
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
N-Heterocyclic carbene–carbodiimide (NHC–CDI) betaine adducts: synthesis, characterization, properties, and applications
Jessica R.
Lamb†
 *,
Christopher M.
Brown
and
Jeremiah A.
Johnson
*
*,
Christopher M.
Brown
and
Jeremiah A.
Johnson
*
Department of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA. E-mail: jrlamb@umn.edu; jaj2109@mit.edu
First published on 19th January 2021
Abstract
N-Heterocyclic carbenes (NHCs) are an important class of reactive organic molecules used as ligands, organocatalysts, and σ-donors in a variety of electroneutral ylide or betaine adducts with main-group compounds. An emerging class of betaine adducts made from the reaction of NHCs with carbodiimides (CDIs) form zwitterionic amidinate-like structures with tunable properties based on the highly modular NHC and CDI scaffolds. The adduct stability is controlled by the substituents on the CDI nitrogens, while the NHC substituents greatly affect the configuration of the adduct in the solid state. This Perspective is intended as a primer to these adducts, touching on their history, synthesis, characterization, and general properties. Despite the infancy of the field, NHC–CDI adducts have been applied as amidinate-type ligands for transition metals and nanoparticles, as junctions in zwitterionic polymers, and to stabilize distonic radical cations. These applications and potential future directions are discussed.
 Jessica R. Lamb | Jessica Lamb received her MS (2014) and PhD (2017) from Cornell University in the lab of Prof. Geoffrey Coates as an NSF Graduate Research Fellow where she worked on developing selective transformations of epoxides using bimetallic catalysts. She then worked as an NIH Ruth L. Kirschstein Postdoctoral Fellow in the group of Prof. Jeremiah Johnson at MIT where she studied photocontrolled radical polymerization and polymeric gel materials. She started as an Assistant Professor at the University of Minnesota in July 2020. The Lamb group works at the interface of catalysis, organic, and polymer chemistry. |
 Christopher M. Brown | Christopher Brown received his MChem degree from Newcastle University in 2011, where he synthesized air-stable primary phosphines under the supervision of Dr Lee Higham. He then moved to the University of British Columbia where he obtained his PhD in 2018 working in the group of Prof. Michael Wolf. His doctoral research focussed on using sulfur oxidation state to tune the electronic and photophysical properties of coordination complexes. Chris started as a Postdoctoral Associate in the group of Prof. Jeremiah Johnson at MIT in 2019, where he is studying N-heterocyclic materials and polymer metal–organic cages. |
 Jeremiah A. Johnson | Jeremiah is a Professor of Chemistry at MIT, where he has been since July 2011. He is a member of MIT's Program in Polymers and Soft Matter, the Koch Institute for Integrative Cancer Research, and the Broad Institute of MIT and Harvard. The Johnson Group's research has been recognized through several awards including a Cope Scholar Award, the Macromolecules-Biomacromolecules Young Investigator Award, and the Nobel Laureate Signature Award for Graduate Education. In recognition of excellence in teaching, Jeremiah received the MIT School of Science Undergraduate Teaching Prize. The Johnson research group develops methods and strategies for macromolecular synthesis. |
1. Introduction
Since the first demonstrations of persistent carbenes,1,2N-heterocyclic carbenes (NHCs) have been used extensively in transition metal and main group chemistries,3–5 as organocatalysts,6 as ligands in organometallic catalysts,7 and in materials chemistry.8,9 NHCs are broadly defined as heterocyclic compounds that contain a divalent carbon and at least one nitrogen within the ring.10 They act as neutral, two-electron species, and are frequently used as phosphine analogues, though with stronger metal–ligand bonds due to their higher σ-donating abilities.11 Kinetic stability is aided by bulky substituents adjacent to the carbene, while the nitrogen substituent(s) electronically stabilize the empty carbene orbital through resonance12 (Fig. 1A).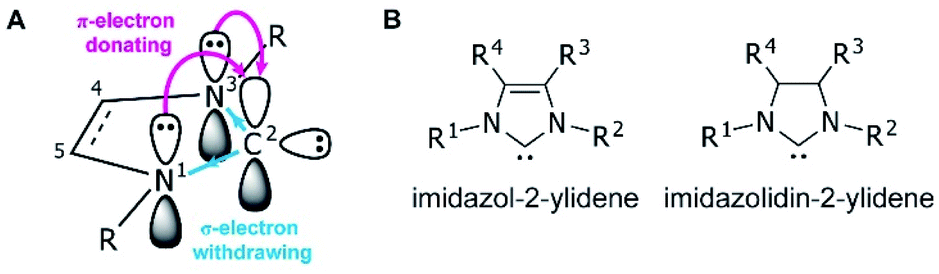 | ||
| Fig. 1 (A) Ground-state electronic structure of imidazol-2-ylidenes with numbering at the heterocycle shown. (B) NHCs of the imidazolidium and imidazolidinium based classes, with unsaturated and saturated backbones, respectively. | ||
NHCs are extremely modular, allowing facile tuning of their electronic and steric properties. Perhaps the most well-studied are the five-membered imidazolium- and imidazolidinium-based NHCs (Fig. 1B).11,13 Variations can be made at the nitrogen substituents (i.e., the wingtip positions) to make either symmetric (R1 = R2) or asymmetric (R1 ≠ R2) species, and likewise the 4- and 5-positions (R3 and R4) can be modified, though with typically a lesser effect on the electronics of the system.13 Consequently, chemists have access to a huge synthetic library of cationic heterocycles to use as NHC precursors.14
Beyond their traditional applications as ligands and organocatalysts, NHCs can add to or insert themselves into a variety of organic species to form stable, neutral adducts, and if a suitable electrophile is used, zwitterionic adducts can be formed.15 Halogens or boranes lead to the formation of ylides, while reactions with allenes, ketenes, or heteroallenes give betaine adducts, which can only be represented by resonance forms with formal charges (Fig. 2). To date, many stable, crystalline NHC–betaine adducts with CS2, CO2, isothiocyanates, and isocyanates have been reported and reviewed elsewhere.16
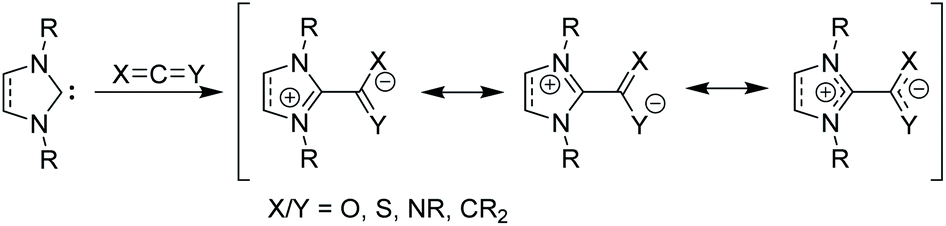 | ||
| Fig. 2 Zwitterionic betaine adducts made from N-heterocyclic carbenes with allenes, ketenes, or heteroallenes. | ||
In this Perspective, we focus on betaine adducts made from NHCs and carbodiimides (CDIs), i.e., zwitterionic amidinates (NHC–CDIs, Fig. 2 X and Y = NR). Amidinates are considered the nitrogen analogues of carboxylates.17 They act as coordinating ligands and feature a wide range of coordination modes; many amidinate complexes of transition metals,18–20 lanthanides,17 actinides,21 and main group elements22 have been reported. One important difference compared to carboxylate ligands is that the nitrogen atoms in amidinates nearly always carry an additional substituent, which allows for fine-tuning of the electronic and steric properties of the ligand system.22 Combined with the similarly modular NHC framework, this feature presents a unique opportunity for tailoring NHC–CDI adducts for specific applications. NHCs and amidinates are also typically both air- and moisture-sensitive, whereas NHC–CDI adducts made from N,N′-diaryl CDIs result in net neutral compounds that are more tolerant to ambient conditions while retaining a strongly Lewis basic character at the amidinyl nitrogens.23 This Perspective will introduce the history, common synthetic and characterization methods, and a summary of the structure–property relationships of the currently known NHC–CDI adducts. Finally, applications to coordination chemistry, nanoparticle functionalization, supramolecular polymers, and distonic radical cation stabilization will be summarized along with future directions of the field.
2. History
To our knowledge, the first zwitterionic NHC–CDI adduct was proposed by Takamizawa and co-workers in 1974.24 These authors found that the addition of a thiazolium iodide species to an N,N′-diaryl CDI in the presence of triethylamine (NEt3) yields a mixture containing a cationic thiazolium–CDI adduct and a spirocycle made via cycloaddition with a second equivalent of CDI. It was proposed that deprotonation of the thiazolium adduct leads to a zwitterionic NHC–CDI intermediate, which can then react with another CDI to give the cycloadduct. While the zwitterionic intermediate could not be isolated, it was noted that analogous zwitterionic adducts formed from the reaction of a phosphorus ylide with an N,N′-diaryl CDI had been previously isolated.25It was not until 25 years later that the first stable, isolable NHC–CDI was reported by Kuhn and co-workers.26 1,3-Diisopropyl-4,5-dimethylimidazol-2-ylidene was allowed to react with diisopropylcarbodiimide, yielding a zwitterionic adduct which was characterized by NMR spectroscopy and X-ray crystallography. The strong Brønsted basicity of the amidinate nitrogen atoms caused decomposition of the adduct upon exposure to moisture, but also indicated the potential for NHC–CDIs as ligands for metallic systems or as organic bases, while remaining electroneutral. Despite this promise, the next report on isolated NHC–CDI adducts did not come, to our knowledge, until bench-stable variants utilizing N,N′-diaryl CDIs were discovered simultaneously by the Johnson and Cámpora groups in 2015.23,27 Since then, the facile synthesis, air- and moisture-stability, and interesting properties of these betaine adducts have spurred new interest in the field.
3. Synthesis and structure
3.1 Methods for NHC–CDI synthesis
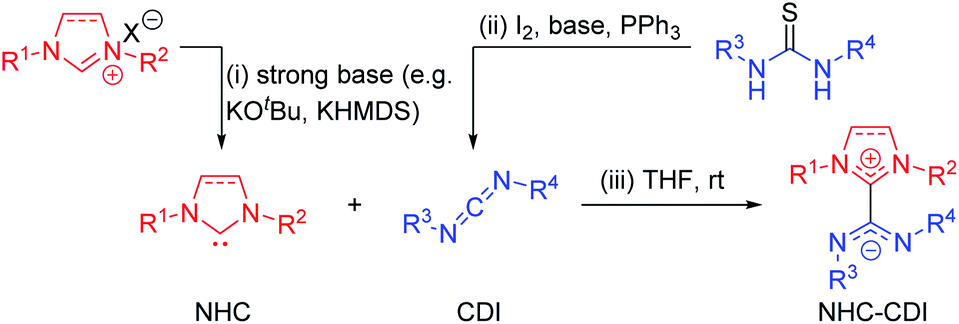 | ||
| Fig. 3 Common synthetic routes to (i) free NHC by deprotonation of an imidazolium salt precursor, (ii) free CDI by desulfurization of a thiourea, and (iii) NHC–CDI adduct. | ||
To avoid the use of very strong bases, light-triggered deprotonation has been achieved using isopropylthioxanthone (ITX) with an azolium salt precursor containing a tetraphenylborate (BPh4−) counteranion.34 Photoexcitation of ITX results in a triplet excited state that can accept a single electron from BPh4− to form ITX˙− which abstracts a proton from the azolium cation to generate the free carbene and ITX–H˙. Heat-sensitive progenitors can also produce free NHCs without the use of a strong base. This strategy is common for latent organocatalysis35 and has been utilized for the synthesis of NHC–CDI adducts by Johnson and co-workers, who first used carboxylate and diphenylphosphine adducts of SIMes23 and later synthesized C6F5-masked NHCs by exposing the corresponding diamines to pentafluorobenzaldehyde in acetic acid at room temperature.33
The most common method for the preparation of NHC–CDI adducts involves directly adding a free carbene to a free CDI in an appropriate solvent (Fig. 3iii).23,27,30,33,41,42 The often brightly-colored products form quickly at room temperature and can in many cases be readily purified by washing with, e.g., cold hexanes. If a thermally-labile NHC precursor is used instead of a free carbene, the masked NHC and free CDI can be heated in an appropriate solvent to form the NHC–CDI adduct.33 It is important to emphasize that the method of preparation is largely personal preference, as any method for releasing a free NHC in the presence of a free CDI will likely result in the formation of the same adduct.
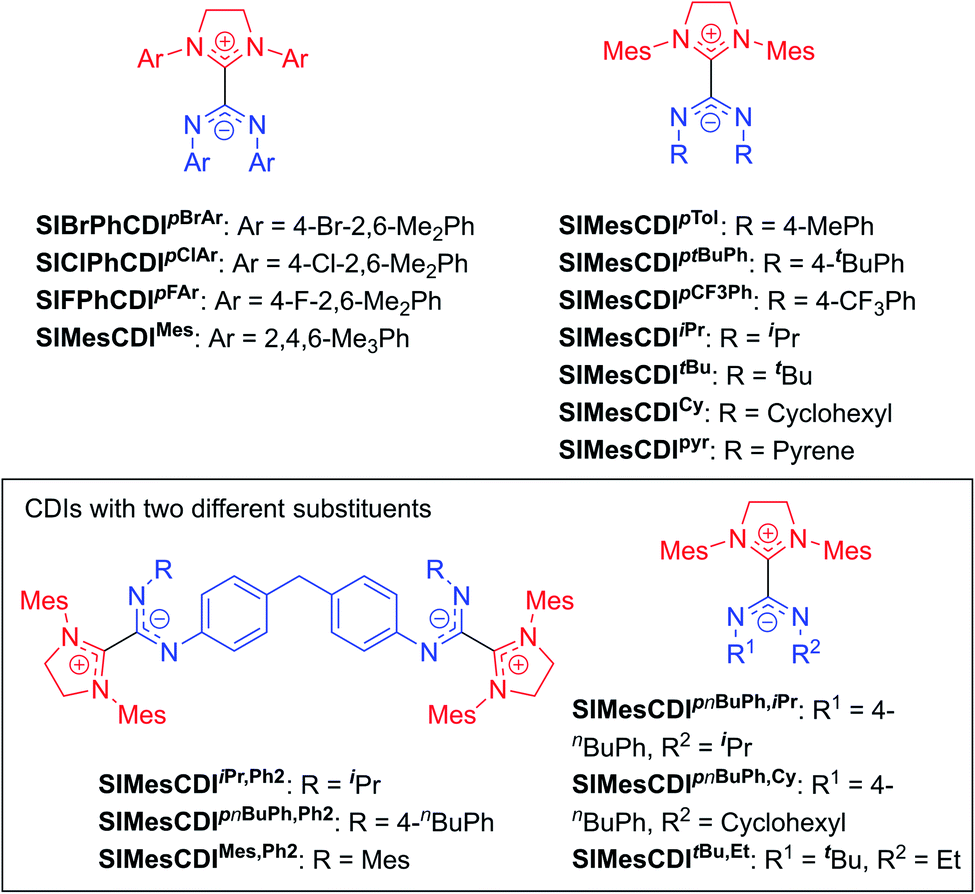
The highly modular nature of both the NHC and the CDI fragments have led to the synthesis of a wide range of NHC–CDI adducts. Previously synthesized imidazol-2-ylidene-based adducts are shown in Fig. 4 and imidazolidin-2-ylidene-based adducts are shown in Fig. 5. The naming convention consists of the NHC backbone (I for imidazol-2-ylidene, MeI for 4,5-dimethylimidazol-2-ylidene, or SI for saturated imidazolidin-2-ylidene) followed by the NHC wingtip substituents, “CDI”, and the amidinate substituents as superscripts.
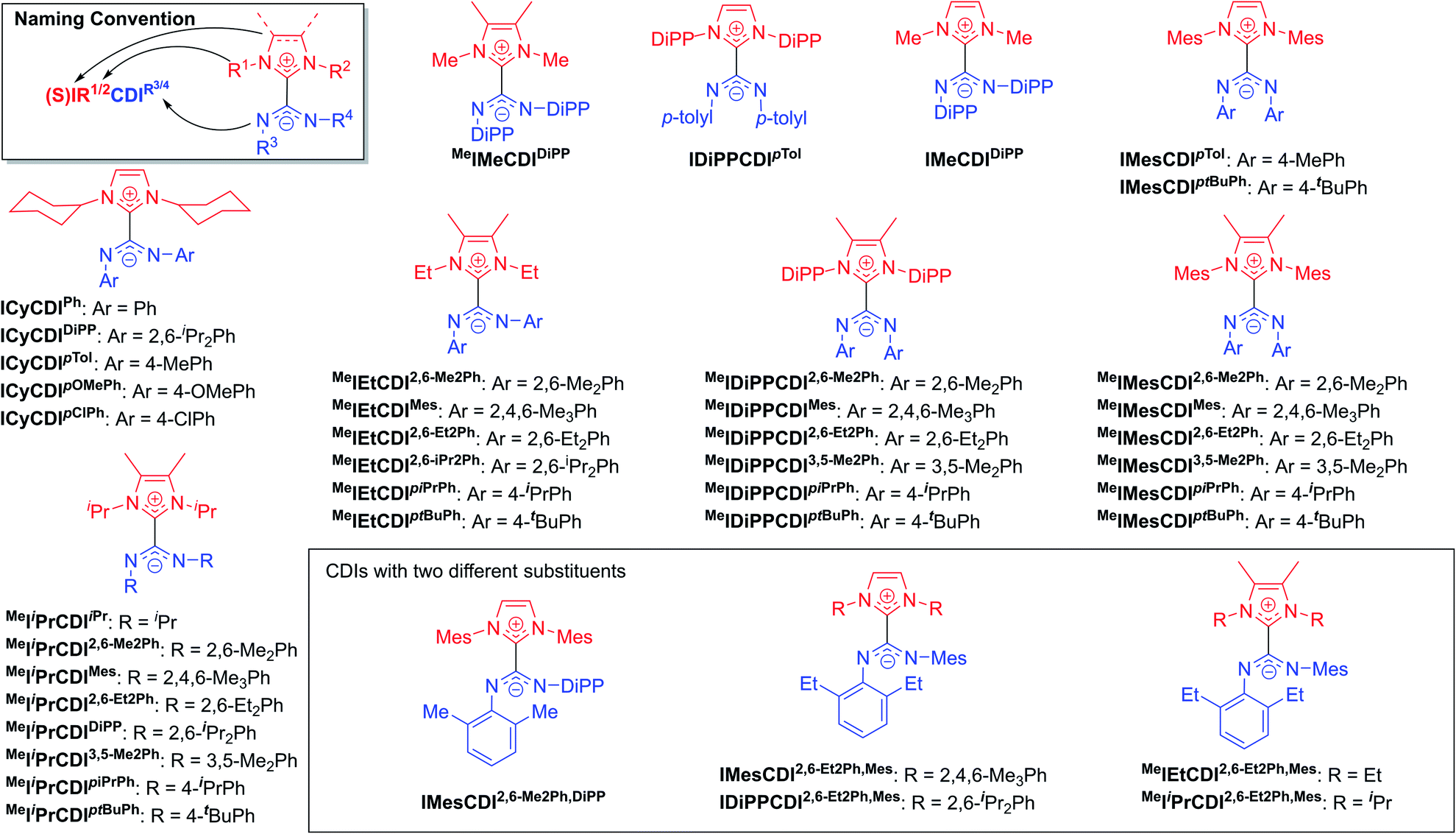 | ||
| Fig. 4 Previously synthesized imidazol-2-ylidene based NHC–CDI adducts. | ||
 | ||
| Fig. 5 Previously synthesized imidazolidin-2-ylidene based discrete NHC–CDI adducts. | ||
Cámpora and co-workers synthesized a variety of imidazol-2-ylidene-based adducts with alkyl substituents (methyls and cyclohexyls) in the wingtip positions.27,30,41,42 Nembenna and co-workers expanded the library utilizing both N,N′-dialkyl and N,N′-diaryl NHCs along with N,N′-diaryl CDIs with either the same or different substituents.43 Variants using saturated NHCs have been made by Johnson and co-workers with N,N′-diaryl, N-aryl-N′-alkyl, and N,N′-dialkyl CDIs, with variable stability (see Section 3.3.1 for more details).23,33,44 Additional adducts were made as repeat units of zwitterionic polymers, which will be discussed further in Section 4.2.
3.2 Characterization of NHC–CDIs
Because of the delocalized double bond character, there is restricted rotation about both amidinate C![[partial double bond, bottom dashed]](https://www.rsc.org/images/entities/char_e00f.gif) N bonds of NHC–CDI adducts, orienting the geometry of each bond as transoid (E) or cisoid (Z) with respect to the other amidinate nitrogen (Fig. 6A). 15N cross polarization magic-angle spinning (CP-MAS) solid-state NMR spectroscopy of ICyCDIPh with one 15N-labeled amidinate nitrogen exhibited two signals at 211 and 218 ppm with approximately equal intensity.30 Density functional theory (DFT) calculations support that these signals correspond to the two non-equivalent nitrogen atoms of the E/Z configuration (Fig. 6B). Many 13C CP-MAS NMR signals are also duplicated due to the non-equivalent phenyl substituents in the E/Z configuration. In contrast, solution-state 1H and 13C{1H} NMR spectra show a single set of aromatic resonances, indicating fast exchange between the E/Z and Z/E configurations in solution. DFT calculations have suggested that the E/E geometry is less stable than the E/Z configuration by 10.2 kcal mol−1, ruling out facile observation of the E/E isomer.30
N bonds of NHC–CDI adducts, orienting the geometry of each bond as transoid (E) or cisoid (Z) with respect to the other amidinate nitrogen (Fig. 6A). 15N cross polarization magic-angle spinning (CP-MAS) solid-state NMR spectroscopy of ICyCDIPh with one 15N-labeled amidinate nitrogen exhibited two signals at 211 and 218 ppm with approximately equal intensity.30 Density functional theory (DFT) calculations support that these signals correspond to the two non-equivalent nitrogen atoms of the E/Z configuration (Fig. 6B). Many 13C CP-MAS NMR signals are also duplicated due to the non-equivalent phenyl substituents in the E/Z configuration. In contrast, solution-state 1H and 13C{1H} NMR spectra show a single set of aromatic resonances, indicating fast exchange between the E/Z and Z/E configurations in solution. DFT calculations have suggested that the E/E geometry is less stable than the E/Z configuration by 10.2 kcal mol−1, ruling out facile observation of the E/E isomer.30
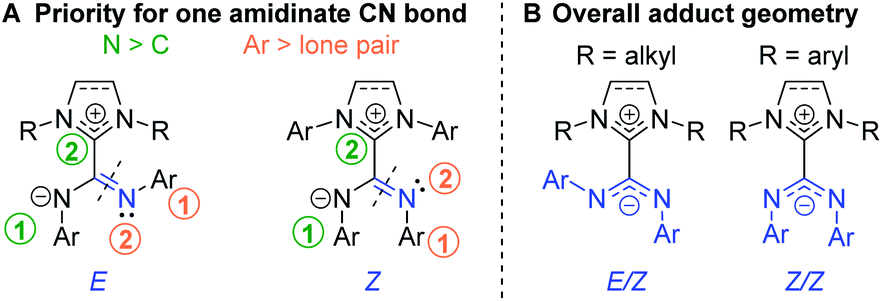 | ||
Fig. 6 (A) Priorities for assigning the geometry of amidinate C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds and (B) overall geometry for NHC–CDI adducts. N bonds and (B) overall geometry for NHC–CDI adducts. | ||
Rotation of the amidinate substituents is highly dependent on the steric bulk of the NHC wingtip substituents. For example, the diastereotopic methyl groups of 2,6-diisopropylphenyl (DiPP) moieties are well-resolved in both the 1H and 13C{1H} NMR spectra for ICyCDIDiPP, indicating a locked configuration on the NMR timescale, but the analogous protons for IMeCDIDiPP show a single resonance in the 1H NMR spectrum and two broad resonances in 13C{1H} NMR spectrum, indicating restricted rotation.41 The NMR spectra of adducts synthesized from CDIs with two different substituents33,43 do not show two sets of peaks for the NHC substituents, indicating that the imidazoyl and amidinyl moieties are effectively orthogonal under the experimental conditions, such that the lack of symmetry of the CDI does not break the symmetry of the NHC.
A remarkable feature revealed by the reported crystal structures of NHC–CDIs is the invariability of the core NHC–CDI C–C and C–N bond lengths. Regardless of the steric or electronic properties of the NHC or amidinate substituents, the reported central C–C bond lengths range from only 1.50–1.52 Å. The NHC CN bond lengths are generally 1.34–1.35 Å for unsaturated imidazol-2-ylidene derivatives41,43 and slightly shorter at 1.32–1.33 Å for saturated imidazolidin-2-ylidene derivatives.23,45 The amidinate CN bond lengths are also fairly uniform for all reported structures at 1.30–1.33 Å. These carbon–nitrogen bond lengths are consistent with delocalized double bonds across the anionic amidinate and cationic carbenic regions of the molecules.
The NHC NCN bond angle is based on the class of NHC used, with unsaturated imidazol-2-ylidenes and saturated imidazolidin-2-ylidenes displaying 106.7–108.3° (ref. 41 and 43) and 111.0–112.2° (ref. 23, 33 and 45) angles, respectively. The amidinate NCN bond angles vary significantly more on the basis of both the class of NHC and the identity of the amidinyl substituents. For imidazol-2-ylidene-based adducts, CDIDiPP yielded amidinate NCN angles of 126.5–127.5°,41,43 bis(dialkylphenyl)carbodiimides (dialkylphenyl = 2,6-dimethylphenyl, 3,5-dimethylphenyl, and 2,6-diethylphenyl/mesityl) displayed amidinate angles of 139.0–140.7°,43 and CDIpTol had amidinate angles of 130.1°, 138.4°, and 141.6° with ICy,41 IMes,23 and IDiPP,23 respectively. For imidazolidin-2-ylidenes, CDIMes and derivatives (methyl, bromo, chloro, or fluoro in the para-position) showed amidinate angles of 139.0–140.0°.23 The solid-state structure of SIMesCDIpyr shows two conformations: one with C2 symmetry and one distorted from C2 symmetry, which show amidinate angles of 136.8° and 137.9°, respectively.45 The dihedral angles between the NHC and amidinate fragments of the adducts vary wildly from 50.2–87.8°, without obvious correlations to either the steric or electronic properties of the nitrogen substituents.
The adduct geometry seems to be dictated by the NHC wingtip substituents, with N,N′-dialkyl NHCs leading to an E/Z configuration41,43 and N,N′-diaryl NHCs yielding a Z/Z configuration23,33,43,45 (Fig. 6B). Notably, MeIiPrCDI2,6-Me2Ph displays a Z/Z geometry despite having isopropyl substituents in the wingtip positions.43
N stretching modes and C–H flexion modes also overlap near 1500 cm−1. An additional asymmetric amidinate NCN stretch can be detected in some systems as a medium-intensity band at ∼1600 cm−1.41
3.3 Properties
Johnson and co-workers further probed this trend by synthesizing a series of adducts containing N,N′-diaryl, N-aryl-N′-alkyl, and N,N′-dialkyl CDIs.23,331H NMR spectroscopy was used to evaluate Keq (Fig. 7A). As expected, N,N′-dialkyl CDIs had relatively low Keq's, even <1 M−1 at room temperature for some adducts, while N,N′-diaryl CDIs gave robustly stable adducts. N-Aryl-N′-alkyl CDIs were predicted to form adducts that strike a balance between these two extremes. While the 1H NMR analysis showed quantitative adduct formation (Keq ≫ 550 M−1) similar to N,N′-diaryl CDIs, competitive binding experiments revealed that N-aryl-N′-alkyl amidinate adducts are indeed more dynamic than their N,N′-diaryl counterparts. N-Aryl-N′-alkyl amidinates could be quantitatively displaced by N,N′-diaryl CDIs to form N,N′,N′′,N′′′-tetraaryl NHC–CDIs, with concomitant evolution of free N-aryl-N′-alkyl CDI (Fig. 7B). Monitoring this process by 1H NMR spectroscopy revealed a first-order rate constant for this transformation as kd (50 °C) = (4.0 ± 0.4) × 10−4 s−1. N,N′,N′′,N′′′-Tetraaryl NHC–CDI adducts do not undergo such CDI exchange, even in the presence of a different N,N′-diaryl CDI (Fig. 7C), indicating that the adduct bond with N-aryl-N′-alkyl amidinates is dynamic at 50 °C, while the bond with N,N′-diaryl amidinates is not.
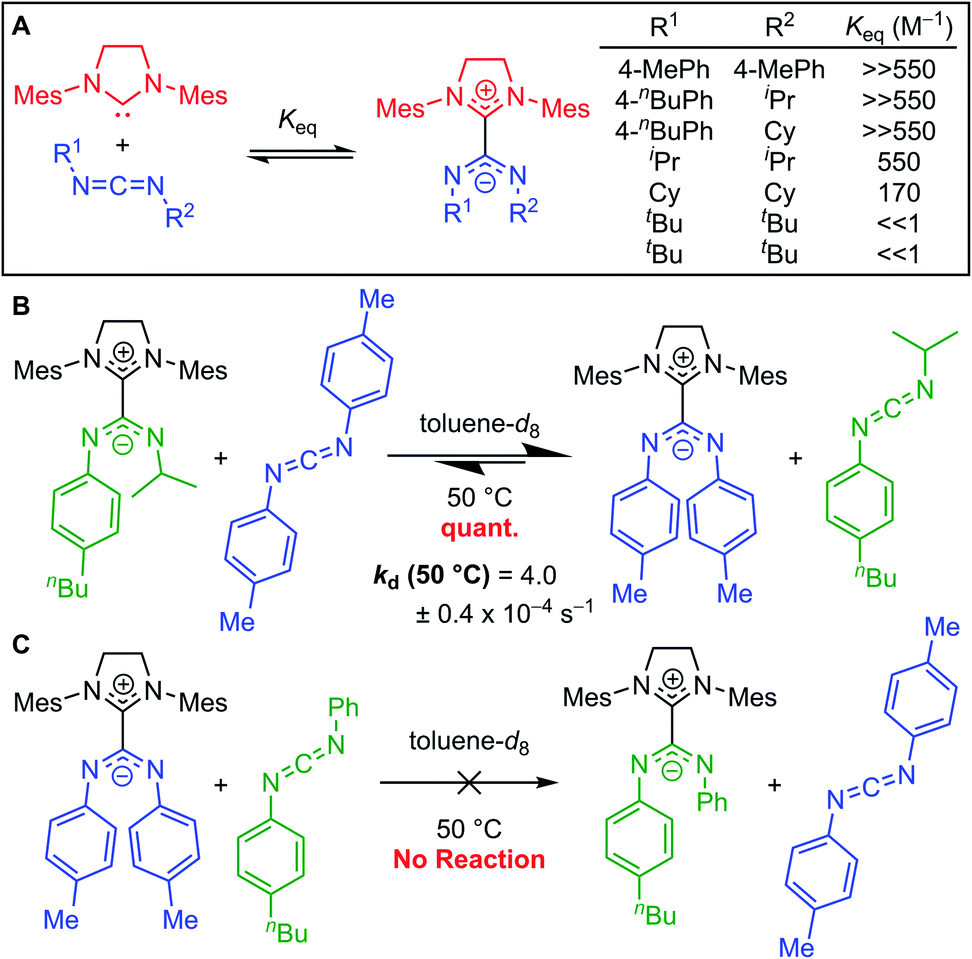 | ||
| Fig. 7 (A) Comparison of binding equilibria of SIMes with N,N′-diaryl, N-aryl-N′-alkyl, and N,N′-dialkyl CDIs. (B) Competitive binding experiment to show relative stability of NHC–CDIs with N-aryl-N′-alkyl and N,N′-diaryl amidinates. (C) Control experiment testing CDI exchange for two N,N′-diaryl CDIs. | ||
The dynamics of the central NHC–CDI bond also play a role in stability. While stable under inert atmospheres, NHC–CDI adducts with N-aryl-N′-alkyl amidinates are less stable to moisture compared to their N,N′-diaryl amidinate counterparts due to a more dynamic central C–C bond (see Section 3.3.1).33 Upon exposure to ambient conditions, the bis-adduct SIMesCDIiPr,Ph2 decomposes over 15 h as measured by changes in its 1H NMR spectra over time. Liquid chromatography mass spectrometry (LCMS) analysis of the resulting solution suggested a complex mixture of degradation products. These data indicate that while there is complete adduct formation according to 1H NMR spectroscopy, a small amount of free NHC is present at equilibrium, which reacts with moisture in the air, similar to MeIiPrCDIiPr. Notably, no such degradation was observed for the N,N′-diaryl amidinate analogues SIMesCDIMes,Ph2 and SIMesCDIpnBuPh,Ph2.
4. Applications
4.1 NHC–CDIs as ligands
Amidinates are π-electron rich chelating ligands, whose coordination chemistry has been extensively studied across the periodic table.17–22 These complexes have seen applications in homogenous catalysis46 (in particular, olefin polymerization19,47). Amidinates are the nitrogen-analogues of carboxylates, containing two nitrogen atoms that can coordinate to metals as monodentate, bidentate, or bridging ligands (Fig. 8).46,48,49 As bridging ligands, amidinates can generate bimetallic “paddlewheel” complexes in both trigonal and tetragonal modes.18,50–52 It is well known that tuning the amidinyl substituents can have a profound effect on the binding modes of these ligands.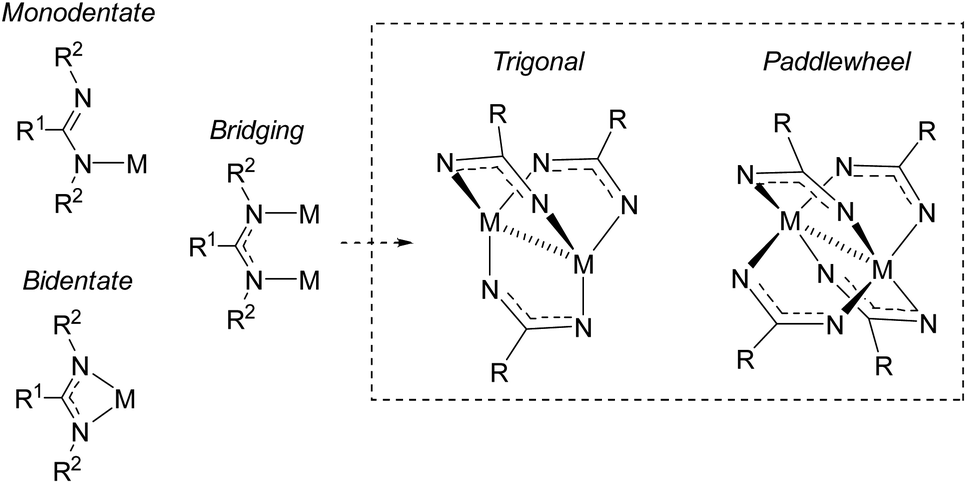 | ||
| Fig. 8 Coordination modes of amindinate ligands. R2 groups omitted from paddlewheel complexes for clarity. | ||
NHC–CDI adducts can act as analogues to amidinate ligands but with an electronically neutral character due to their zwitterionic structure. This neutrality imparts bench-stability to NHC–CDIs which sets them apart from monoanionic amidinate, guanidinate, or carboxylate ligand systems that can be moisture-sensitive. Similar to amidinate ligands, the substituents at the nitrogen atoms of the N2C− moiety of the NHC–CDI can be varied, allowing precise steric and electronic tuning of the binding site. Furthermore, the substituents at the nitrogen atoms of the N2C+ moiety can also be altered, affording further steric and electronic tuning of the ligand. This section will focus on studies of NHC–CDIs as ligands for discrete metal complexes (Section 4.1.1) and as stabilizing ligands for nanoparticle (NP) surfaces (Section 4.1.2).
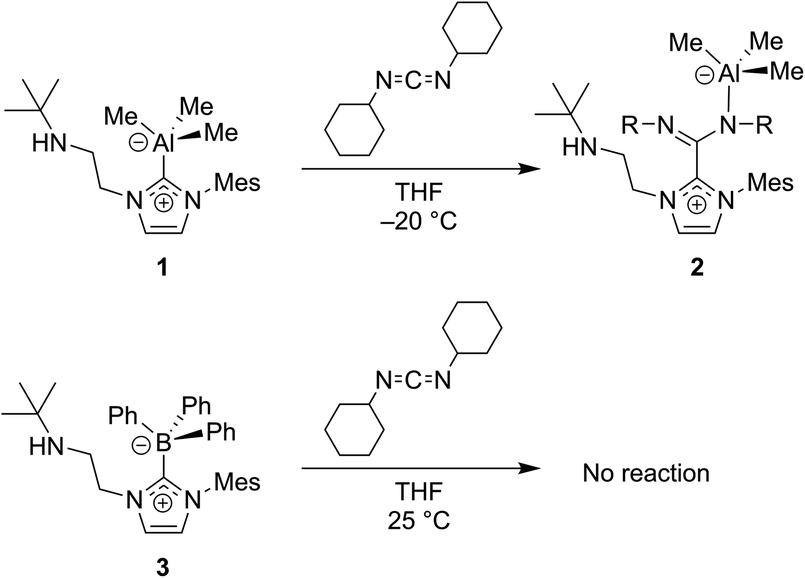 | ||
| Fig. 9 Carbodiimides such as CDICY can be used to probe the non-innocent bonding of NHCs to group 13 atoms.53 | ||
In 2015, Cámpora and co-workers demonstrated the first use of isolated NHC–CDI adducts as ligands, coordinated to Cu(I) metal centers.41 Three different NHC–CDI adducts were designed, containing either ICy or IMe as the NHC and CDIpTol or CDIDiPP as the CDI fragment. Similar to amidinate ligands, the authors show that the steric differences between CDIpTol and CDIDiPP can control the binding modes of the NHC–CDI adducts between terminally bound (monodentate) and bridging (Fig. 8).
Initially, the NHC–CDI adducts were combined with copper(I) acetate with the goal of generating bridged complexes similar to those seen in mixed acetate–amidinate complexes.54 An equimolar mixture of Cu(I) acetate and either DiPP adduct (ICyCDIDiPP or IMeCDIDiPP) lead to pale yellow-green 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (CuOAc)(L) complexes, with the NHC–CDI terminally bound to the copper atom (Fig. 10A, 4a and 4b). The two species were characterized using single crystal XRD and NMR experiments. The 1H and 13C{1H} NMR signals of the NHC–CDI ligands were sharp and defined, suggesting that the monodentate coordination of the amidinate ligands was maintained in solution. The less bulky CDIpTol provided a multinuclear assembly. Upon exposure of ICyCDIpTol to CuOAc, irrespective of reagent ratio, a trigonal paddlewheel complex (5a) formed, consisting of two copper centers bridged by three NHC–CDI ligands. It was also found that changing the counteranion to [BPh4]− had a negligible impact on the solid-state structure (5b). Unlike the yellow-green monometallic Cu(I) species discussed above, this complex exhibited an intense orange color. From the solid-state structure, a short Cu⋯Cu distance of 2.4123 Å was observed, indicating a closed shell (d10–d10) cuprophilic interaction. This interaction results in the strong, red-shifted color, with absorption bands assigned to metal–metal 3d → 4p transitions. The Cu⋯Cu bond distance is comparable to the shortest seen in binuclear Cu(I) complexes bridged by anionic amidinate or guanidinate anions (2.40–2.54 Å).55–57
:1 (CuOAc)(L) complexes, with the NHC–CDI terminally bound to the copper atom (Fig. 10A, 4a and 4b). The two species were characterized using single crystal XRD and NMR experiments. The 1H and 13C{1H} NMR signals of the NHC–CDI ligands were sharp and defined, suggesting that the monodentate coordination of the amidinate ligands was maintained in solution. The less bulky CDIpTol provided a multinuclear assembly. Upon exposure of ICyCDIpTol to CuOAc, irrespective of reagent ratio, a trigonal paddlewheel complex (5a) formed, consisting of two copper centers bridged by three NHC–CDI ligands. It was also found that changing the counteranion to [BPh4]− had a negligible impact on the solid-state structure (5b). Unlike the yellow-green monometallic Cu(I) species discussed above, this complex exhibited an intense orange color. From the solid-state structure, a short Cu⋯Cu distance of 2.4123 Å was observed, indicating a closed shell (d10–d10) cuprophilic interaction. This interaction results in the strong, red-shifted color, with absorption bands assigned to metal–metal 3d → 4p transitions. The Cu⋯Cu bond distance is comparable to the shortest seen in binuclear Cu(I) complexes bridged by anionic amidinate or guanidinate anions (2.40–2.54 Å).55–57
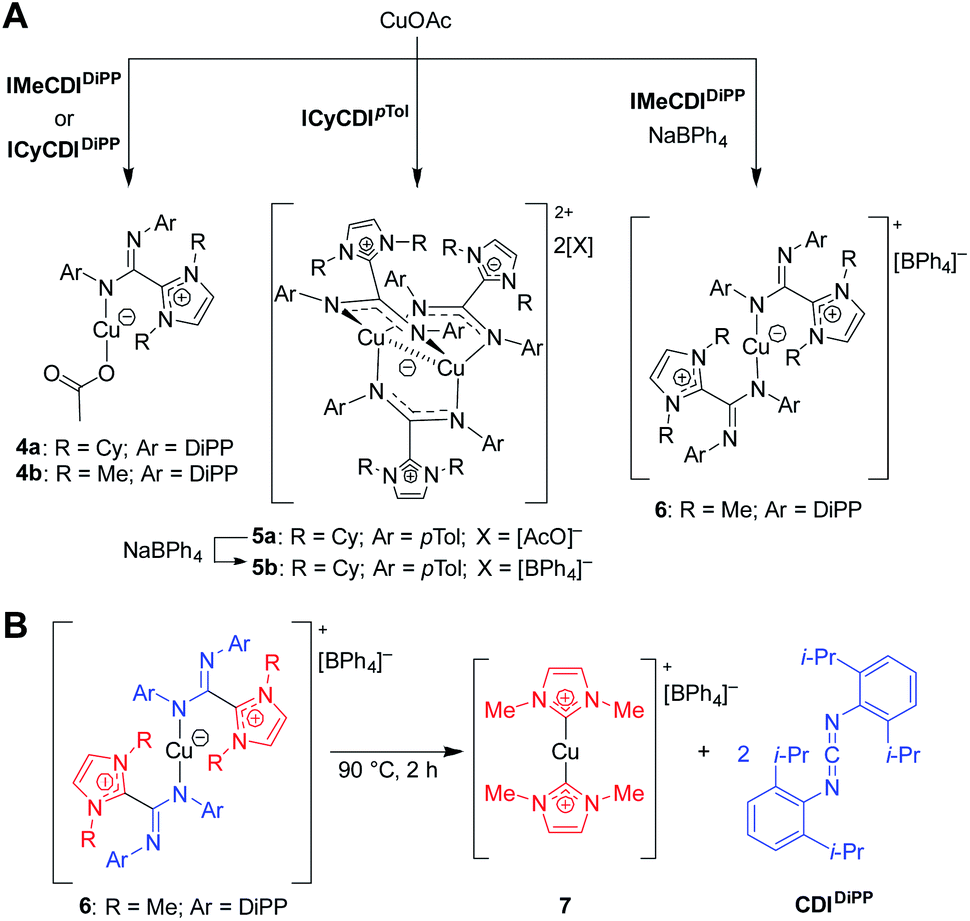 | ||
| Fig. 10 Copper complexes of NHC–CDIs.41 (A) Mono- and bimetallic complexes. (B) Heating complexes to 90 °C causes ejection of CDIDiPP fragment and formation of a bis-NHC Cu(I) complex. | ||
In order to form a binuclear product similar to 5a and 5b with a DiPP-substituted adduct, IMeCDIDiPP was exposed to CuOAc in the presence of NaBPh4. Regardless of whether a 1:1:1 or 2:3:2 ratio of CuOAc:IMeCDIDiPP:NaBPh4 was used, a mononuclear [CuL2][BPh4] species was isolated (6), which contained two terminally-bound NHC–CDI ligands. Interestingly, upon heating a solution of the complex to 90 °C, the CDI fragments were ejected as free carbodiimide, and a bis-NHC Cu(I) complex (7) was formed quantitatively. This thermal decomposition also occurred for the other species discussed, with varying degrees of selectivity.
Expanding on this work, in 2017 Nembenna and co-workers isolated the first N,N′-chelated NHC–CDI adducts as magnesium and zinc complexes.58 Five new complexes were isolated (Fig. 11) based on previously-reported,37 moisture-stable NHC–CDI adducts utilizing MeIEt and either 4-tert-butylphenyl or 4-isopropylphenyl substituents on the CDI. Initially, coordination with group 1 metals (where M = Li, Na, and K) was explored. While complexation was detected by 1H NMR spectroscopy, the desired products could not be isolated.
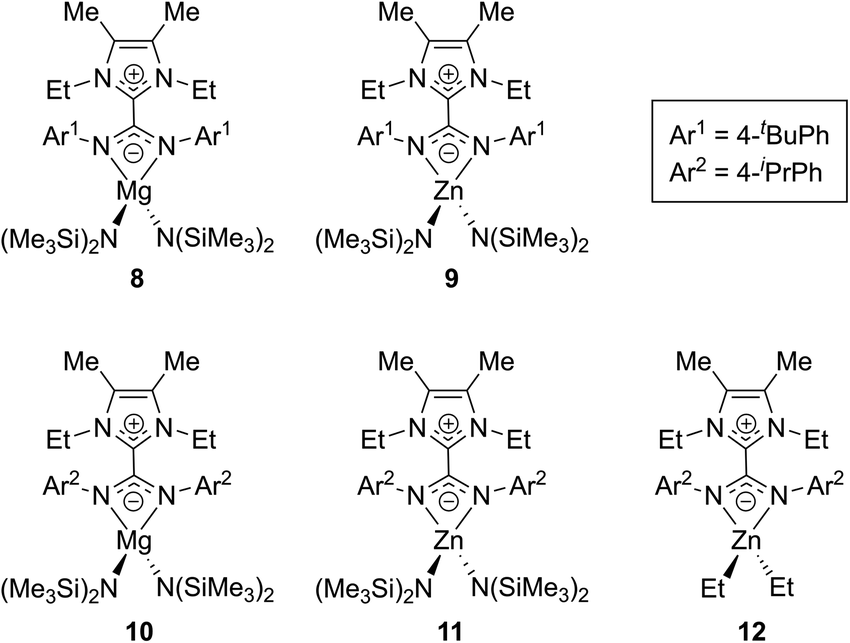 | ||
| Fig. 11 NHC–CDI complexes with Mg and Zn reported by Nembenna and co-workers.58 | ||
Complexes 8–11 were formed by exposure of the appropriate NHC–CDI to M{N(SiMe3)2}2 (M = Mg or Zn) in toluene at room temperature. Species 12 was prepared through the reaction of MeIEtCDIpiPrPh with ZnEt2. The complexes were characterized using multinuclear NMR experiments (1H, 13C, and 29Si) and single crystal XRD analysis, which all indicated N,N′-chelating binding modes for the NHC–CDIs to form four-membered rings with the metal. Ligand displacement reactions of compounds 8 and 9 were probed using neutral-type ligands (NHCs and isocyanides). No displacement of the NHC–CDI was seen, indicating a greater stability of the zwitterionic N,N′-chelate compared to neutral monodentate ligands.
Complex 8 was then tested for its utility as a carbonyl hydroboration catalyst; main-group metal amidinate complexes are known to catalyze this reaction.59 An equimolar ratio of 2-chlorobenzaldehyde and pinacolborane was exposed to 5 mol% of 8 in benzene-d6 at room temperature. Quantitative conversion of the aldehyde to the boronate ester was seen in 2 h, signifying, to our knowledge, the first use of an NHC–CDI metal complex as a catalyst.
In 2018, Cámpora, Mosquera, and co-workers reported a remarkable case of DCM activation when investigating the coordination chemistry of ICyCDIpTol with ZnCl2 (Fig. 12).60 Initially, the complexation reaction progressed very slowly at room temperature despite the strong basicity of the NHC–CDI adduct. After four days, the 1H NMR spectrum of the solution showed a large decrease in ICyCDIpTol, accompanied by the growth of two new species. Some crystalline material was seen; however, the crystal quality was not good enough to produce a solid-state structure. In a case of serendipity while setting up an NMR experiment, ZnCl2 was added to a week-old CD2Cl2 solution of ICyCDIpTol. The 1H NMR spectrum indicated that one of the new species was more prevalent, and crystals were grown from this solution. Single crystal XRD revealed the structure to be a methylene-bridged aminal counterbalanced by the binuclear anion [Zn2Cl6]2− (13), suggesting that 2 equivalents of ICyCDIpTol underwent nucleophilic attack onto DCM in an SN2 fashion with concomitant loss of two equivalents of chloride. Interestingly, both amidine units, N(Ar)–CN(Ar), adopt an E,E-configuration in 13 (Fig. 12B), while the free NHC–CDI adduct prefers a E,Z-geometry. This unusual geometry is due to intramolecular CH⋯N hydrogen bonds between the bridging methylene and CDI nitrogen atoms. In order to determine the second species seen in the 1H NMR spectrum, ICyCDIpTol was exposed to ZnCl2 in a 3:2 ratio in bromobenzene, a solvent that is less susceptible to nucleophilic attack. Through NMR and XRD studies, the species was determined to be a [ZnL3]2+ dication counterbalanced by a [ZnCl4]2− dianion (14, Fig. 12A).
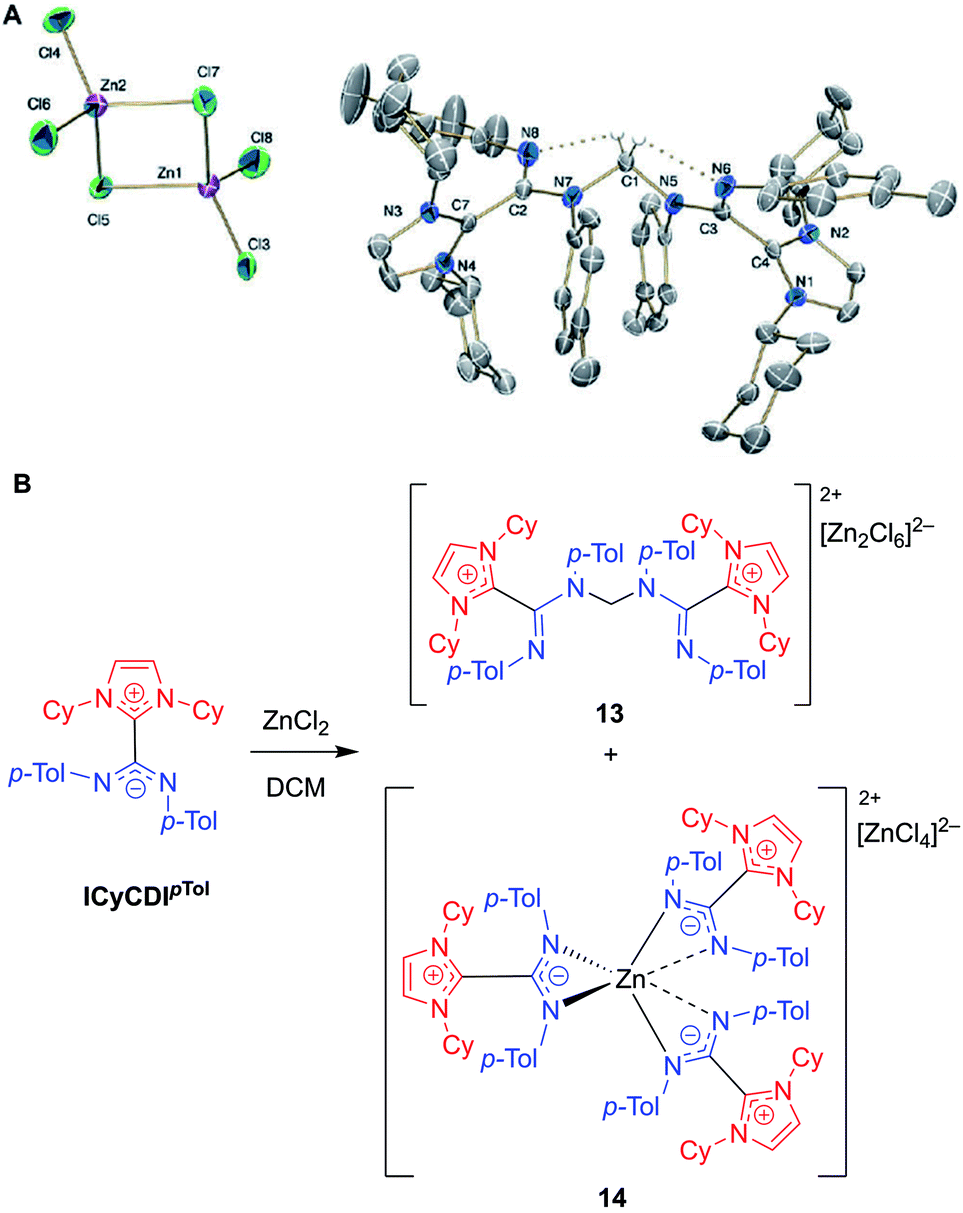 | ||
| Fig. 12 (A) Single crystal structure of methylene-bridged aminal 13. Hydrogen bonds shown in dotted lines. Ellipsoids plotted at 30% probability. Reproduced from ref. 60 with permission from The Royal Society of Chemistry. (B) Reaction of NHC–CDI ICyCDIpTol with ZnCl2 in DCM, as reported by Cámpora and Mosquera.60 (Cy = cyclohexyl, p-Tol = 4-methylphenyl, DCM = dichloromethane). | ||
It was initially thought that the Lewis acidic ZnCl2 was assisting ICyCDIpTol in the nucleophilic activation of dichloromethane; however, the preferential formation of 13 from “aged” solutions of ICyCDIpTol suggested that the ZnCl2 merely acts to trap the formed cation. Monitoring a solution of ICyCDIpTol in CD2Cl2 over the course of a week showed slow formation of the methylene-bridged product with no observed intermediates. Additionally, ICyCDIpTol was shown to react with other polychloroalkane solvents, such as chloroform and 1,2-dichloroethane, eliminating HCl and generating ICyCDIpTolH+˙Cl−, with protonation occurring at one of the amidinyl nitrogen atoms.
Due to the unusual nature of this reaction, a DFT model was built using a simplified version of the NHC–CDI adduct, using Me instead of Cy in the wingtip position. The rate-determining step was ascribed to be the initial SN2 attack of the adduct on dichloromethane, with a 3 kcal mol−1 reduction of the energy barrier if ICyCDIpTol rearranges from the E,Z to E,E configuration. The calculated energy barrier (28.0 kcal mol−1) to the first transition state was found to be in very good agreement with the experimental barrier (27.8 kcal mol−1). The [NHC–CDI–CH2Cl]+ intermediate then undergoes a second SN2 reaction with another NHC–CDI to give the bridged species. This work sets the stage for future work involving NHC–CDI adducts as nucleophiles.
In 2015, Cámpora, Philippot, Chaudret, and co-workers explored the stabilization of ruthenium NPs (RuNPs) using NHC–CDIs as stabilizing ligands with different Ru:ligand ratios.27 It is difficult to control the size of RuNPs in the range of 1 nm, with the vast majority of NPs reported in the 1.5–1.8 nm range, depending on the stabilizer used.82 Metal-to-ligand ratio is commonly used to control NP size, with increased ligand density typically leading to smaller NPs. Thus, the authors hypothesized that the large steric protection afforded by NHC–CDIs could lead to smaller NPs.
Toward this end, ICyCDIpTol was added to THF solutions of Ru(COD)(COT) (COD = 1,5-cyclooctadiene, COT = 1,3,5-cyclooctatriene) at room temperature under 3 bar H2 (Fig. 13A) with ligand:Ru ratios of 0.1, 0.2 and 0.5. L1(0.1)@Ru, L1(0.2)@Ru, and L1(0.5)@Ru had mean particle diameters of 1.3 nm, 1.0 nm, and 1.0 nm, respectively, as determined by transmission electron microscopy (TEM) and radial distribution function (RDF) analysis. Increasing the ligand from 0.1 to 0.2 equiv. resulted in the expected NP size reduction, but a further increase to 0.5 equiv. had no effect on the RuNP diameter due to steric hindrance.83
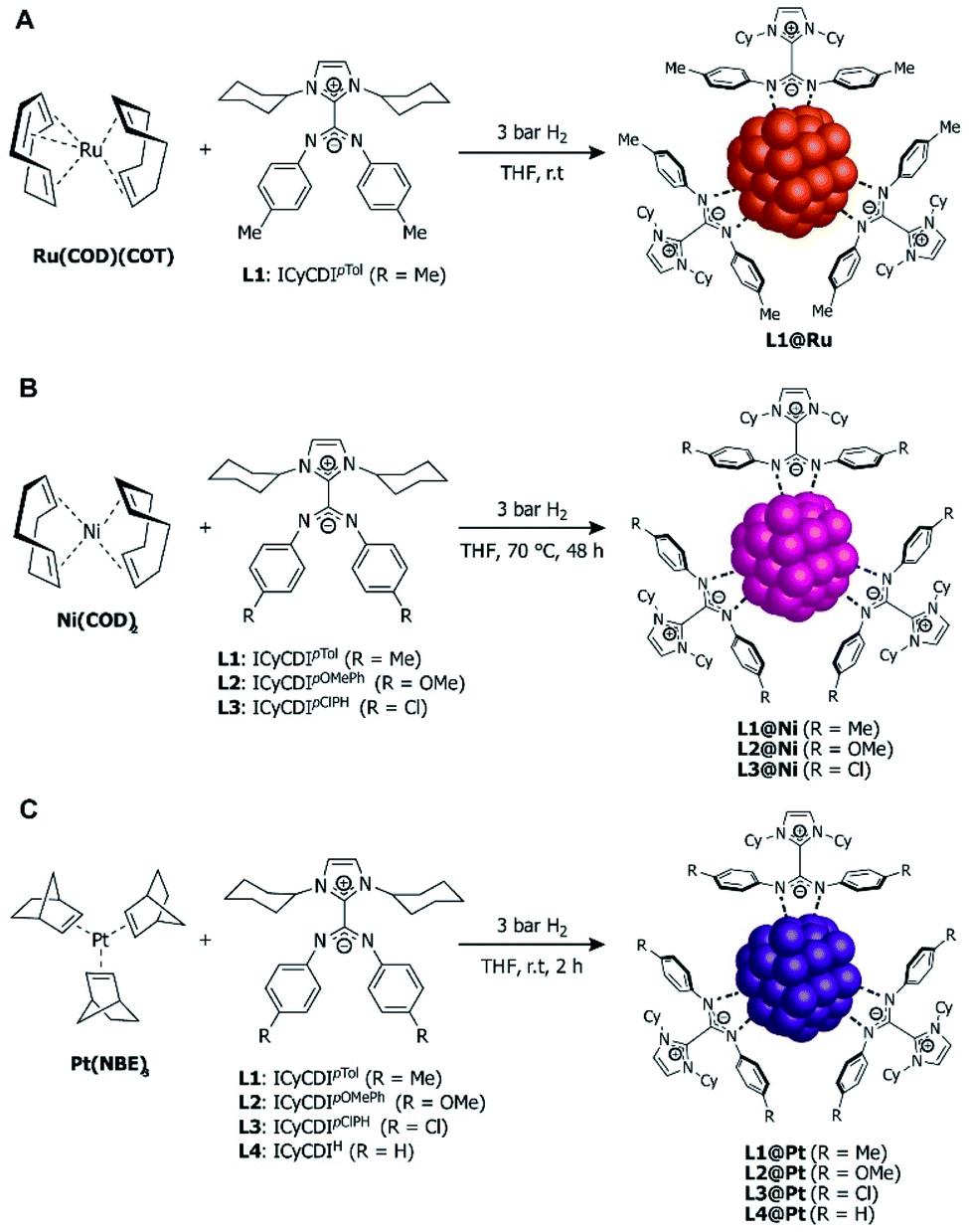 | ||
| Fig. 13 Synthesis of NHC–CDI-stabilized nanoparticles. (A) Ru(COD)(COT) to form RuNPs,27 (B) Ni(COD)2 to form NiNPs,42 and (C) Pt(NBE)3 to form PtNPS.30 | ||
To test the coordination stability of the NHC–CDI ligands, L1(0.1)@Ru was exposed to 10 equiv. of octanethiol and the mixture was heated at 65 °C in C6D6 for 24 h. 1H and 13C{1H} NMR spectroscopy did not show the presence of free NHC–CDI in the solution, indicating that, assuming free NHC–CDI would be stable under the reactions conditions, the strongly coordinating thiol was unable to displace the ligand to a discernible amount on the NMR timescale. Di-n-octyl disulfide, however, was observed, due to the catalytic oxidation of the octanethiol at the metal surface, suggesting that free Ru sites are still accessible as further confirmed by CO binding experiments.
Metal nanoparticles are attractive for catalysis because the facile tuning of both the electronic configuration and surface area of the particle combine the advantages of homogenous and heterogenous catalysts.84 They frequently display high catalytic activities and can often be reclaimed at the end of the reaction. RuNPs are known to catalyze hydrogenation of alkenes and arenes under mild conditions; thus, L1(0.1)@Ru and L1(0.2)@Ru were tested for their performance in styrene hydrogenation to probe the selectivity between vinyl and aromatic moieties. With L1(0.1)@Ru, full hydrogenation to ethylcyclohexane was observed after 24 h, while L1(0.2)@Ru gave a mixture of 26:74 ethylbenzene and ethylcyclohexane over the same time period. Therefore, L1(0.2)@Ru has greater selectively for vinyl hydrogenation over L1(0.1)@Ru, attributed to fewer faces present in L1(0.2)@Ru compared to L1(0.1)@Ru.
In 2020, Martínez-Prieto, van Leeuwen, and co-workers prepared nickel nanoparticles (NiNPs) stabilized by three different NHC–CDI ligands42 featuring ICy and para-substituted phenyl groups at the amidinyl nitrogens (R = Me, OMe, Cl for L1, L2 and L3, respectively). NiNPs were formed by exposing Ni(COD)2 to 0.2 equiv. of the appropriate ligand in THF at 70 °C under H2 at 3 bar (Fig. 13B). The resultant spherical L1@Ni and L2@Ni NPs had sizes of ∼2.8 nm, while L3@Ni NPs were larger (3.4 nm) and considerably more disperse. Structural characterizations were performed using high-resolution TEM, wide-angle X-ray scattering (WAXS), powder X-ray diffraction (pXRD), and X-ray photoelectron spectroscopy (XPS).
In order to screen the catalytic properties of the NiNPs, L1@Ni was tested in the hydrogenation of 8 substrates containing different functional groups (alkenes, alkynes, and carbonyl groups). The hydrogenation reactions proceeded in toluene at room temperature and utilized 3 mol% Ni. Double and triple carbon–carbon bonds both underwent hydrogenation, with 3-hexyne and diphenylacetylene displaying high selectivities for the (Z)-alkenes and only traces of the (E)-alkenes and corresponding alkanes. Carbonyls were not hydrogenated to a significant extent under the same conditions.
Electronic variations at the amidinate moiety (R = Me, OMe, Cl) were tested in the context of 3-hexyne hydrogenation in toluene. At long reaction times (8 h), all three species showed >99% conversion, with selectivity (78–90%) to produce the (Z)-olefin. At shorter reaction times (2–6 h), lower conversions were seen, with the more electron-withdrawing species L3@Ni (R = Cl) displaying the lowest activity. L3@Ni was, however, found to have a slightly higher selectivity towards the formation of (Z)-3-hexene, possibly due to its lower activity. The catalytic activity of these NiNPs are higher than those of other Ni-based systems previously reported.85 Notably, the NiNPs could be easily removed from solution with a magnet and recycled up to 3 times while maintaining good activity.
In 2020, Cámpora, van Leeuwen, and co-workers30 utilized the modularity of NHC–CDIs to synthesize Pt nanoparticles (PtNPs) of varying diameters in order to experimentally probe the effect of NP size on the Knight shift, which is a feature in the NMR frequency of a paramagnetic species caused by an interaction between nuclear spins and the spins of conduction electrons.86,87 Knight shifts occur for metal NPs due to the presence of free electrons on the surface; they can complicate the NMR chemical shifts of both the metallic isotope and the ligands bound to the surface. “Ultra-small” (<1 nm) Pt nanoparticles, however, do not exhibit a Knight shift due to the absence of free electrons on the surface and, thus, behave similarly to molecular species.88
The PtNPs were stabilized using similar NHC–CDI adducts, consisting of ICy as the NHC fragment and para-substituted phenyl groups on the CDI nitrogens (R = Me, OMe, Cl, H for L1, L2, L3 and L4, respectively).27,41 PtNPs were formed by reaction of Pt(NBE)3 (NBE = 2-norbornene) in THF under 3 bar H2 with the appropriate NHC–CDI adduct (Fig. 13C). Pt(NBE)3 is advantageous over typical precursors, such as Pt2(DBA)3 (DBA = dibenzylideneacetone) or Pt(CH3)2(COD), due to faster reaction times and the ability to remove norbornene under vacuum. The PtNPs were synthesized with differing L1:Pt ratios of 0.1, 0.2 and 0.5 molar equiv. (L1(0.1)@Pt, L1(0.2)@Pt, and L1(0.5)@Pt), giving particle diameters of 2.3 nm, 2.1 nm, and 1.9 nm, respectively, as determined by TEM. Crystallinity was confirmed using WAXS. As expected, elemental analysis (EA) indicated that the NHC–CDI adducts remained intact on the NP surface. FTIR studies comparing the strong stretching CN bands of the free adduct L1 at 1530 and 1495 cm−1 and the particle L1(0.5)@Pt at 1630 and 1598 cm−1 suggested that the ligand binds through the amidinate moiety.
Surface properties of the PtNPs were probed using solid state NMR spectroscopy by exposing the nanoparticles to 13CO and then obtaining the 13C MAS NMR spectrum to monitor the Knight shift. The paramagnetic effect caused by surface electrons of the NP cause the 13C signals of the CO to be shifted downfield and broadened considerably (the Knight shift). As the size of the NP is decreased from 2.3 nm to 1.9 nm, a small decrease in the magnitude of the Knight shift was observed. Regardless, in all cases, the large, broad Knight-shifted CO signals (∼100–600 ppm, centered ∼400 ppm) obscure any other resonances in the spectrum. As a correlation was seen between NP size and the magnitude of the Knight shift on the CO band, the authors prepared PtNPs of average ∼1.2 nm diameter using Pt2(DBA)3 as the platinum source in a ligand:Pt ratio of 0.2 molar equiv. in order to suppress the Knight-shifted signal. Following exposure to 13CO, the 13C MAS NMR showed three clear CO resonances between 190–230 ppm. The Knight-shifted CO resonance is heavily suppressed, though still present due to CO coordinated to larger PtNPs present in the sample. Additional FTIR studies showed that CO is adsorbed to the Pt surface in both bridging and terminal coordination modes.
It is important to note here the similarities in NP synthesis between the three reports discussed (Fig. 13). All NHC–CDI adducts were synthesized using methods discussed in 3.1.3 and differ only in the substituent at the para-position of the phenyl groups attached at the CDI nitrogens. Irrespective of the metal precursor used, all NPs were formed by reacting the metal complex with the NHC–CDI in THF under 3 bar H2 and purified by precipitation with pentane. This straightforward methodology, coupled with the subtle structural and electronic effects afforded by NHC–CDIs, allows for fine-tuning of NP size and catalytic activity and may find utility in metal nanoparticle and metal nanocluster applications at the frontier of molecular and solid-state chemistry.
4.2 NHC–CDIs in zwitterionic materials
Zwitterionic polymers comprise macromolecules that contain both cationic and anionic groups either along the main chain or incorporated into the side chain. These polymers have gained interest across a variety of fields due to their biocompatibility, antifouling properties, high ionic conductivity, and ability to stabilize nanoparticles and proteins.89 Zwitterionic polymers can be prepared from mixtures of oppositely charged monomers or from zwitterionic monomers.89,90 The vast majority of the latter materials are made with pendant sulfobetaine, carboxybetaine, or phosphobetaine moieties,89–91 though main-chain polybetaines have also been synthesized from phospholipids,92,93 polysquaraines,94 and NHC-isothiocyanate adducts.95In 2018, Johnson and co-workers reported a new class of polybetaines based on NHC–CDI adducts called poly(azolium amidinate)s (PAzAms, Fig. 14).33 The dative-character of the NHC–CDI adduct bond (see Section 3.3.1) makes PAzAms supramolecular polymers where the equilibrium of adduct formation (Keq) and rate of dissociation (kd) will affect the material properties. NHC–CDIs are good candidates for making such supramolecular polymers due to their high modularity leading to control over Keq, kd, adduct geometry, charge delocalization, air- and moisture-stability, and the potential to use the resulting amidinate-type fragments for metal ion ligation.
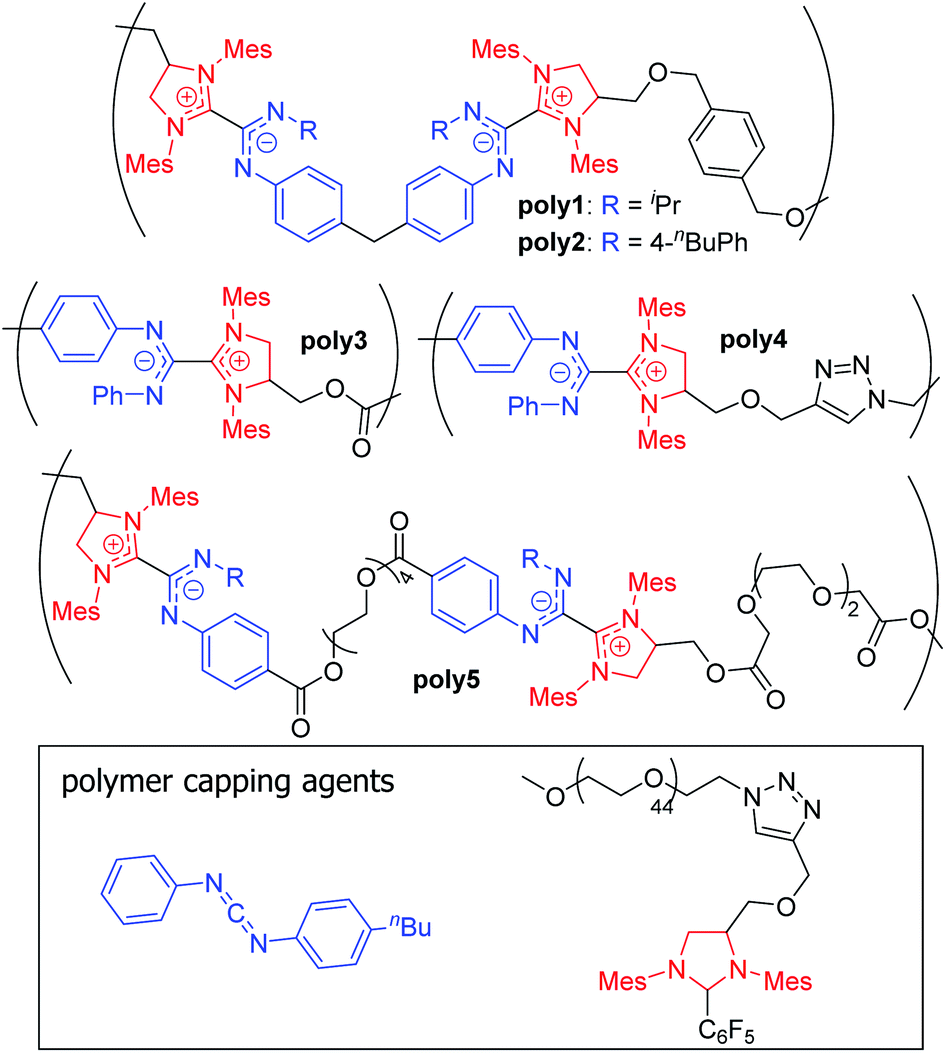 | ||
| Fig. 14 Structures of previously synthesized poly(azolium amidinates) (PAzAms).33 (Mes = 2,4,6-trimethylphenyl). | ||
Initially, two polymers were made via the step-growth polymerization of bis-imidazolidinium salts and bis-CDIs (AA + BB type monomers, poly1 and poly2 in Fig. 14). On the basis of different adduct stabilities in small molecule NHC–CDIs (see Section 3.3.1), PAzAms were synthesized from both bis-(N-aryl-N′-alkyl CDI)s and bis-(N,N′-diaryl CDI)s. Number-average molar masses (Mn) of ∼14.0 kDa for poly1 and ∼6.5 kDa for poly2 were obtained by 1H NMR spectroscopy, which corresponds to degrees of polymerization (DP) of ∼11 and ∼4, respectively. Gel permeation chromatography (GPC) traces exhibited broad dispersities (Đ) expected from a step-growth polymerization. It was found that the preparative procedure for these polymers greatly affected their molar masses; filtering the salt byproduct after deprotonation of the imidazolidinium salt resulted in low DP (DPNMR = 2–4) for both polymers, perhaps due to considerable loss of the bis-NHC during filtration. Removing this filtration step resulted in higher DP for both poly1 (DPNMR = 11) and poly2 (DPNMR = 9), however, the GPC trace of poly2 was irregular and displayed a very broad dispersity (Đ = 2.87), likely due to aggregative effects. When bis-NHC pentafluorobenzene adducts were used as the monomer, DP = 8 poly2 was isolated and displayed a more typical GPC trace, but the polymer was not very soluble in THF. Therefore, the lower DP oligomers of poly2 formed using the filtration procedure were used for initial investigations.
The dynamics of poly1 imparted by the N-aryl-N′-alkyl CDI were probed in many ways. First, depolymerization was instigated through heating to 50 °C in the presence of a free N,N′-diaryl CDI (CDIpTol). First-order kinetics suggested that this process is rate-limited by NHC–CDI adduct dissociation followed by rapid trapping of the free carbene by N,N′-diaryl CDI. Second, poly1 could be converted to poly2 in the presence of CDIpBuPh,Ph2 at 50 °C for 12 h. Complete CDI exchange occurs as the thermally-labile poly1 is converted to the stable poly2. Finally, the stability of poly1 to ambient conditions was tested. While poly1 seems indefinitely stable in solution under inert atmosphere, significant decomposition upon exposure to air was documented via1H NMR spectroscopy, GPC, and LCMS over the course of 18–24 hours. This was explained via degradation of the small fraction of free NHCs present at equilibrium. In contrast, poly2 was very stable to both depolymerization and ambient conditions. Depolymerization could be achieved, but only at 100 °C in molten CDIpTol.
The air- and moisture-stability of poly2 prompted further exploration of N,N′-diaryl CDIs in the context of supramolecular polymers. Heat-activated pentafluorophenyl-masked NHCs were designed to obviate the need for deprotonation of imidazolidinium salts by strong base (see Section 3.1.1). Both AA + BB type and AB type monomers were synthesized, showing the power of this methodology to access both pairwise alternating (+ + − −) and purely alternating (+ −) charge sequences. Monofunctional end caps were utilized to control the molecular weight of the PAzAms and/or introduce end functionality to the chains. Due to perfect 1:1 stoichiometry, AB monomers facilitated higher DP polymers of ≥25 for poly3 and ∼16 for poly4. As expected, increasing the amount of chain-capping agent leads to progressively lower DPs. The DP for AA + BB oligomers could be controlled via monomer ratio, varying between 2 and 8. Generally, hydrophobic linkers led to higher DPs compared to polymers with hydrophilic linkers (poly5), possibly due to residual moisture in hydrophilic monomers leading to unwanted termination events. As expected, all N,N′-diaryl CDI-derived PAzAms displayed excellent stability under ambient conditions, which was tested by heating to 70 °C in 1,2-dichloroethane-d4 under air. The solid material could be annealed at 100 °C for 24 h under N2 with no significant change in Mn; however, in the presence of free CDIpTol, depolymerization takes place under these conditions, underscoring the supramolecular nature of these polymers. Thermogravimetric analysis (TGA) also supports high thermal stability, as mass loss was not observed until ∼175–200 °C.
Most of the reported PAzAms were quite hydrophobic; in order to test the stability of these polybetaines in aqueous environments, a polyethyleneglycol (PEG)-containing capping agent was utilized to increase water solubility. The ratio of hydrophilic to hydrophobic blocks was controlled by the amount of capping agent added. The following samples were prepared: PEG2k-b-(AzAm)4, PEG2k-b-(AzAm)8, and PEG2k-b-(AzAm)25 (AzAm = the repeat unit of poly3) and all exhibited good water solubility (5–30 mg mL−1). 1H NMR spectroscopy in D2O showed evidence of aggregation-induced peak broadening as expected for amphiphilic block copolymers;96 however, the material could be extracted back into organic solvents without showing any significant changes compared to the native sample. These data indicate that PAzAms are stable in aqueous environments for at least several hours at room temperature. The aggregation of these materials was further investigated using dynamic light scattering (DLS) and TEM after dialysis into water from acetone. DLS showed a Gaussian size distributions and average particle diameters ranging from 66–100 nm; no correlation between particle size and hydrophobic block size was observed, perhaps due to the high dispersities (1.47–1.85) of the materials. TEM revealed that most of the aggregates had spherical morphologies of similar sizes to those measured by DLS except for a small amount of elongated worm-like morphologies for PEG2k-b-(AzAm)8. This work lays the foundation for future materials and tunable surface modification based on PAzAm zwitterionic polymers. Recently, Larsen and co-workers were inspired by these dynamic bonds to develop thermal guanidine-based covalent adaptable networks.97
4.3 Distonic radical cation stabilization
Radicals play critical roles in biological and chemical processes, and efforts to harness the reactivity of organic radicals has led to applications in organic synthesis,98,99 energy storage,100,101 biological imaging,102,103 and in polymerization reactions.104,105 It is well studied in the literature that carbenes – in particular NHCs – have been used to stabilize a wide range of main-group radicals106,107 due to their strong σ-donating and π-accepting properties. These species include boryl,108 silyl,109 pnictogenyl,110,111 and aluminium-bound adducts.112 Aminyl radicals that have been stabilized by NHCs113,114 were extensively delocalized into the empty p-orbital of the carbene. Through leveraging the unique electronic properties of neutral, zwitterionic NHC–CDIs, Johnson and co-workers investigated an alternative mode of radical stabilization that does not rely on the π-accepting ability of the carbene in the hopes that these stabilized radical species could be used for a range of redox-active materials.44In this work, pyrene-substituted SIMesCDIpyr is oxidized to generate a “distonic” radical cation (SIMesCDIpyr˙+), i.e., a radical cation where the charge and spin occupy spatially distinct regions within the same molecule (Fig. 15). The positive charge resides on the NHC moiety, while the unpaired electron is stabilized across the N-pyrene substituents. As a comparison, analogous phenyl-derived amidinate species were also synthesized; however, as expected, the generated radical cations of these compounds were too reactive to be observed at room temperature due to insufficient resonance delocalization, causing the compounds to quickly degrade.
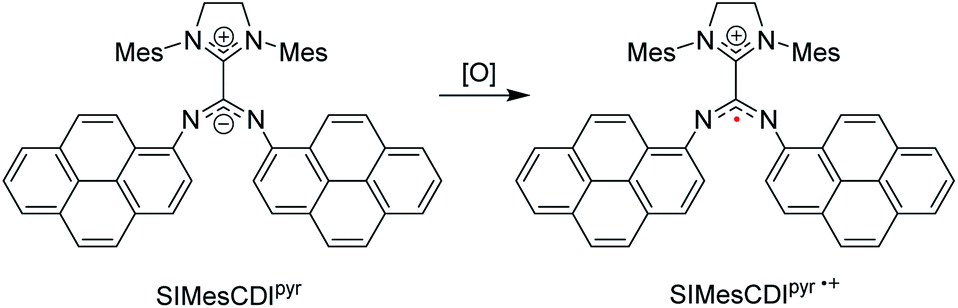 | ||
| Fig. 15 NHC–CDI distonic radical cation reported by Johnson and co-workers.45 (Mes = 2,4,6-trimethylphenyl, [O] = oxidant). | ||
The electrochemical properties of the species were studied using cyclic voltammetry (CV). The SIMesCDIpyr showed a reversible one-electron oxidation, corresponding to the distonic radical cation. The phenyl species, however, displayed irreversible oxidation in their CVs, suggesting a stabilizing influence of the pyrene moieties. Chemical oxidation of the SIMesCDIpyr with silver bistriflimide (AgTFSI) also formed the distonic radical cation, and the extensive delocalization of the unpaired electron across the pyrene groups was observed using electron paramagnetic resonance (EPR) spectroscopy. The EPR spectrum was simulated with the unpaired electron coupled to the two amidinyl nitrogens, which was in good agreement with experimental data and consistent with C2 symmetry in solution. Coupling to 10 protons—corresponding to the bound pyrene groups—was observed, with a significant spin density across each π-system. DFT calculations supported the assignment of a distonic radical cation, with the spin density and singly occupied molecular orbital (SOMO) occupying only the amidinate and pyrene groups on the adduct, with minor contributions from the cationic imidazolidinium moiety. The increased resonance stabilization afforded by the pyrene substituents allowed the distonic radical cation to exist even after exposure to air, with no significant change in the EPR spectral intensity observed. The phenyl-substituted analogues with reduced π-systems, however, showed dimerization after chemical oxidation.
Redox applications are a potential use for this new type of persistent, distonic radical cation. To demonstrate an example, the authors used SIMesCDIpyr as a catholyte (i.e., the electrolyte on the cathode side of an electrochemical cell) in an organic redox-flow battery, with N-methylphthalimide (N-MPI) as the anolyte. During the first charging cycle, the distonic radical showed a flat voltage plateau around 150 mV vs. Ag/Ag+. The species also demonstrated a high reversibility, with an average coulombic efficiency of 94.6% after 20 charging cycles and no performance decay for 100 cycles.
5. Conclusions and outlook
This Perspective is a comprehensive survey of the nascent field of NHC–CDI betaine adducts. While hypothesized as far back as the 1970s, it has only been the recent discovery of robust, air- and moisture-stable variants based on N,N′-diaryl CDIs that has sparked the rapid growth in this field. The highly modular nature of both NHCs and CDIs has enabled the synthesis of many adducts with electronic and steric modifications that allow the probing of structure–property relationships. Although all currently known adducts utilize imidazol-2-ylidene and imidazolidin-2-ylidene NHCs, other NHC classes – such as 1,2,4-triazol-3-ylidene, thiazol-2-ylidene, and cyclic alkylaminocarbenes – could result in even greater variety of properties and applications in the future.Coordination complexes containing NHC–CDIs as net-neutral amidinate-like ligands have been synthesized with terminal, chelating, and bridging binding modes. They can also stabilize a variety of metal nanoparticles. A few of these examples showed catalytic behavior, but given the breadth of reactions traditional amidinate metal complexes can catalyze17,19 – including hydroamination,115 olefin polymerization,116 and lactone polymerization117 – it seems inevitable that NHC–CDIs will become more popular in the future as versatile, electroneutral ligands for catalysts. The basicity that makes these adducts good ligands also contributes to their nucleophilicity, as shown by the reaction of ICyCDIpTol with DCM, hinting at the promise of appending these moieties to organic molecules. Building on these efforts and work done on zwitterionic polymers, NHC–CDIs present vast opportunities for nanoparticle and surface functionalization in the future. These modifications could be done with either functional small molecules or through the polymerization of PAzAms from a surface. Designing hydrophilic monomers for these supramolecular polymers will open up new regimes to explore, such as antibiofouling and biological properties. An example of an application that has already been realized is the bis(pyrene) NHC–CDI that could stabilize a distonic radical to be used as a catholyte in a redox flow battery.
Beyond the fields presented here, there is a remarkable opportunity for NHC–CDIs in the field of organocatalysis. The strong precedence of NHC betaine adducts as latent organocatalysts for Umpolung reactions or lactone polymerization poise NHC–CDI adducts as a more modular alternative to the common carboxylate adducts. It is clear that the NHC–CDI scaffold holds a lot of promise across a variety of fields and the positive attributes of facile synthesis, modularity, and robustness provide a desirable toolbox for exciting research for many years to come.
Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
We acknowledge the National Science Foundation (NSF CHE-1904867) for support of this work. J. R. L. gratefully acknowledges the National Institutes of Health for a postdoctoral fellowship (1F32GM126913-01A1).References
- A. J. Arduengo, R. L. Harlow and M. Kline, A Stable Crystalline Carbene, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS.
- A. Igau, H. Grutzmacher, A. Baceiredo and G. Bertrand, Analogous α,α′-Bis-Carbenoid Triply Bonded Species: Synthesis of a Stable λ3-Phosphinocarbene-λ5-Phosphaacetylene, J. Am. Chem. Soc., 1988, 110, 6463–6466 CrossRef CAS.
- V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, NHCs in Main Group Chemistry, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS.
- L. A. Schaper, S. J. Hock, W. A. Herrmann and F. E. Kühn, Synthesis and application of water-soluble NHC transition-metal complexes, Angew. Chem., Int. Ed., 2013, 52, 270–289 CrossRef CAS.
- M. G. Gardiner and C. C. Ho, Recent advances in bidentate bis(N-heterocyclic carbene) transition metal complexes and their applications in metal-mediated reactions, Coord. Chem. Rev., 2018, 375, 373–388 CrossRef CAS.
- D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Organocatalytic Reactions Enabled by N-Heterocyclic Carbenes, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS.
- K. V. S. Ranganath, S. Onitsuka, A. K. Kumar and J. Inanaga, Recent progress of N-heterocyclic carbenes in heterogeneous catalysis, Catal. Sci. Technol., 2013, 3, 2161–2181 RSC.
- A. V. Zhukhovitskiy, M. J. MacLeod and J. A. Johnson, Carbene Ligands in Surface Chemistry: From Stabilization of Discrete Elemental Allotropes to Modification of Nanoscale and Bulk Substrates, Chem. Rev., 2015, 115, 11503–11532 CrossRef CAS.
- C. A. Smith, M. R. Narouz, P. A. Lummis, I. Singh, A. Nazemi, C. H. Li and C. M. Crudden, N-Heterocyclic Carbenes in Materials Chemistry, Chem. Rev., 2019, 119, 4986–5056 CrossRef CAS.
- P. de Frémont, N. Marion and S. P. Nolan, Carbenes: synthesis, properties, and organometallic chemistry, Coord. Chem. Rev., 2009, 253, 862–892 CrossRef.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, An overview of N-heterocyclic carbenes, Nature, 2014, 510, 485–496 CrossRef CAS.
- In fact, this stabilization is so significant that the lowest energy resonance forms of NHCs have significant double bond character. Thus, NHCs may more appropriately be referred to as ylides.
- D. J. Nelson and S. P. Nolan, Quantifying and understanding the electronic properties of N-heterocyclic carbenes, Chem. Soc. Rev., 2013, 42, 6723–6753 RSC.
- L. Benhamou, E. Chardon, G. Lavigne, S. Bellemin-Laponnaz and V. César, Synthetic routes to N-heterocyclic carbene precursors, Chem. Rev., 2011, 111, 2705–2733 CrossRef CAS.
- W. Kirmse, Carbene complexes of nonmetals, Eur. J. Org. Chem., 2005, 237–260 CrossRef CAS.
- L. Delaude, Betaine adducts of N-heterocyclic carbenes: synthesis, properties, and reactivity, Eur. J. Inorg. Chem., 2009, 1681–1699 CrossRef CAS.
- F. T. Edelmann, Lanthanide amidinates and guanidinates in catalysis and materials science: a continuing success story, Chem. Soc. Rev., 2012, 41, 7657–7672 RSC.
- M. Cortijo, R. González-Prieto, S. Herrero, J. L. Priego and R. Jiménez-Aparicio, The use of amidinate ligands in paddlewheel diruthenium chemistry, Coord. Chem. Rev., 2019, 400, 213040 CrossRef CAS.
- S. Collins, Polymerization catalysis with transition metal amidinate and related complexes, Coord. Chem. Rev., 2011, 255, 118–138 CrossRef CAS.
- C. Sahin, A. Goren and C. Varlikli, Synthesis, characterization and photophysical properties of iridium complexes with amidinate ligands, J. Organomet. Chem., 2014, 772, 68–78 CrossRef.
- C. E. Hayes and D. B. Leznoff, Actinide coordination and organometallic complexes with multidentate polyamido ligands, Coord. Chem. Rev., 2014, 266–267, 155–170 CrossRef CAS.
- T. Chlupatý and A. Růžička, Hybrid amidinates and guanidinates of main group metals, Coord. Chem. Rev., 2016, 314, 103–113 CrossRef.
- A. V. Zhukhovitskiy, J. Geng and J. A. Johnson, Cycloelimination of imidazolidin-2-ylidene N-heterocyclic carbenes: mechanism and insights into the synthesis of stable ‘NHC-CDI’ amidinates, Chem.–Eur. J., 2015, 21, 5685–5688 CrossRef CAS.
- A. Takamizawa, S. Matsumoto and S. Sakai, Addition Reaction of 3-Substituted-4-methyl-5-(2-hydroxy)ethyl-thiazolium Salt with Diaryl Carbodiimide, Chem. Pharm. Bull., 1974, 22, 299–304 CrossRef CAS.
- F. Ramirez, J. F. Pilot, N. B. Desai, C. P. Smith, B. Hansen and N. McKelvie, A New Type of Stable Tetrapolar Phosphorus Ylide, J. Am. Chem. Soc., 1967, 89, 6273–6276 CrossRef CAS.
- N. Kuhn, M. Steimann, G. Weyers and G. Henkelh, 1,3-Diisopropyl-4,5-dimethylimidazolium-2-N,N’-diisopropylamidinat, ein neuartiges Betain [1], Z. Naturforsch., B: J. Chem. Sci., 1999, 54, 434–440 CAS.
- L. M. Martínez-Prieto, C. Urbaneja, P. Palma, J. Cámpora, K. Philippot and B. Chaudret, A betaine adduct of N-heterocyclic carbene and carbodiimide, an efficient ligand to produce ultra-small ruthenium nanoparticles, Chem. Commun., 2015, 51, 4647–4650 RSC.
- M. N. Hopkinson and F. Glorius, in N-Heterocyclic Carbenes in Organocatalysis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2019, pp. 1–35 Search PubMed.
- X. Bantreil and S. P. Nolan, Synthesis of N-heterocyclic carbene ligands and derived ruthenium olefin metathesis catalysts, Nat. Protoc., 2011, 6, 69–77 CrossRef CAS.
- L. M. Martínez-Prieto, I. Cano, A. Márquez, E. A. Baquero, S. Tricard, L. Cusinato, I. Del Rosal, R. Poteau, Y. Coppel, K. Philippot, B. Chaudret, J. Cámpora and P. W. N. M. Van Leeuwen, Zwitterionic amidinates as effective ligands for platinum nanoparticle hydrogenation catalysts, Chem. Sci., 2017, 8, 2931–2941 RSC.
- G. Rivera and R. H. Crabtree, in N-Heterocyclic Carbenes in Synthesis, John Wiley & Sons, Ltd, 2006, pp. 223–239 Search PubMed.
- N. Kuhn and T. Kratz, Synthesis of imidazol-2-ylidenes by reduction of imidazole-2(3H)-thiones, Synthesis, 1993, 561–562 CrossRef CAS.
- N. M. Gallagher, A. V. Zhukhovitskiy, H. V. T. Nguyen and J. A. Johnson, Main-Chain Zwitterionic Supramolecular Polymers Derived from N-Heterocyclic Carbene-Carbodiimide (NHC-CDI) Adducts, Macromolecules, 2018, 51, 3006–3016 CrossRef CAS.
- T. K. H. Trinh, J. P. Malval, F. Morlet-Savary, J. Pinaud, P. Lacroix-Desmazes, C. Reibel, V. Héroguez and A. Chemtob, Mixture of Azolium Tetraphenylborate with Isopropylthioxanthone: A New Class of N-Heterocyclic Carbene (NHC) Photogenerator for Polyurethane, Polyester, and ROMP Polymers Synthesis, Chem.–Eur. J., 2019, 25, 9242–9252 CrossRef CAS.
- S. Naumann and M. R. Buchmeiser, Liberation of N-heterocyclic carbenes (NHCs) from thermally labile progenitors: protected NHCs as versatile tools in organo- and polymerization catalysis, Catal. Sci. Technol., 2014, 4, 2466–2479 RSC.
- A. Williams and I. T. Ibrahim, Carbodiimide Chemistry: Recent Advances, Chem. Rev., 1981, 81, 589–636 CrossRef CAS.
- T. Peddarao, A. Baishya, M. K. Barman, A. Kumar and S. Nembenna, Metal-free access of bulky: N,N′-diarylcarbodiimides and their reduction: bulky N,N′-diarylformamidines, New J. Chem., 2016, 40, 7627–7636 RSC.
- A. R. Ali, H. Ghosh and B. K. Patel, A greener synthetic protocol for the preparation of carbodiimide, Tetrahedron Lett., 2010, 51, 1019–1021 CrossRef CAS.
- F. A. Cotton and P. A. Kibala, Reactions of Iodine with Triphenylphosphine and Triohenvlarsine, J. Am. Chem. Soc., 1987, 109, 3308–3312 CrossRef CAS.
- Y. R. Zhang, S. Aronson and P. G. Mennitt, Ionic dissociation in complexes of iodine with triphenylphosphine and triphenylamine, Can. J. Chem., 1992, 70, 2394–2397 CrossRef CAS.
- A. Márquez, E. Ávila, C. Urbaneja, E. Álvarez, P. Palma and J. Cámpora, Copper(I) Complexes of Zwitterionic Imidazolium-2-Amidinates, a Promising Class of Electroneutral, Amidinate-Type Ligands, Inorg. Chem., 2015, 54, 11007–11017 CrossRef.
- A. M. López-Vinasco, L. M. Martínez-Prieto, J. M. Asensio, P. Lecante, B. Chaudret, J. Cámpora and P. W. N. M. Van Leeuwen, Novel nickel nanoparticles stabilized by imidazolium-amidinate ligands for selective hydrogenation of alkynes, Catal. Sci. Technol., 2020, 10, 342–350 RSC.
- A. Baishya, L. Kumar, M. K. Barman, T. Peddarao and S. Nembenna, Air Stable N-Heterocyclic Carbene-Carbodiimide (“NHC-CDI”) Adducts: Zwitterionic Type Bulky Amidinates, ChemistrySelect, 2016, 1, 498–503 CrossRef CAS.
- N. M. Gallagher, H. Z. Ye, S. Feng, J. Lopez, Y. G. Zhu, T. Van Voorhis, Y. Shao-Horn and J. A. Johnson, An N-Heterocyclic-Carbene-Derived Distonic Radical Cation, Angew. Chem., Int. Ed., 2020, 59, 3952–3955 CrossRef CAS.
- N. M. Gallagher, H. Z. Ye, S. Feng, J. Lopez, Y. G. Zhu, T. Van Voorhis, Y. Shao-Horn and J. A. Johnson, An N-Heterocyclic-Carbene-Derived Distonic Radical Cation, Angew. Chem., Int. Ed., 2020, 59, 3952–3955 CrossRef CAS.
- F. T. Edelmann, Advances in the Coordination Chemistry of Amidinate and Guanidinate Ligands, Adv. Organomet. Chem., 2008, 57, 183–352 CrossRef CAS.
- T. Elkin and M. S. Eisen, Amidinate group 4 complexes in the polymerization of olefins, Catal. Sci. Technol., 2015, 5, 82–95 RSC.
- C. Jones, C. Schulten, R. P. Rose, A. Stasch, S. Aldridge, W. D. Woodul, K. S. Murray, B. Moubaraki, M. Brynda, G. La Macchia and L. Gagliardi, Amidinato- and guanidinato-cobalt(I) complexes: characterization of exceptionally short Co-Co interactions, Angew. Chem., Int. Ed., 2009, 48, 7406–7410 CrossRef CAS.
- L. C. Wu, C. W. Hsu, Y. C. Chuang, G. H. Lee, Y. C. Tsai and Y. Wang, Bond characterization on a Cr-Cr quintuple bond: a combined experimental and theoretical study, J. Phys. Chem. A, 2011, 115, 12602–12615 CrossRef CAS.
- C. M. Zall, D. Zherebetskyy, A. L. Dzubak, E. Bill, L. Gagliardi and C. C. Lu, A combined spectroscopic and computational study of a high-spin S = 7/2 diiron complex with a short iron-iron bond, Inorg. Chem., 2012, 51, 728–736 CrossRef CAS.
- F. A. Cotton, L. M. Daniels, D. J. Maloney, J. H. Matonic and C. A. Muirllo, Dicobalt trigonal lanterns: compounds containing the Co23+ core Co2[RC =(NPh)2]3 (R = H, C6H5) and an oxidized compound {Co2[HC(NPh)2]3(CH3CN)2}PF6, Inorg. Chim. Acta, 1997, 256, 283–289 CrossRef CAS.
- J. P. Krogman and C. M. Thomas, Metal–metal multiple bonding in C3-symmetric bimetallic complexes of the first row transition metals, Chem. Commun., 2014, 50, 5115–5127 RSC.
- C. Ligation, C. C. Tai, Y. T. Chang, J. H. Tsai, T. Jurca, G. P. A. Yap and T. G. Ong, Subtle reactivities of boron and aluminum complexes with amino-linked N-heterocyclic carbene ligation, Organometallics, 2012, 31, 637–643 CrossRef.
- A. Márquez, E. Ávila, C. Urbaneja, E. Álvarez, P. Palma and J. Cámpora, Copper(I) Complexes of Zwitterionic Imidazolium-2-Amidinates, a Promising Class of Electroneutral, Amidinate-Type Ligands, Inorg. Chem., 2015, 54, 11007–11017 CrossRef.
- A. C. Lane, C. L. Barnes, W. E. Antholine, D. Wang, A. T. Fiedler and J. R. Walensky, Di- and Trinuclear Mixed-Valence Copper Amidinate Complexes from Reduction of Iodine, Inorg. Chem., 2015, 54, 8509–8517 CrossRef CAS.
- M. Fan, Q. Yang, H. Tong, S. Yuan, B. Jia, D. Guo, M. Zhou and D. Liu, Synthesis and structural study of an unsymmetrical aliphatic benzamidinato ligand and its use in the formation of selected Cu(I), Mg(II) and Zr(IV) complexes, RSC Adv., 2012, 2, 6599–6605 RSC.
- C. Jones, C. Schulten, L. Fohlmeister, A. Stasch, K. S. Murray, B. Moubaraki, S. Kohl, M. Z. Ertem, L. Gagliardi and C. J. Cramer, Bulky guanidinato nickel(I) complexes: synthesis, characterization, isomerization, and reactivity studies, Chem.–Eur. J., 2011, 17, 1294–1303 CrossRef CAS.
- A. Baishya, L. Kumar, M. K. Barman, H. S. Biswal and S. Nembenna, N-Heterocyclic Carbene-Carbodiimide (‘NHC-CDI’) Adduct or Zwitterionic-Type Neutral Amidinate-Supported Magnesium(II) and Zinc(II) Complexes, Inorg. Chem., 2017, 56, 9535–9546 CrossRef CAS.
- S. Brand, A. Causero, H. Elsen, J. Pahl, J. Langer and S. Harder, Ligand Effects in Calcium Catalyzed Ketone Hydroboration, Eur. J. Inorg. Chem., 2020, 2020, 1728–1735 CrossRef CAS.
- D. Sánchez-Roa, T. G. Santiago, M. Fernández-Millán, T. Cuenca, P. Palma, J. Cámpora and M. E. G. Mosquera, Interaction of an imidazolium-2-amidinate (NHC-CDI) zwitterion with zinc dichloride in dichloromethane: role as ligands and C-Cl activation promoters, Chem. Commun., 2018, 54, 12586–12589 RSC.
- F. Atsushi and P. L. Dhepe, Sustainable green catalysis by supported metal nanoparticles, Chem. Rec., 2009, 9, 224–235 CrossRef.
- T. Ishida, T. Murayama, A. Taketoshi and M. Haruta, Importance of Size and Contact Structure of Gold Nanoparticles for the Genesis of Unique Catalytic Processes, Chem. Rev., 2020, 120, 464–525 CrossRef CAS.
- Y. H. Su, Y. F. Ke, S. L. Cai and Q. Y. Yao, Surface plasmon resonance of layer-by-layer gold nanoparticles induced photoelectric current in environmentally-friendly plasmon-sensitized solar cell, Light: Sci. Appl., 2012, 1, e14 CrossRef.
- F. Wang, R. Deng, J. Wang, Q. Wang, Y. Han, H. Zhu, X. Chen and X. Liu, Tuning upconversion through energy migration in core-shell nanoparticles, Nat. Mater., 2011, 10, 968–973 CrossRef CAS.
- M. Segev-Bar, A. Landman, M. Nir-Shapira, G. Shuster and H. Haick, Tunable touch sensor and combined sensing platform: toward nanoparticle-based electronic skin, ACS Appl. Mater. Interfaces, 2013, 5, 5531–5541 CrossRef CAS.
- W. Wu, Inorganic nanomaterials for printed electronics: a review, Nanoscale, 2017, 9, 7342–7372 RSC.
- O. V. Salata, Applications of nanoparticles in biology and medicine, J. Nanobiotechnol., 2004, 2, 3 CrossRef.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, The golden age: gold nanoparticles for biomedicine, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- X. Li, J. J. Lenhart and H. W. Walker, Aggregation kinetics and dissolution of coated silver nanoparticles, Langmuir, 2012, 28, 1095–1104 CrossRef CAS.
- R. A. Sperling and W. J. Parak, Surface modification, functionalization and bioconjugation of colloidal inorganic nanoparticles, Philos. Trans. R. Soc., A, 2010, 368, 1333–1383 CrossRef CAS.
- C. Xie, Z. Niu, D. Kim, M. Li and P. Yang, Surface and Interface Control in Nanoparticle Catalysis, Chem. Rev., 2020, 120, 1184–1249 CrossRef CAS.
- M. J. MacLeod, A. J. Goodman, H. Z. Ye, H. V. T. Nguyen, T. Van Voorhis and J. A. Johnson, Robust gold nanorods stabilized by bidentate N-heterocyclic-carbene–thiolate ligands, Nat. Chem., 2019, 11, 57–63 CrossRef CAS.
- M. J. MacLeod and J. A. Johnson, PEGylated N-Heterocyclic Carbene Anchors Designed to Stabilize Gold Nanoparticles in Biologically Relevant Media, J. Am. Chem. Soc., 2015, 137, 7974–7977 CrossRef CAS.
- A. V. Zhukhovitskiy, M. G. Mavros, T. Van Voorhis and J. A. Johnson, Addressable carbene anchors for gold surfaces, J. Am. Chem. Soc., 2013, 135, 7418–7421 CrossRef CAS.
- R. W. Y. Man, C. H. Li, M. W. A. MacLean, O. V. Zenkina, M. T. Zamora, L. N. Saunders, A. Rousina-Webb, M. Nambo and C. M. Crudden, Ultrastable gold nanoparticles modified by bidentate N-Heterocyclic Carbene Ligands, J. Am. Chem. Soc., 2018, 140, 1576–1579 CrossRef CAS.
- C. M. Crudden, J. H. Horton, I. I. Ebralidze, O. V. Zenkina, A. B. McLean, B. Drevniok, Z. She, H. B. Kraatz, N. J. Mosey, T. Seki, E. C. Keske, J. D. Leake, A. Rousina-Webb and G. Wu, Ultra stable self-assembled monolayers of N-heterocyclic carbenes on gold, Nat. Chem., 2014, 6, 409–414 CrossRef CAS.
- E. C. Hurst, K. Wilson, I. J. S. Fairlamb and V. Chechik, N-Heterocyclic carbene coated metal nanoparticles, New J. Chem., 2009, 33, 1837–1840 RSC.
- C. Richter, K. Schaepe, F. Glorius and B. Jan Ravoo, Tailor-made N-heterocyclic carbenes for nanoparticle stabilization, Chem. Commun., 2014, 50, 3204–3207 RSC.
- A. Ferry, K. Schaepe, P. Tegeder, C. Richter, K. M. Chepiga, B. J. Ravoo and F. Glorius, Negatively Charged N-Heterocyclic Carbene-Stabilized Pd and Au Nanoparticles and Efficient Catalysis in Water, ACS Catal., 2015, 5, 5414–5420 CrossRef CAS.
- K. V. S. Ranganath, J. Kloesges, A. H. Schäfer and F. Glorius, Asymmetric nanocatalysis: N-heterocyclic carbenes as chiral modifiers of Fe3O4/Pd nanoparticles, Angew. Chem., Int. Ed., 2010, 49, 7786–7789 CrossRef CAS.
- J. Vignolle and T. D. Tilley, N-Heterocyclic carbene-stabilized gold nanoparticles and their assembly into 3D superlattices, Chem. Commun., 2009, 7230–7232 RSC.
- P. Lara, K. Philippot and B. Chaudret, Organometallic Ruthenium Nanoparticles: A Comparative Study of the Influence of the Stabilizer on their Characteristics and Reactivity, ChemCatChem, 2013, 5, 28–45 CrossRef CAS.
- M. Guerrero, Y. Coppel, N. T. T. Chau, A. Roucoux, A. Denicourt-Nowicki, E. Monflier, H. Bricout, P. Lecante and K. Philippot, Efficient ruthenium nanocatalysts in liquid-liquid biphasic hydrogenation catalysis: towards a supramolecular control through a sulfonated diphosphine-cyclodextrin smart combination, ChemCatChem, 2013, 5, 3802–3811 CrossRef CAS.
- L. Liu and A. Corma, Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS.
- A. Reina, I. Favier, C. Pradel and M. Gómez, Stable Zero-Valent Nickel Nanoparticles in Glycerol: Synthesis and Applications in Selective Hydrogenations, Adv. Synth. Catal., 2018, 360, 3544–3552 CrossRef CAS.
- W. D. Knight, Nuclear magnetic resonance shift in metals, Phys. Rev., 1949, 76, 1259–1260 CrossRef CAS.
- J. J. Van Der Klink and H. B. Brom, NMR in metals, metal particles and metal cluster compounds, Prog. Nucl. Magn. Reson. Spectrosc., 2000, 36, 89–201 CrossRef CAS.
- J. S. Bradley, J. M. Millar, E. W. Hill and S. Behal, Surface chemistry on transition metal colloids-an infrared and NMR study of carbon monoxide adsorption on colloidal platinum, J. Catal., 1991, 129, 530–539 CrossRef CAS.
- L. D. Blackman, P. A. Gunatillake, P. Cass and K. E. S. Locock, An introduction to zwitterionic polymer behavior and applications in solution and at surfaces, Chem. Soc. Rev., 2019, 48, 757–770 RSC.
- A. B. Lowe and C. L. McCormick, Synthesis and solution properties of zwitterionic polymers, Chem. Rev., 2002, 102, 4177–4189 CrossRef CAS.
- G. Hu and T. Emrick, Functional Choline Phosphate Polymers, J. Am. Chem. Soc., 2016, 138, 1828–1831 CrossRef CAS.
- T. Nakaya, M. Yasuzawa and M. Imoto, Poly(phosphatidylcholine) Analogues, Macromolecules, 1989, 22, 3180–3181 CrossRef CAS.
- T. Nakaya and Y. J. Li, Phospholipid polymers, Prog. Polym. Sci., 1999, 24, 143–181 CrossRef CAS.
- J. Eldo and A. Ajayaghosh, New low band gap polymers: control of optical and electronic properties in near infrared absorbing π-conjugated polysquaraines, Chem. Mater., 2002, 14, 410–418 CrossRef CAS.
- B. C. Norris and C. W. Bielawski, Structurally dynamic materials based on bis(N-heterocyclic carbene)s and bis(isothiocyanate)s: toward reversible, conjugated polymers, Macromolecules, 2010, 43, 3591–3593 CrossRef CAS.
- Y. Mai and A. Eisenberg, Self-assembly of block copolymers, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- A. J. Melchor Bañales and M. B. Larsen, Thermal Guanidine Metathesis for Covalent Adaptable Networks, ACS Macro Lett., 2020, 9, 937–943 CrossRef.
- F. Dénès, M. Pichowicz, G. Povie and P. Renaud, Thiyl radicals in organic synthesis, Chem. Rev., 2014, 114, 2587–2693 CrossRef.
- K. J. Romero, M. S. Galliher, D. A. Pratt and C. R. J. Stephenson, Radicals in natural product synthesis, Chem. Soc. Rev., 2018, 47, 7851–7866 RSC.
- T. B. Schon, B. T. McAllister, P. F. Li and D. S. Seferos, The rise of organic electrode materials for energy storage, Chem. Soc. Rev., 2016, 45, 6345–6404 RSC.
- Y. Yan, S. G. Robinson, M. S. Sigman and M. S. Sanford, Mechanism-Based Design of a High-Potential Catholyte Enables a 3.2 V All-Organic Nonaqueous Redox Flow Battery, J. Am. Chem. Soc., 2019, 141, 15301–15306 CrossRef CAS.
- H. V. T. Nguyen, A. Detappe, N. M. Gallagher, H. Zhang, P. Harvey, C. Yan, C. Mathieu, M. R. Golder, Y. Jiang, M. F. Ottaviani, A. Jasanoff, A. Rajca, I. Ghobrial, P. P. Ghoroghchian and J. A. Johnson, Triply Loaded Nitroxide Brush-Arm Star Polymers Enable Metal-Free Millimetric Tumor Detection by Magnetic Resonance Imaging, ACS Nano, 2018, 12, 11343–11354 CrossRef CAS.
- G. G. Alvaradejo, H. V. T. Nguyen, P. Harvey, N. M. Gallagher, D. Le, M. F. Ottaviani, A. Jasanoff, G. Delaittre and J. A. Johnson, Polyoxazoline-Based Bottlebrush and Brush-Arm Star Polymers via ROMP: Syntheses and Applications as Organic Radical Contrast Agents, ACS Macro Lett., 2019, 8, 473–478 CrossRef CAS.
- V. Sciannamea, R. Jérôme and C. Detrembleur, In situ nitroxide-mediated radical polymerization (NMP) processes: their understanding and optimization, Chem. Rev., 2008, 108, 1104–1126 CrossRef CAS.
- M. Chen, M. Zhong and J. A. Johnson, Light-Controlled Radical Polymerization: Mechanisms, Methods, and Applications, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS.
- C. D. Martin, M. Soleilhavoup and G. Bertrand, Carbene-stabilized main group radicals and radical ions, Chem. Sci., 2013, 4, 3020–3030 RSC.
- S. Kundu, S. Sinhababu, V. Chandrasekhar and H. W. Roesky, Stable cyclic (alkyl)(amino)carbene (cAAC) radicals with main group substituents, Chem. Sci., 2019, 10, 4727–4741 RSC.
- M. F. Silva Valverde, P. Schweyen, D. Gisinger, T. Bannenberg, M. Freytag, C. Kleeberg and M. Tamm, N-Heterocyclic Carbene Stabilized Boryl Radicals, Angew. Chem., Int. Ed., 2017, 56, 1135–1140 CrossRef CAS.
- K. C. Mondal, H. W. Roesky, M. C. Schwarzer, G. Frenking, I. Tkach, H. Wolf, D. Kratzert, R. Herbst-Irmer, B. Niepötter and D. Stalke, Conversion of a singlet silylene to a stable biradical, Angew. Chem., Int. Ed., 2013, 52, 1801–1805 CrossRef CAS.
- O. Back, B. Donnadieu, P. Parameswaran, G. Frenking and G. Bertrand, Isolation of crystalline carbene-stabilized P2-radical cations and P2-dications, Nat. Chem., 2010, 2, 369–373 CrossRef CAS.
- M. Y. Abraham, Y. Wang, Y. Xie, R. J. Gilliard, P. Wei, B. J. Vaccaro, M. K. Johnson, H. F. Schaefer, P. V. R. Schleyer and G. H. Robinson, Oxidation of carbene-stabilized diarsenic: diarsene dications and diarsenic radical cations, J. Am. Chem. Soc., 2013, 135, 2486–2488 CrossRef CAS.
- S. Kundu, S. Sinhababu, S. Dutta, T. Mondal, D. Koley, B. Dittrich, B. Schwederski, W. Kaim, A. C. Stückl and H. W. Roesky, Synthesis and characterization of Lewis base stabilized mono- and di-organo aluminum radicals, Chem. Commun., 2017, 53, 10516–10519 RSC.
- L. Y. M. Eymann, A. G. Tskhovrebov, A. Sienkiewicz, J. L. Bila, I. Živković, H. M. Rønnow, M. D. Wodrich, L. Vannay, C. Corminboeuf, P. Pattison, E. Solari, R. Scopelliti and K. Severin, Neutral Aminyl Radicals Derived from Azoimidazolium Dyes, J. Am. Chem. Soc., 2016, 138, 15126–15129 CrossRef CAS.
- J. Back, J. Park, Y. Kim, H. Kang, Y. Kim, M. J. Park, K. Kim and E. Lee, Triazenyl Radicals Stabilized by N-Heterocyclic Carbenes, J. Am. Chem. Soc., 2017, 139, 15300–15303 CrossRef CAS.
- N. Kazeminejad, L. Münzfeld, M. T. Gamer and P. W. Roesky, Mono- and bimetallic amidinate samarium complexes-synthesis, structure, and hydroamination catalysis, Dalton Trans., 2019, 48, 8153–8160 RSC.
- T. Elkin, N. V. Kulkarni, B. Tumanskii, M. Botoshansky, L. J. W. Shimon and M. S. Eisen, Synthesis and structure of group 4 symmetric amidinate complexes and their reactivity in the polymerization of α-olefins, Organometallics, 2013, 32, 6337–6352 CrossRef CAS.
- N. Y. Rad’kova, T. A. Kovylina, A. S. Shavyrin, A. V. Cherkasov, G. K. Fukin, K. A. Lyssenko and A. A. Trifonov, Amido rare-earth(III) and Ca(II) complexes coordinated by tridentate amidinate ligands: synthesis, structure, and catalytic activity in the ring-opening polymerization of rac-lactide and ε-caprolactone, New J. Chem., 2020, 44, 7811–7822 RSC.
Footnote |
| † Present address: Department of Chemistry, University of Minnesota, Minneapolis, MN 55455, USA. |
| This journal is © The Royal Society of Chemistry 2021 |