
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and self-assembly of amphiphilic precision glycomacromolecules†
Alexander
Banger
a,
Julian
Sindram
b,
Marius
Otten
b,
Jessica
Kania
a,
Dimitri
Wilms
 a,
Alexander
Strzelczyk
a,
Sean
Miletic
cde,
Thomas C.
Marlovits
cde,
Matthias
Karg
b and
Laura
Hartmann
*a
a,
Alexander
Strzelczyk
a,
Sean
Miletic
cde,
Thomas C.
Marlovits
cde,
Matthias
Karg
b and
Laura
Hartmann
*a
aInstitute of Organic and Macromolecular Chemistry, Heinrich-Heine-University, Universitätsstraße 1, 40225 Düsseldorf, Germany. E-mail: laura.hartmann@hhu.de
bInsitute of Physical Chemistry I: Colloids and Nanooptics, Heinrich-Heine-University, Universitätsstraße 1, 40225 Düsseldorf, Germany
cUniversity Medical Center Hamburg-Eppendorf (UKE), Institute of Structural and Systems Biology, Hamburg, Germany
dCentre for Structural Systems Biology (CSSB), Hamburg, Germany
eDeutsches Elektronen-Synchrotron Zentrum (DESY), Hamburg, Germany
First published on 17th May 2021
Abstract
We present the synthesis of so called amphiphilic glycomacromolecules (APGs) by using solid-phase polymer synthesis. Based on tailor made building blocks, monodisperse APGs with varying compositions are synthesized, introducing carbohydrate side chains and crosslinkable units at defined positions within the macromolecule. Self-assembly of the APGs is characterized via spectroscopic as well as scattering techniques and reveals structural dependencies between APG composition and self-assembly behavior such as the formation of spherical vs. worm-like micelles. Furthermore, polymerizable units within the APGs allow for fixation of the self-assembled structures. Carbohydrate presenting micelles are then evaluated as inhibitors of bacterial adhesion demonstrating their potential for biomedical applications.
Introduction
The self-assembly of amphiphilic structures is one of the key principles in the organization of living matter e.g. the formation of cellular membranes and organelles.1–4 Also in the synthetic world, amphiphiles are ubiquitous and play key roles in our everyday life e.g. in cleaning and emulsification. In general, amphiphiles can be classified by different parameters: their origin – biological or synthetic, their headgroups – cationic, anionic, amphoteric or non-ionic, as well as their size – low or high molecular weight.5 An important class of high molecular weight amphiphiles are blockcopolymers with covalently linked hydrophilic and hydrophobic blocks leading to the formation of polymeric aggregates such as micelles.6–10 Typical low molecular weight amphiphiles are surfactants such as sodium dodecyl sulfate or 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine. In between these two regimes exists the class of peptide amphiphiles as introduced by Stupp and coworkers, consisting of a peptide fragment forming the hydrophilic head group attached to a fatty acid as hydrophobic tail component.11–13 Special focus has been devoted to the development of peptide amphiphiles forming nanofibers and their application in different areas of regenerative medicine. A key feature here is the display of bioactive motifs within the peptide head group that allows for specific interactions e.g. with proteins and cells. Recently a trisulfated monosaccharide has been introduced into a peptide amphiphile with the resulting nanofibers mimicking the natural polysaccharide heparin sulfate and showing advanced biological function e.g. in bone regeneration.14 Previously we have introduced precision glycomacromolecules, a class of biomimetics that allows for the sequence-defined presentation of carbohydrate ligands on a monodisperse oligoamide scaffold.15,16 These molecules have been shown to mimic the multivalent binding of natural oligosaccharides or glycoconjugates such as the glycopeptides but offer the potential to introduce additional functionality such as light switchability or secondary binding motifs.17–19 Furthermore, we could show that precision glycomacromolecules can be used in a bottom-up approach giving access to high molecular weight materials e.g. through polyaddition or grafting-to strategies.20,21 Inspired by the work on peptide amphiphiles, here we explore the possibility to use precision glycomacromolecules as head groups in tailor-made amphiphiles and generate self-assembly structures with potential bioactivity.Results and discussion
Synthesis of amphiphilic precision glycomacromolecules
Our synthetic strategy relies on a library of dimer building blocks comprising a free carboxy group and a Fmoc-protected amine group together with functionalized monosaccharide ligands that we established for their stepwise assembly on solid support. Starting from a commercially available amine-functionalized resin and applying standard Fmoc–peptide coupling protocols, dimer building blocks are assembled in a stepwise fashion giving the scaffold. Using for example building blocks with alkyne side chains and different main chain motifs, alkyne groups are introduced at different positions along the scaffold. These then allow for site selective introduction of carbohydrate motifs via Cu-mediated azide–alkyne conjugation of azido-functionalized carbohydrate derivatives.16 Based on this concept, the head group of the targeted amphiphilic precision glycomacromolecules (APGs) is composed of the carbohydrate units, the building blocks presenting the carbohydrate and the C-terminus of the molecule. The hydrophobic tail can include additional building blocks elongating the macromolecular scaffold and an N-terminal fatty acid (Fig. 1). The straightforward variation of the combination of different building blocks gives access to a library of APGs. In this first study, we focus on APGs presenting only one carbohydrate ligand per molecule but selectively exchanging single building blocks, vary the type of carbohydrate, the linker and the alkyl chain of the tail group (Fig. 2). Making use of our previously established library of building blocks suitable for solid phase polymer synthesis, the following building blocks were chosen for this study: as functional building blocks TDS (triple bond functionalized diethylenetriamine coupled with succinic anhydride) and BADS (benzyl azide functionalized diethylenetriamine coupled with succinic anhydride) were used to introduce carbohydrate ligands via CuAAC reaction.22 Both building blocks will result in a triazol motif within the linker unit, however, BADS will introduce an additional benzyl moiety whereas TDS carries an alkyl spacer. For the hydrophobic tail, two new building blocks, HDM (hexamethylenediamine coupled with maleic anhydride) and HDS (hexamethylenediamine coupled with succinic anhydride), were developed. HDM introduces an alkene functionality into the main chain of the APG, which will be used for post micellization crosslinking while HDS serves as all alkyl derivative for comparison. Additionally, three fatty acids with respective chain lengths of C10, C12 and C15 were introduced as terminal hydrophobic components.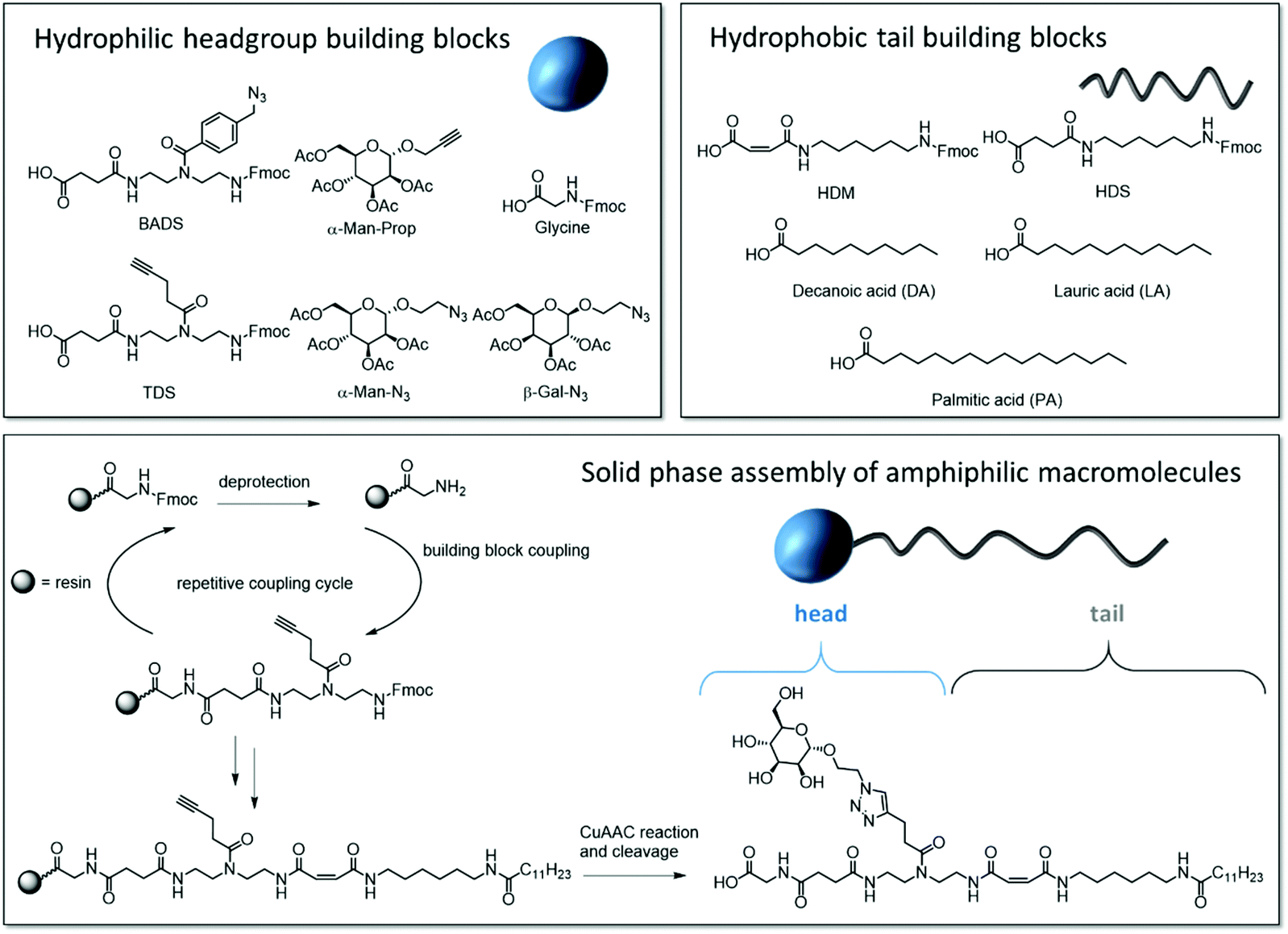 | ||
| Fig. 1 Overview of the utilized building blocks and their position in the APG, general solid phase synthesis of APGs exemplary shown for structure 1 (Gly-TDS(Man)-HDM-C12). | ||
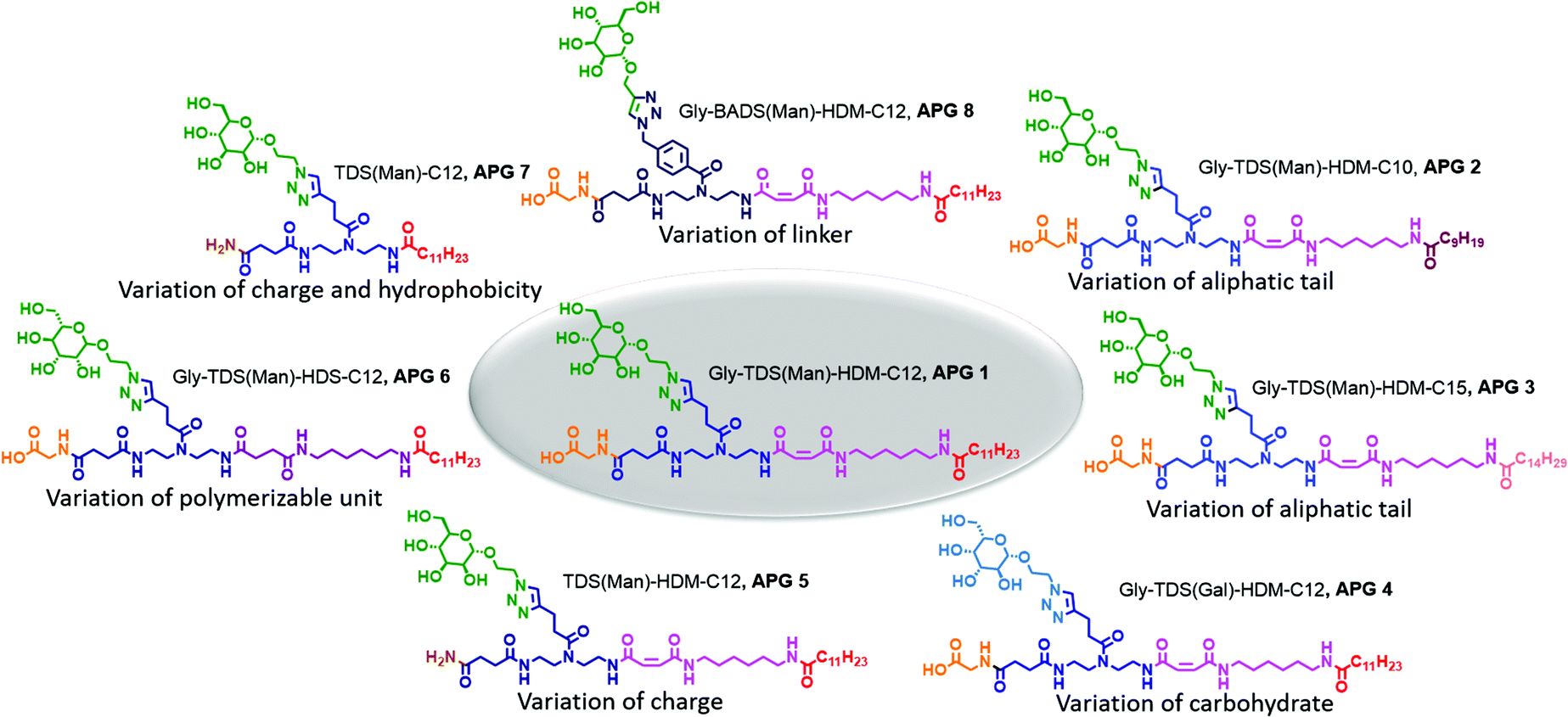 | ||
| Fig. 2 Overview of the synthesized APGs 1–8. | ||
We first synthesized a structure composed of a glycine and a mannose coupled to the functional TDS building block as head group and HDM and lauric acid as tail group (APG 1, Fig. 2). Derived from APG 1, seven more APGs were synthesized by systematically adding, removing or exchanging building blocks in order to later determine the impact on APG self-assembly (Fig. 2). The first two variations, APG 2 and 3, were synthesized to examine the influence of different chain lengths of the terminal fatty acid. Therefore, only the N-terminal fatty acid was altered and all other parts of the molecule were kept the same. Following, for APG 4, only the carbohydrate moiety was exchanged from mannose to galactose. Although galactose and mannose are stereoisomers they exhibit different properties especially considering their interaction with carbohydrate recognizing proteins. Another head group alteration was realized for APG 5, where the C-terminal glycine was omitted. This results in a change of head group charge, where structures with a C-terminal glycine carry a free carboxy group, while removing the amino acid leads to a non-charged amido group. For APG 6, HDS was used instead of HDM, thus removing the alkene moiety within the tail group. For APG 7 both, glycine and HDM, were excluded. For APG 8, the mannose unit was attached to the scaffold via a BADS instead of the TDS building blocks thereby introducing an additional aromatic motif into the head group.
Synthesis of all APGs followed previously established protocols of solid phase polymer synthesis.18 In short, for APG 1–4, 6 and 8, coupling of building blocks on a glycine preloaded Tentagel® resin was performed using benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP) as activation reagent and five equivalents of building block. For APG 5 and 7, a Tentagel® S RAM resin was used. After full assembly of the scaffold including both head and tail group, the carbohydrate units were introduced using Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC). APGs were cleaved from the resin, isolated by precipitation in diethyl ether and further purified by dialysis or if needed by preparative HPLC resulting in APGs with purities >90% (see Table S2†). All structures were verified by 1H-NMR, HPLC-MS and HR-ESI (see Fig. S6–30† for analytical data of all compounds).
Determining the critical micelle concentration (CMC)
In this study we aim at systematically exploring the effect of exchanging single building blocks in the APG structure on the self-assembly properties. Therefore, in a first series of experiments, the CMC was measured. All AGPs showed spontaneous formation of micelles above the CMC. Typically, pyrene is used as fluorescence probe to determine the CMC via the change of the ratio of two vibronic bands (vI at λ = 372 nm and vIII at λ = 383 nm).23–26 However, APGs of this study showed intrinsic fluorescence overlapping with the relevant wavelength range, requiring extensive background corrections and thus rendering this method less suitable for APGs (see Fig. S41–44†). Therefore, Nile red as a highly solvatochromatic dye providing fluorescence in the higher wavelength region of the visible light was used as alternative dye for CMC read-out.27,28 The quantum yield of Nile red in unpolar solvents like n-heptane is 80 times greater than in acetone, thus upon solubilization inside the micelles the fluorescence intensity should markedly increase (see Fig. S45 and 46†).29Table 1 summarizes the data for the Nile red as well as pyrene CMC measurements. Where both methods could be applied, resulting CMC values agree within an error margin of <20%. In a first subset (APG 1 – APG 3), the influence of the length of the terminal fatty acid on the CMC was investigated. As expected, the CMC decreases with increasing chain length of fatty acids (Table 1).30,31 The measured CMC values range between 3.41 mM for the decanoic, 0.64 mM for the dodecanoic and 0.15 mM for the pentadecanoic acid, respectively. Overall, the CMC values are higher compared to peptide amphiphiles of similar size.32–34 Comparing the CMC of APG 1 (0.64 mM) to well-known dodecanoic alkyl tail bearing anionic and non-ionic surfactants like SDS (8.2 mM), pentaethylene glycol monododecyl ether (0.07 mM) and carbohydrate based surfactants like n-dodecyl-β-D-glucopyranoside (0.19 mM), CMCs of the APGs lay in between ionic and non-ionic surfactants. To further investigate this, APG 5 and 7 with a C-terminal non-charged amide instead of the charge bearing glycine moiety were analyzed. APG 5 precipitated in solution over a period of one hour and did not form colloidally stable aggregates, thus no CMC could be determined. This finding highlights the importance of a charged head group to provide sufficient colloidal stability of the formed aggregates. APG 7 was stable and a CMC of 1.7 mM was determined, which is higher than comparable similar non-ionic surfactants such as n-dodecyl-β-D-glucopyranoside. Potentially, not only the carbohydrate moiety but also a part of the oligoamide backbone serves as headgroup, which could lead to this increased CMC. Additionally, the aliphatic carbohydrate linker was exchanged by an aromatic linker for APG 8 leading to a 2 times lower CMC than for APG 1 (see Fig. 3). Since we plan on using self-assembled APGs in biological assays, we also determined the CMC in PBS buffer, a typical buffer system e.g. in lectin binding or bacterial adhesion studies.18,35 It is well known that the salt or ion concentration can strongly influence the CMC, thus differences for APG CMCs in water vs. buffer could be expected.36 Overall, the CMCs of the charged APGs (1, 3, 4, 6, 8) decreased while for the only non-ionic APG 7 the CMC increased (see Table 1). A decrease for the charged APGs was expected as the ions in the PBS buffer reduce the static repulsion between the head groups, thus facilitating the micellization process.36 The CMC of non-ionic surfactants is generally not influenced by the ion concentration but is highly sensitive regarding temperature.37 Maybe here a change in the ambient temperature led to the increased CMC.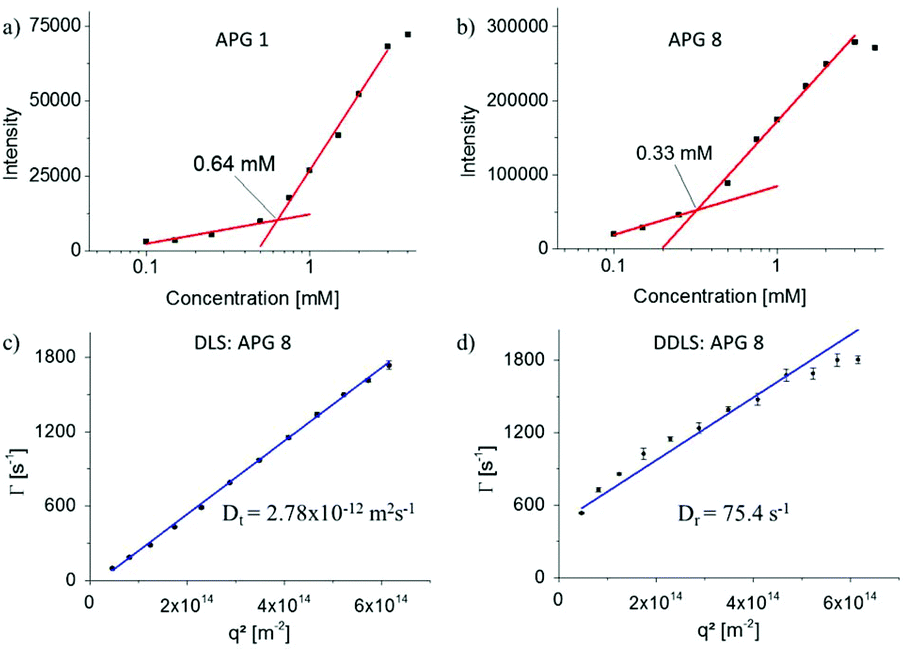 | ||
| Fig. 3 (a and b) Exemplary CMC curves of APG 1 and 8, (c and d) exemplary angle dependent data from cumulant fits of the DLS (c) and DDLS (d) measurements for APG 8. | ||
| Oligomer | CMC in MQ [mM] | CMC in PBS buffer [mM] |
|---|---|---|
| a CMC determined with fluorescence assay using nile red, error values represent standard deviations. b CMC determined with fluorescence assay using pyrene, error values represent standard deviations. c CMC determined using DLS measurements, error values represent standard deviations. n.m. = not measured. | ||
| Gly-TDS(Man)-HDM-C12 (APG 1) | 0.64a ± 0.07–0.95b | 0.59 ± 0.01 |
| Gly-TDS(Man)-HDM-C10 (APG 2) | 3.41a ± 0.08 | n.m. |
| Gly-TDS(Man)-HDM-C15 (APG 3) | 0.15a ± 0.01 | 0.06 ± 0.004 |
| Gly-TDS(Gal)-HDM-C12 (APG 4) | 0.68a ± 0.07–0.74b | 0.63 ± 0.07 |
| TDS(Man)-HDM-C12 (APG 5) | Precipitation | precipitation |
| Gly-TDS(Man)-HDS-C12 (APG 6) | 1.81a ± 0.02–2.36c | 0.49 ± 0.001 |
| TDS(Man)-C12 (APG 7) | 1.70a ± 0.19 | 1.98 ± 0.02 |
| Gly-BADS(Man)-HDM-C12 (APG 8) | 0.33a ± 0.01–0.34b | 0.30 ± 0.01 |
Studying micellar structures with DLS/DDLS
While the largely different CMC values are a first important indication for the change in self-assembly behavior observed for the different APGs, more detailed information is required especially on the size and shape of the assembled structures. Therefore, multiangle dynamic light scattering (DLS) and depolarized dynamic light scattering (DDLS) experiments were performed on selected APGs (Table 2). Exemplary data from DLS and DDLS measurements are shown in Fig. 3c and d. In ordinary, polarized DLS measurements, the mean relaxation rate,![[capital Gamma, Greek, macron]](https://www.rsc.org/images/entities/char_e0ba.gif) , scales linearly with the square of the scattering vector, q2, indicating that only translational motion is probed in the respective time- and q-range. The slope of the linear fit to the data provides the translational diffusion coefficient, Dt. In contrast, the DDLS data show a distinct positive offset of the relaxation rates, which is related to the rotational diffusion coefficient, Dr, while the slope of the linear fit again corresponds to Dt. Surprisingly, all APGs except for the non-charged APG 7 showed formation of cylindrical micelles as indicated by a strong signal in the DDLS data and further supported by atomic force microscopy (AFM) and transmission electron microscopy (TEM) images (see Fig. 5). For single tail surfactants found in literature, mostly spherical micelles are observed unless conditions are drastically altered e.g. to high ion concentrations.38 It has been shown for peptide amphiphiles that intermolecular structure formation such as β-sheet formation can lead to predominantly cylindrical assemblies.39 Hartgerink et al. provided evidence in a systematic study that for peptide amphiphiles intermolecular hydrogen bonding is the key driver in amphiphile packing.39 By systematically exchanging amide bonds by methylated amide bonds they demonstrated that below a certain threshold of amide bonds the micellar shape changes from a rod-like to a spherical shape.
, scales linearly with the square of the scattering vector, q2, indicating that only translational motion is probed in the respective time- and q-range. The slope of the linear fit to the data provides the translational diffusion coefficient, Dt. In contrast, the DDLS data show a distinct positive offset of the relaxation rates, which is related to the rotational diffusion coefficient, Dr, while the slope of the linear fit again corresponds to Dt. Surprisingly, all APGs except for the non-charged APG 7 showed formation of cylindrical micelles as indicated by a strong signal in the DDLS data and further supported by atomic force microscopy (AFM) and transmission electron microscopy (TEM) images (see Fig. 5). For single tail surfactants found in literature, mostly spherical micelles are observed unless conditions are drastically altered e.g. to high ion concentrations.38 It has been shown for peptide amphiphiles that intermolecular structure formation such as β-sheet formation can lead to predominantly cylindrical assemblies.39 Hartgerink et al. provided evidence in a systematic study that for peptide amphiphiles intermolecular hydrogen bonding is the key driver in amphiphile packing.39 By systematically exchanging amide bonds by methylated amide bonds they demonstrated that below a certain threshold of amide bonds the micellar shape changes from a rod-like to a spherical shape.
| Oligomer | D t [m2s−1]a | R H [nm]a | D r [s−1]a | L [nm]a | r [nm]a |
|---|---|---|---|---|---|
| a Translational diffusion coefficient (Dt), hydrodynamic radius (RH), rotational diffusion coefficient (Dr), length of the cylindrical micelle (L), radius of the cylindrical micelle (r). b Not evaluated due to a high dispersity, values were not extracted. c Not measured because micelles were spherical. | |||||
| Gly-TDS(Man)-HDM-C12 (APG 1) | 3.99 × 10−12 | 61.6 | n.e.b | n.e.b | n.e.b |
| TDS(Man)-C12 (APG 7) | 3.97 × 10−12 | 61.9 | n.m.c | n.m.c | n.m.c |
| Gly-BADS(Man)-HDM-C12 (APG 8) | 2.78 × 10−12 | 88.3 | 75.4 | 460 | 24.4 |
Potentially, the oligoamide scaffold of APGs can undergo intermolecular interactions based on the amide groups within the backbone similar to those of peptide structures leading to the formation of cylindrical micelles. Interestingly, omitting the C-terminal charge within the head group and the hydrophobic building block HDM in the tail, results in a different structure of the assembly with APG 7 forming spherical micelles. Based on DLS and DDLS data, spherical micelles of APG 7 have a fairly large hydrodynamic radius, RH, of 61.9 nm (see Table 2). APG 7 has only three amide bonds compared to five amide bonds e.g. in APG 1, supporting the hypothesis that intermolecular interactions of the amide groups in the scaffold lead to cylindrical assemblies. While the non-charged APG 5, which also has five amide bonds, showed insufficient colloidal stability for DLS and DDLS experiments, AFM pictures derived from freshly prepared solutions shows cylindrical micelles (see Fig. S64†). Thus, comparing APG 5 and APG 7 also supports that – similar as was found for the peptide based amphiphiles – the number of amide bonds seem to critically affect the micellar shape.
To further access the cylindrical micelles of APG 1 and APG 8 the light scattering data were fitted using the approach introduced by Garcia de la Torre et al.40,41 For most APGs and also for APG 1 correlation functions indicated a high polydispersity or rather a mix of micelles with strongly different aspect ratios, thus the fit model could not be applied. Interestingly, when exchanging the linker within the head group introducing an additional aromatic motif for APG 8, dispersity of the micelles decreased while the shape remained cylindrical. Presumably, the benzene moiety increases interactions between the headgroups of the micelles via π–π interactions, thus forming more rigid and more uniform micelles as was previously observed for other self-assembly structures.42 The cylindrical micelles were in average round 460 nm long and 49 nm in diameter, which equals an aspect ratio of 9.4.
Covalent crosslinking of micelles
One of the main drawbacks of non-polymeric micelles is their instability e.g. against dilution or high ion concentration.43 We experienced such instability first hand preparing samples for AFM experiments using spin coating at 3000 rpm which effectively destroyed all micelles. Therefore, we additionally explored the possibility to chemically crosslink and thereby fix micellar assemblies of APGs. We performed core-crosslinking experiments for APG 8 containing an alkene motif in the tail group. The core-crosslinking was performed as a free radical polymerisation in water with divinylbenzene (DVB) as crosslinker and 2,2-dimethoxy-2-phenylacetophenone (DMPA) as initiator. Both components have a very low solubility in water and thus crosslinking should only occur upon solubilization inside the core of the APG micelle (see Fig. 4). After irradiation, cross-linked micelles were purified by dialysis in water. To demonstrate crosslinking, solution of APG 8 micelles, cross-linked and non-crosslinked, were dissolved in an ethanol/water mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and the intensities as well as the correlation curves of the DLS measurements were compared.
:1) and the intensities as well as the correlation curves of the DLS measurements were compared.
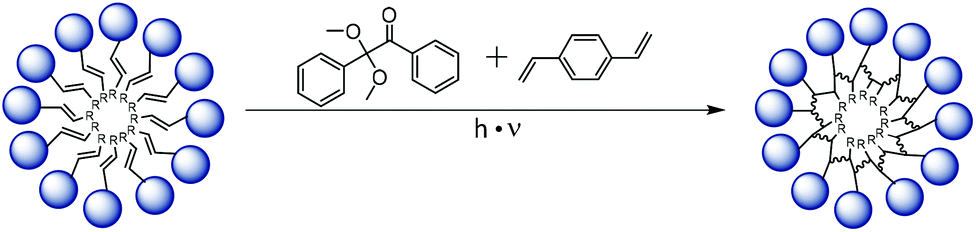 | ||
| Fig. 4 Synthetic scheme of the core-crosslinking procedure for APG 8. | ||
The use of high content of ethanol in this setting should lead to disassembly of micelles that are not covalently crosslinked. Indeed, DLS experiments showed higher intensity of the crosslinked sample of about 5 times in comparison to non-crosslinked sample. Additionally, the y-intercept of correlation curve for the non-crosslinked sample indicates poor data quality, which is expected for non-aggregated APGs (see Fig. S55†). To verify the results from the DLS experiments micellar solutions of APG 7, 8 and crosslinked APG 8 were studied by transmission electron microscopy (TEM) and atomic force microscopy (AFM) (see Fig. 5). For APG 7 spherical micelles with an average diameter of 92 nm and an average height of 12 nm were observed by AFM. Using TEM only very few micelles of APG 7 could be visualized with overall smaller diameters of around 51 nm and greater dispersion (additional TEM images of APG 7 see Fig. S54†). Differences between both methods could be explained due to possible compression of the micelles through the AFM tip while measuring, thus flatting the spherical micelles. Additional evidence gives the average height measured by AFM of only 12 nm, which indicates a flattening.
 | ||
| Fig. 5 (a and b) AFM and TEM images of APG 7, (c and d), AFM and TEM images of APG 8, (e and f) TEM images of crosslinked APG 8 from H2O (e) and EtOH (f). | ||
Furthermore, the high vacuum conditions in the TEM and the overall low contrast for electrons may influence the precise determination of the micelle dimensions. For APG 8, polydisperse, cylindrical micelles with lengths between 60 nm up to 1100 nm and an average core width of 17 nm were observed by TEM. AFM showed similar results but again with a lower dispersity compared to TEM. However, dried cylindrical micelles are expected to have a broad distribution with respect to their length. Since the length of the cylindrical micelles continuously changes upon drying, a broad size distribution can be expected. Interestingly, upon crosslinking of APG 8, the diameter of the micelles reduced to 14 nm, which could indicate a compactization through the crosslinking (see Fig. 5e). Crosslinked APG 8 micelles could also be dispersed in and blotted from ethanol, leading to dense packing of the rods, which resulted bundles of cylinder micelles packed in parallel to each other. Overall, comparing these results to the DLS data, the effective hydrodynamic radii observed for DLS tends to be greater than the radii observed by AFM or TEM. However, DLS is measured in solution and considers the hydration shell of the micelles, while for AFM and TEM the samples are dried and collapsed on a substrate.
Binding studies of APG
The carbohydrate motif in the head group also allows for first testing on the interaction of APG based micelles with carbohydrate recognizing proteins such as the model lectin Concanavalin A (ConA). ConA is a plant lectin derived from jack-bean, which specifically binds to α-D-mannose and α-D-glucose.44,45 For this, rhodamine labeled ConA and APG 8 solutions were mixed, incubated and subsequently investigated using fluorescence microscopy, qualitatively showing ConA binding of Man-functionalized micelles (see Fig. S56–58†).In principle, APGs also allow for the interaction with more complex biological entities such as E. coli bacteria. Many bacterial species including E. coli need to adhere to target cells, in order to cause an infection.46 Therefore bacterial pathogens have developed specialized adhesive organelles, which bind to the outer glycocalyx of the cells. E. coli have type 1 fimbriae presenting at the tip an α-D-mannopyranoside-specific lectin, FimH.47,48 In order to investigate binding efficacy of the carbohydrates presented by the APGs, we assayed binding of a GFP expressing E. coli strain to mannan coated surfaces as a model system.35 Inhibition of E. coli adhesion by APGs is quantified by their half-maximum inhibitory concentration (IC50).49 For all tested APGs 1, 3, 6, 7, 8 and crosslinked APG 8, sigmoidal inhibition curves were obtained and the according IC50 values were derived from this data (see Fig. S62†). Overall, the IC50 values of APGs are in the lower μM region. All APGs show IC50 values below their respective CMC, except for APG 3. Negative control APG 4 carrying a non-binding galactose instead of a mannose moiety showed no inhibition of bacterial adhesion (see Table 3). Therefore, we assume that APGs interact with the bacteria as single molecules rather than as micellar assemblies (see Fig. 6). Indeed, the higher IC50 value, and thus less efficient inhibition, for APG 3 could indicate that only in this case APGs assemble into micelles before interacting with the bacteria.
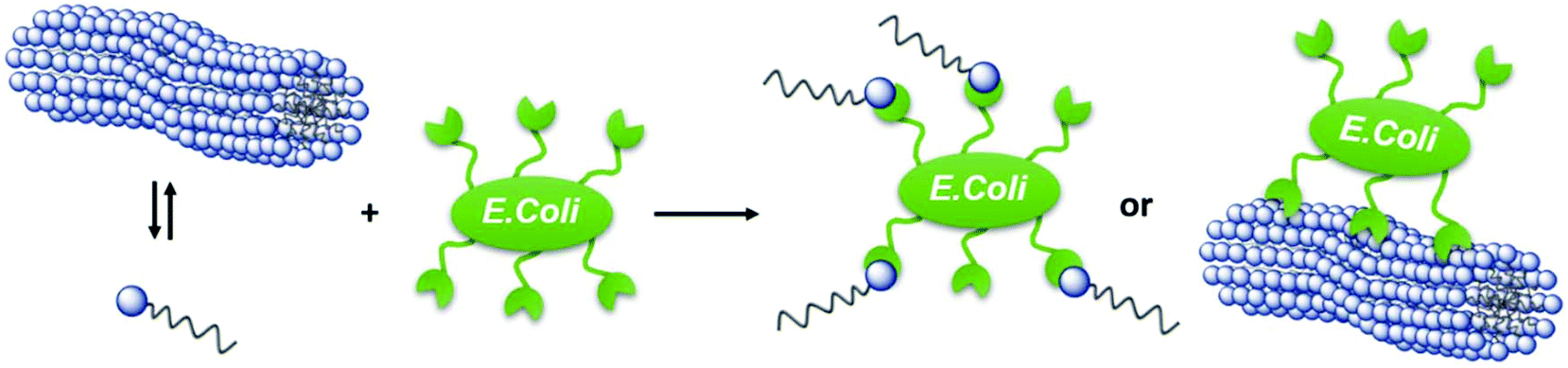 | ||
| Fig. 6 Schematic representation of a single APG binding versus an APG-micelle binding to E. coli bacterium. | ||
| Oligomer | IC50-Value [μM] |
|---|---|
| a For crosslinked APG 8 IC50 values can only be determined in [μg ml−1], for direct comparison, IC50 value of APG 8 is given in the same unit. | |
| APG 1 | 61 ± 20 |
| APG 3 | 503 ± 125 |
| APG 4 | No inhibition |
| APG 6 | 222 ± 21 |
| APG 7 | 135 ± 81 |
| APG 8 | 63 ± 11 |
| Oligomer | IC50-Valuea [μg ml−1] |
| APG 8 | 64 ± 11 |
| Crosslinked APG 8 | 253 ± 27 |
An increase in binding and thereby decrease in IC50 values for multivalent constructs such as the APG assemblies could be expected. However, it is important to keep in mind that at first a high number of molecules is required to form the assembly which can then interact with the bacteria according to our model. Within the assembly, only a fraction of carbohydrates will actually bind to the bacterial receptors, as observed for phase-separated glycopolymer aggregates.50 In the future, micellar assemblies from two types of APGs might be used, combining for example binding and non-binding carbohydrate units in a heteromultivalent fashion. It has been successfully demonstrated e.g. for glycopolymers that such heteromultivalent constructs can maximize the binding efficiency.51
From the Hill plots, we also derived the cooperativity factor n, which is given by the slope of the plot at 50% saturation.52 The cooperativity factor gives information whether the second binding event is potentially more (n > 1) or less favorable (n < 1) than the first initial binding.53–55 Interestingly, crosslinked APG 8 shows a cooperativity factor n = 2. All other APGs show cooperativity of around 1 or smaller, which is typically found for FimH binding also by its natural high mannose type ligands.54 As already discussed by Whitesides and coworkers, multivalent structures can lead to higher binding avidity even if they do not show positive cooperativity.56 However, the fact that we observe positive cooperativity for the crosslinked micelle indicates that binding of the bacteria benefits from this fixed, worm-like multivalent presentation.
Conclusion
In conclusion, we combined solid phase polymer synthesis with tailor made building blocks to synthesize amphiphilic glycomacromolecules, the so-called APGs. Eight different APGs were synthesized varying different structural parameters of the head and tail group. Investigating their structure–property correlations, we could show that both spherical and cylindrical micelles are accessible and can be tuned based on the choice of building blocks installed during APG synthesis. Additionally, the use of the novel building block HDM allowed for core crosslinking of the micelles, locking the structure even at dilutions below the CMC. Carbohydrate motifs in the head groups of the micellar assemblies remain accessible for protein binding as shown by incubation with fluorescently labelled ConA. Inhibition studies demonstrated the potential of APGs to block bacterial adhesion. Interestingly, crosslinked worm-like micelles showed positive cooperativity for the binding to bacteria, indicating a gain in multivalent binding from this type of carbohydrate presentation. Future studies will further explore potential applications of APGs e.g. solubilization of drugs and specific targeting via the carbohydrate ligands including heteromultivalent assemblies.Author contributions
Conceptualization, A.B. and L.H.; methodology, A.B. and M.O.; investigation, A.B., J.S., M.O., J.K., D.W., A.S. and S.M.; formal analysis, A.B. and J.S.; resources, L.H., M.K.; writing – original draft, A.B. and L.H.; writing – review & editing, A.B., L.H. and all coauthors; supervision, L.H., M.K. and T.M.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
LH thanks the DFG for support through the research group ‘Virocarb’ (FOR2327)(HA 5950/5-2). MK acknowledges financial support from the DFG through grant KA 3880/6-1. The authors acknowledge the DFG and the State of NRW for funding the cryo-TEM (INST 208/749-1 FUGG). The authors thank Vadim Kotov for initial TEM experiments and staining advice and Stephan Schmidt for support with the bacterial assays and fruitful discussions.References
- P. A. Monnard and D. W. Deamer, Anat. Rec., 2002, 268, 196–207 Search PubMed.
- D. Philp and J. F. Stoddart, Angew. Chem., Int. Ed. Engl., 1996, 35, 1154–1196 Search PubMed.
- C. G. Palivan, R. Goers, A. Najer, X. Zhang, A. Car and W. Meier, Chem. Soc. Rev., 2016, 45, 377–411 Search PubMed.
- Y. Tu, F. Peng, A. Adawy, Y. Men, L. K. Abdelmohsen and D. A. Wilson, Chem. Rev., 2016, 116, 2023–2078 Search PubMed.
- A. Sorrenti, O. Illa and R. Ortuno, Chem. Soc. Rev., 2013, 42, 8200–8219 Search PubMed.
- Y. Ishihara, H. S. Bazzi, V. Toader, F. Godin and H. F. Sleiman, Chem. – Eur. J., 2007, 13, 4560–4570 Search PubMed.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 Search PubMed.
- S. J. Holder and N. A. Sommerdijk, Polym. Chem., 2011, 2, 1018–1028 Search PubMed.
- A. H. Gröschel, F. H. Schacher, H. Schmalz, O. V. Borisov, E. B. Zhulina, A. Walther and A. H. Müller, Nat. Commun., 2012, 3, 1–10 Search PubMed.
- V. Ladmiral, M. Semsarilar, I. Canton and S. P. Armes, J. Am. Chem. Soc., 2013, 135, 13574–13581 Search PubMed.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 Search PubMed.
- S. Han, S. Cao, Y. Wang, J. Wang, D. Xia, H. Xu, X. Zhao and J. R. Lu, Chem. – Eur. J., 2011, 17, 13095–13102 Search PubMed.
- M. P. Hendricks, K. Sato, L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2017, 50, 2440–2448 Search PubMed.
- S. S. Lee, T. Fyrner, F. Chen, Z. Álvarez, E. Sleep, D. S. Chun, J. A. Weiner, R. W. Cook, R. D. Freshman, M. S. Schallmo, K. M. Katchko, A. D. Schneider, J. T. Smith, C. Yun, G. Singh, S. Z. Hashmi, M. T. McClendon, Z. Yu, S. R. Stock, W. K. Hsu, E. L. Hsu and S. I. Stupp, Nat. Nanotechnol., 2017, 12, 821–829 Search PubMed.
- D. Ponader, F. Wojcik, F. Beceren-Braun, J. Dernedde and L. Hartmann, Biomacromolecules, 2012, 13, 1845–1852 Search PubMed.
- D. Ponader, P. Maffre, J. Aretz, D. Pussak, N. M. Ninnemann, S. Schmidt, P. H. Seeberger, C. Rademacher, G. U. Nienhaus and L. Hartmann, J. Am. Chem. Soc., 2014, 136, 2008–2016 Search PubMed.
- D. Ponader, S. Igde, M. Wehle, K. Märker, M. Santer, D. Bléger and L. Hartmann, Beilstein J. Org. Chem., 2014, 10, 1603–1612 Search PubMed.
- S. Boden, F. Reise, J. Kania, T. K. Lindhorst and L. Hartmann, Macromol. Biosci., 2019, 19, 1800425 Search PubMed.
- T. Freichel, V. Heine, D. Laaf, E. E. Mackintosh, S. Sarafova, L. Elling, N. L. Snyder and L. Hartmann, Macromol. Biosci., 2020, 20, 2000163 Search PubMed.
- C. Gerke, M. F. Ebbesen, D. Jansen, S. Boden, T. Freichel and L. Hartmann, Biomacromolecules, 2017, 18, 787–796 Search PubMed.
- F. Shamout, A. Monaco, G. Yilmaz, C. R. Becer and L. Hartmann, Macromol. Rapid Commun., 2020, 41, 1900459 Search PubMed.
- M. Baier, M. Giesler and L. Hartmann, Chem. – Eur. J., 2018, 24, 1619–1630 Search PubMed.
- A. Nakajima, Bull. Chem. Soc. Jpn., 1971, 44, 3272–3277 Search PubMed.
- K. Kalyanasundaram and J. Thomas, J. Am. Chem. Soc., 1977, 99, 2039–2044 Search PubMed.
- G. B. Ray, I. Chakraborty and S. P. Moulik, J. Colloid Interface Sci., 2006, 294, 248–254 Search PubMed.
- H. Xu, Y. Wang, X. Ge, S. Han, S. Wang, P. Zhou, H. Shan, X. Zhao and J. R. Lu, Chem. Mater., 2010, 22, 5165–5173 Search PubMed.
- M. C. Stuart, J. C. van de Pas and J. B. Engberts, J. Phys. Org. Chem., 2005, 18, 929–934 Search PubMed.
- I. N. Kurniasih, H. Liang, P. C. Mohr, G. Khot, J. r. P. Rabe and A. Mohr, Langmuir, 2015, 31, 2639–2648 Search PubMed.
- P. Greenspan, E. P. Mayer and S. D. Fowler, J. Cell Biol., 1985, 100, 965–973 Search PubMed.
- R. Nagarajan, Langmuir, 2002, 18, 31–38 Search PubMed.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 Search PubMed.
- Q. Meng, Y. Kou, X. Ma, L. Guo and K. Liu, J. Pept. Sci., 2014, 20, 223–228 Search PubMed.
- N. M. Javali, A. Raj, P. Saraf, X. Li and B. Jasti, Pharm. Res., 2012, 29, 3347–3361 Search PubMed.
- D. J. Welsh, P. Posocco, S. Pricl and D. K. Smith, Org. Biomol. Chem., 2013, 11, 3177–3186 Search PubMed.
- M. Hartmann, A. K. Horst, P. Klemm and T. K. Lindhorst, Chem. Commun., 2010, 46, 330–332 Search PubMed.
- E. Fuguet, C. Ràfols, M. Rosés and E. Bosch, Anal. Chim. Acta, 2005, 548, 95–100 Search PubMed.
- M. J. Rosen and J. T. Kunjappu, Surfactants and interfacial phenomena, John Wiley & Sons, 2012 Search PubMed.
- S. Hayashi and S. Ikeda, J. Phys. Chem., 1980, 84, 744–751 Search PubMed.
- S. E. Paramonov, H.-W. Jun and J. D. Hartgerink, J. Am. Chem. Soc., 2006, 128, 7291–7298 Search PubMed.
- M. M. Tirado and J. G. de la Torre, J. Chem. Phys., 1979, 71, 2581–2587 Search PubMed.
- D. La Torre, J. García, M. C. L. Martinez and M. M. Tirado, Biopolymers, 1984, 23, 611–615 Search PubMed.
- B. Song, Z. Wang, S. Chen, X. Zhang, Y. Fu, M. Smet and W. Dehaen, Angew. Chem., Int. Ed., 2005, 44, 4731–4735 Search PubMed.
- Y. Lu, E. Zhang, J. Yang and Z. Cao, Nano Res., 2018, 11, 4985–4998 Search PubMed.
- J. B. Sumner and S. F. Howell, J. Bacteriol., 1936, 32, 227 Search PubMed.
- T. K. Dam, B. S. Cavada, T. B. Grangeiro, C. F. Santos, F. A. De Sousa, S. Oscarson and C. F. Brewer, J. Biol. Chem., 1998, 273, 12082–12088 Search PubMed.
- F. K. Bahrani-Mougeot, E. L. Buckles, C. Lockatell, J. Hebel, D. Johnson, C. Tang and M. Donnenberg, Mol. Microbiol., 2002, 45, 1079–1093 Search PubMed.
- F. G. Sauer, M. Barnhart, D. Choudhury, S. D. Knight, G. Waksman and S. J. Hultgren, Curr. Opin. Struct. Biol., 2000, 10, 548–556 Search PubMed.
- E. H. Beachey, J. Infect. Dis., 1981, 143, 325–345 Search PubMed.
- T. K. Lindhorst, S. Oscarson and J. Bouckaert, Glycoscience and microbial adhesion, Springer, 2009 Search PubMed.
- T. J. Paul, A. K. Strzelczyk, M. I. Feldhof and S. Schmidt, Biomacromolecules, 2020, 21, 2913–2921 Search PubMed.
- J. L. J. Blanco, C. O. Mellet and J. M. G. Fernández, Chem. Soc. Rev., 2013, 42, 4518–4531 Search PubMed.
- J. Wyman Jr., Adv. Protein Chem., 1964, 19, 223–286 Search PubMed.
- A. Whitty, Nat. Chem. Biol., 2008, 4, 435–439 Search PubMed.
- M. M. Sauer, R. P. Jakob, T. Luber, F. Canonica, G. Navarra, B. Ernst, C. Unverzagt, T. Maier and R. Glockshuber, J. Am. Chem. Soc., 2018, 141, 936–944 Search PubMed.
- S. Forsén and S. Linse, Trends Biochem. Sci., 1995, 20, 495–497 Search PubMed.
- M. Mammen, S. K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754–2794 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details including NMR spectra and RP-HPLC elugrams, CMC raw data, DLS and DDLS calculations and bacteria adhesion inhibition curves. See DOI: 10.1039/d1py00422k |
| This journal is © The Royal Society of Chemistry 2021 |