
Crystalline chalcogenidometalate-based compounds from uncommon reaction media
Zhou
Wu†
,
Gina
Stuhrmann†
and
Stefanie
Dehnen
 *
*
Fachbereich Chemie and Wissenschaftliches Zentrum für Materialwissenschaften, Philipps University Marburg, Hans-Meerwein-Straße 4, 35043 Marburg, Germany. E-mail: dehnen@chemie.uni-marburg.de
First published on 15th September 2022
Abstract
Chalcogenides are one of the most versatile inorganic materials families, further subdivided into a large variety of specific groups of compounds, ranging from neat binary or multinary solids and nanoparticles of the same formal compositions, both in crystalline or non-crystalline form, to complicated open-framework structures and cluster compounds, also including organ(ometall)ic derivates of the latter. The large variety regarding both the compositions and the structures is associated with an enormous variety of properties, ranging from simple or high-tech pigments through a multitude of opto-electronic devices and electrolytes to materials for ion separation or high-sophisticated catalysts. Naturally, this also goes hand in hand with a corrosponding breadth of synthesis strategies. Traditionally, chalcogenides have been accessed via high-temperature methods, which continuously have been replaced by lower-temperature approaches for economical and ecological reasons. Moreover, more recent methods also showed that new types of chalcogenide materials can be obtained under such milder conditions that are not accessible via traditional routes. To shed light onto one of the numerous families of chalcogenides, this feature article summarizes current achievements in the generation of multinary chalcogenidometallate-based clusters and networks via non-classical routes, using ionic liquids, surfactants, or hydrazine as reaction media at moderately elevated termperature.
 Zhou Wu | Zhou Wu received his diploma in 2017 from Jiaxing University and his master's degree in 2020 from Soochow University, before he started his PhD studies at Philipps-Universität Marburg under the supervision of Prof. Dr Stefanie Dehnen in the same year. He is currently focusing on the design and controlled synthesis of selenidometalate compounds from ionic liquids, and also on fundamental studies of corresponding cluster and framework compounds. |
 Gina Stuhrmann | Gina Stuhrmann studied chemistry, German language and literature, as well as education at Philipps University Marburg and graduated in 2021. Currently, she is a doctoral research fellow in the Dehnen group. Her research concentrates on the ionothermal synthesis of crystalline sulfidometalates and the selective functionalization of sulfidometalate clusters in and with ionic liquids. |
 Stefanie Dehnen | Stefanie Dehnen obtained her diploma in 1993 and her doctoral degree in 1996 from the University of Karlsruhe (KIT). After a postdoctoral stay in theoretical chemistry (1997) she completed her Habilitation in inorganic chemistry in 2004. As of 2006 she has been Full Professor of Inorganic Chemistry at Philipps University Marburg. She is a member of the European Academy of Sciences (EurASc) and of Leopoldina German National Academy of Sciences, and of several further scientific academies. Her most recent awards include the 2022 Leibniz Prize by the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG) and a 2022 Advanced Grant from the European Research Council (ERC). Current research in the Dehnen Labs is focused on the synthesis and experimental as well as quantum chemical investigation of compounds with multinary, in particular multimetallic, molecular nano-architectures, which possess potential as innovative catalysts, white-light emitters, or battery materials. |
1. Introduction
Compounds based on chalcogenidometalate clusters – either discrete or assembled into networks – have been in the focus of inorganic and material chemists due to their rich structural chemistry and their interesting physical and chemical properties. The unique properties include strong photoluminescence, superion conduction, selective ion exchange, photocatalysis, and specific gas adsorption.1–8 Porous structures are of particular interest as selective ion exchange materials.9–12 The properties of compounds based on chalcogenidometalate structures can be tailored by the building units on which they are based, whereby the pore size is the most variable parameter. By incorporating different types of p- or d-block metal ions into the chalcogenidometalate moieties, the structural diversity is largely increased and the physico-chemical properties are further tuned.13,14 However, also pure p-block chalcogenidometalates, such as the famous structures based on 2D-[Sn3S7]n2− anionic layers, exceptional properties arise, such as efficient ion-exchange for rare-earth element (REE) recovery based on a robust and cheap material.15The optoelectronic properties of chalcogenidometalate-based compounds can be fine-tuned by different approaches. One is the choice of the chalcogenidometalate building unit, for instance, 3D-T4-[Sn20O10S32] or 2D-T3-[Sn10O4S20] clusters.16 Both form the basis of photoconductive compounds, but the current density in the T4-based cluster is higher. The band gaps of both materials also shows differences, being larger in the T3-based compound. As another approach to tune photo-physical or photo-chemical properties is by doping or by changing the elemental combination of a given chalcogenide. The replacement of sulfur with selenium or tellurium generally leads to a reduction in the band gap energy, for instance, and this additionally influences chemical properties like air-sensitivity and solubility. Chalcogenide-based semiconductor quantum dots (QD's) can also be used as photosensitive agents. For example, they improve the catalytic activities of metal oxides and metal–organic frameworks.17,18
The success and outcome of the synthesis of new compounds based on chalcogenidometalate units with interesting structural, physical, and chemical properties depends strongly on the method used. For such compounds, the synthetic access is traditionally divided into different types based on the temperature used. High-temperature syntheses comprise classical solid-state reactions, reaching down to the more moderate temperature regime of molten flux techniques, while low-temperature processes include wet-chemical and solvothermal reactions. Except for the solid-state reactions, these methods require the use of reaction media, which often also act as structure-directing agents and form counterions of the metalate anions in situ (alkali/alkaline earth metal cations, metal complexes, or organic amines).19 The solid-state reactions normally take place at high temperatures (T > 600 °C) to enable diffusion of the components and eventually product formation. Under these conditions, thermodynamically stable phases develop preferably, and the design of the desired products is difficult to influence due to the limited kinetic control of the reactions.20,21 The molten flux techniques apply comparatively low temperatures (T = 150–600 °C), controlled by the melting point of the alkali(ne earth) chalcogenide excess used to form the corresponding (reactive) flux.22,23 Also metal fluxes have been used to synthesize chalcogenidometalate-based compounds. For instance, the reaction of K2Te and HgTe in elemental Hg resulted in the formation of K2[Hg2Te3] in large scale and high purity. The structure consists of a 3D network with corner-linked [HgTe4]-tetrahedra.24
For thermodynamic reasons, higher-temperature approaches typically lead to the formation of extended chalcogenide- or chalcogenidometalate networks (2D or 3D), while compounds prepared by wet-chemical routes, may be regarded as kinetically controlled (‘intermediate’) products that feature lower dimensionalities, down to discrete anionic clusters (“0D”) or 1D-extended chalcogenidometalate substructures. Exceptions to these typical observations exist though. In solvothermal reactions, the reaction takes place under supercritical reaction conditions, thus at temperatures above the boiling point and at elevated pressure owing to the solvent's vapor pressure, which involves a certain safety risk owing to the hazard of explosion. However, the variable selection of solvents ensures that even unusual structures can be accessed by a solvothermal approach. Furthermore, the solvothermal approach enables the preparation of inorganic-organic hybrid compounds containing amine molecules, ammonium counterions derived in situ from them or transition-metal complexes that were generated in situ by combining donor solvent molecules with metal ions from the reaction mixture.25–30 An example of a compound synthesized by using solvothermal methods is (MeNH3)0.75[Cu1.25GeSe3], which features a 3D-{[Cu1.25GeSe3]n0.75n−} open framework with methylammonium cations as counterions.10
The persisting interest in new structures and properties of chalcogenidometalate-based compounds represents a great motivation to develop new synthetic strategies for the preparation of crystalline chalcogenides. In recent years, the interest in the synthesis of corresponding materials from uncommon reaction media has increased. Impressive results emerged, in particular from ionothermal or deep eutectic-based, surfactant-thermal, and hydrazine-thermal processes for the syntheses of crystalline and novel compounds based on chalcogenidometalate substructures.
From the named techniques, the ionothermal and deep eutectic-based reaction conditions enjoy a special role, as they are positioned between the classical syntheses methods: they take place at low temperatures (<200 °C), but in the absence of conventional solvents. Instead of conventional solvents, so-called ionic liquids (ILs, see Fig. 1, left) are used,31–36 or low-melting, eutectic, mixtures.37 Upon having been applied successfully to the synthesis of oxide materials38 and metal nanoparticles,39–42 they were also discovered as excellent reaction medium for the preparation of chalcogenides.43–45
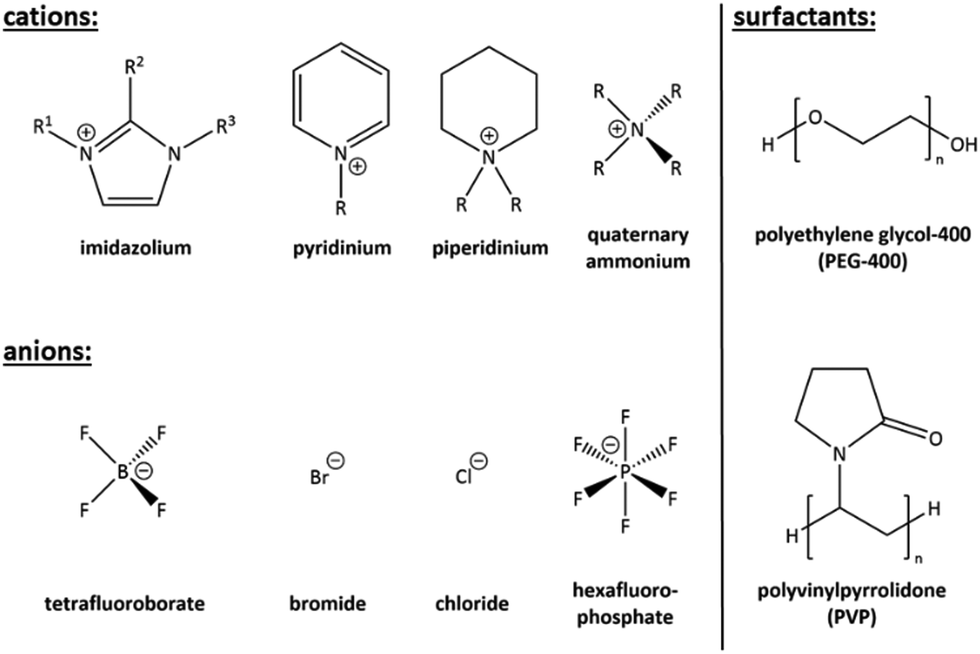 | ||
| Fig. 1 Summary of common cations and anions combined in ionic liquids for ionothermal reactions (left hand side). General formulas of surfactants used for surfactant-thermal syntheses (right hand side). | ||
Surfactants were initially used as templates to vary the size and shape of nanocrystals and the pore size of porous materials.19 Since the successful synthesis of a crystalline chalcogenide by the use of surfactants as reaction media, this method has attracted increasing attention.46 As one of the first compounds produced this way, 3D-{(H3O)2(enH2)[Cu8Sn3S12]} was reported, which was additionally explored for its performance as anode materials for Li-ion batteries (LIB's).47 The quoted chalcogenidometalate-based compound exhibits a promising specific capacity of 563 mA h g−1 at a current density of 0.1 A g−1 after 100 cycles as well as outstanding cyclization properties with a small capacity loss of 7.2% only from the 5th to the 100th cycle.
Besides the negligible vapor pressure, which is also typical for ILs, surfactants have other appealing properties. On the one hand, they combine hydrophilic and hydrophobic groups (see Fig. 1, right), hence creating an amphiphilic behavior and are thus capable of self-assembly into diverse aggregates in solution.19 On the other hand, they have multifunctional properties, such as a cationic, anionic, neutral, basic, or acidic nature. Moreover, surfactants usually show high thermal stability and they are comparatively cheap chemicals.19
Hydrazine has also been used as a reaction medium in recent years because of its unique properties – and in spite of specific risks in the context of its toxicity, which requires extra care. Hydrazine is a basic solvent with a high coordination capability of metal ions. Furthermore, hydrazine can dissolve metal chalcogenides, which makes it attractive for more specific applications.13 Still, the environmental hazards and toxicity make hydrazine not an all-day chemical in the synthesis of chalcogenide compounds and will be mentioned here as a specific medium for purely curiosity-driven research.
This feature article will shed light on some recent developments in the context of the three mentioned less-common contemporary synthetic approaches to chalcogenide compounds. All compounds mentioned herein are summarized in Table 1.
| Compound | Synthetic method | Precursors and reaction medium | Temperature (°C) | Ref. |
|---|---|---|---|---|
| (H+-Me2NH)4/3(H+-Me3N)2/3[Sn3S7]·1.25H2O | Solvothermal | SnCl4·5H2O, S in dimethylamine solution/H2O | 180 | 134 |
| (H+-C3H8N)4[Sn20O10S32] | Solvothermal | SnCl4·5H2O, SnCl2·2H2O, S in propylamine, DMA and ethanol | 160 | 16 |
| (Cat)4[Sn10O4S20] (Cat not specified) | Solvothermal | SnCl4·5H2O, S in dimethylamine and diethylamine | 180 | 16 |
| [(CH3NH3)(C4C1C1Im)][Sn3S7]·0.5H2O | Solvothermal | SnCl4·5H2O, S in (C4C1C1Im)Cl and methylamine aqueous solution | 180 | 135 |
| [(CH3NH3)0.75(C4C1C1Im)]1.25[Sn3S7]·H2O | Solvothermal | Sn, S in (C4C1C1Im)Cl and methylamine aqueous solution | 180 | 135 |
| K2[Hg2Te3] | Hg-flux | K2Te and HgTe in elemental Hg | 350 | 24 |
| (C4C1C1)24[Sn36Ge24Se132], (C4C1Im)24[Sn36Ge24Se132] | Ionothermal | [K4(H2O)3][Ge4Se10], SnCl4·5H2O in (C4C1C1Im)[BF4] or (C4C1Im)[BF4] with dmmp | 150 | 57 |
| (C4C1C1Im)2[Ge4SnSe10] | Ionothermal | [K4(H2O)3][Ge4Se10], SnCl2, in (C4C1C1Im)[BF4] with dmmp | 150 | 60 |
| (C4C1C1Im)2[Ge0.83Sn3.17Se9.06] | Ionothermal | [K4(H2O)3][Ge4Se10], SnCl4·5H2O in (C4C1C1Im)[BF4] with dmmp or en | 150 | 60 |
| (C4C1Im)4[Sn9Se20] | Ionothermal | [K4(H2O)4][SnSe4] in (C4C1Im)[BF4] | 130 (150, 180) | 61 |
| (C4C1C1Im)2[Sn18Se40] | Ionothermal | K2[Sn2Se5] in (C4C1C1Im)[BF4] with dmmp | 165 | 62 |
| (C4C1C1Im)16[Sn24Se56] | Ionothermal | K2[Sn2Se5] in (C4C1C1Im)[BF4] | 120 | 62 |
| (C4C1C1Im)4[Sn6Se14] | Ionothermal | K2[Sn2Se5] in (C4C1C1Im)[BF4] | 150 | 62 |
| (C4C1C1Im)3(H+-dmmp)[Sn6Se14] | Ionothermal | K2[Sn2Se5] in (C4C1C1Im)[BF4] with dmmp | 150 | 62 |
| (C4C1Im)2[HgTe2] | Ionothermal | Na2[HgTe2] in (C4C1Im)[BF4] | 60 | 64 |
| (C4C1Im)2[Hg2Te4] | Ionothermal | Na2[HgTe2] in (C4C1Im)[BF4] | 60 | 64 |
| (CnC1)8[Hg8Te16] | Ionothermal | Na2[HgTe2] in (CnC1Im)[BF4] (n = 10, 12) | 80 | 66 |
| [(CnC1ImTe)4Hg][BF4]2 | Ionothermal | Na2[HgTe2] in (CnC1Im)[BF4] (n = 6, 8), under ambient conditions | 60 | 67 |
| (C4C1C1Im)5[In10Ch16Cl3(C4C1im)] (Ch = S, Se, Te) | Ionothermal | In, Ch in (C4C1C1Im)Cl and methylamine | 160 | 74 |
| (C4C1C1)4[Sn10O4S16(SMe)4] | Ionothermal | K4[SnS4]·4H2O, MnCl2 in (C4C1C1Im)[BF4]/(C4C1C1Im)Cl with en or dmmp | 150/180 | 77 |
| (C4C1C4Im)4+x[Sn10O4S16(SBu)4]Brx | Ionothermal | K4[SnS4]·4H2O in (C4C1C4Im)Br with dmmp | 180 | 80 |
| (C2C1Im)4[Ge4Se10] | Ionothermal | [Na4(H2O)14][GeSe4] in (C2C1Im)[B(CN)4] with dmmp | 150 | 81 |
| (C2C1Im)6[Ge8Se19] | Ionothermal | [Na4(H2O)14][GeSe4] in (C2C1Im)[B(CN)4]/(C2C1Im)[BF4] | 150 | 81 |
| (C2C1Im)8[Ge16Se36] | Ionothermal | [K4(H2O)3][Ge4Se10] in (C2C1Im)[B(CN)4] with dmmp | 150 | 81 |
| (NH3CH3)0.75[Cu1.25GeSe3] | Deep eutectic solvent | Cu, Ge, Se in methylamine hydrochloride, DMU and N2H4·H2O | 140 | 10 |
| [(NH3CH3)0.5(NH2(CH3)2)]0.25[Ag1.25SnSe3] | Deep eutectic solvent | AgOAC, Se, Sn in methylamine hydrochloride, DMU and N2H4·H2O | 160 | 84 |
| [(CH3NH3)3(NH4)3] Cd4Sn3Se13·3H2O | Deep eutectic solvent | CdCl2·2.5H2O, Sn, Se, diethylamine hydrochloride, DMU and N2H4·H2O | 160 | 85 |
| [NH2(CH3)2]2[Sn3Se7]·0.5NH(CH3)2 | Deep eutectic solvent | Sn, Se in dimethylamine hydrochloride, urea and N2H4·H2O | 160 | 20 |
| [NH4]2[Sn4Se9] | Deep eutectic solvent | Sn, Se in ethylamine hydrochloride, urea and N2H4·H2O | 160 | 20 |
| (NH3C2H5)2[Sn3Se7] | Deep eutectic solvent | Sn, Se in ethylamine hydrochloride and N2H4·H2O | 160 | 20 |
| (NH4)3[AgSn3Se8] | Deep eutectic solvent | Sn, AgOAc, Se, trimethylamine hydrochloride, urea and N2H4·H2O | 160 | 20 |
| [(H+-H2O)2(H+-en)2][Cu8Sn3S12] | Surfactant-thermal | Cu2S, Na4SnS4·14H2O, S in ethylenediamine/octylamine | 160 | 47 |
| (NH4)8[Mn2As4S16] | Surfactant-thermal | Mn, As2S3, S, N2H4·H2O in (PVP) | 150 | 94 |
| (NH3)6[Mn2As2S8(N2H4)2] | Surfactant-thermal | Mn, As2S3, S, N2H4·H2O in PEG-400 | 110 | 94 |
| (H+-en)[Cu3As2S5] | Surfactant-thermal | CuI, As2S3, S, en in PEG-400 | 160 | 94 |
| (NH4)[MnAs3S6] | Surfactant-thermal | Mn, As2S3, S, N2H4·H2O and (C16C1Im)Cl | 140 | 94 |
| (H+-DBU)2[Sn3Se7]·PEG | Surfactant-thermal | Sn, Se in DBN, H2O, ethylene glycol and PEG-400 | 120 | 95 |
| (H+-DBU)2[Hg2Sn2Se7] | Surfactant-thermal | HgCl2, Sn, Se in DBU/H2O and PEG-400 | 160 | 96 |
| (H+-DBU)2[Hg2Sn2Se6(Se2)] | Surfactant-thermal | HgCl2, Sn, Se in DBU/H2O and PEG-400 | 160 | 96 |
| (H+-1,3-dap)(H+-pu)[MnGeSe4] | Surfactant-thermal | Mn, GeSe, Se, 1,3-dap and PEG-400 | 160 | 97 |
| [Mn(en)2(H2O)][MnGe3Se9] | Surfactant-thermal | Mn, Se, GeSe, en, PEG-400 | 160 | 97 |
| (H+-1,2-dap)2{[Mn(1,2-dap)2][Ge2Se7]} | Surfactant-thermal | Mn, GeSe, Se, 1,2-dap and PEG-400 | 160 | 97 |
| [Mn3Ge2S7(NH3)4] | Surfactant-thermal | Mn, Se, GeS, N2H4·H2O and PEG-400 | 160 | 97 |
| (N2H4)2[Mn3Sb4S8(μ3-OH)2] | Surfactant-thermal | Mn, Sb2S5, S, N2H4·H2O, HDTBPB | 175 | 99 |
| [(H+)2-TMDP)][As4S6] | Surfactant-thermal | As2S3, S, TMDP, PEG-400 and N2H4·H2O | 180 | 100 |
| [(H+)2-pip)][Mn2As2S6] | Surfactant-thermal | Mn, As2S3, S in pip·6H2O, octylamine and methanol | 180 | 100 |
| Cs[MnAs3S6] | Surfactant-thermal | CsI, MnCl2, As2S3, S, N2H4·H2O in PEG-400 | 180 | 101 |
| Rb[MnAs3S6] | Surfactant-thermal | RbBr, MnCl2, As2S3, S, N2H4·H2O, PEG-400 | 180 | 101 |
| (H+-DBU)6[Ga10S16(SH)4] | Surfactant-thermal | Ga, TA in DBU, PEG-400 | 140 | 104 |
| [Mn2Sb2S5(N2H4)3] | Hydrazine-thermal | Mn, Sb2S3, S, N2H4·H2O | 140 | 114 |
| [Mn(en)3][Mn6(μ6-S)(N2H4)2(μ-N2H4)2(μ3-AsS3)4] | Hydrazine-thermal | Mn, As, S, N2H4/en | 110 | 116 |
| [Mn(dien)2][Mn6(μ6-S)(μ-N2H4)3(μ3-AsS3)4] | Hydrazine-thermal | Mn, As, S, N2H4/dien | 120 | 115 |
| Ba[AgSbS3] | Hydrazine-thermal | Ba(OH)2, Ag, Sb2S3, S, N2H4·H2O | 160 | 73 |
| [Ba(H2O)][AgSbS3] | Hydrazine-thermal | Ba(OH)2, Ag, Sb2S3, S, N2H4·H2O | 160 | 73 |
| Ba[CuSbS3] | Hydrazine-thermal | Ba(CH3COO)2, KOH, Cu, Sb2S3, S and N2H4·H2O | 160 | 117 |
| Ba[CuSbSe3] | Hydrazine-thermal | Ba(OH)2, KOH, Cu, Sb2Se3, Se. N2H4·H2O | 160 | 117 |
| [(H+)2-en)(DMF)(N2H4)0.5][Mn5Sb6S15(N2H4)6] | Hydrazine-thermal | Mn, Sb2S3, S, DMF, en and N2H4·H2O | 75 | 118 |
| [Mn(trien)(N2H4)2]2[Hg2Te4]2 | Hydrazine-thermal | MnCl2·4H2O, HgCl2, Te, N2H4·H2O, trien | 120 | 119 |
| [Mn(tepa)(N2H4)]2[Hg4Te12] | Hydrazine-thermal | MnCl2·4H2O, HgCl2, Te, N2H4·H2O, tepa | 120 | 119 |
| [Mn(trien)][Hg2Te4] | Hydrazine-thermal | MnCl2·4H2O, HgCl2, Te, N2H4·H2O, trien | 120 | 119 |
| [Zn(tepa)]2[Hg5Te12] | Hydrazine-thermal | ZnCl2, HgCl2, Te, N2H4·H2O, tepa | 120 | 119 |
2. Contemporary synthesis strategies
2.1. Crystalline chalcogenides from ionic liquids and deep eutectic solvents
ILs, chemically spoken, are salts that melt at extraordinarily low temperatures owing to a poor match of anions and cations, which causes a very low lattice energy. The first systematically reported – after several earlier studies on low-melting salts – was Paul Walden's seminal work on ethylammonium nitrate (m.p. 12 °C) in 1914.48 However, the compound was not referred as “ionic liquid” in those days; this term was assigned to such low-melting salts as late as in 1943.49 ILs have enjoyed increasing attention in many research fields, in particular in the area of ionothermal synthesis, that is, reactions in ionic liquids under slightly elevated temperatures in sealed reaction containers – yet well below supercritical conditions. This afforded the synthesis of well-defined crystalline materials, especially functional zeolites,50,51 metal organic frameworks,52,53 and crystalline chalcogenides.54As illustrated in Fig. 1, ILs normally are composed of cationic organic moieties (such as imidazolium, pyridinium, piperidinium, and quaternary ammonium) and weakly interacting anionic parts (such as [BF4]− and [PF6]−, but also Br− and Cl−). The ‘salts’ can play the roles of both the reaction medium and a structure-directing agent at the same time.43,55 In comparison with other solvents, the advantages of ionic liquids in terms of lower vapor/reaction pressure, lower toxicity, higher thermal stability, and excellent solvation capabilities, render them excellent environments for effectively and sustainably preparing novel crystalline materials through ionothermal synthesis strategies.56
The use of ILs to synthesize atomically precise chalcogenide clusters in crystalline form has been very fruitful in the past decade.54 Herein we give a survey of the most recent reports of ionothermal approaches to such materials. For the sake of readability, imidazolium-based ionic liquid cations will be abbreviated according to the scheme (Cl(Cm)CnIm)+, with ciphers l, m and n indicating the numbers of carbon atoms in the alkyl chains attached the N1(C2) and N3 atoms of the imidazolium (Im) ring. The underlying neutral rings are abbreviated as ‘im’.
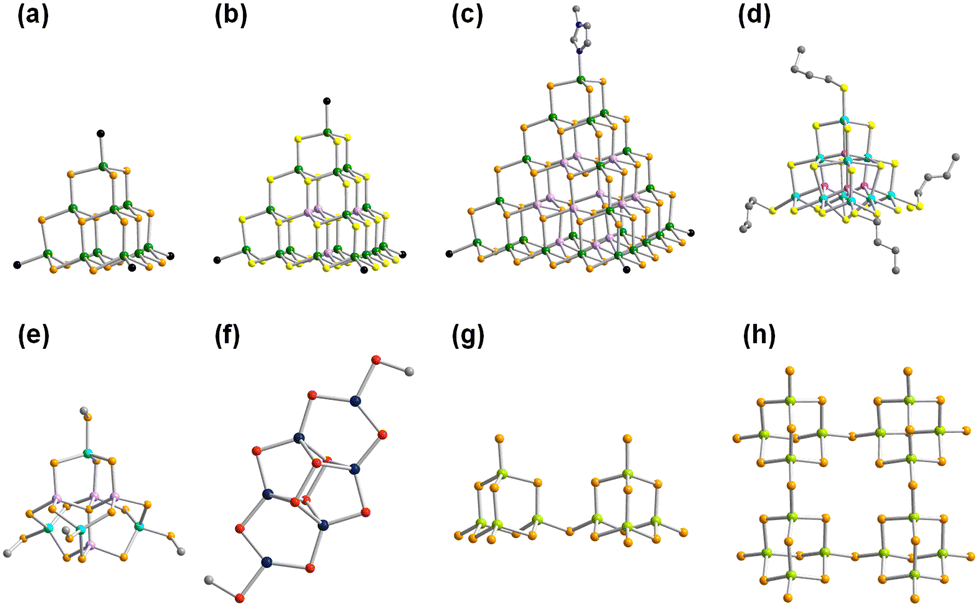
Reactions of salts of the [Ge4Se10]4− anion with SnCl4·5H2O in the ionic liquids (C4C1C1Im)[BF4] or (C4C1Im)[BF4] in the presence of up to 10% (w/w) of 2,6-dimethylmorpholine (dmmp), afforded the first salts of the largest discrete main-group (semi)metal polyanions, (C4C1C1Im)24[Sn36Ge24Se132] and (C4C1Im)24[Sn36Ge24Se132] (Fig. 2a).57
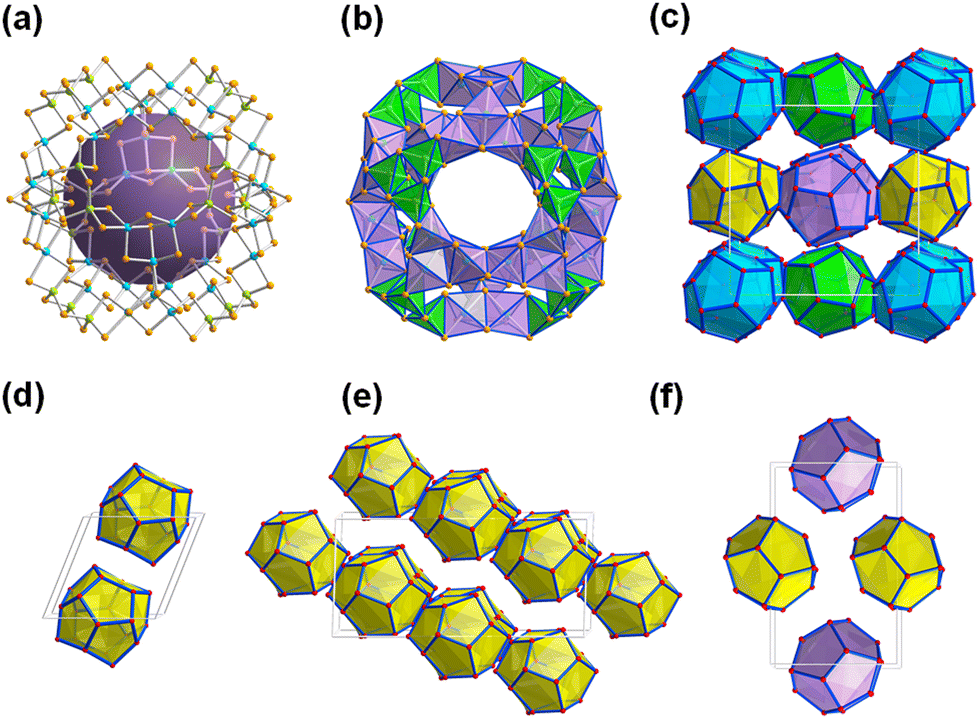 | ||
| Fig. 2 Molecular structure of the superspherical ‘zeoball’ cluster anion [Sn36Ge24Se132]24−, with the purple sphere representing the cavity inside the anion (a), polyhedral representation of the ‘zeoball’ anion (b), crystal packing pattern of the anions with overall pentagonal dodecahedral topology in the prototypical salt (c) and in three further ‘zeoball’ salts (d–f). Counterions are omitted for clarity. Color code: Ge-lime, Sn-turquoise, Se-orange. | ||
As illustrated in Fig. 2b, these ‘zeoball’ polyanions can be simplified into polyhedra connected into a superspherical architecture with a large outer diameter of 2.83 nm (including van der Waals radii) and considerable inner diameter of 1.16 nm (volume of 820 Å, under consideration of van der Waals radii). The compounds are able to trap I2 molecules and activate I–I bond cleavage to eventually form I3−. The influence of counterions on the packing of the ‘zeoball’ [Sn36Ge24Se132]24− anions was systematically explored in subsequent studies (Fig. 2d–f).
Variations in the alkyl chain length of 1-alkyl-(2,)3-(di)methylimidazolium cations (alkyl = butyl, hexyl, or octyl) allowed the crystallization of a series of similar ‘zeoball’ salts with different packing modes.58 It is worth noting, however, that the nature and size of the cations did in no reported case alter the formation of the ‘zeoball’ cluster itself, which seems to enjoy an extraordinary thermodynamic stability in the given reaction space. Moreover, the significance of chloride ions – either from the ionic liquid or from additions – on the reactivity of the tin source and the formation of the superspherical anions was explored and confirmed by 119Sn-NMR spectroscopy. Consequently, the presence of exclusively borate anions ([BF4]− or [B(CN)4]−) in the ionic liquids used yielded other crystalline chalcogenidometalates upon ionothermal reactions.59
In order to further demonstrate the important role of precursors, auxiliaries, and the reaction temperatures used for the preparation of crystalline chalcogenides, corresponding experiments were systematically varied by reacting [SnSe4]4− and [Ge4Se10]4− salts with or without the addition of SnCl4·5H2O in different ionic liquids. This way, novel chalcogenidometalates of various dimensionalities were obtained, from chains in 1D-(C4C1C1Im)2[Ge4SnSe10] and layers in 2D-(C4C1C1Im)2[Ge0.83Sn3.17Se9.06] to extended open frameworks in 3D-(C4C1Im)4[Sn9Se20] and 3D-(C4C1C1Im)2[Sn18Se40]. The different dimensionalities as well as element ratio also caused variations of the photo-optical properties of the crystalline solids.60,61 The studies also served to illustrate that the choice of the specific precursor salts and varied amounts of auxiliaries are of great significance for the modification of the anionic substructures, although the specific roles of these parameters have not yet been fully understood. Regarding the temperature, a single-crystal-to-single-crystal transformation process was observed in the ionothermal reaction mixture affording four crystalline selenidostannates:
The starting phase, 3D-K2[Sn2Se5], was deconstructed into 2D-(C4C1C1Im)16[Sn24Se56] at 120 °C, and further into 1D-(C4C1C1Im)4[Sn6Se14] or 1D-(C4C1C1Im)3(dmmpH)[Sn6Se14] at 150 °C. Upon further increasing the temperature, the latter assemble to another open-framework selenidostannate, 3D-(C4C1C1Im)8[Sn18Se40] at 165 °C.62
This work emphasized the importance of a close temperature control for the reproducible formation and isolation of crystalline chalcogenides from ionothermal reactions. Interestingly, some of the named selenidometalates have also been successfully prepared through the reaction of elemental tin and selenium in the ionic liquid (C4C1C1Im)Cl.63 Hence each set of reactants spans their own reaction space, which needs to be carefully explored.
Besides the exploration of selenidometalates through ionothermal reactions, the expansion towards homologous telluridometalates, especially the family of telluridomercurates, has also been investigated into some detail. By treatment of Na2[HgTe2] in (C4C1Im)[BF4] at relatively low temperature (60 °C), two telluridometalate salts were obtained, (C4C1Im)2[HgTe2] and (C4C1Im)2[Hg2Te4].64 The anionic substructure of the latter represents an infinite zig-zag strand (Fig. 3a). Thanks to the templating effect of the imidazolium cations, its crystal structure differs in details from related compounds {[Mn(en)3]2Cl2}[Hg2Te4] and (N(C2H5)4)2[Hg2Te4].65 The salts obtained from ionic liquids showcased the ability to crystallize telluridometalate salts from ionic liquid at all. Shortly after this initial report, salts of the heaviest known inorganic porphyrin analogue, [Hg8Te16]8− (Fig. 3b), were obtained from corresponding reactions with longer-chain alkyl substituents on the imidazolium cations. The unique molecular anions were formed in reactions of Na2[HgTe2] in the ionic liquids (CnC1Im)[BF4] (n = 10, 12).66 based on the structural relationship of this molecular anionic substructure with the chain of Te-bridged {Hg3Te(Te2)} five rings obtained prior to these compounds, a possible formation pathway of this ‘oligomer’ was proposed; according to this suggestion, an equilibrium of open and closed forms might co-exist under elevated reaction conditions, while the ionic liquid cation present in it will control which of the two forms would crystallize in the end. Notably, the porphyranoid-type anion crystallizes exclusively with imidazolium cations comprising long alkyl chains, thereby forming lamellar crystal structures.
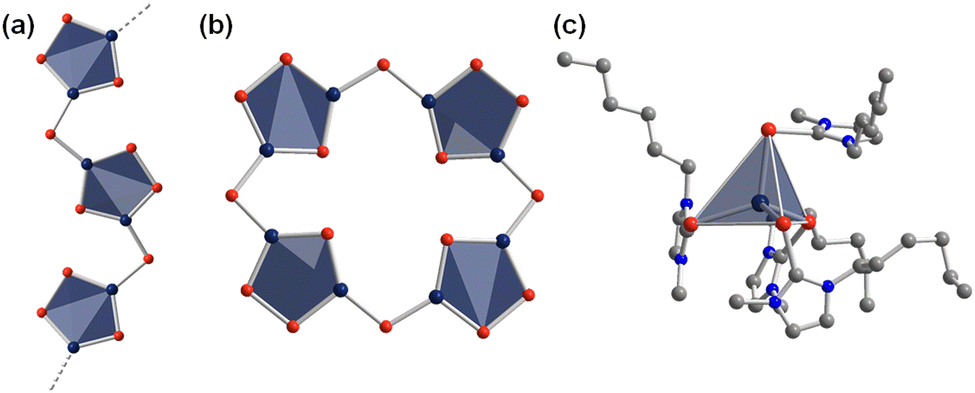 | ||
| Fig. 3 Infinite zig-zag-strand structure of the anion 1D-{[Hg2Te4]2−} (a), molecular structure of the heavy metal porphyranoid anion [Hg8Te16]8− (b), molecular structure of the cationic complex [(C6C1ImTe)4Hg]2+ (c). Hydrogen atoms are omitted for clarity. Color code: Hg-dark blue, Te-red, C-grey, N-blue. | ||
Such considerations and systematic studies may provide guidance for the synthesis of corresponding sulfide- or selenide-based analogues. The high reactivity of the reaction mixtures even upon cooling to room temperature was obvious upon exposure of the mother liquor, consisting of telluridomercurate anions, potassium cations, and ionic liquids, to ambient conditions. This way, two salts comprising exceptions from the collection of compounds presented herein, namely the highly uncommon cationic complexes, [(CnC1ImTe)4Hg]2+ (n = 6, Fig. 3c, and n = 8), were obtained in crystalline form upon in situ formation of the corresponding N-heterocyclic carbene telluride CnC1ImTe.67 It should be noted that this highly sensitive telluride species had not been formed nor captured otherwise prior to this finding.
Beside a number of different chalcogenidometalate-based compounds,68–71 also salts of discrete supertetrahedral chalcogenidometalate clusters were reported to form from ionic liquids. The first of them comprised T5-supertetrahedral clusters with ternary Cu–M–S composition (M = Ga, In).72 The latter were, not only, the first discrete inorganic-organic T5-type clusters, but also showed promising photoluminescence properties. Owing to wide emission bands between 500 nm and 800 nm, they were suggested as good candidates for fluorescent materials. Subsequent ionothermal enables access of a series of further discrete Tn-supertetrahedral clusters (n = 3–5; Fig. 4a–c). Some of the clusters, like [In10Ch16Cl3(C4C1im)]5− (Ch = mixed S, Se, and Te), indicated the ionic liquid to be reactive under the given reaction conditions, as indicated by a hybrid terminal ligand situation:73,74 the corner positions of these supertetrahedra are occupied by three chloride atoms and one 1-butyl-2-methylimidazole ligand each, which formed in situ from the ionic liquid cation and prevented the Tn clusters from aggregation into open framework structures. A control of the element ratio S![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Se:Te served to tune the absorption energies from the UV to the visible region, and thereby control the photodegradation activities toward methyl orange. Subtle modification of the reaction conditions, the imidazole ligands could be replaced by chloride atoms (Fig. 4a),75 yielding the compounds (C4C1C1Im)6[In10Se16Cl4]·(C1im)2. In dimethyl sulfoxide, the crystals could be fully dispersed and degraded to nanoparticles that showed more promising photocatalytic H2 evolution activity than the parent crystals themselves. From reactions under other conditions, it was well-known that T3-type cluster grow to larger Tn-type clusters (n = 4, 5, 6) upon aliovalent incorporation of M+ (Cu+) or M2+ (e.g., Zn2+, Cd2+, Mn2+).76 In a corresponding way, access to a series of discrete T4 and T5 clusters was also achieved in ionic liquids – with or without decoration of the corner with imidazole ligands (Fig. 4b and c).3,17 Like their smaller analogues, these compounds were also capable of inducing photocatalytic H2 generation. Variation of the chalcogenide composition (S versus Se) allowed for fine-tuning of the valence and conduction band profiles as the most important parameter for the photocatalytic activity (see also below for a more detailed discussion).
:Se:Te served to tune the absorption energies from the UV to the visible region, and thereby control the photodegradation activities toward methyl orange. Subtle modification of the reaction conditions, the imidazole ligands could be replaced by chloride atoms (Fig. 4a),75 yielding the compounds (C4C1C1Im)6[In10Se16Cl4]·(C1im)2. In dimethyl sulfoxide, the crystals could be fully dispersed and degraded to nanoparticles that showed more promising photocatalytic H2 evolution activity than the parent crystals themselves. From reactions under other conditions, it was well-known that T3-type cluster grow to larger Tn-type clusters (n = 4, 5, 6) upon aliovalent incorporation of M+ (Cu+) or M2+ (e.g., Zn2+, Cd2+, Mn2+).76 In a corresponding way, access to a series of discrete T4 and T5 clusters was also achieved in ionic liquids – with or without decoration of the corner with imidazole ligands (Fig. 4b and c).3,17 Like their smaller analogues, these compounds were also capable of inducing photocatalytic H2 generation. Variation of the chalcogenide composition (S versus Se) allowed for fine-tuning of the valence and conduction band profiles as the most important parameter for the photocatalytic activity (see also below for a more detailed discussion).
 | ||
| Fig. 4 Molecular structures of supertetrahedral clusters and related species: the chloride-terminated T3-type anion [In10Se16Cl4]6− (a), the chloride-terminated T4-supertetrahedral anion [Cd3In17S31Cl4]9− (b), the core-deficient T5-type anion [Cd6In28Se52Cl3(C1im)]11−, terminated by three chloride and one 1-methylimidazole ligand (c), the supertetrahedral sulfidooxidostannate cluster anion [Sn10O4S16(SBu)4]4−, selectively butylated at its terminal sulfide positions (d), the hetero-bimetallic ternary P1-supertetrahedral cluster anion [Mn4Sn4Se13(SeCH3)4]6−, selectively methylated at its terminal selenide positions (e), the anion [Hg6Te10(TeCH3)2], selectively methylated at its terminal telluride positions (f), the [Ge8Se19]6− cluster dimer (g), the [Ge16Se36]8− cluster tetramer (h). Hydrogen atoms and counterions are omitted for clarity. Color code: In-green, Sn-turquoise, Hg-dark blue, Ge-lime, transition metal-lavender, S-yellow, Se-orange, Te-red, C-grey, N-blue, O-purple. | ||
A breakthrough regarding the non-innocence and potential synthetic use of ionic liquids was reported with the formation and isolation of a series of methylated sulfidooxidostannate T3-supertetrahedra [Sn10O4S16(SMe)4]4− in 2019,77 which had so far only been known as purely inorganic anions [Sn10O4S20]8−.78–80 While the used amount of ionic liquid and the reaction temperature were key factors to modulate the arrangement of such anionic clusters in their corresponding crystalline salts, the main result of this work indeed was the in situ methylation of the terminal sulfide ligands. In comparison with the clusters described above, in which a terminal (chalcogenide or halide) ligand was replaced by an imidazole group that formed in situ under ionothermal conditions, the observation made here represents the counterpart, in which the alkyl group that was obviously released from the ionic liquid cation attacked the cluster structure. The presence of the methylthiolate groups was also confirmed by means of single-crystal Raman spectroscopy. Noteworthy, this represented the first proof of a post-synthetic alkylation of a chalcogenidometalate cluster, which to that date failed to work by using conventional (and much more toxic) alkylation reagents. The reason for this ‘failure’ prior to the quoted work was the extremely low nucleophilicity of such cluster anions, and also a mismatch of reaction conditions and solubilities of the metalate clusters. Besides methylation of the sulfidooxidostannate clusters, also methylation of a P1-supertetrahedral ternary selenidometalate cluster and a telluridometalate cluster were realized under similar conditions in ionic liquids, indicating the universal application potential of this method to selectively methylate terminal chalcogenide ligands (Fig. 4e and f). Whereas (C4(C1)C1Im)+ cations only afforded methylated products upon selective methyl transfer, the method was expanded to other alkyl groups upon using ionic liquids with symmetric substitution of the cations, like butylation with (C4(C1)C4Im)+: The treatment of [K4(H2O)4][SnS4] in (C4(C1)C4Im)Br, allowed for the crystallization of selectively butylated clusters [Sn10O4S16(SBu)4]4− (Fig. 4d) in their corresponding salts.80 In comparison to the more highly charged inorganic analogs or the methylated clusters, the butylated variants are highly soluble in common organic solvents like CH2Cl2 and CH3CN, as confirmed by means of 119Sn-NMR and mass spectra from solutions of the single crystals. Most importantly, the partial alkylation does only slightly change the electronic structure of the supertetrahedral molecules, which thus retain their optoelectronic features. This could create new avenues for the functionalization of chalcogenidometalates without affecting their physical properties. As another means of increased solubility without diminishing opto-electronic features, the formation of finite cluster oligomers was recently shown to work. By treatment of [Na4(H2O)14][GeSe4] or [K4(H2O)3][Ge4Se10] in ionic liquids, dimeric and tetrameric condensation products of the T2-type [Ge4Se10]4− anion, [Ge8Se19]6− (Fig. 4g) and [Ge16Se36]8− (Fig. 4h), were generated and isolated in single-crystalline form as salts.81 The average negative charge per cluster unit was reduced from 4- in the parent cluster to 3- in the dimer and 2- in the tetramer. The correspondingly decreased electrostatic interaction between anions and cations allowed to readily dissolve the salts in DMF. The differently strong cation–anion interaction tendency was also demonstrated by means of electrospray ionization (ESI) mass spectrometry and dynamic light scattering studies of such solutions. Exploration of the optical absorption properties confirmed the excepted decrease of the optical gap in the direction of larger aggregates – now nicely parallel in solution and in the solid state.
In 2021, the spectrum of ionic liquids applied in ionothermal reactions was expanded to the use of pyridinium-based ionic liquids as reaction media.82 These turned out to be equally suitable for the formation of crystalline selenidometalates, which can be understood as the beginning of much more variations to come in this field.
Deep eutectic solvents (DESs), comprising Lewis or Brønsted acids and bases, have also been used as structure-directing agents to allow the formation of chalcogenidometalate-based compounds. DESs do not only possess many advantages already mentioned for ILs, but are also very easy and inexpensive to prepare. Owing to their many similar characteristics, DESs are widely regarded as an emerging class of IL analogues.37
In 2018, reactions of elemental Sn and Se under addition of choline chloride and urea in hydrazine hydrate at 150 °C afforded the formation of a well-known honeycomb-type substructure, 2D-[Sn3Se7]2−. A similar substructure, but with different arrangement of adjacent 2D layers was obtained by replacing choline chloride with choline bromide.83 This work did not only showcase the synthesis of chalcogenidometalate-based compounds from DESs, but also demonstrated the effect of DES anions on structural details.83 By incorporation of Ag+ and use of trimethylamine hydrochloride as the reaction medium, a ternary open-framework compound comprising transition-metal ions formed, [(NH3CH3)0.5(NH2(CH3)2)0.25][Ag1.25SnSe3]. The compound was confirmed to be an excellent inorganic ion-exchanger with a separation factor SFCs/Sr of ∼121.4.84 The lighter homologue, (NH3CH3)0.75[Cu1.25GeSe3], was isolable from the same DES-based synthesis access upon replacement of the reactants AgOAc and Sn with Cu and Ge, respectively.10 Both compounds are stable within a wide pH-range (1–12) and show effective Cs+ exchange properties10,84 due to the strong interactions between the ‘soft’ inner chalcogenide surfaces and the ‘soft’ Cs+ cations.4 Within the M2+/M4+/Ch2− reaction system, a DES-based synthetic approach served to add the compound (CH3NH3)3(NH4)3[Cd4Sn3Se13]·3H2O,85 for which only an (isomorphic) open-framework templated by alkaline metal cations has been previously known.86 Here, a defect-supertetrahedral cluster [Cd4Sn3Se16]12− serves as secondary building unit, forming the anionic open-framework by corner-Se2− sharing. The compound exhibits a good ion-exchange performance toward Cs+ and Sr2+, as shown by experimental studies and by density functional theory (DFT) calculations (see a more detailed discussion on the ion-exchange properties of chalcogenidometalate-based compounds below).85
2.2. Crystalline chalcogenides from surfactant-thermal procedures
Surfactants are regarded as particular interesting organic solvents owing to their integrated hydrophilic properties (causing water-solubility and oil-insolubility) and hydrophobic properties (causing oil-solubility and water-insolubility).87 This amphiphilic behavior makes them suitable platform for self-assembly processes of certain aggregates, especially for the controllable growth of inorganic nanocrystals,88 nanomaterials in general,89 and mesoporous materials.90 In terms of research on chalcogenides, series of luminous mesoporous chalcogenides were generated by reactions of [M4Ch10]4−, [MCh4]4−, or [M2Ch6]4− salts (M = Sn, Ge; Ch = S, Se) with a wide array of metal ions (like Pt2+, Fe2+, Co2+, In3+ and Sn4+) in the presence of surfactant templates.91–93 However, it was not possible to isolate single crystal from such solutions which therefore prohibited to determine atomically precise structural models and derive potential structure–property relations from them. Most probably, the surfactants with long alkyl chain as counterions affect and hamper the crystal growth process owing to the alkyl groups’ large mobility. This was overcome by the additional use of classical templates (like organic amines) in the surfactant-thermal reaction, and eventually the formation of crystalline products. A series of sulfidoarsenates could be crystallized this way from surfactant-thermal reactions, representing either a molecular (0D) anionic cluster (Fig. 5a), a 1D organic-inorganic hybrid strand (Fig. 5b), a 2D layered substructure (Fig. 5c), or a 3D open-framework architecture (Fig. 5d).94 All compounds show semiconductor-like properties according to their energy gaps. In addition, the open framework compound shows a weak antiferromagnetic coupling between the nearest Mn2+ ions, exhibiting an effective magnetic moment of 5.68μB (as expected for Mn2+).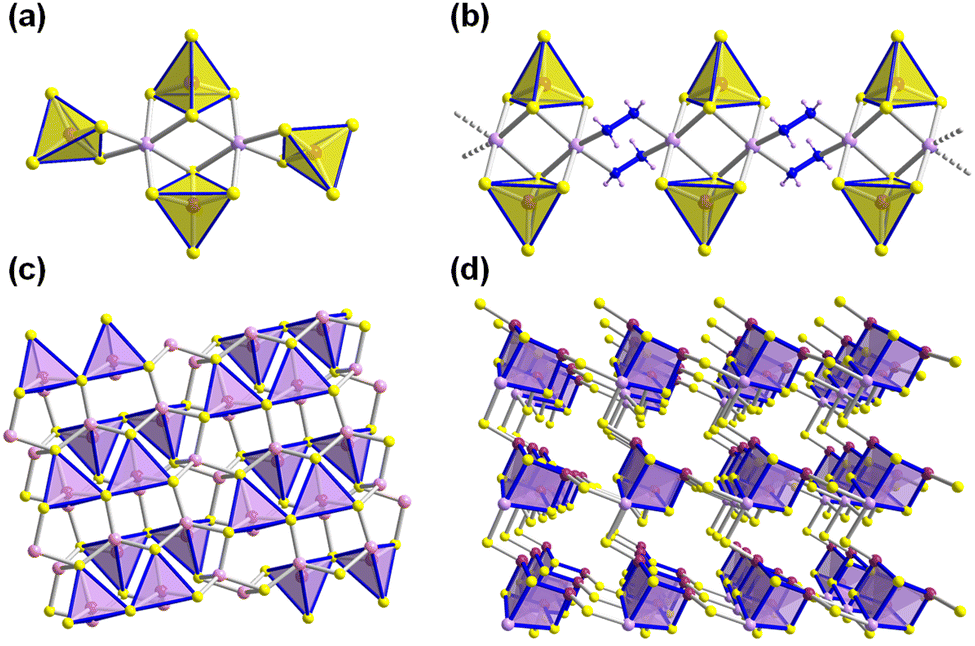 | ||
| Fig. 5 Molecular structure of the [Mn2As4S16]8− cluster anion (a), fragment of the 1D-{[Mn2As2S8(N2H4)2]} strand formed from μ-N2H4-linked [Mn2As2S8]6− clusters (b), view of the 2D-{[Cu3As2S5]−} layer (c) and the open-framework constructed by [MnAs3S6]− clusters (d). Counterions are omitted for clarity. Color code: As-plum, Mn-lavender, Cu-rose, S-yellow, N-blue. | ||
In subsequent work, in situ insertion of polyethylene glycol 400 (PEG-400) polymers into a 2D selenidostannate layer was realized (Fig. 6). Even though this inorganic two-dimensional honeycomb network had been documented in many compounds,43,59 this was the first report on the encapsulation of an organic polymer within the nanochannels, hence opening up a new hierarchy of host–guest chemistry involving selenidometalate hosts.95 Besides, a compound comprising the famous network without a polyethylene glycol polymer was obtained under the same reaction condition by increasing the reaction temperature. Both compounds were determined to be narrow-band-gap semiconductors by means of UV-vis spectra. The semiconductor-like behavior of the former was further investigated by photocurrent–voltage and Mott–Schottky measurements, which indicted an n-type semiconductor nature of the organic-inorganic hybrid compound. An expansion of the synthesis of crystalline chalcogenidometalate compounds by surfactant-thermal methods was also achieved by treatment of HgCl2, Sn, Se in 1,8-diazabicyclo[5.4.0]undec-7-en (DBU)/H2O upon addition of PEG-400. The reaction mixture afforded a product based on one-dimensional anionic strands, 1D-{[Hg2Sn2Se7]2−}. Time-dependent phase transformation was shown to take place from [Hg2Sn2Se6(Se2)]2− to [Hg2Sn2Se7]2−: the former was obtained in pure form during the first 2–4 days, while the latter formed on the surface of the former after reaction times of 5–8 days.96 After a 12 days reaction, the dominant phase changed to comprise [Hg2Sn2Se7]2−, while only a very small amount of the salts based on [Hg2Sn2Se6(Se2)]2− remained. Finally, the pure [Hg2Sn2Se7]2− salt was obtained after 20 days. The use of PEG-400 as reaction medium effectively controlled the single-crystal-to-single-crystal through kinetically induced processes.
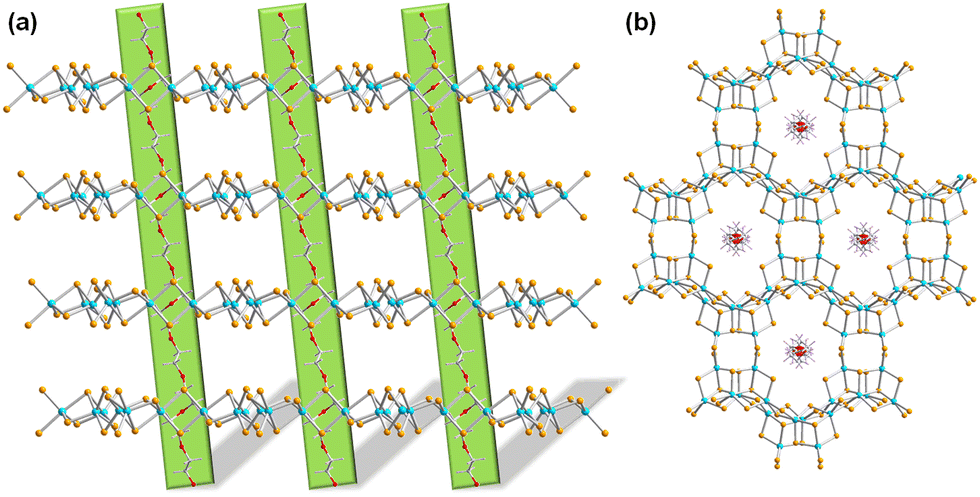 | ||
| Fig. 6 The infinite PEG polymer strands running through the hexagonal nanochannels of 2D-{[Sn3Se7]2−} layers, viewed along different crystallographic direction in (a and b). Hydrogen atoms and counterions are omitted for clarity. Color code: Sn-turquoise, Se-orange, C-grey, O-red. | ||
In a similar M2+/M4+/Ch2− (Ch = S/Se) reaction system, by replacing Hg2+ with Mn2+ and Sn4+ with Ge4+, synergistic effects of surfactants and organic amines for the controllable crystallization of chalcogenidometalates under surfactant-thermal reaction were explored.97 By reacting Mn, Se and GeChx (Ch = S or Se, x = 1 or 2) in PEG-400 in the presence of different amines, a series comprising different binary or ternary anionic substructures were received.
The product spectrum comprised a heterometallic one dimensional substructure 1D-{[MnGeSe6]2−}, a T2 cluster-based 1D-{[Mn(en)2MnGe3Se9]4−} strand, organic-inorganic hybrid 1D-{[Mn(1,2-dap)2][Ge2Se7]} chains, or a two-dimensional 2D-{[Mn2Ge2S8(N2H4)2]6−} layer upon addition of 1,3-propanediamine (1,3-dap), ethylenediamine (en), 1,2-propanediamine (1,2-dap), or hydrazine monohydrate, respectively. This work underscored the significant role of organic amine for the crystallization of chalcogenidometalates under surfactant-thermal conditions. In 2019, the addition of polyethylene glycol 200 (PEG-200) was found suitable for the formation of novel polyselenidostannate compounds. Previously, reactions of TMCl2 (TM = Mn, Zn, Fe, Ni), SnCl4·5H2O, and Se in an en/C2H5OH solvent mixture led to the isolation of a series of compounds featuring 2D-[Sn3Se6(Se2)]2− anionic layers or a 1D-[Sn3Se7.5]2− strand. Upon introduction of PEG-200 to the same reaction mixtures, other compounds formed, in which molecular [Sn(Se)4]2− anions were stabilized by different [TM(en)3]2+ cations. Even though the PEG-200 molecules did not co-crystallize, they effectively modified the reaction environment and allowed the introduction of polyselenide ligands into the anionic substructures.98 Besides the M2+/M4+/Ch2− (Ch = S or Se) combination, the reaction space including M2+/M3+/Ch2− (M2+ = Mn2+, M3+ = Sb3+ or As3+) has also been investigated. A two-dimensional sulfidooxidoantimonate, [(N2H4)2Mn3Sb4S8(μ3-OH)2], was obtained through surfactant-thermal treatment, which allows photocatalytic H2 evolution under visible-light irradiation.99 Upon changing the Sb2S5 source to As2S3 in this reaction system, salts comprising discrete [As4S6]2− anions or a layered substructure, 2D-{[Mn2As2S6]2−}, were prepared.100 Both compounds are semiconductors and exhibit photocurrent with a biased potential applied. The antiferromagnetic properties of the compound comprising the molecular [As4S6]2− anion, and second-harmonic generation (SHG) of the compound based on the layered 2D-{[Mn2As2S6]2−} framework were studied, which further expanded the spectrum of useful properties for such chalcogenidometalates. It should be noted that although the surfactants did not crystallize along with the chalcogenidometalate structures, no crystals were obtained without the addition of surfactants, which further confirmed the key role of the surfactants in the corresponding crystal growth processes. Two more sulfidoarsenates, templated by Mn2+ and Cs+/Rb+ ions, were reported.101 Both CsMnAs3S6 and RbMnAs3S6 show high SHG efficiencies in comparison to the commercially available standard AgGaS2 (AGS; see also below). Upon replacing dications M2+ (mostly Zn2+ or Hg2+) with monocations M+ (Cu+ or Ag+) in the surfactant-thermal approach, a series of quaternary thioantimonate(III) substructures were received, ranging from 2D layers to 3D open-framework structures. As reported for related compounds, such chalcogenidometalates also typically exhibit narrow band gaps. Owing to the different coordination modes of Sb3+ and M+, the various unprecedented chalcogenidometalate networks could be realized this way. Additionally, such compounds were also regarded as good candidates for photocatalytic applications due to highly efficient light absorption capabilities.13,102,103
In 2021, the first isolation of a discrete, ligand-free T3-type sulfidogallate cluster was documented to form in a surfactant-thermal reaction, which further expanded the family of Ga/S clusters.104
2.3. Crystalline chalcogenides from hydrazine-thermal procedures
In recent years, several groups have shown that the solvo(hydro)thermal method is an efficient way to synthesize crystalline compounds comprising chalcogenidometalate substructures.54,105–112 The solvo(hydro)thermal method is based on the selection of solvents suitable to stabilize unusual structures – usually by in situ formation of ‘onium’ cations from solvent molecules. Hydrazine (N2H4) is an excellent solvent and template for preparing chalcogenidometalates, although one needs to be aware of its high toxicity; this is why corresponding reactions are not that frequent and have to be carried out with great care. However, hydrazine is a strong reducing agent and can therefore easily reduce elemental chalcogen to chalcogenide anions Ch2− or polychalcogenide anions Chn2− (Ch = S, Se, Te) according to eqn (1) and (2) shown below113| N2H4 + 2Ch + 4(OH)− → N2 + 2Ch2− + 4H2O | (1) |
| N2H4 + 4Ch + 4(OH)− → N2 + 2Ch22− + 4H2O | (2) |
In the synthesis of chalcogenidometalates, chalcogens can thus be used as precursors instead of pre-formed chalcogenides, which saves a reaction step that in most reported procedures involves the use of liquid ammonia. In addition, hydrazine has a high polarity and a high coordination ability to metal ions with low steric hindrance. Furthermore, hydrazine can act as a structure-directing agent and reaction medium at the same time – as typical for all solvents used in solvothermal reactions. The first example of a ternary Mn/pnictide-hydrazine chalcogenide architecture produced under hydrazine-hydrothermal conditions represents an overall neutral hydrazine-bridged, three-dimensional network, [Mn2Sb2S5(N2H4)3].105 It was prepared by reacting Mn, Sb2S3, and S in N2H4/H2O. In this compound, the roles of hydrazine are a threefold: first, a neutral 2D network is bridged by hydrazine molecules so that these hydrazine molecules act as inter-layer linkers. The second type of hydrazine molecules serve as intra-layer bridges, while the third type of molecules occupy the channels of the compound and thus serves as a crystal structure template. The compound displays visible-light photocatalytic activity for hydrogen-generation without any co-catalyst. Magnetic measurements suggest antiferromagnetic coupling of the Mn2+ ions.114
A series of 1D, 2D and 3D polymeric manganese(II)-sulfidoarsenates were synthesized in mixtures of hydrazine with dien or 1,2-dap (dien = diethylenetriamine) under solvothermal conditions. Among others, compounds with anionic substructures 2D-{[Mn6(μ6-S)(N2H4)2(μ-N2H4)2(μ3-AsS3)4]2−} (Fig. 7a) and 3D-{[Mn6(μ6-S)(μ-N2H4)3(μ3-AsS3)4]2−} (Fig. 7b) were obtained. Both anions consist of heterometallic, cubane-type [Mn6(μ6-S)(μ3-AsS3)4]2− clusters and N2H4 molecules exhibiting different connectivities. In the 3D structure, the [Mn6(μ6-S)(μ3-AsS3)4]2− clusters are connected via three μ-N2H4 bridging ligands, while in the 2D structure, the clusters are connected via two μ-N2H4 bridging ligands (Fig. 7). Hence, hydrazine proved to be a suitable reaction medium for the solvothermal synthesis of sulfidoarsenate networks.115,116 Quaternary sulfidoantimonates(III) were synthesized by a hydrazine-hydrothermal reaction at low temperature. BaAgSbS3 and BaAgSbS3·H2O, despite possessing the same stoichiometry (except for the additional H2O molecule per formula unit in the latter) exhibit different structures. BaAgSbS3 is based on a layered 2D-{[Ag2Sb2S6]4−} network, in which the cavities are occupied by Ba2+ cations. BaAgSbS3·H2O in contrast contains a 3D-{[Ag2Sb2S6]4−} framework, with Ba2+ and H2O molecules accommodated in its channels. Both compounds were shown to be optical semiconductors.73 Upon replacement of Ag with Cu, further quaternary compounds with related structures were obtained, too.117 Another modification of the reaction system involved the replacement of Sb2S3 with As2S3. This way, SrAg4As2S6·2H2O, based on a 2D anionic substructure, and BaAgAsS3, based on 1D-extended anions were obtained in hydrazine-hydrothermal reactions. Experimental and theoretical studies indicated these compounds to be narrow-gap semiconductors.
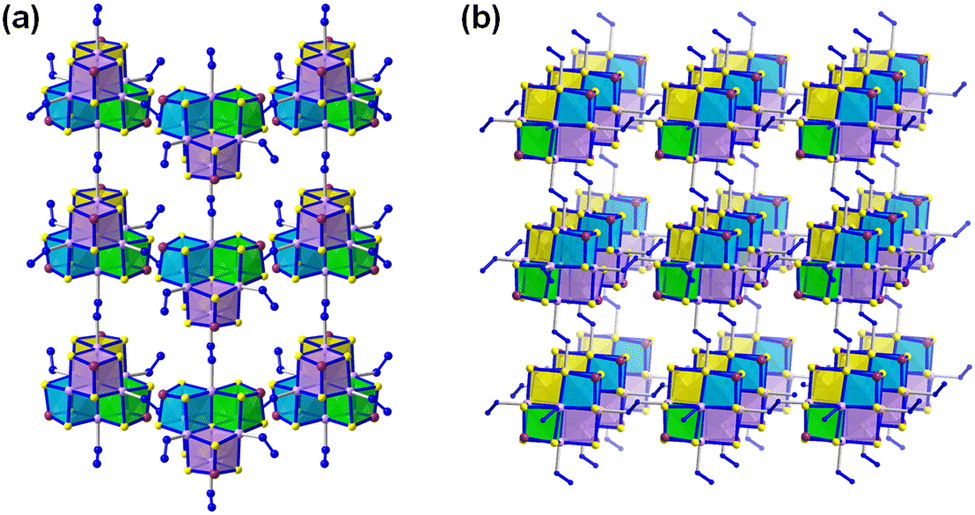 | ||
| Fig. 7 Arrangement of the N2H4-linked [Mn6(μ6-S)(AsS3)4]2− clusters (a) in 2D-{[Mn6(μ6-S)(N2H4)2(μ-N2H4)2(μ3-AsS3)4]2−} and (b) in 3D-{[Mn6(μ6-S)(μ-N2H4)3(μ3-AsS3)4]2−}. Counterions are omitted for clarity. Color code: As-plum, Mn-lavender, S-yellow, N-blue. | ||
In another open-framework chalcogenido metalate, {Sb2S5} building units act as pillars between 2D-{[Mn5S12(N2H4)6]} layers to form the three-dimensional framework 3D-{[Mn5Sb6S15(N2H4)6]2−}.118 Its {Mn12S12} substructure contains 1D hexagonal channels with a diameter of ∼10 Å (disregarding van der Waals radii), in which the counter ions are located (Fig. 8). The compound exhibits high stability in acids and bases and performs well in the electrocatalytic oxygen reduction.118 In 2017, also heavier telluridomercurates were prepared by the hydrazine-solvothermal method, by an one-pot synthesis using tellurium powder as the sole tellurium source. Six new compounds were obtained from this reaction, three of which – the Mn(II) complexes [Mn(trien)(N2H4)2]2[Hg2Te4]2 (trien = triethylenetetramine), [Mn(tepa)(N2H4)][Hg4Te12] (tepa = tetraethylenepentamine), and [Mn(trien){Hg2Te4}] – show photocatalytic activity (see the application section for a detailed disscussion).119
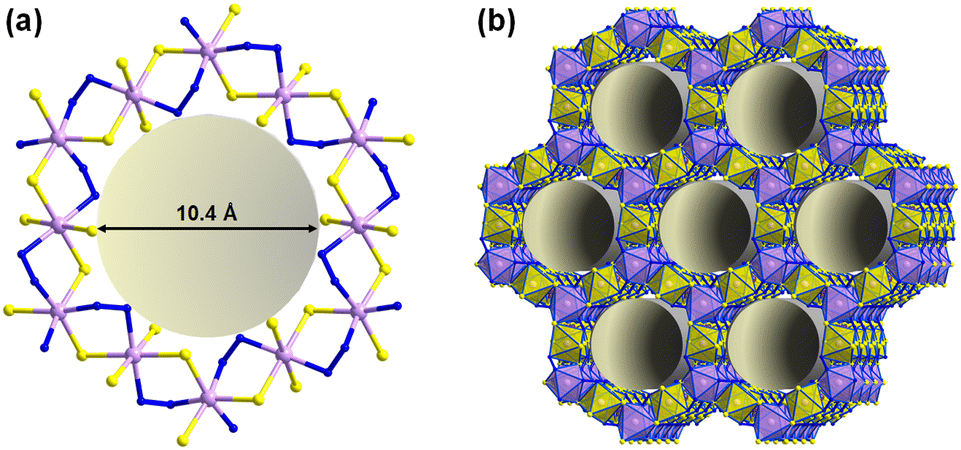 | ||
| Fig. 8 Detail (a) and overall representation (b) of the crystal structure of [Mn5S12(N2H4)6]·(H2en)·DMF·0.5N2H4 with 1D channels running along the crystallographic c-axis. Counterions are omitted for clarity. Color code: Sb-violet, Mn-lavender, S-yellow, N-blue. | ||
3. Application of crystalline chalcogenides obtained from uncommon reaction media
3.1. Photocatalytic activity
Due to the intrinsic semiconducting properties of chalcogenide-based quantum dots (QDs), these have been widely applied as effective photocatalysts for hydrogen evolution from water120 and dye degradation121 in the past. However, the in-depth understanding of their structure–property correlation on the molecular level was hindered by the fact that an atomically-precise structural information of such semiconductor QDs has been elusive. However, crystalline chalcogenidometalates comprising related nanoclusters excellently combined the advantages of a well-defined structure with semiconductor behavior. They have shown possibilities to establish structure–property correlations in regards of photocatalysis.122In this section, we give an overview of the use of crystalline compounds based on supertetrahedral chalcogenide clusters as photocatalysts for the efficient degradation of organic dyes and tunable water splitting and review further investigations on the understanding of photocatalytic mechanisms in this context.
The integration of semiconducting properties and porosity was initiated by application of a series of well-defined open-framework chalcogenidometalate-based compounds as catalysts for visible light-driven photocatalytic H2 generation.123 The reduction of CO2 to CH4 was investigated using noble metal ions incorporated in porous chalcogenidometalate frameworks.5 However, the photocatalytic performances were rather mediocre, probably owing to limited accesses of active sites, as the pores of the open-framework were blocked by structure-directing agents. In order to improve the photocatalytic performance, it thus seemed to be advantageous to access nanomaterials based on discrete cluster units.
Photodegradation properties were reported for crystals comprising discrete T3-type clusters [In10Ch16Cl3(C4C1im)] (Ch = S, Se, Te),74 The photodegradation activity toward methyl orange (MO) was investigated under UV and visible light irradiation on samples with modulated elemental S:Se:Te ratios within the clusters (Fig. 9a). The In/S-based sample displayed the highest UV-photocatalytic activity (nearly 95.4% MO degradation in 80 min). For the In/S/Se-based and the In/Se-based compounds, it took about 120 min to reach the same result. Finally, the In/Se/Te-based material could only degrade 69.4% of the MO in 120 min. Notably, the photodegradation efficiency of these compounds was completely different under visible-light irradiation. As displayed in Fig. 9b, the use of compounds based on In/S, In/S/Se, In/Se, and In/Se/Te elemental compositions led to decompositions of 4.36%, 7.72%, 86.4%, and 94.1% of MO in 3 hours, respectively. Overall, all samples presented better catalytic activities under UV irradiation. The authors attributed their findings to different band structures as a consequence of the different ratios of the involved chalcogenide components. The functional application studies of supertetrahedral chalcogenidometalate clusters were also extended to H2 evolution reactions (HER) from water using three compounds comprising isostructural T4-type clusters [Cd3In17S31−xSexCl4]9− obtained from ionothermal reactions.17 As a finding beside, it was reported that crystals based on the [Cd3In17S31Cl4]9− cluster anion did not dissolve in any common solvent, while the salts of [Cd3In17S13Se18Cl4]9− and [Cd3In17Se31Cl4]9− were highly soluble in DMSO and remained intact in solution according to electrospray ionization mass spectroscopy (ESI-MS) and high-resolution transmission electron microscopy (HRTEM). In solution, [Cd3In17S13Se18Cl4]9− and [Cd3In17Se31Cl4]9− do not only exhibit more reactive sites, but also decrease the transport distance of charge carriers, which altogether enabled a better H2 evolution activity than the undissolved crystals (Fig. 9c). Cycling experiment indicated that the solutions remained unchanged over at least 50 hours (Fig. 9d). As the possible reaction mechanism illustrated in Fig. 9e, the dissolved clusters act as light absorbers. After irradiation, the photo-generated electrons transit from the valence band to the conduction band, before they are further trapped by Pt. Here, H+ can be reduced to H2. In this reaction, triethanolamine serves as a sacrificial agent to consume the photo-generated holes and this way improve the photocatalytic H2 generation. In subsequent studies of the photocatalytic H2-generation ability of the ligand-free T3-type cluster anion [In10Se16Cl4]6− under visible-light illumination, the H2-evolution rate was determined to be 17.9 μmol h−1 g−1 using bulk crystal samples as catalysts (Fig. 9f).75 After dispersion of the crystals in DMSO, thereby forming nanoparticles, the H2 evolution efficiency dramatically increased to 121.0 μmol h−1 g−1. These two works demonstrated how nanomaterials derived from crystalline supertetrahedral chalcogenidometalate cluster compounds can enhance the photocatalytic water splitting activity as compared to the performance of the bulk crystals. Finally, the largest discrete Tn-type selenidometalate clusters, the T5-type cluster anions [Cu5In30Se52-xSxCl3(im)]12− and [Cd6In28Se52-xSxCl3(C1im)]11−, were also studied regarding their catalytic behavior.3 The photocatalytic activity towards crystal violet (CV) degradation by the former and H2 evolution by the latter were probed using the crystalline material. As displayed in Fig. 9g and h, the S-doped cluster [Cu5In30Se48.5S3.5Cl3(im)]12− showed a higher CV-degradation capability than that un-doped one. The photocatalytic H2 evolution experiments using the clusters [Cd6In28Se52−xSxCl3(C1im)]11− showed similar tendencies: the activity increased with increasing sulfur content (Fig. 9i). To understand the potential mechanisms behind the catalytic activity, the band structures of the compounds were explored by means of Mott–Schottky plots and UV-vis spectroscopy. The fine-tuning of the valence band of [Cu5In30Se52−xSxCl3(im)]12− and the conduction band of [Cd6In28Se52−xSxCl3(C1im)]11− by varying the sulfur content are very important to control the photocatalytic performance. Furthermore, transient photocurrent densities of these samples were measured to demonstrate the separation efficiency of photo-excited electron-hole pairs under visible light illumination. Solutions or dispersions of these compounds were not reported, which is most probably due to an inherently poor solubility of salts comprising these highly-charged anions.
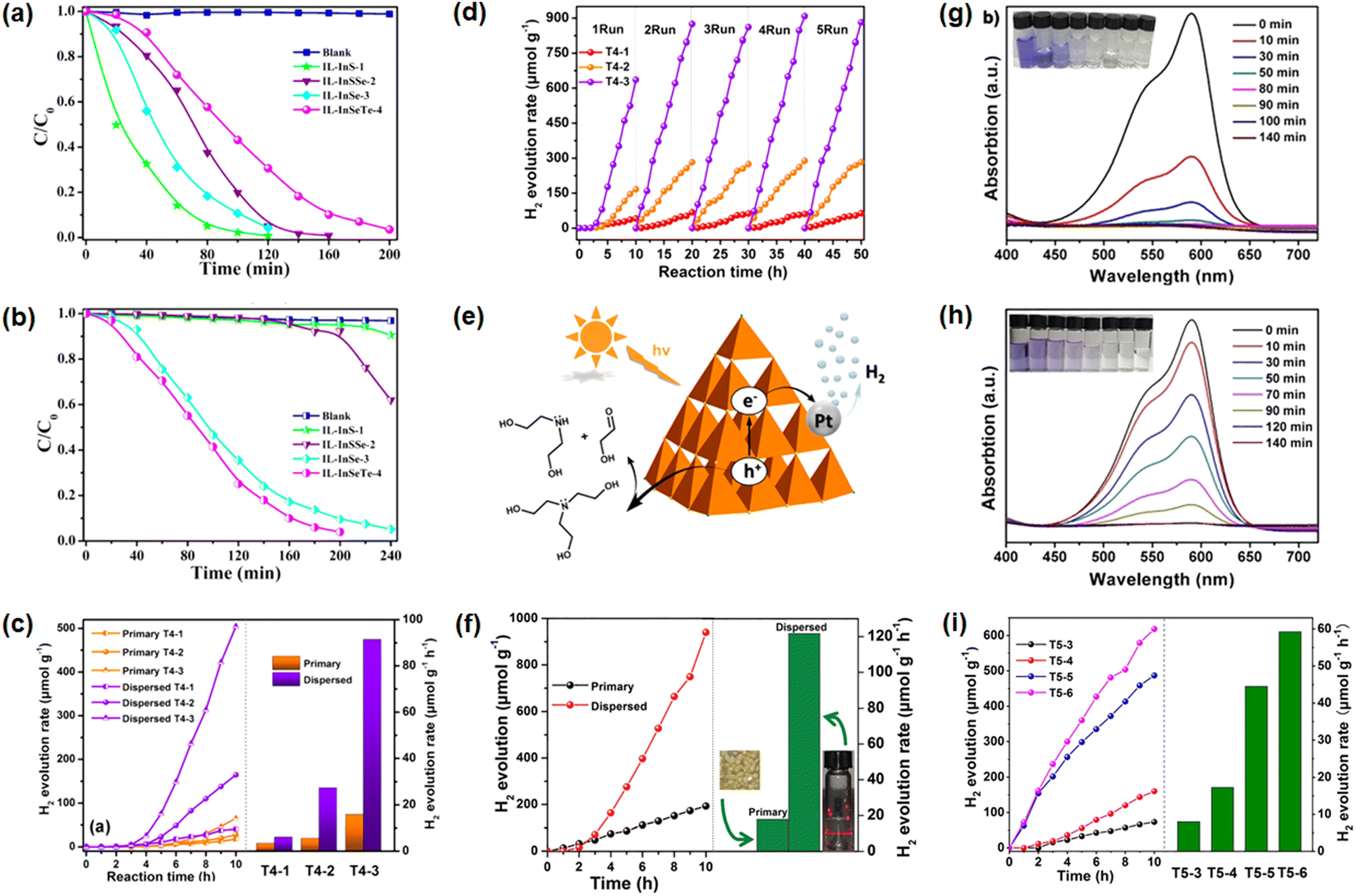 | ||
| Fig. 9 Photodegradation of methyl orange (MO) using photocatalysts (C4(C1)C1Im)[In10S16Cl3(C4C1Im)] (IL-InS-1), (C4(C1)C1Im)[In10S7.12Se8.88Cl3(C4C1Im)] (IL-InSSe-2), (C4(C1)C1Im)[In10Se16Cl3(C4C1Im)] (IL-InSe-3), and (C4(C1)C1Im)[In10Se13.80Te2.20Cl3(C4C1Im)] (IL-InSeTe-4) under UV light (a) and visible light (b) irradiation; photocatalytic H2-evolution performance of crystalline (C4(C1)C1Im)9[Cd3In17S31Cl4] (T4-1), (C4(C1)C1Im)9[Cd3In17S13Se18Cl4] (T4-2), and (C4(C1)C1Im)9[Cd3In17Se31Cl4] (T4-3), and their respective DMSO solutions under visible light illumination (c); cyclization of the H2-evolution experiments using the solutions of (C4(C1)C1Im)9[Cd3In17S31Cl4], (C4(C1)C1Im)9[Cd3In17S13Se18Cl4], and (C4(C1)C1Im)9[Cd3In17Se31Cl4] as photocatalysts (d); proposed photocatalytic mechanism during the water-splitting process involving supertetrahedral clusters (e); photocatalytic H2-evolution performance of crystalline (C4(C1)C1Im)6[In10Se16Cl4](C1im)2 and its dispersion one (f); photodegradation of crystal violet (CV) using (C4(C1)C1Im)12[Cu5In30Se48.5S3.5Cl3(im)] (g) and (C4(C1)C1Im)12[Cu5In30Se52Cl3(im)] (h) as catalysts; comparison of the H2-evolution capability using compounds (C4(C1)C1Im)11[Cd6In28Se52Cl3(C1im)] (T5-3), (C4(C1)C1Im)11[Cd6In28Se28.5S23.5Cl3(C1im)] (T5-4), (C4(C1)C1Im)11[Cd6In28Se16S36Cl3(C1im)] (T5-5), and (C4(C1)C1Im)9[Cd6In28Se8S54Cl(C1im)3] (T5-6) as photocatalysts (i). The figures in (a and b) have been reproduced from ref. 74 with permission from ACS, copyright 2018. The figures in (c–e) have been reproduced from ref. 17 with permission from ACS, copyright 2019. The figure in (f) has been reproduced from ref. 75 with permission from RSC, copyright 2020. The figures in (g–i) have been reproduced from ref. 3 with permission from Wiley-VCH, copyright 2020. | ||
In addition to the photocatalytic studies on sulfidometalates and selenidometalates, also telluridomercurate-based semiconductors have been investigated. Owing to their narrower band gaps, [Mn(trien)(N2H4)2]2[Hg2Te4]2, [Zn(trien)(N2H4)2][Hg2Te4], [Mn(tepa)(N2H4)]2[Hg4Te12], and [Mn(trien)][Hg2Te4] can be used for photocatalytic degradation of CV under visible-light irradiation. As illustrated by powder X-ray diffraction (PXRD) of [Mn(trien)(N2H4)2]2[Hg2Te4]2, [Mn(tepa)(N2H4)]2[Hg4Te12], and [Mn(trien)][Hg2Te4], these compounds show good stability in this process, which is notable with respect to practical applications.119
3.2. Nonlinear optical and magnetic properties
While the application potential of chalcogenidometalate-based compounds for photocatalysis has been discussed for nearly two decades, the exploration of SHG and magnetic properties are still in their infancy. In this section, we briefly comment on corresponding findings reported for crystalline compounds obtained from the uncommon reaction media discussed above.The compound based on discrete [As4S6]2− cluster anions synthesized in surfactant-thermal reaction93 features a strong SHG signal due to its non-centrosymmetric nature (space group type Imm2). Under irradiation by a 1064 nm laser, the pulverized samples generated green light (532 nm) with high efficiency. As the crystal size increases, the SHG intensity increases as well. The intensity of the emitted green light is determined as twice that of standard KH2PO4. Two further chalcogenidometalate-based compounds obtained from surfactant-thermal reactions, AMnAs3S6 (A = Cs, Rb), possess phase-matching properties, too, yielding good efficiency as infrared (IR) second-order nonlinear optical (NLO) materials (Fig. 10).101
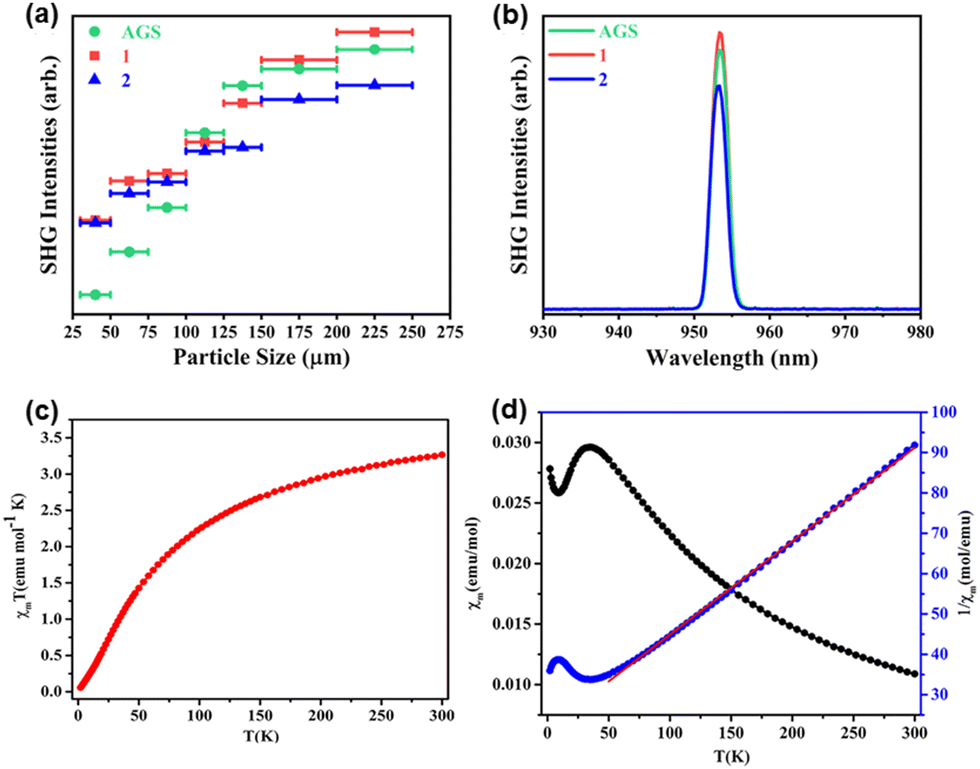 | ||
| Fig. 10 SHG signals of phase-matching compounds CsMnAs3S6 (1), RbMnAs3S6 (2) and AGS within the particle sizes ranging from 200 to 250 μm (a); comparison of SHG intensities of AMnAs3S6 (A = Cs, Rb) with that of AGS (a); temperature-dependence of χmT (c), χm and 1/χm (d) of compound [pipH2][Mn2As2S6]. The figures in (a and b) have been reproduced from ref. 101 with permission from ACS, copyright 2020. The figures in (c and d) have been reproduced from ref. 100 with permission from ACS, copyright 2018. | ||
As shown in Fig. 10a, the SHG intensities of both compounds upon 1910 nm laser light excitation increases as the particle size increases before reaching a plateau within the particle size range from 200 to 250 μm. The SHG intensities of these two solids are nearly the same as the SHG intensity of standard AGS (Fig. 10b), which showcases the advantages of using crystalline chalcogenidometalates as NLO materials.101
Besides nonlinear optical properties, the magnetic behavior of crystalline chalcogenidometalates from uncommon reaction media including open-shell d-block ions was investigated. By measurement of the magnetic susceptibility of the salt comprising [Mn2As2S6]2− anions in the temperature range 2–300 K (see Fig. 10c), antiferromagnetic coupling of the Mn2+ ions were detected.100 The molar susceptibility χm and 1/χmversus T curves (Fig. 10d) indicate a match with the Curie–Weiss law 1/χm = (T − θ)/C between 50 and 300 K. The extrapolated Weiss constant (−405.16 K) confirmed the antiferromagnetic interactions, and the magnetic moment (5.11μB per Mn2+ ion at 300 K) are in agreement with the findings. The magnetic properties of the solids based on the anionic 3D-{[MnAs2S6]−} framework, 1D-{[MnGe3Se9]4−}, 1D-{[Mn(1,2-dap)2Ge2Se7]2−}, and 1D-{[MnGeSe4]2−} were also studied, all of which show antiferromagnetic coupling, too.94,101 These works expanded the properties studies of crystalline chalcogenidometalates. While the Mn2+ compounds did not exhibit extraordinary magnetic properties so far, this might be different for Co- or Ni- containing chalcogenidometalates, the investigation of which will be subject to future work.
3.3. Ion exchange properties
As access to clean water is a key challenge for the coming decades, the removal of heavy metal cations, in particular radioactive cations from drinking and waste water is important for countries all over the world, which motivates the development of new efficient ion exchange materials.124–126 Owing to the relatively ‘soft’ surface of chalcogenidometalate-based open-framework compounds, they have proven to be promising candidates for the removal of corresponding ‘soft’ metal cations like Cd2+, Hg2+, Pb2+ and Cs+ from wastewater.127–133 In this section, we give a brief overview of the use of crystalline chalcogenidometalate-based compounds from uncommon reaction media as ion exchangers.The thiostannate (Me2NH2)4/3(Me3NH)2/3[Sn3S7]·1.25H2O, which was obtained from an solvothermal reaction and is based on the well-known 2D network, was confirmed to be excellent ion-exchanger towards Cs+ and Sr2+ in 2015. The material is not only able to reach the exchange equilibrium within 5 minutes (at 65 °C), but also achieves the maximum ion exchange capacities for Cs+ and Sr2+ at a relatively high level (408.91 mg g−1 and 65.19 mg g−1, respectively) without framework degradation over a wide pH range (0.7–12.7).134 The incorporation of IL cations, which causes a relatively low lattice energy of the solids to result, seems to notably facilitate the ion-exchange process.84,135
A combination of ionothermal and solvothermal synthesis strategies were reported in 2021,135 yielding isomorphic variants of the above-mentioned salts, which were also investigated with regard to their ion-exchange capabilities. The resulting compounds, (CH3NH3)(C4C1C1Im)[Sn3S7]·0.5H2O and (CH3NH3)0.75(C4C1C1Im)1.25[Sn3S7]·H2O, feature a co-existence of ionic liquid and ammonium counter ions. They capture not only Cs+ and Sr2+, but also Eu3+ cations. Both compounds exhibit high capacities and rapid kinetics toward these ions over a wide pH range (2.5–11.8 for Cs+, 3.0–11.9 for Sr2+ and 3.2–9.3 for Eu3+), even in the presence of excess Na+ as competing cations. As shown in Fig. 11, the maximum Cs+-exchange capacities of both according to the Langmuir isotherm model were determined to be 266.54 mg g−1 (Fig. 11a) and 109.68 mg g−1 (Fig. 11b). The maximum Sr2+-exchange capacities were shown to be 59.41 mg g−1 (Fig. 11c), 57.81 mg g−1 (Fig. 11d) according to the Langmuir–Freundlich isotherm model. For Eu3+, the maximum exchange capacities were confirmed as being 58.4 mg g−1 (Fig. 11e) and 61.5 mg g−1 (Fig. 11f), respectively. Notably, the latter compounds showed the highest pH-dependent distribution coefficients for capturing trace amounts of Eu3+ from aqueous solutions comprising excess Na+ loadings. In addition, a proven resistance of both chalcogenidometalate-based open-framework materials towards β and γ radiation is a promising precondition for practical applications.
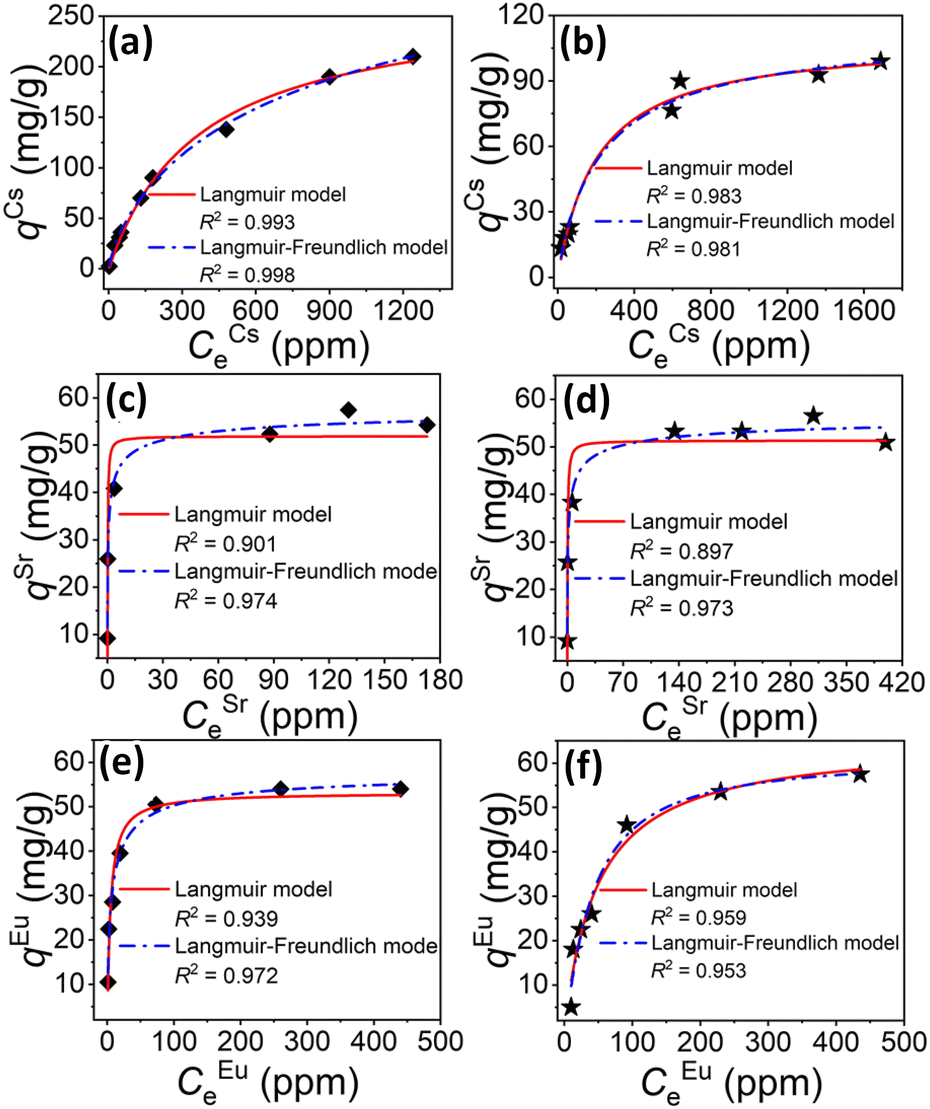 | ||
| Fig. 11 Equilibrium curves for ion exchange processes involving Cs+ (a and b), Sr2+ (c and d), and Eu3+ (e and f) using compounds [CH3NH3][C4C1C1Im]Sn3S7·0.5H2O and [CH3NH3]0.75[C4C1C1Im]1.25Sn3S7·H2O. The red lines and dotted blue lines represent fitting of the data with Langmuir and Langmuir–Freundlich isotherm models, respectively. The images in (a–f) were reproduced form ref. 135 with permission form ACS, copyright 2021. | ||
Besides the investigations of compounds from ionothermal reactions, those that were obtained from DESs have also been explored for ion-exchange applications. The analogous [Cu1.25GeSe3]0.75− and [Ag1.25SnSe3]0.75− networks were confirmed to be highly efficient Cs+ exchangers in the presence of various alkali and alkaline-earth metal cations over a wide pH range (1–12).10,84 The compound based on the [Ag1.25SnSe3]0.75− network even features an SFCs/Sr of ∼121.4.84 In 2022, the ion-exchange properties of (CH3NH3)3(NH4)3[Cd4Sn3Se13]·3H2O were studied in experimental and theoretical investigations. The exchange capacities of Cs+ are similar to those of (Me2NH2)4/3(Me3NH)2/3[Sn3S7]·1.25H2O, but the exchange capacities of Sr2+ are much higher (128.4 mg g−1). The uptake of Cs+ and Sr2+ mainly occurred within 4 hours and remained unchanged after 24 hours. The compounds also showed good selectivity in the presence of various alkali/alkaline earth metal cations. The inclusion of several mineral water, tap water, and lake water sources in the study demonstrated the removal of Cs+ and Sr2+ under real conditions.85 The excellent ion exchange features were attributed to the overall high negative charge density of the [Cd4Sn3Se13]n6−n framework. However, DFT calculations indicated dissimilar efficiencies toward the ion capture in the different voids of the framework structure, which underlines the importance of designing specified voids for the adsorption of different cations.85
Overall, the development of uncommon reaction media for the access of chalcogenidometalate-based compounds did not only serve to expand the spectrum of underlying substructures, but also amplified functional investigations of photocatalysis, SHG, magnetic, and ion-exchange properties for potential future applications.
4. Conclusions
In this feature article, we have briefly reviewed recent advances in the attempts to access atomically precise crystalline chalcogenidometalate-based compounds in uncommon reaction media. This included syntheses under ionothermal conditions, deep eutectic solvothermal and by application of surfactant-thermal as well as hydrazine-thermal strategies. Thanks to the mild reaction conditions, these non-classical reaction conditions allowed the formation and isolation of a variety of compounds featuring uncommon chalcogenidometalate structures. The broad product spectra range from molecular clusters via lower-dimensionality extended structures to open three-dimensional frameworks. The choice of starting materials and auxiliaries turned out to be crucial for the structural diversity accessible via ionothermal approaches, besides the key parameter of the ionic liquid itself: given that the ionic liquid cations are in most cases involved in the crystal structures of the products, their nature obviously plays the most important role for the crystallization process, and they may also play a templating role in the assembly of the metalate structures. Notably, the alkylation of supertetrahedral chalcogenidometalate clusters endows them higher solubility in common solvents, which enhances their meaningfulness for photocatalytic applications. Surfactant-thermal and hydrazine-thermal methods also allowed for the formation of interesting chalcogenidometalate architectures and their respective crystalline salts. Although the surfactants normally do not act as counterions in the crystal structures, they are obviously able to provide a suitable reaction environment – and in this regard are superior innocent reaction media.Although the isolation of chalcogenidometalate-based compounds from these uncommon media was proven very fruitful, the formation mechanisms behind the formation of the underlying, usually complex substructures, still remains elusive. Future work is therefore needed in the direction of time-dependent tracking the assembly of potential subunits, which continues to be a challenging task.
The application potential of compounds obtained from such uncommon reaction media mainly addressed photocatalysis, second-harmonic generation, magnetic, and ion-exchange properties of the products until now. The photocatalytic potential of (mostly soluble) nanoclusters was studied in particular, which allowed to investigate structure–property relationships at molecular scale and further improve the photocatalytic performance this way. Further relevant properties were not yet studied into this detail, but start to be reported in the literature. Overall, potential applications owing to semiconductor characteristics, photoluminescence efficiency, non-linear optical properties, magnetic interactions and also ion-exchange properties found in these chalcogenidometalate-based materials will serve to keep the field highly attractive in the coming years.
Abbrevations
| DMA | N,N-Dimethylacetamide |
| DMU | N,N-Dimethylurea |
| (C4C1C1Im)[BF4] | 1-Butyl-2,3-dimethylimidazolium tetrafluoro-borate |
| (C4C1Im)[BF4] | 1-Butyl-3-methylimidazolium tetrafluoroborate |
| dmmp | 2,6-Dimethylmorpholine |
| en | Ethane-1,2-diamine |
| (C4C1C1Im)Cl | 1-Butyl-2,3-dimethylimidazolium chloride |
| (C2C1Im)[B(CN)4] | 1-Ethyl-3-methylimidazolium tetracyano-borate |
| (C2C1Im)[BF4] | 1-Ethyl-3-methylimidazolium tetrafluoroborate |
| (C4C1C4Im)Br | 1,3-Dibutyl-2-methylimidazolium bromide |
| PVP | Polyvinylpyrrolidon |
| PEG-400 | Polyethylene glycol 400 |
| PEG-200 | Polyethylene glycol 200 |
| SHG | Second-harmonic generation |
| IR | Infrared |
| NLO | Non-linear optical |
| (C16C1Im)Cl | 1-Hexadecyl-3-methylimidazolium chloride |
| DBN | 1,5-Diazabicyclo[4.3.0.]non-5-ene |
| DBU | 1,8-Diazabicyclo[5.4.0.]undec-7-ene |
| 1,3-dap | 1,3-Propanediamine |
| 1,2-dap | 1,2-Propanediamine |
| HDTBPB | Hexadecyltributylphosphonium bromide |
| TMDP | 1,3-Bis(4-piperidyl)propane |
| pip·6H2O | Piperazine hexahydrate |
| TA | Thioacetamide |
| dien | Diethylenetriamine |
| DMF | N,N-Dimethylformamide |
| trien | Triethylenetetramine |
| tepa | Tetraethylenepentamine |
| ESI | Electrospray ionization |
| Cat | Cations |
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG). Z. W. acknowledges a PhD fellowship from the China Scholarship Council (CSC No. 202006920030).References
- X. Xu, W. Wang, D. Liu, D. Hu, T. Wu, X. Bu and P. Feng, J. Am. Chem. Soc., 2018, 140, 888–891 CrossRef CAS PubMed.
- T. Wu, X. Bu, P. Liao, L. Wang, S.-T. Zheng, R. Ma and P. Feng, J. Am. Chem. Soc., 2012, 134, 3619–3622 CrossRef CAS PubMed.
- Y. Wang, Z. Zhu, Z. Sun, Q. Hu, J. Li, J. Jiang and X. Huang, Chem. – Eur. J., 2020, 26, 1624–1632 CrossRef CAS PubMed.
- X.-M. Zhang, D. Sarma, Y.-Q. Wu, L. Wang, Z.-X. Ning, F.-Q. Zhang and M. G. Kanatzidis, J. Am. Chem. Soc., 2016, 138, 5543–5546 CrossRef CAS PubMed.
- K. Sasan, Q. Lin, C. Mao and P. Feng, Nanoscale, 2016, 8, 10913–10916 RSC.
- C. Xu, M.-M. Sheng, H.-T. Shi, M. Strømme and Q.-F. Zhang, Dalton Trans., 2019, 48, 5505–5510 RSC.
- C. Xue, L. Zhang, X. Wang, D. Hu, X.-L. Wang, J. Zhang, R. Zhou, D.-S. Li, H. Su and T. Wu, Inorg. Chem., 2020, 59, 15587–15594 CrossRef CAS PubMed.
- X. Hao, X. Cui, M. Hu, Y. Jia, X. Li, S. Wei and J. Lu, RSC Adv., 2019, 9, 15561–15570 RSC.
- M. J. Manos and M. G. Kanatzidis, Chem. Sci., 2016, 7, 4804–4824 RSC.
- H.-W. Liu, K.-Y. Wang, D. Ding, M. Sun, L. Cheng and C. Wang, Chem. Commun., 2019, 55, 13884–13887 RSC.
- B. Zhang, H.-Y. Sun, J. Li, L.-Z. Li, Y.-L. Deng, S.-H. Liu, M.-L. Feng and X.-Y. Huang, Inorg. Chem., 2019, 58, 11622–11629 CrossRef CAS PubMed.
- K. S. Subrahmanyam, C. D. Malliakas, D. Sarma, G. S. Armatas, J. Wu and M. G. Kanatzidis, J. Am. Chem. Soc., 2015, 137, 13943–13948 CrossRef CAS PubMed.
- L. Nie, G. Liu, J. Xie, T.-T. Lim, G. S. Armatas, R. Xu and Q. Zhang, Inorg. Chem. Front., 2017, 4, 954–959 RSC.
- A. V. Bhaskar Reddy, M. Moniruzzaman, M. A. Bustam, M. Goto, B. B. Saha and C. Janiak, J. Mater. Chem. A, 2020, 8, 15034–15041 RSC.
- X.-H. Qi, K.-Z. Du, M.-L. Feng, Y.-J. Gao, X.-Y. Huang and M. G. Kanatzidis, J. Am. Chem. Soc., 2017, 139, 4314–4317 CrossRef CAS PubMed.
- S.-L. Huang, L. He, E.-X. Chen, H.-D. Lai, J. Zhang and Q. Lin, Chem. Commun., 2019, 55, 11083–11086 RSC.
- M. Hao, Q. Hu, Y. Zhang, M. Luo, Y. Wang, B. Hu, J. Li and X. Huang, Inorg. Chem., 2019, 58, 5126–5133 CrossRef CAS PubMed.
- J. Wen, J. Xie, Z. Yang, R. Shen, H. Li, X. Luo, X. Chen and X. Li, ACS Sustainable Chem. Eng., 2017, 5, 2224–2236 CrossRef CAS.
- W.-W. Xiong and Q. Zhang, Angew. Chem., Int. Ed., 2015, 54, 11616–11623 CrossRef CAS PubMed.
- M. Schwarz, M. Haas and C. Röhr, Z. Anorg. Allg. Chem., 2013, 639, 360–374 CrossRef CAS.
- J. Heine and S. Dehnen, Z. Anorg. Allg. Chem., 2012, 638, 2425–2440 CrossRef CAS.
- S. J. Mugavero, M. Bharathy, J. McAlum and H.-C. zur Loye, Solid State Sci., 2008, 10, 370–376 CrossRef CAS.
- D. Voßwinkel and R. Pöttgen, Z. Naturforsch. B, 2013, 68, 301–305 CrossRef.
- G. Thiele, P. Bron, S. Lippert, F. Nietschke, O. Oeckler, M. Koch, B. Roling and S. Dehnen, Inorg. Chem., 2019, 58, 4052–4054 CrossRef CAS PubMed.
- A. Benkada, H. Reinsch, M. Poschmann, J. Krahmer, N. Pienack and W. Bensch, Inorg. Chem., 2019, 58, 2354–2362 CrossRef CAS PubMed.
- M. Shele, F.-Y. Qi, X.-Y. Tian, Y.-S. Bao and M. Baiyin, J. Solid State Chem., 2021, 296, 121964 CrossRef CAS.
- A. Benkada, H. Reinsch and W. Bensch, Eur. J. Inorg. Chem., 2019, 4427–4432 CrossRef CAS.
- J. Zhou, J. Dai, G.-Q. Bian and C.-Y. Li, Coord. Chem. Rev., 2009, 253, 1221–1247 CrossRef CAS.
- J. Zhou, G.-Q. Bian, Q.-Y. Zhu, Y. Zhang, C.-Y. Li and J. Dai, J. Solid State Chem., 2009, 182, 259–264 CrossRef CAS.
- Y. Lei, Z. Tang, L. Zhu, B. Guo and D. Jia, Polymer, 2011, 52, 1337–1344 CrossRef CAS.
- J. A. Cody, K. B. Finch, G. J. Reynders, G. C. B. Alexander, H. G. Lim, C. Näther and W. Bensch, Inorg. Chem., 2012, 51, 13357–13362 CrossRef.
- M. Watanabe, M. L. Thomas, S. Zhang, K. Ueno, T. Yasuda and K. Dokko, Chem. Rev., 2017, 117, 7190–7239 CrossRef CAS PubMed.
- R. E. Morris, Chem. Commun., 2009, 2990–2998 RSC.
- T. Niu, L. Chao, W. Gao, C. Ran, L. Song, Y. Chen, L. Fu and W. Huang, ACS Energy Lett., 2021, 6, 1453–1479 CrossRef CAS.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS PubMed.
- D. R. MacFarlane, N. Tachikawa, M. Forsyth, J. M. Pringle, P. C. Howlett, G. D. Elliott, J. H. Davis, M. Watanabe, P. Simon and C. A. Angell, Energy Environ. Sci., 2014, 7, 232–250 RSC.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- D. Lu, N. Shomali and A. Shen, Electrochem. Commun., 2010, 12, 1214–1217 CrossRef CAS.
- C. Vollmer and C. Janiak, Coord. Chem. Rev., 2011, 255, 2039–2057 CrossRef CAS.
- A. Schmitz, M. Bülow, D. Schmidt, D. H. Zaitsau, F. Junglas, T.-O. Knedel, S. P. Verevkin, C. Held and C. Janiak, ChemistryOpen, 2021, 10, 153–163 CrossRef CAS PubMed.
- K. Klauke, D. H. Zaitsau, M. Bülow, L. He, M. Klopotowski, T.-O. Knedel, J. Barthel, C. Held, S. P. Verevkin and C. Janiak, Dalton Trans., 2018, 47, 5083–5097 RSC.
- C. Vollmer, E. Redel, K. Abu-Shandi, R. Thomann, H. Manyar, C. Hardacre and C. Janiak, Chem. – Eur. J., 2010, 16, 3849–3858 CrossRef CAS PubMed.
- S. Santner, J. Heine and S. Dehnen, Angew. Chem., Int. Ed., 2016, 55, 876–893 CrossRef CAS PubMed.
- G. Thiele, S. Santner and S. Dehnen, Z. Kristallogr. – Cryst. Mater., 2017, 232, 47–54 CrossRef CAS.
- T. Zhang, T. Doert, H. Wang, S. Zhang and M. Ruck, Angew. Chem., Int. Ed., 2021, 60, 22148–22165 CrossRef CAS PubMed.
- L. Nie, Y. Zhang, W.-W. Xiong, T.-T. Lim, R. Xu, Q. Yan and Q. Zhang, Inorg. Chem. Front., 2016, 3, 111–116 RSC.
- L. Nie, Y. Zhang, K. Ye, J. Han, Y. Wang, G. Rakesh, Y. Li, R. Xu, Q. Yan and Q. Zhang, J. Mater. Chem. A, 2015, 3, 19410–19416 RSC.
- P. T. Walden, Bull. Acad. Imper. Sci., 1914, 405–422 CAS.
- R. M. Barrer, Trans. Faraday Soc., 1943, 39, 59–67 RSC.
- E. R. Cooper, C. D. Andrews, P. S. Wheatley, P. B. Webb, P. Wormald and R. E. Morris, Nature, 2004, 430, 1012–1016 CrossRef CAS PubMed.
- A. Deneyer, Q. Ke, J. Devos and M. Dusselier, Chem. Mater., 2020, 32, 4884–4919 CrossRef CAS.
- M. Teixeira, R. A. Maia, L. Karmazin, B. Louis and S. A. Baudron, CrystEngComm, 2022, 24, 601–608 RSC.
- R. A. Maia, B. Louis and S. A. Baudron, CrystEngComm, 2021, 23, 5016–5032 RSC.
- W.-W. Xiong, G. Zhang and Q. Zhang, Inorg. Chem. Front., 2014, 1, 292–301 RSC.
- Z. Ma, J. Yu and S. Dai, Adv. Mater., 2010, 22, 261–285 CrossRef CAS.
- X. Meng and F.-S. Xiao, Chem. Rev., 2014, 114, 1521–1543 CrossRef CAS PubMed.
- Y. Lin, W. Massa and S. Dehnen, J. Am. Chem. Soc., 2012, 134, 4497–4500 CrossRef CAS PubMed.
- S. Santner, S. Yogendra, J. J. Weigand and S. Dehnen, Chem. – Eur. J., 2017, 23, 1999–2004 CrossRef CAS.
- S. Santner, J. A. P. Sprenger, M. Finze and S. Dehnen, Chem. – Eur. J., 2018, 24, 3474–3480 CrossRef CAS PubMed.
- Y. Lin, W. Massa and S. Dehnen, Chem. – Eur. J., 2012, 18, 13427–13434 CrossRef CAS PubMed.
- Y. Lin and S. Dehnen, Inorg. Chem., 2011, 50, 7913–7915 CrossRef CAS PubMed.
- Y. Lin, D. Xie, W. Massa, L. Mayrhofer, S. Lippert, B. Ewers, A. Chernikov, M. Koch and S. Dehnen, Chem. – Eur. J., 2013, 19, 8806–8813 CrossRef CAS PubMed.
- J.-R. Li, W.-W. Xiong, Z.-L. Xie, C.-F. Du, G.-D. Zou and X.-Y. Huang, Chem. Commun., 2013, 49, 181–183 RSC.
- C. Donsbach and S. Dehnen, Z. Anorg. Allg. Chem., 2017, 643, 14–19 CrossRef CAS.
- S. S. Dhingra, C. J. Warren, R. C. Haushalter and A. B. Bocarsly, Chem. Mater., 1994, 6, 2382–2385 CrossRef CAS.
- C. Donsbach, K. Reiter, D. Sundholm, F. Weigend and S. Dehnen, Angew. Chem., Int. Ed., 2018, 57, 8770–8774 CrossRef CAS.
- C. Donsbach and S. Dehnen, Eur. J. Inorg. Chem., 2018, 4429–4433 CrossRef CAS.
- Q. Zhang, I. Chung, J. I. Jang, J. B. Ketterson and M. G. Kanatzidis, J. Am. Chem. Soc., 2009, 131, 9896–9897 CrossRef CAS PubMed.
- K. Biswas, Q. Zhang, I. Chung, J.-H. Song, J. Androulakis, A. J. Freeman and M. G. Kanatzidis, J. Am. Chem. Soc., 2010, 132, 14760–14762 CrossRef CAS PubMed.
- E. Ahmed, J. Beck, J. Daniels, T. Doert, S. J. Eck, A. Heerwig, A. Isaeva, S. Lidin, M. Ruck, W. Schnelle and A. Stankowski, Angew. Chem., Int. Ed., 2012, 51, 8106–8109 CrossRef CAS.
- E. Ahmed, A. Isaeva, A. Fiedler, M. Haft and M. Ruck, Chem. – Eur. J., 2011, 17, 6847–6852 CrossRef CAS PubMed.
- W.-W. Xiong, J.-R. Li, B. Hu, B. Tan, R.-F. Li and X.-Y. Huang, Chem. Sci., 2012, 3, 1200–1204 RSC.
- C. Liu, Y. Shen, P. Hou, M. Zhi, C. Zhou, W. Chai, J.-W. Cheng and Y. Liu, Inorg. Chem., 2015, 54, 8931–8936 CrossRef CAS PubMed.
- N.-N. Shen, B. Hu, C.-C. Cheng, G.-D. Zou, Q.-Q. Hu, C.-F. Du, J.-R. Li and X.-Y. Huang, Cryst. Growth Des., 2018, 18, 962–968 CrossRef CAS.
- Y. Wang, Q. Hu, J. Jin, J. Li, J. Li and X. Huang, Dalton Trans., 2020, 49, 5020–5023 RSC.
- X. Bu, N. Zheng and P. Feng, Chem. – Eur. J., 2004, 10, 3356–3362 CrossRef CAS PubMed.
- B. Peters, S. Santner, C. Donsbach, P. Vöpel, B. Smarsly and S. Dehnen, Chem. Sci., 2019, 10, 5211–5217 RSC.
- W. Schiwy and B. Krebs, Angew. Chem., Int. Ed. Engl., 1975, 14, 436 CrossRef.
- J. B. Parise, Y. Ko, K. Tan, D. M. Nellis and S. Koch, J. Solid State Chem., 1995, 117, 219–228 CrossRef CAS.
- B. Peters, G. Stuhrmann, F. Mack, F. Weigend and S. Dehnen, Angew. Chem., Int. Ed., 2021, 60, 17622–17628 CrossRef CAS PubMed.
- Z. Wu, I. Nußbruch, S. Nier and S. Dehnen, JACS Au, 2022, 2, 204–213 CrossRef CAS PubMed.
- J.-M. Yu, J.-P. Yu, N. Wang, L.-L. Xiao, H. Wang, Q. Xu, B. Zheng, F.-F. Cheng and W.-W. Xiong, Inorg. Chem., 2021, 60, 4337–4341 CrossRef CAS.
- K.-Y. Wang, D. Ding, S. Zhang, Y. Wang, W. Liu, S. Wang, S.-H. Wang, D. Liu and C. Wang, Chem. Commun., 2018, 54, 4806–4809 RSC.
- D. Ding, L. Cheng, K.-Y. Wang, H.-W. Liu, M. Sun and C. Wang, Inorg. Chem., 2020, 59, 9638–9647 CrossRef CAS.
- J.-Y. Zhu, L. Cheng, Y.-M. Zhao, M.-Y. Li, Z.-Z. Wang, J. Wang, C. Wang and K.-Y. Wang, Inorg. Chem. Front., 2022, 9, 2880–2894 RSC.
- N. Ding, D.-Y. Chung and M. G. Kanatzidis, Chem. Commun., 2004, 1170–1171 RSC.
- D. F. Evans, D. J. Mitchell and B. W. Ninham, J. Phys. Chem., 1986, 90, 2817–2825 CrossRef CAS.
- M.-P. Pileni, Nat. Mater., 2003, 145–150 CrossRef CAS PubMed.
- B. L. Cushing, V. L. Kolesnichenko and C. J. O'Connor, Chem. Rev., 2004, 104, 3893–3946 CrossRef CAS.
- K. K. Rangan, P. N. Trikalitis and M. G. Kanatzidis, J. Am. Chem. Soc., 2000, 122, 10230–10231 CrossRef CAS.
- P. N. Trikalitis, T. Bakas and M. G. Kanatzidis, J. Am. Chem. Soc., 2005, 127, 3910–3920 CrossRef CAS.
- P. N. Trikalitis, K. K. Rangan, T. Bakas and M. G. Kanatzidis, Nature, 2001, 410, 671–675 CrossRef CAS PubMed.
- M. J. MacLachlan, N. Coombs and G. A. Ozin, Nature, 1999, 397, 681–684 CrossRef CAS.
- W.-W. Xiong, E. U. Athresh, Y. T. Ng, J. Ding, T. Wu and Q. Zhang, J. Am. Chem. Soc., 2013, 135, 1256–1259 CrossRef CAS PubMed.
- W.-W. Xiong, J. Miao, K. Ye, Y. Wang, B. Liu and Q. Zhang, Angew. Chem., Int. Ed., 2015, 127, 556–560 CrossRef.
- W.-W. Xiong, P.-Z. Li, T.-H. Zhou, A. I. Y. Tok, R. Xu, Y. Zhao and Q. Zhang, Inorg. Chem., 2013, 52, 4148–4150 CrossRef CAS PubMed.
- G. Zhang, P. Li, J. Ding, Y. Liu, W.-W. Xiong, L. Nie, T. Wu, Y. Zhao, A. I. Y. Tok and Q. Zhang, Inorg. Chem., 2014, 53, 10248–10256 CrossRef CAS PubMed.
- J. Han, L. Zhang, S. Li, W. Zheng, D. Jia and Y. Yuan, CrystEngComm, 2019, 21, 1642–1652 RSC.
- J. Gao, Q. Tay, P.-Z. Li, W.-W. Xiong, Y. Zhao, Z. Chen and Q. Zhang, Chem. – Asian J., 2014, 9, 131–134 CrossRef CAS PubMed.
- D.-D. Yang, Y. Song, B. Zhang, N.-N. Shen, G.-L. Xu, W.-W. Xiong and X.-Y. Huang, Cryst. Growth Des., 2018, 18, 3255–3262 CrossRef CAS.
- R. Ye, B.-W. Liu, X.-M. Jiang, J. Lu, H.-Y. Zeng and G.-C. Guo, ACS Appl. Mater. Interfaces, 2020, 12, 53950–53956 CrossRef CAS.
- Y. Shen, C. Liu, P. Hou, M. Zhi, C. Zhou, W. Chai, J.-W. Cheng, Y. Liu and Q. Zhang, Chem. – Asian J., 2015, 10, 2604–2608 CrossRef CAS PubMed.
- Y. Shen, C. Liu, P. Hou, M. Zhi, C. Zhou, W. Chai, Q. Zhang and Y. Liu, J. Alloys Compd., 2016, 660, 171–177 CrossRef CAS.
- S. Makin and P. Vaqueiro, Molecules, 2021, 26, 5415–5422 CrossRef CAS PubMed.
- Y. Liu, Y. Tian, F. Wei, M. S. C. Ping, C. Huang, F. Boey, C. Kloc, L. Chen, T. Wu and Q. Zhang, Inorg. Chem. Commun., 2011, 14, 884–888 CrossRef.
- M. Yuan and D. B. Mitzi, Dalton Trans., 2009, 6078–6088 RSC.
- D. B. Mitzi, Inorg. Chem., 2005, 44, 3755–3761 CrossRef CAS PubMed.
- M. J. Manos and M. G. Kanatzidis, Inorg. Chem., 2009, 48, 4658–4660 CrossRef CAS PubMed.
- M. Yuan, M. Dirmyer, J. Badding, A. Sen, M. Dahlberg and P. Schiffer, Inorg. Chem., 2007, 46, 7238–7240 CrossRef CAS.
- M. V. Kovalenko, M. Scheele and D. V. Talapin, Science, 2009, 324, 1417–1420 CrossRef CAS.
- D. S. Dolzhnikov, H. Zhang, J. Jang, J. S. Son, M. G. Panthani, T. Shibata, S. Chattopadhyay and D. V. Talapin, Science, 2015, 347, 425–428 CrossRef CAS.
- L. Nie, W.-W. Xiong, P. Li, J. Han, G. Zhang, S. Yin, Y. Zhao, R. Xu and Q. Zhang, J. Solid State Chem., 2014, 220, 118–123 CrossRef CAS.
- S. Li, J. Han, L. Zhang, W. Jiang and D. Jia, J. Solid State Chem., 2019, 269, 341–347 CrossRef CAS.
- Y. Liu, P. D. Kanhere, C. Ling Wong, Y. Tian, Y. Feng, F. Boey, T. Wu, H. Chen, T. J. White, Z. Chen and Q. Zhang, J. Solid State Chem., 2010, 183, 2644–2649 CrossRef CAS.
- C. Tang, Y. Shen, P. Sun, S. Liu, J. Han, Y. Liu, H. Sun and D. Jia, Eur. J. Inorg. Chem., 2016, 3921–3926 CrossRef CAS.
- J. Han, S. Li, C. Tang, W. Zheng, W. Jiang and D. Jia, RSC Adv., 2018, 8, 34078–34087 RSC.
- C. Liu, P. Hou, W. Chai, J. Tian, X. Zheng, Y. Shen, M. Zhi, C. Zhou and Y. Liu, J. Alloys Compd., 2016, 679, 420–425 CrossRef CAS.
- P. Sun, J. Wu, Z. Wang, X. Wang, N. Chen and T. Wu, Dalton Trans., 2021, 50, 16473–16477 RSC.
- P. Sun, S. Liu, S. Li, L. Zhang, H. Sun and D. Jia, Inorg. Chem., 2017, 56, 6152–6162 CrossRef CAS PubMed.
- S. Yu, X.-B. Fan, X. Wang, J. Li, Q. Zhang, A. Xia, S. Wei, L.-Z. Wu, Y. Zhou and G. R. Patzke, Nat. Commun., 2018, 9, 4009–4018 CrossRef PubMed.
- Y. R. Son, M. Kwak, S. Lee and H. S. Kim, J. Nanomater., 2020, 10, 2498–2507 CrossRef CAS.
- X. Chen, X. Bu, Y. Wang, Q. Lin and P. Feng, Chem. – Eur. J., 2018, 24, 10812–10819 CrossRef CAS PubMed.
- N. Zheng, X. Bu, H. Vu and P. Feng, Angew. Chem., Int. Ed., 2005, 44, 5299–5303 CrossRef CAS PubMed.
- B. Hu, H. Wang, R. Liu and M. Qiu, Chemosphere, 2021, 274, 129743 CrossRef CAS PubMed.
- S. Li, L. Dong, Z. Wei, G. Sheng, K. Du and B. Hu, J. Environ. Sci., 2020, 96, 127–137 CrossRef PubMed.
- L. Järup, Br. Med. Bull., 2003, 68, 167–182 CrossRef.
- T. Wu, X. Wang, X. Bu, X. Zhao, L. Wang and P. Feng, Angew. Chem., Int. Ed., 2009, 48, 7204–7207 CrossRef CAS.
- M. J. Manos, K. Chrissafis and M. G. Kanatzidis, J. Am. Chem. Soc., 2006, 128, 8875–8883 CrossRef CAS PubMed.
- S. Bag and M. G. Kanatzidis, J. Am. Chem. Soc., 2010, 132, 14951–14959 CrossRef CAS PubMed.
- P. Feng, X. Bu and N. Zheng, Acc. Chem. Res., 2005, 38, 293–303 CrossRef CAS PubMed.
- M. J. Manos, R. G. Iyer, E. Quarez, J. H. Liao and M. G. Kanatzidis, Angew. Chem., Int. Ed., 2005, 44, 3552–3555 CrossRef CAS PubMed.
- M.-L. Feng, D.-N. Kong, Z.-L. Xie and X.-Y. Huang, Angew. Chem., Int. Ed., 2008, 47, 8623–8626 CrossRef CAS PubMed.
- M.-L. Feng, D. Sarma, X.-H. Qi, K.-Z. Du, X.-Y. Huang and M. G. Kanatzidis, J. Am. Chem. Soc., 2016, 138, 12578–12585 CrossRef CAS PubMed.
- X.-H. Qi, K.-Z. Du, M.-L. Feng, J.-R. Li, C.-F. Du, B. Zhang and X.-Y. Huang, J. Mater. Chem. A, 2015, 3, 5665–5673 RSC.
- W. Ma, T.-T. Lv, J.-H. Tang, M.-L. Feng and X.-Y. Huang, ACS Appl. Mater. Interfaces, 2021, 13, 10191–10201 CrossRef.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |