
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhosphorescent metal complexes as theranostic anticancer agents: combining imaging and therapy in a single molecule
Cai-Ping
Tan
 *,
Yan-Mei
Zhong
,
Liang-Nian
Ji
and
Zong-Wan
Mao
*
*,
Yan-Mei
Zhong
,
Liang-Nian
Ji
and
Zong-Wan
Mao
*
MOE Key Laboratory of Bioinorganic and Synthetic Chemistry, School of Chemistry, Sun Yat-Sen University, Guangzhou, 510275, China. E-mail: tancaip@mail.sysu.edu.cn; cesmzw@mail.sysu.edu.cn
First published on 27th January 2021
Abstract
Phosphorescent metal complexes are a new kind of multifunctional antitumor compounds that can integrate imaging and antitumor functions in a single molecule. In this minireview, we summarize the recent research progress in this field, concentrating on the theranostic applications of phosphorescent iridium(III), ruthenium(II) and rhenium(I) complexes. The molecular design that affords these complexes with tumour- or subcellular organelle-targeting properties is elucidated. The potential of these complexes to induce and monitor the dynamic behavior of subcellular organelles and the changes in microenvironment during the process of therapy is demonstrated. Moreover, the potential and advantages of applying new technologies, such as super-resolution imaging and phosphorescence lifetime imaging, are also described. Finally, the challenges faced in the development of novel theranostic metallo-anticancer complexes for possible clinical translation are proposed.
1. Introduction
At present, cancer research still faces many obstacles, such as tumour heterogeneity, drug resistance and systemic toxicity.1 Although the means of cancer treatment are developing rapidly, traditional chemotherapy still plays important roles in cancer treatment.2 As representatives of metallo-anticancer compounds, cisplatin and its derivatives are widely used in clinic.3 In addition to platinum drugs, non-platinum metallo-anticancer agents have also attracted extensive attention, among which phosphorescent metal complexes show interesting anticancer properties due to their varied anticancer mechanisms different from that of cisplatin, as well as rich photophysical and photochemical properties that endow them with multifunctionalities.4–7The concept “theranostics” combining “diagnosis” and “treatment” is a relatively new research trend for cancer treatment.8 The original concept of “theranostics” comes from nuclear medicine. 123I−, 124I−, and 131I with low activity are used to diagnose the disease, and 131I with high activity is applied for subsequent treatment.9 Now, the term is commonly used to describe multifunctional nano-platforms, in which the therapeutic and diagnostic drugs are physically combined or co-loaded on specially modified nanoparticles to integrate imaging and treatment methods.8,10 Small molecules, including phosphorescent metal complexes, are also applied as “theranostic agents” in some cases.11–13 In the past few years, our group and others demonstrated that phosphorescent metal complexes can not only kill cancer cells effectively, but also provide valuable information on their action mechanisms by monitoring the cellular localization,14 dynamic imaging/tracking of the subcellular organelles,15 and even quantitatively measuring the parameters of the surrounding microenvironment.16 Combined with super-resolution imaging,17,18 two/multi-photon imaging12 and phosphorescence lifetime imaging (PLIM)16 technologies, more accurate and detailed information can be obtained. More importantly, these compounds have the potential to be developed as research tools for life science to accurately monitor the behaviour of subcellular organelles, functions of biomacromolecules, and changes in micro-environmental parameters (e.g., hypoxia, pH, viscosity and polarity) during various biological processes.
Recently, there have been some excellent reviews on metallo-anticancer agents published from different perspectives, such as structure–activity relationship (SAR) and anticancer mechanism analysis,19–21 subcellular organelle targeting design,22 bifunctionality (imaging and therapy)23 and functionalization with nanomaterials.4,24 This review summarizes the advantages and recent research progress in phosphorescent metal complexes as theranostic anticancer agents. The review is focused on the description of Ir(III), Ru(II) and Re(I) complexes integrating imaging and anticancer functions. We also try to clarify the challenges and possible solutions for further clinical translation of these compounds under the background of rapid development of the knowledge on cancer biology as well as fluorescence imaging techniques.
2. Theranostic applications of iridium complexes
Among the iridium complexes investigated for biological applications, cyclometalated Ir(III) complexes are particularly intriguing due to their favourable photophysical properties, e.g., high stability under ambient conditions, tunable emission colours, high emission quantum yields, large Stokes shifts, resistance to photobleaching and long emission lifetime.25,26Lo's group has done a lot of research on the applications of emissive cyclometalated iridium complexes as biological molecular recognition probes, protein labelling agents, bioorthogonal reaction reagents, bioimaging agents and photodynamic therapeutic agents.27–30 They have carried out a variety of targeted modifications on iridium complexes and summarized some important factors that may influence the cellular uptake and localization of these complexes.25 Li's group has also made pilot studies on iridium complexes as cellular imaging agents, particularly for complexes that can form covalent binding with biomolecules.31–33 However, in these early studies, little attention has been paid on their cytotoxic mechanisms, even in some cases, they are highly cytotoxic.34
Recently, the anticancer potential of organometallic iridium complexes has been intensively explored.26 Different from platinum-based drugs, iridium complexes reported mainly act by damaging different subcellular organelles.7,12,16,35 Although a few reports have shown that these complexes can target the protein or DNA in cells, no definite SAR has been reported.14,36,37 Iridium complexes are the most prominent ones among the metal complexes reported as theranostic anticancer agents. The cellular uptake of many drugs is usually related to their lipophilicity, while the dependence of cell penetration capabilities of iridium complexes on lipophilicity is not so obvious, and their cellular uptake mechanisms and intracellular transport pathways are largely unknown.26 In many cases, phosphorescent iridium complexes show an instinct to localize to mitochondria;22 however, through structural modifications, iridium complexes can also target other subcellular organelles, e.g., lysosome,15 lipid droplet38 and endoplasmic reticulum (ER).14 As the emission properties of iridium complexes are affected by the surrounding microenvironment, e.g., pH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 39 and viscosity,16 they can be used to monitor the changes in these parameters in real-time.
39 and viscosity,16 they can be used to monitor the changes in these parameters in real-time.
2.1 Mitochondria-targeting iridium complexes
Mitochondria are the centre of energy and material metabolism in cells, and mitochondrial DNA (mtDNA) and many proteins are potential anticancer targets.40 Due to the high lipophilicity and positive charge of iridium complexes, they can easily aggregate in mitochondria.22 Fluorescence microscopy combined with flow cytometry and inductively coupled plasma mass spectrometry (ICP-MS) can provide valuable information on their cellular uptake levels, localization and membrane penetration mechanisms. Interestingly, mitochondria-targeting iridium complexes induce different types of cell death and varied forms of alterations in mitochondrial morphology, which implies that they may target different biomolecules.16,41–43 Recent studies have given some hints in this regard, but the clear mechanisms and SARs are still not clear.37In our earlier studies, we developed a series of cyclometalated Ir(III) complexes containing different bis-N-heterocyclic carbene (NHC) ligands (1–3; Scheme 1) as photodynamic and theranostic anticancer agents.421–3 can quickly penetrate into human cervical carcinoma (HeLa) cells through energy-dependent pathways. The cytotoxicity of 1–3 is correlated with their lipophilicity and cellular uptake efficiency, and all of them display a higher anti-proliferative activity than cisplatin. Confocal microscopy shows that 1–3 localize in mitochondria and induce apoptosis by inducing mitochondrial damage, reactive oxygen species (ROS) production, cytochrome c release and activation of caspases. As a result, 1–3 can induce and monitor the changes in mitochondrial morphology in a real-time manner. Moreover, a very high photocytotoxicity index (PI) up to 3488 is obtained for 3 in cisplatin-resistant human lung adenocarcinoma (A549R) cells.
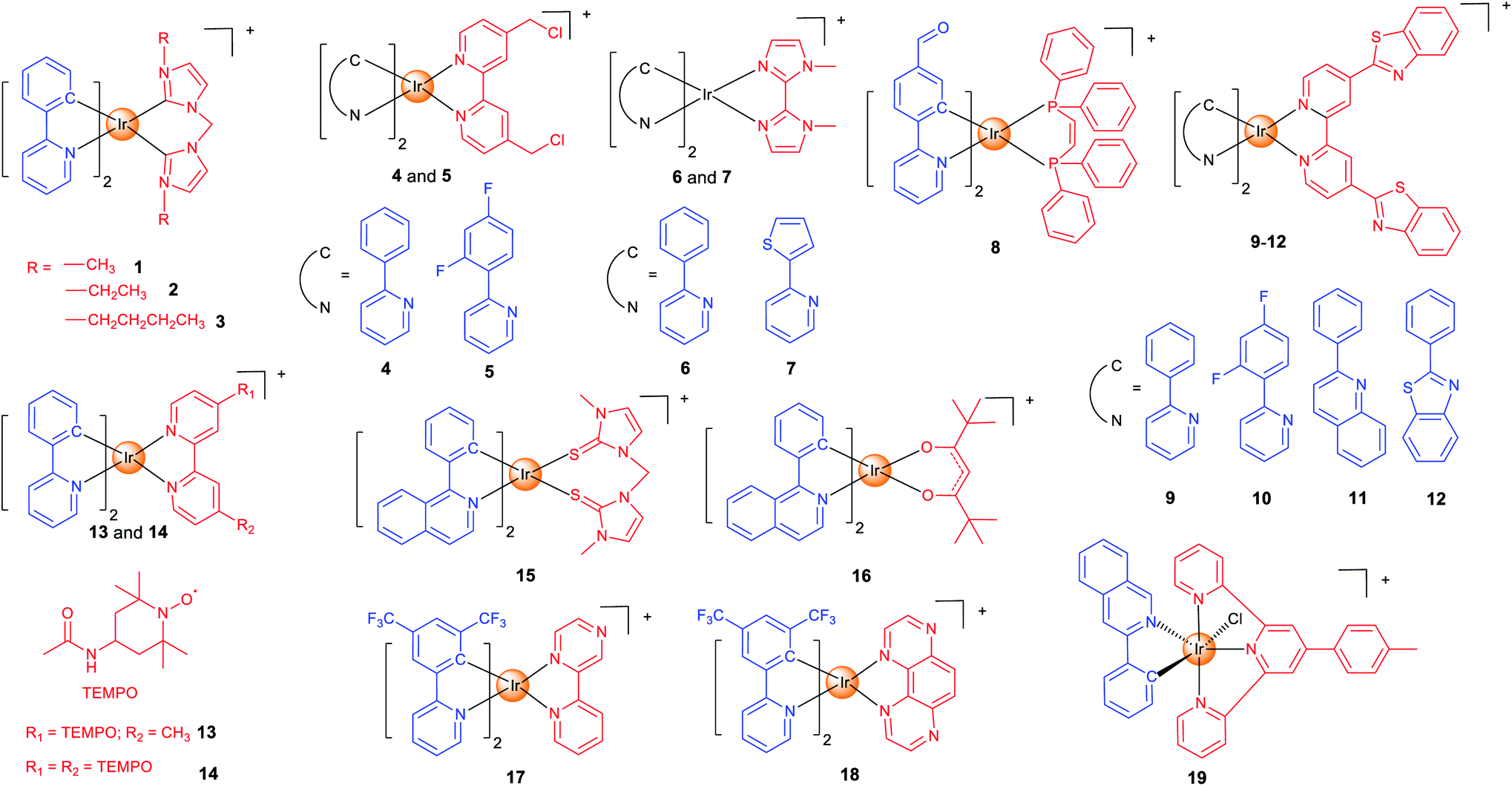 | ||
| Scheme 1 Chemical structures of mitochondria-targeting Ir(III) complexes. | ||
Aiming to increase the retention time of the complexes in mitochondria, we designed two cyclometalated Ir(III) complexes (4 and 5; Scheme 1) containing reactive chloromethyl groups that can react with thiols in mitochondrial proteins.44 The immobilization of 4 and 5 on mitochondria results in an increased cellular penetration capability and cytotoxicity. Genome-wide transcriptional analysis shows that 4 exerts its anticancer activity through metabolism repression and attenuation of multiple cell death signalling pathways. Moreover, 4 causes a collapse of the normal tubular mitochondrial network into mitochondrial aggregates and large perinuclear clusters (a phenomenon observed during mitophagy), which has been subsequently confirmed by transmission electron microscopy.
Specific inducers and imaging agents of mitophagy are rarely reported. Usually, the inducer and imaging agent need to be used at the same time or in sequence to monitor a biological process. Using a single agent that can induce and monitor the biological processes simultaneously can avoid the interference of different reagents and greatly simplify the operation procedures. We identified two phosphorescent Ir(III) complexes (6 and 7; Scheme 1) that can induce and monitor the mitochondrial morphology during mitophagy.456 can be used to study important mitochondria-related events during mitophagy, e.g., autophagosomal engulfment of mitochondria and fusion of mitochondria-engulfed autophagosomes with lysosomes.
The functional integrity of subcellular organelles is correlated with their physical parameters, e.g., viscosity. However, precise quantitative measurement of viscosity at the subcellular level presents great challenges. We designed six Ir(III) complexes as mitochondrial targeting theranostic anticancer agents with viscosity-responsive phosphorescence properties.16 Due to the rotatable aldehyde groups on the cyclometallating ligands and the aromatic rings on diphosphine ligands, 8 (Scheme 1) displays viscosity-dependent phosphorescence intensities and lifetimes. 8 can quantitatively monitor the self-induced changes in mitochondrial viscosity using two-photon phosphorescence lifetime imaging microscopy (TPPLIM). An obvious increase in the emission intensity/lifetime of 8 that reflects an increase in mitochondrial viscosity can be observed along with the incubation time.
Oncosis is a non-apoptotic form of programmed cell death, and oncosis-inducing agents may overcome drug-resistance of the current well-established treatment regimens including platinum-based chemotherapy. Chao et al. presented four mitochondria-targeting cyclometalated Ir(III) complexes that could induce oncosis in drug-resistant cancer cells.359–12 (Scheme 1) exhibit up to ∼30 times selectivity towards cancer cells over normal cells. The localization of the complexes in mitochondria was observed by confocal microscopy and verified by ICP-MS.
As stable free radicals, nitroxides have antioxidant properties mimicking superoxide dismutase and catalase, and they are widely used as spin labelling agents. Sadler et al. reported two luminescent Ir(III) complexes, containing one or two TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl) spin labels (13 and 14; Scheme 1).46 Confocal microscopy shows that 13 and 14 localize in mitochondria. The antiproliferative activity of 14 is ca. 2–30× higher than that of 13 in all the cell lines tested, which may be attributed to the strong spin–spin exchange interaction between two nitroxide radicals and their conformational flexibility. Interestingly, unlike most of the other anticancer Ir(III) complexes reported, 13 and 14 with antioxidant properties show no impact on cellular ROS levels.
At present, most of the anticancer iridium complexes are not designed to target a specific biomolecule in cells. Therefore, the validation of their targets is very important to elucidate the mechanisms of action. Sadler et al. reported two Ir(III) complexes (15 and 16; Scheme 1) with photodynamic therapy (PDT) activities and large two-photon absorption (TPA) properties.37 PLIM experiment shows that the lifetimes of 15 and 16 inside the cells were increased by about 19-fold, which implies that the complexes reside in a more hydrophobic and hypoxic environment that are favourable for efficient photosensitized generation of 1O2. By using the proteomic analysis, the authors demonstrated that 16 can selectively oxidize the histidines in proteins including heat shock protein 70 and aldose reductase.
PDT can mainly function through two pathways: type I process occurs through a photo-induced electron transfer to the biological substrates to form radical species including superoxide (O2˙−), hydroxyl (OH˙), and hydroperoxyl (HO2˙); type II process generates singlet oxygen (1O2) by a direct energy transfer to O2. Most photosensitizers (PSs) act through type II process that requires the participation of O2. Unfortunately, tumour microenvironment (TME) is hypoxic. Elias et al. developed two Ir(III) complexes (17 and 18; Scheme 1) that can exert their PDT effects through type I process independent of O2.4717 and 18 accumulate in mitochondria in the 2D cell monolayer, and both complexes are evenly distributed on slices of 3D multicellular tumour spheroids (MCTSs). However, 17 is more dependent on type II PDT, while type I plays a greater role in 18-mediated photo-initiated cell death, so 18 displays a better PDT effect in MCTSs simulating the hypoxic TME.
Also to overcome the hypoxic TME, Sadler et al. resorted to another strategy.48 Complex 19 (Scheme 1) can catalytically oxidize 1,4-dihydronicotinamide adenine dinucleotide (NADH) to generate NAD˙ radicals under light irradiation. Moreover, it perturbs the electron transfer pathways in mitochondria by reducing Fe3+–cytochrome c, which breaks the redox imbalance in cancer cells. As these photoredox catalyzing reactions are O2-independent, 19 is equipotent towards normoxic and hypoxic adherent cancer cells. Confocal microscopy shows that 19 is localized in mitochondria, and it can induce immunogenic apoptotic cell death upon irradiation with a two-photon light source.
2.2 Lysosome-targeting iridium complexes
Lysosomes contain hydrolytic enzymes that can degrade macromolecules for turnover of the nutrients.49 In addition, lysosomes also participate in regulation of different types of cell death, e.g., apoptosis and autophagy. Especially, lysosomal trafficking and contents are important factors that regulate autophagy.50 Our group developed two iridium(III) complexes (20 and 21; Scheme 2) as one- and two-photon-excited theranostic agents for real-time tracking of autophagic lysosomes.12 The protonation/deprotonation of secondary amines in indole rings on the β-carboline ligands results in the pH-sensitive phosphorescence properties of 20 and 21. The pKa values of 20 and 21 are 5.56 and 5.19, respectively, which are very close to the lysosomal pH. By combining selective autophagy-inducing and lysosomal imaging capabilities in a single molecule, 21 can track the key events during autophagy including autophagosomal–lysosomal fusion in live cells in a real-time manner.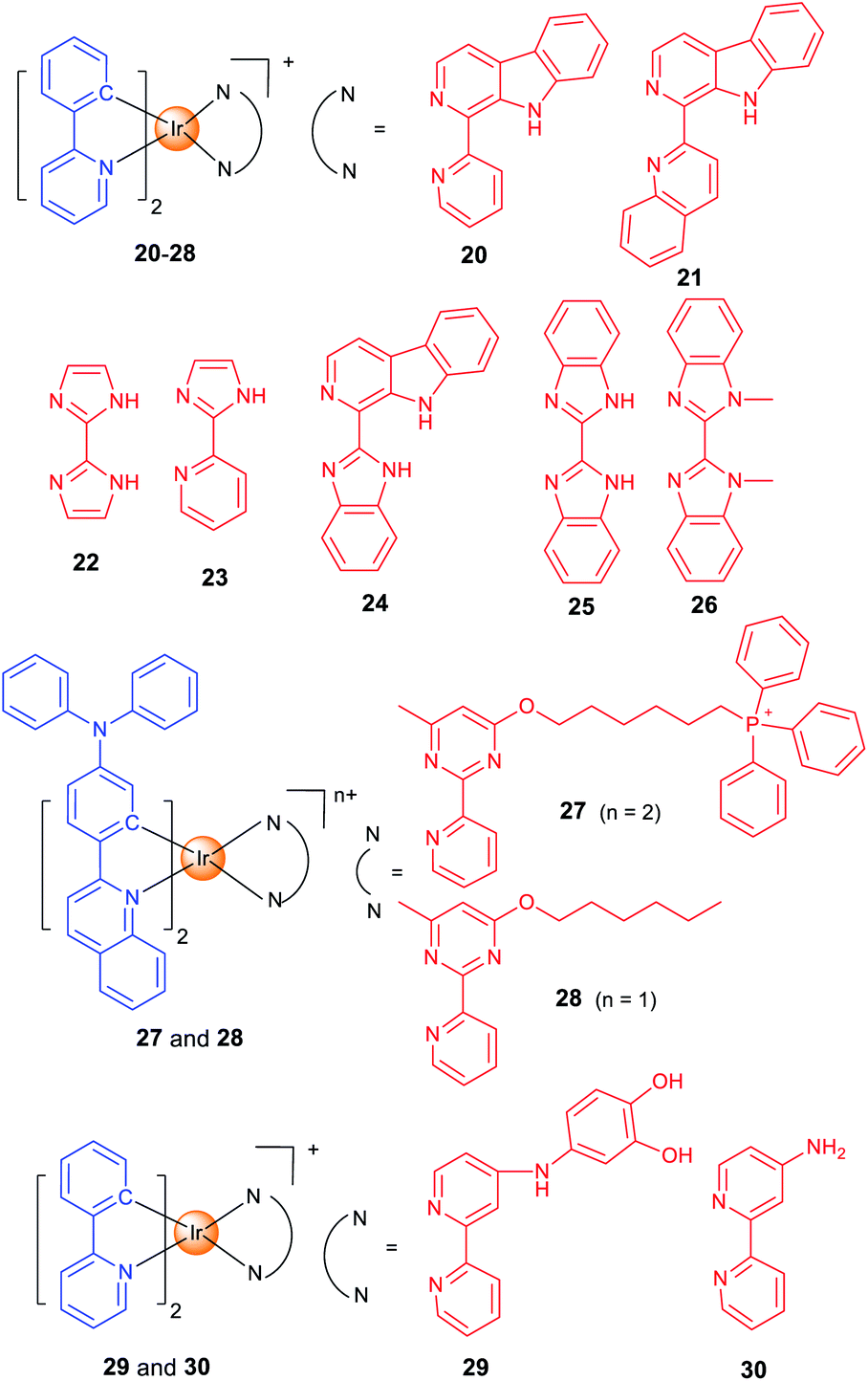 | ||
| Scheme 2 Chemical structures of lysosome-targeting Ir(III) complexes. | ||
The whole process of autophagy, called “autophagic flux”, includes the formation and maturation of autophagosomes, fusion of autophagosomes with lysosomes, the degradation of the cargoes and the release of the degradation products. Based on the previous work,12 we reported two Ir(III) complexes with 2,2′-biimidazole (H2biim, 22) or 2-(1H-imidazol-2-yl)pyridine (Hpyim, 23) as the ancillary ligands (Scheme 2). 22 and 23 are kinetically stable, and no trace of decomposition is observed in human plasma for 72 h. 22 and 23 can image lysosomes as they display pH-dependent phosphorescence caused by the protonation and deprotonation process of the N–H groups on H2biim/Hpyim. Interestingly, both 22 and 23 can act as anion transporters to inhibit autophagic flux by alkalinizing lysosomes.51
By structural optimization, we developed a series of Ir(III) complexes as stimuli-activatable PSs targeting lysosomes.15 Complex 24 (Scheme 2) shows enhanced phosphorescence emission and 1O2 generation in tumour/lysosome-related acidic environments (pH ≤ 6.5) and exhibits a high photocytotoxicity (PI > 833) in human lung adenocarcinoma (A549) cells upon visible light exposure. 24 can photodamage and image lysosomes simultaneously, which provides a reliable and convenient method for real-time monitoring of the outcome of treatment. Later, we designed four cyclometalated Ir(III) complexes containing benzimidazole derivatives as ancillary ligands which exhibit dual anticancer functionalities as effective PSs and metastasis inhibitors.52
Most of the metal complexes applied for PDT utilize UV/Vis light with limited penetration depth; two-photon excitation (TPE) that simultaneously absorbs two lower-energy NIR (near-infrared)-photons can overcome this shortage. Weinstein and Bryant et al. reported two photo-stable Ir(III) complexes (25 and 26) with long-lived triplet excited states as photosensitizers that can be excited under both one- and two-photon light.53
Both 25 and 26 are cell-permeable, and 26 shows higher dark cytotoxicity while 25 possesses more potent PDT activity (lowest PI value >333). 25 localizes in mitochondria and lysosomes sequentially alone with the incubation time, and it can disrupt the functions of both organelles to induce apoptosis upon light irradiation. More importantly, 25 maintains high PDT activity under NIR TPE (760 nm) at low concentrations and light doses.
Targeting different subcellular organelles can achieve different antitumor effects and mechanisms. For example, Huang and Zhao et al. designed two Ir(III) complexes with different substituent groups on the ancillary ligand (27 and 28; Scheme 2).54 Interestingly, HeLa cells treated with the mitochondria-targeting 27 maintains a slower O2 consumption rate and higher intracellular O2 level under hypoxia. So, 27 exhibits an improved PDT effect as compared with the lysosome-targeting 28 under hypoxia.
Some complexes can even change the subcellular localization through chemical reactions and exert their antitumor effect through a sequential localization process. Chao et al. reported a prodrug 29 (Scheme 2) that is firstly localized in lysosomes and subsequently transferred to motochondria.55 The meta-imino catechol group in 29 can be oxidized by Fe3+ inside the cell to release Fe2+, the aminobipyridyl complex 30 and 2-hydroxybenzoquinone. Fe2+ can transform H2O2 into ˙OH through the Fenton reaction, and the benzoquinone compound can interfere with the respiratory chain. Interestingly, the transformation of 29 into 30 leads to an increase in phosphorescence/cytotoxicity, and the migration of subcellular localization from lysosome to mitochondria can be tracked by confocal microscopy.
2.3 Iridium complexes targeting other organelles
Iridium complexes with theranostic functions can also localize in other subcellular organelles, such as ER and nuclei through structural modifications. Lim et al. investigated the PDT mechanisms of four Ir(III) complexes (31–34; Scheme 3) containing different cyclometallating ligands.14 The most active complex 33 is primarily localized in ER and partially in mitochondria. The localization of 33 is also confirmed by lifetime imaging that shows an extended emission lifetime (ca. 500 ns) in the vicinity of ER. Interestingly, upon light irradiation, 33 prompts cell death via photo-cross-linking of proteins, and oxidation of several essential proteins (e.g., TRAP1 and PYCR1) in ER and mitochondria. Moreover, 33 exhibits TPA properties, making them ideal candidates for effective PDT with deep-tissue imaging capabilities.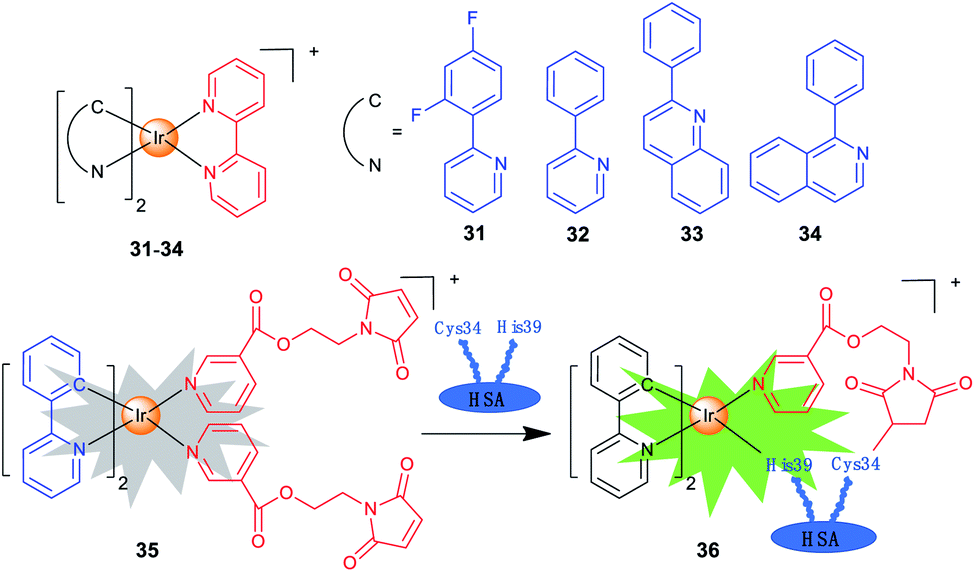 | ||
| Scheme 3 Chemical structures of Ir(III) complexes targeting other subcellular organelles. | ||
Che et al. reported a panel of luminescent Ir(III) porphyrin complexes containing axial NHC ligands.56 The IC50 values obtained for these complexes are in the nanomolar levels upon light irradiation. Confocal microscopy shows that the most active compound acts by targeting ER and catalysing the generation of 1O2 to trigger protein oxidation and apoptosis.
One of the biggest obstacles for the application of iridium complexes in cancer treatment is the lack of selectivity for cancer cells. Human serum albumin (HSA) is an effective, nontoxic, biocompatible and biodegradable drug delivery vehicle. Sadler et al. designed a maleimide-functionalized Ir(III) complex 35 (Scheme 3) that could react with HSA to form a conjugate 36 for PDT.57 The conjugation to HSA greatly enhances the phosphorescence and 1O2 sensitization capability of 35, and the as-formed 36 accumulates mostly in the nuclei of cancer cells. Both 35 and 36 are encouragingly nontoxic toward normal cells, and a high photocytotoxicity (PI = 56.6) is obtained for 36 upon irradiation.
3. Theranostic applications of ruthenium complexes
Ruthenium-based anticancer agents have many intriguing properties, which include good selectivity towards tumour cells, suitable ligand exchange kinetics, and variable biologically accessible oxidation states. Four ruthenium complexes have entered different stages of clinical trials, including NAMI-A, KP1019, NKP-1339 and TLD1433.4,20,58 On the other hand, Ru(II) polypyridyl complexes are the most extensively studied and developed systems among the ruthenium complexes with bioimaging and biosensing applications.59The biological application of luminescent ruthenium complexes originated from the pioneer work done by Barton et al. They found that these molecules could be used as molecular light switches for DNA, and had the ability to distinguish different DNA structures.60,61 In 2007, they first used fluorescence microscopy to study the cellular uptake properties of Ru(II) polypyridyl complexes.62 Since then, the bioimaging and anticancer potential of luminescent polypyridyl ruthenium complexes has been gradually explored.
Ru(II) polypyridyl complexes are known to possess metal-to-ligand charge transfer red emission, large Stokes shift, long emission lifetimes and two/multi-photon absorption properties.21 Due to the phosphorescence quenching effect caused by the energy transfer between the triplet excited state of Ru(II) complexes and the triplet ground state of O2, the luminescence properties of ruthenium complexes are sensitive to O2, and they are used as commercial O2 probes.63 Moreover, Ru(II) polypyridyl complexes can target different subcellular organelles for super-resolution imaging, e.g., stimulated emission depletion imaging.64,65
Although selectivity towards cancer cells and subcellular organelle-targeting properties can be achieved through structural optimizations, e.g., modulation of the molecular charge and lipophilicity, sometimes these strategies turn out to be unsuccessful. In several cases, the conjugation of the Ru(II) polypyridyl moiety with biological vectors, such as tumour-specific recognition groups and subcellular organelle-targeting peptides, has been proved to be very effective.66–68 The intrinsic phosphorescence properties of the complexes can be used to verify the original structural design using fluorescence imaging technologies both in vivo and in vitro.
Biotin is an efficient tumour-targeting ligand that can bind to its surface receptors overexpressed in tumour cells. Chen et al. reported a biotin-modified Ru(II) complex (37; Scheme 4) that can be preferentially ingested by HeLa cells through a receptor-mediated endocytosis pathway for imaging of cancer cells both in vitro and in vivo.66 A similar strategy was adopted by Chao et al., and they designed a biotin-modified Ru(II)-based PS with cancer cell targeting ability in an in vitro co-cultured model (mixed cultivation of A549R and human lung fibroblast (HLF) cells).67
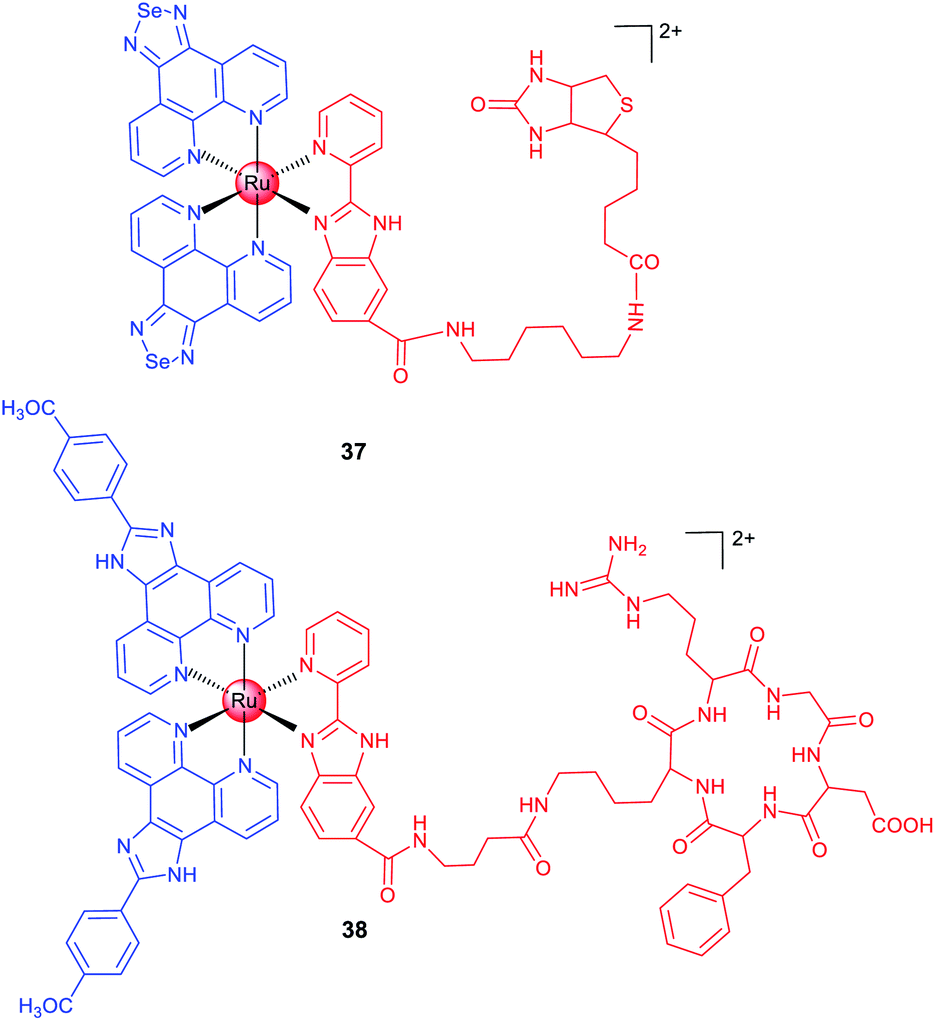 | ||
| Scheme 4 Chemical structures of ruthenium complexes with targeting groups. | ||
In the past few decades, scientists have discovered a variety of peptides to recognize cancer cells, different subcellular organelles and protein targets. Chen et al. conjugated a cRGD peptide with a Ru(II) moiety (38; Scheme 4) for selective targeting of integrin-overexpressed cancer cells.6838 preferentially accumulates at the tumour sites in xenograft mice after intravenous injection and effectively inhibits tumour growth without causing pathological tissue damage or abnormalities. Encouragingly, 38 shows potential for precise tumour diagnosis and therapy in vivo, and exhibits distinct binding capabilities for different grades of cervical carcinoma specimens from clinical patients, supporting its potential applications in rapid tumour diagnosis.
3.1 Subcellular organelle-targeting ruthenium complexes
There are several strategies to optimize the anticancer and imaging performance of ruthenium complexes. Some Ru(II) complexes have instinct subcellular targeting properties, and many Ru(II) complexes tend to accumulate in mitochondria.19 Active targeting can be achieved by structural decoration, such as conjugation with moieties targeting a certain protein, nucleic acids or subcellular organelles.21In many cases, fluorescence imaging technology is used to elucidate the action mechanisms of these complexes. Chao et al. designed a series of Ru(II) polypyridyl complexes containing a ligand with the triphenylphosphine substituent that is widely used as the mitochondrial targeting group.69 As expected, 39 (Scheme 5) localizes in mitochondria and exhibits a 28-fold enhancement in cytotoxicity when exposed to a low dose of visible light in HeLa cells.
 | ||
| Scheme 5 Chemical structures of Ru(II) complexes targeting subcellular organelles. | ||
Peptide-based vectors are also used to modify Ru(II) complexes to endow them with capabilities to target subcellular organelles. Keyes et al. constructed a Ru–dppz complex conjugated with a nuclear localization signal peptide sequence (40; Scheme 5) to direct the complex to the nuclei of HeLa cells successfully.64 Later, they reported a mitochondria-targeting molecular light switch (41; Scheme 5) by peptide vectorization, considering that mtDNA plays crucial roles in cancer.70 By using techniques including confocal fluorescence microscopy, Raman imaging, and luminescence lifetime imaging, they demonstrated that 41 could bind to and image mtDNA. Moreover, in the presence of intense irradiation, 41 can effectively stimulate cell death.
Lysosome may be a more suitable targeting subcellular organelle for PDT, as targeting the mitochondrion can result in high dark cytotoxicity, while targeting the nucleus can easily cause gene mutation. Based on these considerations, Chao et al. reported three highly charged Ru(II) polypyridyl complexes (42–44; Scheme 5) that could accumulate in lysosomes through endocytosis and are basically nontotoxic in the dark.71 Upon two-photon light irradiation, 42–44 show impressive photocytotoxicity in both monolayers and MCTSs. Mechanism investigation shows that 42–44 cause oxidative stress in cells in the presence of light irradiation and induce cell death by necrosis.
Generally, ruthenium complexes containing planar ligands can bind to DNA tightly under physiological conditions. Imaging technology provides clues for target validation of these compounds, which shows that nuclear DNA may not be their primary targets. Thomas et al. designed one mononuclear and three dinuclear complexes with either homoleptic or heteroleptic structures.72 The Ru(II) dppz units usually exhibit a “DNA light switch” effect, while dppn-based complexes commonly have a long lifetime based on the π → π* excited state favourable for 1O2 sensitization. Although complex 45 (Scheme 5) binds to DNA under cell-free conditions, confocal microscopy shows that it localizes in mitochondria and especially lysosomes, which implies damaging nuclear DNA may not be involved in its PDT mechanism. Recently, the same group reported two dinuclear complexes (46 and 47; Scheme 5) containing the Ru(II)-dppz and Re(I)-dppz moieties with different linking ligands.18 Steady-state and time-resolved photophysical studies reveal that the nature of the linkers affects the excited state dynamics of 46 and 47 and their DNA photo-cleavage properties. Their intracellular localizations and photocytotoxicity are significantly affected by the linkers. Notably, 47 initially localizes in mitochondria and lysosomes of live cells, while strong staining of the nucleus and particularly the nuclear membrane can be observed within 5 min.
The coupling of polymetallic compounds can also improve the optical properties and anticancer effects as compared with the mono-nuclear compounds. Based on a Ru(II) polypyridyl donor and a tetramethylammonium-decorated Pt(II) acceptor, Stang and Chao et al. reported a heterometallic Ru–Pt metallacycle (48; Scheme 5) via coordination-driven self-assembly.73 The rigid, planar macrocyclic structure of 48 enhances the favourable photophysical properties including red-shifted emission, enhanced TPA activities and improved intersystem crossing for more efficient ROS generation upon irradiation. Moreover, the highly charged hydrophobic bis(phosphine) Pt(II) building block facilitates the cellular uptake efficiency, and confocal microscopy shows that 48 localizes in both mitochondria and nuclei.
4. Theranostic applications of rhenium complexes
Radioactive 186/188Re has been used in clinical treatment of cancer, and the “cold” organometallic rhenium complexes are mainly used as cell imaging reagents and photocatalysts.25 The most common structural motif applied in biological systems is the stable Re(I) tricarbonyl core, and its facile synthesis can be used to generate a wide range of compounds the properties of which can be tuned to maximize biological activity.6 Recently, more and more studies have shown that organometallic rhenium complexes have potent anticancer activity. Coupled with their excellent phosphorescence properties, the cellular distribution and localization can be monitored by fluorescence microscopy and lifetime imaging.74 Lo's group studied the emissive properties of rhenium complexes and their potential as biological probes, bioorthogonal reaction reagents, cellular imaging agents and anticancer photosensitizers.75–78 Metzler Nolte et al. focused on the conjugation of these complexes with biomolecules and their anticancer activities as well as cytotoxic mechanisms.79,80The [Re(CO)3]+ core can be easily synthesized and modified with functional groups or targeting moieties, and they can also be used for vibrational imaging. The same set of ligands of [Re(CO)3]+ can be coordinated to the [99mTc(CO)3]+ core to synthesize the corresponding radioimaging reagents and radiopharmaceuticals, which offers another opportunity for their in vivo applications.81 Another intriguing possibility is that Re(I) tricarbonyl complexes may deliver CO to the targeted sites and act as photoactivatable CO-releasing molecules.82 Although a considerable number of rhenium complexes have been reported as potent anticancer agents until now, the mechanisms of their cytotoxic effect are barely elucidated.
Alberto et al. reported two organometallic complexes (49 and 50; Scheme 6) formed by the conjugation of the rhenium cyclopentadienyl moiety with doxorubicin (DOX; a topoisomerase II inhibitor).83 Interestingly, the conjugation redirects DOX from the nuclei to mitochondria. Confocal microscopy shows that 49 localizes to mitochondria. ICP-MS also proves that most of the total internalized rhenium of 49 and 50 accumulates in the nuclei (∼60%), and about 20–30% is found in mitochondria. Accordingly, 49 and 50 exert their anticancer effects by disrupting the mitochondrial membrane potential and inhibiting human topoisomerase II simultaneously. It should be pointed out that the emission used for confocal imaging originates from the DOX chromophore rather than the metal center.
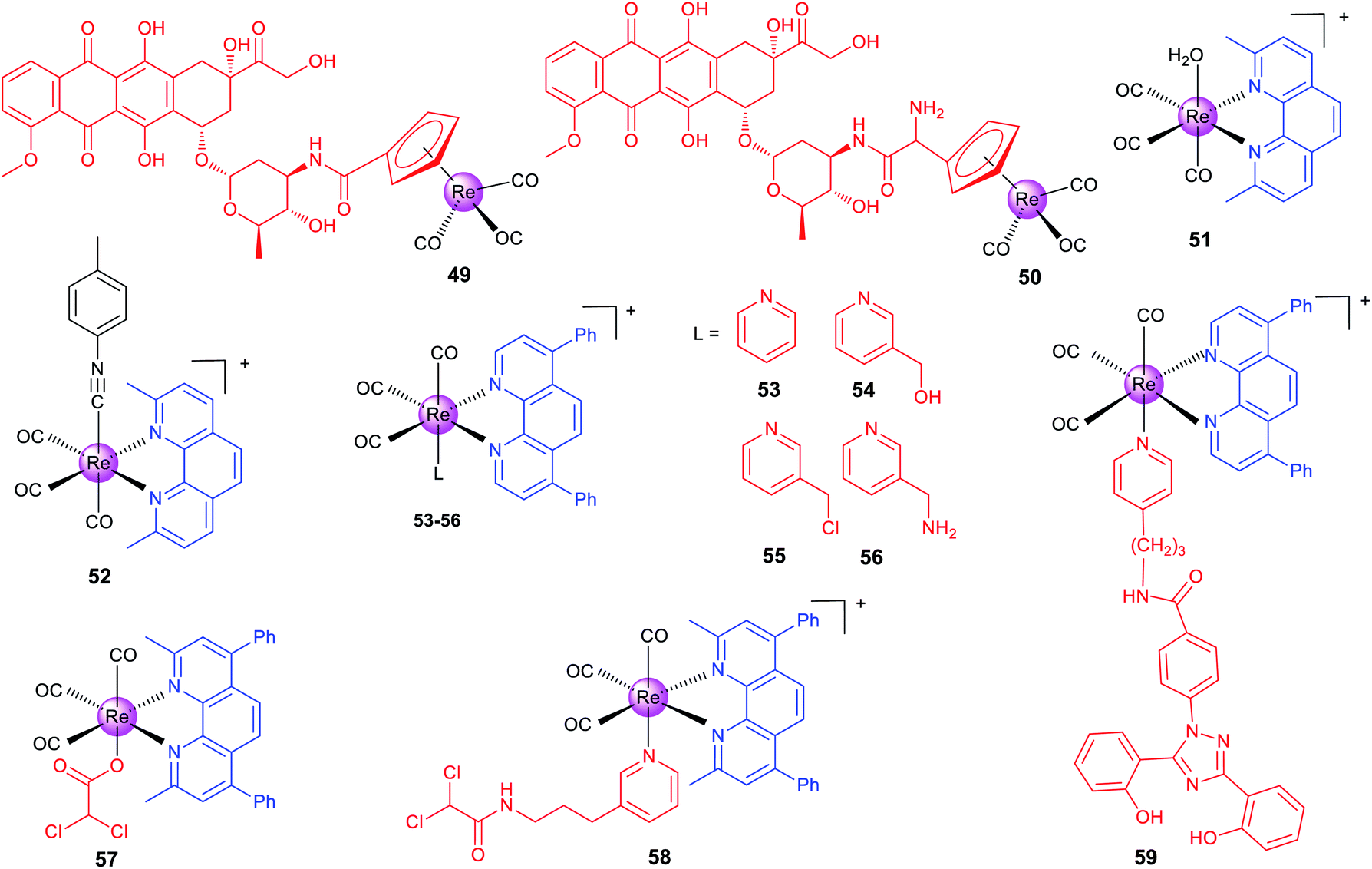 | ||
| Scheme 6 Chemical structures of theranostic Re(I) complexes. | ||
Recently, Wilson's group has made a series of efforts to reveal the SARs, stability and mechanisms of rhenium complexes.81,84–87 The distribution and metabolism of drugs in vivo are very important issues for their potential clinical applications. However, few non-platinum-based metallo-anticancer agents have been studied in this aspect. Wilson et al. reported seven Re(I) complexes of the general formula fac-[Re(CO)3(N–N)(OH2)]+, where N–N represents different bidentate ligands.81 Confocal microscopy indicates that the most active complex (51, Scheme 6) accumulates in some populations of endosomes and lysosomes. Semi-quantitative measurement using flow cytometry shows that 51 penetrates into cancer cells through the endocytotic pathway. Interestingly, 51 acts through a novel mode of action by inducing a non-canonical cell death that cannot be categorized as apoptosis, necrosis, paraptosis, or autophagy.
Importantly, the authors also prepared the 99mTc analogue for in vivo imaging and biodistribution studies, and they found that both 51 and its 99mTc analogue were excreted by the hepatobiliary system. Interestingly, they subsequently used ICP-MS to find that 51 shows similar uptake in both cisplatin-sensitive and -resistant cell lines, and 51 tends to concentrate in mitochondria.84 This result is different from that observed by fluorescence microscopy, because the emission of 51 depends on the pH and coordination environment. At the same time, they also proved that 51 is effective on patient derived ovarian cancer tumour xenografts, which further proves its potential as an anticancer candidate.
Wilson et al. also developed another rhenium(I) tricarbonyl complex (52) containing a chelating polypyridyl ligand and an axial isonitrile ligand as an anticancer candidate.8752 is stable for at least one week in the solid state and aqueous solutions. Moreover, X-ray fluorescence microscopy shows that 52 is highly stable in vitro.8652 shows strong anticancer activity on a variety of cell lines in vitro and can inhibit A2780 ovarian cancer xenografts and prolong mouse survival in vivo. 52 shows partial colocalization with the LysoTracker Red dye and GalT-dsRed fusion protein.87 It triggers unfolded protein response, ER stress, autophagy and apoptotic cell death. Similar in vivo biodistribution patterns were observed for 52 and its 99mTc congener, which suggests that the 99mTc analogue can be used as its diagnostic partner. Later, the authors constructed a 52-resistant ovarian cancer cell line, and identified two genes (the ATP-binding cassette transporter P-glycoprotein and MT1E encoding metallothioneins) involved in its resistance responses.85
Targeting cancer metabolism and reversing its metabolic machine have emerged as promising strategies for cancer therapy. Our group designed a series of phosphorescent Re(I) complexes (53–56; Scheme 6) with a highly lipophilic bidentate ligand as mitochondria-targeting theranostic anticancer agents.7453–56 can quickly and efficiently penetrate into A549 cells and localize in mitochondria. Notably, 55, containing a thiol-reactive chloromethylpyridyl moiety, can be immobilized in mitochondria and induce a cascade of mitochondria-dependent apoptotic events. The O2-sensitive lifetimes of 55 are utilized to track the self-induced mitochondrial respiration repression using PLIM in a real-time manner. Based on this work, our group also synthesized two theranostic Re-DCA (DCA; dichloroacetate; a pyruvatedehydrogenase kinase inhibitor) conjugates (57 and 58; Scheme 6) that can selectively accumulate in mitochondria to reverse the cancer cell metabolism from glycolysis to glucose oxidation.88
Mitochondria are the main sites for ROS production, and glutathione (GSH) in mitochondria is critical for ROS scavenging through GSH reductase and peroxidase systems. Aiming to break the GSH metabolism and redox homeostasis in cancer cells, our group synthesized two mitochondria-accumulating binuclear Re(I) tricarbonyl complexes, in which the azo/disulfide bond in the bridge linkage group could react with GSH.89 These complexes can localize in mitochondria, cause oxidative mitochondrial dysfunction, and induce necroptosis and caspase-dependent apoptosis simultaneously.
The malignancy of cancer is characterized by the changes in the epigenetic modification landscape. Recently, our group reported a mitochondria-targeting Re(I) complex 59 (Scheme 6) that could demolish triple-negative breast cancer cells by influencing their epigenetic status and inducing immunogenic apoptotic cell death.90 By integrating the clinical iron chelating agent deferasirox into its structure, 59 can relocate iron to mitochondria and change the key metabolic species related to the machinery of epigenetic modifications. Confocal microscopy shows that 59 localizes in mitochondria and the emission of 59 is gradually quenched along with the incubation time, which further confirms that Fe is relocated to mitochondria because both Fe2+ and Fe3+ can quench the fluorescence of 59.
5. Conclusions
Phosphorescent metal complexes have shown great potential as multifunctional anticancer compounds that can integrate imaging and therapeutic functionalities. On one hand, the imaging function can provide useful clues for the investigation of their anticancer mechanisms, such as their localization on the subcellular organelles. On the other hand, they can be utilized to track the dynamic behaviors of subcellular organelles during various types of cell death depending on their unique anticancer mechanisms. Of course, the research field is still in its infancy, and there are still many unresolved problems as well as room for improvement for their theranostic applications.In future research, we propose that the following aspects are very important for the development of this field. (1) The excitation and emission wavelengths of most complexes reported are in the visible region with limited penetration depth. More structural optimization is needed to tune the optical properties of the complexes to the NIR region, which is more suitable for in vivo applications. The correlation between the structural design and the function of the complexes is not straightforward with many of the reports displaying certain randomness. Combined with new imaging technologies (e.g., super-resolution imaging), the rational design of phosphorescent metal complexes to more accurately reflect the behaviours of subcellular organelles and biomolecules during the process of therapy is still challenging. (2) Molecular target validation of the complexes needs to be strengthened, which can help to explain their cellular behaviors and more accurately correlate with the specific biological processes monitored by their imaging function. At present, some studies have demonstrated that metallo-compounds can bind with diverse biomolecules in cells, but the biomolecular targets of many complexes in cells are unknown. (3) The mechanism of the uptake, transport, storage and metabolism of the complexes in vitro and in vivo is not clear, and the specific pathways or biomolecules responsible for these processes are unknown. The stability of the complexes under physiological conditions has been rarely investigated, and new in situ research methods are needed to explore their stability both in cells and in vivo. (4) Finally, with regard to the potential clinical transformations of these complexes, it is necessary to construct a much larger library for accurate SAR exploration. The multi-omics approach can be utilized to uncover their global biological effects. Moreover, we should seek cooperation with experts in pharmacology, oncology, clinical research and pharmaceutical industry to explore the possibility of their clinical translation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Natural Science Foundation of China (nos. 22022707, 21778078 and 21837006), the innovative team of Ministry of Education (no. IRT_17R111) and the Fundamental Research Funds for the Central Universities.Notes and references
- I. Dagogo-Jack and A. T. Shaw, Nat. Rev. Clin. Oncol., 2018, 15, 81–94 CrossRef CAS.
- T. Powles, I. Duran, M. S. van der Heijden, Y. Loriot, N. J. Vogelzang, U. De Giorgi, S. Oudard, M. M. Retz, D. Castellano, A. Bamias, A. Flechon, G. Gravis, S. Hussain, T. Takano, N. Leng, E. E. Kadel 3rd, R. Banchereau, P. S. Hegde, S. Mariathasan, N. Cui, X. Shen, C. L. Derleth, M. C. Green and A. Ravaud, Lancet, 2018, 391, 748–757 CrossRef CAS.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS.
- L. L. Zeng, P. Gupta, Y. L. Chen, E. J. Wang, L. N. Ji, H. Chao and Z. S. Chen, Chem. Soc. Rev., 2017, 46, 5771–5804 RSC.
- A. Notaro and G. Gasser, Chem. Soc. Rev., 2017, 46, 7317–7337 RSC.
- L. C. C. Lee, K. K. Leung and K. K. W. Lo, Dalton Trans., 2017, 46, 16357–16380 RSC.
- D. L. Ma, C. Wu, K. J. Wu and C. H. Leung, Molecules, 2019, 24, 2739 CrossRef CAS.
- J. Xie, S. Lee and X. Chen, Adv. Drug Delivery Rev., 2010, 62, 1064–1079 CrossRef CAS.
- L. Filippi, A. Chiaravalloti, O. Schillaci, R. Cianni and O. Bagni, Expert Rev. Med. Devices, 2020, 17, 331–343 CrossRef CAS.
- C. Wang, W. Fan, Z. Zhang, Y. Wen, L. Xiong and X. Chen, Adv. Mater., 2019, 31, 1904329 CrossRef CAS.
- R. Kumar, W. S. Shin, K. Sunwoo, W. Y. Kim, S. Koo, S. Bhuniya and J. S. Kim, Chem. Soc. Rev., 2015, 44, 6670–6683 RSC.
- L. He, C. P. Tan, R. R. Ye, Y. Z. Zhao, Y. H. Liu, Q. Zhao, L. N. Ji and Z. W. Mao, Angew. Chem., Int. Ed., 2014, 53, 12137–12141 CrossRef CAS.
- C. R. Cardoso, M. V. Lima, J. Cheleski, E. J. Peterson, T. Venancio, N. P. Farrell and R. M. Carlos, J. Med. Chem., 2014, 57, 4906–4915 CrossRef CAS.
- J. S. Nam, M. G. Kang, J. Kang, S. Y. Park, S. J. C. Lee, H. T. Kim, J. K. Seo, O. H. Kwon, M. H. Lim, H. W. Rhee and T. H. Kwon, J. Am. Chem. Soc., 2016, 138, 10968–10977 CrossRef CAS.
- L. He, Y. Li, C. P. Tan, R. R. Ye, M. H. Chen, J. J. Cao, L. N. Ji and Z. W. Mao, Chem. Sci., 2015, 6, 5409–5418 RSC.
- L. Hao, Z. W. Li, D. Y. Zhang, L. He, W. Liu, J. Yang, C. P. Tan, L. N. Ji and Z. W. Mao, Chem. Sci., 2019, 10, 1285–1293 RSC.
- Q. X. Chen, C. Z. Jin, X. T. Shao, R. L. Guan, Z. Q. Tian, C. R. Wang, F. Liu, P. X. Ling, J. L. Guan, L. N. Ji, F. S. Wang, H. Chao and J. J. Diao, Small, 2018, 14, 1802166 CrossRef.
- H. K. Saeed, S. Sreedharan, P. J. Jarman, S. A. Archer, S. D. Fairbanks, S. P. Foxon, A. J. Auty, D. Chekulaev, T. Keane, A. J. H. M. Meijer, J. A. Weinstein, C. G. W. Smythe, J. B. de la Serna and J. A. Thomas, J. Am. Chem. Soc., 2020, 142, 1101–1111 CrossRef CAS.
- F. Heinemann, J. Karges and G. Gasser, Acc. Chem. Res., 2017, 50, 2727–2736 CrossRef CAS.
- S. Monro, K. L. Colon, H. M. Yin, J. Roque, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2019, 119, 797–828 CrossRef CAS.
- F. E. Poynton, S. A. Bright, S. Blasco, D. C. Williams, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2017, 46, 7706–7756 RSC.
- Y. Chen, T. W. Rees, L. N. Ji and H. Chao, Curr. Opin. Chem. Biol., 2018, 43, 51–57 CrossRef CAS.
- C. N. Ko, G. D. Li, C. H. Leung and D. L. Ma, Coord. Chem. Rev., 2019, 381, 79–103 CrossRef CAS.
- J. G. Liu, H. Q. Lai, Z. S. Xiong, B. L. Chen and T. F. Chen, Chem. Commun., 2019, 55, 9904–9914 RSC.
- K. K. W. Lo, Acc. Chem. Res., 2015, 48, 2985–2995 CrossRef CAS.
- C. Caporale and M. Massi, Coord. Chem. Rev., 2018, 363, 71–91 CrossRef CAS.
- H. Chen, Q. Zhao, Y. Wu, F. Li, H. Yang, T. Yi and C. Huang, Inorg. Chem., 2007, 46, 11075–11081 CrossRef CAS.
- K. K. Lo, C. K. Chung and N. Zhu, Chem.–Eur. J., 2006, 12, 1500–1512 CrossRef CAS.
- K. K. Lo, C. K. Chung, T. K. Lee, L. H. Lui, K. H. Tsang and N. Zhu, Inorg. Chem., 2003, 42, 6886–6897 CrossRef CAS.
- K. K. Lo, Acc. Chem. Res., 2020, 53, 32–44 CrossRef CAS.
- C. Li, M. Yu, Y. Sun, Y. Wu, C. Huang and F. Li, J. Am. Chem. Soc., 2011, 133, 11231–11239 CrossRef CAS.
- M. Yu, Q. Zhao, L. Shi, F. Li, Z. Zhou, H. Yang, T. Yi and C. Huang, Chem. Commun., 2008, 2115–2117 RSC.
- C. Li, Y. Liu, Y. Wu, Y. Sun and F. Li, Biomaterials, 2013, 34, 1223–1234 CrossRef CAS.
- P. K. Lee, W. H. Law, H. W. Liu and K. K. Lo, Inorg. Chem., 2011, 50, 8570–8579 CrossRef CAS.
- R. Guan, Y. Chen, L. Zeng, T. W. Rees, C. Jin, J. Huang, Z. S. Chen, L. Ji and H. Chao, Chem. Sci., 2018, 9, 5183–5190 RSC.
- J. J. Cao, Y. Zheng, X. W. Wu, C. P. Tan, M. H. Chen, N. Wu, L. N. Ji and Z. W. Mao, J. Med. Chem., 2019, 62, 3311–3322 CrossRef CAS.
- P. Y. Zhang, C. K. C. Chiu, H. Y. Huang, Y. P. Y. Lam, A. Habtemariam, T. Malcomson, M. J. Paterson, G. J. Clarkson, P. B. O'Connor, H. Chao and P. J. Sadler, Angew. Chem., Int. Ed., 2017, 56, 14898–14902 CrossRef CAS.
- L. He, J. J. Cao, D. Y. Zhang, L. Hao, M. F. Zhang, C. P. Tan, L. N. Ji and Z. W. Mao, Sens. Actuators, B, 2018, 262, 313–325 CrossRef CAS.
- D. Y. Zhang, Y. Zheng, H. Zhang, J. H. Sun, C. P. Tan, L. He, W. Zhang, L. N. Ji and Z. W. Mao, Adv. Sci., 2018, 5, 1800581 CrossRef.
- M. P. Murphy and R. C. Hartley, Nat. Rev. Drug Discovery, 2018, 17, 865–886 CrossRef CAS.
- S. Yi, Z. Lu, J. Zhang, J. Wang, Z. Xie and L. Hou, ACS Appl. Mater. Interfaces, 2019, 11, 15276–15289 CrossRef CAS.
- Y. Li, C. P. Tan, W. Zhang, L. He, L. N. Ji and Z. W. Mao, Biomaterials, 2015, 39, 95–104 CrossRef CAS.
- L. He, M. F. Zhang, Z. Y. Pan, K. N. Wang, Z. J. Zhao, Y. Li and Z. W. Mao, Chem. Commun., 2019, 55, 10472–10475 RSC.
- J. J. Cao, C. P. Tan, M. H. Chen, N. Wu, D. Y. Yao, X. G. Liu, L. N. Ji and Z. W. Mao, Chem. Sci., 2017, 8, 631–640 RSC.
- M. H. Chen, F. X. Wang, J. J. Cao, C. P. Tan, L. N. Ji and Z. W. Mao, ACS Appl. Mater. Interfaces, 2017, 9, 13304–13314 CrossRef CAS.
- V. Venkatesh, R. Berrocal-Martin, C. J. Wedge, I. Romero-Canelon, C. Sanchez-Cano, J. I. Song, J. P. C. Coverdale, P. Zhang, G. J. Clarkson, A. Habtemariam, S. W. Magennis, R. J. Deeth and P. J. Sadler, Chem. Sci., 2017, 8, 8271–8278 RSC.
- R. Bevernaegie, B. Doix, E. Bastien, A. Diman, A. Decottignies, O. Feron and B. Elias, J. Am. Chem. Soc., 2019, 141, 18486–18491 CrossRef CAS.
- H. Huang, S. Banerjee, K. Qiu, P. Zhang, O. Blacque, T. Malcomson, M. J. Paterson, G. J. Clarkson, M. Staniforth, V. G. Stavros, G. Gasser, H. Chao and P. J. Sadler, Nat. Chem., 2019, 11, 1041–1048 CrossRef CAS.
- R. E. Lawrence and R. Zoncu, Nat. Cell Biol., 2019, 21, 133–142 CrossRef CAS.
- C. G. Towers and A. Thorburn, Cancer Discov., 2017, 7, 1218–1220 CrossRef.
- M. H. Chen, Y. Zheng, X. J. Cai, H. Zhang, F. X. Wang, C. P. Tan, W. H. Chen, L. N. Ji and Z. W. Mao, Chem. Sci., 2019, 10, 3315–3323 RSC.
- F. X. Wang, M. H. Chen, Y. N. Lin, H. Zhang, C. P. Tan, L. N. Ji and Z. W. Mao, ACS Appl. Mater. Interfaces, 2017, 9, 42471–42481 CrossRef CAS.
- L. K. McKenzie, I. V. Sazanovich, E. Baggaley, M. Bonneau, V. Guerchais, J. A. G. Williams, J. A. Weinstein and H. E. Bryant, Chem.–Eur. J., 2017, 23, 234–238 CrossRef CAS.
- W. Lv, Z. Zhang, K. Y. Zhang, H. Yang, S. Liu, A. Xu, S. Guo, Q. Zhao and W. Huang, Angew. Chem., Int. Ed., 2016, 55, 9947–9951 CrossRef CAS.
- S. Kuang, X. Liao, X. Zhang, T. W. Rees, R. Guan, K. Xiong, Y. Chen, L. Ji and H. Chao, Angew. Chem., Int. Ed., 2020, 59, 3315–3321 CrossRef CAS.
- T. L. Lam, K. C. Tong, C. Yang, W. L. Kwong, X. Guan, M. D. Li, V. Kar-Yan Lo, S. Lai-Fung Chan, D. Lee Phillips, C. N. Lok and C. M. Che, Chem. Sci., 2019, 10, 293–309 RSC.
- P. Zhang, H. Huang, S. Banerjee, G. J. Clarkson, C. Ge, C. Imberti and P. J. Sadler, Angew. Chem., Int. Ed., 2019, 58, 2350–2354 CrossRef CAS.
- S. A. McFarland, A. Mandel, R. Dumoulin-White and G. Gasser, Curr. Opin. Chem. Biol., 2020, 56, 23–27 CrossRef CAS.
- J. Shum, P. K. Leung and K. K. Lo, Inorg. Chem., 2019, 58, 2231–2247 CrossRef CAS.
- K. E. Erkkila, D. T. Odom and J. K. Barton, Chem. Rev., 1999, 99, 2777–2795 CrossRef CAS.
- R. E. Holmlin, E. D. Stemp and J. K. Barton, Inorg. Chem., 1998, 37, 29–34 CrossRef CAS.
- C. A. Puckett and J. K. Barton, J. Am. Chem. Soc., 2007, 129, 46–47 CrossRef CAS.
- G. Di Marco, M. Lanza and S. Campagna, Adv. Mater., 1995, 7, 468–471 CrossRef CAS.
- A. Byrne, C. S. Burke and T. E. Keyes, Chem. Sci., 2016, 7, 6551–6562 RSC.
- S. Sreedharan, M. R. Gill, E. Garcia, H. K. Saeed, D. Robinson, A. Byrne, A. Cadby, T. E. Keyes, C. Smythe, P. Pellett, J. B. de la Serna and J. A. Thomas, J. Am. Chem. Soc., 2017, 139, 15907–15913 CrossRef CAS.
- Z. N. Zhao, P. Gao, Y. Y. You and T. F. Chen, Chem.–Eur. J., 2018, 24, 3289–3298 CrossRef CAS.
- J. Li, L. L. Zeng, K. Xiong, T. W. Rees, C. Z. Jin, W. J. Wu, Y. Chen, L. N. Ji and H. Chao, Chem. Commun., 2019, 55, 10972–10975 RSC.
- Z. Zhao, X. Zhang, C. E. Li and T. Chen, Biomaterials, 2019, 192, 579–589 CrossRef CAS.
- J. P. Liu, Y. Chen, G. Y. Li, P. Y. Zhang, C. Z. Jin, L. L. Zeng, L. N. Ji and H. Chao, Biomaterials, 2015, 56, 140–153 CrossRef CAS.
- C. S. Burke, A. Byrne and T. E. Keyes, Angew. Chem., Int. Ed., 2018, 57, 12420–12424 CrossRef CAS.
- H. Y. Huang, B. L. Yu, P. Y. Zhang, J. J. Huang, Y. Chen, G. Gasser, L. N. Ji and H. Chao, Angew. Chem., Int. Ed., 2015, 54, 14049–14052 CrossRef CAS.
- H. K. Saeed, P. J. Jarman, S. Archer, S. Sreedharan, I. Q. Saeed, L. K. Mckenzie, J. A. Weinstein, N. J. Buurma, C. G. W. Smythe and J. A. Thomas, Angew. Chem., Int. Ed., 2017, 56, 12628–12633 CrossRef CAS.
- Z. X. Zhou, J. P. Liu, T. W. Rees, H. Wang, X. P. Li, H. Chao and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5664–5669 CrossRef CAS.
- J. Yang, J. X. Zhao, Q. Cao, L. Hao, D. Zhou, Z. Gan, L. N. Ji and Z. W. Mao, ACS Appl. Mater. Interfaces, 2017, 9, 13900–13912 CrossRef CAS.
- A. M. Yip, J. Shum, H. W. Liu, H. Zhou, M. Jia, N. Niu, Y. Li, C. Yu and K. K. Lo, Chem.–Eur. J., 2019, 25, 8970–8974 CrossRef CAS.
- M. W. Louie, T. T. Fong and K. K. Lo, Inorg. Chem., 2011, 50, 9465–9471 CrossRef CAS.
- K. K. Lo, M. W. Louie, K. S. Sze and J. S. Lau, Inorg. Chem., 2008, 47, 602–611 CrossRef CAS.
- A. W. Choi, K. K. Tso, V. M. Yim, H. W. Liu and K. K. Lo, Chem. Commun., 2015, 51, 3442–3445 RSC.
- B. Albada and N. Metzler-Nolte, Chem. Rev., 2016, 116, 11797–11839 CrossRef CAS.
- I. Kitanovic, S. Z. Can, H. Alborzinia, A. Kitanovic, V. Pierroz, A. Leonidova, A. Pinto, B. Spingler, S. Ferrari, R. Molteni, A. Steffen, N. Metzler-Nolte, S. Wolfl and G. Gasser, Chem.–Eur. J., 2014, 20, 2496–2507 CrossRef CAS.
- K. M. Knopf, B. L. Murphy, S. N. MacMillan, J. M. Baskin, M. P. Barr, E. Boros and J. J. Wilson, J. Am. Chem. Soc., 2017, 139, 14302–14314 CrossRef CAS.
- I. Chakraborty, J. Jimenez, W. M. C. Sameera, M. Kato and P. K. Mascharak, Inorg. Chem., 2017, 56, 2863–2873 CrossRef CAS.
- S. Imstepf, V. Pierroz, R. Rubbiani, M. Felber, T. Fox, G. Gasser and R. Alberto, Angew. Chem., Int. Ed., 2016, 55, 2792–2795 CrossRef CAS.
- C. C. Konkankit, A. P. King, K. M. Knopf, T. L. Southard and J. J. Wilson, ACS Med. Chem. Lett., 2019, 10, 822–827 CrossRef CAS.
- S. C. Marker, A. P. King, R. V. Swanda, B. Vaughn, E. Boros, S. B. Qian and J. J. Wilson, Angew. Chem., Int. Ed., 2020, 59, 13391–13400 CrossRef CAS.
- C. C. Konkankit, J. Lovett, H. H. Harris and J. J. Wilson, Chem. Commun., 2020, 56, 6515–6518 RSC.
- A. P. King, S. C. Marker, R. V. Swanda, J. J. Woods, S. B. Qian and J. J. Wilson, Chem.–Eur. J., 2019, 25, 9206–9210 CrossRef CAS.
- J. Yang, Q. Cao, H. Zhang, L. Hao, D. Zhou, Z. Gan, Z. Li, Y. X. Tong, L. N. Ji and Z. W. Mao, Biomaterials, 2018, 176, 94–105 CrossRef CAS.
- F. X. Wang, J. H. Liang, H. Zhang, Z. H. Wang, Q. Wan, C. P. Tan, L. N. Ji and Z. W. Mao, ACS Appl. Mater. Interfaces, 2019, 11, 13123–13133 CrossRef CAS.
- Z. Y. Pan, C. P. Tan, L. S. Rao, H. Zhang, Y. Zheng, L. Hao, L. N. Ji and Z. W. Mao, Angew. Chem., Int. Ed., 2020, 59, 18755–18762 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |