
An organotypic model of high-grade serous ovarian cancer to test the anti-metastatic potential of ROR2 targeted Polyion complex nanoparticles†
Nidhi
Joshi‡
a,
Dongli
Liu‡
 b,
Kristie-Ann
Dickson
c,
Deborah J.
Marsh
cd,
Caroline E.
Ford
*b and
Martina H.
Stenzel
*a
b,
Kristie-Ann
Dickson
c,
Deborah J.
Marsh
cd,
Caroline E.
Ford
*b and
Martina H.
Stenzel
*a
aSchool of Chemistry, University of New South Wales, Sydney, 2052, Australia. E-mail: m.stenzel@unsw.edu.au
bSchool of Women's and Children's Health, Faculty of Medicine and Health, University of New South Wales, Australia. E-mail: Caroline.Ford@unsw.edu.au
cTranslational Oncology Group, School of Life Sciences, Faculty of Science, University of Technology Sydney, Ultimo, NSW 2007, Australia
dNorthern Clinical School, Faculty of Medicine and Health, University of Sydney, Camperdown, NSW 2006, Australia
First published on 11th October 2021
Abstract
High-grade serous ovarian cancer (HGSOC) is the most lethal gynaecological malignancy. Most patients are diagnosed at late stages when the tumour has metastasised throughout the peritoneal cavity. The Wnt receptor ROR2 has been identified as a promising therapeutic target in HGSOC, with limited targeting therapeutic options currently available. Small interfering RNA (siRNA)-based therapeutics hold great potential for inhibiting the function of specific biomarkers, however major challenges remain in efficient delivery and stability. The aim of this study was to investigate the ability of nanoparticles to deliver ROR2 siRNA into HGSOC cells, including platinum resistant models, and estimate the anti-metastatic effect via a 3D organotypic model for ovarian cancer. The nanoparticles were generated by conjugating poly[2-(dimethylamino) ethyl methacrylate] (PDMAEMA) of various chain length to bovine serum albumin (BSA), followed by the condensation of ROR2 siRNA into polyplexes, also termed polyion complex (PIC) nanoparticles. The toxicity and uptake of ROR2 siRNA PIC nanoparticles in two HGSOC cell lines, CaOV3 as well as its cisplatin resistant pair (CaOV3CisR), in addition to primary cells used for the 3D organotypic model were investigated. ROR2 knockdown at both transcriptional and translational levels were evaluated via real-time PCR and western blot analysis, respectively. Following 24 h incubation with the nanoparticles, functional assays were performed including proliferation (IncuCyte S3), transwell migration and 3D co-cultured transwell invasion assays. The PICs nanoparticles exhibited negligible toxicity in the paired CaOV3 cell lines or primary cells. Treating CaOV3 and CaOV3CisR cells with ROR2 siRNA containing PICs nanoparticles significantly inhibited migration and invasion ability. The biocompatible ROR2 siRNA conjugated PICs nanoparticles provide an innovative therapeutic option. ROR2 targeting therapy shows potential in treating HGSOC including platinum resistant forms.
Introduction
High-grade serous ovarian cancer (HGSOC) is the most lethal gynaecological malignancy with a 5 year survival rate of just 31%.1 This is largely due to the lack of robust early detection means and effective treatment at advanced stage.2,3 The majority of the HGSOC cases are diagnosed at an advanced stage (stage III and IV) when tumours have metastasised throughout the peritoneal cavity.4The principal treatment for primary ovarian cancer is debulking surgery, with as much tumour as possible removed. As HGSOC tumours often respond well to DNA damaging agents, platinum-based chemotherapy is generally applied following the surgery or for neoadjuvant chemotherapy.5 The combination of carboplatin and paclitaxel has become the standard treatment due to its better tolerability and prognostic outcome compared to the cisplatin-paclitaxel therapy.6,7 Despite the improved clinical response observed in the carboplatin-paclitaxel combination chemotherapy, 25% of patients with early stage and more than 80% patients with advanced stage ovarian cancer suffer relapse.8 Recurrence occurs in most of the advanced patients within 2 years of the initial chemotherapy treatment.9
Chemoresistance remains a significant challenge in advanced ovarian cancer patients, and novel therapies to overcome this resistance are required. The evolutionarily conserved Wnt signalling receptor ROR1 and ROR2 are abnormally expressed in several malignancies including ovarian cancer.10,11 Silencing ROR1 and ROR2 significantly inhibited metastatic features of OVCAR4 in vitro.12 The upregulation of ROR2 was also correlated with platinum resistant cell line models of ovarian cancer.13 While there are numerous ROR1 targeting therapies in development and clinical trials, ROR2 targeting therapies remain limited.14,15
Targeting ROR2 in ovarian cancer has potential as a powerful strategy to overcome one of the major treatment hurdles.16–19 In that regard, gene delivery, which is the successful tool to transfer therapeutic nucleic acids i.e. DNA, siRNA, and oligonucleotides to the target specific site can be explored to assess the therapeutic benefits.20–22 More precisely, siRNA-based therapeutics have been reported to have huge potential due to their ability to suppress the activity of an abnormal gene in a sequence specific manner.23
However, the effectiveness of siRNA therapeutics is compromised as the unprotected drug cannot penetrate the cell membrane alone and is highly susceptible to RNase degradation.24 Therefore, an essential part of siRNA-based therapeutic development is to generate an effective and safe delivery vector to maintain their efficiency and integrity within the bio-environment. An effective and safe delivery vector needs to be constructed in a way that it not only overcomes the body's internal defensive mechanism towards foreign components but also approaches the targeted site without displaying toxic side effects.20,25,26 The efficiency and the integrity of siRNA therapeutics have been reported to be improved when complexed with a delivery vector.26–29 Poly(ethylenimine) (PEI), a versatile and robust gene vector, is considered the gold standard to condense siRNA into polyplexes with high transfection efficiency.30–32 However, the major shortcoming of PEI is its high toxicity. In contrast, the cationic polymer poly[2-(dimethylamino)ethyl methacrylate] (PDMAEMA) presents an attractive alternative to PEI as it can condense siRNA efficiently into polyplexes with negligible toxicity and comparable transfection efficiency.33–35 To further enhance the delivery efficiency, our group previously employed PDMAEMA–albumin conjugates to generate albumin coated nanoparticles, with high efficiency for the delivery of nucleic acid-based therapeutics in cancer treatment.33,36,37 also termed polyion complex (PIC) nanoparticles. Albumin, being a naturally occurring serum protein, possesses an ability to interact with endothelial cell surfaces in a receptor–ligand manner. Among other endothelial receptors, gp60, also known as albondin, and SPARC (secreted protein acidic and rich in cysteine) are albumin binding receptors that are overexpressed in cancer tissues.38–40
The main shortcoming when exploring suitable drug delivery carriers for siRNA, such as the ROR2 silencing siRNA to treat ovarian cancer, is the absence of suitable biological in vitro models that capture the uniqueness of the disease. Modern synthesis techniques make it possible to generate a large library of nanoparticles. However, most nanoparticle-based therapeutic strategies are investigated using a 2D in vitro monoculture cell line model which lacks the relevant physiological characteristics and information compared to in vivo cancer. Many animal models on the other hand are not able to measure the ability of nanoparticles to inhibit metastasis, which in the case of many cancers such as ovarian cancers are more devastating than the primary tumour. Distinct from the hematogenous metastasis observed in most other cancers, ovarian cancer presented a unique metastatic mechanism. In the case of ovarian cancer, cancer cells detach from the primary tumour and disseminate by the physiological movement of peritoneal fluid as single cells or spheroids and spread onto the peritoneum and omentum.3 Then, the cancer cells invade through the mesothelial layer of the omentum and further invade through the extracellular matrix. The interaction between cancer cells and the protective mesothelial layer stands as an essential step for early metastasis and needs to be taken into consideration in pre-clinical models and drug testing.
Therefore, for cancer research, 3D organotypic models present an excellent platform to reconstruct the organ-specific cellular microenvironment which could help to understand the influence of tumour microenvironment on metastatic features.41 Furthermore, 3D organotypic models provide an excellent opportunity to investigate new therapeutics and their response during the multiple stages of cancer progression. An organotypic model based on human omentum derived mesothelial cells and fibroblasts was developed to mimic the microenvironment of ovarian cancer metastasis12,42–45 and has been applied to evaluate nanoparticles previously.45 In short, cancer cells were grown on a layer of human peritoneal mesothelial cells (HPMC) and normal omentum fibroblasts (NOF) that were obtained from fresh omentum samples collected from women with benign or non-metastatic conditions. (Fig. 1). In this work, the HGSOC cell line CaOV3 and its platinum resistant pair CaOV3CisR were incorporated in the model to evaluate the ability of PDMAEMA–albumin conjugates to cargo siRNA into cells and suppress ROR2 expression in HGSOC. The key feature of this study is to investigate the anti-metastatic potential of ROR2 siRNA enclosed in BSA decorated PICs via a patient derived co-cultured 3D organotypic cancer model as depicted by schematic representation in Fig. 1.
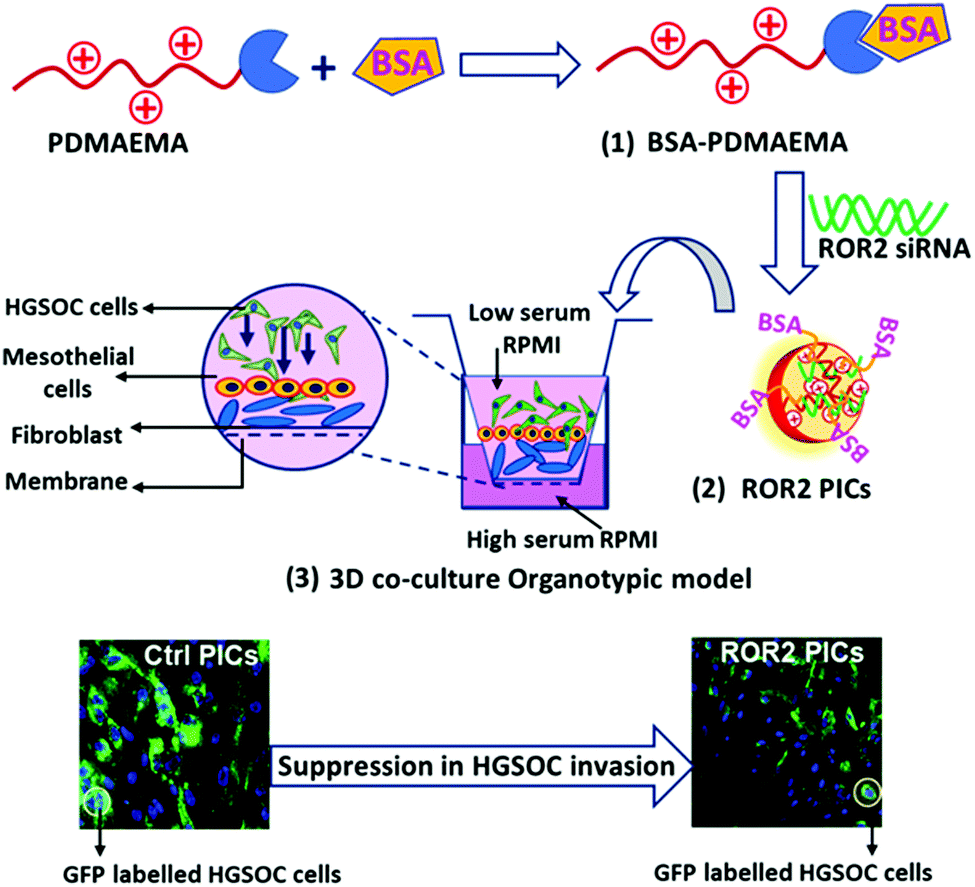 | ||
| Fig. 1 Schematic representation of the synthesis of BSA–PDMAEMA conjugates (1), followed by PICs nanoparticle formation with ROR2 siRNA in HEPES buffer (2). The anti-metastatic ability of PICs nanoparticles was evaluated in 3D co-cultured organotypic cancer model, constructed with primary cells (HPMC and NOF) and GFP labelled high grade serous ovarian cancer (HGSOC) cells (3). | ||
Materials and methods
Materials
All chemicals were of reagent grade and used as received, unless otherwise specified. Cyano-4-[(phenyl carbonothioyl) thiol]-pentatonic acid (CPADB, RAFT agent) was synthesized as described.46 Bovine serum albumin (BSA, Sigma Aldrich, >96%), 4′,4-azobisisobutyronitrile (AIBN), toluene, cyclohexane, N,N-dimethyl amino ethyl methacrylate (DMAEMA, Sigma Aldrich 98%), dimethyl sulphoxide, chloroform, 4-(dimethylamino)pyridine (DMAP, Sigma Aldrich, >99%), ethyl acetate (Ajax, 99%), furan (Aldrich, >99%), hydrochloric acid (Ajax, 31.5% w/w), maleic anhydride (Fluka, >99%), n-hexane (Ajax, >95%), N,N-dimethylformamide (DMF, Ajax, 99.8%), ethanolamine (Ajax, 97%), Silica gel (Sigma-Aldrich, 60 Å, 70–230 mesh), deuterated NMR solvents such as CDCl3 and DMSO, were purchased from Sigma Aldrich. ROR2 siRNA (#s9758) and non-targeting siRNA (#4390844) were purchased from Life Technologies, USA. The synthesis of the protected-maleimide modified RAFT agent (MCPADB) is described elsewhere.33Synthesis and nanoparticle formation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and stirred for 48 hours, and subsequently dialyzed against PBS buffer (10 mM, pH 5) to obtain the BSA conjugated PDMAEMA at pH 5 (isoelectric point of BSA). The BSA conjugated polymer was purified by filtering through a 0.45 μm filter to remove aggregates. Afterwards, BSA–PDMAEMA conjugates were (both filtered and non-filtered) collected and analysed by SDS PAGE and dynamic light scattering (DLS) measurements.
:1 molar ratio of BSA and PDMAEMA of various chain lengths. An aliquot of BSA–PDAEMA conjugate solution (equivalent of 1 mgmL−1) was prepared in PBS (10 mM, pH 5). 20 μL of 20 μM siRNA was dropwise added to the calculated volume of BSA–PDMAEMA solution as represented in Table S2 (ESI†). The solution was incubated for 40 mins to facilitate the condensation of siRNA into nanoparticles. HEPES (10 mM, pH 7) was added to PICs solution to make the final volume of 80 μL (5 μM siRNA).
:1) and stirred for 48 hours, and subsequently dialyzed against PBS buffer (10 mM, pH 5) to obtain the BSA conjugated PDMAEMA at pH 5 (isoelectric point of BSA). The BSA conjugated polymer was purified by filtering through a 0.45 μm filter to remove aggregates. Afterwards, BSA–PDMAEMA conjugates were (both filtered and non-filtered) collected and analysed by SDS PAGE and dynamic light scattering (DLS) measurements.
:1 molar ratio of BSA and PDMAEMA of various chain lengths. An aliquot of BSA–PDAEMA conjugate solution (equivalent of 1 mgmL−1) was prepared in PBS (10 mM, pH 5). 20 μL of 20 μM siRNA was dropwise added to the calculated volume of BSA–PDMAEMA solution as represented in Table S2 (ESI†). The solution was incubated for 40 mins to facilitate the condensation of siRNA into nanoparticles. HEPES (10 mM, pH 7) was added to PICs solution to make the final volume of 80 μL (5 μM siRNA).
Characterization of siRNA loaded PIC nanoparticles
The characterisation and the morphology of siRNA loaded PIC nanoparticles were evaluated by Dynamic Light Scattering and Transmission electron microscopy (TEM) (Fig. 2). Table G in Fig. 2 describes the hydrodynamic diameter (Dh), polydispersity index (PDI) and zeta potential of siRNA loaded PIC nanoparticles.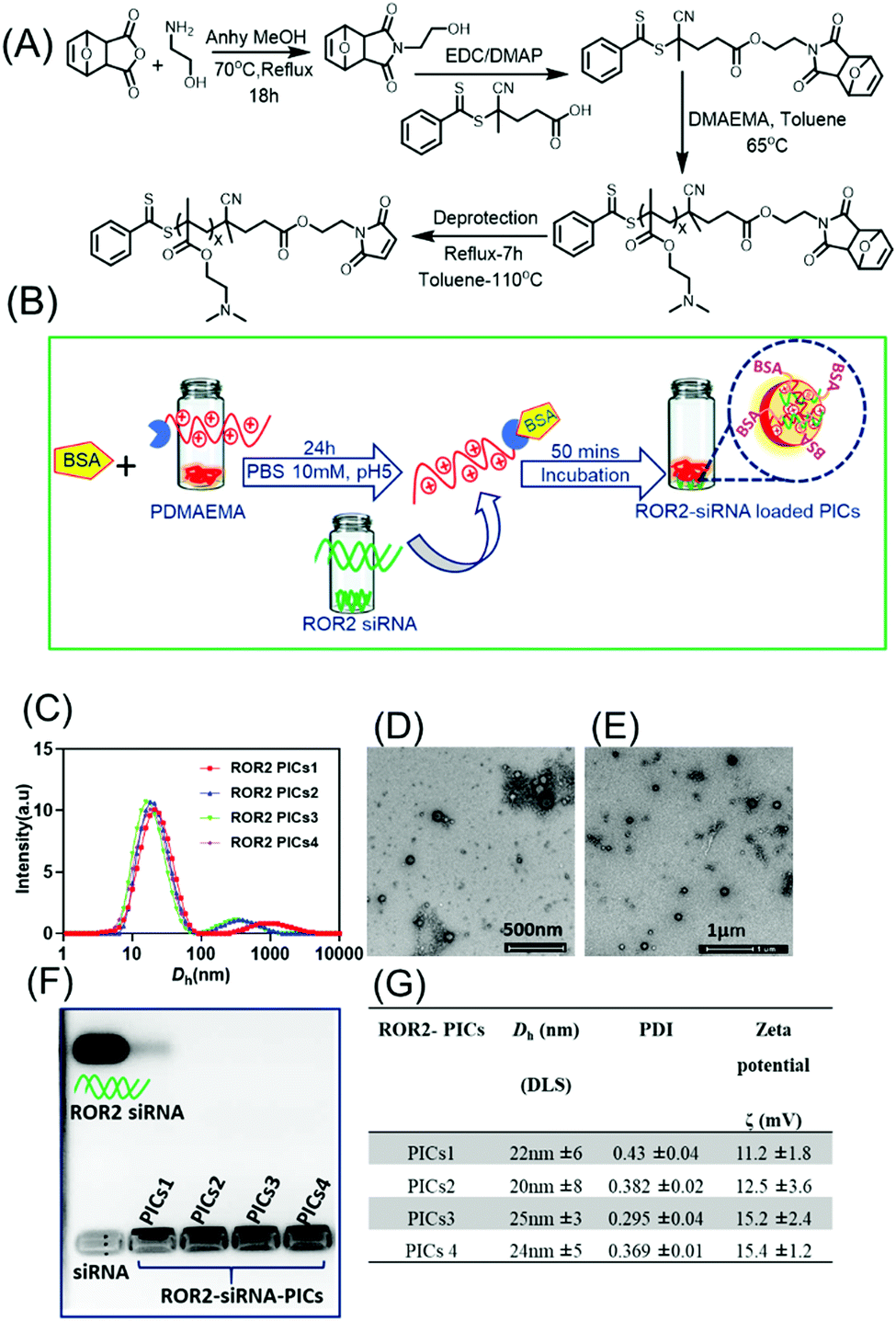 | ||
| Fig. 2 (A) Synthesis of PDMAEMA via RAFT polymerization using the maleimide modified RAFT agent MCPADB. (B) Schematic representation of formation of ROR2 targeting polyion complex nanoparticles (PICs) using BSA conjugated PDMAEMA and ROR2 siRNA in HEPES buffer (10 mM, pH 7). (C) Hydrodynamic size of ROR2 siRNA loaded PICs nanoparticles formed by BSA conjugated polymer with variable chain length at N/P = 10 using Dynamic Light Scattering (DLS). (D–E) TEM images of ROR2 targeting PICs nanoparticles (PICs1; PDMAEMA72 and PICs2; PDMAEMA87) (scale bar 500 nm and 1 μm). (F) Agarose gel electrophoresis of ROR2 siRNA containing PICs nanoparticles (PICs1 to PICs4) with free ROR2 siRNA in lane 1 suggesting that no free siRNA is remaining after entrapment into the nanoparticles (G) Table summarising hydrodynamic size, zeta potential (ζ) and size distribution (PDI) of ROR2 siRNA loaded PICs nanoparticles formed at N/P ratio 10 (mean ±SD, n = 3). | ||
Agarose gel retardation assay
Agarose gel electrophoresis was performed to evaluate the encapsulation of siRNA to BSA conjugated PDMAEMA into polyion complex nanoparticles form. The PICs nanoparticles were prepared between BSA–PDMAEMA conjugates and siRNA at N/P ratio of 10 and loaded with 6 × loading buffer (1 μL (10 mL) in agarose gel (1 wt% agarose gel; 1× TAE buffer). The gel was run for 30 min at 100 V. After 30 mins the gel was stained with ethidium bromide and visualised by an ultraviolet (UV) imaging system (Biored).Cell culture
CaOV3 cells were from the American Type Culture Collection (ATCC, Virginia, USA) and cultured in RPMI 1640 (cat. #42402016, Thermo Fisher Scientific, Mulgrave, VIC, Australia) supplemented with 10% FBS (AusGeneX, Molendinar, QLD, Australia). The CaOV3CisR cell line was generated by exposing CaOV3 cells to 75% of the maximal inhibitory concentration (IC75) of cisplatin (cat. #P4394, Sigma-Aldrich, Sydney, NSW, Australia) for 3 days, then allowing cells to recover. Cisplatin concentrations were gradually increased (IC80, IC85, IC90, then IC95), allowing cells to recover each time over an approximately 6-month period. CaOV3CisR cells were then cultured in cisplatin free media for 5 weeks to ensure complete wash out of any residual drug. CaOV3 and CaOV3CisR cells were treated with increasing concentration of cisplatin and the cell viability was determined by CellTiter 96 Aqueous One Solution Cell Proliferation Assay (cat. #G3581, Promega, Madison, USA) (Fig. S2, ESI†). Each experiment was performed in triplicate and repeated three times, with data reported as the mean ± SEM. As shown in Fig. S2 (ESI†), the IC50 values of Cisplatin for CaOV3 and CaOV3CisR were determined as 1.086 and 5.922 μM respectively. The experimentally observed data suggest 5.45-fold increase in cisplatin resistance of CaOV3CisR cells. Cell line authentication was performed by the Australian Genome Research Facility (AGRF; Melbourne, Victoria, Australia).NOF and HPMC were isolated from fresh omentum samples collected from women with benign or non-metastatic conditions as described in ref. 45 and 48. Ethics approval was obtained from the South Eastern Sydney Local Health District Human Research Ethics Committee (SESLHD HREC approval #16/108) to collect omentum samples from patients at the Royal Hospital for Women and Prince of Wales private Hospital (site specific approval ethics # LNR/16/POWH/236). All the experiments were performed in compliance with the relevant laws and institutional guidelines. Primary cells were maintained in DMEM supplemented with 10% foetal bovine serum (FBS), 1% GlutaMAX and 1% penicillin/streptomycin. All cells were cultured in 5% CO2 at 37 °C and subjected to regular mycoplasma testing.
Cell viability assay
The cell viability of CaOV3 and CaOV3CisR as well as the primary fibroblast cells NOF and mesothelial cells (HPMC) treated with non-targeting siRNA (Ctrl siRNA) loaded PICs nanoparticles (PIC1 and PIC2) were measured by the WST-1 assay. Briefly, non-targeting siRNA conjugated PICs nanoparticles PICs1 and PICs2 were prepared in the biosafety cabinet and sterilized by filtering through a 0.22 μM syringe filters. Cancer cells (CaOV3 and CaOV3CisR) and primary cells (NOF and HPMC) were treated with the PICs nanoparticles (final siRNA = 200 nM) for 24 h before replaced with fresh complete medium. After 48 h of incubation, the WST-1 assay was performed. The absorbance was read at 450 nm with a reference wavelength of 655 nm (Bio-Rad BenchMark).Flow cytometry analysis
Flow cytometry analysis was performed to quantify the internalisation of PICs nanoparticles on CaOV3, CaOV3CisR, as well as on primary cells NOF and HPMC. The cells were seeded into 6 well plates at the concentration of 3 × 105 cells and incubated with fluorescent labelled ROR2 siRNA and PICs nanoparticles (cy3 labelled siRNA and FITC labelled BSA–PDMAEMA) for 4 h at a final concentration of 266 nM siRNA. The transfection was stopped by washing cells three times with cold PBS and collected by trypsinisation. The cell pellet was redispersed in 1 mL HBSS and analysed by measuring the fluorescence intensity of 10000 events per well using BD FACS-Canto TM II Analyser). The data was presented as an average of median fluorescence intensity (MFI) using Flow Jo Software (FlowJO 7.6.1).
Confocal Laser Scanning Microscopy (CLSM)
A Confocal Laser Scanning Microscopy (CLSM) was used to observe the qualitative distribution of siRNA loaded PICs nanoparticles in both CaOV3 and CaOV3CisR cells. Cells were seeded in glass-based 33 mm Fluorodish (2000 cells per mL) and incubated for 24 h before treatment with fluorescent labelled ROR2-siRNA loaded PICs nanoparticles (cy3 labelled ROR2 siRNA and FITC labelled BSA–PDMAEMA for 4 h. The cells were stained with nucleus stain Hoechst 33342 before visualising with the Zeiss LSM 800 instrument. The ZEN blue imaging software (ZEISS) was used for image acquisition and processing.Quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Total RNA was extracted from the cells using RNeasy Mini kit (Qiagen, USA). Up to 1 μg RNA was reverse transcribed with the QuantiTect cDNA synthesis kit (Qiagen, USA) following DNase treatment (Life Technologies, USA). Real time PCR was performed using QuantiNova SYBR Green PCR kit (Qiagen, USA) and was conducted on the AriaMx Real-Time PCR machine (Agilent Technologies, USA). Each sample was repeated in triplicates with non-reverse transcribed RNA samples included as negative controls. The relative expression level of ROR2 was calculated using 2−ΔΔCt method and normalised against the mean of three house-keeping genes (HSPCB, SDHA, RPL13A). Primer sequences were provided in a previous study.10Western blot
Total protein was extracted from the cells using cell lysis buffer (Cell Signalling Technology, USA) with protease inhibitor (Sigma-Aldrich, USA). Western blot analysis was performed as previously described.10 Primary antibodies used in this study were anti-ROR2 (#34045, QED Bioscience, USA) and anti-α-Tubulin (#3873, Cell Signalling, USA).Proliferation assay
The effect of ROR2-siRNA in PICs nanoparticles on cell proliferation was quantified using the IncuCyte S3 Live Cell Analysis system. Briefly, CaOV3 and CaOV3CisR cells were seeded in 6-well plates and incubated with either ROR2-siRNA or non-targeting siRNA loaded PICs nanoparticles for 24 h prior to being placed in the IncuCyte. Phase contrast cell images (9 images per well) were obtained using a 10× objective lens within the instrument every 3 h for 72 h in total. The average confluence of each well was calculated and normalised against the baseline (Time 0).Transwell migration assay
The migration ability of CaOV3 and CaOV3CisR were measured using the Corning transwell inserts according to manufacturer's protocol (Corning Life Sciences, USA). The cells were seeded in 6 well plates (5 × 105 per well) and incubated with ROR2-siRNA or non-targeting siRNA loaded PICs nanoparticles for 24 h. The cells were then trypsinised and plated in the upper chamber of the insert (2 × 105 cells per insert) and incubated for 24 h (CaOV3) or 48 h (CaOV3CisR) before being fixed with methanol and stained with 1% Crystal violet. The membranes were removed from each transwell and mounted onto glass slides for imaging.3D organotypic model: invasion assay
To gain better insight on the impact of siRNA loaded PICs nanoparticles into anti-invasive ability of HGSOC cancer, 3D organotypic model were constructed. HGSOC cells (CaOV3 and CaOV3CisR) were labelled with GFP with pLKO.1-Neo-CMV-tGFP vector (Sigma-Aldrich, USA) and transfected with siRNA loaded PICs nanoparticles and incubated for 24 h prior to being plated on the 3D model. The patient derived organotypic 3D co-culture model of HGSOC was prepared and plated in the Corning transwell inserts as described previously.45 Briefly, 24-well culture plate with transwell inserts (pore size 8 μm) was incubated with 7.5 μg of rat-tail collagen I in PBS (200 μL) overnight then plated with NOF/collagen (4 × 104 mL−1) mixture for 4 h and topped with HPMC cells (4 × 105 mL−1). The co-culture plates were incubated overnight before subsequent analysis. Nanoparticle treated cancer cells were trypsinised, resuspended in low serum RPMI medium (2 × 105 cells), and plated onto the pre-co-cultured inserts. The inserts were placed onto the well filled with high serum media. The co-cultured 3D model was incubated under 37 °C in 5% CO2 for 24 h (CaOV3CisR) and 48 h (CaOV3) respectively. The inserts were washed with PBS followed by 4% paraformaldehyde fixation for 20 mins. The membrane was removed from the inserts and mounted on the glass slide with DAPI mounting. The invaded cells were visualised and counted by Zeiss LSM 800 (ZEISS software) and processed with ZEN blue imaging software (ZEISS).Statistical analysis
At least three sets of PICs were prepared and characterised. Results were presented as mean ± standard deviation (SD). For the in vitro assays, all experiments were repeated three times independently. Paired t-test was performed to analyse the significance. t-Test values below p < 0.05 were considered statistically significant. Correlation between ROR2 knockdown level at transcriptional level and difference in 3D invasion cell amount was performed using nonparametric Spearman correlation coefficient. Statistical significance was defined at p < 0.05. All the analysis and figures were provided with GraphPad Prism (7.04).Results and discussion
Synthesis and characterisation of siRNA loaded PDMAEMA–BSA PIC nanoparticles
To construct the biocompatible polymeric vector to encapsulate and deliver ROR2 targeting siRNA, the cationic polymer PDMAEMA bearing the protected maleimide end group was first synthesized via RAFT polymerisation using procedures reported earlier.36,37 Here, bovine serum albumin (BSA) was used as protein, which has similar properties to human serum albumin (HSA).49 Briefly, we first synthesised the RAFT agent MCPADB carrying the furan protected maleimide group, followed by RAFT polymerisation to synthesise PDMAEMA (Fig. 2A). PDMAEMA with furan protected maleimide group was purified and subsequently deprotected to achieve the free maleimide end group to enable BSA conjugation.33 The deprotection of furan group was confirmed by 1H NMR, evidenced by the maleimide peak at 6.89 ppm (Fig. S1, ESI†). Thereafter, BSA conjugation to PDMAEMA was achieved by Michael addition reaction between the maleimide end group of PDMAEMA and the free thiol group on BSA (Cys34 domain). The quantitative conversion of PDMAEMA and the presence of maleimide group was confirmed by 1HNMR and size exclusion chromatography (SEC) (Fig. S1 and S3, ESI†). PDMAEMAx with repeating units x ranging from 72, 87, 150 and 220 and molecular weights of 12 kDa, 14 kDa, 28 kDa and 36 kDa, respectively, were conjugated to BSA (polymer to BSA ratio was 1:1 molar ratio) as represented in Table S3 (ESI†) and then tested using SDS PAGE (Fig. S4, ESI†). The reader can find in-depth discussions on the conjugation efficiency in earlier work.33,49 The purpose of generating the library of BSA modified polymers differing in cationic block length is to evaluate their binding efficiency with negative siRNA to form PICs complex, and thereafter to investigate the best performing PICs nanoparticles system in terms of their biocompatibility and antimetastatic ability in HGSOC cells.
PICs nanoparticles were generated between the cationic PDMAEMAx (x = 72, 87, 150 and 220) and the negative charged ROR2 targeting siRNA in HEPES buffer (10 mM, pH 7.2) using a previously reported procedure with oligonucleotides.37,49 The calculated amount of ROR2-siRNA (siRNA concentration 20 μM) was dropwise added to the required amount PDMAEMA–BSA at a N/P ratio of 10, followed by 40 mins incubation. The N/P ratio, which is the stoichiometric ratio between amino group of PDMAEMA and phosphate entities of siRNA were chosen to be 10 (N/P = 10). A range of optimization studies (not shown here) revealed this to be the ideal ratio for the generation of PIC nanoparticles with narrow particle size distribution. The formation of ROR2–PICs nanoparticles was confirmed by dynamic light scattering (DLS) analysis (Fig. 2C), transmission electron microscopy (TEM) (Fig. 2D–E), and agarose gel electrophoresis (Fig. 2F). The hydrodynamic diameter of the ROR2–PICs nanoparticles ranged between 20 nm to 50 nm with a polydispersity index, PDI < 0.4. There is a very small fraction of aggregated nanoparticles but considering that the intensity distribution is shown this fraction can be neglected. DLS measurements did not show any significant differences in size between the PICs nanoparticles formed from PDMAEMAx with different chain lengths (x = 72, 87, 150 and 220). TEM images of ROR2–PICs nanoparticles (PICs3 and PICs4, Fig. S5, ESI†) revealed the spherical shape und sizes that are slightly smaller than the ones observed from DLS. Independent from the length of PDMAEMA, all PIC nanoparticles had a positive surface charge (PICs1 = 11.2 mV, PICs2 = 12.5 mV, PICs3 = 15.2 mV and PICs 4 = 15.4 mV). This confirms the successful encapsulation of siRNA, but also the formation of undefined solid nanoparticles with some PDMAEMA located on the surface as this would explain the slightly positive surface charge (Fig. 2G). Agarose gel electrophoresis analysis further validated the formation of PIC nanoparticles by the disappearance of free siRNA band in agarose gel run for 30 mins in 100 mV (siRNA final concentration = 50 nM). ROR2–PIC nanoparticles (PICs1 to PICs4) did not show any band for free ROR2 siRNA (Fig. 2F).
Ability of siRNA loaded nanoparticle to inhibit migration and invasion
Prior to the analysis of the biological activity, we would like to introduce the organotypic models used in the system. As discussed above, the challenge with ovarian cancer is the ability of the cells to metastasize and invade to the other areas in the body. The migration assay model tests the ability of the ovarian cancer cells, CaOV3 and their cisplatin-resistant cell line CaOV3CisR to penetrate through a porous membrane into fresh FBS supplemented media. Drugs that are successful in inhibiting migration will prevent that transfer through the semi-permeable membrane of the transwell plates into the basolateral chamber (Fig. 3A). This model can provide initial results on the ability of cells to translocate, but it does not closely simulate the environment in ovarian cancer. The 3D invasion model (Fig. 3B) considers that for cancer cells to metastasize, they need to be able to invade through the omentum layer in the peritoneal cavity. The cancer cells, which are incubated with drug loaded nanoparticles, are therefore placed on top of a layer of patient derived HPMC. HPMC cells function as a protective layer of omentum and play an important role in the dissemination of ovarian cancer cells such as those responsible for ovarian cancers.50 The HPMC layer is positioned on a layer of patient derived NOF to mimic the omentum barrier. Epithelial ovarian cancer is prone to omentum metastasis as the cancer cells activate the NOFs to contribute to the invasion and adhesion of the cancer cells.51 Again, the absence of cancerous cells in the basolateral chamber is evidence for the successful inhibition of invasion. In both models, good uptake of nanoparticles by the cancerous cells and release of the active payload are prerequisites.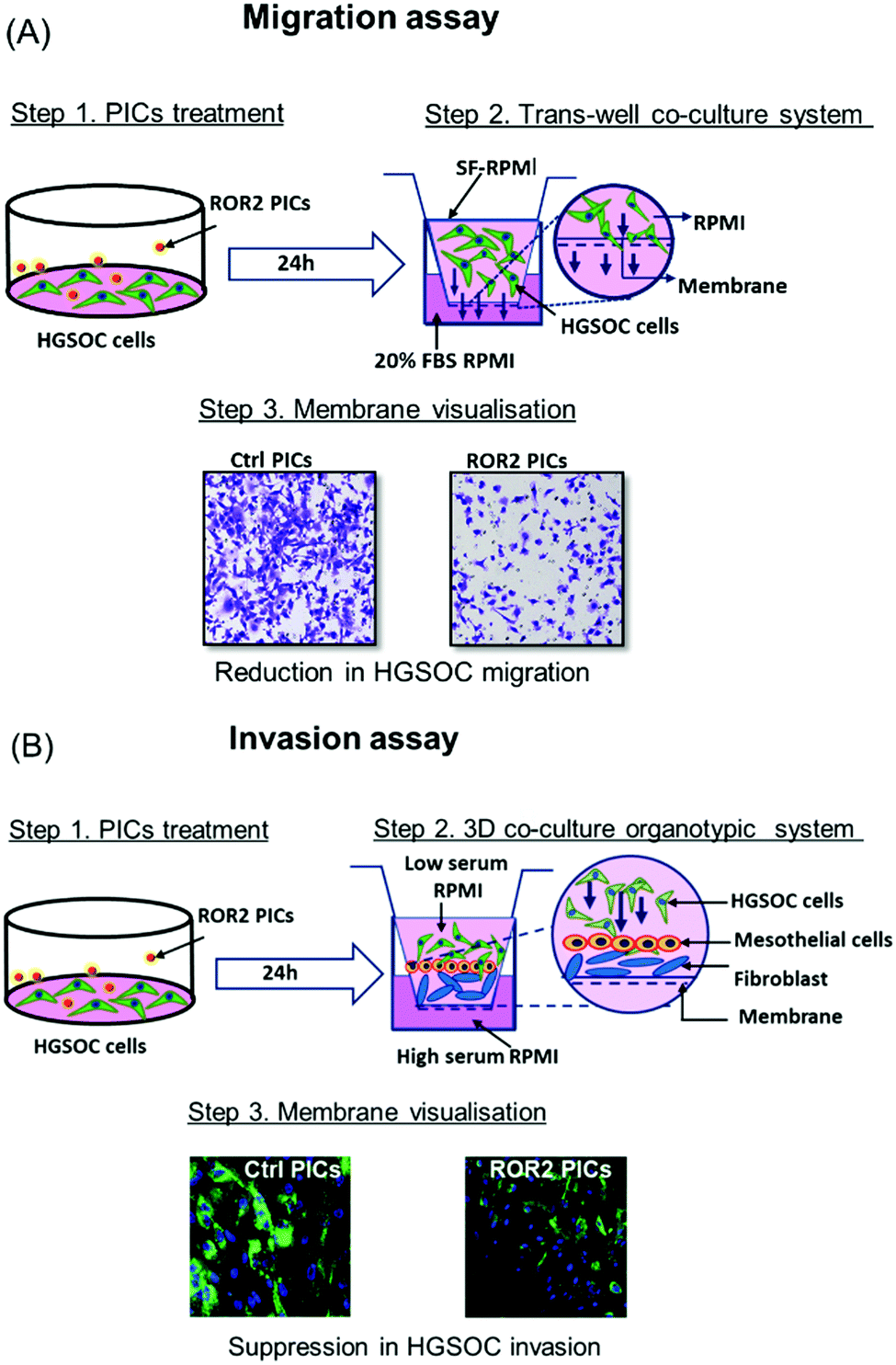 | ||
| Fig. 3 Schematic representation of migration and invasion assay model for HGSOC cells (CaOV3 and CaOV3CisR). (A) Transwell migration of HGSOC treated with ROR2–PICs and Ctrl (control, inactive siRNA) PICs, respectively. Cells migrating through the membrane pores were stained with crystal violet and visualised by CLSM. (B) Invasion assay performed in the 3D organotypic model formed by HGSOC and primary cells. The cells were treated with Ctrl PICs and ROR2 siRNA loaded PICs. The anti-invasive effect was examined by visualising the membrane by CLSM as represented. | ||
Cellular uptake and intracellular distribution of siRNA–PICs micelles to HGSOC cells
Prior to in-depth testing using our organotypic models, we initially investigated the effect of all four PIC nanoparticles summarised in Fig. 2G and Table S2 (ESI†) regarding the ability to reduce the expression of ROR2 when loaded with siRNA. Prerequisite is the non-toxicity of the polymers and the fast uptake of the nanoparticles by the tested cells. The PIC nanoparticles were incubated with CaOV3 and CaOV3CisR for 4 h and the cellular uptake was monitored by confocal laser scanning microscopy and the cell association was studied by flow cytometry (Fig. 4B, Fig. 4(C–F), Fig. S6 and S7, ESI†). All nanoparticles were efficiently taken up by cells, but after incubation for 48 to 72 h, PICs3 and PICs4 showed clear signs of toxicity to both CaOV3 and CaOV3CisR cells (Fig. S8, ESI†) and are therefore omitted from further studies. In contrast, the cytotoxic activity of PICs1 and PICs2 nanoparticles, loaded with scrambled siRNA (Ctrl–PICs1 and Ctrl–PICs2 final concentration 266 nM) as non-toxic drug, were observed to be non-toxic towards cancer cells (CaOV3 and CaOV3CisR) as well as primary mesothelial and fibroblast cells (HPMC and NOF) (Fig. 4A) after 48 h of transfection (Fig. 4A). This chain length-dependent toxicity is well known and shorter polymers are usually less toxic.52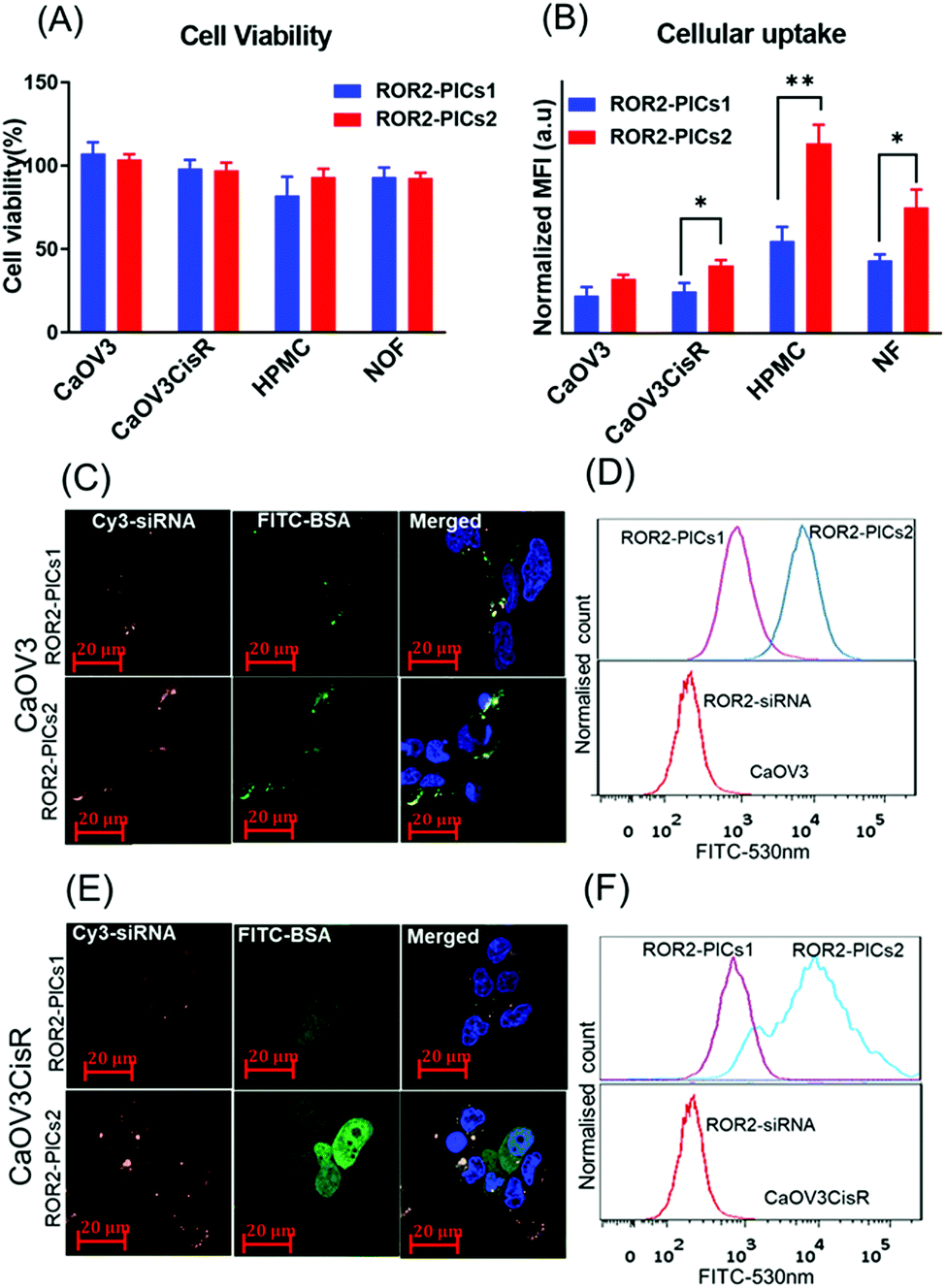 | ||
| Fig. 4 Cell viability and in vitro cellular uptake (A) Cytotoxicity of PICs nanoparticles, PICs1 and PICs2 containing Ctrl siRNA (266 nM) on CaOV3, CaOV3CisR, HPMC and NOF cells. Error bars indicate the standard deviation of quadruplicate wells, n = 1 (B) Flow cytometry analysis on cellular uptake of cy3-labelled ROR2 siRNA alone and ROR2 siRNA enclosed in PICs1 and PICs2 (FITC labelled BSA conjugates) after 4 h of transfection with cancer cells (CaOV3, CaOV3CisR cells) and primary cells (HPMC and NOF) flow cytometry data presented in bar graph (n = 3, mean ± SD); *p = 0.0152 and **p = 0.0026 (paired t-test analysis, p < 0.05) (C–E) Confocal laser scanning microscope visualization of siRNA localization in CaOV3 and CaOV3CisR cells after 4 h of incubation with PICs1 and PICs2 (scale bar, 20 μm; nuclei, blue; nanoparticles, PICs1 & PICs2; FITC–BSA green/cy3–ROR2 siRNA orange) (D–F) Flow cytometry data histogram to represent cellular internalisation of PICs (red: cy3 labelled ROR2 siRNA alone, purple and blue represents ROR2 siRNA enclosed in PICs1 and PICs2. | ||
The cellular uptake efficiency of PICs, now loaded with cy3 labelled ROR2 siRNA, by cancer cells (CaOV3 and CaOV3CisR) as well as primary cells (HPMC and NOF) were evaluated by both flow cytometry and confocal laser scanning measurements. The quantitative flow data suggested that HGSOC cells (both CaOV3 and CaOV3CisR) and primary cells (HPMC and NOF) treated with ROR2–PICs2 (siRNA) demonstrated higher fluorescence intensity than ROR2–PICs1 (FITC labelled BSA conjugates) (CaOV3, p = 0.0152) (Fig. 4B), proposing that ROR2–PICs2 nanoparticles were more efficiently internalised by cells compared to ROR2–PICs1 nanoparticles. It was observed that the uptake of ROR2–PICs2 was significantly higher in both primary cells (HPMC, p ≤ 0.0026 and NOF, p = 0.0255) (Fig. 4B). As BSA is known to be an important drug carrier and can identify many ligands and receptors present in various tissue or cell types including healthy primary cells such as fibroblasts, which show higher affinity to serum albumin.53 Additionally, confocal microscopic observations confirmed the internalisation of ROR2–PICs1 and ROR2–PICs2 (Fig. 4C and D) in both CaOV3 and CaOV3CisR cells after transfection with the ROR2–PICs for 4 h. The orange and green pixels which correspond to cy3 labelled siRNA (ROR2) and FITC labelled BSA–PDMAEMA conjugates, respectively, were observed to be in the cells. It appears that siRNA and drug carrier are co-localized in CaOV3 cells, but drug and drug carriers can be found in parts in different areas in CaOV3CisR, suggesting disassembly after 4 h. The confocal microscopy images confirmed that the cells treated with ROR2–PICs2 displayed higher fluorescence signal for both cy3 labelled siRNA and FITC labelled BSA conjugates compared to ROR2–PICs1. These observations were consistent with the data acquired by flow cytometric analysis (Fig. 4D–F). It is not immediately clear why PICs2 has a higher cellular uptake across all cell lines. Both particles have similar sizes and similar zeta potential, which would suggest a similar degree of translocation into cells. However, analysis of the fluorescence spectra (Fig. S9, ESI†) shows that despite the same fluorescence intensity of the free PDMAEMA polymer that make up PICs1 and PICs2 (Fig. 2G and Table S2, ESI†), the fluorescence in PICs2 is quenched to a greater extend that in PICs1. The reduction in fluoresce intensity can usually be correlated directly to tighter packing of the polymers, which will contribute to hardness of the particles and better cellular uptake.54 The fluorescence analysis of PICs3 and PICs4 display the similar trend as represented by Fig. S10 (ESI†).
Both PIC nanoparticles are efficiently taken up by the cells ensuring high delivery of siRNA into the cells. In the subsequent step, the nanoparticles need to escape the endosomes and unload the siRNA to reach into the cytoplasm. The success of these steps can be observed when measuring the ability of siRNA loaded PICs nanoparticles (PICs1 and PICs2) in reducing the ROR2 expression level (Fig. 5A and B) and (Fig. 6A and B). The upregulation of ROR1 and ROR2 has been linked to metastatic features of ovarian cancer.15,55 As previously shown, targeting these receptors can significantly suppress proliferation and invasion of ovarian cancer cells in vitro.10,56 From the cellular uptake studies, it was observed that PICs2 cargos more siRNA into the cells, so it was expected that PICs2 would display higher knockdown efficiency. However, PICs1 showed a slightly better knockdown efficiency of ROR2 expression level at both transcriptional and translational levels in CaOV3 cells (Fig. 5A and B). This behaviour could be the result of a reduced endosomal escape or reduced release of ROR2-siRNA from the drug carrier, which could have limited their knockdown ability of ROR2 expression. The knockdown assay is usually an endpoint assay, but it cannot provide information on each step in the process such as the cellular uptake, the escape of the nanoparticles from the endosomes and the release of siRNA from the drug carrier.57 PDMAEMA has widely been explored as transfection agent. In homopolymers it was found that longer polymers are more efficient, but this was assigned to the inability of PDMAEMA with short chains to condense the DNA effectively, resulting in large ill-defined particles.52,58–60 In some cases an increased transfection efficiency was observed with longer polymers when the longer polymers are able to provide better protection, they are able to destabilise the cell membrane, or they serve as microenvironmental buffer.52,59,60 At the same time, other systems reported a reduced transfection efficiency with increasing length of the cationic polymer block.61 Reason may be the slow release of the cargo as longer polymers may form stronger electrostatic interactions. It is therefore likely here that despite cellular uptake of the nanoparticles, the longer PDMAEMA does not allow the timely release of siRNA. PICs2 loaded with ROR2-siRNA significantly inhibited proliferation of CaOV3 at 72 h (p < 0.0001, Fig. 5C) while PICs1 did not have any significant effect. The control with scrambled inactive siRNA revealed that the reduced proliferation is indeed the result of the presence of siRNA and not that of the polymer. These standard 2D experiments were only able to reveal the transfection efficiency but cannot confirm if the nanoparticles are indeed able to reduce the ability of these cancerous cells to migrate.
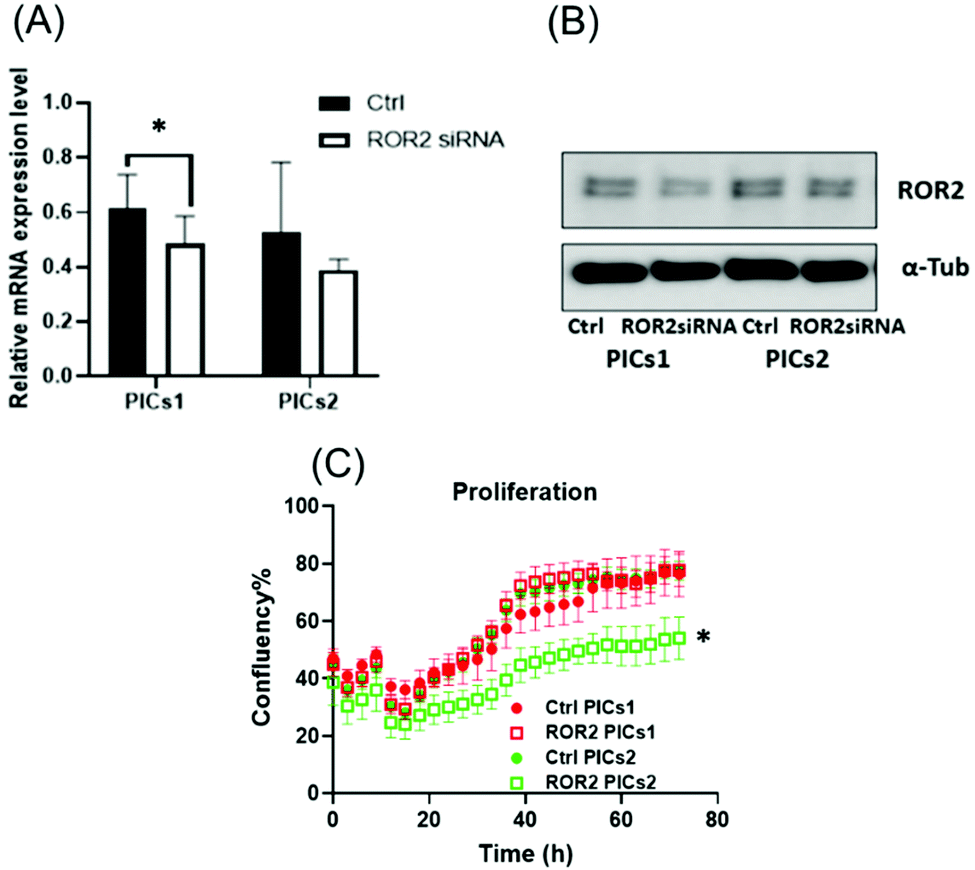 | ||
| Fig. 5 The mRNA and protein expression level of ROR2 in CaOV3 cancer cells. (A and B) mRNA expression of ROR2 and western blot analysis of protein expression in CaOV3 cancer cells after treatment with PICs1 and PICs2. PICs1 performed better than PICs2 in terms of suppressing ROR2 expression level at transcriptional and translational levels (n = 3) (C) ROR2–PICs2 significantly reduced the cell proliferation of CaOV3 at 72 h (p < 0.0001, n = 3). | ||
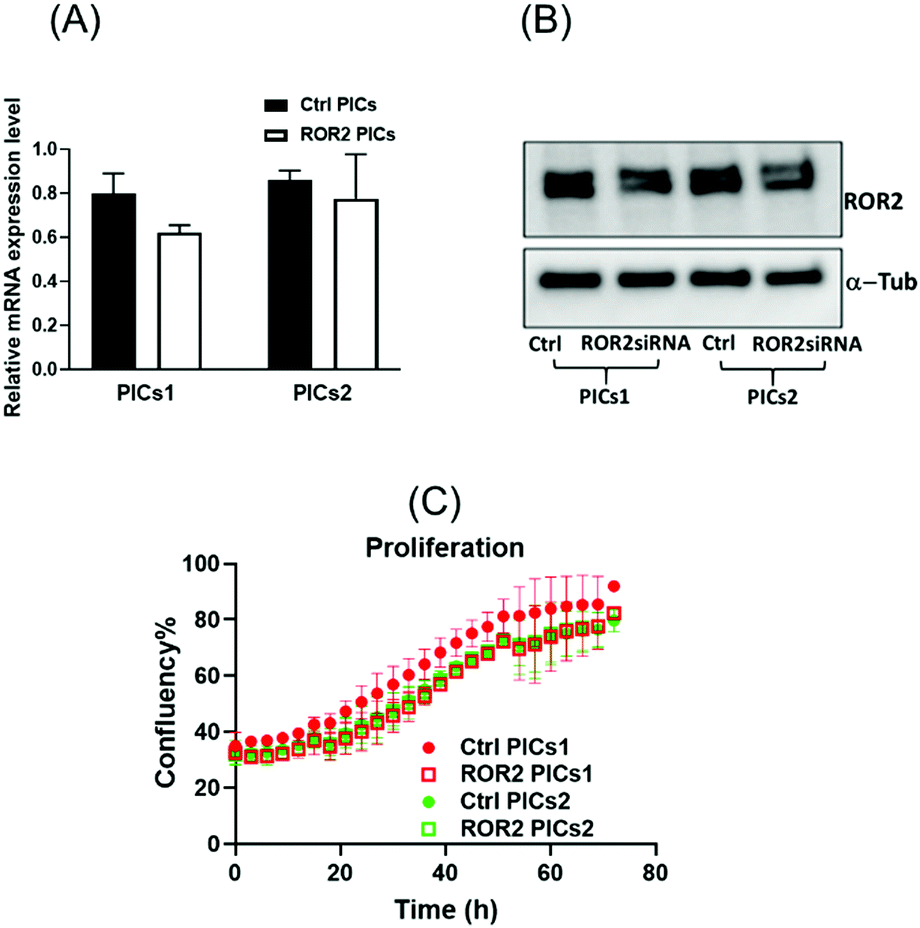 | ||
| Fig. 6 The mRNA and protein expression level of ROR2 in CaOV3CisR cancer cells. (A and B) mRNA expression of ROR2 and western blot analysis of protein expression in CaOV3CisR cancer cells after treatment with PICs1 and PICs2. PICs1 performed better than PICs2 in terms of suppressing ROR2 expression level at transcriptional and translational levels. (C) ROR2 conjugated PICs did not affect cell proliferation of CaOV3CisR at 72 h. | ||
ROR2 siRNA conjugated PICs significantly inhibited the migration and invasion ability of CaOV3
At this point, the models depicted in Fig. 3 can provide additional information. Initially, transwell-based migration assay was used to evaluate if ROR2-siRNA can limit the migration of the cells into the basolateral chamber. The transwell inserts were equipped with a semipermeable membrane which permits the migration of cells through them as represented by Fig. 3A. CaOV3 cells were transfected with PICs nanoparticles containing ROR2-siRNA (PICs1 and PICs2) and their respective controls containing inactive siRNA (Ctrl PICs1 and Ctrl PICs2) were seeded onto transwell inserts containing serum free medium and placed on the chamber with medium (RPMI) containing 20% FBS. The CaOV3 cells treated with ROR2 siRNA loaded PICs (PICs1 and PICs2) have shown significant reduction in the number of migrating cells as observed (Fig. 7A). Analysis of the transwell membrane stained with crystal violet confirmed a significantly reduced number of CaOV3 cells, confirming that the nanoparticle delivered siRNA is indeed able to limit movement (Fig. 7B). In contrast to earlier preliminary experiments that show better uptake and better proliferation inhibition of PICs2 nanoparticles, there is no difference between both nanoparticles. Both nanoparticles were able to deliver siRNA, which then reduces the expression of ROR2. This translates into reduced CaOV3 migration, a sign of potentially reduced metastasis.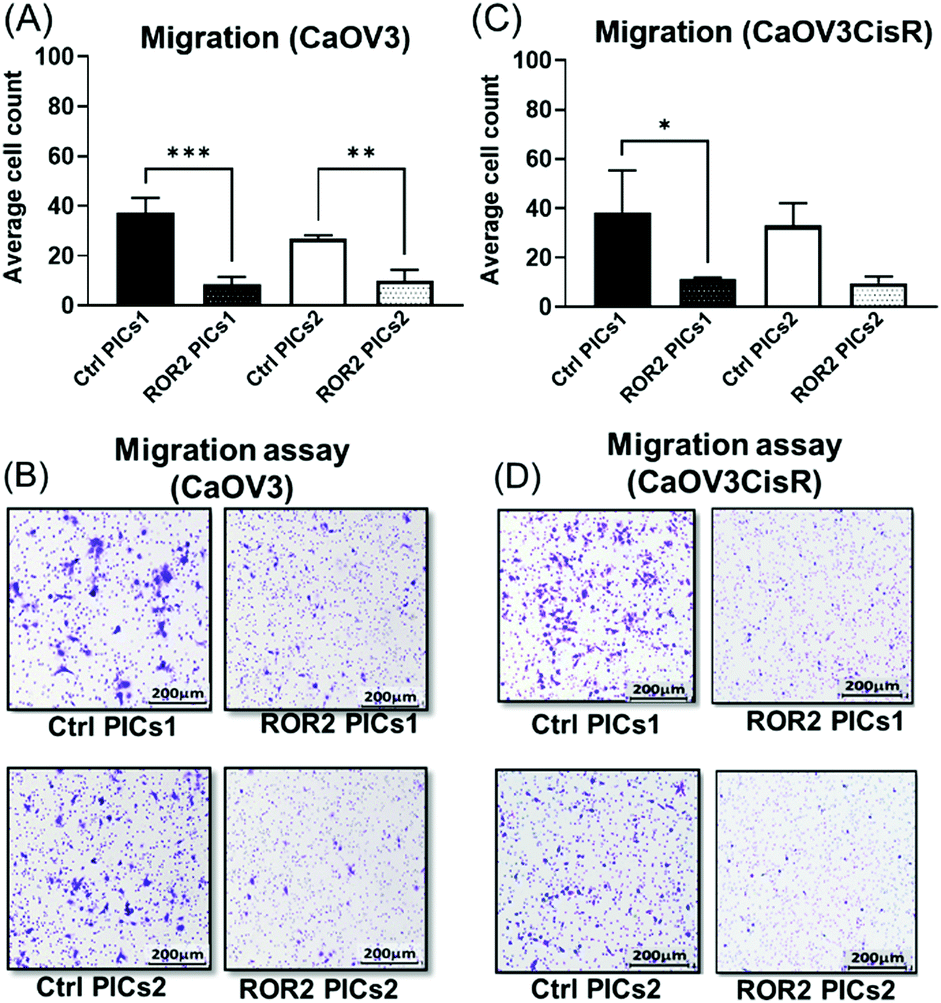 | ||
| Fig. 7 Polyion complex nanoparticles (PICs) loaded with ROR2 siRNA significantly inhibited the migration ability of HGSOC cells. (A) Both PICs1 and PICs2 loaded with ROR2 siRNA significantly decreased the migration ability of CaOV3 (p = 0.0001 and 0.005 respectively). (B) Representative images of transwell membranes stained with crystal violet showed less CaOV3 cells migrated after incubated with PICs1 or PICs2 loaded with ROR2 siRNA (n = 3). (C) ROR2 siRNA loaded PICs1 significantly decreased the migration ability of CaOV3CisR (p = 0.042, n = 3). (D) Representative images of transwell membranes stained with crystal violet showed less CaOV3CisR cells migrated after incubated with PICs1 or PICs2 loaded with ROR2 siRNA. *Significant at p < 0.05 level n = 3. | ||
The nanoparticles were subsequently tested using the organotypic model that captures traits of ovarian cancer Fig. 3(A) and (B). In this 3D invasion assay, the cancerous CaOV3 cells need to migrate through the layers of patient derived NOFs and HPMCs. Effective treatment should be able to limit the invasion. The CaOV3 cells were transfected with PICs nanoparticles containing ROR2-siRNA (PICs1 and PICs2) or their respective controls containing inactive siRNA (Ctrl PICs1 and Ctrl PICs2). The cells were then mounted onto the pre implanted inserts with co-cultured 3D model as described earlier. Compared to the control samples, the 3D invasion ability of CaOV3 was significantly inhibited after treatment with PICs1 and PICs2 containing ROR2 siRNA, (Fig. 8A and B). ROR2 siRNA was reported to inhibit migration of OVCAR3 cells in our previous study.10 ROR2 knockdown prevented the Wnt5a-induced activation of RhoA, which led to reduced migration of the osteosarcoma cells.62 It is therefore well known that ROR2 siRNA can inhibit migration in ovarian cancer, but the organotypic 3D invasion assay used here confirmed now that the ability of ovarian cancer cells to penetrate through the omentum to progress could also be suppressed by ROR2 siRNA. This 3D invasion assay revealed that despite differences in cell uptake and cell proliferation, the ability to invade patient-derived NOFs and HPMCs of both PICs is similar. The correlation analysis showed that inhibition in 3D invasion ability appears not to be correlated with ROR2 knockdown level (Spearman r = −0.257, p = 0.6583).
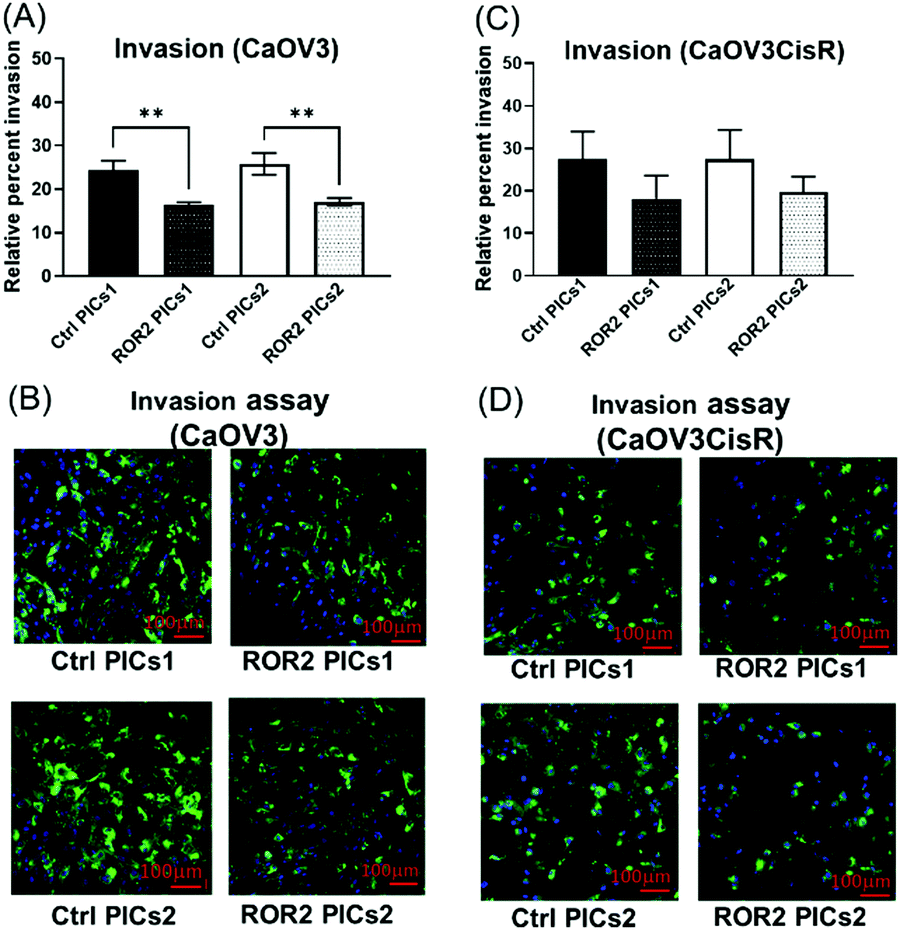 | ||
| Fig. 8 Polyion complex nanoparticles loaded with ROR2 siRNA significantly inhibited the invasion ability of HGSOC cells. (A) Both PICs1 and PICs2 loaded with ROR2 siRNA significantly decreased the 3D invasion ability of CaOV3 (p = 0.003 and 0.001 respectively, n = 3). (B) Representative fluorescent images of 3D invasion transwell membranes showed less CaOV3 cells (GFP labelled) invaded through the membrane after incubated with ROR2 siRNA loaded PICs1 or PICs2 (n = 3). *Significant at p < 0.05 level. **Significant at p < 0.01 level. ***Significant at p < 0.001 level, n = 3. (C) Neither of the PICs (ROR2–PICs1 or ROR2–PICs2) loaded with ROR2 siRNA significantly changed the 3D invasion ability of CaOV3CisR (n = 3). (D) Representative fluorescent images of 3D invasion transwell membranes showed less CaOV3CisR cells invaded through the membrane after incubated with PICs1 or PICs2 loaded with ROR2 siRNA. *Significant at p < 0.05 level n = 3. | ||
ROR2 siRNA conjugated PICs1 significantly reduced the migration potential of CaOV3CisR
The standard care for ovarian cancer involves, next to surgery, the treatment with chemotherapeutic agents, in particular platinum drugs. Patients with resistance to platinum-based drugs, a common form of relapse, have often a poorer cancer prognosis.63 The cisplatin resistant ovarian cancer cell line CaOV3CisR can therefore serve as a model for this scenario.64–69 As observed in the case of CaOV3 cells (Fig. 5A and B), both PICs nanoparticles loaded with ROR2-siRNA were able to suppress ROR2 expression level at both transcriptional and translational levels in CaOV3CisR (Fig. 6A and B). However, no significant effects on proliferation of CaOV3CisR was observed for either of the PICs (Fig. 6C). Still, PICs1 and PICs2 were both able to reduce migration, yet only PICs1 reduced the migration ability of CaOV3CisR significantly (p = 0.042, Fig. 7C) while PICs2 showed only a reduced migration trend (p = 0.102, Fig. 7C). PICs1 or PICs2 containing ROR2-siRNA, both led to a decrease in the 3D invasion ability of CaOV3CisR (Fig. 8C), but this was not statistically significant (p = 0.273 and 0.417, respectively. Compared to CaOV3, the effect of PICs nanoparticles loaded ROR2 siRNA on the cisplatin resistant cell line CaOV3CisR seems to be overall reduced, highlighting the challenges when dealing with more aggressive cancer cells.It can be concluded that ROR2 targeting siRNA, when delivered in nanoparticles, can inhibit the invasion ability of HGSOC. This 3D organotypic model can therefore serve as a link between in vitro and in vivo models and can provide us with valuable information on the anti-metastatic evaluation prior to animal models. In fact, there are currently limited preclinical animal models available that can fully capture metastasis in cancer patients, highlighting the need for new organotypic models that can help to extract additional information.70 These 3D models can help to evaluate novel therapy for intraperitoneal metastatic events in ovarian cancer. It was recently shown that ROR2 could be downregulated via siRNA, which further reduced migration and invasion of ovarian cancer cells.10 However, siRNA alone has no measurable cellular uptake and was generally delivered via the lipofectamine system, which is known to be highly toxic. Despite this, we have used lipofectamine as a control here to show the activity of an efficient transfection agent that however rapidly results in cell death (Fig. S11, ESI†). Nanoparticles made from BSA modified cationic polymers offer a non-toxic alternative to cargo nucleic acid based therapeutics owing to their biocompatibility, negligible toxicity and high circulation time.36 Despite lower initial transfection efficiency of the BSA based nanoparticles, the low toxicity makes this carrier ideal to study the potential of ROR2 siRNA as an antimetastatic agent in HGSOC cells. The ROR2 siRNA loaded PICs nanoparticles were constructed with the series of BSA modified PDMAEMA varying in cationic block length (molecular weight; 12 kDa to 36 kDa). Preliminary studies revealed that very long polymers led to toxic side effects, most likely due to the length of the cationic polymer, and they were therefore omitted. Only the PIC nanoparticles based on shorter polymers were studied using the 3D invasion model. The model depicted in Fig. 3 can be applied with various ovarian cancer cell lines, but in this case CaOV3 and the cisplatin resistant cell line CaOV3CisR were used to model HGSOC and cisplatin resistant context. Overall, it can be observed that the ROR2–PICs1 nanoparticle performed slightly better. Although PICs2 had a noticeable higher cellular uptake (Fig. 4B–F), the anti-metastatic potential of ROR2–PICs1 was observed to be superior in terms of suppressing ROR2 expression level as well as inhibiting metastatic ability of both CaOV3 and CaOV3CisR compared to ROR2–PICs2.
The anti-metastatic abilities of PICs nanoparticles (ROR2–PICs2) formed with higher cationic block could be contributed to by the enhanced binding capabilities of PICs2 to siRNA to form more compact PICs assembly. This could restrain the release of siRNA trapped within the polymeric chain hence influence the anti-tumour activity of ROR2–PICs2 compared to ROR2–PICs1. The reduction in mRNA expression is modest in CaOV3CisR cells, which translates to undetectable changes in proliferation. However, thanks to the models used here, the ability of these nanoparticles to inhibit migration and invasion was evident. Even the CaOV3CisR cells displayed reduced invasion (although not significant in statistical analysis), highlighting the advantage of this 3D invasion assay as it can provide us with information that is otherwise not accessible. Although the number of cells counted in the basolateral chamber appears similar for both cell lines, only PICs1 nanoparticles led to statistically significant reductions in cell numbers compared to the nanoparticles loaded with inactive scampered siRNA.
In summary, the model used here provided us with information on the suitability of the prepared siRNA nanoparticle as nanomedicine against ovarian cancer metastasis. From the originally four formulations, two were found to be suitable for further investigations. However, it should be noted that the structural modification to enhance the stability of BSA decorated PICs nanoparticles carrying the siRNA could be explored further to ensure the maximum release of trapped siRNA from polymeric core network hence enhance the therapeutic ability.
Conclusions
Here, we have studied ROR2 siRNA enclosed BSA modified PICs and investigated their therapeutic potential in HGSOC cells including platinum resistant models via a co-cultured 3D organotypic model. This study highlights the effectiveness of ROR2 siRNA when condensed into BSA decorated micellar formulation in inhibiting the metastatic potential of HGSOC cells. The ROR2 siRNA loaded PICs nanoparticles showed a promising outcome in terms of inhibiting the migration and invasion ability of CaOV3, with less effect on its cisplatin resistant pair. This biocompatible PICs system provides a platform to deliver siRNA into cells with a potential to inhibit gene of interest, with negligible cytotoxic effect. This is of relevance to those biomarkers which have no targeting therapies available.Author contributions
NJ carried out majority of experimental work such as polymer synthesis, nanoparticle characterization, and in vitro studies and drafted first version and the revisions of the manuscript, DL carried out some of the biological evaluations and drafted and revised various versions of the manuscript, KAD established the cell lines and revised the manuscript, DJM established the cell lines and revised the manuscript, CEF designed the studied and edited the manuscript, MHS designed the study and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Research Training Programme (RTP) scholarship from UNSW), a Rivkin Foundation grant to A/Prof. Ford and Prof. Stenzel, and a Translational Cancer Research Network Major Pilot Grant to A/Prof. Ford. We would like to acknowledge all the patients who donated omentum for the study in addition to the surgeons at the Royal Hospital for Women and Prince of Wales Hospital who assisted with the collection, namely Prof. Neville Hacker, Dr Rhonda Farrell, and Dr Vivek Arora. We also would like to thank Dr Claire Henry from University of Otago for 3D model training. The author acknowledges Dr Sandy Wong for the TEM imaging of PICs nanoparticles and Dr Stuart Hamilton for the help on Cy3 labelling of ROR2 siRNA.Notes and references
- K. Lõhmussaar, O. Kopper, J. Korving, H. Begthel, C. P. H. Vreuls, J. H. van Es and H. Clevers, Nat. Commun., 2020, 11, 2660 CrossRef PubMed.
- R. Siegel, D. Naishadham and A. Jemal, Ca-Cancer J. Clin., 2013, 63, 11–30 CrossRef PubMed.
- E. Lengyel, Am. J. Pathol., 2010, 177, 1053–1064 CrossRef PubMed.
- L. A. Torre, B. Trabert, C. E. DeSantis, K. D. Miller, G. Samimi, C. D. Runowicz, M. M. Gaudet, A. Jemal and R. L. Siegel, Ca-Cancer J. Clin., 2018, 68, 284–296 CrossRef.
- D. D. Bowtell, S. Böhm, A. A. Ahmed, P. J. Aspuria, R. C. Bast, V. Beral, J. S. Berek, M. J. Birrer, S. Blagden, M. A. Bookman, J. D. Brenton, K. B. Chiappinelli, F. C. Martins, G. Coukos, R. Drapkin, R. Edmondson, C. Fotopoulou, H. Gabra, J. Galon, C. Gourley, V. Heong, D. G. Huntsman, M. Iwanicki, B. Y. Karlan, A. Kaye, E. Lengyel, D. A. Levine, K. H. Lu, I. A. McNeish, U. Menon, S. A. Narod, B. H. Nelson, K. P. Nephew, P. Pharoah, D. J. Powell, P. Ramos, I. L. Romero, C. L. Scott, A. K. Sood, E. A. Stronach and F. R. Balkwill, Nat. Rev. Cancer, 2015, 15, 668–679 CrossRef CAS PubMed.
- A. du Bois, H. J. Lück, W. Meier, H. P. Adams, V. Möbus, S. Costa, T. Bauknecht, B. Richter, M. Warm, W. Schröder, S. Olbricht, U. Nitz, C. Jackisch, G. Emons, U. Wagner, W. Kuhn, J. Pfisterer, K. von Maillot, W. Lange, D. Berg, E. Schlicht, H. Peterseim, D. Elling, T. Öney, V. Zimmermann, K. Renziehausen, G. Rohrmann, H. J. Bach, H. Müller, W. Jäger, H. Mickan, R. H. Ackermann, K. Wernicke, P. J. Czygan, J. Schulze-Tollert, H. J. Becker, J. Nast, P. Kramb, M. Kröner, E. Petru, M. Carstensen, W. Müller, H. H. Zippel, J. Hilfrich, W. Herchenhein, M. Mesrogli, A. Schneider, G. Deutsch, F. K. Klöck, W. Maurer, S. Sünter, A. Göppinger, R. Strigl, R. Schuhmann, K. Kühndel, D. Fischer, C. Leißner, F. Peters, W. Niedner, K. H. Peschke, T. Silz, D. Schwörer, W. Meinerz, D. Kramer, P. Richter, D. F. Steichele, P. Krieger, M. Lange, T. Beck, K. Friese, D. Rother, L. Heilmann, J. Dietel, E. Petri, J. Meyer-Grohbrügge, V. Jovanovic, K. Robke, E. Merkle, G. Göretzlehner, J. P. Hanker, C. Karg, W. Burkert, A. Grüneberger and S. Flachsenberg, J. Natl. Cancer Inst., 2003, 95, 1320–1330 CrossRef CAS PubMed.
- R. F. Ozols, B. N. Bundy, B. E. Greer, J. M. Fowler, D. Clarke-Pearson, R. A. Burger, R. S. Mannel, K. DeGeest, E. M. Hartenbach, R. Baergen and D. Mackey, J. Clin. Oncol., 2003, 21, 3194–3200 CrossRef CAS.
- S. Manfred, Encyclopedia of Cancer, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011 Search PubMed.
- R. Agarwal and S. B. Kaye, Nat. Rev. Cancer, 2003, 3, 502–516 CrossRef CAS.
- C. Henry, E. Llamosas, A. Knipprath-Meszaros, A. Schoetzau, E. Obermann, M. Fuenfschilling, R. Caduff, D. Fink, N. Hacker, R. Ward, V. Heinzelmann-Schwarz and C. Ford, Oncotarget, 2015, 6, 40310–40326 CrossRef PubMed.
- C. E. Henry, E. Llamosas, B. Daniels, A. Coopes, K. Tang and C. E. Ford, Gynecol. Oncol., 2018, 148, 576–584 CrossRef CAS PubMed.
- C. Henry, N. Hacker and C. Ford, Oncotarget, 2017, 8, 112727–112738 CrossRef PubMed.
- K. Veskimäe, M. Scaravilli, W. Niininen, H. Karvonen, S. Jaatinen, M. Nykter, T. Visakorpi, J. Mäenpää, D. Ungureanu and S. Staff, Transl. Oncol., 2018, 11, 1160–1170 CrossRef PubMed.
- M. Hojjat-Farsangi, A. H. Daneshmanesh, A. S. Khan, J. Shetye, F. Mozaffari, P. Kharaziha, L. S. Rathje, P. Kokhaei, L. Hansson, J. Vågberg, S. Byström, E. Olsson, C. Löfberg, C. Norström, J. Schultz, M. Norin, T. Olin, A. Österborg, H. Mellstedt and A. Moshfegh, Leukemia, 2018, 32, 2291–2295 CrossRef CAS PubMed.
- M. Y. Choi, G. F. Widhopf, E. M. Ghia, R. L. Kidwell, M. K. Hasan, J. Yu, L. Z. Rassenti, L. Chen, Y. Chen, E. Pittman, M. Pu, K. Messer, C. E. Prussak, J. E. Castro, C. Jamieson and T. J. Kipps, Cell Stem Cell, 2018, 22, 951–959.e3 CrossRef CAS.
- Y. N. Loh, E. L. Hedditch, L. A. Baker, E. Jary, R. L. Ward and C. E. Ford, BMC Cancer, 2013, 13, 174 CrossRef CAS PubMed.
- Z. Liang, Z. Lu, Y. Zhang, D. Shang, R. Li, L. Liu, Z. Zhao, P. Zhang, Q. Lin, C. Feng, Y. Zhang, P. Liu, Z. Tu and H. Liu, Curr. Cancer Drug Targets, 2018, 19, 449–467 CrossRef.
- C. Henry, E. Llamosas, A. Knipprath-Meszaros, A. Schoetzau, E. Obermann, M. Fuenfschilling, R. Caduff, D. Fink, N. Hacker, R. Ward, V. Heinzelmann-Schwarz and C. Ford, Oncotarget, 2015, 6, 40310–40326 CrossRef PubMed.
- C. Henry, N. Hacker and C. Ford, Oncotarget, 2017, 8, 112727–112738 CrossRef PubMed.
- R. Kanasty, J. R. Dorkin, A. Vegas and D. Anderson, Nat. Mater., 2013, 12, 967–977 CrossRef CAS PubMed.
- G. Tang, Trends Biochem. Sci., 2005, 30, 106–114 CrossRef CAS PubMed.
- M. Dominska and D. M. Dykxhoorn, J. Cell Sci., 2010, 123, 1183–1189 CrossRef CAS PubMed.
- S. Aghamiri, K. F. Mehrjardi, S. Shabani, M. Keshavarz-Fathi, S. Kargar and N. Rezaei, Nanomedicine, 2019, 14, 2083–2100 CrossRef CAS PubMed.
- K. A. Whitehead, R. Langer and D. G. Anderson, Nat. Rev. Drug Discovery, 2009, 8, 129–138 CrossRef CAS PubMed.
- O. Leng, M. C. Woodle, P. Y. Lu and A. J. Mixson, Drugs Future, 2009, 34, 721–737 CrossRef PubMed.
- J. Yang, H. Liu and X. Zhang, Biotechnol. Adv., 2014, 32, 804–817 CrossRef CAS PubMed.
- S. J. Tan, P. Kiatwuthinon, Y. H. Roh, J. S. Kahn and D. Luo, Small, 2011, 7, 841–856 CrossRef CAS PubMed.
- M. Durymanov and J. Reineke, Front. Pharmacol., 2018, 9, 971 CrossRef PubMed.
- D. E. Large, J. R. Soucy, J. Hebert and D. T. Auguste, Adv. Ther., 2019, 2, 1800091 CrossRef.
- A. Kichler, J. Gene Med., 2004, 6, S3–S10 CrossRef CAS PubMed.
- P. Neuberg and A. Kichler, in Advances in Genetics, ed. E. W. Leaf Huang, D. Liu, Academic Press Inc., 2014, vol. 88, pp. 263–288 Search PubMed.
- B. Newland, H. Tai, Y. Zheng, D. Velasco, A. Di Luca, S. M. Howdle, C. Alexander, W. Wang and A. Pandit, Chem. Commun., 2010, 46, 4698–4700 RSC.
- Y. Jiang, H. Lu, Y. Y. Khine, A. Dag and M. H. Stenzel, Biomacromolecules, 2014, 15, 4195–4205 CrossRef CAS PubMed.
- R. A. Cordeiro, N. Rocha, J. P. Mendes, K. Matyjaszewski, T. Guliashvili, A. C. Serra and J. F. J. Coelho, Polym. Chem., 2013, 4, 3088–3097 RSC.
- U. Stahlschmidt, V. Jérôme, A. Majewski, A. Müller and R. Freitag, Polymers, 2017, 9, 156 CrossRef PubMed.
- K. Taguchi, H. Lu, Y. Jiang, T. T. Hung and M. H. Stenzel, J. Mater. Chem. B, 2018, 6, 6278–6287 RSC.
- Y. Jiang, H. Lu, A. Dag, G. Hart-Smith and M. H. Stenzel, J. Mater. Chem. B, 2016, 4, 2017–2027 RSC.
- E. Schnitzers and P. Oh, J. Biol. Chem., 1994, 269, 6072–6082 CrossRef.
- D. Sleep, Expert Opin. Drug Delivery, 2015, 12, 793–812 CrossRef CAS PubMed.
- L. Noorani, M. Stenzel, R. Liang, M. H. Pourgholami and D. L. Morris, J. Nanobiotechnol., 2015, 13, 25 CrossRef PubMed.
- E. R. Shamir and A. J. Ewald, Nat. Rev. Mol. Cell Biol., 2014, 15, 647–664 CrossRef CAS PubMed.
- H. A. Kenny, S. Dogan, M. Zillhardt, A. K. Mitra, S. D. Yamada, T. Krausz and E. Lengyel, Cancer Treat. Res., 2009, 149, 335–351 CAS.
- P. N. Peters, E. M. Schryver, E. Lengyel and H. Kenny, J. Visualized Exp., 2015, 2015, e53541 Search PubMed.
- P. Harter, Z. M. Muallem, C. Buhrmann, D. Lorenz, C. Kaub, R. Hils, S. Kommoss, F. Heitz, A. Traut and A. Du Bois, Gynecol. Oncol., 2011, 121, 615–619 CrossRef PubMed.
- M. Lu, C. E. Henry, H. Lai, Y. Y. Khine, C. E. Ford and M. H. Stenzel, Biomater. Sci., 2019, 7, 1652–1660 RSC.
- Y. Mitsukami, M. S. Donovan, A. B. Lowe and C. L. McCormick, Macromolecules, 2001, 34, 2248–2256 CrossRef CAS.
- Y. Jiang, H. Lu, Y. Y. Khine, A. Dag and M. H. Stenzel, Biomacromolecules, 2014, 15, 4195–4205 CrossRef CAS PubMed.
- H. A. Kenny, S. Dogan, M. Zillhardt, A. K. Mitra, S. D. Yamada, T. Krausz and E. Lengyel, Cancer Treat. Res., 2009, 149, 335–351 CAS.
- Y. Jiang, C. K. Wong and M. H. Stenzel, Macromol. Biosci., 2015, 15, 965–978 CrossRef CAS PubMed.
- J. Cai, H. Tang, L. Xu, X. Wang, C. Yang, S. Ruan, J. Guo, S. Hu and Z. Wang, Carcinogenesis, 2012, 33, 20–29 CrossRef CAS PubMed.
- X. Y. Zhang, R. Pettengell, N. Nasiri, V. Kalia, A. G. Dalgleish and D. P. J. Barton, J. Soc. Gynecol. Invest., 1999, 6, 333–340 CrossRef CAS PubMed.
- T. M. Hinton, C. Guerrero-Sanchez, J. E. Graham, T. Le, B. W. Muir, S. Shi, M. L. V. Tizard, P. A. Gunatillake, K. M. McLean and S. H. Thang, Biomaterials, 2012, 33, 7631–7642 CrossRef CAS PubMed.
- L. Van de Sande, S. Cosyns, W. Willaert and W. Ceelen, Drug Delivery, 2020, 27, 40–53 CrossRef CAS PubMed.
- J. Zhao, H. Lu, Y. Yao, S. Ganda and M. H. Stenzel, J. Mater. Chem. B, 2018, 6, 4223–4231 RSC.
- S. Zhang, B. Cui, H. Lai, G. Liu, E. M. Ghia, G. F. Widhopf, Z. Zhang, C. C. N. Wu, L. Chen, R. Wu, R. Schwab, D. A. Carson and T. J. Kipps, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17266–17271 CrossRef CAS PubMed.
- C. E. Ford, S. Si, Q. Ma, A. Quadir and R. L. Ward, Int. J. Cancer, 2013, 133, 779–787 CrossRef CAS PubMed.
- S. A. Smith, L. I. Selby, A. P. R. Johnston and G. K. Such, Bioconjugate Chem., 2019, 30, 263–272 CrossRef CAS PubMed.
- P. Van De Wetering, J. Y. Cherng, H. Talsma and W. E. Hennink, J. Controlled Release, 1997, 49, 59–69 CrossRef CAS.
- A. C. Rinkenauer, S. Schubert, A. Traeger and U. S. Schubert, J. Mater. Chem. B, 2015, 3, 7477 RSC.
- T. K. Georgiou, M. Vamvakaki, C. S. Patrickios, E. N. Yamasaki and L. A. Phylactou, Biomacromolecules, 2004, 5, 2221–2229 CrossRef CAS PubMed.
- Y. Wu, M. Wang, D. Sprouse, A. E. Smith and T. M. Reineke, Biomacromolecules, 2014, 15, 1716–1726 CrossRef CAS PubMed.
- B. Dai, T. Yan and A. Zhang, Cancer Cell Int., 2017, 17, 1–9 CrossRef PubMed.
- E. Pujade-Lauraine, S. Banerjee and S. Pignata, J. Clin. Oncol, 2019, 37, 2437–2448 CrossRef CAS PubMed.
- Q. H. Miow, T. Z. Tan, J. Ye, J. A. Lau, T. Yokomizo, J.-P. Thiery and S. Mori, Oncogene, 2014, 34, 1899–1907 CrossRef PubMed.
- P.-N. Teng, G. Wang, B. L. Hood, K. A. Conrads, C. A. Hamilton, G. L. Maxwell, K. M. Darcy and T. P. Conrads, Br. J. Cancer, 2014, 110, 123–132 CrossRef CAS PubMed.
- S. Tsunetoh, Y. Terai, H. Sasaki, A. Tanabe, Y. Tanaka, T. Sekijima, S. Fujiwara, H. Kawaguchi, M. Kanemura, Y. Yamashita and M. Ohmichi, Cancer Biol. Ther., 2010, 10, 1137–1146 CrossRef CAS PubMed.
- J. Haley, S. Tomar, N. Pulliam, S. Xiong, S. M. Perkins, A. R. Karpf, S. Mitra, K. P. Nephew and A. K. Mitra, Oncotarget, 2016, 7, 32810–32820 CrossRef PubMed.
- M. S. Anglesio, K. C. Wiegand, N. Melnyk, C. Chow, C. Salamanca, L. M. Prentice, J. Senz, W. Yang, M. A. Spillman, D. R. Cochrane, K. Shumansky, S. P. Shah, S. E. Kalloger and D. G. Huntsman, PLoS ONE, 2013, 8, e72162 CrossRef CAS PubMed.
- S. Domcke, R. Sinha, D. A. Levine, C. Sander and N. Schultz, Nat. Commun., 2013, 4, 1–10 CrossRef PubMed.
- R. L. Anderson, T. Balasas, J. Callaghan, R. C. Coombes, J. Evans, J. A. Hall, S. Kinrade, D. Jones, P. S. Jones, R. Jones, J. F. Marshall, M. B. Panico, J. A. Shaw, P. S. Steeg, M. Sullivan, W. Tong, A. D. Westwell and J. W. A. Ritchie, Nat. Rev. Clin. Oncol., 2019, 16, 185–204 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary data (instrument description, 1H NMR of RAFT agent and polymer, SEC data of polymer, SDS PAGE of BSA–PDMAEMA conjugates, TEM analysis of PICs3 and PICs4, cellular uptake of PICs3 and PICs4, toxicity of PICs3 and PICs4 and fluorescence study of PICs1 and PICs2). See DOI: 10.1039/d1tb01837j |
| ‡ Joint First Author. |
| This journal is © The Royal Society of Chemistry 2021 |