
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetermination of metallic nanoparticles in biological samples by single particle ICP-MS: a systematic review from sample collection to analysis
Adam
Laycock
 a,
Nathaniel J.
Clark
b,
Robert
Clough
c,
Rachel
Smith
a and
Richard D.
Handy
*bd
a,
Nathaniel J.
Clark
b,
Robert
Clough
c,
Rachel
Smith
a and
Richard D.
Handy
*bd
aCentre for Radiation, Chemical and Environmental Hazards, Public Health England, Harwell Campus, Didcot, OX11 0RQ, UK
bSchool of Biological and Marine Sciences, University of Plymouth, Drake Circus, Plymouth, PL4 8AA, UK. E-mail: r.handy@plymouth.ac.uk
cAnalytical Research Facility, School of Geography, Earth and Environmental Sciences, University of Plymouth, Plymouth, PL4 8AA, UK
dVisiting Professor, Department of Nutrition, Cihan University-Erbil, Kurdistan Region, Iraq
First published on 13th January 2022
Abstract
A systematic review of the use of single particle ICP-MS to analyse engineered nanomaterials (ENMs) in biological samples (plants, animals, body fluids) has highlighted that efforts have focused on a select few types of ENMs (e.g., Ag and TiO2) and there is a lack of information for some important tissues (e.g., reproductive organs, skin and fatty endocrine organs). The importance of sample storage is often overlooked but plays a critical role. Careful consideration of the ENM and matrix composition is required to select an appropriate protocol to liberate ENMs from a tissue whilst not promoting the transformation of them, or genesis of new particulates. A ‘one size fits all’ protocol, applicable to all possible types of ENM and biological matrices, does not seem practical. However, alkaline-based extractions would appear to show greater promise for wide applicability to animal tissues, although enzymatic approaches have a role, especially for plant tissues. There is a lack of consistency in metrics reported and how they are determined (e.g. size limit of detection, and proportions of recovery), making comparison between some studies more difficult. In order to establish standardised protocols for regulatory use, effort is needed to: develop certified reference materials, achieve international agree on nomenclature and the use of control samples, and to create a decision tree to help select the best sample preparation for the type of tissue matrix.
Environmental significanceTo better understand the toxicity, transport and transformation of engineered nanomaterials in biological tissues it is necessary to have the capability of detecting and characterizing these materials in terms of size, number, and size distribution. This presents an analytical challenge which single particle ICP-MS has increasingly been used to address. When applying this technique to biological matrices considerations over sample collection, storage, preparation and analysis are important. The following presents a systematic review of the current state of the art, identifies knowledge gaps and presents suggestions to help advance this area of research and increase confidence in and comparability between future analyses. |
1. Introduction
The determination of engineered nanomaterials (ENMs) in biological samples is needed for several arenas of activity relating to the environmental and human safety of nanomaterials. For example, currently there are no agreed, validated, routine methods for the environmental monitoring of biota in order to understand the fate and health effects of ENMs in ecosystems and for environmental risk assessments. Similarly, national schemes for monitoring the safety of food at the point of sale for consumers with respect to chemical contamination from nano forms are not available, partly because of the difficulty of detecting ENMs in complex matrices such as food.1 Detection methods are also needed to ensure the safety of the agricultural food chain to humans, such as the ability to detect residues of ENMs in crops, farmed animals, and to inform on soil guidelines for ENMs.Engineered nanomaterials, like other new substances, are also subject to a raft of regulatory toxicity tests to ensure their safety with respect to either the environment or human health. Confirming the exposure is an important aspect of experimental design, and the current regulatory environmental toxicity tests use a wide range of organisms from microbes to fish (see Crane, et al.2), sometimes with complex test media that can alter particle behaviour.3 Bioaccumulation testing with fish may also be needed, using a modified version of the Organisation for Economic Cooperation and Development (OECD) technical guidance (TG) 305.4,5 Mammalian toxicity tests are also conducted as part of the safety evaluation of new chemicals, including ENMs, and some of these specifically require the detection of ENMs in tissues. For example, the recently revised OECD inhalation toxicity test guidelines, TG 412 and TG 413, require the determination of lung burdens in circumstances where the material is biopersistent.6,7 The recent revision of the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH) annexes for nanomaterials (regulation (EU) 2018/1881) also asks for consideration of information on the toxicokinetics of ENMs, for example, to assess uptake beyond the organ of entry. The EU guidance on the safety assessment of nanomaterials in cosmetics also requires an investigation of whether material absorbed systemically is in particulate or solubilised/metabolised form.8 The European Food Safety Authority (EFSA) ‘Guidance on Risk Assessment of Nanomaterials to be Applied in the Food and Feed Chain: Human and Animal Health’, requires characterisation and determination of ENMs in regulated food and feed products.9 Consequently, there is a need for standardised methodology for the detection of ENMs in tissue to support the regulatory community with safety testing, as well as environmental monitoring and public health surveillance.
It is possible to use strong acids, such as concentrated nitric acid or aqua regia, to digest tissue samples and then determine the total metal concentrations in the tissue using routine inductively coupled plasma optical emission spectroscopy (ICP-OES) or mass spectrometry (ICP-MS). Such approaches to measuring the total metal in tissue have been applied to aquatic toxicity tests (e.g., TiO2, Federici, et al.10) and dietary bioaccumulation studies (e.g., Ag materials, Clark, et al.11) with fish; as well as to determining total metal in the tissues from ENM exposures in rodents (e.g., Juling, et al.12). However, the concern is that the dissolved versus nano forms of metals may have different bioavailabilities and toxicities.13 It is also becoming apparent that the target organ pathologies from ENMs may differ in both aetiology and magnitude of effect compared to the nearest metal salt (e.g., CuSO4 compared to nano Cu, Al-Bairuty, et al.14). Consequently, it is vital to know how much of the total metal in the tissue is present in the dissolved and nano forms respectively.
Several approaches have been attempted to understand the presence of ENMs on/in tissues. The in situ detection of metallic ENMs might be achieved with scanning electron microscopy (SEM) coupled with energy dispersive X-ray measurements (EDX) for elemental composition, or transmission electron microscopy (TEM) and synchrotron spectroscopy approaches.15,16 Coherent Anti-Stokes Raman Scattering (CARS) microscopy has also been used to identify metallic ENMs in or on fish gills, and enhanced dark-field microscopy has demonstrated utility in detecting metallic nanoparticles in rodent lungs following inhalation.17,18 There has also been effort on developing methods to extract intact ENMs from tissue samples to produce a liquid suspension in which the ENMs could then be quantified. Some of the early approaches include toluene extraction of carbon fullerenes from invertebrates, or acid extraction of acid-resistant ENMs from fish tissues such as TiO2.19,20 However, light scattering methods such as nanoparticle tracking analysis (NTA) or dynamic light scattering (DLS) often cannot be applied to the extracted sample; either because the sample is corrosive, or because of interferences from other colloids or substances in the matrix of the liquid sample. In any event, light scattering methods have a modest detection limit of around 10 mg L−1.15,21
In contrast, the development of single particle inductively coupled plasma mass spectrometry (spICP-MS) has enabled the quantification of ENMs in liquid samples, and with detection limits in the μg L−1 range, or lower. The technique involves aspirating a dispersion of intact particles into the hot plasma of the ICP-MS instrument. Each particle is atomised in the extreme heat of the plasma (5000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 K) creating an ion ‘cloud’ which is detected by the instrument operating in a time resolved mode through the use of short dwell times. The resulting signal is proportional to the size of the particle atomised, and the signal frequency informs on the number of particles of a given size in the sample, as well as aspects of concentration.22 The spICP-MS technique has since been applied to tissue samples from animals and plants (e.g., chicken meat, Peters, et al.;23 rice plants, Deng, et al.24).
000 K) creating an ion ‘cloud’ which is detected by the instrument operating in a time resolved mode through the use of short dwell times. The resulting signal is proportional to the size of the particle atomised, and the signal frequency informs on the number of particles of a given size in the sample, as well as aspects of concentration.22 The spICP-MS technique has since been applied to tissue samples from animals and plants (e.g., chicken meat, Peters, et al.;23 rice plants, Deng, et al.24).
The technique of spICP-MS has been increasingly applied to many different types of tissues from different organisms, using a plethora of potential extraction methods to prepare a liquid suspension (enzymatic, acids, alkali, etc.) (Fig. 1). However, it is unclear which extraction methods are most appropriate for a given ENM and matrix combination. It is also unclear if some digestion methods work better for certain types of metallic ENMs and biological matrices. For regulatory testing, environmental monitoring, food safety, clinical trials with nanomedicines, and public health surveillance, it is desirable that an internationally agreed and validated protocol for the preparation of a biological sample for the determination of ENMs is available. A first step towards this goal is to rationalise the current scientific knowledge on methodologies in order to tease out the most promising approaches, but also to address the utility of each approach in terms of within and between sample variation, detection limit, interferences, and so on.
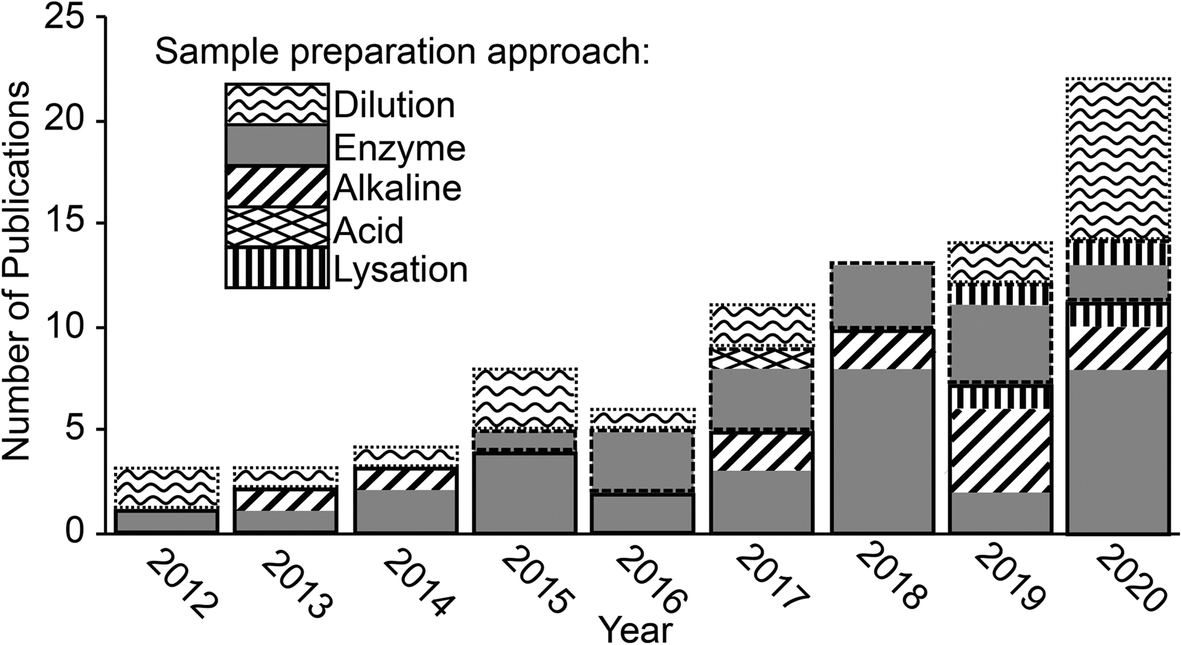 | ||
| Fig. 1 Publication trend showing the number of papers published each year where ENMs were extracted from a biological matrix and analysed by spICP-MS. Bars outlined in solid, dashed and dotted lines indicate studies that looked at animal organs and tissues, plant tissues and biofluids respectively. | ||
The overall aim of the current review was to systematically document the approaches for preparing a biological sample for spICP-MS and to determine which approaches were the most promising. The biological samples included the organs or tissues collected from animals, or from plants, as well as body fluids that might be used as a clinical sample such as whole blood. To achieve this, relevant peer reviewed research papers were systematically identified using Web of Science and the process is summarised in Fig. 2. The review did not consider cell suspensions (i.e., cells already isolated from tissue), or free-living organisms that happen to be individual cells such as microalgae or amoeba, or very small organisms in suspensions such as plankton. Briefly, Web of Science was used to identify all peer-reviewed papers with topics associated with spICP-MS since 2010. This identified a total of 671 papers (search most recently conducted January 2021). These papers were further filtered using the ‘AND’ and ‘OR’ Booleans to identify which of the 671 papers had a topic associated with one of the 10 criteria in boxes 2–11 in Fig. 2, resulting in 346 papers. These papers were further filtered by reading the titles and abstracts to identify 111 potentially relevant papers. These papers were reviewed further and 74 were identified where ENM suspensions had been prepared from biological samples and analysed by spICP-MS. This number was supplemented by 9 additional published materials from various sources such as application notes, agency reports and journal articles that were identified through being referenced in the short listed papers or brought to the authors attention by participants in a technical workshop ran by the EU funded project NanoHarmony (http://www.nanoharmony.eu). The final 83 references are provided in Tables 1–3.
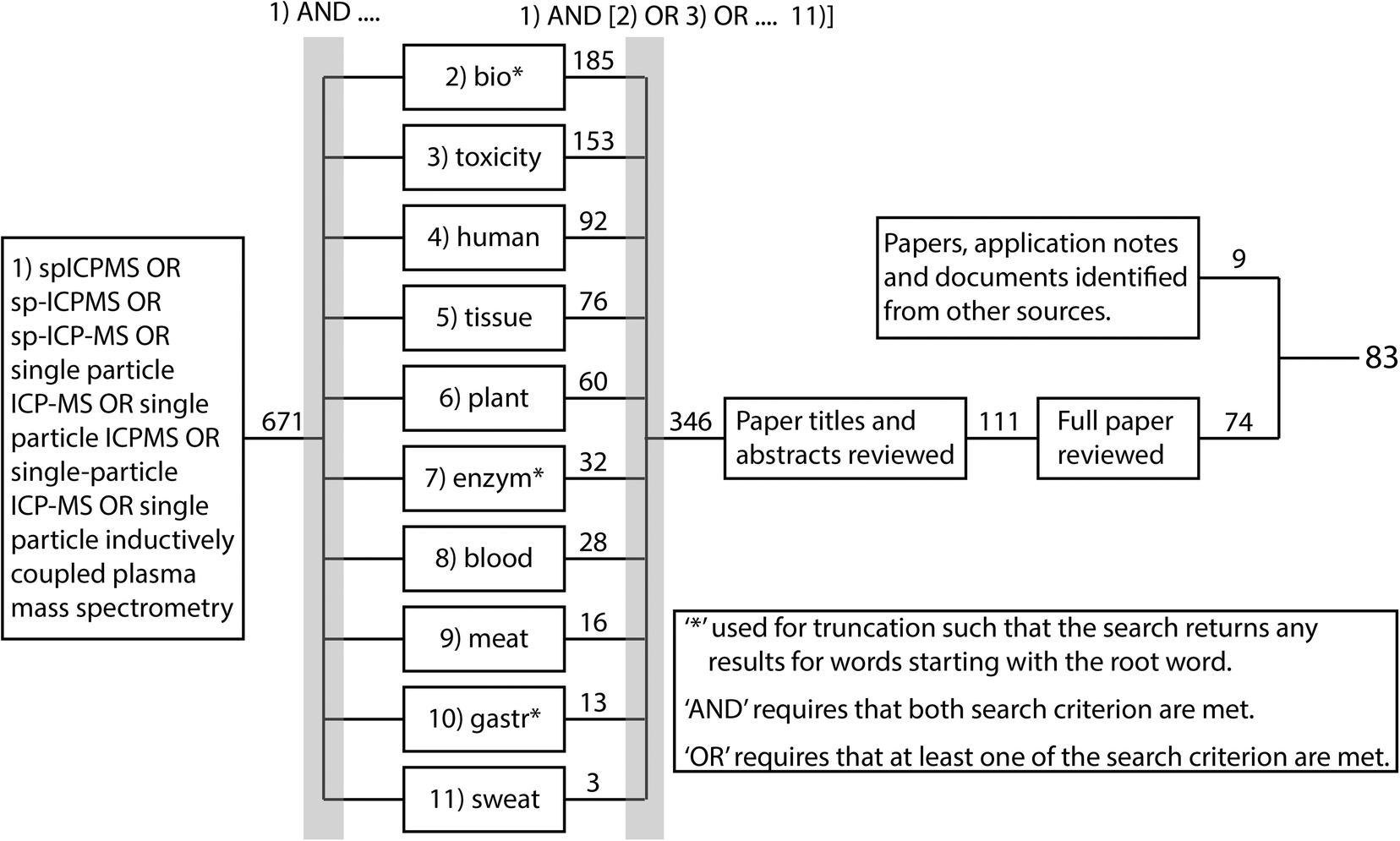 | ||
| Fig. 2 Overview of systematic review process. Web of science was used to identify all peer-reviewed papers associated with the indicated topics published since 2010. The search was conducted January 2021. The numbers show how many papers were identified with each criterion. | ||
| Composition, size & [LODsize] | Matrix | Extraction protocol | Comments | Ref. |
|---|---|---|---|---|
| References denoted with ‘*’ compare multiple extraction methods with the favoured method reported in the table. | ||||
| Enzyme extraction protocols | ||||
| Ag particles | ||||
| 12 & 18 nm [20 nm] | Rat stomach, intestine, liver, spleen kidney & lungs |
1 – 200 mg tissue + 2 mL digestion buffer [0.01 mol L−1 Tris, 1% Triton X-100 and 0.001 mol L−1 calcium acetate (pH 9.5)]. Vortexed for 15 s. 625 μL proteinase K solution (32 U mL−1) added. Vortexed for 10 s. Incubated at 55 °C for 16 hours in a shaking water bath. Vortexed for 60 s. Diluted 40000× with high purity water (HPW) |
ENMs detected in ionic Ag treatment – suggested as originating from the biogenic formation of AgCl, AgS and AgSe | 41 |
| 42 nm [15 nm] | Chicken meat | 2 – 250 mg chicken paste + 5 mL enzyme solution [3 mg mL−1 proteinase K, 5 mg mL−1 sodium dodecyl sulfate (SDS), and 0.2 mg mL−1 sodium azide in 0.05 mol L−1 sodium bicarbonate (pH 7.4–7.7)]. Incubated at 37 °C in a water bath with continuous stirring for 40 min | Mass recoveries of 68% & 80% achieved. Use of AF4 reduced dissolved background | 35, 40 |
| 60 nm | Chicken meat | 3 – 200 mg tissue + 4 mL digestion buffer [0.01 mol L−1 Tris, 1% Triton X-100, 0.001 mol L−1 calcium acetate (pH 9.5)]. Vortexed for 60 s. Ultrasound assisted extraction (UAE) (probe – 4 W) for 5 min on ice. 25 μL proteinase K (822 U mL−1) added then incubated at 35 °C for 3 hours in a shaking water bath | 79–85% recovery of particle mass. Ag ENMs dissolved in chicken meat and AgS particles formed. Extracts stable for up to 3 weeks when stored at 4 °C | 23, 86 |
| <20, 20, 30, 60, 110 nm [20 nm] | Chicken meat & rat liver | Chicken meat – as protocol #3 (ref. 23) | Analysis with sector field ICP-MS offered lower size detection limit for Au, Ag and TiO2 ENMs | 42, 86 |
| Rat liver – adapted from protocol #3 (ref. 23) 2 mL digestion buffer used and incubated at 55 °C for 16 hours | ||||
| 60 nm [20 nm] | Chicken meat | Adapted from protocol #3 (ref. 42) – incubation at 37 °C | Interlaboratory comparison. Samples stored at −130 °C for 6 months. Dissolution occurred during storage and preparation. Suggested that the shorter incubation time and higher enzyme concentration used in protocol #2 resulted in deagglomeration of particles. Average particle number recovery of 19% | 87 |
| 20 nm [10 nm] | Chicken meat, liver & egg yolk | 4 – Adapted from protocol #3 (ref. 42) – 200 mg tissue + 3.09 mL digestion buffer (0.01 mol L−1 Tris buffer, 1% Triton X-100, and 0.001 mol L−1 calcium acetate at [pH 9.5)]. Vigorous stirring for 10 minutes. 0.91 mL 0.75 mg mL−1 proteinase K solution added. Incubated at 37 °C and shaken for 3 hours. Diluted with HPW | Use of a sector field ICP-MS resulted in LODsize | 115 |
| 50 nm [30 nm] | Earthworms |
5 – Individuals snap frozen and ground to a powder in liquid nitrogen. Dispersed at 333 mg mL−1 in 0.02 mol L−1 hydroxyethyl piperazineethanesulfonic acid, 25% glycerol, 0.0015 mol L−1 MgCl2, 0.02 mol L−1 KCl 0.0002 mol L−1 ethylenediaminetetraacetic acid (EDTA), 0.0002 mol L−1 phenylmethylsulfonyl fluoride and 0.0005 mol L−1 dithiothreitol. 0.5 mL 10 mg mL−1 collagenase and 1.5 mL 90 mg mL−1 (300 U mL−1) hyaluronidase added. Incubated for 18 hours at 37 °C with shaking. 0.5 mL 1 mg mL−1 proteinase K added. Incubated for 2 hours at 65 °C. Allowed to cool and layered over 3 mL saturated sucrose (130%). Centrifugation at 21000g for 25 minutes. Lower 1 mL sucrose portion resuspended in 3 mL 0.1% FL-70 and vortexed. Diluted 1:1 with 75% ethanol and centrifugation repeated. Ethanol washed particles resuspended in 0.1% FL–70, 0.01% sodium azide, brought to final volume of 10 mL |
Preliminary sample preparations by enzyme digestion were deemed to show that the method was appropriate (details and data not shown) | 80, 116 |
| High LODsize, worse in samples with high Ag ionic background | ||||
| 40 & 60 nm [13.6–16.2 nm] | Molluscs | 6 – Adapted from protocol #14 (ref. 117) – enzyme solution consisted of 2 g L−1 pancreatin and 2 g L−1 lipase in 0.2 mol L−1 NaH2PO4 and 0.2 mol L−1 NaOH adjusted to pH 7.4 | Particle mass recovery of ∼80% achieved | 106 |
| 60–80 nm [18 nm] | Molluscs | 7 – Adapted from protocols #14 & 15 (ref. 107 and 117) – 1 g mollusc + 5 mL enzyme solution (3 mg L−1 pancreatin and 3 g L−1 lipase in 0.2 mol L−1 phosphate buffer solution (pH 7.4)). Vortexed for 1 minute. UAE (probe) for 10 minutes. Incubated at 35 °C for 12 hours. Centrifugation at 1006g for 15 minutes. Supernatant diluted with HPW | Particle number recoveries of 78–94% achieved | 65 |
| 27 & 34 nm [25 nm] | Human placental tissue | 8 – 2 g tissue homogenised in 2 mL perfusion buffer. Sodium azide added at 200 mg L−1. 2 mL enzyme solution (3 g L−1 proteinase K, 0.5% SDS, 0.05 mol L−1 ammonium bicarbonate and 200 mg L−1 sodium azide) and 0.3 mL HPW added. Vortexed vigorously. Incubated at 37 °C for 1 hour with stirring | Enzyme approach recovered size distribution more accurately than alkaline approach for which agglomeration and particle formation were observed. Particle mass recoveries of 98–124% and 118–133% were achieved for enzyme and alkaline extractions, respectively | 54, 118* |
| 15 nm | Bivalves | As protocol #2 (ref. 40) | 119 | |
| 15 nm | Bivalves | 9 – 10 mL digestion solution (45 μg mL−1 proteinase K, 0.5% SDS, 0.05 mol L−1 NH4HCO3, pH 8.0–8.2) added per 400 mg fresh tissue. Incubated and agitated at 50 °C for 3 hours. Filtered with a 0.45 μm syringe filter | 120 | |
| 15 nm | Freshwater amphipod | 10 – Adapted from protocol #9 (ref. 120) – 10 individuals pooled for 10 mL digestion solution | 121 | |
| TiO2 particles | ||||
| 25, 40, 180 nm [50 nm] | Chicken meat & rat liver | Chicken meat – as protocol #3 (ref. 23) | 42, 86 | |
| Rat liver – adapted from protocol #3 (ref. 23) 2 mL digestion buffer used and incubated at 55 °C for 16 hours | ||||
| 60 nm [27 nm] | Molluscs | As protocol #7 (ref. 65) | 65 | |
| <100 nm | Molluscs | As protocol #4 (ref. 115) | Particle mass recovery of 70–120% | 122 |
| <25 nm | Rat spleen | 11 – Tissue homogenised. UAE in cup horn for 5 minutes at 20% amplitude in 0.05 mol L−1 Tris-HCl buffer and 10% SDS (pH 8). Incubated with mechanical agitation in 2 mg mL−1 proteinase K at 45 °C for 1 hour. UAE at 20% amplitude for 5 minutes, immediately diluted | 123 | |
| 26 nm [100 nm] | Fish intestine, liver, gills, brain |
12 – Tissues homogenised in 1 mL HPW. 1 mL HPW added, UAE (bath) for 10 minutes. 5 mL enzyme solution (50 mg mL−1 proteinase K, 0.050 mol L−1 ammonium bicarbonate, 0.05% SDS) added. Incubated at 50 °C for 10 hours in a water bath with stirring. H2O2 at pH 7.5–8 added at 1:1 ratio in a water bath at 90 °C (H2O2 step repeated three times). 5 mL 0.05% SDS added, UAE (40 W) for 10 minutes, pH then adjusted to 8–8.05 with NaOH |
Particle number and mass recoveries of <25% and 91% respectively. Low number recovery attributed to high LODsize | 71 |
| [85 nm] | Human liver & spleen | 13 – 200 mg ground sample + 4 mL digestion buffer (1.25 mg mL−1 Tris buffer, 0.37 mg mL−1 EDTA, 20 mg mL−1 SDS, 12 mg mL−1 NaCl and 16 mg mL−1 glycine) vortexed for 30 s. Heated at 100 °C for 3 hours, allowed to cool. 0.91 mL 2.5 mg mL−1 proteinase K added. Incubated at 37 °C for 16 hours | Particle mass recovery of 32% | 103 |
| [50 nm] | Human liver & spleen | As protocol #13 (ref. 103) | 105 | |
| 50–500 nm [50 nm] | Human liver, spleen & kidney | As protocol #13 (ref. 103) | Particle mass recovery of 70–76% | 124 |
| 50, 100 nm [29/34 nm] | Molluscs | 14 – 1 g homogenised tissue + 7.5 mL enzyme solution (3 g L−1 pancreatin and 3 g L−1 lipase in 0.2 mol L−1 NaH2PO4 and 0.2 mol L−1 NaOH adjusted to pH 7.4). UAE for 10 minutes at 60% amplitude. Centrifugation at 3900 rpm for 25 minutes at 8 °C. Diluted with 1% glycerol | Particle number recovery of 95%. Ti contamination from ultrasonic probe | 117 |
| 50, 100, 300 nm [31.3/37.1 nm] | Crab | 15 – Adapted from protocol #14 (ref. 117) 1 g homogenised tissue + 7.5 mL enzyme solution (8 g L−1 pancreatin and 8 g L−1 lipase in 0.2 mol L−1 NaH2PO4 and 0.2 mol L−1 NaOH adjusted to pH 7.4). Incubated at 37 °C for 12 hours. Centrifugation at 3900 rpm for 10 minutes at 8 °C. diluted with 1% glycerol | ‘Quantitative recovery’ | 107 |
| 30 nm | Bivalves | As protocol #2 (ref. 40) | 119 | |
| 30 nm | Bivalves | As protocol #9 (ref. 120) | 120 | |
| Au particles | ||||
| 10, 30, 60 nm [20 nm] | Chicken meat & rat liver | Chicken meat – as protocol #3 (ref. 23) | 42, 86 | |
| Rat liver – adapted from protocol #3 (ref. 23) 2 mL digestion buffer used and incubated at 55 °C for 16 hours | ||||
| CeO2 particles | ||||
| 13 nm [18 nm] | Mice lung and liver | 16 – Adapted from protocol #2 (ref. 40) – 25 mg unhomogenised lung or homogenised liver + 3 mL enzyme solution [3 mg mL−1 proteinase K, 5 mg mL−1 SDS, 0.2 mg mL−1 sodium azide in 0.05 mol L−1 sodium bicarbonate (pH 7.4–7.7)] and 1 mL HPW. Incubated overnight at 40 °C in water bath with continuous stirring | Low mass recovery of 6–40%, majority of particles being below the LODsize | 72 |
| 20 nm [30–40 nm] | Fish intestine, liver, gills, brain | As protocol #12 (ref. 71) | Particle number and mass recoveries of 91% and 98% respectively | 71 |
| 30–50 nm | Rat liver | 17 – Liver tissues lyophilised and ground to a powder. 1.95 mL enzyme solution (0.02% (w/v) proteinase K, 0.5% (w/v) SDS and 0.08% (w/v) EDTA in 0.01 mol L−1 Tris-HCl buffer adjusted to pH 7.4). Vortexed. Incubated overnight at 37 °C, while shaking in a water bath | 125 | |
| SiO2 particles | ||||
| 150–850 nm | Human liver, spleen & kidney | As protocol #13 (ref. 103) | 124 | |
| 100 nm–10 μm | Chicken meat & rat liver | Chicken meat – as protocol #3 (ref. 23) | 42, 86 | |
| Rat liver – adapted from protocol #3 (ref. 23) 2 mL digestion buffer used and incubated at 55 °C for 16 hours | ||||
| 13–45 nm [350 nm] | Rat liver | As protocol #11 (ref. 123) | LODsize determined from procedural blanks. Particles agglomerated – possibly as a consequence of sample preparation | 108 |
| Other particle compositions | ||||
| CuO – 50 nm [18 nm] | Molluscs | As protocol #7 (ref. 65) | 65 | |
| ZnO – 80 nm [23 nm] | ||||
| HgSe – natural | Whale liver & brain | 18 – 20 mg freeze-dried tissue defatted. Enzyme solution [1 mg mL−1 protease and 5 mg mL−1 SDS in 0.05 mol L−1 ammonium bicarbonate (pH 7.4)] added. Incubated overnight at 37 °C. Dissolved Hg and Se washed out using centrifugal filter with 50 kDa cut off | 64 | |
| Pb 40–750 nm [40–80 nm] | Game meat |
19 – Tissue mixed 1:1 with water, homogenised, 200 mg aliquot taken. 2 mL enzyme solution added [3 mg mL−1 proteinase K, 5 mg mL−1 SDS, 0.2 mg mL−1 sodium azide in 0.05 mol L−1 sodium bicarbonate (pH 7.4–7.7)] and 0.3 mL water added. Incubated at 37 °C for 1 hour with stirring. Samples iced to stop enzyme activity. Diluted with HPW |
Rinsing the sample introduction system between with a surfactant containing acid between samples analyses was found to reduce carryover. Storage for 3 days at 4 °C resulted in dissolution of particles. Longer incubation times of 16 hours resulted in particle dissolution | 66 |
| Se 50, 100 nm [18 nm] | Se rich yeast | 20 – 200 mg yeast suspended in 5 mL water. UAE for 1 hour followed by centrifugation at 4500g for 10 minutes. Pellet resuspended in 5 mL enzyme solution (4% driselase in 0.03 mol L−1 Tris at pH 7.5). Incubated at 25 °C for 17 hours then centrifugation as above. Pellet resuspended in 5 mL second enzyme solution (4 mg mL−1 protease in 0.03 mol L−1 Tris at pH 7.5). Incubated at 37 °C for 17 hours then centrifugation as above. Pellet resuspended in 5 mL 4% SDS, UAE (bath) for 1 hour, centrifugation as above. Supernatant analysed | 126 | |
| Alkaline extractions protocols | ||||
| Ag particles | ||||
| 60, 100 nm [15–20 nm] | Ground beef, freshwater crustacean, earthworms | 21 – 0.5 g tissue [or 2 mg in the case of freshwater crustacean (Daphnia magna)] in 10 mL 20% TMAH. UAE (bath) for 1 hour. Incubated at room temperature for 24 hours. Diluted to ≤1% TMAH prior to analysis | Particle number and mass recoveries in the range of 83–106% and 104–121% respectively | 67 |
| 18–20 nm [13 nm] | Rat lungs, liver, spleen, embryo & placenta |
22 – 20% TMAH added to samples, 1:20 w/v ratio. UAE (probe) for 5 minutes, pulse mode (6 s/2 s). Mechanically shaken and incubated overnight at room temperature. Diluted with HPW |
Number based and mass-based recovery of 87% and 102% respectively | 127 |
| 50 nm [20 nm] | Earthworms |
23 – 0.4 g homogenised powdered tissue + 20% TMAH (1:20 w/v). UAE (bath) for 30 minutes. Incubated overnight at room temperature. UAE for 30 minutes. Diluted with HPW |
Particle mass recovery of 92%. Particles detected in samples exposed to ionic Ag | 34 |
| 30 & 80 nm | Fish liver, intestine & gill | 24 – Tissue + 1 mL 25% TMAH. UAE (probe) for 5 minutes. Filtered with a 0.45 μm cellulose acetate filter | Au ENMs used to assess recovery, see below | 53* |
| 60 nm [11 nm] | Shrimp, mussel, clam, snail, fish | 25 – 0.1 g homogenised, freeze dried sample + 5 mL 10% TMAH. Vortexed for 30 s. Agitated at room temperature for 2 hours. Overnight settling. Supernatant diluted with HPW | Particle mass and number recoveries in the range of 93–102% and 73–127% respectively | 70 |
| 50 nm [16 nm] | Fish liver | 26 – 50 mg dry liver + 0.4 mL 0.025 mol L−1 CaCl2 + 1.6 mL 25% TMAH. Vortexed for 60 s. Incubated in the dark at room temperature for 24 hours | Particle mass and number recovery was 100 ± 5%. Proteinase K digestion approach not as effective, potentially due to high lipid content of matrix | 52* |
| 30, 70, 100 nm | Mouse liver, heart, lung, spleen & kidney |
27 – Tissue homogenised in phosphate buffered saline at 1:10 w/v ratio. Treated at 1:1 ratio with 0.1 mol L−1 NaOH or 25% TMAH. Incubated at 37 °C for 3 hours |
Particle dissolution observed for acid and enzyme methods. Aggregation observed in presence of TMAH. Alkaline method offered best recovery | 51* |
| 100 nm | Mouse skin | As protocol #27 (ref. 51) | 128 | |
| TiO2 particles | ||||
| 60 nm [27 nm] | Shrimp, mussel, clam, snail, fish | As protocol #25 (ref. 70) | Particle mass and number recoveries in the range of 89–94% and 6–23% respectively | 70 |
| Au particles | ||||
| 100 nm [15–20 nm] | Ground beef freshwater crustacean, earthworms | As protocol #21 (ref. 67) | Particle number and mass recoveries in the range of 90–95% and 88–95% respectively | 67 |
| 60 nm [44 nm] | Rat spleen | 28 – Spleen homogenised. UAE for 1 hour. 0.2 mL homogenate aliquot, TMAH added to a concentration of 5%. Bovine serum albumin (BSA) solution added at 300 BSA molecules per Au ENM (calculated from mass and assuming 60 nm diameter). UAE for 1 hour then mechanically rotate overnight | Recovery using alkaline extraction was 4× higher compared to enzyme protocol | 43* |
| 30, 60 nm | Soil nematodes | 29 – 0.1 mg lyophilised nematodes + 1 mL 7% TMAH. Vortexed, 30 s. Incubated, RT, 2 hours. Diluted with HPW | 48 | |
| 30, 80 nm | Fish liver, intestine & gill | As protocol #24 (ref. 53) | Particle mass recovery using alkaline method was 102% compared to 74% for enzymatic method | 53* |
| 23 nm [18 nm] | Human breast cancer cells | 30 – 1 mL 5% TMAH with 10 μL 1.5 mg mL−1 BSA added, incubated overnight at room temperature. Diluted five-fold | Higher TMAH concentrations resulted in loss of ENMs, likely by dissolution or aggregation | 49 |
| 50 nm | Clams & oysters | 31 – 0.1 g tissue + 2 mL 20% TMAH. Vortexed. UAE for 1 hour at 37 °C. Incubated at room temperature for 24 hours whilst shaking. Filtered using a 0.45 μm cellulose acetate filter. Diluted to ≤1% TMAH with 0.1% Triton X-100 | Alkaline method was determined to be faster and more effective than enzyme method. 96% and 104% particle mass recovery when using the alkaline and enzyme approach respectively | 50* |
| Other particle compositions | ||||
| Y, La, Ce, Pr, Gd, Nd – natural | Clams & oysters | As protocol #31 (ref. 50) | 50* | |
| Ag2S 20 nm | Earthworms | As protocol #23 (ref. 34) | 34 | |
| Other extraction protocols | ||||
| Au 40 nm [18 nm] | Rat liver |
32 – Livers snap frozen and homogenised in a mortar cooled by liquid nitrogen. Samples suspended in lyse buffer (0.15 mol L−1 NaCl, 0.001 mol L−1 EDTA, 0.02 mol L−1 Tris-HCl (pH 7.5), 1% Triton X-100). UAE in an ice bath for 15 minutes. Centrifugation for 15 minutes at 13000g and 4 °C. Diluted 20× in a 10% methanol mix |
Dilution in methanol resulted in higher sensitivity. 48.3% transport efficiency from total consumption nebuliser | 56 |
| ZnO [95 nm] | Chicken meat | 33 – 1 g chicken breast cut into small pieces + 5 mL Tris-HCl at neutral pH. UAE (probe) for 2 minutes at 40% amplitude. Diluted with HPW | LODsize was 26 nm, this increased to 95 nm in the presence of sample matrix. ‘60% extraction efficiency’ | 57* |
| Composition, size & [LODsize] | Matrix | Extraction protocol | Comments | Ref. |
|---|---|---|---|---|
| References denoted with ‘*’ compare multiple extraction methods with the favoured method reported in the table. | ||||
| Enzyme extraction protocols | ||||
| Ag particles | ||||
| 15 nm | Wheat, rape seed & barley | As protocol #38 (ref. 129) | Particle mass recovery of 12%. Increase in observed particle sizes suggests aggregation | 73 |
| 10 nm | Cress | 34 – Tissue homogenised in 0.002 mol L−1 citrate buffer (pH 3.5–7). 5% macerozyme R-10 added. Incubated at 37 °C for 24 hours | 102 | |
| 35 nm | Cress roots & shoots | As protocol #41 (ref. 130) | Isotopically labelled particles used | 131 |
| 17 nm [14 nm] | Soybean & rice plant leaves |
35 – 0.1 g leaf tissue cut to small pieces. 8 mL 0.002 mol L−1 citrate buffer (pH 6) added. Macerozyme R-10 enzyme powder added at tissue:enzyme powder ratio (w/w) of 1:3. Incubated at 37 °C for 36 hours |
Particle number recovery of 93–102%. A TMAH approach achieved a particle number recovery of 53–58% and resulted in a shift to lower particle sizes | 55* |
| 15 nm [14 nm] | Wheat | As protocol #35 (ref. 55) | Ag particles not seen in exposure with ionic Ag | 132 |
| 15 nm [14 nm] | Wheat | 36 – Tissues digested in macerozyme R-10 buffer at 37 °C for 36 hours. Settling for 1 hour. Supernatant diluted | Spiked recovery test showed no particle dissolution or aggregation | 133 |
| 60, 75, 100 nm | Lettuce | 37 – Cut to small pieces with scissors. Homogenised in 8 mL 0.002 mol L−1 citrate buffer (pH range 3.5–7.0). 2 mL 50 mg mL−1 macerozyme R-10 solution added. Shaken in incubator at 37 °C for 24 hours. Settled for 1 hour, gravity filtered, 0.45 μm cellulose acetate. Diluted to 20 mL with HPW | 134 | |
| Au particles | ||||
| 40–100 nm [20 nm] | Tomato plants & shoots | 38 – Tissue homogenised in 8 mL 0.002 mol L−1 citrate buffer (pH 3.5–7). 2 mL 50 mg mL−1 macerozyme R-10 solution added. Samples shaken, 37 °C, 24 hours. Settled for 1 hour, 0.1 mL supernatant removed and diluted with HPW | Particle number recovery of 88%. No dissolution or aggregation observed | 129 |
| 13 nm [12 nm] | Wheat | As protocol #35 (ref. 55) | 132 | |
| CeO2 particles | ||||
| 29 nm | Wheat, rape seed & barley | As protocol #38 (ref. 129) | Particle mass recovery of 70%. Increase in observed particle sizes suggests aggregation | 73 |
| 30–50, 50–100 nm [23–25 nm] | Tomato, soybean, pumpkin, cucumber | 39 – Tissues homogenised in 9 mL 0.02 mol L−1 2-(N-morpholino)ethanesulfonic acid (MES) buffer (pH 5). 1 mL 30 mg mL−1 macerozyme R-10 solution added. Shaken at 37 °C for 24 hours. Settled for 30 minutes. 0.1 mL aliquot taken, diluted 100× in 0.020 mol L−1 MES buffer. Passed through 5 kDa filter | Sample preparation method does not impact size distribution. Approximately 90% recovery | 135 |
| 30–50 nm | Radish | As protocol #41 (ref. 130) | Enzyme digestion and presence of plant material shown to have no impact on particles | 136 |
| Cu & CuO particles | ||||
| Cu 79 nm | Cress roots & shoots | As protocol #41 (ref. 130) | 131 | |
| CuO 20–100 nm | Lettuce, kale cabbage | 40 – Circular pieces of leaf cut (6.35 mm Ø), 2 mL 50 mg mL−1 macerozyme R-10 added. Shaken at 25 °C for 24 hours | 137 | |
| Zn & ZnO particles | ||||
| Zn 69 nm | Cress roots & shoots | As protocol #41 (ref. 130) | 131 | |
| ZnO 80–200 nm | Lettuce | ‘Macerozyme R-10 was used’ | No particles found in plant tissues due to rapid dissolution of ZnO | 138 |
| Other particle compositions | ||||
| Ag2S [20–25 nm] | Cucumber & wheat | As protocol #38 (ref. 129) | 139 | |
| Pt 70 nm | Cress & white mustard | 41 – 25 mg plant tissue homogenised in 8 mL 0.002 mol L−1 citrate buffer (pH 4.5) with UAE (probe) for 5 minutes. 2 mL macerozyme R-10 enzyme solution added [5 mg mL−1 (roots) and 25 mg mL−1 (shoots)]. Shaken at 37 °C for 24 hours in a water bath. Filtered with a 0.45 μm filter | Enzyme only extraction did not impact particle size distribution. Particle number recoveries of 87% and 98% for roots and shoots respectively | 130 |
| Pd 77 nm [25–30 nm] | White mustard | As protocol #41 (ref. 130) | Enzyme treated particle size distribution comparable to pristine particles | 104 |
| TiO2 30, 100 nm [20 nm] | Radish | 42 – Leaves and stems combined; roots processed separately (approx. 20 mg). 7 mL 0.002 mol L−1 citrate buffer added (pH 4.5) and homogenised. 1.5 mL enzyme solution (0.01 g (roots) or 0.04 g (leaves and stems) macerozyme R-10 powder in 1.5 mL HPW added). Incubated at 37 °C for 24 hours with shaking. Samples left to stand for 1 hour | Ultrasonication probe was not used as it was a source of TiO2 particles | 140 |
| Other extraction protocols | ||||
| TiO2 19–37 nm [47 nm] | Rice plant roots & leaves |
43 – Tissues + 12 mL 3:1 HNO3:HCl. Microwave digested (ramp to 180 °C in 5.5 minutes, hold for 9.5 minutes) diluted with HPW |
Approximately 6× higher particle mass recovery with acid relative to enzyme extraction. Broader size distribution in acid extraction deemed to be from more efficient extraction or changes due to acid treatment | 24* |
| CuO 38 nm [20 nm] | Lettuce | 44 – Tissue homogenised in 0.010 mol L−1 buffer solution (pH 8, KOH adjusted). UAE for 3 minutes. 0.5 mL aliquot taken, 3.75 mL 50% MeOH added, agitated for 1 hour. 1.25 mL 1% TWEEN 20 added. UAE for 3 minutes | Methanol based approach developed to minimise dissolution during extraction | 141 |
| CuOH2 [55 nm] | ||||
| Au 50 nm, CuO 37 nm, ZnO 80–200 nm | Lettuce, corn, kale | 45 – 1 g leaf tissue homogenised in 20 mL 10 mM CAPSO buffer solution (pH 9, KOH adjusted). UAE for 3 minutes. 0.5 mL aliquot taken, 3.75 mL 50% MeOH added and agitated for 1 hour. 1.25 mL 1% TWEEN 20 added. UAE for 3 minutes | 44 | |
| Composition, size & [LODsize] | Matrix | Extraction protocol | Comments | Ref. |
|---|---|---|---|---|
| Dilution preparation protocols | ||||
| Ag | ||||
| 12, 18 nm [20 nm] | Rat whole blood |
46 – Diluted 40000× with HPW water |
41 | |
| 60 nm | Gastric fluids | 47 – Simulated saliva, gastric juice, duodenal juice and bile juice, with and without proteins diluted with HPW | 142 | |
| 100 nm × 500 nm | Crustacean hemolymph |
48 – Carapace punctured with needle, contents extracted. 5 extracts combined. Diluted 100–10000× with HPW. UAE in a water bath for 10 minutes |
143 | |
| 40 nm | Gastric fluids | As protocol #47 (ref. 142) | 144 | |
| 40, 60 nm | Whole blood | 49 – Diluted 20× with TMAH and 0.1% Triton X | 145 | |
| 40, 80 nm | Whole blood & urine | 50 – Blood – diluted 20× with 0.5% NH4OH & 0.1% Triton-X | 146 | |
| 51 – Urine – diluted 20× with 0.5% HNO3 | ||||
| 20, 40, 60, 100 nm [16 nm] | Plasma & whole blood | 52 – Diluted 20× with 0.1% TMAH and 0.1% Triton X and 2.8% NH4OH | 147 | |
| 30, 50, 100 nm [10–30 nm] | Whole blood | 53 – 0.75 mL whole blood, 0.15 mL 10% Triton-X, 1.5 mL 25% TMAH diluted to a final volume of 15 mL with HPW | 148 | |
| 40, 60 nm | Urine |
54 – UAE for 5 minutes. Diluted 1:10 with 1% glycerol. UAE for 5 minutes |
149 | |
| 50 nm | Gastric fluids | ‘Suspensions analysed’ | 150 | |
| 20, 50, 100 nm [19–21 nm] | Artificial sweat |
55 – Diluted 10000× with HPW |
151 | |
| 20, 60, 100 nm [10–14 nm] | Urine, serum, whole blood | 56 – Urine & serum – diluted 10× with HPW | Particle number recovery of 82–105% | 152 |
|
57 – Whole blood – 25% TMAH solution added to blood (5:1 v/v TMAH:blood). UAE for 1 hour in a cooled water bath. Incubated at RT for 24 hours. Diluted with 0.1% Triton-X |
||||
| 10–100 nm [20 nm] | Gastric fluid | 58 – Diluted with HPW water to approximately 105 particles per mL | 153 | |
| TiO2 | ||||
| 50, 100 nm [8–15 nm] | Urine |
59 – UAE for 5 minutes. Diluted in a 1:10 ratio with 1% glycerol. UAE for 5 minutes |
149 | |
| 30, 70, 100, 115 nm | Urine & whole blood | 60 – 300 μL biofluid diluted to 10 mL with HPW | 154 | |
| 71 nm [44–50 nm] | Urine | 61 – 125 μL diluted to 10 mL with 0.1% HNO3 | Particle mass recoveries of 67–84% achieved | 155 |
| Au | ||||
| 10, 60 nm | Whole blood | As protocol #49 (ref. 145) | 145 | |
| 30, 60 nm | Whole blood & urine | As protocol #50 & 51 (ref. 146) | 146 | |
| 45 nm | Rat whole blood | Au ENMs dosed directly to fresh whole blood, diluted 50-fold with HPW | 156 | |
| 30, 50, 100 nm [10–30 nm] | Whole blood | As protocol #53 (ref. 148) | 148 | |
| 5, 20, 40, 60 nm [7–11 nm] | Urine, serum, whole blood | As protocol #56 & 57 (ref. 152) | Particle number recovery of 76–122% | 152 |
| 10–80 nm [15 nm] | Gastric fluid | As protocol #58 (ref. 153) | 153 | |
| 50 nm [19 nm] | Whole blood | 63 – 20 μL diluted to appropriate concentrations with HPW | 157 | |
| Other ENMs | ||||
| Fe3O4 27–30 nm | Whole blood | 64 – Diluted with de ionised water | 158 | |
| Cr, Co | Hip aspirate | As protocol #2 (ref. 40) | 63 | |
| CeO2 30–50 nm CuO 25–55 nm | Gastric fluids | ‘Suspensions analysed’ | 159 | |
| CeO2 30–50 nm [25 nm] | Gastric fluids | As protocol #58 (ref. 153) | 153 | |
| ZnO 80–200 nm [35 nm] | ||||
| CeO2 30–50 nm | Urine & Plasma |
65 – Urine – filtered, 0.45 μm nylon syringe filter. Diluted 500–25000× with HPW |
125 | |
|
66 – Plasma – diluted 500–25000× with HPW |
||||
The specific objectives were to: (i) identify key aspects of sample collection, storage and shelf life so that a sample may remain suitable for spICP-MS; (ii) critically evaluate the available extraction methods for preparing a biological sample for spICP-MS, and determine the prospect of moving towards a standardised method that has utility for a variety of ENMs and tissue matrices; (iii) outline the requirements for quality assurance including the use of spike recovery tests and other approaches in the current absence of certified reference materials for tissues; and (iv) provide brief guidance on instrument set up and key aspects of spICP-MS that are relevant for tissue samples.
2. Sample collection, storage and shelf life
The key purpose of any tissue, or whole organism, sample collection is to ensure that: (i) a representative sample is taken; (ii) that the sample preservation method and storage occurs without post-mortem change, so the specimen is preserved as close to the in vivo condition as possible and without deterioration that might invalidate any subsequent chemical analysis; and ideally, (iii) that the sample is preserved in a way that enables a variety of chemical analyses or observations so that the sample can be used for several purposes within an experiment (i.e., the stored sample has utility). These principles have been applied in eco/toxicology, clinical and forensic studies for many years;25,26 and are also clearly important to the determination of ENMs in biological samples.Many aspects of sample collection are not nano-specific, for example, the type of sample will depend on the size of the organism. The whole body may be collected and analysed as one sample for a whole body burden determination, or in the case of small invertebrates the whole body samples may be pooled to obtain enough biomass, or when the organism is large enough to dissect, individual organs or tissues can be collected (e.g., fish, rodents, plants). For a typical total trace metal analysis in a tissue sample, a biomass of at least 30–100 mg wet weight (e.g., invertebrates, zebrafish embryos) is needed, and more usually it is convenient to collect 0.5–2 g of tissue for acid digestion methods (e.g., organs from fish/mammals, plant tissue). The minimum biomass required for a reliable spICP-MS measurement is not yet agreed, but with extra difficulties regarding procedural recoveries and the efficiency of detection for ENMs, a similar amount, or slightly more tissue would be prudent to collect for spICP-MS work. Ideally, appropriate sample sizes should be taken in order to obtain a representative sample. Here the concerns are the same as for a traditional metal analysis, and regardless of how the tissue is subsequently digested, homogenised, or otherwise extracted to make a liquid sample. The issues of technical replicates (within tissue sample variation) and between organism or sample variation (variation within organisms within a treatment), and variation between experimental treatments, also apply to ENM exposures. So far, the evidence suggests these sources of variation are not larger than those usually encountered in total metal determination from ENM exposures (e.g., TiO2 ENMs in trout, Shaw, et al.20), or for spICP-MS (e.g., Ag ENMs in gut sacs, Clark, et al.27).
It is also important that the biological sample is not contaminated or compromised by excess ENMs from the external media, especially where the purpose is to determine the internalised metal, or true fraction of bioaccumulated ENMs. The separation of very small organisms such as microalgae or plankton from suspension of ENMs is reviewed elsewhere (Petersen, et al.28), with suggestions of using sucrose gradients and similar centrifugation approaches to separate such organism from the ENMs in the external media. For whole organisms, tissues and organs, rinsing procedures are important to remove excess material and/or determine any surface-adsorbed fraction. For traditional dissolved metals, rinsing the tissue or organ with copious amounts of ultrapure water or saline as appropriate, followed by an ethylenediaminetetraacetic acid (EDTA) wash is usually sufficient to remove the external fraction of excess or loosely associated total metal. However, the extra concern for ENMs is their propensity to agglomerate and settle onto the surface of the tissue, and the potentially strong mechanisms by which nanoscale particles ‘stick’ to the tissue (steric hindrance with secreted mucus, aggregation in the ionic strength at the tissue surface, electrostatic attraction, etc., see Handy, et al.29 for fish gills). For mucous epithelia such as the gut, the surface-associated fraction of ENMs can be substantial (e.g., a third of the total metal, TiO2, Al-Jubory and Handy30). However, the surface-associated fraction will also depend on the exposure dose and type of material. For instance, double washing in saline is effective in removing 95% of surface bound Ag ENMs from the gut of trout.27 So far, for fish tissue and the gut of rodents at least, the experience is that ultrapure water or saline rinses of tissue samples, followed by an EDTA wash seems to be effective for removing most (90% or more) of the apparent adsorbed total metal from the surface of the tissue. This type of washing works for exposures to metal-containing ENMs, as well as for dissolved metals.27,30,31
Another concern for whole body burden determination is whether it takes longer to purge organisms of their gut contents. For regulatory tests on bioaccumulation potential there is the option to remove the gut entirely, so the whole-body burden is just on the remaining carcass, or to analyse the gut separately (e.g., OECD TG 305 using fish). However, for the body burden determination of small invertebrates (e.g., Daphnia, earthworms), it is customary to purge the gut contents by placing the animals in clean water/media (e.g., for 24–48 h) prior to total metal analysis, and such approaches also seem to work for total metals from ENMs (e.g., CuO in earthworms, Tatsi, et al.32). However, the transit of particulate material in the gut lumen is not the same as dissolved metals and also depends on particle size.33 So, any purging of gut contents may show a time-dependent influence on any particle sizes remaining in the lumen; with a bias of smaller particle sizes tending to remain. Regardless, the overall aim is often just to make this external background of ENMs low enough to measure the true body burden, so that aspects such as uptake kinetics can be explored.34 Another source of ‘external contamination’ includes the implements used for dissection, and the usual procedures of acid-washing implements and the laboratory wares, being careful to use different implements for control animals or to dissect controls first, are also sufficient for experiments with ENMs.
Ideally a biological sample would be prepared and analysed immediately after it is collected. However, this is often not practical or feasible, and samples will require some form of storage. Appropriate storage needs to ensure the ENMs will not degrade through dissolution, or irreversibly aggregate; both of which will result in changes to particle sizes, size distributions and number concentrations; and potentially cause misleading data when samples are analysed. From the viewpoint of preserving a specimen as quickly and closely as possible to the in vivo state, snap freezing in liquid nitrogen and storage at −80 °C has been routinely used for biochemistry, trace metal analysis, and aspects of histopathology with great success. However, the effect of storage conditions on ENMs in biological matrices has not been thoroughly investigated. Loeschner, et al.35 demonstrated that Ag ENMs in chicken meat could not be recovered after long-term (10 months) storage at −80 °C, likely as a consequence of dissolution, chemical transformation and agglomeration/aggregation. Repeated defrosting of frozen samples is always best avoided, and this can readily be achieved by aliquoting the tissue into sub-samples prior to freezing. Freeze-drying is another way of preparing biological samples for storage and indeed this process is often used in the manufacture of ENMs,27,36 but its potential to alter ENMs when freeze dried in a biological sample has also not been properly investigated. Samples stored by freezing can simply be thawed immediately prior to sample preparation and analysis. For total dissolved metals, the tissue digestion and extraction methods are well known (e.g., Subramanian37). However, alternative preparation methods typically need to be applied for samples to be analysed by spICP-MS where the metal-containing particles must be preserved and extracted into a suspension appropriate for analysis, techniques that have been applied in the literature to date are in section 3.
Storage duration is an important aspect for ENMs preservation within a biological sample. The shelf life of a biological sample will typically depend on the physiological state of the organism when the sample was collected (e.g., respiratory distress/systemic hypoxia during euthanasia leading to oxidative damage to the tissue), how quickly the sample was collected and preserved, the media used for long-term storage and the storage conditions (temperature, light, humidity, etc.). The choice of container (e.g. plastic versus glass) used for storage has been identified as an important consideration for ENM recovery both in terms of mass and particle size distribution.22 For example, Shaw, et al.20 found that storage in plastic containers resulted in nearly a two-fold better mass recovery for TiO2 compared to glassware.
Despite the importance of sample storage, aspects of storage condition have not been systematically investigated for a range of ENMs and biological matrix types, therefore technical guidance on storing legal samples for forensics or regulatory purposes, or similar activities is some way off. Clinical samples may be required to be stored for weeks to years, depending on the type of study. In the case of standards or certified materials, a shelf life of months or years is desirable, and such long-term studies with tissue samples containing ENMs are yet to be reported. Given the potential for ENMs to undergo transformations during storage it would be prudent to include spiked controls with stored samples which may be used for quality assurance, these considerations are discussed in section 4.
3. Extractions from biological samples
In order to analyse a biological sample by spICP-MS it is necessary to present the sample to the instrument as a suspension of the ENMs. Therefore, a sample preparation method is required that can disintegrate, digest or dissolve the biological components (i.e., the proteins, fats and carbohydrates that make plant or animal tissue) into a liquid phase, but at the same time preserving the integrity of ENMs of interest. It would be advantageous if one unified method could be developed that could extract ENMs from a broad range of different types of tissue samples, ranging from the soft parenchyma in internal organs such as the spleen or liver, to very fatty tissues such as endocrine organs, through to the hard carapaces of some invertebrate species. However, in practice, so far, methods have been developed on a tissue-by-tissue basis, and for a particular type of ENM (Fig. 3, Tables 1–3).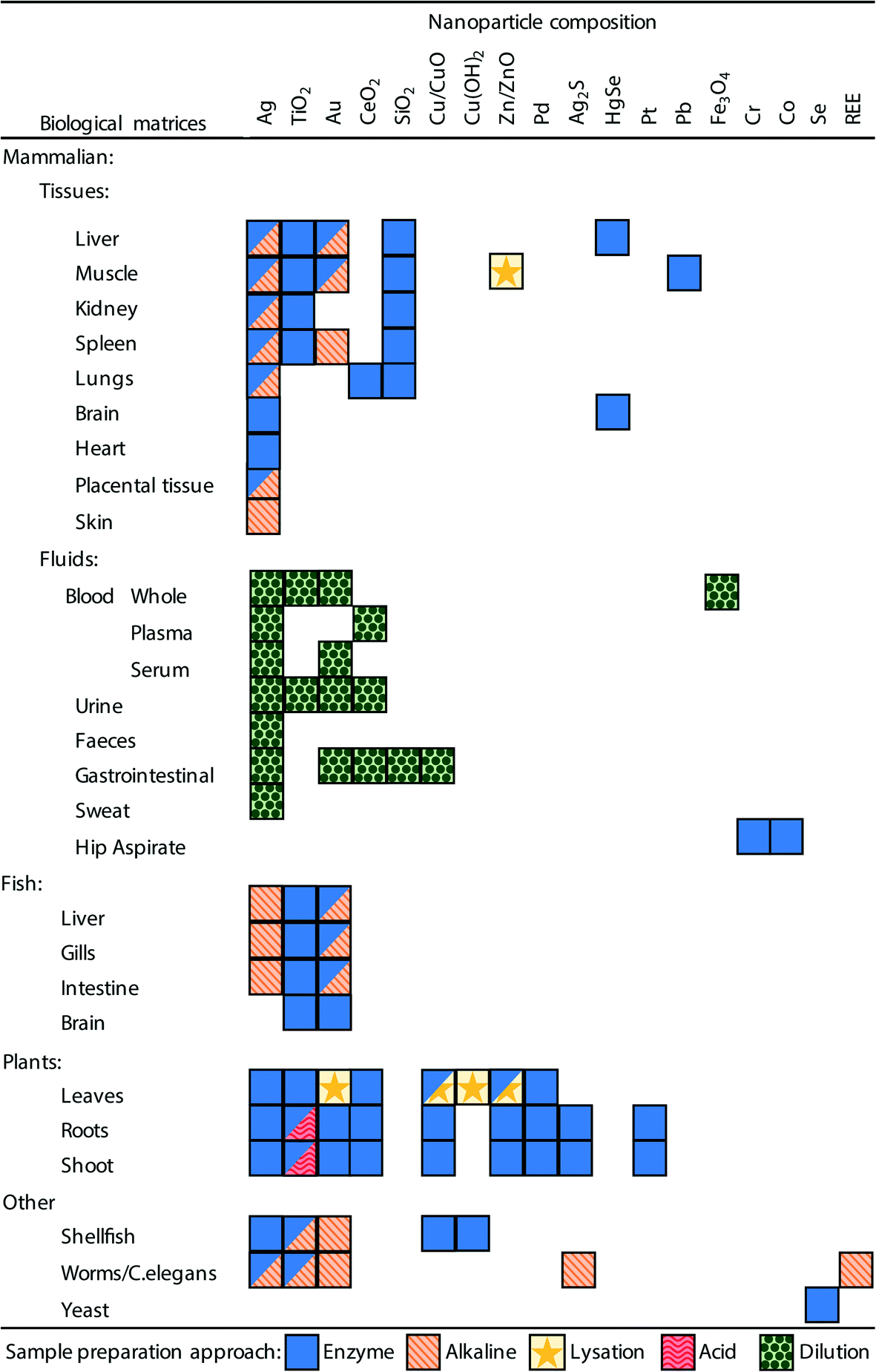 | ||
| Fig. 3 A summary of the sample preparation approaches used in the literature and the biological matrices and ENM combinations to which they have been applied. Boxes with multiple symbols indicate combinations where different approaches have been used in different studies, full details can be seen in Tables 1–3. | ||
It is also critical that the preparation method does not create conditions whereby particle aggregation/agglomeration occurs, leading to the loss of the analyte from the dispersion, or indeed promote any chemical reactions that might cause particle formation leading to false positives. The properties and reactivity of the ENM may also limit the utility of any extraction method. For example, a strong acid digest may be considered appropriate for acid-resistant ENMs, such as TiO2 particles,20,24 but would be inappropriate for a material that readily dissolves in acid such as CuO ENMs. So far, attempts at extractions from biological matrices include the organs from mammals and some fish, plant tissues, and limited work on invertebrates; as well as studies on (mammalian) blood, urine and faeces. Fig. 3 and Tables 1–3 demonstrate the range of biological matrices and ENM combinations that have been studied, and the extraction approaches that have been applied to those situations. It can be seen that, although quite a large range of ENM compositions have been investigated, by far the most commonly used ENM composition is Ag, followed by TiO2 and Au. For the other types of ENM compositions, only a very limited number of matrices have been investigated. Similarly, there is lack of studies that have looked at certain biological matrices such as skin, reproductive organs, bone, etc.
Inevitably, all of these methods have been intended for metal-containing ENMs that might be detected by spICP-MS. For the internal organs of animals and plant tissue, the most prevalent approaches make use of enzymes or alkaline-based extraction methods (Fig. 3, Tables 1 and 2). For biological fluids simple dilution is typically applied (Fig. 3, Table 3), and depending on the matrix (e.g., whole blood, plasma or urine) the dilution has been performed with ultrapure water, acid or alkali media. Clearly, all the different facets of a desirable extraction method for biological samples presents a technical challenge and a number of different approaches have been explored including enzymatic digestions, the use of acids or alkali, solvent extraction, physical break down and lysation (Tables 1 and 2). The main approaches are discussed below.
3.1 Enzyme-based extraction protocols
Enzyme-based extraction protocols have been the most widely applied approach for the preparation of particle suspensions from animal and plant tissues (Fig. 3). These methods are attractive because they can break down organs or tissues to a liquid form without transforming/altering the ENMs, at least in the case of solid metal particles that do not have organic coatings. Typically, for the enzymatic digestion of a tissue, the appropriate enzyme should be selected (e.g., proteases from muscle, lipases for fatty tissue, etc.) and presented at the appropriate specific activity in conditions that promote the enzyme activity (e.g., optimum pH, presence of Mg and ATP, substrate not limiting, etc.). Two enzymatic methods of note are emerging from the literature. Of the 31 publications identified in Table 1 where enzymes have been used to break down animal organs or specific tissues, 27 of them made use of proteinase K, while the remaining 4 used pancreatin (a commercial cocktail of various digestive enzymes derived from the pancreas) mixed with a lipase to break down fat. In the case of plant tissues, macerozyme R-10 (a mixture of pectinase, cellulase and hemicellulase) was applied in all cases (Table 2).Proteinase K is active in a pH range of 7.5–12.0 (ref. 38) and therefore has utility for a wide variety of tissues. It has an optimum specific activity at 37 °C, but will work at higher temperatures and is also a Ca-dependent enzyme.39 Typically for the digestion of tissues exposed to ENMs, activation with a reaction mixture containing 1–5 mmol L−1 Ca2+ has been used; and often in conjunction with 1% Triton X-100 and/or 0.5% sodium dodecyl sulphate (SDS) to help disperse organic matter of the tissue homogenate (Table 1). Loeschner, et al.40 found that the addition of Ca2+ made no difference to the digestion efficiency, likely because sufficient intracellular free Ca2+ was already present in the chicken meat used, as only <0.1 μmoles of free Ca2+ ions are needed to activate proteinase K.39 For enzyme digests of animal tissues to enable subsequent determination of ENMs, up to 1 g of wet tissue is generally used (Table 1). Considering the examples for ENMs (Table 1), a typical digestion protocol using proteinase K will involve: (i) physical break down of the tissue sample using an homogenizer or ultrasonication in a buffer solution, (ii) addition of the enzyme solution typically resulting in a final proteinase K concentration of 0.4–8 mg mL−1 resulting in between 0.6 and 360 μg enzyme per mg tissue being applied, (iii) incubation periods ranging between 40 minutes to 18 hours at 35–55 °C with the sample undergoing agitation, (iv) filtration or centrifugation is sometimes used to ‘clean’ the undigested debris from the tissue homogenate, and (v), the resulting ‘supernatant’ is usually sonicated and diluted to an appropriate particle number concentration prior to analysis by spICP-MS. When detergents are used in the initial tissue homogenisation, it would be important to use sufficient dilution immediately prior to analysis by spICP-MS to ensure any residual detergent is very low (e.g., <0.1%) to minimise the risk of it altering the behaviour of the particles or the instrument (e.g., detergent displacing debris from the peristaltic tubing). Of particular note is the fact that, of the studies where enzyme digestion is applied, the mass concentration of enzyme used is nearly always reported. However, the more relevant metric to be reported is the enzyme activity, which is reported in only a small number of studies.41–43 This is likely due to the researchers involved in the studies being more familiar with reporting units as mass per volume units. Enzyme activity is a measure of the moles of substrate converted per unit time and usually reported as enzyme units (U) where U = 1 μmol min−1 and therefore is an indication of the enzyme efficacy at breaking down specific tissues.
In the case of plant tissues, enzymatic digestion protocols have focused on the use of macerozyme R-10 (Table 2) which has an operating pH range of 3.5–7.0. A low pH media (e.g., pH 5.5 and lower) should be used with caution as the acidity causes dissolution of ENMs such as Ag, ZnO and CuO.44 Wojcieszek, et al.45 provide some useful guidance on best practices for studies investigating metal ENM uptake in plants, including on how to prepare and perform spICP-MS. A typical protocol (Table 2) with this enzyme mixture for the extraction of ENMs will involve: (i) homogenization of leaf, shoot or root tissue in a buffer solution (pH 3.5–7), (ii) addition of macerozyme R-10 enzyme solution resulting in a final volume and enzyme concentration of 1–50 mL and 1–50 mg mL−1 respectively, and (iii) incubation for 24–36 hours at 37 °C. As above, the enzyme solutions used were reported as mass concentrations of powder used as opposed to enzyme activity. In addition, the mass of plant tissues in the digests usually went unreported, making it unclear how much enzyme was being used per unit mass of tissue.
Concerns about interferences from ENMs, or toxic metal ions derived from ENMs, with the enzyme activity are not specifically investigated in the studies reviewed here. It has been demonstrated that the application of Ag ENMs to soils at high doses can reduce soil enzyme activity.46 In those studies where control tissues or organs were included there were no reports of decreased digestion efficacy relative to those dosed with ENMs, suggesting there was no or little impact on enzyme activity. The binding of CuO ENMs to the tertiary structure of proteinase K has been shown to lead to increased enzyme activity,47 but whether or not such binding would lead to poorer recoveries is not clear. It has also been suggested that the presence of enzymes may contribute to the accelerated dissolution of ENMs.44 Also of concern is the potential for transformation or dissolution of material as a consequence of long incubation times, and the loss of ENMs during any filtration or centrifugation steps used to clean the tissue homogenates of debris.
3.2 Alkaline extraction methods
Alkaline extractions, mainly using tetramethylammonium hydroxide (TMAH) have been widely applied to the preparation of animal tissues for spICP-MS (Table 1). A typical protocol involves the addition of TMAH to give a final concentration of 10–20% (w/v) and at a volume of 20–50 μL per mg biological tissue resulting in a final volume in the range of 0.25–10 mL. The exception to this is where the samples consist of only low amounts of biomass (e.g. nematodes and cell cultures) so a larger volume of TMAH per mg biomass was used.48,49 Digests are then often agitated or homogenised using physical or sonication methods and allowed to incubate for as little as 2 hours,48 but more typically for 16–25 hours and usually at room temperature. Before analysis, samples are diluted with ultrapure water so that the final TMAH concentration is 1%. Any further dilution of the digest is typically performed with 1% TMAH or ultrapure water. A strong alkaline based protocol was identified as being favourable over a proteinase K based extraction protocol in instances where the organs of interest had a high lipid content as proteinase K will not break down the fatty tissues.50–52 A number of studies raised concerns about the agglomeration of particles in the presence of TMAH which could potentially result in the loss of particles and change the size distribution of particles in the sample.49,513.3 Enzyme versus alkaline
Of the studies summarised in Tables 1 and 2 there are seven publications that compare alkaline- and enzyme-based approaches, of these five concluded that an alkaline-based extraction approach was favourable over the enzyme method studied. Ishizaka, et al.51 compared five extraction approaches (2 acid, 1 enzyme, and 2 alkaline extractions) for mouse livers containing Ag ENMs. The study found that particle dissolution by the acid and enzyme-based approaches made them impractical, but the two alkaline approaches, which used either TMAH or NaOH solutions, enabled particle size distributions to be determined, although some agglomeration was seen with the TMAH approach. Clark, et al.52 applied enzyme digestion and alkaline digestion, both with and without the addition of CaCl2, to fish livers containing Ag ENMs. This work found that the enzyme digestion was not effective at breaking down the liver tissue and suggests that proteinase K may have not been appropriate because of the high lipid content of the matrix. The optimal approach was the alkaline digestion of fish liver using TMAH, with the addition of CaCl2 to prevent the formation of AgOH and/or Ag2O species, and a recovery of 103% was achieved.52 Zhou, et al.50 digested clam and oyster samples containing Au ENMs and achieved high recoveries for both alkaline- and enzyme-based approaches (96% and 104% respectively); however, the alkaline digestion method was favoured for being faster and more effective. Sung, et al.53 successfully digested fish liver samples containing Au and Ag ENMs using both enzyme and alkaline based digestion methods, however the higher recovery (102% vs. 74%) with the alkaline method made it preferable over an enzyme-based approach. Loeschner, et al.43 came to a similar conclusion when digesting rat spleens containing Au ENMs, where it was found that a four-fold higher recovery was achieved with an alkaline method over the enzyme approach.Interestingly, a study by Vidmar, et al.,54 where human placental tissue containing Ag ENMs was digested, found that by using a ten-fold higher concentration of proteinase K than Loeschner, et al.,43 a much higher recovery of 98–124% was achieved. Vidmar, et al.54 concluded that the enzyme method was favourable over the alkaline-based approach where particle agglomeration or formation was observed. These authors demonstrated that Ag particles formed, likely as chlorides or sulphides, from dissolved Ag when in the presence of tissue and TMAH. This observation was also made by Clark, et al.52 where it was demonstrated that the inclusion of CaCl2 prevented the formation of particles from ionic Ag. There is only one study, by Li, et al.,55 that compared an enzyme- and alkaline-based digestion in plant tissue containing Ag ENMs. These authors found a higher recovery for the enzyme-based approach (93–102% vs. 53–58%) and that there was no observable shift in the size distribution for the enzyme digest, but there was a shift to smaller particle sizes when using a TMAH based digestion protocol.
Overall, these comparative studies favour an alkaline-based approach, primarily because the extractions were more effective, quicker and offered higher recoveries relative to the enzyme digestion approaches. However, it should be noted that, to date, the range of ENM compositions where TMAH has been applied is limited, mainly to Au and Ag (Fig. 3), and its applicability to a wider range of ENM types requires further testing. In some cases particle agglomeration and/or formation was noted when the TMAH approach was applied. This highlights the importance of using high purity reagents, and they are recommended where possible. Although TMAH is available at a purity of 98% or greater, there may be significant impurities present which could result in particle formation from the metal ions present in a sample, or affect the measurement by other means. Enzyme products will inevitably have some incidental trace metals associated with the biological material the enzyme was derived from, and these too must be carefully assessed. It is therefore important to include appropriate procedural blanks and ionic controls when processing samples; this is discussed in detail in section 4. In the case of Ag at least, using TMAH rather than proteinase K also decreased the background signal of dissolved Ag thus reducing the achievable size limit of detection (LODsize).52
3.4 Other extraction methods
Other methods of extraction have been applied to a lesser extent, these include strong acid digestion, solvent-based extraction method and use of a lysis buffer. Deng, et al.24 used reverse aqua regia (3:1 ratio of nitric acid to hydrochloric acid) acid to extract TiO2 ENMs from the roots and leaves of rice plants and compared that to an extraction using macerozyme R-10. These workers found that the acid digest approach recovered approximately fifty-fold more particles, and recovered the expected particle size distribution more reliably, compared to the enzyme method. Laughton, et al.44 demonstrated that a macerozyme R-10 extraction approach was inappropriate for ENMs susceptible to dissolution, such as CuO and ZnO, due to the low pH required for enzyme activity. This work also reported an extraction method using 50% methanol in ultrapure water and demonstrated the successful extraction of CuO and ZnO ENMs from a range of leaf materials whilst successfully preserving the particle size distribution. Tris(hydroxymethyl) aminomethane (Tris) to lysate samples using a Tris-HCl approach with ultrasonication has been used for the extraction of Au ENMs from rat liver and ZnO ENMs from chicken meat respectively.56,57 Using this approach particle size distribution of ZnO ENMs was preserved, as determined by comparing to the distribution determined from TEM analysis of the freshly synthesised particles. The ‘extraction efficiency’ for these same particles was reported to be 60%.57 The aim here was to lyse the tissue using a solution that is very hypotonic and hypo-osmotic relative to the body fluids of the organism, thus ‘osmotic shock’ begins to break up the tissue without the use of aggressive chemicals. Consequently, hypotonic Tris-HCl extractions can be undertaken at neutral pH and at room temperature (or even on ice). Provided the hypotonic solution is in excess, the disruption of the tissue is rapid, within minutes, and homogenization or 2–15 minutes of ultrasonication has been used to disintegrate the tissue (Table 1). Interestingly, testing of a Tris-HCl extraction with and without the presence of (unspecified) protease found that hypotonic Tris-HCl likely inhibited the enzyme activity.57 This is no surprise, given that enzymes are also easily denatured by osmotic shock. However, hypotonic Tris buffers can be made from very pure ingredients, and this approach might therefore limit the dissolved metal background observed during analysis by spICP-MS.
A range of biofluid samples have also been prepared for analysis by spICP-MS including, blood, urine, artificial sweat, gastric fluids and hip aspirate (Table 3). Whole blood and blood plasma samples have been prepared by diluting twenty-fold with 0.1–2.5% TMAH and 0.1% Triton-X100, or directly with ultrapure water with dilution factors of 20–40000 fold. Urine has been analysed by diluting 10, 20 or 33-fold with 1% glycerol, 5% HNO3 or ultrapure water respectively. Simulated gastric fluids and sweat have been prepared by simple dilution with water prior to analysis. Hip aspirate has also been prepared using the enzyme digestion approach developed by Loeschner, et al.40 for chicken meat. These approaches for bodily fluids simply attempt to lyse any cells or other components during the dilution, and at the same time resolve interferences due to the sample matrix by dilution. However, the concerns are that excessive dilution may also begin to drive dissolution, but on the other hand, may also favour dispersion of the particles.
4. Method validation
The validation of any extraction method and subsequent analysis is an important part of quality assurance in analytical chemistry, and should include the estimation of the limit of detection (LOD) and quantification, the linearity/working range, trueness/recovery, precision, selectivity and ruggedness/robustness.58,59 Few if any papers report on all of these parameters, partly because the methods or approaches needed to address these metrics have yet to be agreed for tissue samples with ENMs. For example, agreement is needed on how to establish the LOD when the natural particulate background may vary in the tissue samples. However, discussions and working groups at the International Organization for Standardisation (ISO), the OECD, and other bodies, are beginning the process of standardisation with the goal of a universal approach for sample preparation in mind, as well as the analysis and quality control measures for the technique of spICP-MS for regulatory purposes. While there is some guidance on spICP-MS of media samples,60 this has not yet been developed for tissues. However, the general guidance on method validation for analytical chemistry (Magnusson and Örnemark58 and Thompson, et al.59) has also been interpreted for the determination of ENMs in foodstuffs by Linsinger, et al.61 The principles described therein could also be applied to the determination of ENMs in tissues.Nonetheless, the analysis step could be validated by using ENMs of known size distributions, synthetic solutions and suspensions of ENMs in the extractants, and the likely extractable material. In that circumstance, the recovery of the ‘expected’ particle sizes and metal composition could be confirmed in the analysis. However, one should be mindful that the ‘expected’ particle size will depend on what method was used to establish the particle size distribution for the stock of ENMs used in the experiments, as this may also alter the calculated particle number concentration in those stocks of ENMs.62 Thus, also the interpretation of how many particles are used in the exposure of the organism. The extraction step would remain to be validated, but if the mass balance could be calculated, some indication of the mass recovery might be possible.
There are currently no certified reference materials (CRMs) for ENM particle number concentration, particle mass concentration, particle size and the particle size distribution in a tissue matrix, so alternative approaches to validation need to be used. Currently, a common approach in use for partial method validation is to determine the overall ‘procedural recovery’ from the biological sample. This approach explores the amounts of the analyte that are recovered, or, conversely lost in the entire process from sample collection, preparation and analysis. By comparing what is ‘known’ to be in the sample to what is detected a procedural recovery can be determined, and for most chemicals a procedural recovery of 100 ± 10% would typically be deemed acceptable. In the absence of a CRM for ENMs in a biological matrix, the approach of spiking an unexposed biological matrix with a known amount of ENM is often adopted and some guidance for this is given for food.61 The use of this approach to determine the procedural recovery is heavily influenced by two crucial factors: (i) the metric measured by spICP-MS and (ii) how any spiking of the sample with ENMs is performed. Ideally the procedural recovery would be independently validated or benchmarked against alternative analytical approaches. Unfortunately, there is a lack of appropriate analytical approaches that are able to quantitatively detect ENMs in complex biological matrices. Attempts have been made to determine procedural recoveries and validate protocols in the literature. In the following the above problems and potential solutions are discussed.
4.1 Particle metrics for validation by spICP-MS
There are several particle metrics that may be used for validation of ENM extractions from biological samples including: particle number concentration, particle mass concentration, particle size and the particle size distribution. Of the studies included in Table 1 the favoured metric to report recovery is particle mass concentration, with 42% of studies doing so, while 18% use particle number concentration, and 11% report both particle mass and number. The remaining 51% of the studies do not report on recovery, instead often referring to previous work. Within this same group of studies 60% made efforts to check for changes to the particle size distribution as a consequence of sample preparation, for example by comparing ENM suspensions extracted from exposed tissues to pristine particle suspensions. Most of the studies in Table 1 made some efforts to assess changes in particle size distribution. However, these metrics are not predictive of one another. For example, particle aggregation and agglomeration would decrease particle number concentration, increase the mean particle size thus shifting the size distribution to larger particles while the particle mass concentration would remain unchanged. Whereas particle dissolution would reduce particle mass concentration, particle number concentration, mean particle size and shift the size distribution towards smaller particles. Thus, it is important to consider these inter-relationships when interpreting results, it is therefore recommended that multiple particle metrics are considered for validation purposes.524.2 Spike recovery tests
If the scientific objective is simply to establish the presence/absence of particles, then detailed validation with multiple metrics may not be needed. In some instances, spICP-MS has been used to establish the presence of particles from ‘unknown’ samples, including hip aspirate, whale brain tissue, mollusks and game meat.63–66 However, while such explorations are appropriate for academic research, a more comprehensive approach is generally needed for regulatory purposes. Validation by measuring the procedural recovery from spiked samples is currently the best available approach given the absence of CRMs for ENMs in biological matrices. If samples are spiked at different stages of the procedure it is possible to determine which part of the process is not working or problematic, and so a series of spike recovery experiments are often done for ENMs.Typically, a stock suspension of ENMs diluted in ultrapure water or relevant saline is spiked into the extraction matrix and/or onto control tissues, and either analysed immediately or left to reflect the incubation time in the intended protocol. While this approach can identify which steps are giving poor recoveries, for example, loss of particles due to dissolution in an acidification step, it is important to add enough ‘spike’ to get a clear result. Adding too little may result in poor detection because the spike does not sufficiently exceed the particle background already in the sample, and adding too much can erroneously inflate the recovery to 100% or more because the spike is swamping the matrix of the tissue sample and is too accessible. These considerations have been investigated and discussed for total metal spikes into tissues where it has been identified that it is the ratio of spike: tissue mass that is important.20 Where there is uncertainty over the metal burden in real samples it may be useful to determine this to inform on the appropriate level of spiking.52
Acceptable recoveries from spike recovery tests for traditional chemicals are typically 100 ± 10%. This has been shown to be readily achievable for total metals from an ENM-exposed tissue.20 However, the additional complexities of working with colloids, such as ENMs, rather than aqueous solutions, can mean that quantitative recovery is more challenging in terms of cumulative errors and therefore slightly less arduous targets for variation should be used.3 For spICP-MS, some authors have suggested a widening of the procedural recovery to ±25% for metallic ENMs.67 This is also consistent with the recommended spike recovery criteria for the analysis of lung fibre number burdens,68 which represents a similarly challenging analytical exercise involving tissue digestion. What may be considered an acceptable recovery for ENMs extracted from biological matrices and analysed by spICP-MS is likely to depend on the characteristics of the particle of interest (size, composition etc.) and the biological matrix, and if the data is to be used qualitatively or quantitatively. For example, the extraction and measurement of carbon-based ENMs (by methods other than spICP-MS) is particularly problematic, and spike recovery can be as low as 50%.69 Some authors report very low spike recoveries in tissues (e.g., 7%, Xiao, et al.70) and this would not generally be acceptable for research or regulatory use. In many instances recovery of the full particle size distribution may not be feasible due to analytical limitations, primarily due to when a portion of the particles are below the LODsize. This limitation might, at least in part, explain low procedural recoveries where the ENMs of interest have a size range below the LODsize.70–73 This and other analytical considerations and limitations are discussed in more detail in section 6.
In addition, for a regulatory toxicity test or research experiment, such as a bioaccumulation study, it is highly desirable to include a ‘negative tissue control’ (i.e., tissue from an organism in the unexposed control group), so that the incidental particle background or any naturally occurring biogenic particles in the tissue can be assessed. In the hazard assessment of ENMs, it is also important in some cases to understand the behaviour and toxicity of the nearest equivalent metal salt or bulk (micron-sized) material.3,13 In such circumstances, the procedural recovery protocol could include the reagent/extraction matrix without tissue and spiked with the necessary ionic form or bulk material control. Similarly, control tissue could be spiked with an ionic or bulk form of the substance of interest. Crucially, the ionic controls enable some understanding of how the evolution of sparingly soluble metal species in the sample matrix can promote particle formation (e.g., formation of Ag particles from Ag2+, Clark, et al.52). When spiking the extraction reagents with an ionic form of the analyte, e.g. Ag2+, an increased baseline signal should be expected, but no particle-like events where clouds of ions from the ionization of particles reach the detector of the ICP-MS, should be observed. If particle-like events are identified in the data, this would indicate that the extraction reagents are promoting particle formation from ionic species. This then suggests that particles are likely to be formed from ionic or elemental species of the analyte co-extracted with ENMs from the sample matrix giving false positive results.52 When conducting analysis of ‘bulk’ (i.e. non-nano) particles (i.e. >100 nm) by spICP-MS the upper size limit of detection needs to be carefully considered. This will be influenced by many factors, and is not fully understood, but will be reached when the particle of interest is too large for complete ionization to occur in the plasma. The effect of the sample matrix loading on the plasma, which can reduce the overall ionization efficiency, may also be important.74 Adjusting the operating mode of the ICP-MS to less sensitive setting can extend the upper size limit by another 100 nm for gold particles,75 but whether this would work for other ENMs is not clear. A further difficulty in analysing bulk particles is that conventional modern spray chambers, such as cyclonic spray chambers, have low internal volumes (approximately 50 mL) and are designed to reduce washout and increase signal stability by allowing only the finest aerosols, with an upper droplet size limit of approximately 1–10 μm, to reach the plasma. This upper limit may be even smaller for bulk particles than droplets, which will likely have a higher density and therefore the upper size limit for particles is likely to be smaller than for droplets. Thus, ‘bulk’ particles above this size do not pass through to the plasma and cannot be detected. Recently, sample introduction systems for single-cell ICP-MS have been developed. These systems include syringe-driven sample introduction and micro-flow low-pressure nebulisers with a more direct pathway to the plasma for the aerosol, which is done in an effort to keep the fragile and ‘large’ cells intact when they enter the plasma. These systems potentially offer an alternative sample introduction for some spICP-MS applications, however there is currently insufficient information to assess its suitability to various types of ENMs and biological matrix types.76 A further restriction on the detectable upper size limit is detector saturation due to the much greater ion signal generated from ‘bulk’ particles compared with that from ENMs.
Overall, in the studies reviewed there is a lack of consistency in the types of control samples included for validation and how they are reported. Of the eighty-three studies in Tables 1–3, only twenty-four reported on the use of a procedural blank (as defined in Table 4). These studies typically state that no particle signals were observed in the analyses and/or a plot of the raw data signal intensities was provided. A further ten studies stated procedural blanks were conducted, but they were not further reported on. Inconsistencies in the use of terminology and description in the literature complicates attempts to compare between studies. For example, there is often a lack of clarity on whether the spiking of control samples is performed before or after the extraction procedure. In addition, the term ‘blank’ has been used to mean the blank for dissolved metal standards (not always matrix matched) in calibrations, or with reference to unexposed and/or spiked tissues as a matrix control. Clearly, for regulatory use, including aspects such as food safety, it is vital that there is confidence in the results of any procedural recovery measurements, as these will be a foundation stone in the overall confidence in the measurement of test samples.
| Sample type | Procedure | Scientific purpose | Notes |
|---|---|---|---|
| 1. Pristine ENM suspension | Analysis of freshly prepared suspension of the ENMs | To characterise the ENMs in terms of size, size distribution and number concentration | This data can be used to confirm/determine the characteristics of the material being used in the experiment and can be compared to data from other analytical techniques |
| 2. Procedural blank | Extraction protocol alone (in the absence of ENMs and biological matrix) | To test for background levels of the analyte in extraction protocol reagents | The presence of particles, or elevated baseline signals indicate that the extraction reagents are not of a pure enough grade and will produce false positives in the data and/or hinder the detection of ENMs |
| 3. Procedural blank with ENMs | Extraction protocol in the presence of pristine ENMs | Characterisation data of the pristine ENMs processed through the extraction protocol | This characterisation data should be indistinguishable from that for the pristine ENM suspension. Any differences indicate transformations (i.e. dissolution) induced by the extraction process |
| 4. Procedural blank with ionic form | Extraction protocol in the presence of a dissolved species | To confirm particle formation is not induced by the extraction process | An increase in the baseline signal should be expected, however the presence of particles would indicate their formation from the ionic species under the extraction protocol conditions |
| 5. Control non-spiked biological sample | Extraction protocol performed on non-exposed control sample | To identify background level of any natural and/or biogenic particles in samples | Particles may inherently be in the sample; in which case these may be indistinguishable from the ENMs added and will need to be taken into consideration |
| 6. Control biological sample spiked with ENMs | Extraction protocol performed on non-exposed control sample spiked with ENMs | To demonstrate that ENMs can successfully be processed in the presence of the biological matrix without undergoing transformation | As with #3 above, this characterisation data should be indistinguishable from that for the pristine ENM suspension. Spiking non-exposed samples gives an indication of losses and best-case scenario recoveries |
| 7. Control biological sample spiked with ionic form | Extraction protocol performed on non-exposed control sample spiked with ionic form | To confirm particle formation is not induced by the extraction process when in the presence of the biological sample | As with #4, an increase in the baseline signal should be expected, however the presence of particles would indicate their formation from the ionic species under the extraction protocol conditions and in the presence of a biological matrix |
| 8. Certified reference material | Extraction protocol | To demonstrate that the sample preparation and analytical protocols can recover the certified values from a CRM that is comparable to the samples of interest | This is the best way to validate the methods applied, however in the absence of appropriate CRM the above control samples can be included to give confidence in the data |
From the approaches described in the literature and our experiences at the bench using spike recovery for ENMs with spICP-MS, the following are recommended: (i) that a freshly prepared ‘pristine ENM suspension’ of known composition is analysed and used for spike recovery; (ii) that a ‘procedural blank’ of the extraction matrix only, i.e. without any biological matrix, is used to monitor possible contamination in the procedure; (iii) that procedural blanks spiked with a dissolved/ionic form and the pristine ENM suspension of the ENM under study is used to monitor for any particle (trans)formation by the action of the conditions and reagents used; (iv) that a control non-spiked tissue, which could also be termed a ‘sample blank’,77 is processed and extracted to identify the presence of any indigenous particles; and (v) that control non-spiked tissues spiked with a dissolved/ionic form and the pristine ENM suspension to monitor for any particle (trans)formation by the extraction in the presence of the tissue matrix. We recommend that terminology should be clearly defined in relevant publications and suggest the adoption of an agreed set of terms, such as those outlined above and summarised in Table 4.
4.3 Matrix effects and interferences with procedural recovery experiments
While the spike recovery in the presence of a tissue/biofluid approach was the most common method of validation in forty-nine of the extraction protocols reported in Tables 1–3, very few explored the details of how the recipe of the extraction reagent or the matrix of the tissue homogenate affected the apparent recovery of ENMs from the sample. In the case of Ag materials, the presence/absence of tissue and the composition of the extraction reagent has been shown to affect the recovery in terms of particle number concentration.52 High mmol L−1 concentrations of cations in the extraction reagent is a concern for particle aggregation, while the presence of up to 5 g L−1 NaCl has shown no effect on the nebulization efficiency during aspiration of the sample into the ICP-MS.42 The high salt concentration will affect the sensitivity of the instrument through signal suppression due to the easily ionizable element effect, and there may be salt deposition on the sampler, skimmer cones, and possibly the first extraction lens. This could lead to a negative bias with respect to the particle size and mass concentration estimates if calibration standards were prepared in a different matrix to that of the samples.The presence of endogenous, natural or biogenic (or combination thereof) metal-containing particles is likely to be expected in some tissue samples.34,52,78 In particular this may be expected in instances where the element of interest is ubiquitous and/or occurs in natural colloids.79 Where possible, in such instances it may be necessary to include appropriate control samples, such as negative control tissues, to show relative contributions of incidental particles and ENMs.
4.4 Comparative approaches for assessing the trueness of the size distribution and mass recovery
Recovery may be considered in terms of both the particle mass and number recovery, and particle size distribution. All are important because the mass recovery alone does not indicate if there is any size effect. One approach to assess this is to compare the size distribution of the extracted ENMs in the presence and absence of the biological matrix with that of the original pristine particle suspension. Assuming that the particles are not transformed or modified in any way by the tissue, or sample preparation process, the particle size distributions of each of the above should be identical. For example Makama, et al.80 showed that Ag ENMs extracted from earthworms had a comparable size distribution to the pristine particles and determined their sample preparation protocol to be appropriate. Modrzynska, et al.72 also applied this comparative approach to CeO2 ENMs, following an enzyme-based extraction protocol in the absence of biological matrix, it was found that the median particle size reduced from 50 to 35 nm when compared to the pristine suspension. An approach that has been used to quantify the mass recovery is to calculate the total mass of the element of interest in both particulate and dissolved form from spICP-MS analysis, and compare this to the total mass of the element of interest in the same sample when determined using conventional ICP-MS.34 Agreement between the two analyses demonstrates quantitative mass recovery for the method, however this does not give an indication of if the ENMs of interest were preserved.One concern for the regulatory acceptance of a standardised protocol for detecting ENMs in biological samples by spICP-MS is that both the extraction techniques and the measurement method are relatively new applications. Although there are several technical documents that provide that guidance regarding the analysis of samples by spICP-MS (e.g. ISO/TS 19590:2017),60 there is limited guidance regarding sample preparation, especially in regard to complex solid samples such as biological tissues. The guidance document CEN/TS 17273:2018 suggests using an enzymatic digest approach for sample preparations of foods and biological samples prior to analysis by spICP-MS and outlines that a stepwise evaluation should be applied to evaluate the appropriateness of a protocol.81 The weight of evidence for the use of spICP-MS for detecting ENMs in complex biological matrices has increased quickly through active research (Fig. 1, Tables 1–3). To increase regulatory acceptance with international bodies involved in standardisation, such as the OECD, any results would ideally be confirmed by using an independent analytical method to determine the presence and composition of particles in the extractant.
Transmission electron microscopy is frequently used to establish the mean particle size and size distribution, and these typically compare favourably with spICP-MS. The advantage of TEM over spICP-MS is that it is one of the few methods capable of providing information on the morphology of particles, the LODsize is only a few nm, and if coupled with X-ray methods (e.g., XRD) information on the chemical composition can also be obtained. The drawback to TEM is that it can be a time and labour-intensive method that will typically include only a relatively small number of particles with the potential for user bias to play a role. Because only small areas of samples can be imaged by TEM it is not well suited to imaging complex biological samples (or extracts) containing ENMs which will be difficult to locate and identify unless present at extremely high concentrations.82 Therefore, while such independent methods of validation were used in pioneering papers establishing the technique of spICP-MS analysis of water samples,83 it is not generally considered for studies on tissue samples, this is reflected by the lack of TEM images used to directly identify and characterise ENMs in biological tissues in the studies reviewed here.
5. Moving towards CRMs for ENMs in biological matrices
For regulatory testing purposes, it is usual to include a CRM in the sample run to confirm the entire procedure is working as expected. Such CRMs are widely available for different types of biological matrices for total metals and for some organic chemicals. Certification of reference materials typically starts with a certifying body assessing market need, then collecting and preparing a sample, which is then distributed to a range of participating laboratories that will use their own in-house methods for preparation and analysis. This will ideally result in the reference material being certified for the metrics of interest with an associated uncertainty. It is recognised that the matrix of the tissue can influence the measurement and a CRM should ideally be matched as closely as possible to the experimental sample being analysed. So, for example, for a shellfish sample, one might use a mussel or oyster CRM. The use of CRMs in new method development is to be recommended in order to give confidence in the data and demonstrate the validity of said method. If the results obtained by the approach used are in statistical agreement with the values of the CRM, then it can be argued that the method is validated for that tissue, and the specific analyte. In addition to, or in the absence of a CRM, in-house reference materials are sometimes used. In-house reference materials are not certified by an approved body but are from a sample that has been measured numerous times by the same laboratory and therefore the workers have a high degree of confidence in the expected data, although any in-built laboratory method bias is not accounted for. For quality assurance purposes it is therefore good practice for a CRM and/or an in-house reference material to be analysed alongside real samples at regular intervals.Unfortunately, there are currently very few CRMs for ENMs, and these are only for pristine particle suspensions or powders. For instance, gold ENMs are available from the Laboratory of the Government Chemist (LGC) in the UK and the National Institute of Science and Technology (NIST) in the USA, with guidance on storage, how measurements were made (e.g., spICP-MS), the uncertainty of the measurement, and instructions for use. These materials can, of course, be used to validate the spICP-MS measurement process, which should include mean particle size, particle number concentration and particle mass concentration, but this does not confer any validity on any extraction procedure used for sample preparation of ENMs in a biological matrix.
Thus, there is a need for tissue-based CRMs for ENMs. Certifying a tissue sample for the above ENM parameters would involve at least three steps: (i) the production of a biological material containing the ENM(s) of interest, (ii) conducting inter-laboratory comparisons on the materials for the ENM(s) using expert laboratories with knowledge of how ENMs behave in tissues and spICP-MS, and (iii) assessment of the data realised for validity. All of these steps should be overseen by an organization accredited to ISO 17034,84 and therefore meeting the general requirements for the competence of reference material producers. Inter-laboratory testing of test suspensions (Montoro Bustos, et al.85), and complex matrices (chicken meat, Grombe, et al.,86 Weigel, et al.;87 confectionary, Geiss, et al.88) have previously been undertaken. However, these have had varying results, with some complications. For example, only around 25% of the particulate Ag could be recovered following spiking onto chicken meat, suggesting it is dissolving in the tissue matrix.85
The production of a CRM is therefore not a trivial matter, in addition to being comprehensively characterised, an ideal CRM must be homogenous, and reasonably plentiful and stable over long periods of time (years) to ensure its longevity and acceptance. The fact that long-term storage of ENMs in biological tissues has been shown to be problematic is of particular concern.35,85 The production of a CRM for ENMs in a biological matrix is complicated further by the fact that in an ideal situation a wide range of ENM and matrix types would be available, each with a range of particle sizes and particle concentrations. Each of these factors may impact on final results, for example it would be inappropriate to use a chicken meat CRM with a high concentration of 60 nm Au ENMs when the samples of interest might be oysters containing low levels of 20 nm Ag ENMs. One possibility is that existing CRMs may already inherently contain particles, so could serve as a CRM for ENMs in a biological matrix. However, this has yet to be investigated.
The production of a comprehensive range of CRMs to cover the wide range of ENM compositions and biological matrices, such as those in Fig. 3, and for a range of particle sizes and concentrations is not feasible at the present time. However, the research community could benefit from a limited number of CRMs, for example for a select few particles that may exemplify compositions susceptible or resistant to chemical changes (e.g., Ag and TiO2) and within a select tissue (e.g., liver). The possibility that existing CRMs for metals may inherently contain a particle population that may be used is an option that has not been explored. For example, decapod crustaceans and marine bivalves are known to make endogenous metal-containing granules as part of their defences for metal sequestration.89 Therefore, the detection of particles in existing CRMs for shellfish is an interesting prospect, such as ‘ERM-CE278k mussel tissue’ available from the EU Joint Research Centre. This type of material would fit with the recent proposal for representative test materials,90 with the possibility of certification for any natural ENMs at a later date, if these were detected and found to be stable within a homogenous matrix. Approaches to reference materials for nano have been discussed.91,92 In addition to any CRMs that may become available, appropriate control and spiked samples should be included, in line with the recommendations in section 4 above.
6. Practical considerations for nanomaterial measurement by spICP-MS
There is much guidance published elsewhere on the practical application of spICP-MS.60,74,81,93–98 The following provides an overview of the key considerations particularly in the context of analysing biological samples by spICP-MS.6.1 Precision and accuracy for tissue samples by spICP-MS
In the routine analysis of tissues using a validated method for total trace metal concentration, it is expected that repeated sub-sampling, preparation and analysis of a homogenised sample will yield values that are in statistical agreement. For total dissolved metals in aqueous samples, the precision is often reported as the coefficient of variation of the sample measurement and is typically only a few percent and should certainly be <5% variation. In such circumstances, factors such as analyte stability in the sample and small amounts of instrumental drift are the causes of variation. However, from a biological perspective, it is the precision obtainable from extractions of sub-samples of the bulk tissue rather than a single digest that is important. The within tissue variability of the tissue sample is determined by taking multiple sub-samples of the tissue (e.g., six sub-samples from one piece of meat) and digesting or extracting each one separately, then analysing each sample by ICP-MS. This measurement represents the precision of the entire procedure and the sources of variation could be the heterogeneous nature of the tissue itself, random and systematic errors in the digestion/extraction protocol and any instability from the ICP-MS instrument. The procedural precision for a tissue sample has been reported for repeated measures of total Ti from TiO2 in trout skeletal muscle and is typically <5% and within acceptable limits.20 However, preparing ENM suspensions from tissue homogenates for analysis by spICP-MS is more challenging, as is the measurement method compared to a conventional total dissolved metal concentration measurement by ICP-MS. It therefore raises the concern that <5% variation could be too stringent for within tissue sample precision for spICP-MS. The between tissue sample variation (i.e., differences between the same tissues in individual animals in an experimental treatment), reflects the biological variation within a treatment. This variation is widely reported in terms of standard deviations or standard errors on mean values for a treatment, and of course, provided the within tissue sample precision is very good, the variation between animals in a treatment can be revealed.Accuracy refers to how close the measurements are to the ‘true’ values for the sample and the closeness of agreement between these measurements, or the trueness and precision respectively.99 The use of spike recovery tests as outlined in section 4 do not confer accuracy but provide information on how much of the added ENM can be recovered. However, recovery from a freshly spiked tissue is likely to be more readily achieved than from a real sample due to the ENM being added to the sample, rather than being biologically incorporated into it. The efficiency of the extraction procedure may therefore be lower for real samples. Thus, the need is for tissue CRMs to become available to allow an estimation of accuracy to be more firmly reported (as discussed in section 5). It is possible that analyses can be very precise (all the values cluster together), and yet not be accurate (e.g., all the samples over-reading or under-reading). For example, a positive bias due to the extraction protocol causing particle formation. For spICP-MS the under- or over-estimation of ENMs in tissue samples, due to the extraction protocol being inefficient and/or causing transformation and/or formation of the ENMs, is of major concern. A further problem is that of the particle LODsize which is discussed in the next section.
6.2 spICP-MS limits of detection
The limit of detection (LOD) for spICP-MS can be considered in terms of the particle size (LODsize), particle number concentration (LODnum), and the particle mass concentration (LODmass). The LODsize of spICP-MS is a product of several factors, principally these are the inherent properties of the particle of interest such as elemental composition and density which will determine how much mass of analyte is present in a particle of a given size. The other factors that will determine if the mass in a given particle is sufficient for detection includes the instrumental sensitivity, instrumental settings (such as dwell time and sampling position),74 and baseline signal intensity. The latter arises from dissolved ions of the element of interest in the samples as well as isobaric and polyatomic interferences that are not uncommon in samples with complex matrices, and from the electronic noise of the detection system. It is reasonable for the baseline signal intensity to vary from sample to sample, and therefore the LODsize is not expected to be a single value for all samples. Similarly, it is misleading to determine the LODsize based on a ‘clean’ matrix such as ultrapure water as this is unlikely to be achievable in real samples. A comprehensive study by Lee, et al.100 calculated the theoretical instrumental LODsize for 40 different ENMs in deionised water under ideal conditions (Fig. 4). New data suggests improvements in the LODsize of up to an order of magnitude have been made for some elements (e.g., TiO2 and Se). These improvements in LODsize are likely through new and more sensitive instruments with a higher signal to noise ratio, the advent of triple quadrupole ICP-MS instruments which can further reduce the effects of polyatomic interferences, and the ability to use shorter dwell times. Regardless, the LODsize in best case situations remains around 10 nm. Fig. 4 shows the theoretical LODsize reported by Lee, et al.100 and those reported in the studies summarised in Tables 1–3 for a range of particle compositions, it can be seen that overall there is a good agreement between these values.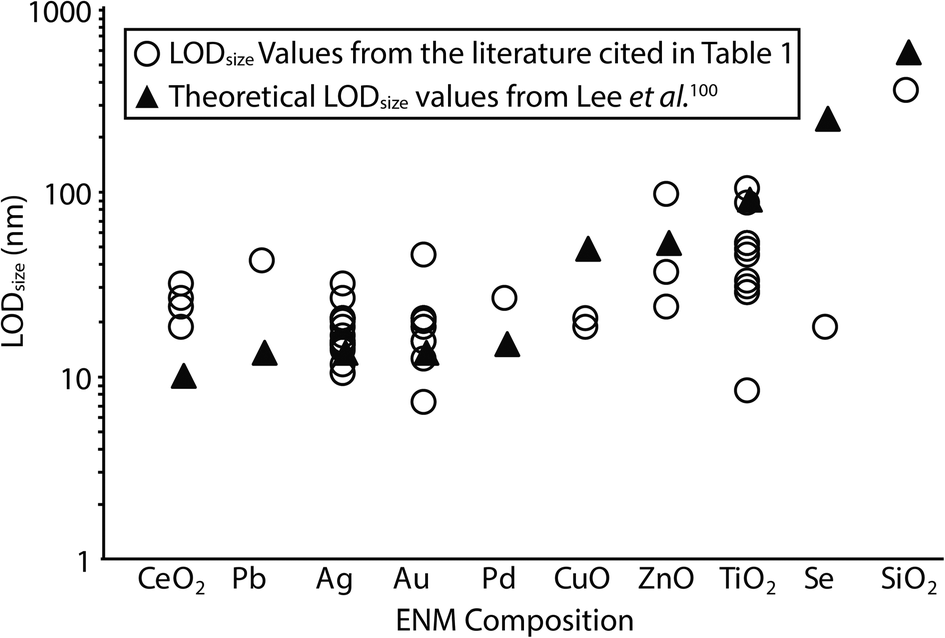 | ||
| Fig. 4 A comparison of spICP-MS LODsize determined theoretically by Lee et al.100 to those reported in the literature cited in Tables 1–3. | ||
There are several approaches used for the determination and reporting of the LODsize in the existing literature. Essentially, the LODsize is related to a threshold value whereby if a signal intensity is above this value it is determined to be a signal from an ion cloud produced by the ionization of a particle in the plasma, whereas below this value the signal makes up the baseline. The duration of a particle signal is typically 0.2–0.5 ms, therefore when short dwell times of <1 ms are used consecutive events that exceed the threshold value are assumed to be from the same ion cloud (and therefore particle) and must be summed to determine the total signal intensity of each particle event.22 Where longer dwell times, typically of 3–10 ms are used, it is assumed that the total particle event is captured in a single dwell time. The threshold value is typically determined using one of two approaches: (i) user selection, or (ii) using an iterative approach as reported by Laborda, et al.101 and Tuoriniemi, et al.94 This data processing has been performed online using data processing software available from several instrument manufacturers or offline by exporting the data into third party programs including Microsoft Excel™, MatLab™, R etc. where the user can manipulate the raw data.
Determination of the detection limit threshold value will be partly influenced by the user and how they (subjectively) read the baseline signal to differentiate it from the particle signal. In situations where there is a clear distinction between the full particle size distribution and the baseline signal, as is the case with 60 nm Au ENMs in ultrapure water, this may not pose a problem. However, in many situations, the full particle size distribution will overlap with the baseline signal and selection of the threshold value is not so clear cut. A more systematic approach, where there is a continuous baseline signal, would be to use an iterative method. Here the average and standard deviation of the signal intensity is calculated for all the data. Then a threshold value is calculated by the average plus n times the standard deviation, n could be any value, but 3 or 5 is often used. All signal intensities above this threshold value are omitted and a new average and standard deviation is calculated from the remaining data, the process is repeated until there is no change in the calculated threshold value. The LODsize is then determined as the size of a particle required to produce a signal intensity that is equal to the threshold value minus the average baseline signal. Using this more systematic approach is desirable as it means all data is processed using the same criteria and this limits user bias. The iterative method assumes that the baseline signal is stable and continuous, and thus has a normal distribution. Therefore this approach cannot be readily applied to situations where the baseline does not exhibit a normal distribution, i.e., when the baseline is very low such as where there is no or very little dissolved background signal and/or where very fast (sub microsecond) dwell times are used.
There is a lack of consistency and clarity in the literature in what is being reported as the ‘LODsize’. In some instances the LODsize is simply reported as the theoretical values determined by Lee, et al.48,55,100,102–107 In many ways this value alone is meaningless, as it does not take into account the specific instrument settings used in the individual studies and there is no consideration to determine the LODsize in the sample and extraction matrix used.108 In some cases a LODsize is determined using the iterative approach in a clean reagent mix or control tissue, this again lacks relevance as it does not take into account the baseline signal and noise that is likely to be higher in an exposed sample and will likely vary between samples depending on dosage used, exposure length and the degree of dissolution. This is why some studies report a range of values for the LODsize, depending on the origin of the sample.66 For example, comparing the LODsize values determined for ideal clean matrices to those in exposed biological matrices often results in at least a 10 nm difference between them.52
The LODnum is determined by the frequency of particle events that reach the detector. This is a function of the particle number concentration in the sample, the uptake rate of the sample, the transport efficiency, and the proportion of particles above the LODsize. The maximum acceptable number of particle events observed in blanks should not exceed 10 particles per minute of analysis, while the minimum number of particle events should not be less than 100 per minute. In situations where only low particle numbers are counted it may be necessary to analyse a sample multiple times or for a longer period. There is also an upper limit to the particle frequency as it is essential that particle events can be resolved from one another, ISO/TS 19590:2017 recommends that the number of observed particle events should not exceed 10% of the maximum number than is theoretically possible.60 For example, when using a 3 ms dwell time 20000 data points shall be collected in 60s, therefore the maximum number of particle events recorded should not exceed 2000. Laborda, et al.109 provide further guidance and considerations for the determination of nanoparticle number, size and size distribution.
The LODmass will largely be a function of the particle size, size distribution, particle number concentration, and particle composition. For example, a spherical particle with a 10× larger diameter will have 1000× more mass, therefore the LODmass of 100 nm particles will be 125× higher than for 20 nm particles. The particle composition will dictate the density of the particle and the proportion of which comprises of the analyte of interest. Some guidance on the LODmass is provided in IOS/TS 19590:2017, this ranges from 0.2–20 ng L−1 depending on the particle size and composition.
6.3 Instrumental settings and the sample run
Analysis by spICP-MS involves certain differences compared to using an ICP-MS for total metal concentrations. In addition to ISO 19590,60 previous reviews (e.g., Mozhayeva and Engelhard, Goenaga-Infante and Bartczak, and Meermann and Nischwitz)74,98,110 have given a detailed tutorial on how to analyse a sample by spICP-MS; considering the effects of calibration (e.g., matrix matched standards and particle standards), instrumental settings, distinguishing the ionic background from the particles, upper size detection limits and calculating histograms from raw data. There have also been discussions over appropriate sample introduction systems.22,74 Due to the time intensive nature of making the measurements and handling the data, the aim here is to provide our experiences to help bench scientists identify and troubleshoot some of the common problems encountered.The user will need to define the instrumental settings to be used for data acquisition. Arguably the two main settings defined by the user will be the dwell time, as discussed above, and acquisition time. The acquisition time is the duration that the signal shall be continuously monitored for, this is typically 60 s, however longer times may be used, for example in situations where the particle number concentration is low. From the authors' experiences, when shorter dwell times (<0.5 ms) are used it is important to confirm that continuous acquisition is achieved, as some older instruments are not capable of this. This can be done by simply confirming the expected number of data points (dwell time × acquisition time) matches the actual number. Fig. 5 provides a typical example of the types of signal and data that can be anticipated from spICP-MS analysis.
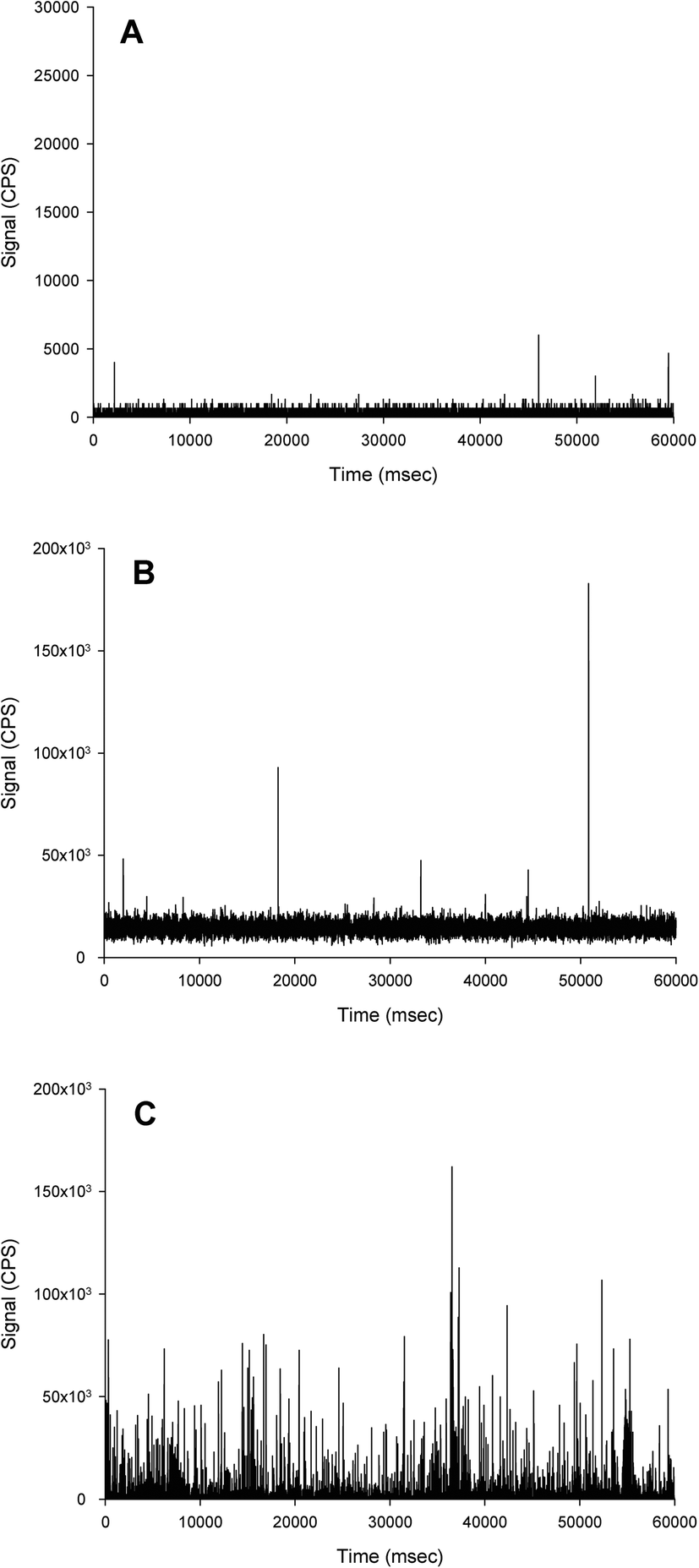 | ||
| Fig. 5 Example time scans of Ag extracted from the mucosa of the mid intestine of rainbow trout following exposure to no added Ag (control, A), 1 mg L−1 AgNO3 (B) or 1 mg L−1 Ag ENMs (C). Note, the Ag from the AgNO3 treatment remains dissolved, and the Ag extracted from the Ag ENM treatment remains particulate. Also note the control sample (A) is on a different scale. The extraction was performed according to Clark et al.,27 using 20% TMAH + 5 mM CaCl2. Note the absence of particles in (B) indicates the Ag has remained in the dissolved form. | ||
Prior to the analysis of samples, appropriate calibration of the instrument must first be performed. As with total dissolved metal analysis, a calibration with a series of dissolved standards, matrix matched to the samples, must be performed to determine the instrumental response. In addition to dissolved standards, it is also necessary to analyse one, or a series of ENM standard suspensions. Typically, Au ENMs are used for this purpose, RM 8013 60 nm Au nanoparticles from NIST have been a popular reference material and are certified for size, but unfortunately this reference material is no longer available. Similar Au ENMs from several suppliers that have been well characterised, but not certified, are frequently used and readily available. Although reliable for particle size, the number concentration provided with these suspensions is often calculated from the total mass of Au divided by the mass of Au in the primary particle size of the suspension. This metric should therefore not be considered as reliable and care must be taken if it is used in the calibration steps discussed next. To our knowledge, so far, only one CRM, LGCQC5050 produced by LGC, has been made available for an Au nanoparticle suspension that has been certified for both particle size and number concentration.
Analysis of the standard ENM suspension(s) allows for the transport efficiency to be determined. The transport efficiency can be determined using one of several approaches. Pace, et al.93 assessed three approaches using: ‘particle frequency’, ‘particle mass’ and ‘waste collection’. More recently a ‘dynamic mass flow’ approach has been proposed.97 The transport efficiency allows the dissolved calibration to be used to determine the instrumental response for particles and therefore particle mass to be determined. The transport efficiency also demonstrates the proportion of injected particles that reach the detector and therefore is needed to calculate sample particle number concentrations.
For conventional ICP-MS it is good practice to monitor an internal standard during sample and standard measurements to quantify instrumental drift and ion suppression from high matrix loads. The continuous monitoring of a single mass to charge ratio is usually necessary for spICP-MS, as this is typically done using single detector ICP-MS instrumentation, where only one mass can be continuously monitored with time it is not possible to monitor both the analyte and internal standard signal during sample analysis. Therefore, check standards must be included within a sample run to confirm instrument performance. This should include both a dissolved ion and a particle suspension standard to monitor for instrumental drift and transport efficiency respectively. The frequency of running standard checks is not routinely reported, but experience suggests every 10–15 samples, similar to total metal analysis, is sufficient. Notably, advances in new quadrupole ICP-MS instruments enable very short dwell times, such that it is possible to almost simultaneous measure the ratio of the analyte and the internal standard signal intensities, and could be used to more accurately correct for instrument drift.111
There has been a lack of discussion in the literature on what is a clean enough system to start the measurement of samples. It is recommended that the first solutions analysed are matrix-matched blanks to monitor for carryover from previous analyses. The aim should be for as low a baseline as possible with minimal to no particle like events present. Low reagent grades can have trace analyte contamination, thus increasing the dissolved background, and should be evaluated before use. A peristaltic pump is usually used to introduce the sample into the instrument, and it can be helpful to keep the pump tubing as short as possible to reduce the risk of adsorption of the particles to this tubing. The pump tubing should be changed frequently to avoid the build-up of adsorbed material, with time allowed for any new tubing to ‘bed in’ on the pump, so that a constant flow rate is assured. Notably, any adsorbed particles on the peristaltic tubing and other internal surfaces of the instrument, or particles formed from dissolved ions adsorbed to the tubing, may be dislodged by a change in the properties of the liquid being pumped. For example, switching from an acidic to a basic solution. Consequently, it is important to thoroughly flush the instrument prior to analysis with, for example, an acid wash, ultrapure water, then with several washings with the sample matrix to be used in the sample run. The effects of washing and the sample matrix on noise and drift in the instrument should be established before any experimental samples are analysed.
It is possible for a small number of false positive signals to be observed in particle-free ‘clean’ wash or blank sample matrix solutions. To demonstrate this, an example of raw data plots from a measurement session looking at Ag ENMs can be seen in Fig. 5. In Fig. 5A a few signal pulses of up to 5000 counts per second (cps) can be discerned, although the baseline signal is less than 500 cps. In this example a threshold value of 2000 cps to distinguish particle signals might not be unreasonable, in which case 3 false negatives particle events would be identified in the 60 s acquisition. Fig. 5B shows the raw data plot from a sample containing only dissolved Ag, similar to Fig. 5A, only a small number of peaks can be observed. However the signal baseline is significantly higher (about 10000 cps), this is typical of a sample containing a dissolved species as the ions are homogenous in the liquid sample introduced into the ICP-MS instrument. In comparison the raw data plot of a sample containing particles (Fig. 5C) contains several thousand signal peaks of up to 100000 cps. Therefore, the small number of expected false positives will not impact on the final data in any significant way. A 2% HNO3 wash solution is generally accepted as standard for ICP-MS analyses (single particle or otherwise), but in some instances the washing procedure was found to be more effective when a more aggressive acid, such as 2% HNO3 and 4% HCl, or a surfactant-containing acid solution in order to help flush particles from the tubing and sample introduction.52,66 In instances where particle signals persist despite washing, this can occur if the sample introduction system is ‘overloaded’ by a sample with a very high particle number concentration, it may be necessary to cleaning the spray chamber (soaking it in 10–30% nitric acid has proved to be very efficient) and possibly replace the uptake and/or pump tubing.
7. Conclusions and recommendations
This review describes the current state of the art for the determination of ENMs in biological matrices using spICP-MS from sample collection, storage, preparation and analysis. The key findings are summarised below and the recommendations that have been identified from the reviewed literature are summarised in Fig. 6.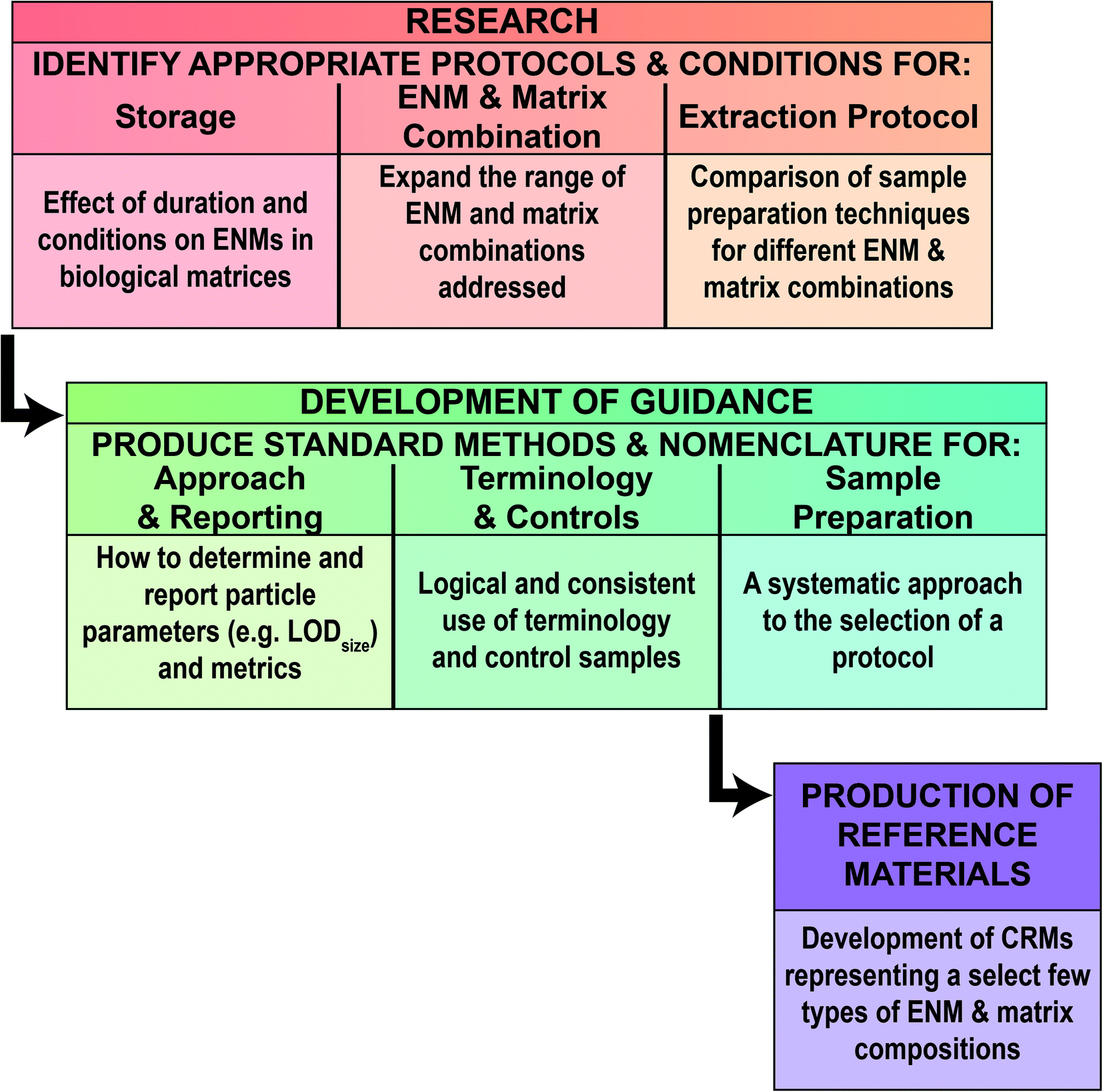 | ||
| Fig. 6 Summary of key findings and recommendations. | ||
When collecting tissues/organs containing ENMs the same considerations and good practices that are used for any other chemical should be applied to ensure the sample is clean of excess substance in the external media. For example, washing the exposure medium off the epithelium of the gut by rinsing with water or saline followed by EDTA. Concerns over the preservation of ENMs whilst being stored have been raised. However, there is a lack of data to assess the most appropriate conditions for long-term storage for different ENM and biological matrix combinations. The ideal scenario is therefore to prepare and analyse samples immediately, or soon after, collection. In situations where this may not be practical or feasible, the inclusion of spiked control samples with real stored samples may help to assess particle preservation whilst in storage.
From a regulatory and standardisation perspective, a common protocol for ENM extraction from biological samples would be beneficial. However, a ‘one size fits all’ protocol that may be applied to all possible types of ENM in any type of biological matrix does not seem practical. For acid-resistant ENMs, such as TiO2, acid-based extractions may be appropriate, where the biological matrix is rapidly degraded, and the particles liberated into a suspension for analysis. However, this approach is not suitable for ENMs that dissolve in acidic conditions such as CuO, ZnO or Ag ENMs; and instead a strong alkaline extraction is best. Alkaline extraction has been used on a wide range of animal tissues, without promoting dissolution, and this relatively simple approach has good prospects for standardisation and use in the regulatory environment. A few studies reported particle formation or agglomeration under strong alkaline conditions, and it is recommended that control samples are included in the analysis to assess this.
Of the possible enzymatic methods of extraction, proteinase K is the main technique for animal tissue. The conditions for optimal enzyme activity (pH 7.5–12 and 37 °C) are consistent with the preservation of particles and avoiding dissolution, but proteinase K does not degrade lipids or carbohydrates in tissue. The latter problem causes incomplete extraction and/or the need for other enzymes to breakdown the tissue. Overall, the alkaline-based extraction approach is less problematic, and more practical for many internal organs, including liver. The strong alkaline approach is not appropriate for the degradation of plant tissues, but the routine use of the commercially available enzyme mix, macerozyme R-10, has proven to be effective, although some ENMs could be susceptible to dissolution or transformation under the optimum conditions for enzyme activity (pH 3.5–7.0). Macerozyme R-10 is suitable for chemically resistant particles (e.g. Au and SiO2). Where dissolution is the concern, lysation with a solvent may be more effective at preserving particles during extractions from plant tissue (e.g. CuO and ZnO). The preparation of biological fluids, such as urine or blood, is relatively straightforward, where a ‘dilute and measure’ approach is commonly applied. Care must still be taken here, for example, high dilution factors with ultrapure water may drive dissolution. The inclusion of dispersing agents, such as a few percent of SDS or Triton-X100, is a sensible step to aid in the stabilization of particles in suspension made of biological fluids.
The extraction approaches above, have been performed on mainly a few types of ENMs (e.g., Ag ENMs) and biological matrices (e.g., lung and liver). It would be beneficial for a wider range of ENMs to be addressed, including materials such as ZnO that are prone to dissolution, and more advanced composite ENMs such as those used in medical devices. Data is also lacking or sparse on some types of biological samples including skin, very fatty endocrine organs and reproductive organs. Notably, hard tissues with high mineral backgrounds, such as bone, present more challenges in terms of sample preparation and have yet to be investigated, despite indications that they may be a significant deposition site in mammalian studies.112
A recently novel development to spICP-MS is the technique of laser ablation spICP-MS (LA-spICP-MS).113 This approach makes use of the latest generation of laser ablation systems with high repetition rates and necessities a dwell time of 0.1 ms or less be used. This technique offers a promising alternative to solution spICP-MS as solid samples can be analysed and therefore the extraction of metal ENMs from tissues would not be necessary and information on particle distribution within a tissue can be gleaned. Some limitations to this approach are likely to be that only a limited area of tissue section(s) may be sampled, analysis will be more time intensive, and the technique will not be as widely available due to the requirement of the use of the newer generation of laser ablation units. Existing software for spICP-MS data processing will not be appropriate for use with LA-spICP-MS. There are currently only a limited number of publications where LA-spICP-MS has been used, so its applicability to a wider range of ENM and tissue types has not yet been extensively investigated.114
Whilst the strength of spICP-MS is its ability to detect particles in complex matrices, a commonly cited shortcoming is that its LODsize is higher than other methods (e.g., TEM). There are several approaches that may be used to determine the LODsize for an analysis by spICP-MS An iterative approach is recommended where the mean and n standard deviations of the signal for the sample of interest is used to distinguish the particle signals from the baseline. This approach is systematic and does not depend on the opinion of the analyst. As the LODsize can vary on a sample to sample basis it would be appropriate to report the LODsize as a range of values. This contrasts with the single values reported in studies so far, that may not consider how the baseline signal may have varied between samples and thus the effect of this variation on the LODsize.
As highlighted in Table 4, the inclusion of a range of spiked and non-spiked procedural blank and control samples are a valuable tool for quality assurance and a careful choice of nomenclature is required. The procedural recovery of a given approach must be reported, and while recoveries of 100 ± 20% or better are highly desirable for standardisation in a regulatory context, further optimization of some methods will be needed. When recoveries are less than expected, one feature may be the preferential recovery of one particular particle size over another, leading to a false impression of the particle size distribution and/or number concentration in the sample. It is recommended to include spiked controls to calculate the recovery, both in terms of the mass of analyte recovered but also the size and size distribution. This necessitates the use of freshly spiked samples, with the ENMs of interest likely more accessible than those incorporated into the biological matrix, and therefore can only be considered as a best-case scenario.
The current lack of CRMs for ENMs in biological matrices presents a problem for quality assurance and control. The development of a limited number of CRMs for this application would likely be greatly beneficial. For example, an ENM representative of those susceptible to dissolution and transformation (e.g. Ag, Cu or Zn) and a chemically resistant particle composition (e.g. Au or TiO2) available within both a lean and fatty tissue matrix. It is possible that some pre-existing CRMs may inherently contain a particle population that could be exploited as a CRM for ENMs and this requires further investigation.
This systematic review has identified the most promising extraction approaches for ENMs in biological matrices, and aspects of good practice such as control samples and quality assurance, and what particle parameters to report (particle size, size distributions, and number and mass concentrations). The next step for the scientific community is a consensus-based approach to arrive at standardised methods for the most promising approaches, and also technical guidance documents on how best to apply the methods, with a decision tree to help select the most appropriate method for the type of sample or ENM, and also agreed nomenclature and technical definitions for the extractions and subsequent measurements by spICP-MS.
Author contributions
Adam Laycock: conceptualisation, writing – original draft, review & editing, visualisation. Nathaniel J. Clark: conceptualisation, writing – original draft, review & editing. Robert Clough: conceptualisation, writing – original draft, review & editing. Rachel Smith: conceptualisation, writing – review & editing, supervision, project administration, funding acquisition. Richard, D. Handy: conceptualisation, writing – review & editing, supervision, funding acquisition.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This study was conducted as part of the NanoHarmony project under grant agreement No. 885931 of the European Union's Horizon 2020 research and innovation programme. This work was also supported by the EU H2020 NanoFASE project, grant agreement no. 646002, with core funding from the National Institute for Health Research's Health Protection Research Unit in Environmental Exposures. The views expressed are those of the authors and not necessarily those of the National Institute for Health Research, the Department of Health and Social Care, or Public Health England.References
- G. Singh, C. Stephan, P. Westerhoff, D. Carlander and T. V. Duncan, Measurement methods to detect, characterize, and quantify engineered nanomaterials in foods, Compr. Rev. Food Sci. Food Saf., 2014, 13, 693–704 CrossRef CAS PubMed.
- M. Crane, R. D. Handy, J. Garrod and R. Owen, Ecotoxicity test methods and environmental hazard assessment for engineered nanoparticles, Ecotoxicology, 2008, 17, 421 CrossRef CAS PubMed.
- R. D. Handy, G. Cornelis, T. Fernandes, O. Tsyusko, A. Decho, T. Sabo-Attwood, C. Metcalfe, J. A. Steevens, S. J. Klaine and A. A. Koelmans, Ecotoxicity test methods for engineered nanomaterials: practical experiences and recommendations from the bench, Environ. Toxicol. Chem., 2012, 31, 15–31 CrossRef CAS PubMed.
- OECD, Test No. 305: Bioaccumulation in Fish: Aqueous and Dietary Exposure, OECD Guidelines for the Testing of Chemicals, Section 3, OECD Publishing, Paris, 2012, DOI:10.1787/9789264185296-en.
- R. D. Handy, J. Ahtiainen, J. M. Navas, G. Goss, E. A. Bleeker and F. von der Kammer, Proposal for a tiered dietary bioaccumulation testing strategy for engineered nanomaterials using fish, Environ. Sci.: Nano, 2018, 5, 2030–2046 RSC.
- OECD, Test No. 412: Subacute Inhalation Toxicity: 28-Day Study, OECD Guidelines for the Testing of Chemicals, Section 4, OECD Publishing, Paris, 2018, DOI:10.1787/9789264070783-en.
- OECD, Test No. 413: Subchronic Inhalation Toxicity: 90-day Study, OECD Guidelines for the Testing of Chemicals, Section 4, OECD Publishing, Paris, 2018, DOI:10.1787/9789264070806-en.
- U. Bernauer, L. Bodin, Q. Chaudhry, P. Coenraads, M. Dusinska, E. Gaffet, I. Panderi, V. Rogiers, C. Rousselle, M. Stepnik, T. Vanhaecke, S. Wijnhoven, N. Goetz, W. De Jong and A. Simonnard, The SCCS guidance on the safety assessment of nanomaterials in cosmetics, Regul. Toxicol. Pharmacol., 2020, 112, 104611 CrossRef CAS PubMed.
- EFSA Scientific Committee, S. More, V. Bampidis, D. Benford, C. Bragard, T. Halldorsson, A. Hernandez-Jerez, S. H. Bennekou, K. Koutsoumanis, C. Lambre, K. Machera, H. Naegeli, S. Nielsen, J. Schlatter, D. Schrenk, V. S. Deceased, D. Turck, M. Younes, J. Castenmiller, Q. Chaudhry, F. Cubadda, R. Franz, D. Gott, J. Mast, A. Mortensen, A. G. Oomen, S. Weigel, E. Barthelemy, A. Rincon, J. Tarazona and R. Schoonjans, Guidance on risk assessment of nanomaterials to be applied in the food and feed chain: human and animal health, EFSA J., 2021, 19, e06769 Search PubMed.
- G. Federici, B. J. Shaw and R. D. Handy, Toxicity of titanium dioxide nanoparticles to rainbow trout (Oncorhynchus mykiss): gill injury, oxidative stress, and other physiological effects, Aquat. Toxicol., 2007, 84, 415–430 CrossRef CAS PubMed.
- N. J. Clark, D. Boyle, B. P. Eynon and R. D. Handy, Dietary exposure to silver nitrate compared to two forms of silver nanoparticles in rainbow trout: bioaccumulation potential with minimal physiological effects, Environ. Sci.: Nano, 2019, 6, 1393–1405 RSC.
- S. Juling, G. Bachler, N. von Götz, D. Lichtenstein, L. Böhmert, A. Niedzwiecka, S. Selve, A. Braeuning and A. Lampen, In vivo distribution of nanosilver in the rat: The role of ions and de novo-formed secondary particles, Food Chem. Toxicol., 2016, 97, 327–335 CrossRef CAS PubMed.
- B. J. Shaw and R. D. Handy, Physiological effects of nanoparticles on fish: a comparison of nanometals versus metal ions, Environ. Int., 2011, 37, 1083–1097 CrossRef CAS PubMed.
- G. A. Al-Bairuty, B. J. Shaw, R. D. Handy and T. B. Henry, Histopathological effects of waterborne copper nanoparticles and copper sulphate on the organs of rainbow trout (Oncorhynchus mykiss), Aquat. Toxicol., 2013, 126, 104–115 CrossRef CAS PubMed.
- R. D. Handy, N. van den Brink, M. Chappell, M. Mühling, R. Behra, M. Dušinská, P. Simpson, J. Ahtiainen, A. N. Jha and J. Seiter, Practical considerations for conducting ecotoxicity test methods with manufactured nanomaterials: what have we learnt so far?, Ecotoxicology, 2012, 21, 933–972 CrossRef CAS PubMed.
- C. Guo, S. Robertson, R. J. Weber, A. Buckley, J. Warren, A. Hodgson, J. Z. Rappoport, K. Ignatyev, K. Meldrum and I. Römer, Pulmonary toxicity of inhaled nano-sized cerium oxide aerosols in Sprague–Dawley rats, Nanotoxicology, 2019, 13, 733–750 CrossRef CAS PubMed.
- B. D. Johnston, T. M. Scown, J. Moger, S. A. Cumberland, M. Baalousha, K. Linge, R. van Aerle, K. Jarvis, J. R. Lead and C. R. Tyler, Bioavailability of nanoscale metal oxides TiO2, CeO2, and ZnO to fish, Environ. Sci. Technol., 2010, 44, 1144–1151 CrossRef CAS PubMed.
- R. R. Mercer, J. F. Scabilloni, L. Wang, L. A. Battelli, J. M. Antonini, J. R. Roberts, Y. Qian, J. D. Sisler, V. Castranova and D. W. Porter, The fate of inhaled nanoparticles: detection and measurement by enhanced dark-field microscopy, Toxicol. Pathol., 2018, 46, 28–46 CrossRef CAS PubMed.
- K. Tervonen, G. Waissi, E. J. Petersen, J. Akkanen and J. V. Kukkonen, Analysis of fullerene-C60 and kinetic measurements for its accumulation and depuration in Daphnia magna, Environ. Toxicol. Chem., 2010, 29, 1072–1078 CAS.
- B. J. Shaw, C. S. Ramsden, A. Turner and R. D. Handy, A simplified method for determining titanium from TiO2 nanoparticles in fish tissue with a concomitant multi-element analysis, Chemosphere, 2013, 92, 1136–1144 CrossRef CAS PubMed.
- V. Filipe, A. Hawe and W. Jiskoot, Critical evaluation of Nanoparticle Tracking Analysis (NTA) by NanoSight for the measurement of nanoparticles and protein aggregates, Pharm. Res., 2010, 27, 796–810 CrossRef CAS PubMed.
- M. D. Montaño, J. W. Olesik, A. G. Barber, K. Challis and J. F. Ranville, Single particle ICP-MS: advances toward routine analysis of nanomaterials, Anal. Bioanal. Chem., 2016, 408, 5053–5074 CrossRef PubMed.
- R. J. Peters, Z. H. Rivera, G. van Bemmel, H. J. Marvin, S. Weigel and H. Bouwmeester, Development and validation of single particle ICP-MS for sizing and quantitative determination of nano-silver in chicken meat, Anal. Bioanal. Chem., 2014, 406, 3875–3885 CAS.
- Y. Q. Deng, E. J. Petersen, K. E. Challis, S. A. Rabb, R. D. Holbrook, J. F. Ranville, B. C. Nelson and B. S. Xing, Multiple method analysis of TiO2 nanoparticle uptake in rice (Oryza sativa L.) plants, Environ. Sci. Technol., 2017, 51, 10615–10623 CrossRef CAS PubMed.
- S. P. Elliott, D. W. Stephen and S. Paterson, The United Kingdom and Ireland association of forensic toxicologists forensic toxicology laboratory guidelines, Sci. Justice, 2018, 58, 335–345 CrossRef PubMed.
- R. Handy and M. Depledge, Physiological responses: their measurement and use as environmental biomarkers in ecotoxicology, Ecotoxicology, 1999, 8, 329–349 CrossRef CAS.
- N. J. Clark, D. Boyle and R. D. Handy, An assessment of the dietary bioavailability of silver nanomaterials in rainbow trout using an ex vivo gut sac technique, Environ. Sci.: Nano, 2019, 6, 646–660 RSC.
- E. J. Petersen, M. Mortimer, R. M. Burgess, R. Handy, S. Hanna, K. T. Ho, M. Johnson, S. Loureiro, H. Selck, J. J. Scott-Fordsmand, D. Spurgeon, J. Unrine, N. van den Brink, Y. Wang, J. White and P. Holden, Strategies for robust and accurate experimental approaches to quantify nanomaterial bioaccumulation across a broad range of organisms, Environ. Sci.: Nano, 2019, 6, 1619–1656 RSC.
- R. D. Handy, T. B. Henry, T. M. Scown, B. D. Johnston and C. R. Tyler, Manufactured nanoparticles: their uptake and effects on fish - a mechanistic analysis, Ecotoxicology, 2008, 17, 396–409 CrossRef CAS PubMed.
- A. R. Al-Jubory and R. D. Handy, Uptake of titanium from TiO2 nanoparticle exposure in the isolated perfused intestine of rainbow trout: nystatin, vanadate and novel CO2-sensitive components, Nanotoxicology, 2013, 7, 1282–1301 CrossRef CAS PubMed.
- N. J. Clark, W. Woznica and R. D. Handy, Dietary bioaccumulation potential of silver nanomaterials compared to silver nitrate in wistar rats using an ex vivo gut sac technique, Ecotoxicol. Environ. Saf., 2020, 200, 110745 CrossRef CAS PubMed.
- K. Tatsi, B. J. Shaw, T. H. Hutchinson and R. D. Handy, Copper accumulation and toxicity in earthworms exposed to CuO nanomaterials: Effects of particle coating and soil ageing, Ecotoxicol. Environ. Saf., 2018, 166, 462–473 CrossRef CAS PubMed.
- M. van der Zande, A. Jemec Kokalj, D. J. Spurgeon, S. Loureiro, P. V. Silva, Z. Khodaparast, D. Drobne, N. J. Clark, N. W. van den Brink, M. Baccaro, C. A. M. van Gestel, H. Bouwmeester and R. D. Handy, The gut barrier and the fate of engineered nanomaterials: a view from comparative physiology, Environ. Sci.: Nano, 2020, 7, 1874–1898 RSC.
- M. Baccaro, A. K. Undas, J. de Vriendt, J. Van Den Berg, R. Peters and N. W. van den Brink, Ageing, dissolution and biogenic formation of nanoparticles: how do these factors affect the uptake kinetics of silver nanoparticles in earthworms?, Environ. Sci.: Nano, 2018, 5, 1107–1116 RSC.
- K. Loeschner, J. Navratilova, R. Grombe, T. P. J. Linsinger, C. Kobler, K. Molhave and E. H. Larsen, In-house validation of a method for determination of silver nanoparticles in chicken meat based on asymmetric flow field-flow fractionation and inductively coupled plasma mass spectrometric detection, Food Chem., 2015, 181, 78–84 CrossRef CAS PubMed.
- G. Chen and W. Wang, Role of freeze drying in nanotechnology, Drying Technol., 2007, 25, 29–35 CrossRef CAS.
- K. S. Subramanian, Determination of metals in biofluids and tissues: sample preparation methods for atomic spectroscopic techniques, Spectrochim. Acta, Part B, 1996, 51, 291–319 CrossRef.
- W. Ebeling, N. Hennrich, M. Klockow, H. Metz, H. D. Orth and H. Lang, Proteinase K from Tritirachium album limber, Eur. J. Biochem., 1974, 47, 91–97 CrossRef CAS PubMed.
- J. Bajorath, W. Hinrichs and W. Saenger, The enzymatic activity of proteinase K is controlled by calcium, Eur. J. Biochem., 1988, 176, 441–447 CrossRef CAS PubMed.
- K. Loeschner, J. Navratilova, C. Købler, K. Mølhave, S. Wagner, F. von der Kammer and E. H. Larsen, Detection and characterization of silver nanoparticles in chicken meat by asymmetric flow field flow fractionation with detection by conventional or single particle ICP-MS, Anal. Bioanal. Chem., 2013, 405, 8185–8195 CrossRef CAS PubMed.
- M. van der Zande, R. J. Vandebriel, E. Van Doren, E. Kramer, Z. Herrera Rivera, C. S. Serrano-Rojero, E. R. Gremmer, J. Mast, R. J. B. Peters, P. C. H. Hollman, P. J. M. Hendriksen, H. J. P. Marvin, A. A. C. M. Peijnenburg and H. Bouwmeester, Distribution, elimination, and toxicity of silver nanoparticles and silver ions in rats after 28-day oral exposure, ACS Nano, 2012, 6, 7427–7442 CrossRef CAS PubMed.
- R. Peters, Z. Herrera-Rivera, A. Undas, M. van der Lee, H. Marvin, H. Bouwmeester and S. Weigel, Single particle ICP-MS combined with a data evaluation tool as a routine technique for the analysis of nanoparticles in complex matrices, J. Anal. At. Spectrom., 2015, 30, 1274–1285 RSC.
- K. Loeschner, M. S. J. Brabrand, J. J. Sloth and E. H. Larsen, Use of alkaline or enzymatic sample pretreatment prior to characterization of gold nanoparticles in animal tissue by single-particle ICPMS, Anal. Bioanal. Chem., 2014, 406, 3845–3851 CrossRef CAS PubMed.
- S. Laughton, A. Laycock, G. Bland, F. von der Kammer, T. Hofmann, E. A. Casman and G. V. Lowry, Methanol-based extraction protocol for insoluble and moderately water-soluble nanoparticles in plants to enable characterization by single particle ICP-MS, Anal. Bioanal. Chem., 2021, 413, 299–314 CrossRef CAS PubMed.
- J. Wojcieszek, J. Jimenez-Lamana, L. Ruzik, J. Szpunar and M. Jarosz, To-do and not-to-do in model studies of the uptake, fate and metabolism of metal-containing nanoparticles in plants, Nanomaterials, 2020, 10, 1480 CrossRef CAS PubMed.
- B. Asadishad, S. Chahal, A. Akbari, V. Cianciarelli, M. Azodi, S. Ghoshal and N. Tufenkji, Amendment of agricultural soil with metal nanoparticles: effects on soil enzyme activity and microbial community composition, Environ. Sci. Technol., 2018, 52, 1908–1918 CrossRef CAS PubMed.
- M. Hosseini-Koupaei, B. Shareghi, A. A. Saboury, F. Davar, V. A. Sirotkin, M. H. Hosseini-Koupaei and Z. Enteshari, Catalytic activity, structure and stability of proteinase K in the presence of biosynthesized CuO nanoparticles, Int. J. Biol. Macromol., 2019, 122, 732–744 CrossRef CAS PubMed.
- M. E. Johnson, S. K. Hanna, A. R. M. Bustos, C. M. Sims, L. C. C. Elliott, A. Lingayat, A. C. Johnston, B. Nikoobakht, J. T. Elliott, R. D. Holbrook, K. C. K. Scoto, K. E. Murphy, E. J. Petersen, L. L. Yu and B. C. Nelson, Separation, sizing, and quantitation of engineered nanoparticles in an organism model using inductively coupled plasma mass spectrometry and image analysis, ACS Nano, 2017, 11, 526–540 CrossRef CAS PubMed.
- J. Noireaux, R. Grall, M. Hullo, S. Chevillard, C. Oster, E. Brun, C. Sicard-Roselli, K. Loeschner and P. Fisicaro, Gold nanoparticle uptake in tumor cells: Quantification and size distribution by sp-ICPMS, Separations, 2019, 6, 3 CrossRef CAS.
- Q. Zhou, L. Liu, N. Liu, B. He, L. Hu and L. Wang, Determination and characterization of metal nanoparticles in clams and oysters, Ecotoxicol. Environ. Saf., 2020, 198, 110670 CrossRef CAS PubMed.
- T. Ishizaka, K. Nagano, I. Tasaki, H. Tao, J. Q. Gao, K. Harada, K. Hirata, S. Saito, H. Tsujino, K. Higashisaka and Y. Tsutsumi, Optimization and evaluation of pretreatment method for sp-ICP-MS to reveal the distribution of silver nanoparticles in the body, Nanoscale Res. Lett., 2019, 14, 1–7 CrossRef CAS PubMed.
- N. J. Clark, R. Clough, D. Boyle and R. D. Handy, Development of a suitable detection method for silver nanoparticles in fish tissue using single particle ICP-MS, Environ. Sci.: Nano, 2019, 6, 3388–3400 RSC.
- H. K. Sung, E. Jo, E. Kim, S. K. Yoo, J. W. Lee, P. J. Kim, Y. Kim and I. C. Eom, Analysis of gold and silver nanoparticles internalized by zebrafish (Danio rerio) using single particle-inductively coupled plasma-mass spectrometry, Chemosphere, 2018, 209, 815–822 CrossRef CAS PubMed.
- J. Vidmar, T. Buerki-Thurnherr and K. Loeschner, Comparison of the suitability of alkaline or enzymatic sample pre-treatment for characterization of silver nanoparticles in human tissue by single particle ICP-MS, J. Anal. At. Spectrom., 2018, 33, 752–761 RSC.
- C. C. Li, F. Dang, M. Li, M. Zhu, H. Zhong, H. Hintelmann and D. M. Zhou, Effects of exposure pathways on the accumulation and phytotoxicity of silver nanoparticles in soybean and rice, Nanotoxicology, 2017, 11, 699–709 CrossRef CAS PubMed.
- R. Á.-F. García, N. Fernández-Iglesias, C. López-Chaves, C. Sánchez-González, J. Llopis, M. Montes-Bayón and J. Bettmer, Complementary techniques (spICP-MS, TEM, and HPLC-ICP-MS) reveal the degradation of 40 nm citrate-stabilized Au nanoparticles in rat liver after intraperitoneal injection, J. Trace Elem. Med. Biol., 2019, 55, 1–5 CrossRef PubMed.
- B. Gomez-Gomez, M. T. Perez-Corona and Y. Madrid, Using single-particle ICP-MS for unravelling the effect of type of food on the physicochemical properties and gastrointestinal stability of ZnONPs released from packaging materials, Anal. Chim. Acta, 2020, 1100, 12–21 CrossRef CAS PubMed.
- B. Magnusson and U. Örnemark, The fitness for purpose of analytical methods: A laboratory guide to method validation and related topics, LGC, Teddington, Middlesex, UK, 2014 Search PubMed.
- M. Thompson, S. L. Ellison and R. Wood, Harmonized guidelines for single-laboratory validation of methods of analysis (IUPAC Technical Report), Pure Appl. Chem., 2002, 74, 835–855 CAS.
- ISO:TS19590:2017, Nanotechnologies - Size Distribution and Concentration of Inorganic Nanoparticles in Aqueous Media via Single Particle Inductively Coupled Plasma Mass Spectrometry, 2017 Search PubMed.
- T. P. J. Linsinger, R. Peters and S. Weigel, International interlaboratory study for sizing and quantification of Ag nanoparticles in food simulants by single-particle ICPMS, Anal. Bioanal. Chem., 2013, 406, 3835–3843 CrossRef PubMed.
- E. J. Petersen, A. R. Montoro Bustos, B. Toman, M. E. Johnson, M. Ellefson, G. C. Caceres, A. L. Neuer, Q. Chan, J. W. Kemling, B. Mader, K. Murphy and M. Roesslein, Determining what really counts: modeling and measuring nanoparticle number concentrations, Environ. Sci.: Nano, 2019, 6, 2876–2896 RSC.
- K. Loeschner, C. F. Harrington, J. L. Kearney, D. J. Langton and E. H. Larsen, Feasibility of asymmetric flow field-flow fractionation coupled to ICP-MS for the characterization of wear metal particles and metalloproteins in biofluids from hip replacement patients, Anal. Bioanal. Chem., 2015, 407, 4541–4554 CrossRef CAS PubMed.
- Z. Gajdosechova, M. M. Lawan, D. S. Urgast, A. Raab, K. G. Scheckel, E. Lombi, P. M. Kopittke, K. Loeschner, E. H. Larsen, G. Woods, A. Brownlow, F. L. Read, J. Feldmann and E. M. Krupp, In vivo formation of natural HgSe nanoparticles in the liver and brain of pilot whales, Sci. Rep., 2016, 6, 34361 CrossRef CAS PubMed.
- L. Xu, Z. Wang, J. Zhao, M. Lin and B. Xing, Accumulation of metal-based nanoparticles in marine bivalve mollusks from offshore aquaculture as detected by single particle ICP-MS, Environ. Pollut., 2020, 260, 114043 CrossRef CAS PubMed.
- B. Kollander, F. Widemo, E. Agren, E. H. Larsen and K. Loeschner, Detection of lead nanoparticles in game meat by single particle ICP-MS following use of lead-containing bullets, Anal. Bioanal. Chem., 2017, 409, 1877–1885 CrossRef CAS PubMed.
- E. P. Gray, J. G. Coleman, A. J. Bednar, A. J. Kennedy, J. F. Ranville and C. P. Higgins, Extraction and analysis of silver and gold nanoparticles from biological tissues using single particle inductively coupled plasma mass spectrometry, Environ. Sci. Technol., 2013, 47, 14315–14323 CrossRef CAS PubMed.
- D. M. Bernstein and J. Riego-Sintes, Methods for the determination of the hazardous properties for human health of man made mineral fibers (MMMF), European Chemicals Bureau, 1999 Search PubMed.
- F. A. Monikh, N. Grundschober, S. Romeijn, D. Arenas-Lago, M. G. Vijver, W. Jiskoot and W. J. Peijnenburg, Development of methods for extraction and analytical characterization of carbon-based nanomaterials (nanoplastics and carbon nanotubes) in biological and environmental matrices by asymmetrical flow field-flow fractionation, Environ. Pollut., 2019, 255, 113304 CrossRef PubMed.
- B. Xiao, Y. Zhang, X. Wang, M. Chen, B. Sun, T. Zhang and L. Zhu, Occurrence and trophic transfer of nanoparticulate Ag and Ti in the natural aquatic food web of Taihu Lake, China, Environ. Sci.: Nano, 2019, 6, 3431–3441 RSC.
- F. A. Monikh, L. Chupani, E. Zusková, R. Peters, M. Vancová, M. G. Vijver, P. Porcal and W. J. Peijnenburg, Method for extraction and quantification of metal-based nanoparticles in biological media: number-based biodistribution and bioconcentration, Environ. Sci. Technol., 2018, 53, 946–953 CrossRef PubMed.
- J. Modrzynska, T. Berthing, G. Ravn-Haren, K. Kling, A. Mortensen, R. R. Rasmussen, E. H. Larsen, A. T. Saber, U. Vogel and K. Loeschner, In vivo-induced size transformation of cerium oxide nanoparticles in both lung and liver does not affect long-term hepatic accumulation following pulmonary exposure, PLoS One, 2018, 13, e0202477 CrossRef PubMed.
- S. Wagener, H. Jungnickel, N. Dommershausen, T. Fischer, P. Laux and A. Luch, Determination of nanoparticle uptake, distribution, and characterization in plant root tissue after realistic long-term exposure to sewage sludge using information from mass spectrometry, Environ. Sci. Technol., 2019, 53, 5416–5426 CrossRef CAS PubMed.
- D. Mozhayeva and C. Engelhard, A critical review of single particle inductively coupled plasma mass spectrometry – A step towards an ideal method for nanomaterial characterization, J. Anal. At. Spectrom., 2020, 35, 1740–1783 RSC.
- J. Liu, K. E. Murphy, R. I. MacCuspie and M. R. Winchester, Capabilities of single particle inductively coupled plasma mass spectrometry for the size measurement of nanoparticles: a case study on gold nanoparticles, Anal. Chem., 2014, 86, 3405–3414 CrossRef CAS PubMed.
- M. Corte-Rodríguez, R. Álvarez-Fernández, P. García-Cancela, M. Montes-Bayón and J. Bettmer, Single cell ICP-MS using on line sample introduction systems: Current developments and remaining challenges, TrAC, Trends Anal. Chem., 2020, 132, 116042 CrossRef.
- Blanks in Method Validation - Supplement to Eurachem Guide the Fitness for Purpose of Analytical Methods, ed. H. Cantwell, 1st edn, 2019, Available from http://www.eurachem.org Search PubMed.
- B. E. Brown, The form and function of metal-containing 'granules' in invertebrate tissues, Biol. Rev., 1982, 57, 621–667 CrossRef CAS.
- M. D. Montaño, F. von der Kammer, C. W. Cuss and J. F. Ranville, Opportunities for examining the natural nanogeochemical environment using recent advances in nanoparticle analysis, J. Anal. At. Spectrom., 2019, 34, 1768–1772 RSC.
- S. Makama, J. Piella, A. Undas, W. J. Dimmers, R. Peters, V. F. Puntes and N. W. van den Brink, Properties of Silver Nanoparticles Influencing their Uptake in and Toxicity to the Earthworm Lumbricus rubellus Following Exposure in Soil, Environ. Pollut., 2016, 218, 870–878 CrossRef CAS PubMed.
- CEN (European Committee for Standardisation), Nanotechnologies - Guidance on Detection and Identification of Nano-objects in Complex Matrices, European Committee for Standardisation, 2018 Search PubMed.
- J. R. Lead, G. E. Batley, P. J. J. Alvarez, M. N. Croteau, R. D. Handy, M. J. McLaughlin, J. D. Judy and K. Schirmer, Nanomaterials in the environment: Behavior, fate, bioavailability, and effects-An updated review, Environ. Toxicol. Chem., 2018, 37, 2029–2063 CrossRef CAS PubMed.
- F. Laborda, J. Jiménez-Lamana, E. Bolea and J. R. Castillo, Selective identification, characterization and determination of dissolved silver(I) and silver nanoparticles based on single particle detection by inductively coupled plasma mass spectrometry, J. Anal. At. Spectrom., 2011, 26, 1362–1371 RSC.
- ISO:17034:2016, General Requirements for the Competence of Reference Material Producers, 2016 Search PubMed.
- A. R. Montoro Bustos, E. J. Petersen, A. Possolo and M. R. Winchester, Post hoc interlaboratory comparison of single particle ICP-MS size measurements of NIST gold nanoparticle reference materials, Anal. Chem., 2015, 87, 8809–8817 CrossRef CAS PubMed.
- R. Grombe, G. Allmaier, J. Charoud-Got, A. Dudkiewicz, H. Emteborg, T. Hofmann, E. H. Larsen, A. Lehner, M. Llinàs and K. Loeschner, Feasibility of the development of reference materials for the detection of Ag nanoparticles in food: neat dispersions and spiked chicken meat, Accredit. Qual. Assur., 2015, 20, 3–16 CrossRef CAS.
- S. Weigel, R. Peters, K. Loeschner, R. Grombe and T. P. J. Linsinger, Results of an interlaboratory method performance study for the size determination and quantification of silver nanoparticles in chicken meat by single-particle inductively coupled plasma mass spectrometry (sp-ICP-MS), Anal. Bioanal. Chem., 2017, 409, 4839–4848 CrossRef CAS PubMed.
- O. Geiss, J. Ponti, C. Senaldi, I. Bianchi, D. Mehn, J. Barrero, D. Gilliland, R. Matissek and E. Anklam, Characterisation of food grade titania with respect to nanoparticle content in pristine additives and in their related food products, Food Addit. Contam., Part A, 2020, 37, 239–253 CrossRef CAS PubMed.
- P. S. Rainbow, Trace metal bioaccumulation: models, metabolic availability and toxicity, Environ. Int., 2007, 33, 576–582 CrossRef CAS PubMed.
- G. Roebben, K. Rasmussen, V. Kestens, T. P. J. Linsinger, H. Rauscher, H. Emons and H. Stamm, Reference materials and representative test materials: the nanotechnology case, J. Nanopart. Res., 2013, 15, 1455 CrossRef.
- G. Roebben, V. A. Hackley and H. Emons, Reference nanomaterials to improve the reliability of nanoscale measurements, Metrology and Standardization of Nanotechnology: Protocols and Industrial Innovations, ed. E. Mansfield, D. L. Kaiser, D. Fujita and M. Van de Voorde, Wiley-VCH, Germany, 2017, pp. 307–322 Search PubMed.
- G. Roebben, V. Kestens, Z. Varga, J. Charoud-Got, Y. Ramaye, C. Gollwitzer, D. Bartczak, D. Geißler, J. Noble, S. Mazoua, N. Meeus, P. Corbisier, M. Palmai, J. Mihály, M. Krumrey, J. Davies, U. Resch-Genger, N. Kumarswami, C. Minelli, A. Sikora and H. Goenaga-Infante, Reference materials and representative test materials to develop nanoparticle characterization methods: the NanoChOp project case, Front. Chem., 2015, 3, 56 Search PubMed.
- H. E. Pace, N. J. Rogers, C. Jarolimek, V. A. Coleman, C. P. Higgins and J. F. Ranville, Determining transport efficiency for the purpose of counting and sizing nanoparticles via single particle inductively coupled plasma mass spectrometry, Anal. Chem., 2011, 83, 9361–9369 CrossRef CAS PubMed.
- J. Tuoriniemi, G. Cornelis and M. Hassellov, Size discrimination and detection capabilities of single-particle ICPMS for environmental analysis of silver nanoparticles, Anal. Chem., 2012, 84, 3965–3972 CrossRef CAS PubMed.
- F. Laborda, E. Bolea and J. Jiménez-Lamana, Single particle inductively coupled plasma mass spectrometry for the analysis of inorganic engineered nanoparticles in environmental samples, Trends Environ. Anal. Chem., 2016, 9, 15–23 CrossRef CAS.
- C. Stephen and R. Thomas, Single-particle ICP-MS: a key analytical technique for characterizing nanoparticles, Spectroscopy, 2017, 32, 12–25 Search PubMed.
- S. Cuello-Nuñez, I. Abad-Álvaro, D. Bartczak, M. E. del Castillo Busto, D. A. Ramsay, F. Pellegrino and H. Goenaga-Infante, The accurate determination of number concentration of inorganic nanoparticles using spICP-MS with the dynamic mass flow approach, J. Anal. At. Spectrom., 2020, 35, 1832–1839 RSC.
- H. Goenaga-Infante and D. Bartczak, Single Particle Inductively Coupled Plasma Mass Spectrometry (spICP-MS), in Characterization of Nanoparticles, ed. V.-D. Hodoroaba, W. E. S. Unger and A. G. Shard, Elsevier, Amsterdam, 2020, pp. 65–77 Search PubMed.
- ISO5725-4:2020, Accuracy (Trueness and Precision) of Measurement Methods and Results, 2020 Search PubMed.
- S. Lee, X. Bi, R. B. Reed, J. F. Ranville, P. Herckes and P. Westerhoff, Nanoparticle size detection limits by single particle ICP-MS for 40 elements, Environ. Sci. Technol., 2014, 48, 10291–10300 CrossRef CAS PubMed.
- F. Laborda, E. Bolea and J. Jimenez-Lamana, Single particle inductively coupled plasma mass spectrometry: a powerful tool for nanoanalysis, Anal. Chem., 2014, 86, 2270–2278 CrossRef CAS PubMed.
- D. P. Bao, Z. G. Oh and Z. Chen, Characterization of silver nanoparticles internalized by arabidopsis plants using single particle ICP-MS analysis, Front. Plant Sci., 2016, 7, 32 Search PubMed.
- M. B. Heringa, R. J. B. Peters, R. L. A. W. Bleys, M. K. van der Lee, P. C. Tromp, P. C. E. van Kesteren, J. C. H. van Eijkeren, A. K. Undas, A. G. Oomen and H. Bouwmeester, Detection of titanium particles in human liver and spleen and possible health implications, Part. Fibre Toxicol., 2018, 15, 1–9 CrossRef PubMed.
- K. Kinska, J. Jimenez-Lamana, J. Kowalska, B. Krasnodebska-Ostrega and J. Szpunar, Study of the uptake and bioaccumulation of palladium nanoparticles by Sinapis alba using single particle ICP-MS, Sci. Total Environ., 2018, 615, 1078–1085 CrossRef CAS PubMed.
- R. J. Peters, A. K. Undas, J. Memelink, G. van Bemmel, S. Munniks, H. Bouwmeester, P. Nobels, W. Schuurmans and M. K. van der Lee, Development and validation of a method for the detection of titanium dioxide particles in human tissue with single particle ICP-MS, Current Trends in Analytical Bioanalytical Chemistry, 2018, vol. 2 Search PubMed.
- M. V. Taboada-Lopez, N. Alonso-Seijo, P. Herbello-Hermelo, P. Bermejo-Barrera and A. Moreda-Pineiro, Determination and characterization of silver nanoparticles in bivalve molluscs by ultrasound assisted enzymatic hydrolysis and sp-ICP-MS, Microchem. J., 2019, 148, 652–660 CrossRef CAS.
- M. V. Taboada-Lopez, P. Herbello-Hermelo, R. Dominguez-Gonzalez, P. Bermejo-Barrera and A. Moreda-Pineiro, Enzymatic hydrolysis as a sample pre-treatment for titanium dioxide nanoparticles assessment in surimi (crab sticks) by single particle ICP-MS, Talanta, 2019, 195, 23–32 CrossRef CAS PubMed.
- F. Aureli, M. Ciprotti, M. D'Amato, E. do Nascimento da Silva, S. Nisi, D. Passeri, A. Sorbo, A. Raggi, M. Rossi and F. Cubadda, Determination of total silicon and SiO2 particles using an ICP-MS based analytical platform for toxicokinetic studies of synthetic amorphous silica, Nanomaterials, 2020, 10, 888 CrossRef CAS PubMed.
- F. Laborda, J. Jiménez-Lamana, E. Bolea and J. R. Castillo, Critical considerations for the determination of nanoparticle number concentrations, size and number size distributions by single particle ICP-MS, J. Anal. At. Spectrom., 2013, 28, 1220–1232 RSC.
- B. Meermann and V. Nischwitz, ICP-MS for the analysis at the nanoscale – a tutorial review, J. Anal. At. Spectrom., 2018, 33, 1432–1468 RSC.
- H. El Hadri, E. J. Petersen and M. R. Winchester, Impact of and correction for instrument sensitivity drift on nanoparticle size measurements by single-particle ICP-MS, Anal. Bioanal. Chem., 2016, 408, 5099–5108 CrossRef CAS PubMed.
- J. Tentschert, P. Laux, H. Jungnickel, J. Brunner, I. Estrela-Lopis, C. Merker, J. Meijer, H. Ernst, L. Ma-Hock and J. Keller, Organ burden of inhaled nanoceria in a 2-year low-dose exposure study: dump or depot?, Nanotoxicology, 2020, 14, 554–576 CrossRef CAS PubMed.
- D. Metarapi, M. Sala, K. Vogel-Mikus, V. S. Selih and J. T. van Elteren, Nanoparticle analysis in biomaterials using laser ablation-single particle-inductively coupled plasma mass spectrometry, Anal. Chem., 2019, 91, 6200–6205 CrossRef CAS PubMed.
- D. Metarapi, J. T. van Elteren, M. Šala, K. Vogel-Mikuš, I. Arčon, V. S. Šelih, M. Kolar and S. B. Hočevar, Laser ablation-single-particle-inductively coupled plasma mass spectrometry as a multimodality bioimaging tool in nano-based omics, Environ. Sci.: Nano, 2021, 8, 647–656 RSC.
- F. Gallocchio, G. Biancotto, V. Cibin, C. Losasso, S. Belluco, R. Peters, G. van Bemmel, C. Cascio, S. Weigel and P. Tromp, Transfer study of silver nanoparticles in poultry production, J. Agric. Food Chem., 2017, 65, 3767–3774 CrossRef CAS PubMed.
- S. Makama, R. Peters, A. Undas and N. W. van den Brink, A novel method for the quantification, characterisation and speciation of silver nanoparticles in earthworms exposed in soil, Environ. Chem., 2015, 12, 643–651 CrossRef CAS.
- M. V. Taboada-Lopez, S. Iglesias-Lopez, P. Herbello-Hermelo, P. Bermejo-Barrera and A. Moreda-Pineiro, Ultrasound assisted enzymatic hydrolysis for isolating titanium dioxide nanoparticles from bivalve mollusk before sp-ICP-MS, Anal. Chim. Acta, 2018, 1018, 16–25 CrossRef CAS PubMed.
- J. Vidmar, K. Loeschner, M. Correia, E. H. Larsen, P. Manser, A. Wichser, K. Boodhia, Z. S. Al-Ahmady, J. Ruiz, D. Astruc and T. Buerki-Thurnherr, Translocation of silver nanoparticles in the ex vivo human placenta perfusion model characterized by single particle ICP-MS, Nanoscale, 2018, 10, 11980–11991 RSC.
- C. Schlechtriem, B. Knopf, S. Kühr, B. Meisterjahn and N. Schröder, Development of a method to determine the bioaccumulation of manufactured nanomaterials in filtering organisms (Bivalvia), German Environment Agency, 2020 Search PubMed.
- S. Kuehr, B. Meisterjahn, N. Schröder, B. Knopf, D. Völker, K. Schwirn and C. Schlechtriem, Testing the bioaccumulation of manufactured nanomaterials in the freshwater bivalve Corbicula fluminea using a new test method, Environ. Sci.: Nano, 2020, 7, 535–553 RSC.
- S. Kuehr, J. Klehm, C. Stehr, M. Menzel and C. Schlechtriem, Unravelling the uptake pathway and accumulation of silver from manufactured silver nanoparticles in the freshwater amphipod Hyalella azteca using correlative microscopy, NanoImpact, 2020, 19, 100239 CrossRef.
- F. Gallocchio, G. Biancotto, A. Moressa, F. Pascoli, T. Pretto, A. Toffan, G. Arcangeli, F. Montesi, R. Peters and A. Ricci, Bioaccumulation and in vivo formation of titanium dioxide nanoparticles in edible mussels, Food Chem., 2020, 323, 126841 CrossRef CAS PubMed.
- R. Tassinari, F. Cubadda, G. Moracci, F. Aureli, M. D'Amato, M. Valeri, B. De Berardis, A. Raggi, A. Mantovani and D. Passeri, Oral, short-term exposure to titanium dioxide nanoparticles in Sprague-Dawley rat: focus on reproductive and endocrine systems and spleen, Nanotoxicology, 2014, 8, 654–662 CrossRef CAS PubMed.
- R. J. B. Peters, A. G. Oomen, G. van Bemmel, L. van Vliet, A. K. Undas, S. Munniks, R. Bleys, P. C. Tromp, W. Brand and M. van der Lee, Silicon dioxide and titanium dioxide particles found in human tissues, Nanotoxicology, 2020, 14, 420–432 CrossRef CAS PubMed.
- Y. Huang, J. Tsz-Shan Lum and K. Sze-Yin Leung, Single particle ICP-MS combined with internal standardization for accurate characterization of polydisperse nanoparticles in complex matrices, J. Anal. At. Spectrom., 2020, 35, 2148–2155 RSC.
- J. Jiménez-Lamana, I. Abad-Álvaro, K. Bierla, F. Laborda, J. Szpunar and R. Lobinski, Detection and characterization of biogenic selenium nanoparticles in selenium-rich yeast by single particle ICPMS, J. Anal. At. Spectrom., 2018, 33, 452–460 RSC.
- L. Campagnolo, M. Massimiani, L. Vecchione, D. Piccirilli, N. Toschi, A. Magrini, E. Bonanno, M. Scimeca, L. Castagnozzi and G. Buonanno, Silver nanoparticles inhaled during pregnancy reach and affect the placenta and the foetus, Nanotoxicology, 2017, 11, 687–698 CrossRef CAS PubMed.
- H. Tao, K. Nagano, I. Tasaki, T. Q. Zhang, T. Ishizaka, J. Q. Gao, K. Harada, K. Hirata, H. Tsujino, K. Higashisaka and Y. Tsutsumi, Development and evaluation of a system for the semi-quantitative determination of the physical properties of skin after exposure to silver nanoparticles, Nanoscale Res. Lett., 2020, 15, 187 CrossRef CAS PubMed.
- Y. Dan, W. Zhang, R. Xue, X. Ma, C. Stephan and H. Shi, Characterization of gold nanoparticle uptake by tomato plants using enzymatic extraction followed by single-particle inductively coupled plasma-mass spectrometry analysis, Environ. Sci. Technol., 2015, 49, 3007–3014 CrossRef CAS PubMed.
- J. Jimenez-Lamana, J. Wojcieszek, M. Jakubiak, M. Asztemborska and J. Szpunar, Single particle ICP-MS characterization of platinum nanoparticles uptake and bioaccumulation by Lepidium sativum and Sinapis alba plants, J. Anal. At. Spectrom., 2016, 31, 2321–2329 RSC.
- J. Nath, I. Dror, P. Landa, T. Vanek, I. Kaplan-Ashiri and B. Berkowitz, Synthesis and characterization of isotopically-labeled silver, copper and zinc oxide nanoparticles for tracing studies in plants, Environ. Pollut., 2018, 242, 1827–1837 CrossRef CAS PubMed.
- W. Y. Zhang, Q. Wang, M. Li, F. Dang and D. M. Zhou, Nonselective uptake of silver and gold nanoparticles by wheat, Nanotoxicology, 2019, 13, 1073–1086 CrossRef CAS PubMed.
- F. Dang, Q. Wang, W. P. Cai, D. M. Zhou and B. S. Xing, Uptake kinetics of silver nanoparticles by plant: relative importance of particles and dissolved ions, Nanotoxicology, 2020, 14, 654–666 CrossRef CAS PubMed.
- L. Torrent, M. Iglesias, E. Marguí, M. Hidalgo, D. Verdaguer, L. Llorens, A. Kodre, A. Kavčič and K. Vogel-Mikuš, Uptake, translocation and ligand of silver in Lactuca sativa exposed to silver nanoparticles of different size, coatings and concentration, J. Hazard. Mater., 2020, 384, 121201 CrossRef CAS PubMed.
- Y. B. Dan, X. M. Ma, W. L. Zhang, K. Liu, C. Stephan and H. L. Shi, Single particle ICP-MS method development for the determination of plant uptake and accumulation of CeO2 nanoparticles, Anal. Bioanal. Chem., 2016, 408, 5157–5167 CrossRef CAS PubMed.
- J. Wojcieszek, J. Jimenez-Lamana, K. Bierla, L. Ruzik, M. Asztemborska, M. Jarosz and J. Szpunar, Uptake, translocation, size characterization and localization of cerium oxide nanoparticles in radish (Raphanus sativus L.), Sci. Total Environ., 2019, 683, 284–292 CrossRef CAS PubMed.
- A. A. Keller, Y. Huang and J. Nelson, Detection of nanoparticles in edible plant tissues exposed to nano-copper using single-particle ICP-MS, J. Nanopart. Res., 2018, 20, 1–13 CrossRef CAS.
- J. Wojcieszek, J. Jimenez-Lamana, K. Bierla, M. Asztemborska, L. Ruzik, M. Jarosz and J. Szpunar, Elucidation of the fate of zinc in model plants using single particle ICP-MS and ESI tandem MS, J. Anal. At. Spectrom., 2019, 34, 683–693 RSC.
- P. Wang, E. Lombi, S. K. Sun, K. G. Scheckel, A. Malysheva, B. A. McKenna, N. W. Menzies, F. J. Zhao and P. M. Kopittke, Characterizing the uptake, accumulation and toxicity of silver sulfide nanoparticles in plants, Environ. Sci.: Nano, 2017, 4, 448–460 RSC.
- J. Wojcieszek, J. Jiménez-Lamana, L. Ruzik, M. Asztemborska, M. Jarosz and J. Szpunar, Characterization of TiO2 NPs in radish (Raphanus sativus L.) by single-particle ICP-QQQ-MS, Front. Environ. Sci., 2020, 8, 100 CrossRef.
- S. Laughton, A. Laycock, F. von der Kammer, T. Hofmann, E. A. Casman, S. M. Rodrigues and G. V. Lowry, Persistence of copper-based nanoparticle-containing foliar sprays in Lactuca sativa (lettuce) characterized by spICP-MS, J. Nanopart. Res., 2019, 21, 1–13 CrossRef CAS.
- A. P. Walczak, R. Fokkink, R. Peters, P. Tromp, Z. E. Herrera Rivera, I. M. Rietjens, P. J. Hendriksen and H. Bouwmeester, Behaviour of silver nanoparticles and silver ions in an in vitro human gastrointestinal digestion model, Nanotoxicology, 2012, 7, 1198–1210 CrossRef PubMed.
- L. D. Scanlan, R. B. Reed, A. V. Loguinov, P. Antczak, A. Tagmount, S. Aloni, D. T. Nowinski, P. Luong, C. Tran, N. Karunaratne, D. Pham, X. X. Lin, F. Falciani, C. P. Higgins, J. F. Ranville, C. D. Vulpe and B. Gilbert, Silver nanowire exposure results in internalization and toxicity to Daphnia magna, ACS Nano, 2013, 7, 10681–10694 CrossRef CAS PubMed.
- K. Ramos, L. Ramos and M. M. Gomez-Gomez, Simultaneous characterisation of silver nanoparticles and determination of dissolved silver in chicken meat subjected to in vitro human gastrointestinal digestion using single particle inductively coupled plasma mass spectrometry, Food Chem., 2017, 221, 822–828 CrossRef CAS PubMed.
- K. Neubauer, C. Stephan and C. Shelton, Determination of gold and silver nanoparticles in blood using single particle ICP-MS, Perkin Elmer Application Note, 2014 Search PubMed.
- C.-M. Cirtiu, N. Fleury, C. Stephan and C. Shelton, Assessing the fate of nanoparticles in biological fluids using SP-ICP-MS, PerkinElmer application note, 2015 Search PubMed.
- M. Roman, C. Rigo, H. Castillo-Michel, I. Munivrana, V. Vindigni, I. Mičetić, F. Benetti, L. Manodori and W. R. L. Cairns, Hydrodynamic chromatography coupled to single-particle ICP-MS for the simultaneous characterization of AgNPs and determination of dissolved Ag in plasma and blood of burn patients, Anal. Bioanal. Chem., 2016, 408, 5109–5124 CrossRef CAS PubMed.
- M. Witzler, F. Küllmer and K. Günther, Validating a single-particle ICP-MS method to measure nanoparticles in human whole blood for nanotoxicology, Anal. Lett., 2017, 51, 587–599 CrossRef.
- K. Badalova, P. Herbello-Hermelo, P. Bermejo-Barrera and A. Moreda-Pineiro, Possibilities of single particle-ICP-MS for determining/characterizing titanium dioxide and silver nanoparticles in human urine, J. Trace Elem. Med. Biol., 2019, 54, 55–61 CrossRef CAS PubMed.
- A. Abdelkhaliq, M. van der Zande, A. K. Undas, R. J. B. Peters and H. Bouwmeester, Impact of in vitro digestion on gastrointestinal fate and uptake of silver nanoparticles with different surface modifications, Nanotoxicology, 2020, 14, 111–126 CrossRef CAS PubMed.
- D. M. Peloquin, E. J. Baumann, Jr. and T. P. Luxton, Multi-method assessment of PVP-coated silver nanoparticles and artificial sweat mixtures, Chemosphere, 2020, 249, 126173 CrossRef CAS PubMed.
- B. Bocca, B. Battistini and F. Petrucci, Silver and gold nanoparticles characterization by SP-ICP-MS and AF4-FFF-MALS-UV-ICP-MS in human samples used for biomonitoring, Talanta, 2020, 220, 121404 CrossRef CAS PubMed.
- X. He, H. Zhang, H. Shi, W. Liu and E. Sahle-Demessie, Fates of Au, Ag, ZnO, and CeO2 nanoparticles in simulated gastric fluid studied using single-particle-inductively coupled plasma-mass spectrometry, J. Am. Soc. Mass Spectrom., 2020, 31, 2180–2190 CrossRef CAS PubMed.
- S. Salou, C. M. Cirtiu, D. Lariviere and N. Fleury, Assessment of strategies for the formation of stable suspensions of titanium dioxide nanoparticles in aqueous media suitable for the analysis of biological fluids, Anal. Bioanal. Chem., 2020, 412, 1469–1481 CrossRef CAS PubMed.
- S. Salou, D. Lariviere, C. M. Cirtiu and N. Fleury, Quantification of titanium dioxide nanoparticles in human urine by single-particle ICP-MS, Anal. Bioanal. Chem., 2021, 413, 171–181 CrossRef CAS PubMed.
- S. V. Jenkins, H. Qu, T. Mudalige, T. M. Ingle, R. Wang, F. Wang, P. C. Howard, J. Chen and Y. Zhang, Rapid determination of plasmonic nanoparticle agglomeration status in blood, Biomaterials, 2015, 51, 226–237 CrossRef CAS PubMed.
- Y. Sun, N. Liu, Y. Wang, Y. Yin, G. Qu, J. Shi, M. Song, L. Hu, B. He, G. Liu, Y. Cai, Y. Liang and G. Jiang, Monitoring AuNP dynamics in the blood of a single mouse using single particle inductively coupled plasma mass spectrometry with an ultralow-volume high-efficiency introduction system, Anal. Chem., 2020, 92, 14872–14877 CrossRef CAS PubMed.
- K. C. Nwoko, A. Raab, L. Cheyne, D. Dawson, E. Krupp and J. Feldmann, Matrix-dependent size modifications of iron oxide nanoparticles (Ferumoxytol) spiked into rat blood cells and plasma: Characterisation with TEM, AF4-UV-MALS-ICP-MS/MS and spICP-MS, J. Chromatogr., B, 2019, 1124, 356–365 CrossRef CAS PubMed.
- M. Hayder, J. Wojcieszek, M. Asztemborska, Y. Zhou and L. Ruzik, Analysis of cerium oxide and copper oxide nanoparticles bioaccessibility from radish using SP-ICP-MS, J. Sci. Food Agric., 2020, 100, 4950–4958 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |