
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFlow biocatalysis 101: design, development and applications
Ana I.
Benítez-Mateos†
 ,
Martina L.
Contente†
,
David
Roura Padrosa†
and
Francesca
Paradisi
*
,
Martina L.
Contente†
,
David
Roura Padrosa†
and
Francesca
Paradisi
*
Department of Chemistry and Biochemistry, University of Bern, Freiestrasse 3, Bern, Switzerland. E-mail: francesca.paradisi@dcb.unibe.ch
First published on 3rd February 2021
Abstract
The integration of enzyme-catalyzed reactions in flow systems has been boosted during the last few years. Nowadays, biocatalysis is officially recognized as a tool to increase reaction specificity and sustainability, however applications are sometimes characterized by low productivity. A logical step to improve the performance of biocatalytic reactions is represented by the combination of enzymes and flow facilities. This tutorial review aims at introducing the key concepts of flow biocatalysis, guiding the reader through its advantages and highlighting the current trends in the field to encourage innovative applications of enzymes in flow reactors. Experimental cases are also presented and discussed as a troubleshooting guide for development of flow biocatalytic processes.
 Ana I. Benítez-Mateos | Ana I. Benítez-Mateos is a Postdoctoral researcher (SELF Fellowship) in the University of Bern. She got a MSc from the University of Seville before she joined the group of Prof. Fernando López-Gallego to work in Synthetic Biology and Biocatalysis. She earned the PhD from the University of the Basque Country (UPV/EHU) in 2019. Her research interests include novel strategies and materials for protein immobilization, protein engineering, biocatalysis, and continuous-flow biosynthesis of pharmaceuticals. |
 Martina Letizia Contente | Martina Letizia Contente is a senior Postdoctoral researcher at the University of Nottingham. She graduated in Pharmacy at the University of Milan, where she obtained also her PhD in Medicinal Chemistry. During her years as post doc she had the opportunity to work in different international environments increasing her expertise in the development of intensified and sustainable flow-biobased processes for the preparation of high value molecules. Among her research interests enzyme discovery, protein immobilization and stabilization for continuous processing are the most important. |
 David Roura Padrosa | David Roura Padrosa is a Postdoctoral researcher in the University of Bern. He earned his MSc in Molecular Biology and Medicine at the University of Girona, before undertaking the PhD studies at the University of Nottingham under the supervision of Prof. Francesca Paradisi. His research has been focused on the discovery of new biocatalysts and their application to the synthesis of valuable chemicals, combining biocatalysis and flow chemistry, as well as the development of bioinformatic tools to be applied in the field of biocatalysis. |
 Francesca Paradisi | Francesca Paradisi joined the University of Bern as the Chair of Sustainable Pharmaceutical Chemistry in 2019. Trained as a synthetic organic chemist (University of Bologna), she joined the group of Prof. Engel (University College Dublin) where she learnt first about biocatalysis. The use of enzymes as a sustainable approach to the synthesis of valuable products has been the focus of her research group since she became an independent academic in 2006. Over the last few years, her team developed a number of enzyme-based processes in continuous flow, reducing the gap between academic discovery and industrial application. |
1. Introduction
1.1 Process in continuous: flow chemistry
Cost-efficiency, in any process, is the major driving force of innovation but nowadays, other factors must also be considered. Public awareness on the importance to minimize the impact of our ever-growing product demand on the environment has reached a turning point during the last two decades. Consequently, not only the costs are considered, but also the process sustainability and compliance with the green chemistry principles.1,2 It is not a coincidence that for more than 10 years now, continuous processing has found its niche in chemical production. This technology was already used for petrochemical and bulk chemical production, for example, but it is not as common in fine chemicals and pharma industry.3 It is through the synergy between green process development and the design of an efficient flow-based approach that continuous biocatalytic processing and production, and its benefits compared to batch-based strategies can be truly implemented.4Batch processes are commonly used in pharmaceutical and synthetic chemistry due to their flexible production planning, fast implementation as well as process and product traceability. Although companies favor batch processes due to the availability of sunk capital for this technique and accumulated knowledge, they require significant investment in material, large storage facilities for chemicals, solvents and, noteworthy, process intermediates. Also, the scalability is never straightforward as heat and mass transfer are a challenge.
In contrast, continuous flow setups normally require smaller equipment footprint and their automation reduces the need for human manipulation. As per the scalability, while scale up approaches would face the same heat and mass transfer issues, scale out approaches, (connecting different flow reactors in series or parallel) are a good alternative. Initially, microflow scale out gained attention but there has been a shift in the later years towards meso-reactors as their productivity matches better with industrial needs. Continuous flow strategies are also safer than their batch counterparts, allowing the use of conditions (such as pressure, temperature or reactivities) that would be nearly impossible to apply safely in batch. In addition, continuous operations can improve the cost-efficiency and sustainability through process intensification and feedback loop strategies (increased yields and decreased solvent/energy waste) opening the possibility of continuous monitoring. These are the fundamental principles that drive the expansion of flow chemistry.5
Early on, flow chemistry was applied in the field of organometallic chemistry as a strategy for heterogeneous catalysis to shorten reaction times or when hazardous compounds were utilized. More recently, scientists are taking advantage of flow chemistry not only in ‘process intensification’ but also in ‘discovery’. As an example, packed-bed reactors (PBR) can provide information about the performance of a new catalyst while scalability and stability are tested at a laboratory scale with very high catalyst to substrate ratios. In this way, emerging and interconnected scientific disciplines have been developed through innovative approaches which integrate flow technologies in chemistry, biotechnology, biomedicine, and photocatalysis among others.
The core of flow reactors is very simple: it requires pumps to feed solvents and reagents, different size channels, and the vessel where the reaction takes place. Besides those essential elements, several types of junctions, mixers, and pressure regulators are typically part of flow machines. Temperature controllers can be also added. In more advanced set-ups, in-line separation devices (extractors), product purification accessories (scavenger columns), as well as gas, UV-light or microwave suppliers complement the flow process design. Even analytical components (LC-MS, GC-MS, spectrophotometers, bench-top NMR) may be connected to the continuous flow line to provide real-time reaction monitoring. Such a set-up may seem of a high complexity but there is a broad variety of customized flow reactors which are commercially available and can cater for the specific need of a research lab in both academia and industry (Vapourtec Ltd, Ehrfeld, Syrris, AM Technology, Corning, ThalesNano Nanotechnology Inc., Advion Inc., Future Chemistry Holding BV, YMC Co. Ltd, Accendo, Uniqsis Ltd and Chemtrix BV). Nevertheless, fabrication of DIY-equipment is a customary trend in many labs around the world, especially when affordability or availability become an issue. Interestingly, 3D-printers are expanding the possibilities of tailored flow chemistry reactors for many researchers in the field.6
1.2 What is biocatalysis?
Biocatalysis, is defined as the use of whole cells or their components as catalysts for a certain chemical reaction. Although sometimes biocatalysis is perceived as something new, it comes from very ancient times. Our group recently published an almost unique review on the history of enzymes and their affirmation as powerful synthetic tools.7The use of whole cells, also referred as fermentations, present the major drawback of possible cross-reactivity with other metabolites and naturally expressed enzymes. In addition, cell-wall permeability may affect the substrate diffusion hampering its transformation, while its stability may lead to cell-lysis and loss of compartimentalization.8
As an alternative, purified enzymes can be used as catalysts, which solves in one shot the two problems stated before: diffusion and side-reactivity. In this case, the main drawbacks are the costs for the preparation of the pure catalyst, its stability and reusability. Enzyme immobilization can however come to the rescue and significantly reduce these limitations.9–11 In fact, once the enzymes are immobilized, their removal from the reaction bulk, reuse, and incorporation in continuous-mode reactors is straight-forward. This, greatly simplifies the work-up, reducing the produced waste and impacting the cost-effectiveness of the process.
Biocatalyzed reactions in flow can benefit from improved productivity thanks to enhanced mass transfer and better control of the reaction parameters. Better process control makes the bioreaction more efficient minimizing waste and energy consumption. In addition, the absence of harsh mixing prolongs the biocatalysts lifetime, thus reducing the costs associated with their preparation.
In this review, our intention is to stimulate the interest of chemists and biochemists so that they may expand their toolbox and knowledge and embrace flow biocatalysis, which has the potential to become a widespread tool to move chemical transformation to a more sustainable, productive and cost-efficient way.
It must be noted early on that not all processes are (yet) suitable for flow, but when flow is an option it may offer not just an alternative approach, but a system with benefits beyond expectations.
2. Terminology
As any other field, flow biocatalysis uses a specific terminology that might seem a highly technical jargon for a newcomer in this area and be off-putting. For this reason, the parameters used to define and evaluate performance and efficiency of flow biocatalysis are described in this section. In addition, it will help specialists to standardize their results and transmit the content more effectively.2.1 Flow chemistry parameters
Chemistry reactions for the preparation of valuable commercial products need to be operated in an efficient way with respect to the starting reagents, catalyst and equipment, in a reactor compatible with the characteristics of the reaction: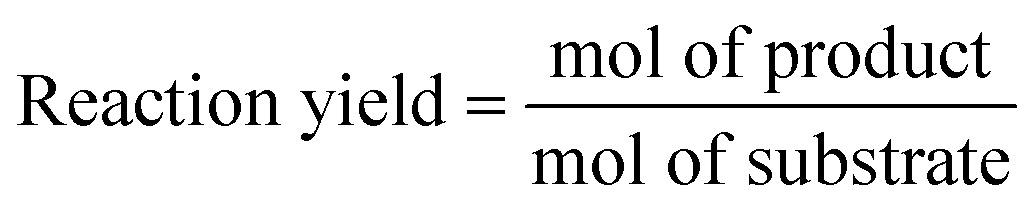
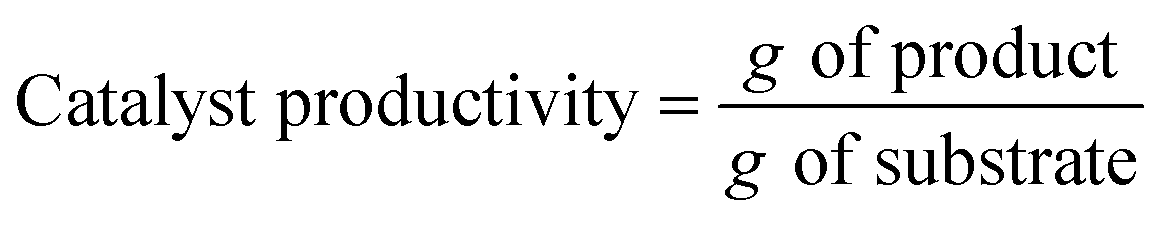
The schematic representation of a flow process and the typical equipment are reported in Fig. 1 and 2. It consists of pumps: HPLC, syringe, peristaltic or gear centrifugal pumps used to deliver solvents and reagents in a controlled and reproducible way; loops: for the introduction of small volume of reagents; mixing tubes: first point of mixing between different reagents; reactors typically temperature controlled: mesoreactors (coils or column reactors), microreactors (chips); back pressure regulator (BPR): controlling the pressure of the whole system; downstream units: work-up operations, purifications and analysis.
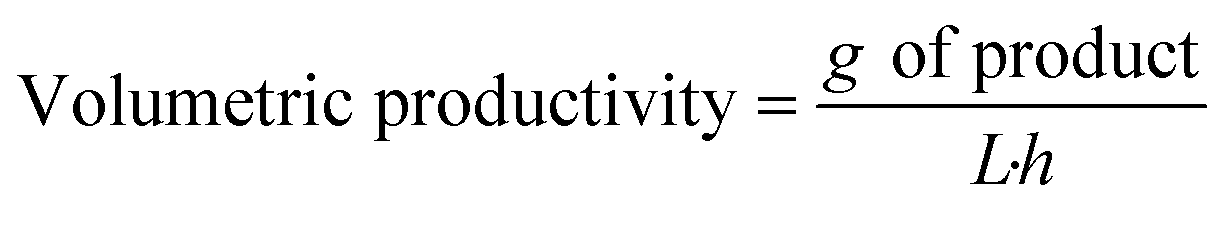
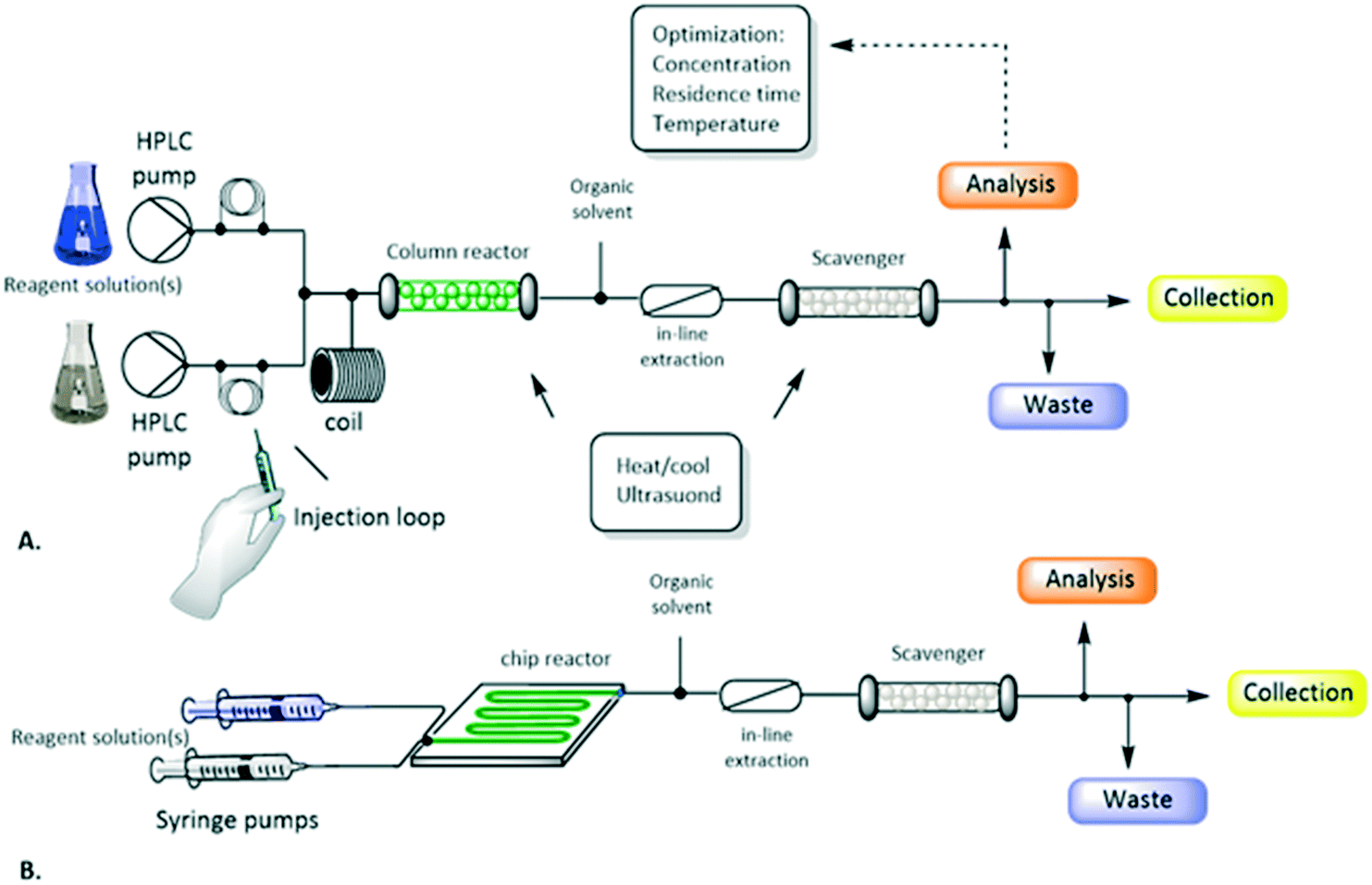 | ||
| Fig. 1 Schematic representation of a flow reactor configuration involving mesoreactors (A) or microreactors (B). Figure adapted from Tamborini et al.59 | ||
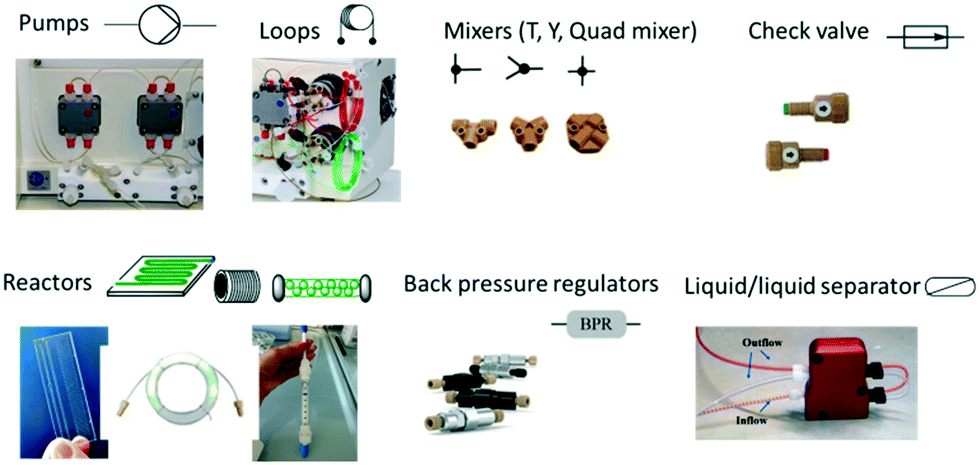 | ||
| Fig. 2 Flow equipment and representation. | ||
Miniaturization for flow reactors is associated with devices typically designed with channels or tubes with different internal diameters (i.d.) from μm (microreactors) to mm (mesoreactors) (Fig. 1 and 2). The first ones, typically microfluidic reactors, present an internal diameter <500 μm and a volume in the μL range. They are classified as chips or microtube devices where the reaction environment is a microchannel made of metal, glass or plastic. Among the advantages, effective heat/mass transfer and temperature control are noteworthy. On the other hand, mixing is limited by diffusion while high pressure drops (decreasing of pressure between the 2 lengths of the channel due to unbalanced pressure and viscosity forces) and channel obstruction may occur, especially in the presence of suspensions.
Mesoreactors have an internal diameter >500 μm to several mm, and a volume in the mL range. They present high flow capability and low pressure drops but less efficient heat transfer. While microreactors are characterized by laminar flow, mesoreactors exceeding 1 mm can present a turbulent flow, especially at high flow rates. Between 500 μm and 1 mm i.d. the type of flow generated depends on the flow conditions. To overcome issues related to mixing efficiency, mesoreactors are available with different designs; coil reactors providing homogeneous mixing are preferable for liquid–liquid interaction while column reactors are typically packed with immobilized catalysts.
To compare the performance of different biotransformations or of the same biotransformation in different flow reaction conditions, key parameters need to be properly reported:
- Residence time: time (min) the reagents take to go across the reactor (reaction time).
- Biocatalyst loading: amount of biocatalyst added to the reactor (for immobilized biocatalysts is better to specify also the matrix loading: mgbiocatalyst gmatrix−1).
- Specific biocatalyst activity: U mgenzyme−1 before the beginning of the process.
- Biocatalyst activity: specific activity per biocatalyst loading expressed per gram of matrix (U gmatrix−1) before starting the reaction.
- Reagent/substrate concentration: concentration of the reagent/substrate entering the reactor.
- Reactor size: available reactor volume (channel size or void volume need also to be reported).
- Bioreactor productivity: space–time yield (STY) normalized to the available reactor volume (product quantity (mol)/catalyst volume (L) time (h)).
- Biocatalyst productivity: g of product synthesized per g of biocatalyst employed in the flow reaction.
- Bioreactor stability: biocatalyst activity over time.
Reaction rate of a flow reaction is usually calculated as reported in the following equation:
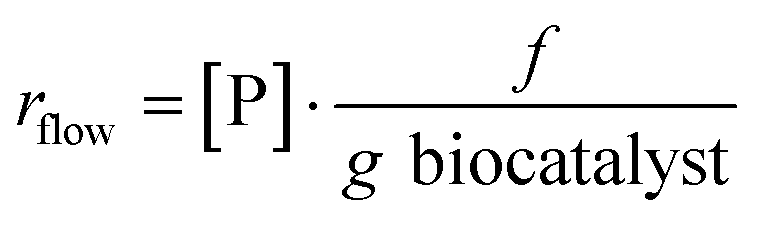
2.2 Metrics of sustainability
The contribution of flow biocatalysis to more eco-friendly synthetic processes, promotes the need for the evaluation of its “greenness” to be able to compare with both chemical catalysis and batch biocatalytic processes. Atom economy, the E factor and process mass intensity (PMI) are the most widely used metrics in green chemistry.12Atom economy (AE) is a theoretical value very useful as a prediction tool to rapidly assess the waste that will be produced within individual steps. AE is expressed as a percentage and calculated as follows, where MW is the molecular weight:
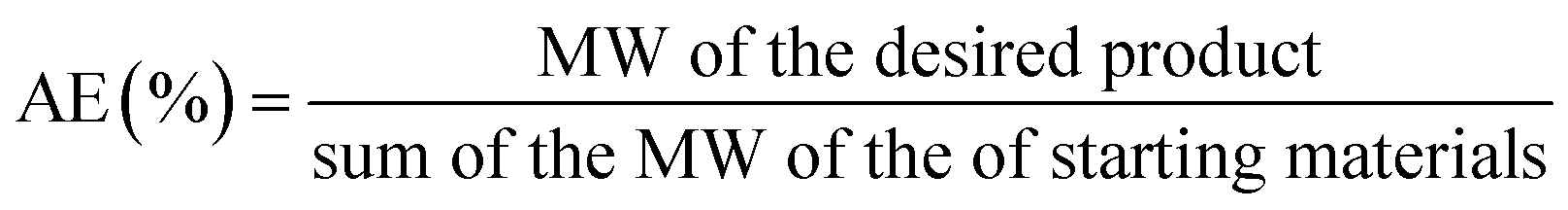
The E factor is an experimental parameter that refers to the actual waste produced and normalized to the product:
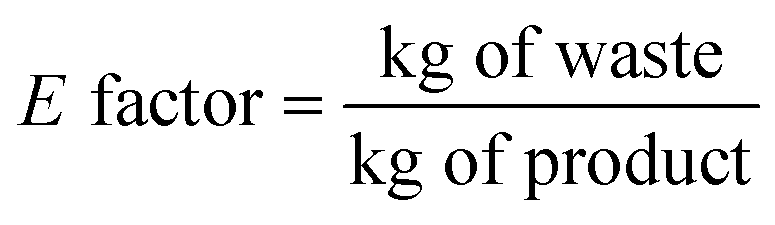
Another mass-based metrics is the process mass intensity (PMI) which is tracked during the lifecycle of pharmaceutical compounds.13 To evaluate the PMI, the roundtable tool developed by the ACS Green Chemistry Institute Pharmaceutical can be used.14
3. Key enabling technologies for enzyme stability
Enzymes are designed by nature to fulfil the requirements of life sustainment. Clearly, the natural environment is often quite different from the industrial conditions required for large-scale processes. An ideal biocatalyst for industrial applications should be stable for a long period of time, resistant to harsh (non-physiological) reaction conditions, eco-friendly, and economically viable. Several technologies such as enzyme immobilization, protein engineering, and computational tools are employed during the design and development of biocatalysts to achieve the highest efficiency.153.1 Enzyme immobilization
During the immobilization process, enzyme molecules are typically attached to, or confined into, solid materials. The immobilized enzymes acquired advantages in terms of stability against inactivating agents (i.e. temperature, pH, organic solvents), separation of the products from the biocatalyst, reuse/recycle of the biocatalyst for a new reaction, and improved reaction rates.16 These features are paramount for the integration of enzymes in flow reactors.11 However, no universal immobilization methodology has yet been proven suitable for any given enzymes.10 Therefore, enzyme immobilization is still an empirical process trying to achieve the best equilibrium between activity and stability of the biocatalyst of interest.17Two main immobilization strategies are adopted in flow biocatalysis: attachment of enzymes on the reactor wall or immobilization onto a solid carrier material suitable to be integrated into a PBR (Fig. 3). Enzyme immobilization in wall-coated reactors is often applied for microfluidic systems where solid carriers could cause clogging issues.18 Experimentally, the enzyme solution in the appropriate buffer is incubated with the solid support under gentle shaking for a certain time (usually, hours) until all the offered protein has been immobilized or the maximum binding capacity has been reached.
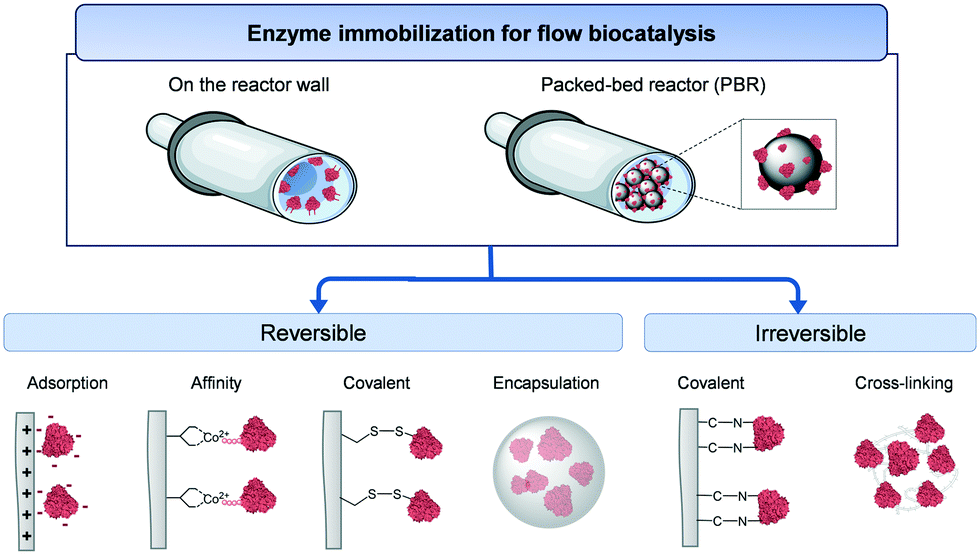 | ||
Fig. 3 Scheme of the enzyme immobilization strategies in flow biocatalysis. An example of each type of interaction is also depicted: ionic interaction (adsorption); metal coordination bond (affinity); disulfide bond (covalent reversible); sol–gel matrix (encapsulation); C–N bond (covalent irreversible); CLEAs (crosslinking). Flow reactor:  , enzyme: , enzyme:  , carrier: , carrier:  . . | ||
Enzyme immobilization can be performed by either reversible or irreversible interactions. We start testing irreversible immobilization techniques which offer a higher stability and avoid leaching of the enzyme while operating flow reactors. These advantages increase the operational lifespan of the biocatalysts promoting one of the key goals of flow biocatalysis: a high accumulated STY relative to the amount of enzyme used. Enzymes can be irreversibly immobilized on a carrier material by forming covalent bonds between selected residues of the protein and the functional groups of the carrier surface. Unfortunately, irreversible immobilization strategies compromise the recyclability of the reactor/carrier when the enzyme activity tends to zero. Alternatively, cross-linked enzyme aggregates (CLEAs) are a carrier-free immobilization technique which relies on the irreversible binding of enzymes mediated by crosslinking agents such as glutaraldehyde and biominerals.16 Yet, the mechanical stability, the mass transfer issues, and the wide distribution of particle size of the CLEAs limit their use in flow biocatalysis. Moreover, irreversible immobilization promotes sometimes a dramatic loss of enzyme activity upon immobilization.
As a reversible immobilization technique, enzyme adsorption (i.e. through ionic and hydrophobic interactions) onto the carrier usually maintains the activity of the immobilized enzyme. Another reversible immobilization protocol is accomplished by genetic fusion of peptide tags (i.e. (6×) His-tag, Halo-tag, Spy-tag) to the enzymes, enabling their attachment to either a carrier or a reactor wall through affinity interactions. This technique allows the purification and specific immobilization of the enzyme in just one step as well as the control of the enzyme orientation. Additionally, physical entrapment/encapsulation is used by covering the enzymes with polymers or sol–gel matrices which result into a milder distortion of the enzyme structure. All these reversible procedures can be reverted, which allow the recycling and reuse of the costly carriers once the enzyme is inactive by reloading it with fresh enzyme. Nevertheless, the stability of the immobilized enzymes is often pretty low due to the lack of rigidification of the enzyme structure and leaching of the enzyme while working in continuous flow conditions.
The most used materials for enzyme immobilization are either methacrylate or agarose microbeads. From an industrial perspective, methacrylate is a robust premade support with high resistance to flow conditions such as high pressure and presence of solvents. Agarose is a more hydrophilic support which is chosen to tackle issues of poor enzyme activity and stickiness of substrates/products on hydrophobic supports. Other supports like silica particles, lignin, magnetic nanoparticles, etc. can be used for integration of immobilized enzymes in flow reactors. In case of enzyme immobilization on microreactor walls, polymeric tubing (i.e. polytetrafluoroethylene, polystyrene, polydimethylsiloxane) are commercially available.9
3.2 Protein engineering and computational tools
Evolution of native enzymes to become efficient industrial biocatalysts is addressed by protein engineering, often aided by computational tools (i.e. Chimera, Pymol). In general terms, there are two approaches to improve the functional properties of enzymes: directed evolution and rational design.Directed evolution rely on random combinations of mutations in the protein sequence that may potentially generate improved enzymes.19 While the probability of success of this very time-consuming strategy is quite low, it is an appealing option when no structural information of the enzyme or no prior knowledge of the catalytic mechanism is available. The rational design depends on the analysis of the sequence and the structure of proteins to further introduce mutations in specific positions.20 For these reasons, a combined strategy using rational design and directed evolution is usually the best approach.
Nowadays, the increasing number of protein libraries and in silico tools such as protein modelling and dynamic simulations is speeding up the optimization and development of process-specific biocatalysts.21 Moreover, new computational tools that take advantage of bioinformatic analysis specifically for protein immobilization (i.e. CapiPy)22 are opening new possibilities to apply semi-rational approaches to enhance the performance of immobilized biocatalysts.
4. A never-ending reaction: versatility, combination and automation
4.1 Cofactor recycling in flow mode
Most of the examples we find in flow biocatalysis are single step transformations, but this technology can also be applied for multi-enzyme-mediated cascade reactions. Very recently, John M. Woodley discussed the hurdles present in the multi-step design and implementation of such enzymatic cascades for industry.23 Due to the versatility and compartmentalization of flow reactors some of the mentioned problems can indeed be tackled by continuous operations. Initial examples about immobilized enzymes in continuous mode, where limited to the use of packed-bed reactors containing only the biocatalyst for a single biotransformation, facing since the beginning the issue of expensive cofactor requirement especially for redox enzymes. Further research allowed for the development of different strategies for flow mode cofactor recycling. For example, the continuous addition of a co-substrate (e.g., EtOH) as a sacrificing substrate to recycle the cofactor was reported by Contente & Paradisi in cascades involving the reduction of aldehydes into the corresponding alcohols through the horse liver alcohol dehydrogenase (HLADH).24 This technique allows for the use of only one biocatalyst able to carry out both the redox reaction and the cofactor regeneration, positively impacting on process economy. On the other hand, the required co-substrate large excess to push the equilibrium towards product formation, can affect the biocatalyst performance. The enhanced catalyst stability achieved through the combination of immobilization techniques and flow facilities deeply mitigated this phenomenon. Other strategies involved the (co)-immobilization of two different enzymes: the principal one for the main reaction and the ancillary enzyme for the cofactor recycling. When two or more biocatalysts are simultaneously co-immobilized onto the same support, a high catalyst compatibility is required. When this condition is not achievable, individual biocatalyst immobilization followed by mixing of the resins (mixed-bed reactor),25 or by sequential immobilization onto the same support are pursued. A further implementation of the latter technique was achieved by the group of Prof. López-Gallego. Through a spatial organized immobilization involving also the co-factors, they gave rise to self-sufficient systems, increasing the process cost-efficiency and avoiding the addition of exogenous expensive molecules.264.2 Telescoped reactions
While in batch the use of sequential reactions is tedious and requires manual adjustment as well as work-up and purification between the different steps, in flow, they can be combined in a modular way. Theoretically, if a sequence of stepwise reactions can be set up using the same solvent or simply applying an exchanging solvent mechanism, the optimized reactions could be processed in tandem. In this way the reaction mixture for the first step becomes the reagent for the next one creating a telescoped sequence. This versatility allows cascade design using different reaction conditions, as some flow system allows different reactors to be maintained in different working conditions; temperature can be individually controlled as well as pH, ionic strength, etc. For example, pH can be changed by the addition of an acid or a base, a second reagent can be added between the reactors, and concentrations modulated by adding a diluting factor. If on one side this ideal scenario is not so easy to achieve due to the need to quench reactions, work-up intermediates, and consequently purify the stream between the different transformations, on the other side it is possible to integrate other enabling technologies to obtain in-line filtration, extraction, distillation, and purification to maintain a continuous flowing sequence. All these steps can be automated through dedicated connected software. With a careful process design, continuous flow allows a fine control with minimum manual operator intervention and manipulation.4.3 Downstream unit operations and automation
The concept of “automated flow” in organic chemistry was extensively developed by Prof. Ley and his research group in order to obtain more efficient, sustainable and innovative processes.One of the main aspects concerns work-up procedures and purification steps, which are often considered a bottleneck in continuous synthesis. Solid supported reagents have widely dominated product purification in flow multi-step processes. They mainly consist in reactive species associated with heterogeneous support material.27 Ideally, the use of such reagents should trap the impurities from the flow stream giving a pure product without any traditional work-up procedure (i.e. chromatography, crystallization, distillation) fulfilling either electrostatic or covalent interactions between the solid matrix and the undesired species. Due to their universal application, a wide range of functionalized scavenger columns are now available on the market. However, while these materials are extensively used, they present some limitations, especially in terms of cost and reduced lifetime. Typically, solid-supported reagents cannot be recycled continuously requiring an interruption of the flow sequence. This is the case for example of catch and release strategies through ion-exchange resins where the addition of diluted solutions of acids or bases let the recovery of the trapped molecules as salt form.28,29 Moreover, increasing the amount of the scavenger during the reaction scale-up, leads to undesired scale-depended dispersion/diffusion phenomena. Consequently, a thorough evaluation of the flow design is necessary before using them. Despite the development of numerous tools to increase the automation of work-up procedures considered time- and cost-consuming, and to reduce the environmental burden, continuous synthesis is usually followed by “discontinuous” purification because the limited number of available options.30,31 Often chemists necessitate to resort to old fashion batch chromatography methods for the separation of complex mixture of products, particularly when they present same functional groups.32 So far, high purity can be achieved by in-line procedures through multi-column chromatography,33 or simulated moving bed (SMB) chromatography.34,35 O'Brien and coworkers36 developed the first example of in-line SMB (simulated moving-bed) chromatography for the continuous production of a clean product. This concept, born in the 60s is still used in many industrial applications (e.g., petrochemical, food, and pharmaceutical industries) and consist basically in the simulation of the movement of the stationary phase in the opposite direction of the fluid to achieve a counter-current flow, rather than flowing fluids through a static bed. While SMB chromatography has a great potential for the purification of different organic compounds, it showed also several drawbacks in terms of costs (high initial investment and maintenance related expenses) as well as higher complexity when compared to single column chromatography. Moreover, it lacks the versatility as no solvent gradient purification can be performed. Notably, the pressure generated by the SMB system can be significant and has to be taken into account before integrating it with flow reactors.
Liquid–liquid extractions is another fundamental separation strategy in any chemistry reaction. Although extractions are very common purification strategies also at industrial scale,37 this process is considered one the most manually intensive and time/space lab consuming one. Moreover, the massive use of organic solvents makes it also unsustainable. It is not surprising, therefore, that significant efforts have been put into the development of devices to automatically perform such procedure minimizing the amount of solvent. Commercially available gadgets (i.e., Zaiput liquid–liquid separator) typically rely on a fast mixing of the immiscible solvents using PTFE tubes and a separation due to the interactions with a hydrophobic membrane (one phase will have an affinity for the membrane and fill the pores – wetting phase – while the other one will be repelled not filling the pores – non-wetting phase). Once the membrane pores are filled with the wetting phase, a pressure differential is applied between the two sides of the membrane to push the wetting phase without forcing the non-wetting phase through the pores. They are usually plug-and-play modular units.
When a flow tandem process is designed, one of the major considerations is the solvent compatibility between the different reactions. In an ideal multi-step process, both solvent and reagent concentration should be kept constant throughout. In reality, this is almost never the case. Solvent switching is a very time-consuming procedure and often it has to be performed by manual intervention stopping the flow continuous process. Few examples of in-line evaporation and distillation have been reported,38 and distillation in particular can allow for both in-line purification, solvent exchange, and solvent recovery and recycling.
Despite the promising results of flow chemistry technology, there are still many hurdles to overcome for the implementation of continuous processes particularly regarding the handling of solids in flow reactors. One of the most important elements to monitor clogging events is the pressure which increases when suspensions are formed, even though in an ideal design of a multi-step process, precipitation should be avoided. Among the common strategies, flushing the reactor with an appropriate solubilizing solvent could be an option.39 Alternatively, sonication while the reagents are flowing through the reactor can prevent the formation of particulate.40 While a number of solutions have been developed to manage slurries41 in continuous mode, these strategies are tailored to specific systems (some of them are reported in the below section) and a common technology is not available yet. In a reaction involving precipitation event, it is necessary to separate the solid from the liquid phase. There are two possible outcomes: the collection of the solid (filter cake residue) or of the liquid (filtrate). In the paper reported by Mascia et al.42 which aims at the collection of the solid for downstream processing, two different in-line crystallization and filtration process are performed, both based on low temperature and vacuum supported devices.
5. Troubleshooting: selected examples of flow biocatalysis
5.1 Combining enzymes with chemical catalysts
Although the focus of this review is the use of biocatalysis in flow, continuous techniques can take advantage of typical catalysts too. When combining both chemical and biocatalysts the main challenge is to find conditions that satisfy both requirements. While enzymes normally prefer aqueous environments, most chemicals are incompatible with this solvent and not always intermediate conditions are compatible due to poisoning or deactivation of one of the catalysts.One of the options to surpass these limitations is the use of enzymes which do not suffer inactivation in organic solvents, either tailoring them to be used in such conditions43 or taking advantage of their intrinsic characteristics.44 The perfect example of such biocatalysts are lipases. While in aqueous media lipases catalyze the cleavage of ester or amide bonds, in organic solvents they can catalyze the opposite reaction.
For example, Farkas et al. in 201845 overcame the limitation of typical dynamic kinetic resolutions of amines combining the acylation of the racemic starting material via a stereo-selective enzymatic reaction with the racemization of the remaining enantiomer through a palladium-based catalyst in 2-methyl-2-butanol (Fig. 4). The authors developed a system using a commercially available sol–gel encapsulated version of Candida antarctica lipase B (CALB-TDP-10) which showed selectivity for the (S)-enantiomer of the benzylic amines. After the first conversion, the palladium catalyst is used to interconvert the remaining (R) enantiomer into the (S) which is in turn acetylated by the enzyme present in the same packed bed reactor with excellent yields (67–96%) and enantiopurity (>99%) for at least three of the tested amines.
Another option to combine both strategies is the compartmentalization, operating each reactor at different conditions taking advantage of the modularity of flow machines. This strategy was applied by Sperl et al.,46 in 2016 for the synthesis of 2-keto-3-deoxy sugar acids (Fig. 4). In their work, implementing a fed-batch continuous flow hybrid process, the incompatibility between gold catalyzed sugar oxidation with enzymatic dehydration could be surpassed. This is also an excellent example of the step-by-step process optimization, solving issues that might appear during the process assembly, such as pH incompatibility or side-product removal. In their case, removal of hydrogen peroxide from the gold-catalyzed oxidation was key to ensure the correct operation of the immobilized dihydroxy acid dehydrogenase (SsDDH). Continuous operation of their system over 100 mL resulted in very good yields (69–91%) and good recovered yields after purification for two of the tested sugars (58 and 86%).
5.2 Overcoming insolubility in flow
Flow biocatalysis cannot be regarded as a simple translation of the batch reactions in continuous. Most of the times, the reaction conditions and design have to be optimized when translated into continuous.In this sense, in the work of Farkas et al. (Fig. 4), a very common issue in flow had to be addressed: substrate insolubility. Previous attempts to perform similar strategies in flow faced a critical problem as ammonium formate, while necessary to avoid side reactions in the racemization, was insoluble in such solvents. This is one of the major problems when working in continuous flow and, if it cannot be solved, often imposes the use of semi-continuous approaches (with intermediate filtrations) to avoid system clogging, detected specially by an increase on the back pressure of the system. In their work, the change of solvent to 2-methyl-2-butanol avoided the precipitation of the ammonium formate.
In other cases, the solvent choice is not so broad as most enzymes do not perform well in non-aqueous media. Even if peristaltic pumps are suitable to pump slurry solutions into the reactor, normally, poor solubility decreases the efficiency of the process. To prevent it, the use of surfactants to increase the water solubility of organic molecules ensure an efficient mass transfer in the reactor, but bear in mind that they can also have an effect on the enzymatic preparation.47
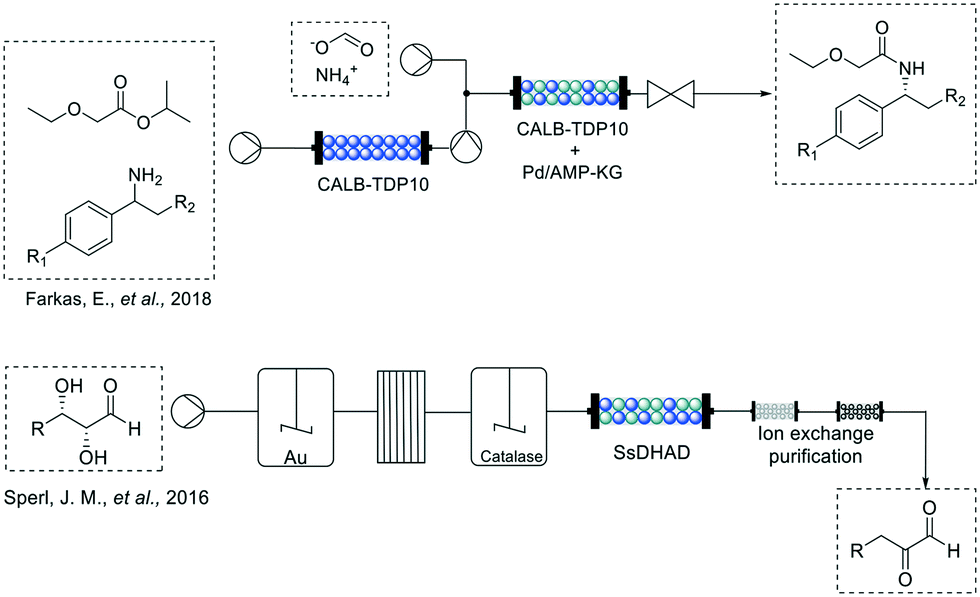 | ||
| Fig. 4 Schematic representation of chemoenzymatic dynamic resolution of benzylic amines developed by Farkas et al. and the fed-batch continuous approach for the chemo enzymatic synthesis of 2-keto-3-deoxy sugar acids by Sperl et al. | ||
Related to this and especially in microfluidic systems, if protein leakage occurs during operation due to reversible immobilization techniques or the presence of multimeric enzymes, the denatured protein can also cause a block. In this case, it would normally be a good idea to set up regular washes with cleaning solutions (such as NaOH 1 M or guanidinium chloride 4 M) to ensure the denature protein does not have a negative effect on the system.
5.3 The impact of biocatalysis and flow facilities on sustainability: a self-sustaining closed-loop continuous reaction
Despite the benefits of biocatalytic processes in flow chemistry reactors listed above, their application on large scale poses a significant challenge regarding the recovery of the main solvent:water.48 Common methodologies such as distillation and ultra-filtration are not economically-feasible49 and are energy and time consuming. Moreover, the solvents collected downstream of the process typically contain traces of unreacted reagents, side-products or additives that are usually discarded, deeply impacting on the economy of the system. An excellent example about a zero-waste reaction was described by Contente & Paradisi, in 2018.24 The multi-enzymatic process was firstly improved with respect to batch methods by using catalytic amounts of cofactors and applied for the synthesis of valuable products as hydroxytyrosol from commercially available amines at 10 mM scale. The system was further implemented with in-line work-up and purification steps making it fully automated. The major leap forward was represented by the design of a closed-loop reaction for the recovery of both by-products and cofactors (Fig. 5).Standard reactions have been used to prove the feasibility and the efficiency of the system. The ultra-efficiency concept reported here for the first time, was achieved using trapping columns downstream the process to separate the pure products and the benign by-products (at the end also these ones recovered through a catch and release strategy). The partially purified waste waters containing the cofactors in catalytic amounts have been extensively recirculated allowing the obtainment of a closed-loop system virtually generating no waste.
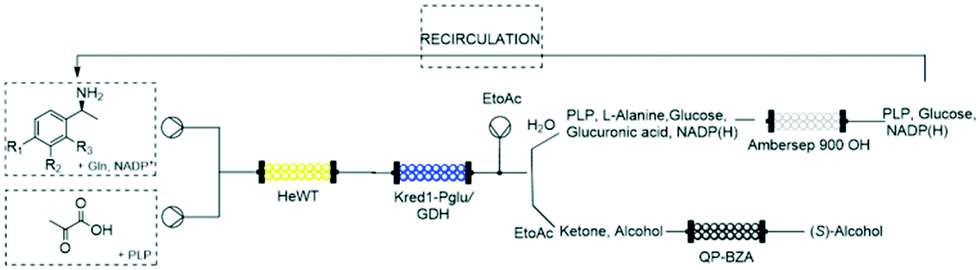 | ||
| Fig. 5 Multi-enzymatic closed-loop reaction. | ||
5.4 Pressurized flow reactors: free versus immobilized enzymes
Oxygen (O2) is a powerful oxidant required in many biocatalytic processes, however, its low solubility in aqueous phase restricts its application in flow (bio)reactors. The application of a gas–liquid segmented flow can be used to increase the O2 transfer from the gas to the liquid phase. By increasing the gas flow rate, various flow regimes (from the slowest to the fastest: bubble, slag and annular flow) are achieved for the gas–liquid mixing.50 When free enzymes are the catalysts, tube-in-tube reactors may be operated by flowing the enzymes and substrates in the inner tube (liquid phase) that is separated by a membrane from the outer tube (gas phase). Unfortunately, the two-phase strategies may cause denaturation of enzymes in the gas–liquid interface, especially of free enzymes.Pressurized flow-reactors are an attractive alternative that facilitates automation and intensification of oxidative flow-biocatalytic processes while protecting the enzymes against denaturing contact. The pressurized flow (bio)reactor (≤34 bar) can be operated in a single liquid phase because the gas–liquid O2 transfer is decoupled in space and time from the O2-dependent biocatalytic reaction (Fig. 6).51 In case that free enzymes are employed, the gas delivery (1–10 mL min−1) is firstly adjusted by a mass-flow controller until it mixes with the substrate (Fig. 6A). Then, the free enzymes can be pumped (0.1–0.4 mL min−1) for mixing with the substrate-O2 flow. Finally, the reaction happens in a coiled reactor whose input and out pressure are controlled. This system proved an increase of up to 6-fold on the reaction rate.51 Moreover, immobilized enzymes can be integrated into the pressurized flow-reactors (Fig. 6B). In this case, substrates and O2 are mixed and pumped to the PBR where the enzymatic reaction happens (Fig. 6B). As one may expect expected, TON, catalyst productivity and STY of the PBR are superior to the liquid reactor operated with free enzymes.
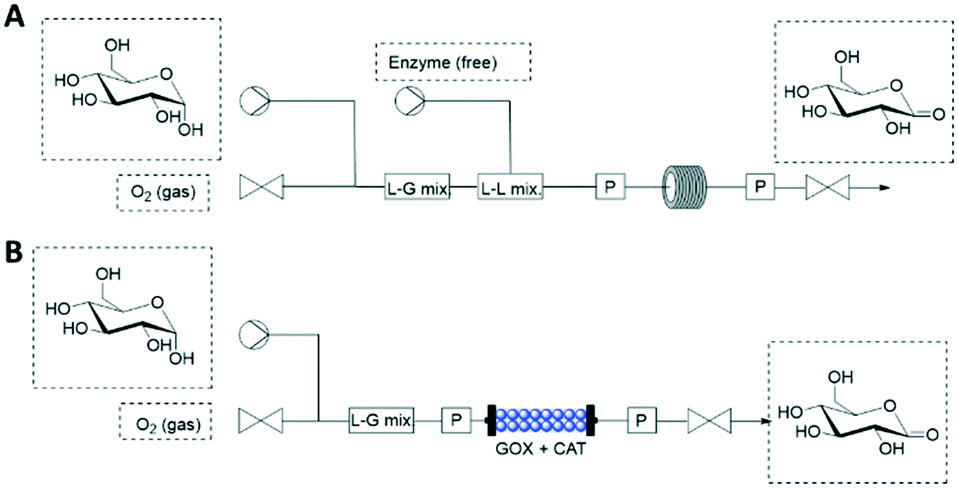 | ||
| Fig. 6 Flow biocatalysis using pressurized reactors for the O2 supply. (A) Free enzymes. (B) Packed-bed reactor with co-immobilized enzymes (GOX: glucose oxidase; CAT: catalase). | ||
5.5 Microfluidics and modelling as reactor engineering tools
Microfluidics enable the miniaturization, integration and automation of biotransformations while maintaining benefits like increased reaction rates, large surface-to-volume ratios, and reduced process costs due to the little volumes needed.52 For these reasons, microfluidic reactors may be preferred for instance, to carry out screenings, and to produce pharmaceuticals that are demanded in small scale (i.e. personalized drugs). The microreactor device is typically a few centimeters size containing micro-tubing in which the reaction will happen. 3D-printing is an innovative tool that allows to shape microreactors on-demand as required by the reaction conditions.53Mathematical modelling is a powerful prediction tool for reactor/reaction engineering.54 A quick optimization and assessment of a complex flow bioreactor can be performed in a short period of time. Then, the mathematical model is experimentally evaluated, usually in combination with microfluidics. As an example, Miložič et al.55 utilized mathematical models to facilitate the description of convective and diffusion mass transfer in a flow reaction kinetics. The model was tested for the microfluidic reaction where a ω-transaminase was immobilized on the inner microreactor walls by ionic interactions (Fig. 7). The substrate solution was pumped (2–32 μL min−1) to the silicon/glass microreactor and the resulting product mixture was diluted with NaOH for further in-line analysis by HPLC. Microfluidic enabled to test various inlet substrates, enzyme concentrations, and residence times to verify the proposed models. Finally, the experimental data can validate the mathematical models for the biotransformation processes even within consecutively connected microreactors.
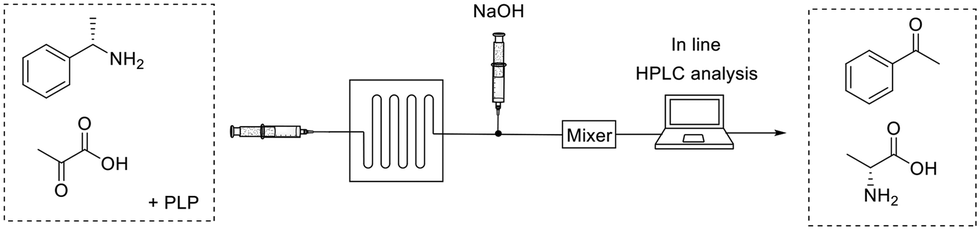 | ||
| Fig. 7 Transamination in a microflow reactor containing surface immobilized enzymes. | ||
Noteworthy, automation of biocatalytic processes is one of the main advantages of its integration in flow systems. In this sense, machine learning for self-optimization of processes must be taken into consideration for both flow chemistry56 and enzyme engineering.57 Although the application of machine learning for flow biocatalysis is still in its infancy.
5.6 How to meet industrial demands? An example of multi-gram scale production
While scale up of flow reactions is generally easier than batch, many processes have been reported as a proof of concept, with low substrate concentration and productivity, clearly highlighting a gap between the lab scale and the industrial demand. A very good example of high productivity is represented by the biosynthesis of a commercially relevant product on a multi-gram scale via an intensified process in flow reported by Contente, et al.,58 in 2019.As it can be seen in Fig. 8, via a single step enzymatic transformation using Mycobacterium smegmatis acyl transferase (MsAcT), melatonin could be synthesized from 5-methoxy tryptamine among others. In this example, the enzyme was covalently immobilized onto glyoxyl-agarose beads with a loading of only 1 mg of enzyme per g of resin, dramatically increasing the catalyst stability. The chemoselectivity of the enzyme as well as the absence of hydrolysis side reaction due to the flow mode prevented the formation of any by-products obtaining the desired amide with a very high purity. Moreover, the molar conversions observed were good with ethyl acetate (>60%) but have been further improved using a more activated acyl donor such as vinyl acetate (>90%).
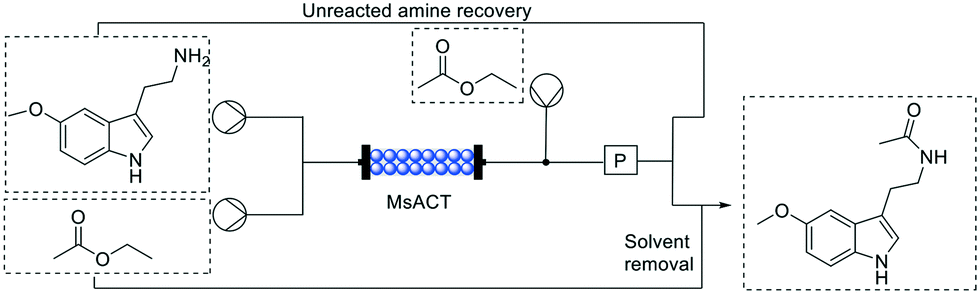 | ||
| Fig. 8 Schematic representation of production of melatonin. | ||
In this case, particular attention must be paid on the substrate loading: the amine was supplied at a concentration of 0.5 M (95 g L−1) through a liquid–liquid segmented flow. That means that the amine was dissolved in aqueous solution while the acetyl donors, were added via a T junction creating a biphasic stream to the column. The residence time in this case was only 5 minutes, exemplifying the shortened reaction times that can be achieved in flow due to the increased mass transfer and control over the reaction conditions. To exemplify the extraordinary productivity, the system was left running for 24 h with the automated work-up described below with isolated yields up to 36.9 g.
Apart from its very high productivity, the system was designed to be as efficient as possible. Not only the space–time yield and atom economy were excellent, but also through an in-line separation, the ethyl acetate containing the final product was evaporated, condensed and recirculated yielding the pure N-acetyl molecules. As reported above, also the aqueous phase with the unreacted amine was recovered and reused. In this way, the reaction has negligible waste contribution, excellent atom economy and ensures no loss of substrate or organic solvent.
6. Conclusions
Global climate change made us aware that a more sustainable way of working and living is necessary. The innovative combination of biocatalysis and flow chemistry facilities has the power to address this requirement. Flow biocatalysis with its efficient and eco-friendly production of various organic molecules applicable to food, cosmetic, chemical and pharmaceutical industry is promoting green chemistry with a significant economic and environmental impact on the society.This tutorial review outlines how biocatalytic processes are dramatically improved by continuous flow technology fulfilling also industrial needs such as short reaction times, increased performance and productivity. Key concepts and detailed definitions, the “corner stone” of flow biocatalysis, have been described. In addition, to help newcomers to the field, the main challenges are discussed, and practical specific solutions are presented.
Although flow biocatalysis may be considered at an early stage of its development, we feel that there is a bright future, especially for multi-enzymatic cascade reactions, overcoming the market, regulatory, technical, and cultural barriers for its implementation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the SNSF for financial support (grant number 200021_192274/1).References
- R. A. Sheldon and J. M. Woodley, Role of Biocatalysis in Sustainable Chemistry, Chem. Rev., 2018, 118(2), 801–838, DOI:10.1021/acs.chemrev.7b00203.
- P. Anastas and N. Eghbali, Green Chemistry: Principles and Practice, Chem. Soc. Rev., 2010, 39(1), 301–312, 10.1039/b918763b.
- C. Badman, C. L. Cooney, A. Florence, K. Konstantinov, M. Krumme, S. Mascia, M. Nasr and B. L. Trout, Why We Need Continuous Pharmaceutical Manufacturing and How to Make It Happen, J. Pharm. Sci., 2019, 108(11), 3521–3523, DOI:10.1016/j.xphs.2019.07.016.
- R. L. Hartman, Flow Chemistry Remains an Opportunity for Chemists and Chemical Engineers, Curr. Opin. Chem. Eng., 2020, 42–50, DOI:10.1016/j.coche.2020.05.002.
- J. Britton and C. L. Raston, Multi-Step Continuous-Flow Synthesis, Chem. Soc. Rev., 2017, 46(5), 1250–1271, 10.1039/c6cs00830e.
- J. M. Neumaier, A. Madani, T. Klein and T. Ziegler, Low-Budget 3D-Printed Equipment for Continuous Flow Reactions, Beilstein J. Org. Chem., 2019, 15(1), 558–566, DOI:10.3762/bjoc.15.50.
- C. M. Heckmann and F. Paradisi, Looking Back: A Short History of the Discovery of Enzymes and How They Became Powerful Chemical Tools, ChemCatChem, 2020, 12(24), 6082–6102, DOI:10.1002/cctc.202001107.
- T. Ishige, K. Honda and S. Shimizu, Whole Organism Biocatalysis, Curr. Opin. Chem. Biol., 2005, 174–180, DOI:10.1016/j.cbpa.2005.02.001.
- R. A. Sheldon and S. Van Pelt, Enzyme Immobilisation in Biocatalysis: Why, What and How, Chem. Soc. Rev., 2013, 42(15), 6223–6235, 10.1039/c3cs60075k.
- J. M. Guisan, F. López-Gallego, J. M. Bolivar, J. Rocha-Martín and G. Fernandez-Lorente, The Science of Enzyme Immobilization, in Methods in Molecular Biology, Humana Press Inc., 2020, vol. 2100, pp. 1–26, DOI:10.1007/978-1-0716-0215-7_1.
- M. Romero-Fernández and F. Paradisi, Protein Immobilization Technology for Flow Biocatalysis, Curr. Opin. Chem. Biol., 2020, 55, 1–8, DOI:10.1016/j.cbpa.2019.11.008.
- R. A. Sheldon, Metrics of Green Chemistry and Sustainability: Past, Present, and Future, ACS Sustainable Chem. Eng., 2018, 6(1), 32–48, DOI:10.1021/acssuschemeng.7b03505.
- J. Li, E. M. Simmons and M. D. Eastgate, A Data-Driven Strategy for Predicting Greenness Scores, Rationally Comparing Synthetic Routes and Benchmarking PMI Outcomes for the Synthesis of Molecules in the Pharmaceutical Industry, Green Chem., 2017, 19(1), 127–139, 10.1039/c6gc02359b.
- C. Jiménez-González, C. Ollech, W. Pyrz, D. Hughes, Q. B. Broxterman and N. Bhathela, Expanding the Boundaries: Developing a Streamlined Tool for Eco-Footprinting of Pharmaceuticals, Org. Process Res. Dev., 2013, 17(2), 239–246, DOI:10.1021/op3003079.
- A. S. Bommarius and M. F. Paye, Stabilizing Biocatalysts, Chem. Soc. Rev., 2013, 42(15), 6534, 10.1039/c3cs60137d.
- R. A. Sheldon, R. Schoevaart and L. M. Van Langen, Cross-Linked Enzyme Aggregates (CLEAs): A Novel and Versatile Method for Enzyme Immobilization (a Review), Biocatal. Biotransform., 2005, 23(3–4), 141–147, DOI:10.1080/10242420500183378.
- J. M. Bolivar and F. López-Gallego, Characterization and Evaluation of Immobilized Enzymes for Applications in Flow Reactors, Curr. Opin. Green Sustain. Chem., 2020, 100349, DOI:10.1016/j.cogsc.2020.04.010.
- H. Wang and L. Shiyou, Immobilization of Enzymes and Cells, 1997, DOI:10.1385/0-89603-386-4:319.
- F. H. Arnold, Directed Evolution: Bringing New Chemistry to Life, Angew. Chem., Int. Ed., 2018, 57(16), 4143–4148, DOI:10.1002/anie.201708408.
- M. L. Contente, D. Roura Padrosa, F. Molinari and F. Paradisi, A Strategic Ser/Cys Exchange in the Catalytic Triad Unlocks an Acyltransferase-Mediated Synthesis of Thioesters and Tertiary Amides, Nat. Catal., 2020, 3(12), 1020–1026, DOI:10.1038/s41929-020-00539-0.
- M. Ali, H. M. Ishqi and Q. Husain, Enzyme Engineering: Reshaping the Biocatalytic Functions, Biotechnol. Bioeng., 2020, 117(6), 1877–1894, DOI:10.1002/bit.27329.
- D. Roura Padrosa, V. Marchini and F. Paradisi, CapiPy: Python Based GUI-Application to Assist in Protein Immobilization, Bioinformatics, 2021 DOI:10.1093/bioinformatics/btab030.
- J. M. Woodley, Accelerating the Implementation of Biocatalysis in Industry, Appl. Microbiol. Biotechnol., 2019, 103(12), 4733–4739, DOI:10.1007/s00253-019-09796-x.
- M. L. Contente and F. Paradisi, Self-Sustaining Closed-Loop Multienzyme-Mediated Conversion of Amines into Alcohols in Continuous Reactions, Nat. Catal., 2018, 1(6), 452–459, DOI:10.1038/s41929-018-0082-9.
- F. Dall'Oglio, M. L. Contente, P. Conti, F. Molinari, D. Monfredi, A. Pinto, D. Romano, D. Ubiali, L. Tamborini and I. Serra, Flow-Based Stereoselective Reduction of Ketones Using an Immobilized Ketoreductase/Glucose Dehydrogenase Mixed Bed System, Catal. Commun., 2017, 93, 29–32, DOI:10.1016/j.catcom.2017.01.025.
- S. Velasco-Lozano, A. I. Benítez-Mateos and F. López-Gallego, Co-Immobilized Phosphorylated Cofactors and Enzymes as Self-Sufficient Heterogeneous Biocatalysts for Chemical Processes, Angew. Chem., Int. Ed., 2017, 56(3), 771–775, DOI:10.1002/anie.201609758.
- S. V. Ley, I. R. Baxendale, R. N. Bream, P. S. Jackson, A. G. Leach, D. A. Longbottom, M. Nesi, J. S. Scott, R. I. Storer and S. J. Taylor, Multi-Step Organic Synthesis Using Solid-Supported Reagents and Scavengers: A New Paradigm in Chemical Library Generation, J. Chem. Soc., Perkin Trans. 1, 2000, 23, 3815–4195, 10.1039/b006588i.
- V. De Vitis, F. Dall’oglio, F. Tentori, M. L. Contente, D. Romano, E. Brenna, L. Tamborini and F. Molinari, Bioprocess Intensification Using Flow Reactors: Stereoselective Oxidation of Achiral 1,3-Diols with Immobilized Acetobacter Aceti, Catalysts, 2019, 9(3), 208, DOI:10.3390/catal9030208.
- M. Planchestainer, M. L. Contente, J. Cassidy, F. Molinari, L. Tamborini and F. Paradisi, Continuous Flow Biocatalysis: Production and in-Line Purification of Amines by Immobilised Transaminase from Halomonas Elongata, Green Chem., 2017, 19(2), 372–375, 10.1039/C6GC01780K.
- F. J. Agostino and S. N. Krylov, Advances in Steady-State Continuous-Flow Purification by Small-Scale Free-Flow Electrophoresis, TrAC, Trends Anal. Chem., 2015, 72, 68–79, DOI:10.1016/j.trac.2015.03.023.
- P. Bana, R. Örkényi, K. Lövei, Á. Lakó, G. I. Túrós, J. Éles, F. Faigl and I. Greiner, The Route from Problem to Solution in Multistep Continuous Flow Synthesis of Pharmaceutical Compounds, Bioorg. Med. Chem., 2017, 25(23), 6180–6189, DOI:10.1016/j.bmc.2016.12.046.
- S. Newton, C. F. Carter, C. M. Pearson, C. De, L. Alves, H. Lange, P. Thansandote and S. V. Ley, Accelerating Spirocyclic Polyketide Synthesis Using Flow Chemistry, Angew. Chem., Int. Ed., 2014, 53(19), 4915–4920, DOI:10.1002/anie.201402056.
- K. Gilmore, D. Kopetzki, J. W. Lee, Z. Horváth, D. T. McQuade, A. Seidel-Morgenstern and P. H. Seeberger, Continuous Synthesis of Artemisinin-Derived Medicines, Chem. Commun., 2014, 50(84), 12652–12655, 10.1039/c4cc05098c.
- E. Horosanskaia, S. Triemer, A. Seidel-Morgenstern and H. Lorenz, Purification of Artemisinin from the Product Solution of a Semisynthetic Reaction within a Single Crystallization Step, Org. Process Res. Dev., 2019, 23(9), 2074–2079, DOI:10.1021/acs.oprd.9b00175.
- B. Sreedhar, B. Shen, H. Li, R. Rousseau and Y. Kawajiri, Optimal Design of Integrated SMB-Crystallization Hybrid Separation Process Using a Binary Solvent, Org. Process Res. Dev., 2017, 21(1), 31–43, DOI:10.1021/acs.oprd.6b00294.
- A. G. O'Brien, Z. Horváth, F. Lévesque, J. W. Lee, A. Seidel-Morgenstern and P. H. Seeberger, Continuous Synthesis and Purification by Direct Coupling of a Flow Reactor with Simulated Moving-Bed Chromatography, Angew. Chem., Int. Ed., 2012, 51(28), 7028–7030, DOI:10.1002/anie.201202795.
- G. Towler and R. Sinnott, Chemical Engineering Design, Principles, Practice and Economics of Plant and Process Design, 2008 Search PubMed.
- B. J. Deadman, C. Battilocchio, E. Sliwinski and S. V. Ley, A Prototype Device for Evaporation in Batch and Flow Chemical Processes, Green Chem., 2013, 15(8), 2050–2055, 10.1039/c3gc40967h.
- C. B. Kelly, C. Lee and N. E. Leadbeater, An Approach for Continuous-Flow Processing of Reactions That Involve the in Situ Formation of Organic Products, Tetrahedron Lett., 2011, 52(2), 263–265, DOI:10.1016/j.tetlet.2010.11.027.
- J. Sedelmeier, S. V. Ley, I. R. Baxendale and M. Baumann, KMnO4-Mediated Oxidation as a Continuous Flow Process, Org. Lett., 2010, 12(16), 3618–3621, DOI:10.1021/ol101345z.
- C. Amador, A. Gavriilidis and P. Angeli, Flow Distribution in Different Microreactor Scale-out Geometries and the Effect of Manufacturing Tolerances and Channel Blockage, Chem. Eng. J., 2004, 101(1–3), 379–390, DOI:10.1016/j.cej.2003.11.031.
- S. Mascia, P. L. Heider, H. Zhang, R. Lakerveld, B. Benyahia, P. I. Barton, R. D. Braatz, C. L. Cooney, J. M. B. Evans, T. F. Jamison, K. F. Jensen, A. S. Myerson and B. L. Trout, End-to-End Continuous Manufacturing of Pharmaceuticals: Integrated Synthesis, Purification, and Final Dosage Formation, Angew. Chem., Int. Ed., 2013, 52(47), 12359–12363, DOI:10.1002/anie.201305429.
- M. D. Truppo, H. Strotman and G. Hughes, Development of an Immobilized Transaminase Capable of Operating in Organic Solvent, ChemCatChem, 2012, 4(8), 1071–1074, DOI:10.1002/cctc.201200228.
- P. Berglund, Hydrolases in Organic Synthesis: Regio- and Stereoselective Biotransformations. By Uwe T. Bornscheuer and Romas J. Kazlauskas, ChemBioChem, 2006, 7(8), 1280–1280, DOI:10.1002/cbic.200600269.
- E. Farkas, M. Oláh, A. Földi, J. Kóti, J. Éles, J. Nagy, C. A. Gal, C. Paizs, G. Hornyánszky and L. Poppe, Chemoenzymatic Dynamic Kinetic Resolution of Amines in Fully Continuous-Flow Mode, Org. Lett., 2018, 20(24), 8052–8056, DOI:10.1021/acs.orglett.8b03676.
- J. M. Sperl, J. M. Carsten, J. K. Guterl, P. Lommes and V. Sieber, Reaction Design for the Compartmented Combination of Heterogeneous and Enzyme Catalysis, ACS Catal., 2016, 6(10), 6329–6334, DOI:10.1021/acscatal.6b01276.
- D. Roura Padrosa, V. De Vitis, M. Contente, F. Molinari and F. Paradisi, Overcoming Water Insolubility in Flow: Enantioselective Hydrolysis of Naproxen Ester, Catalysts, 2019, 9(3), 232, DOI:10.3390/catal9030232.
- D. Romano, F. Bonomi, M. C. de Mattos, T. de Sousa Fonseca, M. da C. F. de Oliveira and F. Molinari, Esterases as Stereoselective Biocatalysts, Biotechnol. Adv., 2015, 33(5), 547–565, DOI:10.1016/j.biotechadv.2015.01.006.
- P. D. de María and F. Hollmann, On the (Un)Greenness of Biocatalysis: Some Challenging Figures and Some Promising Options, Front. Microbiol., 2015, 6, 6–10, DOI:10.3389/fmicb.2015.01257.
- P. De Santis, L.-E. Meyer and S. Kara, The Rise of Continuous Flow Biocatalysis – Fundamentals, Very Recent Developments and Future Perspectives, React. Chem. Eng., 2020, 5(12), 2155–2184, 10.1039/d0re00335b.
- J. M. Bolivar, A. Mannsberger, M. S. Thomsen, G. Tekautz and B. Nidetzky, Process Intensification for O 2 -dependent Enzymatic Transformations in Continuous Single-phase Pressurized Flow, Biotechnol. Bioeng., 2019, 116(3), 503–514, DOI:10.1002/bit.26886.
- Y. Zhu, Q. Chen, L. Shao, Y. Jia and X. Zhang, Microfluidic Immobilized Enzyme Reactors for Continuous Biocatalysis, React. Chem. Eng., 2020, 5(1), 9–32, 10.1039/c9re00217k.
- D. H. Ko, K. W. Gyak and D. P. Kim, Emerging Microreaction Systems Based on 3D Printing Techniques and Separation Technologies, J. Flow Chem., 2017, 7(3–4), 72–81, DOI:10.1556/1846.2017.00013.
- D. Vasić-Rački, Z. Findrik and A. Vrsalović Presečki, Modelling as a Tool of Enzyme Reaction Engineering for Enzyme Reactor Development, Appl. Microbiol. Biotechnol., 2011, 845–856, DOI:10.1007/s00253-011-3414-0.
- N. Miložič, M. Lubej, M. Lakner, P. Žnidaršič-Plazl and I. Plazl, Theoretical and Experimental Study of Enzyme Kinetics in a Microreactor System with Surface-Immobilized Biocatalyst, Chem. Eng. J., 2017, 313, 374–381, DOI:10.1016/j.cej.2016.12.030.
- D. E. Fitzpatrick, T. Maujean, A. C. Evans and S. V. Ley, Across-the-World Automated Optimization and Continuous-Flow Synthesis of Pharmaceutical Agents Operating Through a Cloud-Based Server, Angew. Chem., Int. Ed., 2018, 57(46), 15128–15132, DOI:10.1002/anie.201809080.
- S. Mazurenko, Z. Prokop and J. Damborsky, Machine Learning in Enzyme Engineering, ACS Catal., 2020, 10(2), 1210–1223, DOI:10.1021/acscatal.9b04321.
- M. L. Contente, S. Farris, L. Tamborini, F. Molinari and F. Paradisi, Flow-Based Enzymatic Synthesis of Melatonin and Other High Value Tryptamine Derivatives: A Five-Minute Intensified Process, Green Chem., 2019, 21(12), 3263–3266, 10.1039/c9gc01374a.
- L. Tamborini, P. Fernandes, F. Paradisi and F. Molinari, Flow Bioreactors as Complementary Tools for Biocatalytic Process Intensification, Trends Biotechnol., 2018, 36(1), 73–88, DOI:10.1016/j.tibtech.2017.09.005.
Footnote |
| † These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2021 |