytic activity, but much lower than those of the modified silica gels: the three modified silica gels containing basic units (Si-TBD, Si-GUA, Si-TEA) showed excellent catalytic activities and selectivities (96–100% PET conversions, 83–92% BHET yields) with relatively low standard deviation. Applying the Si-THU catalyst, high deviation was observed supposedly because of the catalyst's instability at high temperature. As the aim was to recycle the catalyst in as many cycles as possible, Si-TEA was chosen as the best catalyst for further reactions because of its relatively high catalytic activity and good thermal stability based on the TG-DSC measurements (see Table 1).
 | ||
| Scheme 2 PET glycolysis with EG using different catalysts. | ||
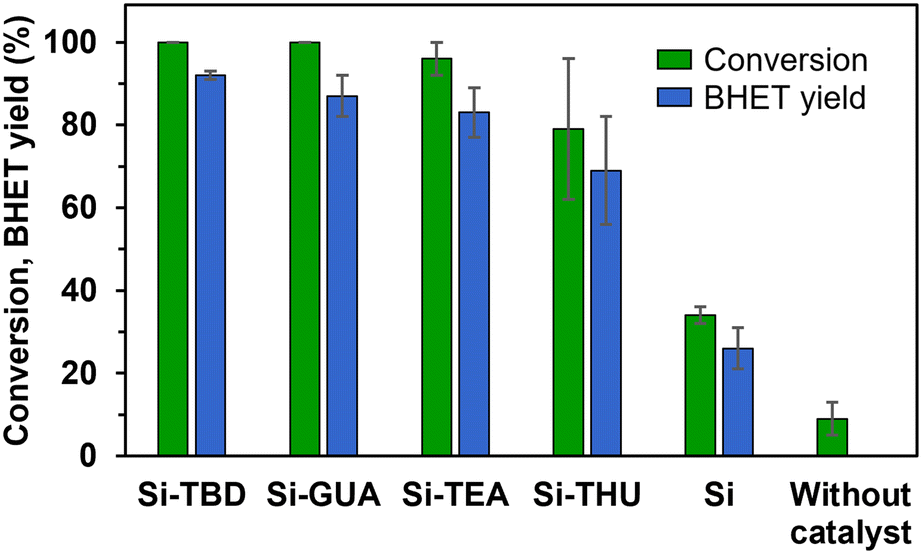 | ||
| Fig. 3 The catalytic activities of different catalysts in PET glycolysis. Reaction conditions: 190 °C, 10 mol% (or 40 wt% in the case of Si) catalyst (if used), EG:PET ratio of 16, 2 h. BHET yield was determined by HPLC. | ||
To confirm the advantages of the Si-TEA modified silica gel catalyst, triethylamine (TEA) was also applied as a catalyst. TEA should only be used in a closed reaction vessel; otherwise, it will evaporate from the reaction mixture. The latter catalyst also gave an excellent non-isolated yield (91%), but the remaining triethylamine traces in the reaction mixture probably hindered the crystallization of BHET, as a poorer isolated yield (69%) was obtained than with the heterogeneous catalysts. To demonstrate the difference, in one case when Si-TEA was used, a non-isolated yield of 90% was observed with an isolated yield of 85%.
3.3 Regression model based on experimental design
The effects of four independent variables (i.e., reaction temperature, catalyst:PET ratio in mol%, EG:PET molar ratio, and reaction time) were investigated as quantitative factors most frequently studied in the literature on glycolysis of PET. The aim was to set up a simple model with relatively few experiments, so a 24-1 experimental design was developed to describe the variation of the dependent variable, non-isolated BHET yield, as a function of the independent parameters using a linear regression model. Si-TEA catalyst was used during the experiments because of its highest thermal stability among the investigated modified silica gels and appropriate activity. The lower and upper levels of the parameters were chosen based on preliminary experiments by changing the parameters one by one (see Fig. S13–S15 in the ESI†) to obtain an approximately linear relationship between the dependent variable and the parameters in the chosen parameter ranges. Their coded and uncoded values are shown in Table 2. The experimental design matrix and the observed BHET yields and PET conversions from 20 experiments are presented in Table 3.
| Independent variables | Factor | Coded level | ||
|---|---|---|---|---|
| −1 | 0 | +1 | ||
| Temperature (°C) | A | 170 | 180 | 190 |
| Catalyst:PET ratio (mol%) |
B | 5 | 12.5 | 20 |
| EG:PET molar ratio (−) |
C | 11 | 13.5 | 16 |
| Reaction time (h) | D | 1 | 1.5 | 2 |
| Run | Temperature (°C) | Catalyst:PET ratio (mol%) |
EG:PET molar ratio (−) |
Reaction time (h) | Conversion (%) | BHET yield (%) |
|---|---|---|---|---|---|---|
| C: centre point. | ||||||
| 1 | 170 | 5.0 | 11.0 | 1.0 | 1 | 0 |
| 2 | 190 | 5.0 | 11.0 | 2.0 | 100 | 88 |
| 3 | 170 | 20.0 | 11.0 | 2.0 | 51 | 45 |
| 4 | 190 | 20.0 | 11.0 | 1.0 | 94 | 80 |
| 5 | 170 | 5.0 | 16.0 | 2.0 | 1 | 0 |
| 6 | 190 | 5.0 | 16.0 | 1.0 | 34 | 28 |
| 7 | 170 | 20.0 | 16.0 | 1.0 | 1 | 0 |
| 8 | 190 | 20.0 | 16.0 | 2.0 | 100 | 90 |
| 9 (C) | 180 | 12.5 | 13.5 | 1.5 | 38 | 31 |
| 10 (C) | 180 | 12.5 | 13.5 | 1.5 | 58 | 51 |
| 11 | 170 | 5.0 | 11.0 | 1.0 | 5 | 0 |
| 12 | 190 | 5.0 | 11.0 | 2.0 | 97 | 85 |
| 13 | 170 | 20.0 | 11.0 | 2.0 | 37 | 29 |
| 14 | 190 | 20.0 | 11.0 | 1.0 | 100 | 87 |
| 15 | 170 | 5.0 | 16.0 | 2.0 | 7 | 0 |
| 16 | 190 | 5.0 | 16.0 | 1.0 | 21 | 16 |
| 17 | 170 | 20.0 | 16.0 | 1.0 | 8 | 0 |
| 18 | 190 | 20.0 | 16.0 | 2.0 | 99 | 89 |
| 19 (C) | 180 | 12.5 | 13.5 | 1.5 | 46 | 41 |
| 20 (C) | 180 | 12.5 | 13.5 | 1.5 | 45 | 40 |
In the fractional design, the triple interaction between temperature, catalyst:PET ratio and EG:PET ratio is confounded with the fourth factor (reaction time). In this case, the main effects are not mixed with each other or with two-factor interactions, while the two-factor interactions are confounding with each other (A by B with C by D; A by C with B by D; A by D with B by C). Since out of the interactions, only A by D (and its linear combination, B by C) were found to be significant, the B by C interaction was chosen as the only significant interaction based on practical considerations (B by C interaction is the interaction of catalyst quantity and EG:PET ratio, which influences the catalyst concentration in the reaction mixture). The results of ANOVA for the regression model are shown in Table 4. A factor is considered significant if its p-value is less than 0.05, and the higher the F-value, the greater the significance of its effect on the model response. Thus, all the main effects and the B by C interaction effect were found to be significant. The Pareto charts of standardised effects in the case of BHET yield or PET conversion as the dependent variable can be found in Fig. S16 and S17 in the ESI.†
| Factor | Sum of squares | DF | Mean square | F-Value | p-Value | Significant |
|---|---|---|---|---|---|---|
| DF: degree of freedom. | ||||||
| Curvature | 2.81 | 1 | 2.81 | 0.07 | 0.7935 | No |
| (A) Temperature (°C) | 14945.06 |
1 | 14945.06 |
382.09 | <0.0001 | Yes |
| (B) Catalyst:PET ratio (mol%) |
2575.56 | 1 | 2575.56 | 65.85 | <0.0001 | Yes |
| (C) EG:PET ratio (−) |
2280.06 | 1 | 2280.06 | 58.29 | <0.0001 | Yes |
| (D) Reaction time (h) | 2889.06 | 1 | 2889.06 | 73.86 | <0.0001 | Yes |
| A by B | 189.06 | 1 | 189.06 | 4.83 | 0.0502 | No |
| A by C | 115.56 | 1 | 115.56 | 2.95 | 0.1136 | No |
| B by C | 280.56 | 1 | 280.56 | 7.17 | 0.0215 | Yes |
| Error | 430.25 | 11 | 39.11 | |||
| Total sum of squares | 23708.00 |
19 | ||||
After model reduction by removing the statistically insignificant factors, a new linear regression model was fitted to the experimental data, and it proved to be adequate based on the curvature check with a high value of R2 (0.969) and adjusted R2 (0.955). Analysis of variance for the reduced linear regression model can be seen in Table S3 in the ESI.† Comparing the yields estimated from the model and the observed ones, it was found that the measured points are approximately along a line of slope 1 through the origin (Fig. 4), therefore the regression model was adequate for predicting the BHET yield in the design space, especially at high yields. The homogeneity of variances was checked comparing the predicted and residual values of BHET yield (see Fig. S18 in the ESI†), and using a Cochran's C test (see ESI†). Based on the latter, the assumption that the random error variance is constant can be accepted. Taking this into account, the reduced model can be used to determine the optimal conditions.
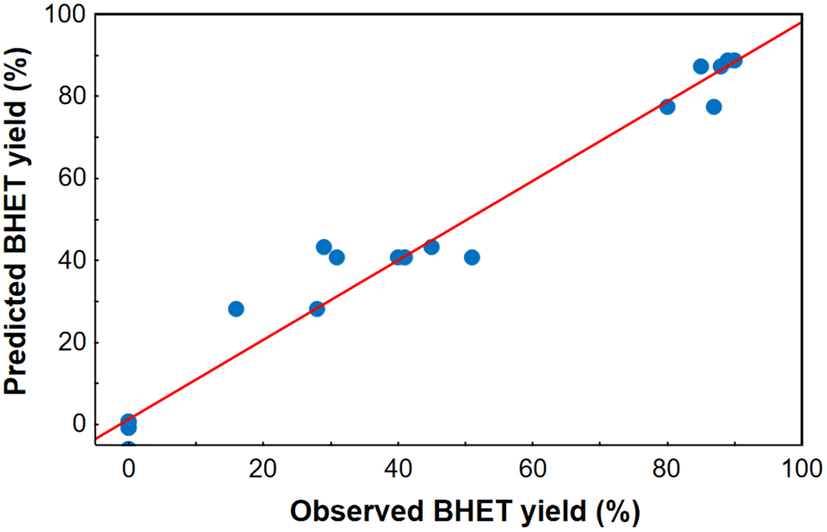 | ||
| Fig. 4 Comparison of observed and predicted BHET yields. | ||
The reduced linear regression model representing the relationship between the BHET yield response Y and the coded values of the four independent factors was obtained as
| Y = 39.8 + 30.6A + 12.7B − 11.9C + 13.4D + 4.2BC | (4) |
Based on this model, the temperature (A) has the strongest effect on the BHET yield, while the interaction of catalyst ratio and EG ratio (BC) has low significance.
The interaction between catalyst:PET ratio and EG:PET ratio at a constant temperature of 190 °C and with a reaction time of 2 h was plotted in Fig. 5 on a response surface plot (a function of two factors with the others at constant values). At a fixed catalyst ratio, the yield decreases with increasing EG ratio in this investigating range. It should be noted, however, that in our preliminary experiments, we found that the yield has a maximum at a certain EG ratio value. For this, the explanation could be that the transformation of dimer into monomer is a reversible process. Therefore, prolonging the reaction after reaching equilibrium can cause a backward shift, increasing the amount of dimer at the expense of the BHET monomer.50 In this case, the higher ratio of EG presumably shifted the equilibrium faster towards the dimer. In the design of experiments, however, the factor values were chosen so that there was an approximately linear relationship between yield and EG ratio. On the surface plot, the interaction can be observed that at a lower catalyst ratio, the EG ratio has a greater negative effect on yield than at a higher catalyst ratio, and similarly, at a higher EG ratio, the catalyst ratio has a greater positive effect on yield than at a lower EG ratio.
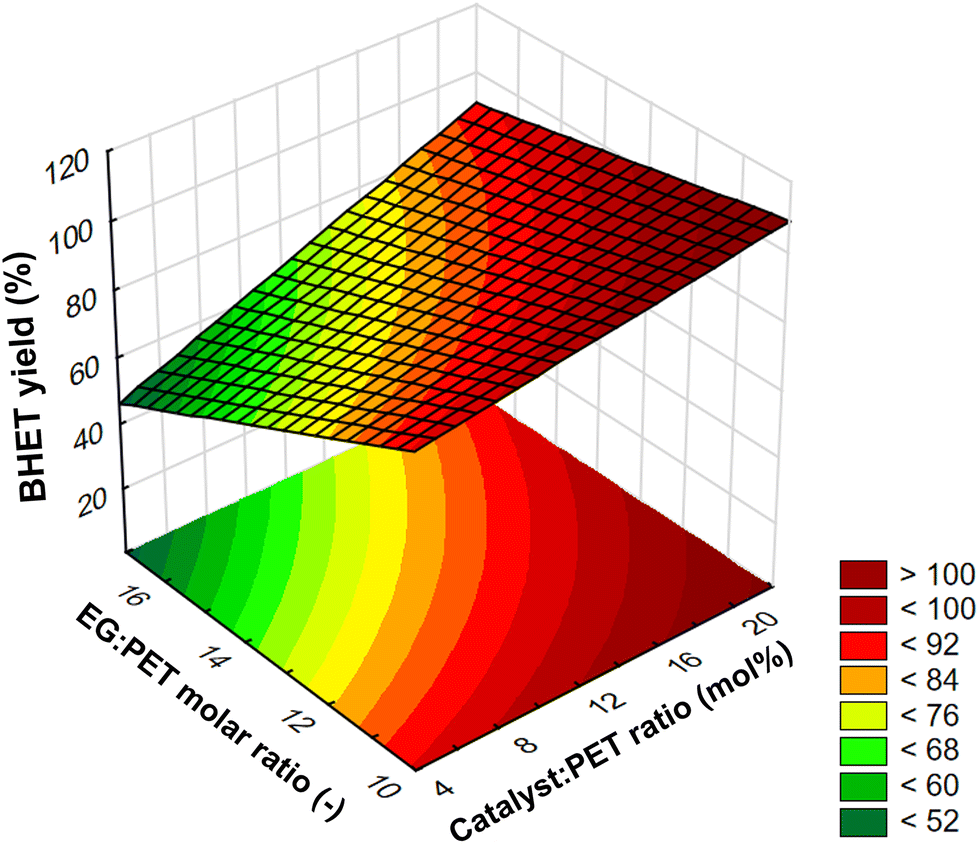 | ||
| Fig. 5 Response surface plot for the interaction effect between catalyst:PET ratio and EG:PET ratio at a constant temperature of 190 °C and with a reaction time of 2 h. | ||
3.4 Optimisation of reaction conditions
The optimisation was implemented using response surface methodology with gradient method. Accordingly, with the step intervals chosen adequately, steps are taken from the centre point of the design space towards the local maximum along the gradient of the reduced model because this is the direction of steepest ascent. The temperature step interval was chosen as the determining one (since this is the most difficult to set to a certain value), which is 2.5 °C in all cases. The step intervals for the other parameters can be calculated from this. The actual parameter value at a given point was obtained by adding the step interval to the parameter value in the previous step, the starting point being the centre point (experiment point 0), the value of which is known from the experimental design. The calculated step intervals and coded factor values at each point are shown in Table S4.† The parameter settings for the different optimisation points are presented in Table 5, and the estimated and measured yields are shown in Fig. 6.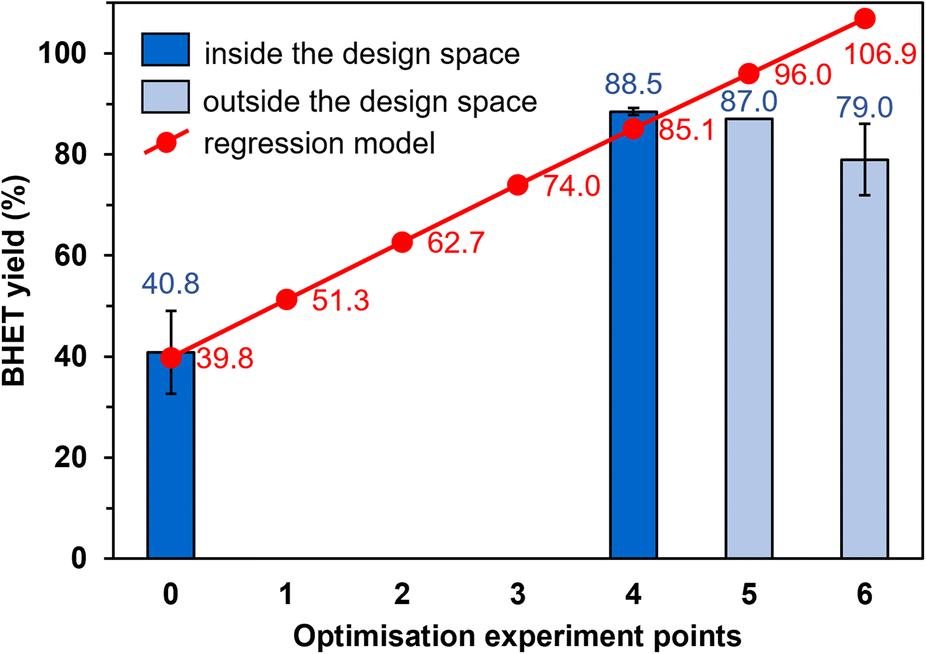 | ||
| Fig. 6 Observed and predicted yields in the optimisation experiments. | ||
| Experiment point | Temperature (°C) | Catalyst:PET ratio* (mol%) |
EG:PET molar ratio* (–) |
Reaction time* (h) |
|---|---|---|---|---|
| *Given to two decimal places to indicate monotonic growth of values. | ||||
| 0 | 180.0 | 12.50 | 12.50 | 1.50 |
| 1 | 182.5 | 13.28 | 12.26 | 1.55 |
| 2 | 185.0 | 14.03 | 12.02 | 1.61 |
| 3 | 187.5 | 14.76 | 11.80 | 1.66 |
| 4 | 190.0 | 15.47 | 11.58 | 1.72 |
| 5 | 192.5 | 16.15 | 11.37 | 1.77 |
| 6 | 195.0 | 16.81 | 11.17 | 1.83 |
Strictly speaking, the reduced model is only valid within the limits of the experimental design, while outside the design space, there may be a larger difference between the estimated and observed yields. For this reason, the optimisation experiments outside the design space (experiment points 5 and 6, Fig. 6) were conducted first. The BHET yield measured at point 5 is 9% lower than the estimated value (87.0% instead of 96.0%, Fig. 6), and at point 6, the yield is 8% lower (79.0%) than at point 5, so the yield decreases outside the design space. Returning to the limits of the experimental design, we performed the reaction at point 4, during which we measured high BHET yield (88.5%). This was accepted as the optimal setting because the model is valid inside the design space, so experiments in points 1–3 are not needed to be conducted. Thus, the most favourable reaction conditions were 190 °C, 15.5 mol% Si-TEA catalyst, EG:PET molar ratio of 12.6, and 1.7 h reaction time in point 4.
Comparing the results achieved under these optimal conditions with literature ones also attained by experimental design (Table 6), excellent PET conversion (100%), BHET yield and selectivity (88.5%) were achieved with an easily recyclable, heterogeneous organocatalyst instead of inorganic salt catalysts with costly51 recycling. This was achieved by performing relatively few experiments but by testing four factors using a simple linear model. It can be observed that compared to inorganic metal salts, organocatalysts require a high catalyst loading, but this can be acceptable by recycling the catalyst (see later in Section 3.5).
| Ref. | Catalyst | Design | Temperature (°C) | Reaction time (h) | Catalyst:PET molar ratio (%) |
EG:PET molar ratio (−) |
Conversion (%) | Yield (%) |
|---|---|---|---|---|---|---|---|---|
| Ref. 36 | Co(OAc)2 | 23 | 190 (130–190) | 2.0 (0.5–2) | 1.0 (0–1) | 6.2 (fixed) | 100 | n.d. |
| Ref. 37 | Mn(OAc)2 | 23 | 190 (130–190) | 1.5 (0.5–2) | 0.5 (0.05–0.5) | 6.2 (fixed) | 100 | n.d. |
| Ref. 38 | Zn(OAc)2 | Taguchi L9 | 208 (195–220) | 2.5 (2.5–3.5) | 0.2 (0.2–1) | 18.6 (6.2–18.6) | n.d. | 85 |
| Ref. 41 | Zn(OAc)2 | Taguchi L20 | 250 (MW) (fixed) | 0.5 (fixed) | 1.0 (1–4) | 9.3 (3.1–15.5) | n.d. | 65 |
| Ref. 40 | NaHCO3 | 4 factor Box–Behnken design | 192 (192–200) | 3.0 (3–5) | 1.9 (1.4–2.3) | 18.8 (7.7–23.2) | n.d. | 75.7 |
| This work | Si-TEA | 24-1 | 190 (170–195) | 1.7 (1–2) | 15.5 (5–20) | 12.6 (11–16) | 100 | 88.5 |
3.5 Catalyst recycling
The immobilisation of various organic compounds on silica gels is well known in the literature,52 so the best-performing catalyst could be produced on a larger scale more cheaply if required. In any case, if the catalyst recycling is successful and straightforward, the use of these catalysts can be economical, even if they have high prices.After determining the optimal reaction conditions, we aimed to test the recyclability of Si-TEA, which has the highest thermal stability among the modified silica gels, and Si-TBD, too, because this catalyst has the highest catalytic activity and a fairly good thermal stability. During the recycling experiments, catalyst mass decreased from cycle to cycle (see Table S5†), in the case of Si-TEA, by 6.5 ± 3.3%, and in the case of Si-TBD, by 7.0 ± 3.2% per cycle compared to the original catalyst mass. Thus, to maintain the optimal ratio of the parameters, the mass of PET and the amount of ethylene glycol were reduced in proportion to the mass of the recycled catalyst. The Si-TEA catalyst showed >89% BHET yield in the first four cycles of repeated PET glycolysis reactions, which slightly decreased (73%) in the fifth cycle (Fig. 7(a)). The Si-TBD catalyst resulted in only 78% BHET yield in the first cycle but >91% in the next 3, while this catalyst also showed a slightly lower yield (79%) in the fifth cycle (Fig. 7(b)). The latter experience may be explained by stirring problems due to the reduction in reaction size (because of the catalyst loss) during recycling. The slightly lower yield in the first cycle in the case of Si-TBD could be attributed to the lack of end-capping of the catalyst, so for larger scales, it is worth considering end-capping. Taking all five reaction cycles into account, the cumulative BHET yields are 89% (1.74 g) for Si-TEA and 88% (1.70 g) for Si-TBD. Consequently, there was no significant catalytic activity loss during the catalyst recycling experiments.
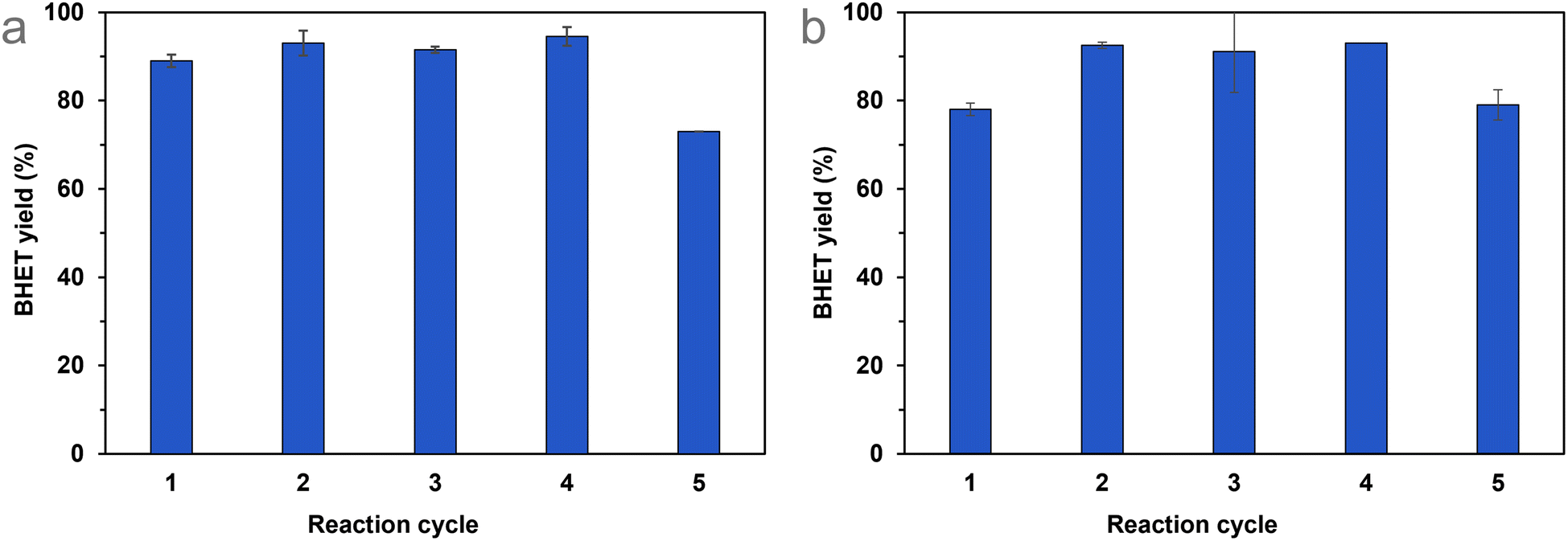 | ||
| Fig. 7 Catalyst recycling in PET glycolysis applying Si-TEA (a) and Si-TBD (b) catalysts. | ||
Stabilities of Si-TEA and Si-TBD catalysts were further investigated by HS-GC-MS, and only a small amount of amines were detected as decomposition products only in the case of Si-TEA. After a 4-hour glycolysis reaction under optimised conditions, we detected a very low amount of different amines (triethylamine, diethylmethylamine, diethylamine) compared to a triethylamine reference, which contained 5% of the TEA amount grafted onto the silica gel (Fig. S19 and 20†). Their peak areas can be seen in Table S6† (those fragments of the detected compounds were depicted on the chromatogram, which had approximately the same intensities considering the mass spectra of the amines; thus, it gives a relative quantitative estimation for the decomposition products). Besides amines, acetone and methyl 1,3-dioxolane (product of the reaction of ethylene glycol and acetaldehyde – the latter is a by-product of PET degradation reactions53) were detected. In the case of Si-TBD, no decomposition products were detected by HS-GC-MS. All reaction mixtures were also analysed by HPLC-MS, and no decomposition products were detected in the case of Si-TEA. However, in the case of Si-TBD, trace amounts of TBD were detected, but only by the first application of the catalyst. After catalyst recycling, it could not be detected.
Thus, we assume that the reason for the slight catalyst mass loss (see Table S5†) is that smaller-sized particles separate from the irregular shaped silica gel at high temperature while stirring in the polar ethylene glycol. These particles go through the G3 porosity glass filter, thus causing the catalyst mass loss. To examine this, we recovered some of the lost catalyst when a large amount of water was added to the filtrate (see Section 2.3), and the precipitate was found to contain Si-TEA. A catalyst recycling experiment was conducted with this recovered smaller-sized Si-TEA, which remained active (64% non-isolated BHET yield in the case of 5 mol% Si-TEA). It should be noted, however, that the lost catalyst was difficult to recover and still contained BHET dimer after washing with a methanol/ethyl acetate mixture. Consequently, future studies should focus on examining the stability of spherical silica gels and other types of catalyst carriers.
Based on the work of Thielemans and co-workers,9 the environmental energy impact (ξ, eqn (5)) value equals the quotient of the environmental factor (Efactor) and the energy economy factor (ε). This green chemistry metric has allowed for quantitative comparison between different studies and for determining their relative feasibility. The lower the environmental energy impact, the more favourable the technology is, since the Efactor is equal to the mass of waste divided by the mass of product, and the energy economy factor is equal to the yield divided by the temperature times the reaction time (see Section 8 in the ESI†). Comparing Si-TEA and Si-TBD to other organocatalysts (Table 7, and Table S7† in more detail), their environmental energy impacts, environmental factors, and energy economy factors are slightly better, so these catalysts can be feasible alternative catalysts in PET glycolysis. The cumulative BHET yields (for five reaction cycles) were applied in the calculations, and the catalysts were not included in the waste mass because of their recyclability (at a certain reaction cycle number, the catalyst mass can be neglected) (see ESI†), as also reported in the work of Thielemans.
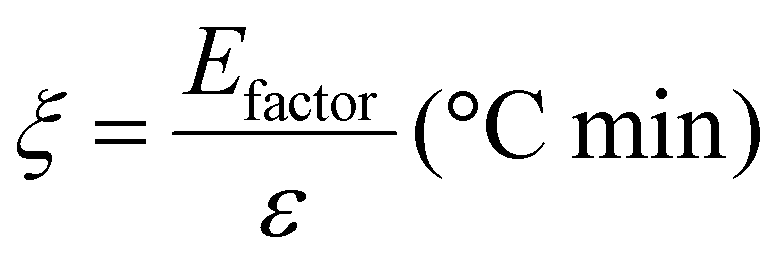 | (5) |
While most heterogeneous catalysts applied in PET glycolysis do not often maintain high BHET selectivity and require higher temperatures than most other catalysts,51 in this work, high selectivity was maintained in 5 reaction cycles at only 190 °C. In the future, catalyst recycling in more cycles will be performed on a larger scale.
3.6 Preliminary scale-up of PET glycolysis process
To examine the achievable yield and how much longer it takes to depolymerise PET on a larger scale under the previously optimised conditions at small scale, a 6-fold scale-up was applied using Si-TEA catalyst at 190 °C, and the catalyst:PET ratio and the EG:PET ratio were adjusted to the optimal values as previously determined (see detailed description of the scale-up in Section 2.5). The reaction time was increased to 2.5 h, as the reaction mixture still visibly contained PET particles after 1.7 h, which was the optimal time for the smaller size. Full conversion was achieved, and the preparative BHET yield was 74% (meaning that the BHET dimer was still present in the reaction mixture after the reaction time of 2.5 h). The slightly lower preparative BHET yield could probably be attributed to the non-optimal reaction time because of the sub-optimal stirring on the larger scale, suggesting that further optimisation of the scale-up reaction time or stirring speed is required in order to obtain higher monomer selectivity and yield. Although this reaction size is still far from the industrial scale, the results suggest that the method may be suitable for the degradation of PET waste on larger scale.
Using the unmodified silica gel (Si) as a catalyst, 34% PET conversion and 26% non-isolated BHET yield were achieved in 2 h reaction time during the catalyst screening experiments (see Fig. 3). Since silica gel is cheaper and thermally more stable than functionalized silica gels, its application in the glycolysis of PET was further investigated. Silica gel was used as a catalyst in a 6-fold scale-up reaction at 50 wt%. The reaction was carried out at 190 °C, using an EG:PET ratio of 12.6 in 10 h reaction time since the PET particles were broken down in the reaction mixture during this time. Under these conditions, complete conversion and 72% BHET yield were obtained, i.e., the silica gel could be successfully used as a catalyst for the process. Nevertheless, the reaction took significantly more time, showing that silica gels functionalized with organic moieties have a much higher catalytic activity, and thus their application on an industrial scale may be more advantageous in terms of energy savings.
To prove the feasibility of scaling-up by using mechanical stirring, an 18-fold scale-up experiment was performed with mechanical stirring for 5 hours. The glycolysis reaction gave full conversion, 83% HPLC yield of BHET, and 72% isolated BHET yield by crystallization. The isolated BHET yield could be further increased by 7% (79% in sum) by column chromatography. Then, further side-products besides dimer of BHET were identified by HPLC-MS: BHET trimer, 2-hydroxyethyl terephthalic acid (in a higher proportion than the rest), and 2-(2-hydroxyethoxy)ethyl (2-hydroxyethyl) terephthalate (Fig. S12 and Table S2†). As only one experiment was performed, which was stopped based on the disappearance of PET flakes, the reaction time of the scaled-up reaction needs to be further optimised in the future.
These findings serve as preliminary studies for future scale-up optimisation. Glycolytic PET depolymerisation is already applied on pilot and even commercial scales,5 however, these processes have limitations: using catalysts is not preferred because of the issues associated with their often difficult recovery and high cost, and in the case of mixed PET waste processes, the removal of pigments and dyes is problematic.54 In our work, catalyst recovery is a crucial aspect, and by using solid-supported organocatalysts, their reuse is made more straightforward as they can be recycled by simple filtration. An issue is the catalyst loss, which will be addressed in the future, and investigating the depolymerisation of mixed PET waste streams is also planned. Later, the nature of the support can also be optimised to address, e.g., issues of the catalyst recovery or the cost of the catalyst. The recovery of ethylene glycol was reported in earlier studies,23,44 this will be also investigated in the future scale-up optimisation.
4. Conclusions
In this work, three commercially available functionalized silica gels and a newly synthesised one, namely solid-supported TBD, were tested as heterogeneous organocatalysts in PET glycolysis. Si-TEA was found to have the highest thermal stability based on TG-DSC and good catalytic activity, while Si-TBD had the highest activity. The effects of four parameters were investigated by experimental design, and a simple linear model was set up to represent the relationship between the BHET yield and the coded values of the four independent factors. The reaction temperature had the highest effect on BHET yield at 170–190 °C. The optimisation of PET glycolysis was implemented using response surface methodology with gradient method applying the most stable catalyst, Si-TEA. The optimal reaction conditions are 190 °C, 15.5 mol% Si-TEA catalyst, EG:PET molar ratio of 12.6, and 1.7 h reaction time. This method compared favourably with other methods applying experimental design in PET glycolysis optimisation. The best catalysts, Si-TEA and Si-TBD can be easily recycled by filtration while preserving high BHET yields in five reaction cycles. The cumulative yields were similarly high for both catalysts (89% for Si-TEA and 88% for Si-TBD), and the methods applying these catalysts have low environmental energy impact, which can be a revealing indicator of sustainability. Consequently, both catalysts can be good alternatives to organocatalysts or other heterogeneous catalysts to be applied in PET glycolysis with high yield and selectivity. This work serves as a “proof of concept” for using solid-supported organocatalysts in PET glycolysis. A 6-fold and an 18-fold scale-up (the latter by mechanical stirring) were applied using Si-TEA catalyst, which showed that further optimisation of the scale-up reaction time or stirring speed is needed to reach a higher yield, which is planned for the future.
Author contributions
Zsuzsanna Fehér: conceptualisation, methodology, investigation, data curation, formal analysis, visualisation, and writing – original draft; Johanna Kiss: conceptualisation, and investigation. Péter Kisszékelyi: conceptualisation, investigation, and writing – review & editing; János Molnár: data curation, formal analysis, and writing – review & editing; Péter Huszthy: funding acquisition, and writing – review & editing; Levente Kárpáti: conceptualisation, and writing – review & editing; József Kupai: conceptualization, supervision, funding acquisition, project administration, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the New National Excellence Program of the Ministry of Human Capacities, grant numbers ÚNKP-22-2-I-BME-146 (J. Kiss), ÚNKP-21-3-I-BME-311 (Z. F.), and ÚNKP-20-5-BME-322 (J. Kupai), and the János Bolyai Research Scholarship of the Hungarian Academy of Science (J. Kupai). It was also supported by the National Research, Development, and Innovation Office (grant numbers FK138037 (J. Kupai) and K128473 (P. H.)), the Richter Gedeon Excellence PhD Scholarship of Gedeon Richter Talentum Foundation (Gedeon Richter Plc.) (Z. F.), and Richter Gedeon Research Student Scholarship of Gedeon Richter's Centenary Foundation (J. Kiss). The authors are grateful to Diana Daicu from Budapest University of Technology and Economics (BUTE) for her assistance with the preliminary experiments related to PET glycolysis, and to József Balla and Judit Mátyási from BUTE for the HS-GC-MS measurements.References
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- PlasticsEurope, Plastics—the facts 2019. An analysis of European plastics production, demand and waste data, https://plasticseurope.org/wp-content/uploads/2021/10/2019-Plastics-the-facts.pdf.
- R. L. Smith, S. Takkellapati and R. C. Riegerix, ACS Sustainable Chem. Eng., 2022, 10, 2084–2096 CrossRef CAS PubMed.
- G. P. Karayannidis and D. S. Achilias, Macromol. Mater. Eng., 2007, 292, 128–146 CrossRef CAS.
- S. Hann and T. Connock, Chemical Recycling: State of Play Report for CHEM Trust, 2020 Search PubMed.
- H. Nishida, Polym. J., 2011, 43, 435–447 CrossRef CAS.
- N. George and T. Kurian, Ind. Eng. Chem. Res., 2014, 53, 14185–14198 CrossRef CAS.
- E. Gubbels, T. Heitz, M. Yamamoto, V. Chilekar, S. Zarbakhsh, M. Gepraegs, H. Köpnick, M. Schmidt, W. Brügging, J. Rüter and W. Kaminsky, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA., Weinheim, Germany, 2018, pp. 1–30 Search PubMed.
- E. Barnard, J. J. R. Arias and W. Thielemans, Green Chem., 2021, 23, 3765–3789 RSC.
- R. López-Fonseca, I. Duque-Ingunza, B. de Rivas, L. Flores-Giraldo and J. I. Gutiérrez-Ortiz, Chem. Eng. J., 2011, 168, 312–320 CrossRef.
- M. Khoonkari, A. H. Haghighi, Y. Sefidbakht, K. Shekoohi and A. Ghaderian, Int. J. Polym. Sci., 2015, 124524 Search PubMed.
- K. R. Delle Chiaie, F. R. McMahon, E. J. Williams, M. J. Price and A. P. Dove, Polym. Chem., 2020, 11, 1450–1453 RSC.
- R. Esquer and J. J. García, J. Organomet. Chem., 2019, 902, 120972 CrossRef CAS.
- C. N. Hoang, T. T. N. Le and Q. D. Hoang, Polym. Bull., 2019, 76, 23–34 CrossRef CAS.
- L. Kárpáti, F. Fogarassy, D. Kovácsik and V. Vargha, J. Polym. Environ., 2019, 27, 2167–2181 CrossRef.
- J. T. Du, Q. Sun, X. F. Zeng, D. Wang, J. X. Wang and J. F. Chen, Chem. Eng. Sci., 2020, 220, 115642 CrossRef CAS.
- M. R. Nabid, Y. Bide and M. Jafari, Polym. Degrad. Stab., 2019, 169, 108962 CrossRef CAS.
- L. Bartolome, M. Imran, K. G. Lee, A. Sangalang, J. K. Ahn and D. H. Kim, Green Chem., 2014, 16, 279–286 RSC.
- M. Imran, D. H. Kim, W. A. Al-Masry, A. Mahmood, A. Hassan, S. Haider and S. M. Ramay, Polym. Degrad. Stab., 2013, 98, 904–915 CrossRef CAS.
- F. Scé, I. Cano, C. Martin, G. Beobide, O. Castillo and I. De Pedro, New J. Chem., 2019, 43, 3476–3485 RSC.
- F. R. Veregue, C. T. P. da Silva, M. P. Moisés, J. G. Meneguin, M. R. Guilherme, P. A. Arroyo, S. L. Favaro, E. Radovanovic, E. M. Girotto and A. W. Rinaldi, ACS Sustainable Chem. Eng., 2018, 6, 12017–12024 CrossRef CAS.
- P. Fang, B. Liu, J. Xu, Q. Zhou, S. Zhang, J. Ma and X. lu, Polym. Degrad. Stab., 2018, 156, 22–31 CrossRef CAS.
- C. Jehanno, I. Flores, A. P. Dove, A. J. Müller, F. Ruipérez and H. Sardon, Green Chem., 2018, 20, 1205–1212 RSC.
- M. Blümel, J. M. Noy, D. Enders, M. H. Stenzel and T. V. Nguyen, Org. Lett., 2016, 18, 2208–2211 CrossRef PubMed.
- K. Fukushima, D. J. Coady, G. O. Jones, H. A. Almegren, A. M. Alabdulrahman, F. D. Alsewailem, H. W. Horn, J. E. Rice and J. L. Hedrick, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1606–1611 CrossRef CAS.
- I. Cano, C. Martin, J. A. Fernandes, R. W. Lodge, J. Dupont, F. A. Casado-Carmona, R. Lucena, S. Cardenas, V. Sans and I. de Pedro, Appl. Catal., B, 2020, 260, 118110 CrossRef CAS.
- Y. Liu, X. Yao, H. Yao, Q. Zhou, J. Xin, X. Lu and S. Zhang, Green Chem., 2020, 22, 3122–3131 RSC.
- J. Sun, D. Liu, R. P. Young, A. G. Cruz, N. G. Isern, T. Schuerg, J. R. Cort, B. A. Simmons and S. Singh, ChemSusChem, 2018, 11, 781–792 CrossRef CAS PubMed.
- A. M. Al-Sabagh, F. Z. Yehia, G. Eshaq and A. E. ElMetwally, Ind. Eng. Chem. Res., 2015, 54, 12474–12481 CrossRef CAS.
- R. Wang, T. Wang, G. Yu and X. Chen, Polym. Degrad. Stab., 2021, 183, 109463 CrossRef CAS.
- E. Sert, E. Yılmaz and F. S. Atalay, J. Polym. Environ., 2019, 27, 2956–2962 CrossRef CAS.
- B. Liu, W. Fu, X. Lu, Q. Zhou and S. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 3292–3300 CrossRef CAS.
- Q. Wang, X. Yao, Y. Geng, Q. Zhou, X. Lu and S. Zhang, Green Chem., 2015, 17, 2473–2479 RSC.
- L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Román-Leshkov, N. Wierckx and G. T. Beckham, Nat. Catal., 2021, 4, 539–556 CrossRef CAS.
- H. Sardon, M. Valle, X. Lopez de Pariza, M. Ximenis and J. M. W. Chan, Angew. Chem., Int. Ed., 2022, e202203043 Search PubMed.
- C.-H. Chen, C.-Y. Chen, Y.-W. Lo, C.-F. Mao and W.-T. Liao, J. Appl. Polym. Sci., 2001, 80, 956–962 CrossRef CAS.
- C.-H. Chen, J. Appl. Polym. Sci., 2003, 87, 2004–2010 CrossRef CAS.
- A. Aguado, L. Martínez, L. Becerra, M. Arieta-araunabeña, S. Arnaiz, A. Asueta and I. Robertson, J. Mater. Cycles Waste Manage., 2014, 16, 201–210 CrossRef CAS.
- D. M. Scremin, D. Y. Miyazaki, C. E. Lunelli, S. A. Silva and S. F. Zawadzki, Macromol. Symp., 2019, 383, 1800027 CrossRef.
- D. T. Van-Pham, Q. H. Le, T. N. Lam, C. N. Nguyen and W. Sakai, Polym. Degrad. Stab., 2020, 179, 109257 CrossRef CAS.
- R. Park, V. Sridhar and H. Park, J. Mater. Cycles Waste Manage., 2020, 22, 664–672 CrossRef CAS.
- A. Stoski, M. F. Viante, C. S. Nunes, E. C. Muniz, M. L. Felsner and C. A. P. Almeida, Polym. Int., 2016, 65, 1024–1030 CrossRef CAS.
- S. Katoch, V. Sharma, P. P. Kundu and M. B. Bera, ISRN Polym. Sci., 2012, 2012, 1–9 CrossRef.
- Q. Wang, X. Yao, S. Tang, X. Lu, X. Zhang and S. Zhang, Green Chem., 2012, 14, 2559–2566 RSC.
- M. Ferré, R. Pleixats, M. Wong Chi Man and X. Cattoën, Green Chem., 2016, 18, 881–922 RSC.
- A. Chuma, H. W. Horn, W. C. Swope, R. C. Pratt, L. Zhang, B. G. G. Lohmeijer, C. G. Wade, R. M. Waymouth, J. L. Hedrick and J. E. Rice, J. Am. Chem. Soc., 2008, 130, 6749–6754 CrossRef CAS PubMed.
- K. Fukushima, O. Coulembier, J. M. Lecuyer, H. A. Almegren, A. M. Alabdulrahman, F. D. Alsewailem, M. A. McNeil, P. Dubois, R. M. Waymouth, H. W. Horn, J. E. Rice and J. L. Hedrick, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1273–1281 CrossRef CAS.
- R. Srivastava, J. Mol. Catal. A: Chem., 2007, 264, 146–152 CrossRef CAS.
- R. D. Allen, K. M. Bajjuri, G. Breyta, J. L. Hedrick and C. E. Larson, WO2015/056377Al, 2015.
- A. Sheel and D. Pant, in Recycling of Polyethylene Terephthalate Bottles, ed. S. Thomas, A. V. Rane, K. Kanny, V. K. Abitha and M. G. Thomas, William Andrew Publishing, 2019, pp. 61–84 Search PubMed.
- S. C. Kosloski-Oh, Z. A. Wood, Y. Manjarrez, J. P. de los Rios and M. E. Fieser, Mater. Horiz., 2021, 8, 1084–1129 RSC.
- P. T. Nguyen, B. Nohair, N. Mighri and S. Kaliaguine, Microporous Mesoporous Mater., 2013, 180, 293–300 CrossRef CAS.
- A. M. Al-Sabagh, F. Z. Yehia, G. Eshaq, A. M. Rabie and A. E. ElMetwally, Egypt. J. Pet., 2016, 25, 53–64 CrossRef.
- T. Thiounn and R. C. Smith, J. Polym. Sci., 2020, 58, 1347–1364 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc02860c |
| This journal is © The Royal Society of Chemistry 2022 |